

Abstract
MLS1082 is a structurally novel pyrimidone-based D1-like dopamine receptor positive allosteric modulator. Potentiation of D1 dopamine receptor (D1R) signaling is a therapeutic strategy for treating neurocognitive disorders. Here, we investigate the relationship between D1R potentiation and two prominent structural features of MLS1082, namely the pendant N-aryl and C-alkyl groups on the pyrimidone ring. To this end, we synthesized 24 new analogues and characterized their ability to potentiate dopamine signaling at the D1R and the closely related D5R. We identified structure–activity relationship trends for both aryl and alkyl modifications and our efforts afforded several analogues with improvements in activity. The most effective analogues demonstrated an approximately 8-fold amplification of dopamine-mediated D1R signaling. These findings advance the understanding of structural moieties underlying the activity of pyrimidone-based D1R positive allosteric modulators.
Keywords: Cognitive decline therapeutics, Copper-mediated amidation, GPCR modulation
The D1 receptor (D1R) is a G protein-coupled receptor activated by the endogenous agonist dopamine (DA) and a key regulator of dopaminergic signaling, As such, it is associated with numerous neurological processes and is an attractive drug target for the treatment of a wide range of neuropsychiatric disorders, including cognitive decline associated with Parkinson’s disease, schizophrenia and Alzheimer’s disease. D1R activation has demonstrated promising results for improving cognition and working memory in mouse models.1 Work by Goldman-Rakic and colleagues has shown that an optimum level of D1R activity in the prefrontal cortex (PFC) is required for peak performance in learning and memory.1–4 This has led to the inverted U hypothesis for the relationship of D1R activity in the PFC and cognitive function.5 At low levels of D1R signaling, such as in diseased states, cognitive function is depressed. At extremely high levels of D1R activity, as observed during stress, cognitive function is also at suboptimal levels. Suboptimal levels of D1R stimulation have been suggested to underlie age-associated learning deficits and contribute to the decreased cognition observed in various pathophysiological states. Modulation of D1R signaling has therefore become an attractive strategy for developing therapeutics to treat cognitive decline.
The traditional approach to influencing D1R signaling is through development of D1R orthosteric compounds, which act at the DA binding site, however, this approach has proven therapeutically problematic. D1R orthosteric agonists suffer from numerous drawbacks, including: (1) a narrow therapeutic window for attaining optimal D1R activation, (2) rapid onset of tolerance and desensitization,6 (3) increased potential for seizures,7 and (4) therapeutically limiting hypotensive side effects.8 In contrast to orthosteric D1R agonists, D1R positive allosteric modulators (PAMs) could deliver the therapeutic benefits of increased D1R activation while avoiding many of the pitfalls that have hindered the therapeutic development of D1R agonists. PAMs have the potential to be a more useful approach since, rather than directly activating the D1R, a PAM will potentiate the action of endogenous DA. As mentioned, the inverted U-shaped curve describing the relationship between cortical D1R signaling and cognitive function may be rather narrow,1,7,8 resulting in a restricted therapeutic window for an agonist. Thus, a PAM can be advantageous as the available DA level provides a natural ceiling effect to PAM activity and the endogenous spatial and temporal regulation of DA-mediated stimulation is maintained.9 Thus, D1R PAMs have recently emerged as an alternative drug development strategy with the potential of enhanced clinical utility.10
To date, seven D1R PAM structural classes have been discovered (Fig. 1).11–15 This includes two (MLS1082 and MLS6585) recently reported and characterized by our group utilizing a high-throughput screening (HTS) discovery approach.16 MLS1082 and MLS6585 are structurally unique from each other16 and the D1R PAMs described in the research and patent literature11–15 (Fig. 1).
Fig. 1.

Structures of the D1 dopamine receptor positive allosteric modulators MLS1082, MLS6585, BMS Compound B, DETQ, BMS Compound A, UCB patent lead and Astellas patent lead.
MLS1082 and MLS6585 are effective D1R PAMs, potentiating both agonist-stimulated G protein- and β-arrestin-mediated signaling as well as increasing DA’s binding affinity for the D1R. Neither compound has any intrinsic agonist activity in the absence of a D1R agonist (e.g., DA). Importantly, we have demonstrated that MLS1082 and MLS6585 act at two distinct binding sites on the D1R.16 MLS1082 shares a similar binding site with at least two other D1R PAMs (Compound B and DETQ, Fig. 1), which engage a pocket in the second intracellular loop (IL2) of the D1R.17 As this IL2 interaction is shared by at least three of the known D1R PAM scaffolds, we sought to develop a structure–activity relationship (SAR) underlying D1R allosteric modulation around this PAM interaction site. To this end we investigated structural modifications of MLS1082 and their effects on D1-like receptor potentiation. We focused efforts on the two prominent side chains on the Eastern side of MLS1082 (C-2 and N-3, Fig. 1), with the aim of establishing the importance of these moieties to D1R PAM engagement as well as the potential to improve efficacy and affinity of the parent (MLS1082) compound. A late-stage diversification strategy enabled the independent replacement of pendant groups at both the N-3 and C-2 positions which were subsequently tested for activity.
To access new MLS1082 analogues, we had envisioned that acylation of an appropriate intermediate (e.g., quinolinone 4) could provide the penultimate precursor 2 that, following cyclization, would afford the target analogues (Fig. 2). However, all our initial efforts to access the target precursor 2 via intermediates 3, 4 or 5 were unsuccessful. We read with interest the reported copper-mediated nitrile to amide conversion developed by Zhou and coworkers18 and initially sought to determine if this method would be applicable to our scaffold. Though the application of the literature reaction conditions to nitrile 5 afforded only trace amounts of desired product, minor modifications led directly to pre-paratively useful yields of the final, cyclized pyrimidones 1, likely through the unisolated intermediate 2. Our modified conditions used increased equivalents of both nucleophile (aniline) and copper catalyst (Cu(OAc)2), while eliminating the need for any added ligands (Scheme 1). Though we screened numerous combinations of solvent (e.g., DMF, EtOH, DMSO, 1,2-dimethoxyethane, 1,4 dioxane, dichloromethane or HFIP), ligand (e.g., 2-piperidinecarboxylic acid, propylenediamine, ethylene glycol, proline, cis- or trans-1,2-cyclohexanediol, pyrido[3,2-g] quinoline and 2-indolecarboxylic acid) and copper catalyst (e.g., CuCl2, CuI, Cu(OTf)2, Cu(BF4)2 or Cu(PF6)(MeCN)4), the preferred conditions remained very similar to those initially reported. We used this modified route to resynthesize the HTS hit compound, MLS1082 (1a), and construct 24 diverse analogues. The requisite nitrile substrates 5a–5e for this reaction sequence was prepared by the addition of malonontrile to N-methylisatoic anhydride19,20 and subsequent acylation of the amine group with an appropriate acid chloride (Scheme 1). The analogues explored the effect of replacing both pendant groups on the pyrimidone ring of the scaffold (i.e., the C-2 cyclohexyl and N-3 phenyl in the HTS hit compound 1a (MLS1082, Table 1)).
Fig. 2.

Proposed retrosynthetic cyclization strategy to pyrimidone analogues and potential synthetic precursors to access key intermediate 2.
Scheme 1.

Reagents and conditions: (a) malononitrile, K2CO3, Et3N, DMF, 120 °C (MW), 45 min, 76% yield; (b) R1COCl (2.0 equiv), pyridine, rt, 22–81% yield; (c) H2NR2 (5.0 equiv), Cu(OAc)2 (0.5 equiv), water, 100 °C (MW), 2 h, 1–26% yield.
Table 1.
Structure–activity summary of pyrimidone analogues and D1R potentiation.
| Compound ID | structure | DA-stimulated G protein signalinga | DA-stimulated β-airestin recruitmentb | ||||
|---|---|---|---|---|---|---|---|
| D1 EC50 (nM) [95% CI] | D1 Emax (%) ± SEM | D1 EC50 (μM) [95% CI] | D1 Emax (%) ± SEM | D5 EC50 (nM) [95% CI] | D5 Emax (%), ± SEM | ||
| fold potentiation vs. DA alone | |||||||
| control | DA | 214 [160–286] | 100 ± 3.4 | 1.4 [0.9–2.1] | 99.4 ± 0.5 | 81.7 [45.6–146] | 102 ± 1.6 |
| — | — | — | — | — | — | ||
| MLS1082 1a | 123 [84.3–180]** | 104 ± 4.7 | 0.32 [0.18–0.55]* | 118 ± 5.7** | 28.8 [16.9–49.0]**** | 152 ± 9.1*** | |

|
1.74 | 1.04 | 4.34 | 1.19 | 2.84 | 1.50 | |
| 1b |

|
163 [90.0–298] | 104 ± 4.7 | 0.51 [0.29–0.90]* | 89.4 ± 3.1* | 27.4 [8.9–84.1]* | 92.4 ± 11.1 |
| 1.31 | 1.04 | 2.73 | 0.90 | 2.98 | 0.91 | ||
| 1c |

|
158 [92.7–270] | 97.4 ± 3.8 | 0.80 [0.63–1.01] | 110 ± 6.4 | 44.1 [5.8–336] | 93.9 ± 1.8* |
| 1.35 | 0.97 | 1.74 | 1.11 | 1.85 | 0.92 | ||
| 1d |

|
255 [181–361] | 104 ± 2.8 | 1.19 [0.58–2.42]* | 113 ± 8.4 | 80.2 [23.0–280] | 140 ± 12.9* |
| 0.84 | 1.04 | 1.17 | 1.13 | 1.02 | 1.38 | ||
| 1e |

|
185 [110–309] | 90.9 ± 3.8 | 0.41 [0.10–1.75] | 87.5 ± 3.3* | 62.1 [18.0–214] | 155 ± 3.0 |
| 1.16 | 0.91 | 3.39 | 0.88 | 1.32 | 1.53 | ||
| 1f |

|
199 [111–356] | 96.9 ± 4.3 | 0.83 [0.10–7.0] | 109 ± 9.9 | 58.5 [22.1–154] | 101 ± 6.7 |
| 1.08 | 0.97 | 1.67 | 1.10 | 1.40 | 0.99 | ||
| 1g |

|
55.9 [34.1–91.5]**** | 103 ± 3.3 | 0.18 [0.14–0.23]** | 92.1 ± 9.6 | 103. [36.6–290] | 119 ± 10.3 |
| 3.83 | 1.03 | 7.72 | 0.93 | 0.79 | 1.17 | ||
| 1h |

|
185 [96.1–356] | 95.6 ± 4.7 | 0.37 [0.17–0.82]* | 103 ± 5.9 | 43.9 [4.8–703] | 88.0 ± 1.3 |
| 1.16 | 0.96 | 3.76 | 1.03 | 1.86 | 0.87 | ||
| 1i |

|
150 [84.2–268] | 113.7 ± 4.7 | 0.38 [0.28–0.53]** | 86.2 ± 6.1 | 56.1 [16.0–197] | 63.4 ± 1.8* |
| 1.43 | 1.14 | 3.66 | 0.87 | 1.46 | 0.62 | ||
| 1j |

|
423 [241–745] | 101 ± 4.8 | 0.63 [0.32–1.26] | 106 ± 4.2 | 63.1 [30.0–133] | 77.1 ± 7.0 |
| 0.51 | 1.01 | 2.21 | 1.06 | 1.29 | 0.76 | ||
| 1k |

|
83.0 [31.7–218] | 122 ± 7.4 | 0.15 [0.05–0.47]* | 92.6 ± 5.0 | 33.7 [2.6–437] | 145 ± 37.8 |
| 2.58 | 1.22 | 9.27 | 0.93 | 2.42 | 1.42 | ||
| 1l |

|
233 [109–468] | 104 ± 6.0 | 0.51 [0.32–0.81]** | 95.7 ± 2.6 | 57.7 [6.0–552] | 131 ± 17.4 |
| 0.92 | 1.04 | 2.73 | 0.96 | 1.42 | 1.29 | ||
| 1m | 43.1 [29.0–64.0]** | 98.2 ± 2.7 | 0.32 [0.23–0.45]** | 109 ± 14.6 | 80.4 [17.5–370] | 157 ± 15.0 | |

|
4.97 | 0.98 | 4.34 | 1.09 | 1.02 | 1.54 | |
| 1n |

|
150 [68.7–324] | 102 ± 5.1 | 0.20 [0.07–0.47]* | 116 ± 10.1 | 26.5 [3.4–209] | 192 ± 20.0* |
| 1.43 | 1.02 | 6.95 | 1.16 | 3.08 | 1.89 | ||
| 1o |
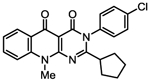
|
137 [71.1–263]* | 94.4 ± 4.8 | 0.32 [0.24–0.43]** | 116 ± 10.3 | 42.5 [17.1–105]* | 196 ± 23.9* |
| 1.57 | 0.94 | 4.34 | 1.17 | 1.92 | 1.93 | ||
| 1p |

|
167 [109–256] | 104 ± 3.5 | 0.16 [0.09–0.29]** | 93.5 ± 7.2 | 43.0 [18.1–102] | 130 ± 11.1* |
| 1.28 | 1.04 | 8.69 | 0.94 | 1.90 | 1.28 | ||
| 1q |

|
122 [51.4–291] | 107 ± 6.1 | 0.15 [0.09–0.25]*** | 116 ± 9.1 | 25.4 [7.5–86.1] | 192 ± 11.7** |
| 1.75 | 1.07 | 9.27 | 1.17 | 3.21 | 1.89 | ||
| 1r |

|
117 [61.0–225] | 106 ± 4.9 | 0.58 [0.49–0.70] | 92.5 ± 3.5 | 27.5 [5.7–133] | 124 ± 11.2* |
| 1.83 | 1.06 | 2.40 | 0.93 | 2.97 | 1.22 | ||
| 1s |

|
196 [123–313] | 100 ± 3.5 | 1.6 [0.37–6.7] | 120 ± 10.3 | 49.1 [22.8–106] | 134 ± 19.5 |
| 1.09 | 1.00 | 0.89 | 1.21 | 1.66 | 1.32 | ||
| 1t |

|
276 [180–422] | 102 ± 3.5 | 1.4 [0.6–2.3] | 95.3 ± 5.8 | 168 [57.0–496] | 120 ± 15.4 |
| 0.77 | 1.02 | 1.00 | 0.96 | 0.49 | 1.18 | ||
| 1u |

|
431 [334 −556]* | 111 ± 2.4 | 1.5 [1.2–1.8] | 96.9 ± 9.2 | 73.5 [36.8–147] | 98.3 ± 10.4 |
| 0.5 | 1.11 | 0.95 | 0.97 | 1.11 | 0.97 | ||
| 1v |
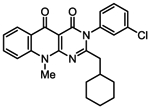
|
245 [178–337] | 103 ± 2.8 | 1.5 [0.53–4.12] | 115 ± 10.5 | 94.8 [42.5–212] | 110 ± 11.1 |
| 0.87 | 1.03 | 0.94 | 1.16 | 0.86 | 1.08 | ||
| 1w |
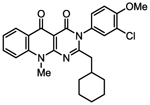
|
295 [202–430] | 105 ± 3.2 | 1.8 [0.94–3.4] | 111 ± 2.7* | 94.4 [44.9–199] | 108 ± 8.7 |
| 0.73 | 1.05 | 0.79 | 1.11 | 0.86 | 1.06 | ||
| 1x | 170 [104 −273] | 101 ± 4.1 | 0.65 [0.41–1.0] | 87.8 ± 5.7 | 81.9 [39.6–167] | 122 ± 15.2 | |

|
1.27 | 1.01 | 2.14 | 0.88 | 1.00 | 1.20 | |
| 1y |

|
163 [115–231] | 101 ± 2.8 | 0.84 [0.42–1.67]* | 105 ± 3.5 | 95.1 [23.8–383] | 126 ± 16.2 |
| 1.32 | 1.01 | 1.65 | 1.06 | 0.86 | 1.24 | ||
Concentration-response curves for DA-stimulated D1R signaling were generated in the absence (DA only) or presence of 50 μM test compound.
G protein signaling measured by cAMP accumulation using the DiscoverX HitHunter assay.16
β-arrestin recruitment measured using the DiscoverX Pathhunter assay.16
DA vs. DA + PAM via paired Student’s t-test,
p < 0.05,
p < 0.01,
p < 0.001,
p < 0.0001.
Importantly, many of the MLS1082 analogues retain measurable PAM activity, validating this overall scaffold as a D1-like receptor modulator. From this defined analogue set, we established several general SAR trends and discovered analogues that showed enhanced D1R potentiation. Potential PAM activities of the analogues were tested in two assays measuring D1R activation and signaling: cAMP accumulation (a measure of G protein activation), and β-arrestin recruitment to the receptor. For illustrative purposes the potentiation of DA-mediated recruitment of β-arrestin to the D1R by the parent compound, MLS1082, is depicted in Fig. 3. Cells expressing the D1R are incubated with increasing concentrations of the endogenous agonist DA (black circles). Co-incubation with a single, maximally effective concentration (50 μM) of MLS1082 potentiates the DA response, shifting the DA concentration–response curve to the left, demonstrating an increase in potency (black arrow), and increasing the maximum response (white arrow). These potentiation assays were performed for each of the novel analogues and their effects on DA signaling at D1-like receptors are summarized in Table 1. In addition to DA EC50 and Emax values in the presence of each analogue, we calculated the fold change in values (vs. DA alone, blue-shaded rows) for these parameters to provide a clearer comparison of analogue PAM activity. DA EC50 fold change was calculated by dividing the control DA EC50 by the DA EC50 observed in the presence of each given analog. Fold change for Emax was calculated similarly, with the Emax in the presence of PAM divided by Emax of the control dopamine curve. These values are reported in Table 1.
Fig. 3.

Depiction of the potentiation of D1R signaling by MLS1082. Increasing concentrations of dopamine (black circles) stimulate recruitment of β-arrestin to the D1R. Inclusion of a single, maximally effective (50 μM) concentration of MLS1082 potentiates this response by causing an increase in DA’s potency (EC50; black arrow) and efficacy (Emax; white arrow).
We prepared an array of analogues derived from the combination of four aliphatic sidechains (R1) of various steric bulk at the C-2 position and six substituted phenyl groups (R2) at the N-3 position as well as the two singleton analogues 1x and 1y. These efforts afforded 24 new analogues, which possessed a range of D1R-like potentiation activities and revealed notable SAR trends. The strongest correlation related to the steric bulk at the C-2 position, where replacing the cyclohexyl moiety found in the screening hit 1a with the slightly smaller cyclopentyl (1m to 1r) afforded many of the most active analogues in this study with these analogues either equally or more active than the corresponding cyclohexyl analogues. We used fold potentiation of DA-stimulated β-arrestin recruitment as the primary SAR metric (except where otherwise noted) as the DiscoverX Pathhunter assay provides a reliable read out for D1R activation over a wide activity range. The isobutyl analogues (1g to 1l) were also more active than the cyclohexyl analogues, to varying degrees. Comparison of the cyclopentyl and isobutyl analogues showed modestly better PAM activity for the former with the most significant potentiation improvement for the cyclopentyl analogues 1n and 1p over their isobutyl counterparts 1h and 1j. To survey the effect of increased steric bulk, the cyclohexyl in 1a was replaced with a methylene-linked cyclohexyl group (1s to 1w). This replacement was uniformly detrimental to the desired PAM activity with no analogues showing any appreciable D1R potentiation. We examined a single cyclopropyl analogue (1y) and found only marginal D1R potentiation, and thus this and other less bulky R1 groups were not explored further.
We surveyed six phenyl side chains at the R2 position, including unsubstituted phenyl, electron-donating phenyl (4-methyl and 4-methoxy), electron-withdrawing phenyl (4-chloro and 3-chloro) and an electronically mixed phenyl (3-chloro-4-methoxy) with the activity patterns for these R2 analogues being less straightforward to interpret than for the R1 analogues. While for the cyclohexyl R1 series (1a–1f), the unsubstituted phenyl (1a) showed the greatest D1R potentiation (4.3-fold for β-arrestin recruitment by dopamine), for both the cyclopentyl (1m–1r) and isobutyl (1g–1l) analogues, it was the 4-methoxyphenyl side chain at the R2 position that afforded the greatest potentiation of β-arrestin recruitment (analogues 1k and 1q, 9.3-fold for both). The analogue 1q displayed barely measureable D1R potentiation of G protein signaling (1.8-fold) though it was an active potentiator of D5R β-arrestin recruitment (3.2-fold). In the cyclopentyl series, the most active G protein signaling (cAMP production) potentiation remained the unsubstituted phenyl R2 group (4.9-fold, analogue 1m). Interestingly, in the cyclopentyl series, several R2 sidechains (4-methylphenyl, 4-chlorophenyl, 3-chlorophenyl and 4-methoxyphenyl) afforded D1R potentiators for β-arrestin recruitment (1n to 1q). The electronically mixed 3-chloro-4-methoxyphenyl analogues often demonstrated the lowest D1R potentiation across each R2 series, with the exception where R2 was cyclopentyl, which afforded an analogue (1r) of modest D1R potentiation for both β-arrestin recruitment and G protein signaling (2.4- and 1.8-fold, respectively). The requirement that R1 even be an aryl group at all is countered by the modest activity of the desphenyl analogue 1x. Comprehensive SAR exploration at this position may therefore be critical to afford a more efficacious and selective potentiator of the D1R.
Our lead analogues illustrate the overall SAR trends identified using this compound set. The potentiation of dopamine-stimulated β-arrestin recruitment to the D1R by a single, high concentration of analogues 1k and 1q is depicted in Fig. 4. Both analogues have a group with smaller steric bulk than the parent compound at the R1 position, either a cyclopentyl (1q) or an isobutyl group (1k) and the electron donating 4-methoxy group at the R2 position. Both 1k and 1q showed a more than two-fold increase in EC50 potentiation over the parent PAM 1a (9.3-fold for 1k and 1q vs. 4.3-fold for 1a). These data suggest that the smaller group at R1 and additional electron donation at R2 increase the potentiation of agonist potency. Interestingly, neither analogue increased the potentiation of dopamine’s Emax (1a = 1.2-fold increase, 1k = 0.9-fold and 1q = 1.2-fold). This may indicate that compound contributions to Emax potentiation occur through different moieties of the scaffold with separate structure–activity relationships, as allosteric modulators can affect agonist efficacy and potency through discrete mechanisms of action. Both analogues have a smaller effect on the potentiation of the G-protein-mediated signaling assay, cAMP accumulation. This type of assay can be confounded by signal amplification, due to both spare receptor phenomena and signal amplification of second messenger (cAMP) generation, as evident by the significantly higher potency observed with DA in the cAMP vs. β-arrestin recruitment assays. This tends to narrow the potentiation windows, making the observed fold shifts in the cAMP responses relatively smaller than those observed for β-arrestin recruitment.
Fig. 4.

Potentiation of DA-stimulated β-arrestin recruitment by analogues 1k (left) and 1q (right). Increasing concentrations of dopamine (black circles) stimulate recruitment of β-arrestin to the D1R.This effect is potentiated by including a single, high (50 μM) concentration of the analogues 1k (left, blue triangles) and 1q (right, blue triangles). Both analogues potentiated dopamine’s potency to a similar or greater extent than the parent compound MLS1082 (1a, red squares).
The largest improvement on the parent compounds was enhancement of the fold potentiation of potency (EC50) for both D1-like receptors (D1R and D5R) rather than an increase in subtype selectivity. The lack of subtype selectivity of the scaffold may not be surprising given that the interaction site for this scaffold lies at least partially within the IL2 region of the receptor.15,16 Comparing the D1R with the D5R sequence in IL2 and the portions of transmembrane regions 3 and 4 (TM3 or TM4, respectively) closest to IL2 shows that the two receptors are nearly identical with many of the sequence differences representing conservative changes (e.g., an isoleucine residue instead of a valine). Only three of the fifteen residues in TM4 closest to IL2 substantially differ between D1R and D5R, while sequence alignment for TM3 and ICL2 shows only one amino acid difference in each region. This sequence homology may underly the difficulty in improving D1R–D5R receptor selectivity on the MLS1082 PAM scaffold. One notable trend, however, was that the methylenecyclohexyl analogues 1s and 1u demonstrated enhanced D5R potentiation. It is also of note that a number of compounds lost PAM activity all together. These findings underlie important implications for the homology of allosteric sites between closely related receptors.
Overall, we have established a number of SAR trends for this series of PAMs, most notably concerning the steric bulk of the aliphatic group on the pyrimidone ring. During the course of the SAR investigation, we identified several analogues with modestly improved potentiation of dopamine-induced D1R activation and highlight the importance of groups on the pyrimidone ring in maintaining PAM activity. Together these structural criteria can be applied to the future refinement of this class of D1R PAMs and enhance our understanding of this allosteric site on the D1R.
Acknowledgements
We thank Benjamin Neuenswander at the University of Kansas for performing mass-directed HPLC purification, purity analysis and HRMS determination of final analogues. We acknowledge financial support from the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, the Molecular Libraries Initiative funding to the University of Kansas Specialized Chemistry Center (U54HG005031) and the generous support of the Eshelman Institute for Innovation at the UNC Eshelman School of Pharmacy.
Footnotes
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: The authors are inventors on U.S. and European patent applications covering the compounds in this report.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2020.127696. These data include MOL files and InChiKeys of the most important compounds described in this article, as well as synthesis protocols and characterization for all compounds.
References
- 1.Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology. 2004;174:3–16. [DOI] [PubMed] [Google Scholar]
- 2.Castner SA, Goldman-Rakic PS. Enhancement of working memory in aged monkeys by a sensitizing regimen of dopamine D1 receptor stimulation. J Neurosci. 2004;24: 1446–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins TW, Arnsten AFT. The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annu Rev Neurosci. 2009;32:267–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnsten AFT, Vijayragavan S, Wang M, Gamo NJ, Paspalas CD. Dopamine’s Influence on Prefrontal Cortical Cognition: Actions and Circuits in Behaving Primates, in Dopamine Handbook 2009, (Iversen LL ISD, Dunnett SB, Bjorklund A ed) pp 230–248, Oxford University Press, New York, NY. [Google Scholar]
- 5.Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AFT. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376–384. [DOI] [PubMed] [Google Scholar]
- 6.Lewis MM, Watts VJ, Lawler CP, Nichols DE, Mailman RB. Homologous desensitization of the D1A dopamine receptor: efficacy in causing desensitization dissociates from both receptor occupancy and functional potency. J Pharmacol Exper Therapeut. 1998;286:345–353. [PubMed] [Google Scholar]
- 7.O’Sullivan GJ, Dunleavy M, Hakansson K, et al. Dopamine D1 vs. D5 receptor-dependent induction of seizures in relation to DARPP-32, ERK1/2 and GluR1-AMPA signaling. Neuropharmacology. 2008;54:1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng C, Zhang M, Asico LD, Eisner GM, Jose PA. The dopaminergic system in hypertension. Clin Sci. 2007;112:583–597. [DOI] [PubMed] [Google Scholar]
- 9.Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov. 2013;12:630–644. [DOI] [PubMed] [Google Scholar]
- 10.Hall A, Provins L, Valade A. Novel strategies to activate the dopamine D1 receptor: recent advances in orthosteric agonism and positive allosteric modulation. J Med Chem. 2019;62:128–140. [DOI] [PubMed] [Google Scholar]
- 11.Beadle CD, Coates DA, Hao J et al. , 3,4-Dihydroisoquinolin-2(1H)-yl compounds.2014, WO 2014/193781A1, 1–90. [Google Scholar]
- 12.Lewis MA, Hunihan L, Watson J, et al. Discovery of D1 dopamine receptor positive allosteric modulators: characterization of pharmacology and identification of residues that regulate species selectivity. J Pharmacol Exp Ther. 2015;354:340–349. [DOI] [PubMed] [Google Scholar]
- 13.Ates A, Jnoff E, Provins L, Valade A, Hall A. Tetrahydroisoquinoline derivatives. PCT Int Appl. 2017. WO2017178377. [Google Scholar]
- 14.Kawahami S, Imaizumi T, Masuda N, Kunikawa S, Morita M, Yarimizu J. Imidazodiazepine compounds. U.S. Patent2018, 20180185383.
- 15.Hao J, Beck JP, Schaus JM, et al. Synthesis and pharmacological characterization of 2-(2,6-dichlorophenyl)-1-((1S,3R)-5-(3-hydroxy-3-methylbutyl)-3-(hydroxymethyl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (LY3154207), a potent subtype selective, and orally available positive allosteric modulator of the human dopamine D1 receptor. J Med Chem. 2019;62:8711–8732. [DOI] [PubMed] [Google Scholar]
- 16.Luderman KD, Conroy JL, Free RB, et al. Identification of positive allosteric modulators of the D1 dopamine receptor that act at diverse binding sites. Mol Pharmacol. 2018;94:1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Heinz BA, Qian Y-W, et al. Intracellular binding site for a positive allosteric modulator of the dopamine D1 receptor. Mol Pharmacol. 2018;94:1232–1245. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Li Z, Deng H, Zhou X. An efficient protocol for the preparation of amides by copper-catalyzed reactions between nitriles and amines in water. Tetrahedron Lett. 2013;54:2212–2216. [Google Scholar]
- 19.Coppola GM, Hardtmann GE, Pfister OR. Chemistry of 2H-3,1-benzoxazine-2,4-(1H)-dione (isatoic anhydride). 2. Reactions with thiopseudoureas and carbanions. J Org Chem. 1976;41:825–831. [Google Scholar]
- 20.Khoramdelan F, Davoodnia A, Bozorgmehr MR, Ebrahimi M. Comparative study on conventional heating, ultrasonication and microwave assisted synthesis of 2-amino-1-alkyl-4-oxo-1,4-dihydroquinoline-3-carbonitriles. Heterocyclic Lett. 2017;7: 947–952. [Google Scholar]