

Abstract
The Central Dogma of Biology does not allow for the study of glycans using DNA sequencing. We report a “Liquid Glycan Array” (LiGA) platform comprising a library of DNA ‘barcoded’ M13 virions that display 30–1500 copies of glycans per phage. A LiGA is synthesized by acylation of phage pVIII protein with a dibenzocyclooctyne, followed by ligation of azido-modified glycans. Pulldown of the LiGA with lectins followed by deep sequencing of the barcodes in the bound phage decodes the optimal structure and density of the recognized glycans. The LiGA is target agnostic and can measure the glycan-binding profile of lectins such as CD22 on cells in vitro and immune cells in a live mouse. From a mixture of multivalent glycan probes, LiGAs identifies the glycoconjugates with optimal avidity necessary for binding to lectins on living cells in vitro and in vivo; measurements that cannot be performed with canonical glass slide-based glycan arrays.
Introduction
The Central Dogma of Biology, DNA → RNA → Protein, facilitates the study of these biopolymers using a unified toolbox of next-generation DNA sequencing1. This ability has revolutionized and transformed all areas of biomedical and life science. In contrast, investigating the biological roles of carbohydrates cannot rely on DNA sequencing directly. Akin to the DNA microarrays used in the 1990s2, glycan arrays – made by printing carbohydrates on distinct locations on a glass surface3 – are a high throughput approach for identifying carbohydrates that bind to glycan binding proteins (GBPs)4–8. The information provided by glycan arrays, a glycan binding profile for a receptor, is a critical starting point for downstream fundamental applications such as improved design of inhibitors, vaccines and therapeutics9–16. DNA arrays have been largely replaced by de novo analysis of DNA by deep sequencing. A technology that allows the one-to-one correspondence between a DNA sequence and a carbohydrate structure would enable application of powerful deep sequencing approaches to identify the binding profiles of GBPs in cells in vitro and in vivo.
Valency and spatial presentation of glycans are critical variables in glycan–GBP interactions17–19. Traditional ‘solid’ glycan arrays mimic the multivalent nature of these interactions, and the valency of glycans on the surface can be adjusted20–24. Indeed, the density of the multivalent display influences both the affinity and specificity of binding for lectins, carbohydrate processing enzymes, and antibodies3, 24, 25. For example, systematic variation of glycan density on microarrays enables differentiating subpopulations of serum antibodies that are not detectable using a single glycan density24. The solid-phase nature of canonical glycans arrays, however, fundamentally limits the ability to study the cross talk and dynamic competition between multiple glycans with a GBP. The format has also demonstrated limited utility for investigating cell-surface GBPs and is incompatible with in vivo studies. Ligation of glycans to DNA26–32 or peptide-nucleic-acid (PNA)33 is possible, but the monovalent display on DNA cannot mimic the multivalent presentation of glycans, a critical requirement because the very large majority of GBPs recognize their biologically relevant ligands with weak affinity. Tri- and tetravalent displays of glycans on RNA34 and recently DNA35, have been reported. However, investigating protein-glycan interactions in a cellular milieu and in vivo would place such platforms at risk of degradation by nucleases, which are ubiquitous.
One of the most successful platforms for investigating interactions between glycans and cell surface GBPs ex vivo and in vivo are glycan-decorated liposomal nanoparticles36, 37 and viral capsids38. However, neither technology permits encoding or tracking of different glycan structures. The multivalent presentation of different glycans was reported on micron-size beads encoded via the Luminex® strategy39–41 or encoded chemical tags42, but cell-binding assays and in vivo applications, which would necessitate injection of microbeads, have not been reported. This summary of these state-of the art technologies leads to the ideal platform for glycan arrays: it should display glycans on a multivalent, monodisperse carrier of sub-micron size equipped with an internal DNA barcode. This carrier must be stable and non-interfering with binding assays in vitro, in cell culture or in vivo. The internal location of the barcode would protect the DNA from nucleases and would also prevent undesired interactions between the coding element and other biological species. To satisfy these design elements, we describe here a liquid glycan array (LiGA) format built on M13 phage particles with silent DNA barcodes43 inside the phage genome. The LiGA technology combines the multivalent presentation of traditional ‘solid’ or bead-based arrays and liposome constructs with a soluble format and DNA encoding of both the composition and density of glycans. The components of a LiGA—a multivalent display of chemically-defined synthetic glycans—are fundamentally different from another reported glycan-functionalized phage: ‘glycophage display’44–46, which was obtained via the heterologous expression of the Campylobacter jejuni glycosylation machinery in E. coli. LiGAs retain most of the benefits of other M13 phage-displayed libraries, importantly genetic-encoding and robust protection of the DNA message inside the virion, which has allowed successful profiling of ligand–receptor interactions on the surface of cells, organs in live animals47, 48, and in humans49, 50.
Results
Construction of the LiGA building blocks: glycan–phage conjugates
Although many types of phage and viruses have been used for genetically-encoded display, we selected the M13 platform51 as a scalable, stable platform, compatible with a wide range of cloning methods, as well as chemical and enzymatic conjugation strategies. As we show here, while the M13 virion is not naturally glycosylated, desired glycans can be chemically conjugated to a subset of the 2700 copies of the major coat protein pVIII to produce a multivalent display of ~200–1500 glycans on the monodisperse 700×5 nm virion, creating a phage population with a single glycan composition. For DNA-encoding, the previously reported silent barcode43 cloned near the pIII cloning site of vector M13KE can be used (Supplementary Fig. S1a,b). This strategy allows the assembly of a glycan library of phage with distinct DNA encoded barcodes, each with a different glycan. Cloning of degenerate nucleotide sequences with 1010 theoretically-possible combinations into the N-terminal region of protein pIII yielded a library of ‘silently’ barcoded vectors that encode chemically-identical phage particles. After serial propagation of the library to remove the unfit clones, we subcloned and amplified separate populations of phage particles that contained DNA barcode sequences separated by least three nucleotide substitutions (Supplementary Table S1). This design allowed us to correct any mistakes during DNA deep sequencing (Supplementary Fig. S1).43
As the glycan source, we used synthetic glycans with an alkyl-azido linker, which are common intermediates in oligosaccharide synthesis; 63 glycans functionalized in this way were obtained as part of the public Consortium for Functional Glycomics (CFG) collection52, and an additional 12 azido-glycans were synthesized in this report (Supplementary Schemes S1–S6) or previous reports (see Supplementary Table S2 for list of glycans). We first tested a reported38 ligation of azido glycans by Cu-Activated Azido Alkyne Cycloaddition (CuAAC) to virus particles acylated by N-succinimidyl 4-pentynoate; however, CuAAC reduced the number of infective M13 phage particles indicating damage to either DNA or the capsid of phage (Supplementary Fig. S2). We tested Strain Promoted Azido Alkyne Cycloaddition53 (SPAAC) as an alternative (Fig. 1a) and, importantly, SPAAC ligation did not decrease the number of infectious particles (Supplementary Fig. S2c,d). Acylation of the N-terminus of the pVIII with dibenzocyclooctyne-N-hydroxysuccinimidyl ester (DBCO-NHS) followed by SPAAC ligation could be monitored by MALDI-TOF mass spectrometry (Fig. 1b). As the spacing of pVIII on the surface of M13 phage is well defined, the degree of conjugation would be expected to influence binding to antibodies or glycan binding proteins (Fig. 1c). The peak intensities in MALDI yielded a semi-quantitative estimate of the densities of the glycans on phage; such analysis was reliable down to ~107 phage particles (Supplementary Fig. S3). Control of the concentration of DBCO–NHS (Fig. 1d) and reaction conditions (Supplementary Fig. S4) enabled modification from 1% to 50% of pVIII proteins, yielding phage particles with 30–1400 copies of glycan per virion. For example, 0.5–1.5 mM of DBCO led to acylation of 15–50% of the pVIII population (Fig. 1d). Subsequent addition of 2 mM azido-glycan quantitatively consumed the alkyne and ligated the glycan to pVIII as evidenced by the pVIII–DBCO–glycan signal and concomitant disappearance of pVIII–DBCO signal (Fig. 1d). Quality control by MALDI confirmed the conjugation of diverse glycan structures (Fig. 1e) and detected rare problems in reactivity: for example, the use of glycosyl azides in the SPAAC yielded suboptimal coupling leaving unreactive pVIII-DBCO even after 24 h reaction time (Fig. 1b). This problem was resolved by introducing a linker between the glycan and the azido group (Fig. 1d,e). When the density of modification exceeded 50%, we observed that ~5% of pVIII proteins contained two modifications per protein (Supplementary Fig. S5). To assess the regioselectivity of the acylation, we took advantage of the known spontaneous cleavage of the Asp7–Pro8 bond in pVIII by TFA in a sinapinic acid matrix54. We observed that the amide bond is formed predominantly at an N-terminal amine and not at the Lys10 of the pVIII sequence (Supplementary Fig. S6).
Fig. 1: Synthesis and characterization of LiGA components.

a, Representation of two-step chemical glycosylation of phage. b, MALDI mass spectrometry characterization of starting material (protein pVIII), alkyne-functionalized product (DBCO-pVIII, P1), and glycoconjugate product (P2). c, Spacing of glycans on M13 virion (PDB: 2MJZ and 2C0W) compared to spacing of binding sites of IgG antibody and lectin ConA. d, Control of the density of the modification by controlling the concentration of DBCO-NHS. Spectra in top panel represent before βMan addition and bottom panel shows after βMan addition. e, Representative spectra of chemical modification of coat protein pVIII with glycans of different structural complexity. In MALDI conditions, sialoglycans exhibited a loss of sialic acid during ionization in acidic matrix; therefore MALDI of phage particles decorated with glycans containing terminal Neu5Ac contained two peaks (intact glycoconjugate and that with cleaved Neu5Ac).
Functional validation of LiGAs
We produced ~140 glycan-modified phage conjugates and characterized the density of glycans on phage by MALDI (summarized in Data/Maldi.pdf in the Supplementary Information). Mixing a subset of these differentially barcoded glycan-labeled phages produces a LiGA library that contains any desired combinations of glycans and glycans at different densities. Here, we employed several related LiGA libraries by mixing of 50–80 conjugates that display different glycan types (Supplementary Table S3). An ELISA confirmed the functional integrity of glycans on individual glycosylated phage (Supplementary Fig. S7); however, analysis by ELISA was incompatible with optimization of assays that contained mixtures of phage clones. We thus employed a LiGA composed of phage that transduce galactosidase, mNeonGreen55, or mCherry56 reporters within host cells (Fig. 2a–b). Composition of the mixture of these phage clones can be measured as plaque forming units (PFU). Plating of the input and the output of the assay on a bacterial agar overlay enabled measurement of blue, white, green-fluorescent and red-fluorescent plaques (Fig. 2c) and define recovery of each subpopulation of phage as PFUoutput/PFUinput. This recovery tracked the binding of the associated glycans: such as binding the Manα-[green] to ConA but not Galβ1–4Glc1β-[red] and unmodified wild type [white] present in the same mixture (Fig. 2c). It further demonstrated that binding of α-Man-decorated clone to ConA either alone, or as part of the complex mixture LiGA YY was statistically indistinguishable (Fig. 2c); LiGA YY contained competing ligands β-Man, α-Man, and α-Man3 (Manα1–6(Manα1–3)Manα1) (see Table S3 for LiGA composition). Similarly, interaction of G3c with LNT-NAc-[green] (Galβ1–3GlcNAcβ1–3Galβ1–4GlcNAcβ1) was comparable in three distinct LiGA mixtures mixtures that contained different number of competing ligands (Supplementary Fig, S8). Both observations suggest that in an assay with super-stoichiometric GBP, the binding of the glycosylated particles is not influenced by the composition of the LiGA (for calculations of stoichiometry see Methods 1.11.3). The PFU assay validated that ConA or Galectin-3 (G3C) proteins immobilized on agarose beads can enrich clones decorated by Manα1–6(Manα1–3)Manα1-[red]-phage (Man3-[red]-phage) and Galβ1–3GlcNAcβ1–3Galβ1–4GlcNAcβ1-phage (LNT-NAc-[green]-phage) from a complex mixture of 75 glycosylated clones. Enrichment of LNT-NAc by G3C was specifically inhibited by soluble lactose (Fig. 2d, Supplementary Fig. S8f). The PFU-assay is target agnostic and allows optimization of both protein and cell-binding assays. For example, it confirmed that Chinese hamster ovary (CHO) cells engineered to express murine CD22 (mCD22) preferentially bound phage with Neu5Gcα2–6LacNAc over those with Neu5Acα2–6LacNAc, reflecting the known specificity of mCD22 for sialosides with Neu5Gc. In contrast, CHO cells that expressed human CD22 (hCD22) enriched both glycans, consistent with the previous observations that hCD22 has similar affinity for α2–6 sialosides with NeuAc or NeuGc68. As expected, the parental CHO cell line exhibited no enrichment of either sialoglycan (Fig. 2f).
Fig. 2: A colormetric PFU assay for rapid validation of LiGA.
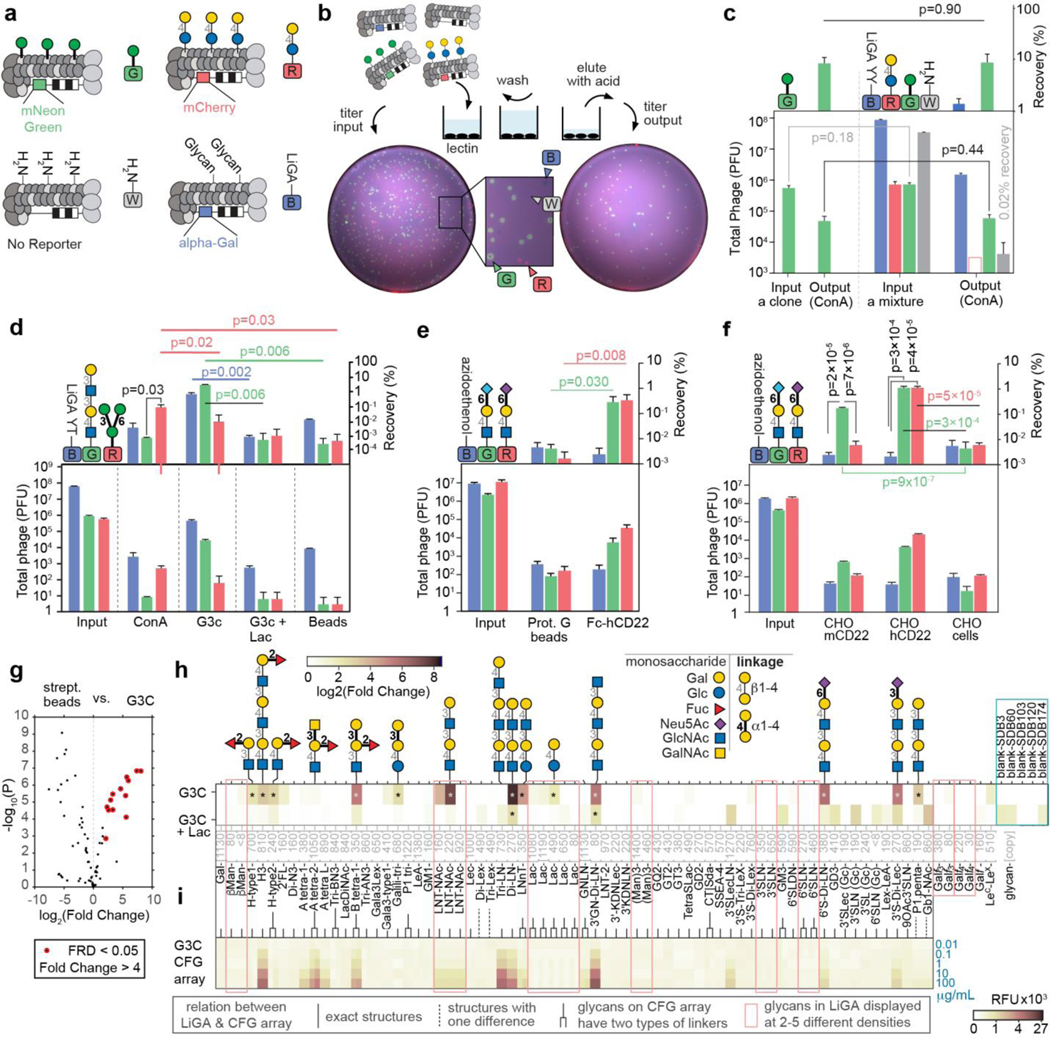
a, Clonal phage containing mNeonGreen reporter gene modified with mannose (α-Man), clonal phage containing mCherry gene with lactose (Lac), and LiGA mixture, constructed with LacZ reporter gene. b, Scheme of screenings against an immobilized GBPs and a fluorescent image of agarose plate used in plaque forming assay. c, Recovery of α-Man (mNeonGreen) in complex mixture (LiGA YY) and alone. n=3 input, n=4 output, Student’s t-test. (LiGA YY contained competing ligands β-Man, α-Man, and α-Man3, see Table S3). d, The titer results describe specific binding of reporter phage decorated by G3C binding glycan and ConA binding glycan to ConA and G3C; reporter cloners were part of LiGA YT. Composition of LiGA YT is detailed in panel h and supporting Fig. S8); n=4, Student’s t-test. e, hCD22 retains α2–6 sialoside, Neu5Gc (mNeonGreen) and Neu5Ac (mCherry) indiscriminately and has significantly higher recoveries compared to agarose beads. n=2 inputs, n=6 other, Student’s t-test. f, mCD22 expressing CHO cells have preference for recognizing α2–6 sialoside, Neu5Gc while hCD22 on cells do not have preference for Neu5Gc and Neu5Ac. n=5 CHO-hCD22, n=4 others, Student’s t-test. g, Bioconductor EdgeR differential enrichment (DE) analysis of deep-sequencing of LiGA YT binding to G3C and streptavidin beads (n=3) as described in (d). h-i, comparison of the (h) Fold Change (FC) values observed in DE analysis. n=3 for G3c and G3C+Lac (i) data observed in glass-based glycan array (n=6); public microarray data, CFG request #2564; searchable as “primscreen_6003” to “primscreen_6011” at CFG website52. Data in (c-f) are presented as mean + s.d. In (g-h) the FC, FDR and p-values were calculated using negative binomial model, TMM-normalization and BH-correction for FDR (* designate FDR<0.05). Recoveries in (c-f) are calculated as (PFUOutput/PFUInput)×100. Hollow bar in c indicate that no plaques were detected; the value represents the upper limit of detection.
We used the PFU assay to optimize the input and wash stringency of LiGA binding assays, to check the quality of the biotin-labeled protein (Supplementary Fig. S9) or to detect whether a complex LiGA mixture contained target-binding clones (Supplementary Fig. S10). The PFU assay was a critical proxy for downstream PCR and sequencing. For example, robust PCR-based preparation of phage DNA for sequencing requires >103 copies of phage DNA. The PFU assay identified the optimal conditions that minimized non-specific binding and yielded >104 phage particles (Fig. 2d), which were then used as DNA templates for deep-sequencing and further analysis of glycan-protein interaction (Fig. 2g–h).
LiGA characterizes the glycan recognition profile of glycan binding proteins (GBPs)
Next we analyzed the interactions of several GBPs with LiGAs (Fig. 2g,h). Here we used LiGA-75, which contains 75 glycoconjugates produced from 62 different glycans with nine of these glycans displayed at 2–5 different densities. Differential enrichment (DE) analysis was employed to examine phage DNA sequences associated with the control streptavidin agarose (SA) beads and phage DNA sequences associated with biotinylated GBP immobilized on SA-beads. Significantly enriched glycans were identified by Biocondutor EdgeR DE analysis57, 58 using negative binomial model Trimmed Mean of M-values (TMM) normalization59, Benjamini–Hochberg (BH)60 correction to control the false discovery rate (FDR) at α = 0.05 (Supplementary Fig, S11). This analysis identified 15 glycans associating significantly with Galectin-3 (G3C)-SA-beads and not SA-beads alone (Fig. 2g). The enrichment pattern was consistent between independently conducted experiments (Supplementary Fig, S12) and it was ablated in the presence of soluble lactose (Fig. 2h,). The results for glycan recognition by G3C using LiGA-75 (Fig. 2h) is consistent with results obtained using a glass slide-based printed microarray deposited to the CFG database52 (Fig. 2i). To expand on these observations, we tested binding of a panel of biotinylated GBP and antibodies to LiGA-65 comprising 65 glycoconjugates produced from 56 different glycans with four of the glycans (βMan, Lac, 3’SLN and Galf4) displayed at 2–4 different densities. Fig. 3a and Supplementary Fig. S13 describes enrichment by FLAG monoclonal antibody (Flag) and anti-galactofuranose monoclonal antibody (Galf4) and nine lectins: Cholera Toxin B subunit (CTb), Lens culinaris agglutinin (LCA), Pisum sativum agglutinin (PSA), Soybean agglutinin (SBA), Agrocybe cylindracea lectin (ACG), Narcissus pseudonarcissus lectin (NPL), Maackia amurensis leucoagglutinin (MAL I), Ulex europaeus agglutinin (UEA), and Sambucus nigra lectin (SNA). Fig. 3b describes public CFG microarray data for eight of the aforementioned lectins.
Fig. 3: Summary of lectin binding to LiGA composed of 65 different glycophage constructs.

a, Heat map describing the binding of 11 proteins to LiGA-65. Glycan notations, color-code of α/β linkages is shown in the legend, n=4, the assays were performed in PBS and HEPES (20 mM HEPES, 150 mM NaCl, 2 mM CaCl2) as working buffers (see details in Supplementary Fig. S13). b, Binding specificity of the glycan binding proteins to glass-based glycan array. The data is an aligned subset of multiple publicly available CFG array data, n=6. Color of the bars represents the CFG request numbers or experiment date (see details in Supplementary Fig. S21). The lines between (a) and (b) connect either identical or structural related glycans in LiGA and CFG. FC was calculated by EdgeR DE analysis using negative binomial model, TMM-normalization and BH-correction for FDR; asterisks designate FC>3.8, whereas circles designate 3.8>FC>2 with FDR≤0.05 in both cases.
There was general agreement between glycan-GBP interactions measured by LiGAs and binding of lectins to printed glycan microarrays. Similarly to previous reports that evaluated the binding of lectins to glycans presented on different platforms30, 61, lectin binding to glycans in LiGAs and printed arrays highlighted some cross-platform differences. We noted several glycans that exhibited binding to GBP in LiGA experiments and in the published literature but not in printed arrays. For example (Neu5Acα2–8)nNeu5Acα2–3Galβ1–4Glcβ-Sp glycans (n=3: TetraSLac, n=1: GD3) do not bind to MAL-I in printed array (Fig. 3b); however, these glycans bind to this lectin as part of a LiGA (Fig. 3a) and when printed on glass as a multivalent BSA conjugate61. Binding of Galili-tri (Galα1–3Galβ1–4Glcβ-Sp) to SBA was not observed in printed array but this glycan bound to SBA as part of a LiGA and interaction of this glycan with SBA was confirmed by lectin frontal affinity chromatography62 (entry LfDB0166 at Lectin Frontier Database63). Interactions of cholera toxin B (CTB) with blood group B antigens on type 2 chains (2’F-B type 2) is not detected in a printed array but is detected in a LiGA. Such interactions have been confirmed by X-ray crystallography to occur at the non-primary binding site of CTB with low mM monovalent affinity64. We also note that some glycans observed to bind to lectins in printed arrays did not exhibit enrichment in pull down of a LiGA by lectins. Notable examples were: (i) phage decorated with Manα1–6[Manα1–3]Manα1- was not enriched by NPL; (ii) Phage decorated LeY was not recognized by neither UEA nor CTB; in the same LiGA mixture, both UEA and CTB enriched LeY-(LeX)1–2 glycans but not the LeX-repeats alone; and (iii) Phage clones decorated by tri-LN and group A type-2 glycans were not recognized by G3C (Fig. 2h). Diminished recognition of glycan by lectins may result from suboptimal display of glycans on phage. Indeed, we noted that that when the same glycan was presented to G3C at several different densities on phage (e.g., LNT-NAc Fig. 2h), enrichment was the most optimal at one of those densities. Controlling the valency of the displayed glycans over several orders of magnitude is a critical technological benefit of LiGAs when compared to a monovalent DNA-glycan platform or low-valency phage-displayed glycan platforms. Building on this advantage, we further tested the effect of density of display of glycans on phage virion on recognition by some lectins.
To more systematically investigate the importance of density, a LiGA-9×6 was produced that contains nine glycans displayed at six different densities (Fig. 4a). Binding of LiGA-9×6 components to tetravalent ConA, Galectin-3 and mAb-Galf4 target uncovered bimodal density-dependent response in addition to the expected affinity-dependent response (Fig. 4b–d). Within the same glycan type, we observed a bimodal response: phage decorated with high copy of Manα1–6(Manα1–3)Manα1- (>300 per phage) exhibited weaker binding when compared to particles that contained intermediate (130–300) copies (Fig. 4b, Supplementary Fig. S14). We observed similar bimodal binding at different densities of Galf-glycans to anti-Galf IgG and the binding of LNT-NAc to biotinylated Galectin-3 displayed on streptavidin beads (Fig. 4c–d, Supplementary Fig. S14). The LiGA technology decoupled affinity-based and avidity-based observations, highlighting both optimal recognition units and their density. Similar observations have been made in prior studies that employed arrays of glycan-BSA conjugates printed on a glass slide24. Steric occlusion (the inhibition of binding of a protein to one glycan by neighboring glycans) is one of the mechanisms that may explain the decrease in binding of multivalent ligands to proteins at high density of ligands69. To test this hypothesis, we produced a hybrid phage that contained both productive glycan (Man3) and unproductive glycan (Galf4) on the same phage virion (Supplementary Fig. S15). We observed decreased binding to productive glycan in the presence of increasing density of unproductive glycan present on the same phage. This observation confirmed that steric occlusion is one of the factors that decreases the binding of proteins to phages modified with high density of glycans (Supplementary Fig. S15). Critical to both approaches is the quality control of every synthesized glycoconjugate or LiGA components by MALDI to create arrays with predefined, consistent valency of glycans.
Fig. 4: LiGA measures affinity and avidity-based responses in purified GBPs.

a, LiGA 9×6 is composed of nine glycans displayed at up to six densities. b, Binding of LiGA 9×6 to ConA. n=3 test, n=4 control. c, Galectin-3, n=4. d, mAb-Galf4, n=3. Measurements by deep-sequencing confirms enrichment of specific glycans; for each glycan-GBP pair, the phage that display high density of the glycan showed significantly lower enrichment than those that display intermediate density. MALDI spectra of the phage glycoconjugates of low and high density. LiGA components b1, b2, b3, c1, c2, d1 and d2 described in (b-d). FC was calculated by Bioconductor EdgeR DE analysis using negative binomial model, TMM-normalization and BH-correction for FDR. Error bars represent s.d. propagated from the variance of the TMM-normalized sequencing data. In b-c, biotinylated proteins were immobilized on streptavidin beads; in d on protein A beads and after the binding assay, the beads were boiled for 10 min to release the bound phage prior to PCR and deep sequencing. Binding of LiGA 9×6 to streptavidine and protein A beads was used as a control in DE analysis.
LiGAs characterize the glycan recognition profile of cell-surface lectins
The previously mentioned PFU assay highlighted the possibility of interrogating specific recognition of glycans on the cell surface. Therefore, we probed two important immunomodulatory receptors, hCD22 and DC-SIGN, expressed on the cell surface with LiGA-71 produced from 66 different glycans with five of these glycans displayed at two different densities. To initially dissect hCD22 recognition, we immobilized hCD22-Fc on protein G-resin and tested with LiGA-71. Deep-sequencing confirmed known recognition of α2–6-linked sialic acid glycans but not the α2–3-linked regioisomers or asialo-glycans (Fig. 5a). We incubated the same LiGA-71 with CHO cells expressing hCD22 or control CHO cells, and sequenced the associated phage (Fig. 5b). Differential enrichment analysis uncovered indistinguishable recognition profile within LiGA-71 for hCD22 on the surface of CHO cells compared to recombinant soluble hCD22 immobilized on agarose beads.
Fig. 5: LiGAs measure affinity and avidity-based responses in cell-displayed GBPs.

a, Binding of LiGA-71 to construct hCD22-Fc normalized by binding of the same LiGA to protein-G beads, n=3. b, Binding of LiGA-71 to hCD22(+) CHO cells normalized by binding of the same LiGA to CHO cells, n=4. c, Binding of LiGA-71 to DC-SIGN(+) fibroblasts normalized by binding of the same LiGA to fibroblasts, n=4. d, Binding of LiGA 9×6 to DC-SIGN(+) fibroblasts performed analogously to (c), n=3. Phage virions that display 300 copies (d) or 460 copies (c) of core trimannoside exhibit significant binding to DC-SIGN clusters on the cell surface, virions with <150 copies or >1200 copies exhibit insignificant enrichment. Calculations of FC, FDR and error bars are identical to those described in Fig. 2h, Fig. 3a, and 4b–d.
Next, we incubated LiGA-71 with rat fibroblasts expressing DC-SIGN or isogenic fibroblasts without DC-SIGN and compared the barcoded DNA associated with the DC-SIGN(+) and DC-SIGN(–) cell pellets. The resulting binding profile from DC-SIGN (+) cells revealed enrichment of αMan3 (Manα1–6(Manα1–3)Manα1-), glycans with LeX motifs and other fucosylated (α1–3 or α1–2-linked) glycans (Fig. 5c). We observed a statistically significant but modest fold change (FC) enrichment of blood-group glycans, which was considerably lower than FC in interaction of these glycans with blood-group antibodies (Supplementary Fig. S16). Binding of LiGAs to DC-SIGN on cells was qualitatively similar to the glycan-binding profile of purified DC-SIGN measured by canonical printed glycan arrays deposited to CFG website52 (Supplementary Fig. S17), with only minor differences. For example, αMan3 (Manα1–6(Manα1–3)Manα1-(CH2)6-phage) was reproducibly enriched in LiGA-71 as well as in LiGA mixtures of other composition (Supplementary Fig. S17a), whereas the CFG glycan microarray only detected binding only to penta- and nonamannose glycans. Recognition of alpha-linked Man3-motifs by DC-SIGN was postulated based on structural studies65 and specific binding of Manα1–6[Manα1–3]Manα1-(CH2)6-N3 to DC-SIGN was measured66. Thus, lack of detection of the Manα1–6(Manα1–3)Manα1-(CH2)9-amino glycan on the CFG glycan microarray may be the result of differences in the presentation of the glycan in the LiGA and printed array formats (Supplementary Fig. S17b–c). Deploying the density-scanning LiGA-9×6 array with DC-SIGN(+)/(−) fibroblasts not only confirmed the specific recognition of αMan3 but also uncovered pronounced bell-shaped dependence on density of αMan3 (Fig. 5d, Supplementary Fig. S14 and S18). Phage particles that displayed 300 copies exhibited binding significantly higher than those with <150 or >1000 copies of αMan3. This density scan further suggests that presentation of the αMan3 on the slide-based arrays may not have been optimal for the recognition by DC-SIGN. These observations highlight the ability of the LiGA platform to robustly and reproducibly measure both affinity-based and avidity-based effects on glycan-GBPs interactions and in a complex biological milieu on the surface of mammalian cells.
LiGAs, like other M13-display platforms, makes it possible to detect interactions between displayed ligands and receptors on the surface of organs and cells in live animals47, 48. To demonstrate this capacity, we tracked the biodistribution of a LiGA to different organs in mice and its association with two subpopulations of B cells. We carried out a mixed bone marrow chimera to generate mice that contain a 50:50 mixture of B cells expressing transgenic hCD2267 or lacking hCD22 expression, both of which are on a mCD22−/− background (Fig. 6a and Methods for details). We used a red-green-blue-white (RGBW) LiGA, which was a 1:1 mixture of LiGA-70 linked to Lac-reporter phage (‘blue’) and non-glycosylated phage associate with reporter-free phage (‘white’) supplemented with ~1% of two phage clones displaying Neu5Gcα2–6LacNAc and Neu5Acα2–6LacNAc linked to NeonGreen and mCherry transducing phage. The latter ‘red’ and ‘green’ clones previously demonstrated significant enrichment by cell surface-displayed hCD22 protein (Fig. 2f). We injected this RGBW-LiGA in the tail vein of mice (n=3), recovered the organs after one hour, sorted the B cell populations from the spleen and measured the composition of the library associated with each organ or B cell type by PFU assay (Fig. 6b). Gratifyingly, we observed significant enrichment of the phage displaying the NeuAcα2–6Gal sialosides only with B cells that expressed hCD22 sorted from the same spleen of the same mouse (Fig. 6c). We observed a similar level of enrichment of Neu5Ac and Neu5Gc sialosides, consistent with the previous observations that hCD22 has similar affinity for both analogues of sialic acid68. No enrichment of the phage displaying the NeuAcα2–6Gal sialosides was found in the bulk organs, including the lungs, heart, liver, kidney. This experiment demonstrates the feasibility of studying glycan-GBP interactions on the surface of cells in vivo using the LiGA technology.
Fig. 6:

a, Lethally irradiated mice were reconstituted with a 50:50 mixture of bone marrow from the two indicated recipient bone marrow to generate chimeric mice that have a mixture of hCD22+ and hCD22- B cells both lacking mCD22 and with different CD45 congenic markers for sorting purposes. b, The LiGA was injected into mice via the tail vein. One hour later, the mice were euthanized, and organs collected. The B cells were stained with the appropriate antibodies and sorted for differential expression of hCD22. Sorted cells and cells from organs were analyzed for phage by PFU assay. c, Analysis of types of phage from the PFU assay expressed as PFU enrichment (top) or total PFU (bottom). Tissues from n=3 mice, p-values calculated by Student’s t-test. In c, Neu5Gcα2–6LacNAc and Neu5Acα2–6LacNAc were supplemented at ~1% ratio to simulate the contribution of a clone in a typical LiGA library.
Discussion
Multivalent interactions are ubiquitous in biological systems69. At a cell surface interface, multivalent receptor–ligand interactions are central to signal transduction, cell–cell recognition and the interpretation of external signals by cells11. The LiGA technology makes interrogation of these interactions possible using a mixture of monodisperse multivalent probes19: each probe with a DNA barcode and well-defined glycan density. Modern advances in DNA barcoding, deep-sequencing and bioorthogonal modification enabled high-throughput manufacturing of LiGA components via a parallel conjugation of the glycans to phage virion. Like the majority of arrays with glycans printed on glass, the linkage between the glycan and the carrier (phage) is not natural. However, chemical conjugation affording 30–1500 copies of glycan per phage particle was key to addressing two critical limitations of prior glycophage: density and glycan diversity. The pioneering reports of glycophage display by groups of DeLisa44 and Aebi45 employed M13 “3+3” phagemid particles displaying a D/E-X1-N-X2-S/T acceptor motif on pIII protein. Like all 3+3 display systems, it is a monovalent display of one copy of engineered pIII fusion and four copies of wild type pIII (on average). Importantly, both groups observed that only 1 in 100 phagemid particles carried a complete glycan biosynthesized by the bacterial host. The display of truncated glycopeptides was common. The latter problem was subsequently resolved by DeLisa and co-workers46 by employing truncated pIII instead of full-length pIII. However, even in the optimized construct, the authors could not quantify the density of glycans on the different glycophages due to the low level of phage glycosylation46. The biosynthesis of a DNA-encoded bona fide multivalent display of diverse glycan structures on phage remains an interesting bioengineering challenge. Recent advances in this area include biosynthesis of multivalent display of glycoproteins on mammalian cell surface70 and multivalent display of biosynthesized glycans on DNA-free virus-like particles71. Unlike these and other display systems that employ the biosynthesis of glycans, LiGA decouples DNA encoding and glycan display from biosynthesis. Chemical manufacturing of LiGA components allows the repurposing of many chemical approaches previously employed in the construction of traditional slide-based glycan arrays20. Chemical conjugation of small SPAAC linkers to the N-terminus of the pVIII protein is likely to yield a random distribution of conjugation sites and glycans along the length of the virion. Although glycan-to-glycan distances in such system are not monodisperse, the distribution of distances can be modeled based on existing NMR structure of M13 virion (Supplementary Fig. S19).
Although SPAAC is a convenient linking strategy, other linking strategies may also work. For example, ligation of glycans with a carboxy linker to lysine side chains of bovine serum albumin is routinely employed in microarray manufacturing24 (reviewed in20). This approach is straightforward to adapt to LiGA manufacturing via acylation of N-termini of the pVIII protein instead of BSA. A growing palette of site-specific chemical conjugation strategies can inspire new and improved variants of LiGAs. The source of glycans in this paper is chemical, but it should also be possible to develop LiGAs using glycans isolated from natural sources. For example, ongoing efforts in our laboratory are focused on exploring the chemoenzymatic synthesis of LiGAs from biantennary N-linked glycan precursors.
We have shown that the multivalent LiGA platform effectively measured the glycan-binding preferences of purified GBPs and GBPs displayed on cells, including cells in organs in live animals. A typical preparation of one of the individual LiGA components requires only 50–100 μg of glycan and yields 1012 PFU of glycosylated phage particles. This amount is sufficient to perform on the order of 100,000 lectin-binding or cell-binding assays using 106–107 copies of each glycosylated clone as input. LiGA, like other DNA-encoded reagents, are simple to distribute in the scientific community and analyze in any laboratory using readily available DNA deep-sequencing. We anticipate that LiGA-like reagents will find application in diverse glycan recognition assays in vitro, on the cell surfaces and in vivo. The deployment of the LiGA technology further benefits from accurate knowledge of copy number, composition, and valency of LiGA components. This critical information will facilitate the investigation and modeling of complex interactions between a multivalent display of glycans and multivalent display of receptors on the cell surface. The possibility to DNA-encode and study the density of glycans makes it possible to systematically study the role of multivalency and avidity in these assays. The format of the data—DNA sequencing—is poised for standardization, sharing and systematization of knowledge; such formats will help integrating knowledge from the glycobiology community with other sequence-centric knowledge bases. The LiGA platform has the potential to uncover critical glycan binding information of between glycans and cell-displayed GBP that has remained elusive due to limitation of other glycan array platforms.
Methods
1. Biochemical Methods
1.1. Materials and general information.
Sanger sequencing and deep sequencing was performed at the molecular biology service unit (University of Alberta) using Illumina NextSeq500. DNA primers were ordered from Integrated DNA Technologies. Biochemical reagents were purchased from Thermo Fisher Scientific (TFS) unless noted otherwise. Human Galectin-3 residues 107−250 (G3C) was a gift of Christopher Cairo (University of Alberta). Biotinylated Cholera Toxin pentamer B subunit was a gift of Bruce Turnbull (University of Leeds). Murine monoclonal anti-A and anti-B IgM were a gift of Lori West (University of Alberta). Azido-glycans are described in Supplementary Methods and Table S2. MS-MALDI-TOF spectra were recorded on AB Sciex Voyager Elite MALDI, mass spectrometer equipped with MALDI-TOF pulsed nitrogen laser (337 nm) (3 ns pulse up to 300 μJ/pulse) operating in full scan MS in positive ionization mode.
1.2. Construction of silent double barcoded (SDB) M13 phage library.
A library of degenerate codons, termed, silent double barcodes (SDB) was created in the phage genome at a position proximal to the gIII cloning site starting from a M13KE vector containing the stuffer sequence CAGTTTACGTAGCTGCATCAGGGTGGAGGT. The insert fragment was PCR amplified using the primers P1 and P2 and the vector fragment was PCR amplified using primers P3 and P4. Sequences:
P1 GAGATTTTCAACGTGAAAAAACTNCTNTTYGCNATHCCNCTNGTGGTACCTTTCTATTCTCA
P2 TTAAGACTCCTTATTACGCAGTA; P3 TTGCTAACATACTGCGTAATAAG;
P4 TTTTTTCACGTTGAAAATCTC; P5 GTGGTACCTTTCTATTCTCACTCGAGYGTNGARAAR-AAYGAYCARAARACNTAYCAYGCNGGNGGNGGNTCGGCCGAAACTGTTGAAAG;
P6 CGAGTGAGAATAGAAAGGTAC
PCR was performed using 50 ng phage dsDNA with 1 mM dNTPs, 0.5 μM primers, 0.5 μL Phusion High Fidelity DNA polymerase in 1x PCR buffer (NEB #B0518S) in a total volume of 50 μL. PCR amplified fragments were treated with restriction enzyme DPN 1 (NEB #R0176S) and then gel purified. NEBuilder Hifi DNA assembly was then carried out following the manufacturer protocols by mixing 100 ng of vector, 4 ng insert, 10 μL of NEBuilder Hifi DNA assembly master mix (NEB #E2621S), and deionized H2O up to a total volume of 20 μL. The resulting ligated DNA was transformed into E.coli K12 ER2738 and propagated overnight at 37 °C. Into the resulting “silent barcoded” (SB) vector SB1-QFT*LHQ, we cloned a degenerate codon library as follows: The vector was PCR amplified using primers P4 and P6, the insert was amplified from annealed primers P5 and P2. PCR fragments were processed using NEBuilder Hifi DNA assembly kit (NEB #E2621S) using steps described above. The resulting ligated DNA was transformed into electrocompetent cells E.coli SS320 (Lucigen) and propagated overnight. The culture was centrifuged to remove host cells and incubated with 5% PEG-8000, 0.5 M NaCl for 8 h at 4 °C, followed by 15 min centrifugation at 13000 g. PEG precipitated phage were re-suspended in PBS-Glycerol 50% and stored at −20 °C. Resulting M13-SDB-SVEKY is a library of chemically identical phage with 6,144 and 2.1×106 theoretical redundant sequence combinations in two cloned regions (Supplementary Fig. S1). Sequence of the M13-SDB-SVEKY vector is available on GeneBank (#MN865131). Deep sequencing of the M13-SDB-SVEKY and SB1-QFT*LHQ libraries is available at: https://48hd.cloud/file/20161215–67OOooOO-NB and https://48hd.cloud/file/20161105–68OOooIC-NB.
1.3. SDB clone isolation and amplification.
The M13-SDB-SVEKY library described in the previous section was used to isolate monoclonal silently encoded phage and purify each clone by a PEG-precipitation (see Supplementary Methods). Phage clones were characterized by Sanger Sequencing (Supplementary Table S1) and Illumina sequencing (Fig. S1G–H). Illumina sequencing of the isolated clones is available at http://ligacloud.ca/ by searching for specific SDB number. Example of searching for “SDB50”: http://ligacloud.ca/searchDB?search=SDB50
1.4. Construction of phage that transduce mCherry and mNeonGreen fluorescent proteins.
Vectors pBAD-mCherry and pBAD-mNeonGreen (gift from Robert Campbell, University of Alberta) were PCR amplified using primers P7 (GCGGATAACAATTTCACACAGGAAACAGCTATGGT-GAGCAAGGGCGAG) and P8 (TTAAATTTTTGTTAAATCAGCTCATTTTTTACTTGTACAGCT-CGTCCA) and cloned into M13-SDB-SVEKY vectors following the protocol described in section 1.3 and Supplementary Methods. The sequences of the resulting M13 mNeonGreen SDB SVEKY and M13 mCherry SDB SVEKY vectors are available as GeneBank MN865132 and MN872303
1.5. Monitoring phage viability under CuAAC reaction.
A solution of 50 mL of phage clone in Tris-borate (200 mM pH 8.0) combined with NHS-4-pentynoate (50 mM in DMF) was reacted for 1 h and purified on Zeba Spin Column (40K MWCO, TFS# 87766). To the filtered phage solution, azidomethyl-1-thio-β-D-mannopyranoside (Fig. 1d) was added to afford the 2 mM concentration followed by ~1 mg of copper powder. The CuAAC reaction was initiated by adding 3 μL of Bathophenantroline/Cu+ catalyst prepared as described72. After incubation for 8 h at 4 °C under argon atmosphere, the mixture was purified on Zeba Spin column. Plaque forming units (PFU) assay of solution sampled throughout the reaction was used to monitor the infectivity of phage (Fig. S2C–D).
1.6. Monitoring phage viability under SPAAC reaction
A solution of 50 μL of phage clone (50 μL, 3.4 × 1013 PFU/mL PBS pH 7.4) combined with DBCO-NHS (2.5 μL, 20 mM in DMF) was incubated for 1 h and purified on Zeba Spin column. To the filtrate, solution of azidomethyl-1-thio-β-D-mannopyranoside was added to afford the 2 mM concentration. The solution was incubated for 8 h at 4 °C, purified on Zeba Spin column and analyzed by PFU assay (Fig. S2C–D).
1.7. Analysis of glycosylation of phage samples by MALDI-TOF MS.
The sinapinic acid matrix (Sigma, #D7927) was formed by deposition of two layers: layer 1 (10 mg/mL sinapinic acid in acetone-methanol, 4:1) and layer 2 (10 mg/mL sinapinic acid in acetonitrile:water, 1:1, with 0.1% TFA). In a typical sample preparation, 2 μL of phage solution in PBS was combined with 4 μL of layer 2, then a mixture of 1:1 layer 1 : layer 2+phage was deposited in that order onto the MALDI inlet plate ensuring that layer 1 is completely dry before adding layer 2+phage. The spots were washed with 10 μL of water with 0.1% TFA to remove salt ions from the PBS.
1.8 & 1.9. Quantification of phage-surface chemical modifications and Modification of phage clones with glycans to build components of LiGA.
A solution of each SDB phage clone (1012-1013 PFU/mL in PBS) was combined with DCBO-NHS (20 mM in DMF) to afford 0.1–1.5 mM concentration of DCBO-NHS in reaction mixture, which typically yields 15–50% of pVIII modification after 45 min incubation. Solutions of glycan-azide (10 mM in H2O) were added directly to the reaction mixture to afford a 2–3 mM concentration of glycan-azide and the solutions were further incubated overnight at 4 °C. The reaction mixture was purified on Zeba Spin column and analyzed by MALDI-TOF.
1.10. Preparation of LiGA from glycosylated clones.
In a typical protocol, LiGA was prepared by mixing 108 PFU of desired glycan-phage conjugates. Each unique LiGA mixture was assigned a two-letter identifier (e.g., “YZ”) and a “dictionary” (e.g., YZ.xlsx), a table that describes the correspondence between the DNA barcodes and the glycans in the LiGA mixture (Supplementary Table S3). These dictionaries were used to translate from DNA in the deep-sequencing files to the corresponding glycans and their density. Dictionaries for LiGA mixtures are available in Data/LiGA Dictionaries/ folder. References to the deep-sequencing composition of these libraries on http://ligacloud.ca are available in Supplementary Table S3. Composition of LiGA mixtures, as determined by deep-sequencing did not change significantly after storage for 4 months at 4 °C and one year in −20 °C as a glycerol stock (Supplementary Fig. S20).
1.11. Methods that measure binding of LiGA components (details are in Supplementary Methods).
1.11.1. Binding of glycosylated clones measured by ELISA.
96 well plate (Corning®, #CLS3369) was coated by ConA (Sigma-Aldrich #C2010, 1 mg/mL) overnight at 4 °C, washed 3x by PBS-T (PBS+0.1%Tween), blocked with PSB, 2% BSA for 1 h, and incubated with LiGA clones with concentrations ranging from 106 to 1010 PFU/mL (50 μL in PBS) for 1 h. After wash (3x, PBS-T), wells were probed with anti-M13-HRP.
1.11.2. Binding of LiGA to ConA or Antibodies immobilized on Plate.
ConA-coated plates were blocked (see above) and LiGA mixture was added to the wells. After 1 h, the wells were washed 2x with PBS-T, 1x with PBS, eluted with 50 μL of HCl (pH 2.0) for 9 min at rt, and the content of each well was transferred to an Eppendorf tube containing 25 μL of 5x Phusion HF buffer (NEB, #M0530S). The neutralized solution was used for titer and as DNA template for PCR and Illumina sequencing. Binding of LiGA to anti-A and anti-B antibodies was performed in similar fashion.
1.11.3. Binding of LiGA to CD22-Fc immobilized on Protein G Magnetic Beads.
Protein G magnetic beads (Promega #G7472) were washed and blocked per manufacturer instructions and coated by CD22-Fc protein (1 mg/mL in PBS) overnight at 4 °C. Binding to LiGA (108 PFU/mL in HEPES Buffer) was processed using KingFisher™ Duo Prime Purification System using automated washes with HEPES+1% Tween (2x) and HEPES (1x). At the end of the program, the beads with bound phage were transferred to individual Eppendorf™ tubes, resuspended in nuclease-free H2O (30 μL); 2 μL of the suspension solution was sampled for titering. The remaining solution was incubated at 90 °C for 15 min and 25 μL of the supernatant was used as template for PCR reaction and Illumina Sequencing.
1.11.3. Binding of LiGA to Lectins immobilized on Streptavidin Magnetic Beads
Same method as above using Streptavidin magnetic beads (Promega #Z5481)+biotinylated ConA or other biotinylated lectins described in Fig. 3 or S13 using either PBS or HEPES (20 mM HEPES, 150 mM NaCl, 2 mM CaCl2) as working buffers.
1.11.4. Binding of LiGA YZ to CHO cells expressing CD22.
Confluent CHO-CD22(+) or CHO-wt cells were detached using PBS+5 mM EDTA and washed with PBS (2×5 mL). Suspension of at 2 ×106 cells in HEPES-1% BSA (20 mM HEPES, 150 mM NaCl, 2 mM CaCl2, pH 7.4, 1% BSA) in round bottom 3 mL tube (Corning, #352054) was combined with LiGA (108 PFU) and incubated for 1 h at 4 °C. The cells were then washed HEPES-0.1% BSA in (3×3mL) and HEPES buffer (1×1mL). Washed cell pellet was resuspended in 30 μL Nuclease Free H2O; 2 μL of the suspension solution was sampled titering. The remaining solution was incubated at 90 °C for 15 min, centrifuged at 21,000×g for 10 min, and 25 μL of the supernatant was used for PCR and sequencing.
1.11.5. Binding of LiGA to Fibroblasts cells expressing DC-SIGN.
The Rat-6 fibroblast DC(+) and DC(−) cell lines73 (gift of Kurt Drickamer Imperial College London) were maintained in DMEM-F12 medium (TFS #11330057) supplemented with 10% Fetal Bovine Serum (TFS #26140079) and Penicillin-Streptomycin (TFS #15140122). The DC-SIGN(+) and DC-SIGN(−) fibroblast cells were detached from culture flask using TrypLE (TFS #12605036) and resuspended in HEPES-1% BSA at 2×106 cells/mL. Binding of cells to LiGA was performed as outlined in “Binding of LiGA YZ to CHO cells expressing CD22”.
1.12. Production and maintenance of CHO cells expressing CD22
Full length human CD22, in the pcDNA5/FRT vector, was stably transfected into CHO cell line through Flp-In™ (TFS #K601001) system under selection with 0.5 mg/mL hygromycin-B for two weeks. The cells were passaged using 1 mM EDTA/PBS (TFS #13151014) as a dissociation solution and maintained in DMEM-F12 medium (TFS, #11330057) supplemented with 10% Fetal Bovine Serum (TFS #26140079) and Penicillin-Streptomycin (TFS #15140122). Untransfected CHO cells were maintained in the same conditions and used as control.
1.15. PCR Protocol and Illumina sequencing.
The 25 μL of DNA template solution in Nuclease free water was amplified in total volume of 50 μL with 1x Phusion® buffer, 50 μM of each dNTPs, 500 μM MgCl2, 1 μM forward barcoded primer, 1 μM reverse barcoded primer and one unit of Phusion® High-Fidelity DNA Polymerase (NEB, #M0530S). The volumes and PCR cycling parameters are further detailed in the in Supplementary methods. The PCR products were quantified by 2% agarose gel in Tris-Borate-EDTA buffer. PCR products that contain different indexing barcodes were pooled allowing 10 ng of each product in the mixture. The mixture was purified by eGel, quantified by Qubit (Thermo Fisher) and sequenced using the Illumina NextSeq paired-end 500/550 High Output Kit v2.5 (2×75 Cycles). Data was automatically uploaded to BaseSpace™ Sequence Hub. Processing of the data is described in section “2.2 Processing of Illumina data”.
1.16. Generation of Monoclonal anti-Galf4 antibody
Azido-Galf4 was conjugated to Tetanus toxoid (TT, Statens Serum Institute) using CuAAC as detailed in “1.16.1 Conjugation of Galf4 to tetanus toxoid” in Supplementary methods. The degree of Galf4 units incorporated on the TT was 14/protein as determined by MALDI-TOF MS. Female balb/c mice (5 mice, Charles River, Canada, 6–8 weeks old) were used to raise the monoclonals. All the procedures and experiments involving animals were carried out using a protocol approved by Animal Care Committee: Biosciences, University of Alberta. The protocol was approved as per Canadian Council on Animal Care (CCAC) guidelines. Animals were immunised three times (10.0 μg of TT-Galf4/mouse/injection) at intervals of 21 days as detailed in “1.16.3 Immunizations” in Supplementary methods. Antibody levels in the murine sera were studied using enzyme linked immuno-sorbent assay (ELISA) following a published protocol74 and hybridoma fusions were performed by following earlier published protocols75; all are detailed in sections “1.16.6 Hybridoma fusion and cloning” and “1.16.7 Isotyping and specificity testing of the hybridomas” in Supplementary methods. Anti anti-Galf4 clones were found to be IgG1 type
1.17. Panning of LiGA in mice.
All the procedures and experiments involving animals were carried out using a protocol approved by the Health Sciences Laboratory Animal Services (HSLAS), University of Alberta. The protocol was approved as per the Canadian Council on Animal Care (CCAC) guidelines. All mice were maintained in pathogen-free conditions at the University of Alberta breeding facility. A mixed bone marrow chimera mouse was created by lethally irradiating the recipient mice and transplanting them with a 50:50 mixture of bone marrow collected from the femurs of mCD22KO (mCD22−/− hCD22- CD45.2+/+) and hCD22 transgenic (mCD22−/− hCD22+ CD45.2+/+) mice. Four weeks after transplantation, mice were bled to verify that the desired chimera had been established. Six weeks after irradiation, animals were injected with LiGA (0.2 m, 1 × 1011 PFU/mL in PBS). One hour post-injection mice were euthanized with CO2. Internal organs (heart, liver, kidney, lung and spleen) were collected and stored in cold DMEM.
Tissues were homogenized by grinding between 75 mm frosted microscope slides. Homogenized tissues was transferred into 5 mL of RBC lysis buffer (155 mM ammonium chloride, 10 mM, EDTA, 100 mM Tris, pH 7.4), incubated for 7 min, centrifuged at 300 rcf for 5 min, and the resulting pellet was resuspended in 0.4 mL PBS + 0.1% BSA. Cell suspension was combined with E.coli K12 ER2738 and plated in agar overlay to determine the amount of phage particles in specific organ by PFU assay. To sort the B cells, the spleen was homogenized, RBC-lysed as described above and RBC-depleted splenocytes were stained with a cocktail of fluorescent antibodies (Biolegend): anti-mouse/human CD45R/B220-APC-Cy7 (1 μg/mL), Brilliant anti-mouse CD19-BV605 (1 μg/mL), anti-mouse CD22-AF647 (2 μg/mL), anti-human CD22-BV421 (2 μg/mL), anti-mouse CD45.1- PE/Cy7 (1 μg/mL), and anti-mouse CD45.2-BV421 (1 μg/mL). Dead cells were excluded with propidium iodide (1 μg/mL, Thermo Fisher). The stained cells were sorted on FACS Aria cell sorter. Viable singlet B cells (CD19+B220+) were divided into CD45.1+/+ (mCD22−/−hCD22-) and CD45.2+/+ (mCD22−/−hCD22+). Both populations were verified to be lack mCD22 expression and the CD45.2+/+ cells were verified to express hCD22. One million cells from each population were collected and these cells were analyzed using PFU-assay.
2. Data Processing Methods
2.1. General data processing methods.
Data analysis was performed in Python, Matlab or R/Bioconductor. Core scripts are available as part of the Supplementary Data.Zip or https://github.com/derdalab/liga/. Data storage cloud http://ligacloud.ca/ was implemented in Linux-Apache-MySQL-Python (LAMP) architecture and details of this implementation are beyond the scope of this report. Analysis of enrichment of LiGA and its significance was performed as differential enrichment (DE) analyses similar to our previous reports.12–13 We used Bioconductor EdgeR14–15 DE analysis with modeling of the observed counts using a negative binomial model, Benjamini–Hochberg (BH) adjustment to control the FDR at α = 0.0516 and normalization of data across multiple samples using the TMM normalization17. Prior to DE-analysis, “test” and “control” data sets were retrieved from the http://ligacloud.ca/ server as tables of glycans, DNA, and raw sequencing counts. Tables of raw sequencing counts and DA values and for every Figure in this manuscript are available in Supplementary Data.zip folder.
2.2. Processing of Illumina data.
The Gzip compressed FASTQ files were downloaded from BaseSpace™ Sequence Hub. The files were converted into tables of DNA sequences and their counts per experiment, as previously described18–20 and detailed in Supplementary Methods. Briefly, FASTQ files were parsed based on unique multiplexing barcodes within the reads discarding any reads that contained a low quality score. Mapping the forward (F) and reverse (F) barcoding regions, mapping of F and R priming regions allowing no more than one base substitution each and F-R read alignment allowing no mismatches between F and R reads yielded DNA sequences located between the priming regions. Using an SDB-lookup table, we mapped DNA sequences to SDB and translated them to glycans using a LiGA-specific lookup table (“LiGA dictionary”). Any reads with one substitution away from the expected SDB sequence were assigned to the parent SDB sequence. The files with DNA reads, raw counts, and mapped glycans were uploaded to http://ligacloud.ca/ server. Each experiment has a unique alphanumeric name (e.g., 20180711–87YOrdRB-JM) and unique static URL: for example http://ligacloud.ca/searchLibInfo?f=0&b=0&d=20180711–87YOrdRB-JM
3. Synthetic Methods
Details of the synthetic methods, procedures for synthesis of Galf1, Galf2, Galf3, Galf4, of DiN3, TriAN3, TriBN3 and azido-peg-betagalactoside and described in the Supplementary Information Sections 3.1–3.6 and Supplementary Schemes S1–S6.
Other Materials and Methods / Supplementary Information:
Supplementary Information document contains Supplemental Figures S1–S13, Supplemental Schemes S1–S4, Supplemental Tables S1–S3 and expanded descriptions of Biochemical Methods 1.1–1.17, Data Processing Methods 2.1–2.6, and Synthetic Methods 3.1–3.6 detailed the synthesis of LiGA components, protein- and cell-binding assays, animal experiments, the synthesis of the glycans, and data processing methods describing the analysis of the DNA sequencing data, and statistical methods. Availability of Reagents: DNA-barcoded clones of M13 phage used for preparation of LiGA, limited amounts of chemically glycosylated phage clones (LiGA components), limited amounts of the Galf4 monoclonal antibody and CD22(+)-CHO cells are available upon request from the corresponding author.
Source data:
submitted as “data.zip” contain files describing (i) MALDI spectra of the LIGA components, (ii) *.xlsx tables describing the correspondences between DNA and glycan structures (“dictionaries”); (iii) *.xlsx tables describing raw sequencing data and statistical analysis used to generate Figures 2h, 3a, 4b–d, 5a–d, S9c, S10, S11a, S12; (iv) CFG glass array data used to generate Figures 3b, S11b, S13.
Data Availability:
All raw deep-sequencing data is publically available on http://ligacloud.ca/ with data-specific URL listed in Table S3. DNA sequences of the three LiGA phage constructs have been deposited to GeneBank (#MN865131, MN865132, MN872303). MatLab, Python and R scripts have been deposited to https://github.com/derdalab/liga
Supplementary Material
Acknowledgments:
We thank the staff at the University of Alberta mass spectrometry facility (Chemistry Department) for help with MALDI analysis and Sophie Dang at the molecular biology service unit for assistance with Illumina sequencing. Cell sorting was performed at the University of Alberta, Faculty of Medicine and Dentistry Flow Cytometry Facility with financial support from the Faculty of Medicine and Dentistry and Canada Foundation for Innovation (CFI) awards to contributing investigators. We thank Kurt Drickamer (Imperial College, London), Bruce Turnbull (University of Leeds), David Bundle, Christopher Cairo and Lori West (University of Alberta) for provision of critical reagents. The authors acknowledge funding from NSERC (RGPIN-2018-04365 to T.L.L., RGPIN-2018-03815 to M.S.M., and RGPIN-2016-402511 to R.D.) and NSERC Accelerator Supplement (to R.D.), GlycoNet (SD-1 to T.L.L., TP-22 to R.D.), Alberta Innovates Strategic Research Project to R.D., and NIH projects (AI118842 to M.S.M., and GM062116 and AI050143 to J.C.P.). Infrastructure support was provided by CFI New Leader Opportunity (to R.D. and M.M.). J.M. acknowledges summer research fellowship from GlycoNet and Alberta Innovates Health Solutions. Many compounds were prepared by the Consortium for Functional Glycomics supported by NIH GM061126.
Footnotes
Competing Interests:
R.D and N.J.B. are shareholders of the start-up company 48Hour Discovery Inc. (Edmonton, AB), that licensed the patent application (WO2018141058A1) describing LiGA technology. R.D, N.J.B., and S.Sa are co-inventors on the aforementioned application.
Dedication: The paper is dedicated to Laura L. Kiessling on the occasion of her 60th birthday.
References
- 1.Shendure J. & Ji H. Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Lander ES Array of hope. Nat. Genet. 21, 3–4 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Blixt O. et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proceedings of the National Academy of Sciences of the United States of America 101, 17033–17038 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Kooyk Y. & Rabinovich GA Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 9, 593–601 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 446, 1023–1029 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Stevens J. et al. Structure and Receptor Specificity of the Hemagglutinin from an H5N1 Influenza Virus. Science 312, 404–410 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Stevens J, Blixt O, Paulson JC & Wilson IA Glycan microarray technologies: tools to survey host specificity of influenza viruses. Nat. Rev. Microbiol. 4, 857–864 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raman R, Raguram S, Venkataraman G, Paulson JC & Sasisekharan R. Glycomics: an integrated systems approach to structure-function relationships of glycans. Nat. Methods 2, 817–824 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Geissner A. & Seeberger PH Glycan Arrays: From Basic Biochemical Research to Bioanalytical and Biomedical Applications. Annu. Rev. Anal. Chem. (Palo Alto Calif.) 9, 223–247 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Smith DF, Cummings RD & Song X. History and future of shotgun glycomics. Biochem. Soc. Trans. 47, 1–11 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Bertozzi CR & Kiessling LL Chemical glycobiology. Science 291, 2357–2364 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Li Q, Rodriguez LG & Gildersleeve JC An array-based method to identify multivalent inhibitors. J. Am. Chem. Soc. 132, 9653–9662 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang CC et al. Glycan microarray of Globo H and related structures for quantitative analysis of breast cancer. Proc. Natl. Acad. Sci. U. S. A. 105, 11661–11666 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia L, Schrump DS & Gildersleeve JC Whole-Cell Cancer Vaccines Induce Large Antibody Responses to Carbohydrates and Glycoproteins. Cell Chem. Biol. 23, 1515–1525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacob F. et al. Serum antiglycan antibody detection of nonmucinous ovarian cancers by using a printed glycan array. Int. J. Cancer 130, 138–146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukui S, Feizi T, Galustian C, Lawson AM & Chai W. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat. Biotechnol. 20, 1011–1017 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Demetriou M, Granovsky M, Quaggin S. & Dennis JW Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409, 733–739 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Cecioni S, Imberty A. & Vidal S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 115, 525–561 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Kiessling LL, Gestwicki JE & Strong LE Synthetic multivalent ligands as probes of signal transduction. Angew. Chem. Int. Ed. 45, 2348–2368 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park S, Gildersleeve JC, Blixt O. & Shin I. Carbohydrate microarrays. Chemical Society Reviews 42, 4310–4326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rillahan CD & Paulson JC Glycan Microarrays for Decoding the Glycome. Annu. Rev. Biochem. 80, 797–823 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godula K, Rabuka D, Nam KT & Bertozzi CR Synthesis and microcontact printing of dual end-functionalized mucin-like glycopolymers for microarray applications. Angew. Chem. Int. Ed. Engl. 48, 4973–4976 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Godula K. & Bertozzi CR Density variant glycan microarray for evaluating cross-linking of mucin-like glycoconjugates by lectins. J. Am. Chem. Soc. 134, 15732–15742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oyelaran O, Li Q, Farnsworth D. & Gildersleeve JC Microarrays with Varying Carbohydrate Density Reveal Distinct Subpopulations of Serum Antibodies. Journal of Proteome Research 8, 3529–3538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dam TK & Brewer CF Lectins as pattern recognition molecules: The effects of epitope density in innate immunity. Glycobiology 20, 270–279 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Kwon SJ et al. Signal amplification by glyco-qPCR for ultrasensitive detection of carbohydrates: applications in glycobiology. Angew. Chem. Int. Ed. Engl. 51, 11800–11804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tom JK, Mancini RJ & Esser-Kahn AP Covalent modification of cell surfaces with TLR agonists improves & directs immune stimulation. Chem. Commun. (Camb.) 49, 9618–9620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J. et al. Specific recognition of lectins by oligonucleotide glycoconjugates and sorting on a DNA microarray. Chem. Commun. (Camb.), 6795–6797 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Ciobanu M. et al. Selection of a synthetic glycan oligomer from a library of DNA-templated fragments against DC-SIGN and inhibition of HIV gp120 binding to dendritic cells. Chem. Commun. (Camb.) 47, 9321–9323 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Yan MM et al. Next-Generation Glycan Microarray Enabled by DNA-Coded Glycan Library and Next-Generation Sequencing Technology. Anal. Chem. 91, 9221–9228 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Chevolot Y. et al. DNA-based carbohydrate biochips: A platform for surface glyco-engineering. Angew. Chem.-Int. Edit. 46, 2398–2402 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Thomas B. et al. Application of Biocatalysis to on-DNA Carbohydrate Library Synthesis. Chembiochem 18, 858–863 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Novoa A, Machida T, Barluenga S, Imberty A. & Winssinger N. PNA-encoded synthesis (PES) of a 10 000-member hetero-glycoconjugate library and microarray analysis of diverse lectins. ChemBioChem 15, 2058–2065 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Horiya S, Bailey JK, Temme JS, Schippe YVG & Krauss IJ Directed Evolution of Multivalent Glycopeptides Tightly Recognized by HIV Antibody 2G12. Journal of the American Chemical Society 136, 5407–5415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondengaden SM et al. DNA Encoded Glycan Libraries as a next-generation tool for the study of glycan-protein interactions. bioRxiv, doi: 10.1101/2020.1103.1130.017012 (2020). [DOI] [Google Scholar]
- 36.Macauley MS et al. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J. Clin. Invest. 123, 3074–3083 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen WC et al. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood 115, 4778–4786 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaltgrad E. et al. On-virus construction of polyvalent glycan ligands for cell-surface receptors. J. Am. Chem. Soc. 130, 4578–4579 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pochechueva T. et al. Comparison of printed glycan array, suspension array and ELISA in the detection of human antiglycan antibodies. Glycoconj. J. 28, 507–517 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pochechueva T. et al. Multiplex suspension array for human anti-carbohydrate antibody profiling. Analyst 136, 560–569 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Purohit S. et al. Multiplex glycan bead array for high throughput and high content analyses of glycan binding proteins. Nat. Commun. 9, 258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang R. et al. Parallel synthesis and screening of a solid phase carbohydrate library. Science 274, 1520–1522 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Tjhung KF et al. Silent Encoding of Chemical Post-Translational Modifications in Phage-Displayed Libraries. J. Am. Chem. Soc. 138, 32–35 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Celik E, Fisher AC, Guarino C, Mansell TJ & DeLisa MP A filamentous phage display system for N-linked glycoproteins. Protein Science 19, 2006–2013 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durr C, Nothaft H, Lizak C, Glockshuber R. & Aebi M. The Escherichia coli glycophage display system. Glycobiology 20, 1366–1372 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Celik E. et al. Glycoarrays with engineered phages displaying structurally diverse oligosaccharides enable high-throughput detection of glycan-protein interactions. Biotechnol. J. 10, 199–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pasqualini R. & Ruoslahti E. Organ targeting In vivo using phage display peptide libraries. Nature 380, 364–366 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Kolonin MG, Saha PK, Chan L, Pasqualini R. & Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nature Medicine 10, 625–632 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Krag DN et al. Selection of Tumor-binding Ligands in Cancer Patients with Phage Display Libraries. Cancer Research 66, 7724–7733 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Arap W. et al. Steps toward mapping the human vasculature by phage display. Nat. Med. 8, 121–127 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Scott JK & Smith GP Searching for peptide ligands with an epitope library. Science 249, 386–390 (1990). [DOI] [PubMed] [Google Scholar]
- 52. http://www.functionalglycomics.org.
- 53.Jewett JC & Bertozzi CR Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 39, 1272–1279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crimmins DL, Mische SM & Denslow ND Chemical Cleavage of Proteins in Solution. Current Protocols in Protein Science 41, 11.14.11–11.14.11 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Shaner NC et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaner NC et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Robinson MD & Smyth GK Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23, 2881–2887 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Robinson MD & Smyth GK Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Robinson MD & Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benjamini Y. & Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. B 57, 289–300 (1995). [Google Scholar]
- 61.Wang LL et al. Cross-platform comparison of glycan microarray formats. Glycobiology 24, 507–517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tateno H, Nakamura-Tsuruta S. & Hirabayashi J. Frontal affinity chromatography: sugar-protein interactions. Nat. Protoc. 2, 2529–2537 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Hirabayashi J, Tateno H, Shikanai T, Aoki-Kinoshita KF & Narimatsu H. The Lectin Frontier Database (LfDB), and Data Generation Based on Frontal Affinity Chromatography. Molecules 20, 951–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heggelund JE et al. High-Resolution Crystal Structures Elucidate the Molecular Basis of Cholera Blood Group Dependence. PLoS Pathog. 12, e1005567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feinberg H, Mitchell DA, Drickamer K. & Weis WI Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 294, 2163–2166 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Ng S. et al. Genetically-encoded fragment-based discovery of glycopeptide ligands for DC-SIGN. Bioorganic & Medicinal Chemistry 26, 5368–5377 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Bednar KJ et al. Human CD22 Inhibits Murine B Cell Receptor Activation in a Human CD22 Transgenic Mouse Model . J Immunol 199, 3116–3128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Macauley MS et al. Unmasking of CD22 Co-receptor on Germinal Center B-cells Occurs by Alternative Mechanisms in Mouse and Man. Journal of Biological Chemistry 290, 30066–30077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mammen M, Choi SK & Whitesides GM Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 37, 2755–2794 (1998). [DOI] [PubMed] [Google Scholar]
- 70.Narimatsu Y. et al. An Atlas of Human Glycosylation Pathways Enables Display of the Human Glycome by Gene Engineered Cells. Mol. Cell 75, 394–407 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tytgat HLP et al. Cytoplasmic glycoengineering enables biosynthesis of nanoscale glycoprotein assemblies. Nat. Commun. 10, 5403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lipinski T, Kitov PI, Szpacenko A, Paszkiewicz E. & Bundle DR Synthesis and immunogenicity of a glycopolymer conjugate. Bioconjug. Chem. 22, 274–281 (2011). [DOI] [PubMed] [Google Scholar]
- 73.Guo Y. et al. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat. Struct. Mol. Biol. 11, 591–598 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Bundle DR et al. Oligosaccharides and Peptide Displayed on an Amphiphilic Polymer Enable Solid Phase Assay of Hapten Specific Antibodies. Bioconjug. Chem. 25, 685–697 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Bundle DR, Gidney MA, Kassam N. & Rahman AF Hybridomas specific for carbohydrates; synthetic human blood group antigens for the production, selection, and characterization of monoclonal typing reagents. J. Immunol. 129, 678–672 (1982). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw deep-sequencing data is publically available on http://ligacloud.ca/ with data-specific URL listed in Table S3. DNA sequences of the three LiGA phage constructs have been deposited to GeneBank (#MN865131, MN865132, MN872303). MatLab, Python and R scripts have been deposited to https://github.com/derdalab/liga