

Abstract
The cell cycle is a highly regulated and evolutionary conserved process that results in the duplication of cell content and the equal distribution of the duplicated chromosomes into a pair of daughter cells. Histones are fundamental structural components of chromatin in eukaryotic cells, and their post-translational modifications (PTMs) benchmark DNA readout and chromosome condensation. Aberrant regulation of cell cycle associated with dysregulation of histone PTMs is the cause of critical diseases such as cancer. Monitoring changes of histone PTMs could pave the way to understanding the molecular mechanisms associated with epigenetic regulation of cell proliferation. Previously, our lab established a novel middle-down workflow using porous graphitic carbon (PGC) as a stationary phase to analyze histone PTMs which utilizes the same reversed phase chromatography for gradient separation as canonical proteomics coupled with on-line MS. Here, we applied this novel workflow for high-throughput analysis of histone modifications of H3.1 and H3.2 during cell cycle. Collectively, we identified 1133 uniquely modified canonical histone H3 N-terminal tails. Consistent with previous findings, histone H3 phosphorylation increased significantly during M phase. Histone H3 variant-specific and cell cycle-depend expressions of PTMs were observed, underlining the need to not combine H3.1 and H3.2 together as H3. We confirmed previously known H3 PTM crosstalk (e.g. K9me-S10ph) and revealed new information in this area as well. These findings imply that the combinatorial PTMs play a role in cell cycle control and they may serve as markers for proliferation.
Keywords: Histone modifications, H3 variants, Cell cycle, Middle-down, Quantitative proteomics
Introduction
The cell cycle is a highly regulated and evolutionary conserved process. In eukaryotic cells, the cell cycle is a 4-stage process consisting of gap 1 (G1), synthesis (S), gap 2 (G2), and mitosis (M) where G1, S, and G2 are known collectively as the interphase [1, 2]. During interphase, the cell grows (G1 and G2), and genetic material is duplicated (S phase). During M phase, the replicated chromosomes, organelles, and cytoplasm separate into two new cells. Regulation of the cell cycle involves processes that are crucial to the survival of a cell, including the detection and repair of DNA damage as well as the prevention of uncontrolled cell division [3]. Aberrant regulation of cell cycle is the root cause of many diseases such as cancer [1, 4].
Histones are fundamental structural components of chromatin in eukaryotic cells [5] and are subject to multiple post-translational modifications (PTMs), including acetylation (ac), methylation (me) and phosphorylation (ph) [6, 7]. These histone PTMs benchmark DNA readout and play initial roles in epigenetic regulation [8, 9]. Emerging evidence has suggested that individual histone PTMs rarely function independently; combinations of histone PTMs appear to correlate better with a particular chromatin structure or function [8, 10]. Pre-existed modifications can promote or block the deposition of another modification, which is known as PTM crosstalk [10–12]. Distinct patterns of histone PTMs distinguish chromatin elements and recruit the proper regulator proteins to those regions, constituting the ‘histone code’ regulation mechanisms [12]. Their dysregulation has been noted in cancer and other diseases, and are associated with progression, aggressiveness, and metastasis [13–15]. Thus, a deep understanding of histone PTMs is needed to comprehend both normal cellular function and disease. Due to its high-throughput, accuracy, and flexibility, mass spectrometry (MS)-based proteomics strategies have emerged as powerful tools in the histone modification field, allowing the comprehensive and unbiased analysis of histone PTMs [16–18].
The most common workflow to characterize and quantify histone PTMs is bottom-up proteomics with chemical derivatization [19, 20], where histones are digested into short peptides (5–20 AA). This approach is highly efficient and well-utilized, providing the most reliable and accurate quantification of histone PTMs with good reproducibility. However, the short tryptic peptides are not suitable for the study of marks that co-occur on the same histone (i.e. the combinatorial PTMs). Other workflows, namely middle-down and top-down, have been implemented to analyze the combinatorial modifications [21–23], especially the crosstalk between long-distance modified sites such as H3K9 and K27 [24]. In particular, middle-down proteomics has proved to be high-throughput and feasible, reaching the similar level of reliability as bottom-up [25–27]. However, despite the achievements in accuracy and sensitivity, the middle-down strategy has not been widely applied for biological insights [28], partly because of the high complexity of this method, e.g. special chromatography with a different buffer system compared to reversed-phase liquid chromatography (RPLC) [29].
Here, we applied a high-throughput middle-down workflow using porous graphitic carbon (PGC) as a stationary phase to analyze histone PTMs during cell cycle [26]. This workflow used the same RPLC buffer setup which is convenient for proteomics laboratories that have C18 columns coupled with MS. Asynchronized HeLa S3 cells and synchronized cells in G1, S and M phase were harvested separately. Histone H3.1 and H3.2 were isolated using off-line C18 separation. A detailed analysis of H3.1 and H3.2 PTMs were performed using high resolution MS/MS with ETD fragmentation. By using this workflow, we identified 1133 histone H3 codes. In consistence with previous findings, histone H3 phosphorylation increased significantly during M phase. Different expression patterns of PTMs on H3.2 compared to H3.1 were observed during cell cycle, underlining the need to not combine H3.1 and H3.2 together as a single H3 sample. Our analyses validated known H3 PTM crosstalks (e.g. K9me3 and S10ph) and discovered novel ones (e.g. S10ph and T11ph) as well. Our data proved that the interdependencies of histone modifications could distinguish minor differences between cell cycle states rather than any single or binary histone marks.
Experimental
Chemicals and Reagents
All reagents were acquired from MilliporeSigma, unless otherwise noted. Solvents, including HPLC grade and MS grade water (H2O), and acetonitrile (ACN) were purchased from Thermo Fisher Scientific.
Cell culture and synchronization
HeLa S3 cells were cultured and synchronized as described previously [18, 30]. In brief, HeLa cells were cultured in 15 cm plate in Dulbecco’s Modified Eagle Medium (DMEM, CORNING) with 10% Gibco newborn calf serum (Thermo Fisher Scientific), 1*GlutaMAX ™ Supplement (Thermo Fisher Scientific) and 1*penicillin-streptomycin (CORNING).
The cell synchronization procedure is illustrated in Figure 1A. HeLa cells were synchronized in S phase by double thymidine block starting with 60% confluency: 2.5 mM thymidine (Acros Organics) for 19 hours, release for 9 hours, followed by a second thymidine treatment for 15 hours. For thymidine nocodazole block, cells were treated with thymidine for 23 hours and subsequently released for 3 hours. 100 ng/ml Nocodazole (EMD Millipore) was added to the media and cells were harvested in M phase after 12 hours treatment. To synchronize in G1 phase, HeLa cells were released into fresh media for 6 hours after thymidine nocodazole block. Every treatment was performed in triplicate. Asynchronized (asy) HeLa cells and cells synchronized in G1/S/M phase were analyzed by flow cytometry or mass spectrometry.
Figure 1.

Middle-down proteomics workflow for histone tail characterization. (A) Model system adopted in the current work. HeLa S3 cells were synchronized at S phase by double thymidine block, and M or G1 phase by thymidine nocodazole block, respectively. Asynchronized cells were also harvested. (B) MS-based proteomics was performed using the middle-down strategy. Histone H3.1 and H3.2 were purified by C18 separation, then cleaved by Glu-C. H3 N-terminal tails (AA 1–50) were separated through PGC column before high-resolution MS-MS/MS analysis.
Histone Extraction
Histones were acid-extracted from HeLa cells as previously described [31]. In brief, cell pellets were resuspended in nuclear isolation buffer (NIB, 15mM Tris, pH7.5, 60 mM KCL, 15 mM NaCl, 5 mM MgCl2,1 mM CaCl2, 250 mM sucrose, 1 mM DTT) with 1 * protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific) and 0.2% NP-40, incubated for 10 min on ice, and centrifuged at 1000g to collect the nuclei pellet. Pellets were washed with NIB two times to completely remove NP-40. Nuclei were resuspended in 0.2 M H2SO4 and rotate at 4 °C for 2 hours. Samples were centrifuged at 14,000 rpm for 10 min and the supernatant was collected as the histone containing faction. Histones were precipitated with 25% trichloroacetic acid (TCA) overnight and washed with cold acetone. Protein concentration was measured using the Bradford assay (Bio-Rad).
Immunoprecipitation and immunoblotting
For immunoprecipitation (IP), protein A agarose beads (Thermo Fisher Scientific) were pre-incubated with anit-K9me2 antibody (Abcam, AB1220) in blocking solution (1% BSA in PBS) for 6 hours at 4°C. After three washes with blocking solution, acid-extracted histones were incubated with the conjugated beads and rotated overnight at 4°C (IP buffer: 10 mM Tris, pH7.5, 100 mM NaCl, 10 mM KCl, 1 mM MgCl2, 0.1% NP-40). The beads were extensively washed with IP buffer containing 1% NP-40, then eluted with SDS-PAGE loading buffer.
Immunoblotting (also named western blotting, WB) was carried out according to standard protocol. In brief, elution samples were separated through NuPAGE 12% Bis-Tris gel (Thermo Fisher Scientific) and electrotransferred to 0.2 μm nitrocellulose membrane (Bio-Rad). After blocking in TBST with 5% BSA, the membrane was incubated with anti-H3K9me2, anti-H3K27me3 (MilliporeSigma, 07–449) or anti-H3 (Abcam, AB1791) overnight at 4°C. After rinsing with TBST, membrane was incubated with HRP-conjugated second antibody for 1 hour at room temperature. Finally, immunoreactive bands were revealed using ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences) and detected by Amersham Imager 600 (GE Healthcare Life Sciences).
Histone H3 isolation and Glu-C digestion
Acid-extracted histones (100 μg) were resuspended in 0.1% trifluoroacetic acid (TFA) and loaded on a 260 * 4.6 mm Vydac 5-μm C18 column using an off-line Beckman Coulter (System Gold) HPLC system (Buffer A: 0.2% TFA, 5% ACN; Buffer B: 0.2% TFA, 5% H2O). HPLC-UV separation was performed using a gradient of 35–47% in 15 min, followed by 47%−60% B in 50 min at flow rate of 0.8 ml/min with UV detection at 220 nm. Histone H3.2 and H3.3 co-eluted and were pooled into a single fraction. H3.1 separated well from H3.2 and H3.3 and were collected separately (Figure S2).
Purified histone H3 were dried in a Savant SpeedVac and resuspended in 5 mM ammonium acetate (pH 4.0). Samples were digested overnight at room temperature by adding 1 ug of Glu-C to generate 50 residue N-terminal tails (AA 1–50). Resulting peptides were desalted using in-house C18 stage tips and subjected to nLC-MS/MS.
nLC-MS/MS analysis
Peptides were separated using an Easy-nLC 1000 (Thermo Fisher Scientific) equipped with an in-house packed PGC column. The gradient was set to 19–21% B over 60 min at 400 nl/min (Buffer A: 0.1% formic acid; Buffer B: 0.1% formic acid / 80% ACN). MS acquisition was performed using an Orbitrap Fusion (Thermo Fisher Scientific) in data-dependent mode as previously described [29]. A full mass spectrum was acquired at a narrow scan range (665–705 m/z) with a resolution of 120,000. Only charge state 8+ was selected for MS/MS fragmentation. The top10 most intense ions were isolated with isolation window of 2 m/z, and fragmentated using ETD with a reaction time of 20 ms and a reagent target of 1e5. The MS/MS resolution was set to 30,000 in the Orbitrap. The AGC target was set to 1e5 and maximum injection time of 400 ms. Three microscans were averaged for each MS/MS spectrum.
Data processing
Database search was performed in Proteome Discoverer 2.2 using the Mascot (v2.5, Matrix Science) search engine. Spectra were deconvoluted with Xtract and search against human histones H3 (UniProt). Enzyme was set to Glu-C with 0 missed cleavages allowed. Variable modifications were acetylation (K), mono- and di-methylation (KR), tri-methylation (K) and phosphorylation (STY). Mass tolerance was set 2.1 Da for precursor and 0.01 for fragment ions.
Mascot output files were exported as CSV files and processed with our in-house software ProteoformQuant for confident identification and quantification [32]. Peptides without sufficient fragment ions to unambiguously localize PTM were automatically discarded by the software. Co-eluting peptides were assigned abundances based on the ratio of unique fragments. The relative abundance of a given modified peptide was calculated by dividing the total ion intensity of this peptide by the sum of all peptides sharing the same sequence.
Results and Discussion
Workflow for analyzing histone H3 PTMs during cell cycle
We applied a middle-down proteomics strategy for large-scale analysis of histone H3 PTMs during cell cycle, including asynchronized HeLa S3 cells, and cells synchronized in G1, S and M phase (Figure 1). The cell synchronization procedure is illustrated in Figure 1A, and flow cytometry results show that we were able to enrich for cells in specific cell cycle phases (Figure S1). Histone H3.1 differs from H3.2 in only one amino acid (AA 96: cysteine/serine, respectively) [33]. Thus, most studies tend to group these two variants as one H3 [24]. However, previous bottom-up MS results do indicate that histone H3.1 and H3.2 differ in PTM patterns: H3.2 is enriched in PTMs associated with gene silencing (e.g. H3K27me2, H3K27me3), and H3.1 is enriched in both active (e.g. H3K14ac) and repressive PTMs (e.g. H3K9me2), suggesting H3 variant-specific biological functions [20, 30]. Here, histone variants were fractionated using off-line C18 separation (Figure S2). Histone H3.1 separated well from the other histone variants and were collected separately. H3.2 partially co-eluted with H3.3 and were pooled into a single fraction. Purified histones were cleaved at E50 by Glu-C, resulting in longer H3 N-terminal tails where most of the modifications reside.
Middle-down proteomics has been commonly adopted using weak cation exchange-hydrophilic interaction chromatography (WCX-HILIC) to provide the most sensitive characterization of histone combinatorial PTMs [16, 27, 28]. However, WCX-HILIC uses a different buffer system compared to the commonly used RPLC in most proteomics laboratories and usually requires tedious work in buffer preparation and gradient optimization [29]. Thus, despite its excellent performance in peptide separation, it is not widely accessible to most proteomics laboratories. Several attempts have been made to make RPLC more suitable for middle-down analysis, including chemical derivatization [34], alternative protease [35], and the use of ion mobility [36, 37]. Our group present a new workflow utilizing PGC as a novel stationary phase for histone analysis using the same RPLC C18 buffer setup [26]. The new workflow overcomes the limitations of WCX-HILIC and achieved a good correlation of 0.85 between PGC-RPLC and WCX-HILIC methods, indicating that using PGC for middle-down analysis does not compromise the quality of the data [26].
We hypothesized that our recently reported middle-down proteomics workflow using PGC could reveal changes of histone PTMs during cell cycle thereby providing insights into epigenetic regulatory mechanisms. False positive modification localization is a common problem encountered when analyzing heavily modified histone peptides and no backbone cleavage occurs between two possible modification sites to unambiguously assign the accurate location. ProteoformQuant addresses this issue by ensuring at least one site determining fragmentation ion before and after the assigned PTM site [32]. This tool includes filtering of ambiguously assigned PTMs and quantification. In case of mixed MS/MS spectra generated as a result of co-fragmentation co-eluted isobaric peptides, the software calculates the fragment ion relative ration of such peptides and divided the MS2-TIC for this ratio. Representative ETD product ion spectra for the deconvoluted spectrum of K9me2S10phK23acK27ac (4 modification sites) and K9me2S10phT11ph (3 modification sites) were found to show sufficient peptide coverage with assigned fragment ions that bracket the modification site (Figure S3).
Middle-down can be a little overwhelming with more layers of information compared to bottom-up analyses. Thus, the output identification and quantification results of histone codes were further sorted and transformed into single mark, binary marks, and interplay scores for better decoding and mining of the dataset [28, 29]. In brief, the histone code represents various distinct combinations of PTMs (e.g. acetylation, methylation, and phosphorylation) at distinct amino acid residues. The relative abundance of a given modified peptide (histone code) was calculated by dividing the total ion intensity of this peptide by the sum of all peptides sharing the same sequence. Histone single mark or binary mark is the peptide carrying indicated one or two PTMs respectively; other residues can either be modified or unmodified. The relative abundances of individual PTMs were calculated by summing the relative abundance of all peptides that contained the particular PTM. The interplay score was calculated using the relative abundance of a given binary mark (Fab) and single marks (Fa and Fb) using the equation in Figure 1B [11]. A negative interplay score means the observed abundance of the binary modification (Fab) is smaller than the predicated abundance (Fa*Fb), indicating that the two PTMs are mutually exclusive. Modifications that likely coexist on the same N-terminal tail yield a positive interplay score indicating that one PTM triggers the addition of another PTM at a different residue either directly or indirectly.
Characterization of histone codes on H3 N-terminal tails
In total, we identified 1133 histone H3 codes from 24 samples (4 cell cycle stages, 2 histone H3 variants, 3 biological replicates, Table S1A). A maximum detection of 298 and a minimum of 102 various histone codes were achieved in a single MS analysis (Figure S4A), providing sufficient results for further analysis of PTM crosstalk [29]. Notably, an average of 20% of histone code types were reproducibly quantified in all three replicates and 61% were only detected in one replicate (Figure S4B). However, quantitative analysis showed that the 20% consistently detected histone codes accounted for 63–93% of all combinatorial PTMs, while the 61% replicate-unique histone codes accounted for <20% of the H3 N-terminal tails by abundance (Figure S4C). One of the possible reasons is that the low abundant histone codes can co-eluted with other high abundant forms and therefore can not be selected for MS2. Even when the parent ion is selected for MS2, it may not have backbone cleavage occurring between two possible modification sites to unambiguously assign the accurate location. Peptides without sufficient fragment ions to unambiguously localize PTM were automatically discarded by ProteoformQuant, leading to poor identification of these ultra-low abundant histone codes. Pearson correlation analysis showed relatively high reproducibility between replicates (Table S2), indicating that the missing identification of these ultra-low abundant histone codes did not compromise the quality and reliability of the data. These data illustrate the great complexity of combinatorial PTM patterns (also referred to histone codes) present on the H3 N-terminal tails, indicating that the vast majority of chromatin is decorated by a complex combinatorial PTMs network.
The N-terminal tail of H3 (AA 1–50) can be heavily modified, reaching a maximum of seven modified residues (Figure 2, Table S1A), e.g. K9me2S10phK14me1K18me1K23me1K27me1K36me1. The majority of histone codes contain 2–3 modification sites (by counts, Figure 2a) and most of the histone H3 N-terminal tails carry 2–4 modification sites (by relative abundance, Figure 2b), highlighting the fact that analyzing only single marks in genomics experiments or targeted experiments (e.g. immunoblotting) could not provide sufficient information to unravel histone code regulation mechanisms in depth. Table 1 exhibits the 15 most abundant histone H3 codes that were detected. Among those, K9me2K27me3 is the most abundant combinatorial PTM in all conditions (relative abundance ranging from 7% to 32%). This observation would not be uncovered with bottom-up MS because the analysis of short peptides does not allow characterization of long-distance PTMs.
Figure 2.

Characterization of histone H3 codes. Sum of the (A) counts and (B) relative abundance of H3 N-tails containing indicated number of modified amino acid residues. Average ± standard deviation of three replicates.
Table 1.
Top 15 most abundant histone codes quantified in each condition (average ± standard deviation of three replicates). The figure (the upper-right corner) showed the changes of relative abundance for the most abundant histone code K9me2K27me3 in distinct cell cycle phases.
| Histone code | H3.1_Asy | Histone code | H3.2_Asy |
|
|
|
| |||||
| K9me2K27me3 | 7% ± 1.4% | K9me2K27me3 | 11% ± 3.0% | ||
| K9me2K27me1K36me1 | 7% ± 1.5% | K9me1K14me1K27me2 | 11% ± 2.6% | ||
| K9me2R42me2 | 5% ± 5.4% | K9me2K27me1K36me1 | 6% ± 1.2% | ||
| K9me1K14me1K27me2 | 4% ± 1.0% | R17me2K27me2 | 4% ± 0.7% | ||
| K9me2K23acK27me1K36me1 | 4% ± 3.4% | K9me2K14acK27me3 | 3% ± 1.0% | ||
| R17me2K27me2 | 3% ± 1.0% | K9me2K23acK27me3 | 3% ± 1.2% | ||
| K9me1K14me1K23acK27me2 | 3% ± 0.8% | K9me1K14me1K23acK27me2 | 3% ± 0.3% | ||
| K9me2K14acK27me3 | 2% ± 1.2% | K9me1K14me1K27me2K36me2 | 3% ± 1.0% | ||
| K9me2K23acK27me3 | 2% ± 1.9% | K9me1K14me1K18acK27me2 | 2% ± 1.0% | ||
| K9me1K14me1K18acK27me2 | 2% ± 0.8% | K9me2K18acK27me3 | 2% ± 0.5% | ||
| K9me1R17me1K27me2 | 2% ± 1.2% | K14me2K27me3 | 2% ± 0.4% | ||
| K9me2K18acK27me3 | 2% ± 1.5% | K9me1R17me1K27me2 | 2% ± 0.6% | ||
| K9me2K18acK23acK27me1K36me1 | 2% ± 0.8% | K27me3 | 2% ± 1.0% | ||
| K14me2K27me3 | 2% ± 0.6% | K9me2K23acK27me1K36me1 | 1% ± 0.9% | ||
| K9me2K23acK27ac | 2% ± 0.6% | K9me2K27me3K36me1 | 1% ± 0.1% | ||
|
| |||||
| Histone code | H3.1_G1 | Histone code | H3.1_S | Histone code | H13.1_M |
|
| |||||
| K9me2K27me3 | 29% ± 5.1% | K9me2K27me3 | 32% ± 5.4% | K9me2K27me3 | 10% ± 5.5% |
| K9me2K23acK27me3 | 6% ± 1.2% | K9me2K23acK27me3 | 7% ± 0.7% | K9me2S10phK23me1K27me1 | 6% ± 1.7% |
| K9me2K23acK27me1K36me1 | 4% ± 2.3% | K9me2K27me3K36me1 | 5% ± 1.2% | K9me2S10phK27me3 | 6% ± 1.3% |
| K9me2K27me1K36me1 | 4% ± 0.5% | K9me2K23acK27me1K36me1 | 4% ± 0.9% | K9me2S10phK27me1K36me1 | 3% ± 1.2% |
| K9me2K27me3K36me1 | 3% ± 1.9% | K9me2K27me1K36me1 | 3% ± 1.0% | K9me1K14me1K27me2 | 3% ± 2.2% |
| K9me2K36me3 | 3% ± 1.4% | K9me1K14me1K27me2K36me2 | 3% ± 1.7% | K9me2S10phK23acK27ac | 3% ± 1.8% |
| K9me1K14me1K27me2 | 3% ± 1.1% | K9me1K14me1K27me2 | 3% ± 0.9% | K9me1K14me1K23acK27me2 | 3% ± 2.0% |
| K9me2K27me1K36me3 | 3% ± 1.2% | K9me1K14me1K27me3 | 2% ± 0.9% | K9me2K23acK27me1K36me1 | 3% ± 1.5% |
| K9me1K14me1K23acK27me2 | 3% ± 1.4% | K9me2K27me1K36me3 | 2% ± 1.5% | K9me2K27me3K36me1 | 2% ± 1.0% |
| K9me2K18acK27me3 | 2% ± 0.6% | K9me2K14acK18acK27me3 | 2% ± 0.3% | K9me3S10phK23me2K27me2 | 2% ± 1.2% |
| K27me3 | 2% ± 1.3% | K9me2K36me3 | 2% ± 0.7% | K9me2K36me3 | 2% ± 1.7% |
| K9me1K14me1K27me2K36me2 | 2% ± 0.7% | K9me2K18acK27me3 | 2% ± 0.7% | K9me2K27ac | 2% ± 0.6% |
| K9me3K27me2K36me2 | 1% ± 0.5% | K27me3 | 2% ± 0.9% | K9me3S10phK27me2 | 2% ± 1.1% |
| K9me1K14me1K27me3 | 1% ± 0.6% | K9me2K18acK23acK27me1K36me1 | 2% ± 0.5% | K9me2S10phR26me2 | 2% ± 0.9% |
| K9me2K27me1R40me2 | 1% ± 1.0% | K9me2K18acK23acK27me3 | 2% ± 0.4% | K9me2K18acK27me1K36me1 | 2% ± 0.8% |
|
| |||||
| Histone code | H3.2_G1 | Histone code | H3.2_S | Histone code | H3.2_M |
|
| |||||
| K9me2K27me3 | 29% ± 3.7% | K9me2K27me3 | 30% ± 3.2% | K9me2K27me3 | 14% ± 1.4% |
| K9me1K14me1K27me2 | 6% ± 3.6% | K9me2K23acK27me3 | 5% ± 1.1% | K9me2K27me3K36me1 | 5% ± 1.2% |
| K9me2K23acK27me3 | 5% ± 0.2% | K9me2K27me3K36me1 | 5% ± 0.5% | K9me2K36me3 | 5% ± 2.5% |
| K9me2K27me3K36me1 | 5% ± 0.9% | K9me1K14me1K27me2 | 4% ± 0.8% | K9me1K14me1K27me2 | 4% ± 2.9% |
| K9me2K23acK27me1K36me1 | 5% ± 1.8% | K9me2K23acK27me1K36me1 | 3% ± 0.4% | K9me1K14me1K27me2K36me2 | 4% ± 0.9% |
| K9me2K27me1K36me1 | 5% ± 1.2% | K9me2K27me1K36me1 | 3% ± 1.7% | K9me2K27me1K36me1 | 3% ± 2.3% |
| K9me2K36me3 | 3% ± 0.1% | K9me3K27me2K36me2 | 3% ± 0.8% | K9me2S10phK23me1K27me1 | 3% ± 2.3% |
| K9me1K14me1K27me2K36me2 | 3% ± 1.7% | K9me2K36me3 | 3% ± 0.6% | K9me2K27me1K36me3 | 3% ± 0.7% |
| K9me1K14me1K27me3 | 2% ± 1.1% | K9me1K14me1K27me3 | 2% ± 0.5% | K9me2S10phR26me2 | 2% ± 1.3% |
| K9me2K27me1K36me3 | 2% ± 0.4% | K9me2K27me1K36me3 | 2% ± 0.8% | K9me2S10phK27me3 | 2% ± 1.3% |
| K9me2K18acK27me3 | 2% ± 0.7% | K27me3 | 2% ± 0.4% | K27me3 | 2% ± 1.9% |
| K27me3 | 2% ± 0.8% | K9me1K14me1K23acK27me2 | 2% ± 0.4% | K9me2S10phK27me1K36me1 | 2% ± 2.6% |
| K9me1K14me1K23acK27me2 | 2% ± 0.4% | K9me2K18acK27me3 | 2% ± 0.4% | K9me1K14me1K23acK27me2 | 2% ± 0.9% |
| K9me2K23acK27ac | 1% ± 0.2% | K9me1K14me1K27me2K36me2 | 2% ± 0.4% | K9me2K23acK27me1K36me1 | 2% ± 1.1% |
| K9me3K27me2K36me2 | 1% ± 0.3% | K14me2K27me3 | 1% ± 1.2% | K9me3S10phK27me2 | 2% ± 0.6% |
Dynamic changes of single histone marks
To further deconvolute the data for quantitative analysis, the dataset was transformed into relative abundance for single marks. In total, relative abundances for 48 single marks were quantified (Table S1B). The abundance levels of the majority of histone H3 modifications were stable throughout the cell cycle (Figure 3) [38]. Phosphorylation of H3 is a well-known marker of cells in M phase [39, 40]. As expected, phosphorylation (H3S10ph, T11ph, and T22ph) increased exclusively in M phase. In addition to phosphorylation, we also observed that H3K9me3, K23me1 and H3.1K27ac were elevated in M phase. H3K9me3 is an established hallmark of constitutive heterochromatic regions that are likely maintained across division (M phase) [41]. Oppositely, H3K9me2 and K27me3 were more abundant in G1/S phases. The abundance of H3K9me2 and K27me3 were further validated by Western blot where a good correlation was obtained with the MS quantification results (Figure S5), proving the reliability of our dataset.
Figure 3.

Heatmap of hierarchical clustering for the relative abundance for histone H3 single marks
Distributions of PTMs per amino acid residue were reported in Figure 4 where it helps to find the different predominant histone PTMs. High abundance of acetylation (K14, K18, K23), mono-methylation (K9, K14, K27, K36), di-methylation (K9, K27, K36), and tri-methylation (K27) were the predominant PTMs in all cell cycle phases while phosphorylation (S10, T11, T22) were exclusively found in M phase. Previous results indicate that H3.2 contains mostly silencing modifications, and H3.1 is enriched in both active and repressive PTMs [42]. Therefore, we investigated the changes of single mark relative abundance between H3.1 and H3.2 in asynchronized and synchronized cells (Figure S6). Cell cycle phase-dependent modification expression differences were observed between H3.1 and H3.2. For example, the abundance of K27ac on H3.1 was significantly higher in M phase (H3.1/H3.2 fold change: 6.86, p-value: 0.001), and lower in G1 phase (H3.2/H3.1 fold change: 3.03, p-value: 0.009). Histone H3K27ac is an active enhancer mark that is highly associated with activation of transcription [6, 10]. The difference in expression patterns between H3.1 and H3.2 across cell cycle indicate H3 variant-specific biological functions in distinct processes, underlining the need to not combine H3.1 and H3.2 together as H3 in study of specific mammalian models.
Figure 4.

Comparison of the distribution of PTMs per amino acid residue of (A) H3.1 and (B) H3.2 between the cell cycle phases. Average ± standard deviation of three replicates. Color code represents different PTMs: mono-methylation (me, green), di-methylation (me2, blue), tri-methylation (me3, red), acetylation (ac, yellow), and phosphorylation (ph, pink).
Co-existing of PTMs on the same N-terminal tail
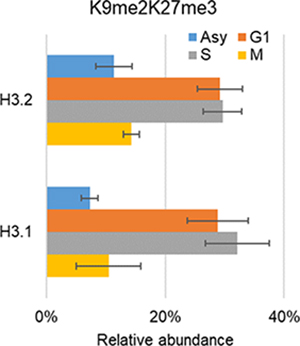
In addition to the relative abundance of single marks, we are more interested in the co-occurrence frequencies of PTM pairs residing on the same N-terminal tail as they provide more divers aspects of chromatin biology [8]. Thus, the co-frequencies (also referred to relative abundances) were determined by summing up the relative abundance of histone codes that have a certain combination of two PTMs, resulting in 589 binary marks (Table S1C). Histone PTMs that do not co-occur by chance. The more abundant a modification, the more likely it is to co-occur with other PTMs. Histone H3K9me2 and K27me3 are the most intense PTMs concluded from quantitative analysis results of single marks (Figure 3 and Table S1B). Here, co-existing of K9me2K27me3 in G1/S is the most abundant mark among all detected binary marks (Table S1C). To confirm the co-existing, we performed an immunoprecipitation (IP) using anti-K9me2. In consistent with MS outputs, the WB results showed a less intense co-existing pattern for K9me2_K27me3 in G1 phase than S phase (Figure 5).
Figure 5.

(A) Relative abundance of single marks K9me2 and K27me3 and binary mark K9me2_K27me3. Average ± standard deviation of three replicates. (B) WB results of anti-K9me2 IP of cells synchronized in G1 and S phases.
Selected binary marks containing K9 methylation and the corresponding relative abundance were listed in Table 2. Histone H3K9 methylation is a crucial epigenetic mark that is corelated with gene silencing [41]. Previous studies have demonstrated that H3K9 methylation usually occurs in combination with its adjacent modified sites (e.g. S10ph, K14ac/me) [43]. Here, we quantified stable expression of not only short-distance binary marks (e.g. K9me1_K14me1, K9me2_K14ac) confirming the already reported histone PTMs, but also long-distance binary marks (e.g. K9me1_K23ac, K9me2_K36me3) where currently there is a lack of knowledge on their molecular functions in gene regulation. How they cooperate to regulate molecular mechanisms throughout the cell cycle remains open and require further research.
Table 2.
Relative abundance for selected binary marks containing K9 methylation (average ± standard deviation of three replicates). Binary marks listed here were quantified in all LC-MS/MS analysis.
| Binary mark | H3.1_Asy | H3.1_G1 | H3.1_S | H3.1_M | H3.2_Asy | H3.2_G1 | H3.2_S | H3.2_M |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| K9me1_K14me1 | 14% ± 4.8% | 12% ± 1.2% | 12% ± 5.3% | 13% ± 3.3% | 24% ± 2.0% | 16% ± 1.2% | 15% ± 0.5% | 15% ± 2.8% |
| K9me1_K18ac | 6% ± 1.2% | 2% ± 0.6% | 3% ± 0.3% | 1% ± 1.7% | 4% ± 1.5% | 1% ± 0.4% | 3% ± 0.4% | 2% ± 0.9% |
| K9me1_K23ac | 5% ± 0.3% | 5% ± 0.8% | 4% ± 1.7% | 5% ± 4.4% | 5% ± 0.9% | 3% ± 0.4% | 5% ± 0.9% | 2% ± 1.1% |
| K9me1_K27me2 | 15% ± 4.1% | 11% ± 1.7% | 11% ± 3.7% | 14% ± 3.5% | 23% ± 3.2% | 13% ± 3.0% | 12% ± 1.7% | 17% ± 3.4% |
| K9me1_K36me2 | 5% ± 2.6% | 5% ± 1.0% | 6% ± 2.3% | 2% ± 0.2% | 6% ± 0.5% | 5% ± 1.9% | 6% ± 0.3% | 7% ± 0.7% |
| K9me2_K14ac | 9% ± 1.9% | 3% ± 0.5% | 5% ± 1.1% | 1% ± 1.0% | 6% ± 0.9% | 2% ± 0.3% | 4% ± 0.3% | 4% ± 1.3% |
| K9me2_K18ac | 9% ± 2.4% | 7% ± 1.3% | 9% ± 1.1% | 3% ± 0.3% | 5% ± 1.4% | 5% ± 0.8% | 7% ± 0.2% | 1% ± 0.2% |
| K9me2_K23me1 | 3% ± 1.7% | 1% ± 0.7% | 1% ± 0.5% | 11% ± 1.2% | 2% ± 0.6% | 2% ± 0.8% | 2% ± 1.5% | 5% ± 2.4% |
| K9me2_K23ac | 14% ± 0.7% | 14% ± 4.9% | 17% ± 2.1% | 8% ± 3.9% | 7% ± 2.9% | 15% ± 1.0% | 15% ± 1.0% | 4% ± 1.1% |
| K9me2_K27me1 | 18% ± 1.6% | 16% ± 1.4% | 13% ± 0.9% | 20% ± 2.7% | 12% ± 3.5% | 15% ± 3.0% | 13% ± 1.8% | 16% ± 3.2% |
| K9me2_K27me3 | 19% ± 4.0% | 45% ± 2.6% | 52% ± 5.4% | 22% ± 5.7% | 24% ± 2.0% | 45% ± 4.1% | 48% ± 3.2% | 26% ± 3.7% |
| K9me2_K27ac | 6% ± 1.9% | 1% ± 0.6% | 2% ± 0.6% | 9% ± 2.3% | 4% ± 0.9% | 4% ± 0.5% | 2% ± 0.3% | 1% ± 0.3% |
| K9me2_K36me1 | 21% ± 2.3% | 17% ± 3.0% | 16% ± 3.3% | 16% ± 2.8% | 14% ± 4.6% | 19% ± 3.5% | 15% ± 2.5% | 15% ± 2.5% |
| K9me2_K36me3 | 4% ± 1.7% | 7% ± 2.1% | 6% ± 1.8% | 3% ± 1.1% | 3% ± 0.9% | 6% ± 0.9% | 7% ± 1.3% | 8% ± 2.4% |
Crosstalk between histone H3 PTMs
To reveal underlaying rules for the histone modification crosstalk, the histone code dataset was further converted into interplay scores (Table S1D). Representative interplay graphs show how H3S10ph is interconnected with other PTMs during M phase (Figure 6). The network provides an overview of the expression patterns of two PTMs, showing the relative abundance of the binary mark as well the interplay type and score of the indicated two PTMs. The color of PTM nodes represents the relative abundance of binary marks containing S10ph. Line thickness represents the absolute of average interplay scores of three replicates. Positive interplay (blue) indicates two PTMs likely coexist rather than being isolated from each other, while negative interplay (red) indicated mutual exclusion.
Figure 6.

Histone PTM crosstalk between S10ph and other H3 PTMs in M phase. Binary marks listed here were detected in all three replicates in both H3.1 and H3.2.
Crosstalk occurring between H3S10ph and adjacent modified sites has been well characterized in previous studies. These data suggest that binary mark of K9 methylation and S10 phosphorylation is required for mitosis (the so-called ‘methylation/phosphorylation switch’) [40, 43, 44]. Here, we observed weak interplays either positive or negative between K9me1/me2 and S10ph and a strong positive crosstalk between K9me3 and S10ph, which are consistent with previous findings [18]. Phosphorylation of H3S10 can enhance acetylation of H3K14 [39, 45], indicating a positive crosstalk of S10ph_K14ac. In our dataset, we detected a strong positive interplay between K14ac and S10ph in G1/S phase, and a negative interplay in M phase (Table S1D). Dual phosphorylation of S10ph_T11ph was reported in vitro where phosphorylation of S10 by kinase Ataurora1 is decreased by T11 phosphorylation [45]. Our data showed that dual-phosphorylation (S10ph_T11ph) can be found on the same H3 N-terminal tail in vivo and the likelihood of co-existence is very low giving its negative interplay scores.
In general, the expression pattern of S10ph with other PTMs are similar between H3.1 and H3.2 at both the co-frequencies and interplay type levels except for S10ph_K27ac. The relative abundances of binary mark S10ph_K27ac are 4.0% and 1.3% in H3.1 and H3.2, respectively, and interplay scores are negative 0.13, and positive 1.79. As we described earlier, K27ac is more abundant in H3.1 (relative abundance of single mark: 11.4%) than H3.2 (1.7%) in M phase and no significant changes were observed between the levels for S10ph in M phase (fold change H3.1/H3.2: 1.6, p-value: 0.065). It is interesting to detect a strong positive crosstalk on H3.2 and a relative weak negative crosstalk on H3.1 between S10ph and K27ac during M phase. The mechanism and distinct functions behind the histone variant-specific combinatorial PTMs remain largely unaddressed.
Correlation analysis of histone PTMs
Finally, we examined the correlation between distinct cell stages and different histone H3 variants by performing Pearson correlation coefficient (Figure S7) and Principal Component Analysis (PCA, Figure 7). As expected, a high correlation of a minimum of 0.9 were achieved between histone H3.1 and H3.2 (Figure S7), indicating that in general, function similarity of H3 variants exists. The PTMs during M phase were less correlated with other cell cycle phases and a correlation of 0.99 between G1 and S phase was obtained which are inconsistence with flow cytometry results (Figure S1). By PCA analysis, we observed the cluster groups as expected only when using the interplay scores (Figure 7C) where G1 phase was separated from S phase. This suggests that the relative abundances of single/binary mark do not provide sufficient confidence in distinguishing minor differences between G1 and S phase. In other words, the changes of combinatorial marks patters (interplay) seem to be a more complex, but also a more informative, to define cell cycle status. The interplays would make novel suggestions about how cells propagate different chromatin domains and epigenetic information across the cell cycle.
Figure 7.

Principal Component Analysis (PCA) of different phases by investigating (A) relative abundance for single marks, (B) relative abundance for binary marks, and (C) calculated interplay score.
Conclusions
Understanding the dynamics and stability of histone PTMs in different histone variants during the cell cycle is crucial in revealing their molecular mechanisms in gene regulation. Here, we present the first global analysis of histone H3 variants PTMs during the cell cycle using a high throughput middle-down proteomics. Histone PTMs of H3.1 and H3.2 from asynchronized and synchronized (G1, S, and M phase) HeLa S3 cells were analyzed by MS. We detected histone H3 variant-specific and cell division-dependent expressions of PTMs (e.g. K27ac), indicating H3 variant-specific biological functions in distinct processes. Enrichment of M phase-specific phosphorylation was quantified which is in consistent with previous reports [18, 40]. Phosphorylation of H3S10 appears to have both a positive and a negative connotation and only become meaningful in combination with other histone marks. In addition to the already reported crosstalk (e.g. K9me3_S10ph, S10ph_K14ac), we were able to reveal novel interplays (e.g. S10ph_T11ph), providing novel insights into distinct functions of different combinatorial PTMs. The interdependencies of histone modifications could define more accurately the cell cycle states rather than histone single mark or binary mark relative abundances. Further research is required to fully understand the mechanisms through which these combinatorial PTMs might exert an effect.
Collectively, we demonstrated the application of PGC-based chromatography to decipher how different PTMs are interconnected. Our study identified peculiar PTM patterns for each phase of the cell cycle. This knowledge will be used to understand the molecular mechanisms associated with epigenetic regulation of cell proliferation and investigate treatment for uncontrolled proliferating cells, e.g. cancer.
Supplementary Material
Acknowledgements
This work was supported by NIH grants CA196539, AG031862 and NS111997. Additionally, we acknowledge support from the St. Jude Children’s Research Hospital Chromatin Consortium.
Footnotes
Supporting Information: 7 supplemental figures (results of cell synchronization, histone purification, representative spectrum, and additional data analysis of histone H3 PTMs); 2 supplemental tables (results of quantification of histone H3 PTMs and Pearson correlation coefficient analysis).
References
- 1.Kastan MB, Bartek J: Cell-cycle checkpoints and cancer. Nature 432, 7015, 316–323 (2004) [DOI] [PubMed] [Google Scholar]
- 2.Osley MA: The regulation of histone synthesis in the cell cycle. Annu Rev Biochem 60, 1, 827–861 (1991) [DOI] [PubMed] [Google Scholar]
- 3.Ma Y, Kanakousaki K, Buttitta L: How the cell cycle impacts chromatin architecture and influences cell fate. Front Genet 6, 19 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Otto T, Sicinski P: Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 2, 93 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ: Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 6648, 251–260 (1997) [DOI] [PubMed] [Google Scholar]
- 6.Bowman GD, Poirier MG: Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev 115(6), 2274–2295 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Y, Garcia BA: Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol 7(9), a025064 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latham JA, Dent SYR: Cross-regulation of histone modifications. Nat. Struct. Mol. Biol 14(11), 1017–1024 (2007) [DOI] [PubMed] [Google Scholar]
- 9.Zentner GE, Henikoff S: Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol 20, 3, 259 (2013) [DOI] [PubMed] [Google Scholar]
- 10.Holt MV, Wang T, Young NL: Recent Advances in Understanding Histone Modification Events. Curr Mol Bio Rep 3(1), 11–17 (2017) [Google Scholar]
- 11.Schwämmle V, Aspalter C-M, Sidoli S, Jensen ON: Large scale analysis of co-existing post-translational modifications in histone tails reveals global fine structure of cross-talk. Mol. Cell. Proteom 13, 7, 1855–1865 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strahl BD, Allis CD: The language of covalent histone modifications. Nature 403, 6765, 41–45 (2000) [DOI] [PubMed] [Google Scholar]
- 13.Nacev BA, Feng L, Bagert JD, Lemiesz AE, Gao J, Soshnev AA, Kundra R, Schultz N, Muir TW, Allis CD: The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567(7749), 473–478 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esteller M: Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet 8, 4, 286–298 (2007) [DOI] [PubMed] [Google Scholar]
- 15.Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S: Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat. Rev. Clin. Oncol 17(2), 91–107 (2020) [DOI] [PubMed] [Google Scholar]
- 16.Jiang T, Hoover ME, Holt MV, Freitas MA, Marshall AG, Young NL: Middle-down characterization of the cell cycle dependence of histone H4 posttranslational modifications and proteoforms. Proteomics 18, 11, 1700442 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noberini R, Sigismondo G, Bonaldi T: The contribution of mass spectrometry-based proteomics to understanding epigenetics. Epigenomics 8, 3, 429–445 (2016) [DOI] [PubMed] [Google Scholar]
- 18.Lin S, Yuan Z-F, Han Y, Marchione DM, Garcia BA: Preferential Phosphorylation on Old Histones during Early Mitosis in Human Cells. J Biol Chem 291(29), 15342–15357 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia BA, Mollah S, Ueberheide BM, Busby SA, Muratore TL, Shabanowitz J, Hunt DF: Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc 2, 4, 933 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Hu Y, Yu Y, Zhang L, Yang P, Jin H: Quantitative analysis of post-translational modifications of histone H3 variants during the cell cycle. Anal Chim Acta 1080, 116–126 (2019) [DOI] [PubMed] [Google Scholar]
- 21.Moradian A, Kalli A, Sweredoski MJ, Hess S: The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 14, 4–5, 489–497 (2014) [DOI] [PubMed] [Google Scholar]
- 22.Toby TK, Fornelli L, Kelleher NL: Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem 9(1), 499–519 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cristobal A, Marino F, Post H, van den Toorn HWP, Mohammed S, Heck AJR: Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem 89(6), 3318–3325 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska-Wrzesinska A, Jensen ON: Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 45(16), 9272–9289 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sidoli S, Lin S, Karch KR, Garcia BA: Bottom-up and middle-down proteomics have comparable accuracies in defining histone post-translational modification relative abundance and stoichiometry. Anal. Chem 87(6), 3129–3133 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janssen KA, Coradin M, Lu C, Sidoli S, Garcia BA: Quantitation of Single and Combinatorial Histone Modifications by Integrated Chromatography of Bottom-up Peptides and Middle-down Polypeptide Tails. J Am Soc Mass Spectrom 30(12), 2449–2459 (2019) [DOI] [PubMed] [Google Scholar]
- 27.Sidoli S, Lu C, Coradin M, Wang X, Karch KR, Ruminowicz C, Garcia BA: Metabolic labeling in middle-down proteomics allows for investigation of the dynamics of the histone code. Epigenet. Chromatin 10(1), 34 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidoli S, Garcia BA: Middle-down proteomics: a still unexploited resource for chromatin biology. Expert Rev. Proteomics 14(7), 617–626 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coradin M, Mendoza MR, Sidoli S, Alpert AJ, Lu C, Garcia BA: Bullet points to evaluate the performance of the middle-down proteomics workflow for histone modification analysis. Methods (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hake SB, Garcia BA, Duncan EM, Kauer M, Dellaire G, Shabanowitz J, Bazett-Jones DP, Allis CD, Hunt DF: Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem 281(1), 559–568 (2006) [DOI] [PubMed] [Google Scholar]
- 31.Shechter D, Dormann HL, Allis CD, Hake SB: Extraction, purification and analysis of histones. Nat. Protoc 2, 6, 1445 (2007) [DOI] [PubMed] [Google Scholar]
- 32.Greer SM, Sidoli S, Coradin M, Schack Jespersen M, Schwämmle V, Jensen ON, Garcia BA, Brodbelt JS: Extensive characterization of heavily modified histone tails by 193 nm ultraviolet photodissociation mass spectrometry via a middle–down strategy. Anal. Chem 90, 17, 10425–10433 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maze I, Noh K-M, Soshnev AA, Allis CD: Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet 15(4), 259–271 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao R, Zheng D, Nie A, Zhou S, Deng H, Gao Y, Yang P, Yu Y, Tan L, Qi W, Wu J, Li E, Yi W: Sensitive and Precise Characterization of Combinatorial Histone Modifications by Selective Derivatization Coupled with RPLC-EThcD-MS/MS. J. Proteome Res 16(2), 780–787 (2017) [DOI] [PubMed] [Google Scholar]
- 35.Schräder CU, Ziemianowicz DS, Merx K, Schriemer DC: Simultaneous Proteoform Analysis of Histones H3 and H4 with a Simplified Middle-Down Proteomics Method. Anal. Chem 90(5), 3083–3090 (2018) [DOI] [PubMed] [Google Scholar]
- 36.Shliaha PV, Baird MA, Nielsen MM, Gorshkov V, Bowman AP, Kaszycki JL, Jensen ON, Shvartsburg AA: Characterization of Complete Histone Tail Proteoforms Using Differential Ion Mobility Spectrometry. Anal. Chem 89(10), 5461–5466 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shliaha PV, Gorshkov V, Kovalchuk SI, Schwämmle V, Baird MA, Shvartsburg AA, Jensen ON: Middle-Down Proteomic Analyses with Ion Mobility Separations of Endogenous Isomeric Proteoforms. Anal. Chem 92(3), 2364–2368 (2020) [DOI] [PubMed] [Google Scholar]
- 38.Zee BM, Britton L-MP, Wolle D, Haberman DM, Garcia BA: Origins and formation of histone methylation across the human cell cycle. Mol Cell Biol 32(13), 2503–2514 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cerutti H, Casas-Mollano JA: Histone H3 phosphorylation: universal code or lineage specific dialects? Epigenetics 4(2), 71–75 (2009) [DOI] [PubMed] [Google Scholar]
- 40.Jeong YS, Cho S, Park JS, Ko Y, Kang Y-K: Phosphorylation of serine-10 of histone H3 shields modified lysine-9 selectively during mitosis. Genes Cells 15(3), 181–192 (2010) [DOI] [PubMed] [Google Scholar]
- 41.Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T: Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299, 5607, 721–725 (2003) [DOI] [PubMed] [Google Scholar]
- 42.Hake SB, Allis CD: Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc. Natl. Acad. Sci 103, 17, 6428–6435 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD: Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438(7071), 1116–1122 (2005) [DOI] [PubMed] [Google Scholar]
- 44.Duan Q, Chen H, Costa M, Dai W: Phosphorylation of H3S10 blocks the access of H3K9 by specific antibodies and histone methyltransferase. Implication in regulating chromatin dynamics and epigenetic inheritance during mitosis. J. Biol. Chem 283(48), 33585–33590 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demidov D, Hesse S, Tewes A, Rutten T, Fuchs J, Karimi Ashtiyani R, Lein S, Fischer A, Reuter G, Houben A: Aurora1 phosphorylation activity on histone H3 and its cross-talk with other post-translational histone modifications in Arabidopsis. Plant J 59, 2, 221–230 (2009) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.