

Abstract
Aims
PCSK9 is secreted into the circulation, mainly by the liver, and interacts with low-density lipoprotein receptor (LDLR) homologous and non-homologous receptors, including CD36, thus favouring their intracellular degradation. As PCSK9 deficiency increases the expression of lipids and lipoprotein receptors, thus contributing to cellular lipid accumulation, we investigated whether this could affect heart metabolism and function.
Methods and results
Wild-type (WT), Pcsk9 KO, Liver conditional Pcsk9 KO and Pcsk9/Ldlr double KO male mice were fed for 20 weeks with a standard fat diet and then exercise resistance, muscle strength, and heart characteristics were evaluated. Pcsk9 KO presented reduced running resistance coupled to echocardiographic abnormalities suggestive of heart failure with preserved ejection fraction (HFpEF). Heart mitochondrial activity, following maximal coupled and uncoupled respiration, was reduced in Pcsk9 KO mice compared to WT mice and was coupled to major changes in cardiac metabolism together with increased expression of LDLR and CD36 and with lipid accumulation. A similar phenotype was observed in Pcsk9/Ldlr DKO, thus excluding a contribution for LDLR to cardiac impairment observed in Pcsk9 KO mice. Heart function profiling of the liver selective Pcsk9 KO model further excluded the involvement of circulating PCSK9 in the development of HFpEF, pointing to a possible role locally produced PCSK9. Concordantly, carriers of the R46L loss-of-function variant for PCSK9 presented increased left ventricular mass but similar ejection fraction compared to matched control subjects.
Conclusion
PCSK9 deficiency impacts cardiac lipid metabolism in an LDLR independent manner and contributes to the development of HFpEF.
Keywords: PCSK9, Heart, Cholesterol, LDLR, HFpEF
Graphical Abstract
See page 3091 for the editorial comment on this article (doi:10.1093/eurheartj/ehab480)
Translational perspective
PCSK9 inhibitors target circulating PCSK9, increase hepatic LDL receptor expression, and reduce LDL-cholesterol levels and ischaemic heart disease. On the contrary, genetic PCSK9 deficiency results in increased risk of developing diabetes or ectopic fat accumulation. Here, we extend this line of evidence showing that systemic PCSK9 deficiency results in heart failure with preserved ejection fraction, a finding independent of the modulation of LDL receptor and of circulating PCSK9. This observation supports the safety profile emerged so far with therapies targeting PCSK9, which are directed towards liver-derived circulating PCSK9.
Introduction
The heart largely uses aerobic metabolism for its energetic needs with the majority of adenosine triphosphate (ATP) being produced following fatty acid (FA) oxidation.1
Unlike the liver, the heart is not able to synthesize high amounts of FAs and, therefore, FA demand is primarily supported by the uptake from the circulation, where FAs are transported either as free FAs bound to albumin or as triglycerides in lipoproteins.2 FAs are then dissociated from albumin or released from lipoproteins through the activity of lipoprotein lipase (LPL), and delivered to cardiomyocytes through FA transport proteins or FA translocases such as CD36. Lipoproteins deliver FAs, and cholesterol to cardiomyocytes by interacting with specific receptors on cell surface including the very low-density lipoprotein receptor (VLDLR).3 , 4
Cardiac lipid demand is a finely tuned process that balances lipid uptake and mitochondrial beta-oxidation to support cardiac metabolism with the need to prevent excessive lipid accumulation which on the contrary induces cardiomyocyte dysfunction.5 In patients with heart failure, cardiomyocytes switch to glycolysis as preferred pathway for ATP generation, thus promoting cellular triacylglycerol (TAG) accumulation.6 Lactate, ketone bodies, or amino acids7 are used as source of energy alternative to FAs7 in subjects with metabolic syndrome and diabetes.8 At the molecular level, on one hand TAG accumulation and FA overload promote mitochondrial dysfunction and oxidative phosphorylation uncoupling in cardiomyocytes while on the other hand contribute to the production of lipotoxic species including diacylglycerol, long-chain acyl-CoA, acylcarnitines and lyso-phospholipids, which contribute to heart failure.5
Several experimental observations have provided a connection between lipid accumulation and cardiac dysfunction. Mice lacking adipose triglyceride lipase present a marked cardiac accumulation of TAG paralleled by heart failure and premature death.9 Similarly, the overexpression of both peroxisome proliferator-activated receptor alpha (PPARα) and gamma (PPARγ) increases FA oxidation but causes an imbalance in cardiac lipid metabolism as a consequence of FA uptake exceeding oxidation, thus leading to lipid accumulation.10 On the same line, mice expressing glycosylphosphatidylinositol (GPI)-anchored human LPL (αMHC-LpLGPI) selectively in cardiomyocytes have increased cardiac uptake and accumulation of lipids that are derived from circulating lipoproteins including triglycerides, FAs, and cholesterol.11
All together these data are consistent with the concept that promoting the activity of pathways involved in lipid uptake might contribute to cardiac lipid overload and toxicity and suggest that factors increasing lipoprotein receptor expression in the heart might result in cardiac dysfunction as was shown also for hypoxia inducing VLDLR-mediated cardiac lipotoxicity.12
Proprotein convertase subtilisin/kexin type 9 (PCSK9), a 692-amino acid glycoprotein, is known to control low-density lipoprotein receptor (LDLR) recycling.13 , 14 In addition to LDLR, PCSK9 targets LDLR homologous and non-homologous receptors, including VLDLR,15 ApoER2 (LRP8),16 LRP1,17 and CD36,18 and, disrupts their recycling by inhibiting the dissociation from the lipoprotein in the endosome–lysosome compartment, thus promoting their degradation. As such, increased PCSK9 levels result in reduced recycling of these receptors, a finding extensively described in the liver. PCSK9, however, exerts extra-hepatic effects19; its deficiency is associated with impaired pancreatic beta-cell function20 and increased risk of developing diabetes in humans.20–22 Of note Pcsk9 KO mice present increased body weight and visceral adipose tissue deposition, a finding confirmed in carriers of a PCSK9 loss-of-function variant.23 The observation that these subjects also present increased epicardial adipose tissue,23 coupled to the key role of PCSK9 in controlling key receptors involved in cardiac lipoprotein uptake provides the rationale for investigating whether PCSK9 deficiency impacts cardiac lipid metabolism and function. To this end, we tested the impact of PCSK9 deficiency on heart metabolism and function in an experimental setting where insulin response is preserved. Pcsk9 KO mice were indeed fed standard fat diet for 20 weeks and we observed that PCSK9 deficiency affected lipid metabolism and energy production in the heart that lead to the thickening of the left ventricular wall and the development of heart failure with preserved ejection fraction (HFpEF). To further investigate the impact of this observation in the clinical setting, cardiac function was investigated in subjects carrying a loss-of-function polymorphism of PCSK9.
Methods
A detailed description of mice, echocardiographic analysis, exhaustion test, forelimb grip test, oxygen consumption rate, metabolomics, proteomics, western blot analysis, analysis of cholesterol accumulation in cardiac tissue, plasma dosages, bioinformatics, and statistical analysis is presented in the Supplementary material online.
Our study complies with the Declaration of Helsinki, the local ethics committee approved the research protocol, and informed consent was obtained from all subjects (or their legally authorized representative).
Results
Pcsk9 deficiency is associated with heart failure with preserved ejection fraction
To investigate whether the impact of PCSK9 on systemic and cellular lipid and lipoprotein metabolism might affect cardiac function, we evaluated heart morphology in Pcsk9 KO and wild-type (WT) male mice fed for 20 weeks with standard fat diet. Electrocardiographic analysis (Figure 1A) of the heart of Pcsk9 KO mice showed an increased thickness of the left ventricular posterior wall (LVPW) during both systole and diastole as compared to WT mice (Figure 1B and C) despite no difference in total body weight between the two groups (Supplementary material online, Figure S1A). This was coupled with an increased relative wall thickness (Figure 1D) in spite of similar left ventricular mass normalized on body weight ratio (Figure 1E) and ejection fraction (Figure 1F) in Pcsk9 KO compared to WT mice, thus suggesting a concentric remodelling of the heart. To evaluate whether this profile associates with impaired cardiac functionality, exercise intolerance and running resistance in fatigue test were performed in WT and Pcsk9 KO mice. The latter experienced a significant reduction of the running distance and the running time observed in the exhaustion test compared to WT mice (Figure 1G and H).
Figure 1.

Pcsk9 deficiency is associated with heart failure with preserved ejection fraction. (A) Representative image of echocardiographic analysis for wild-type and Pcsk9 KO mice. (B, C) Left ventricular posterior wall thickness during systole (P = 0.049) and diastole (P = 0.03). (D–F) relative wall thickness (RWT) (P = 0.02), the left ventricular mass/weight (P = 0.37) and ejection fraction (%) (P = 0.95) are shown. (G, H) Running endurance following exhaustion test for wild-type and Pcsk9 KO mice is presented as running distance (P = 0.0003) and running time (P = 0.0003). (I) Results from the forelimb grip test are presented (P = 0.33). Data are shown as mean ± SD; n = 8 mice per group. Non-parametric t-test was used to compare each group. *P < 0.05 and ***<0.001.
Of note, this phenotype was not the consequence of a reduced skeletal muscle performance as similar results were observed in the forelimb grip test (Figure 1I) coupled with no differences in oxygen consumption rate in soleus muscle between Pcsk9 KO and WT mice (Supplementary material online, Figure S1B). These data indicate that Pcsk9 KO mice present a profile suggestive of HFpEF.
Cardiac mitochondrial metabolism is altered in Pcsk9-deficient mice
Given the key role of PCSK9 in cellular lipid biology, we next evaluated the impact of PCSK9 deficiency on cardiac lipid metabolism and its relevance for heart energetic demand. Freshly isolated hearts from Pcsk9 KO mice exhibited a significant reduction in oxygen consumption rate compared to control mice under maximal coupled and uncoupled respiration (Figure 2A). This finding, coupled with the reduced activity of the complex II of the electron transport chain (ETC) observed in Pcsk9 KO mice (Supplementary material online, Figure S2A and B), prompted us to test whether ATP production was affected. ATP levels (Figure 2B) and ATP energy charge (Figure 2C) were reduced in the heart of Pcsk9 KO mice compared to WT littermates together with other cofactors involved in energy production (Supplementary material online, Figure S2D–K). Moreover, a detailed proteomic analysis of cardiac tissue showed that the expression of several mitochondrial proteins was affected in Pcsk9 KO heart compared to WT (Supplementary material online, Figure S2A), including those of key structural components of ETC complexes (Figure 2D). This finding was further confirmed by western blot analysis of proteins representative of each mitochondrial complex, including NADH ubiquinone oxidoreductase subunit B8 (Ndufb8—Complex I), succinate dehydrogenase complex subunit B (Sdhb—Complex II), and ubiquinol-cytochrome C reductase core protein 2 (Uqcrc2—Complex III) that were significantly reduced in the heart of Pcsk9 KO mice (Figure 2E and F) compared to WT littermates. This finding together with the reduction of complex II activity (Supplementary material online, Figure S2B) supports the possibility that FADH2 could undergo a less efficient entry of into the ETC, a process that could impact FA catabolism. In parallel with these findings, also mitochondrial DNA copy number was reduced (Supplementary material online, Figure S2C), suggesting that mitochondrial function and content are affected in the heart of Pcsk9 KO mice.
Figure 2.

Pcsk9 deficiency is associated with mitochondrial dysfunction. (A) The oxygen consumption rate was investigated in the heart of Pcsk9 KO mice and oxygen consumption was measured at basal, maximal coupled, uncoupled and maximal uncoupled conditions (Basal, P = 0.39; ADP, P = 0.04; Oligo, P = 0.24; CCCP, P = 0.04). Data are shown as mean ± SD; n = 7 mice per group. (B and C) Adenosine triphosphate (P = 0.01) quantification and energy charge (P = 0.01) of the heart. Data are shown as fold of change ± SD; n = 7 mice per group. (D) Relevant proteins of ETC are displayed. (E) Representative image of western blot of ETC complexes on heart lysate is showed. (F) Proteins quantification is normalized to beta-actin expression (CI, P = 0.01; CII, P = 0.02; CIII, P = 0.008; CIV, P = 0.08, CV, P = 0.59). Data are shown as mean ± SD; n = 3 mice per group. Non-parametric t-test was used to compare each group (*P < 0.05).
We next asked whether this translates into altered cardiac metabolism. The combied analysis of metabolomic, proteomic, and lipidomic profiles in the heart of Pcsk9 KO and of WT mice (Figure 3A and Supplementary material online, Figure S3) revealed that impaired mitochondrial activity results from: (i) impaired FA oxidation as supported by the increase of acyl-carnitines carrying medium-chain FA (C8 and C10) coupled to the reduction of β-oxidation enzymes, such as sterol carrier protein 2 (SCP2), enoyl-CoA hydratase, and short chain 1 (ECHS1) (Figure 3A and Supplementary material online, Figure S3C and D), and (ii) decreased tricarboxylic acid (TCA) cycle flux (Figure 3A and Supplementary material online, Figure S3A and E) as indicated by the reduced levels of TCA intermediates and enzymes catalyzing key enzymatic reactions. The reduced levels of glucose-6P (Figure 3A and Supplementary material online, Figure S3B and F) and the increased levels of lactate dehydrogenase coupled with increased lactate plasma levels (Figure 3A) further confirmed a switch to anaerobic metabolism, which, however, is not sufficient to support heart energy demand and indeed a net reduction in energy charge is observed in the heart of Pcsk9 KO mice compared to WT (Figure 2C). Concordantly, functional pathway analysis of cardiac proteome showed increased cardiac dysfunction related to the inability of sustaining cellular energy demand (Figure 3B) and the electron microscopy analysis of mitochondria from both WT and Pcsk9 KO hearts showed less dense and organized mitochondrial cristae in cardiomyocytes from Pcsk9 KO heart compared to WT (Figure 3C).
Figure 3.

Metabolic profile of failing Pcsk9 KO heart. (A) Results from combined metabolomic, proteomic and lipidomic profile in the heart of Pcsk9 KO mice compared to wild-type mice are shown. Metabolites that are significantly modulated (belonging to glycolysis, pentose phosphate pathway, Krebs cycle, or carnitines for beta-oxidation) (G6-P, P = 0.012; R5-P, P = 0.01; E4-P, P = 0.02; acetyl-CoA, P = 0.02; citrate, P = 0.04; α-ketoglutarate, P = 0.01; succinyl-CoA, P = 0.03; fumarate, P = 0.003; malate, P = 0.003; C8, P = 0.048; C10, P = 0.048) are shown in bar graph as fold of change ± SD; n = 7 mice per group. Proteins that were significantly modulated following proteomics analysis are shown as coloured dots. Plasma levels of lactate are shown (P = 0.02). Data are shown as mean ± SD; n = 7 per group. (B) Functional pathway analysis of proteomics data is presented. Hierarchical clustering is based on Pearson’s correlation and heatmap showing relative protein expression values (z-score-transformed LFQ protein intensities) of n=65 proteins corresponding to ETC mitochondrial complexes in GO analysis (FDR < 0.05). Non-parametric t-test was used to compare each group. (C) Representative photomicrographs of myocardium mitochondria by transmission electron microscopy are shown. (*P < 0.05 and **<0.01)
PCSK9 deficiency results in increased LDLR and CD36 expression in the heart coupled with lipid accumulation
To get further insights into the molecular mechanisms underlying the dysfunctional cardiac phenotype, we investigated the expression of key PCSK9 targets in the heart. Both the expressions of LDLR and CD36 were increased in Pcsk9 KO mice compared to WT littermates (Figure 4A and B). Of note, transmission electron microscopy analysis of the hearts showed longitudinally oriented myofibrils and regular intercalated discs in both WT and Pcsk9 KO mice (Figure 4C and D). Pcsk9 KO hearts, however, presented abundant lipid droplets tightly associated with mitochondria (Figure 4D), a characteristic barely appreciated in WT mice (Figure 4C). This observation further supports the presence of lipid accumulation in the heart of Pcsk9 KO mice. The quantitative analysis of ultrathin sections showed that Pcsk9 KO mice presented a statistically significant increase in the number of lipid droplets (Figure 4E and Supplementary material online, Figure S4A) as well as of lipid droplets diameter (Figure 4F). Moreover, increased total cholesterol (Figure 4G) and arachidonic acid levels (Supplementary material online, Figure S4B) but similar total triglyceride content (Figure 4H) were observed in the heart of Pcsk9 KO mice compared to WT littermates. As expected, plasma cholesterol and triglyceride levels were lower in Pcsk9 KO mice compared to WT20 (Supplementary material online, Figure S3G and H).
Figure 4.

PCSK9 deficiency results in increased lipids and lipoprotein receptor expression coupled with cholesterol accumulation and the increase of lipid droplets in the heart. (A) Representative image and quantification of immunoblotting analysis for LDLR in cardiac tissue from Pcsk9 KO and wild-type mice (P = 0.03). Data are shown as mean ± SD; n = 6 mice for the wild-type group and n = 7 mice for the Pcsk9 KO group. (B) Representative image and quantification of immunoblotting analysis for CD36 in cardiac tissue from Pcsk9 KO and wild-type mice (P = 0.04). Data are shown as mean ± SD; n = 7 mice for group. (C and D) Representative photomicrographs of myocardium by transmission electron microscopy in wild-type and Pcsk9 KO mice are shown. Black arrows indicate lipid droplets. The inset panels show a magnification of wild-type mice (C) and of Pcsk9 KO mice (D). Bars 1 µm. (E and F) Lipid droplets number (P < 0.0001) and diameter (P = 0.008) obtained from transmission electron microscopy analysis are shown as violin plot. (G) Intracardiac total cholesterol (P = 0.04) and (E) triglycerides levels (P = 0.77) are shown. Data are presented as mean ± SD; n = 7 mice for group. Non-parametric t-test was used to compare each group (*P < 0.05, **P < 0.01 and ***<0.001).
The observation of increased lipid droplets number and cholesterol content, coupled with increased LDLR receptor expression in the heart of Pcsk9 KO mice, points to a possible role of LDLR in promoting the increased cardiac lipid accumulation. To test this hypothesis, we characterized heart function, morphology, and metabolism in mice lacking both PCSK9 and LDLR (DKO).
DKO mice presented a significant reduction compared to Ldlr KO mice in the distance and the time of running observed in the exhaustion test (Figure 5A and B); this effect was not dependent on differences in skeletal muscle strength, as the results from the forelimb grip test were similar (Figure 5C). Notably heart left ventricular mass/weight and heart LVPW thickness in systole were significantly lower in Ldlr KO mice compared to WT (left ventricular mass/weight: WT 5.31 ± 0.98 vs. Ldlr KO 4.10 ± 0.91, P = 0.04) (LVPW: WT 1.23 ± 0.13 vs. Ldlr KO 1.10 ± 0.08, P = 0.04) (Supplementary material online, Table S1), a finding that fits with their reduced performance on running endurance compared to WT (running distance: WT 331.1 ± 46.20 vs. Ldlr KO 245.3 ± 40.93 m, P = 0.049) (Supplementary material online, Figure S5A–C). On the other hand, the echocardiographic profile of DKO was very similar to that observed in Pcsk9 KO mice (Figures 1B and 5C), suggesting that the impact of PCSK9 on heart function does not depend on the modulation of LDLR expression and on changes in plasma cholesterol levels (Supplementary material online, Figure S5D and E). Indeed, echocardiographic analysis showed that DKO mice presented a significant left ventricular thickening during systole (Figure 5D) without alterations in ejection fraction (Figure 5E) compared to Ldlr KO mice. In addition, metabolic analysis showed that ATP levels and energy charge were still significantly reduced in DKO mice as compared to Ldlr KO mice (Figure 5F and G). This profile is consistent with a reduced flux of TCA cycle coupled with mitochondrial dysfunction (Figure 5H) in the heart, and with increased plasma levels of lactate (Figure 5I and Supplementary material online, Figure S5F). These data strongly support the concept that the increased cardiac LDLR expression induced by PCSK9 deficiency is not involved in the phenotype observed. To get further insights into the connection between PCSK9 and cardiomyocytes physiology, we performed a series of studies in cardiomyocytes differentiated from human pluripotent stem cells (iPSC-CMs). The acquisition of cardiomyocyte phenotype was documented by the increase, up to 300 folds, of troponin T expression (Supplementary material online, Figure S6A). VLDL supplementation leads to a significant increase in the expression of LDLR, CD36, LPL, glucose transporter type 4 (GLUT4), and FA synthase mRNA expression in iPSC-CMs compared to cells grown in control medium (Supplementary material online, Figure S6B). Moreover, VLDL treatment resulted in decreased mitochondrial mass (Supplementary material online, Figure S6C) and increased accumulation of neutral lipids (Supplementary material online, Figure S6D). These effects were reverted when cardiomyocytes were pre-treated with PCSK9 (Supplementary material online, Figure S6E and F) confirming a role for PCSK9 in regulating lipids uptake and mitochondrial function in cardiomyocytes. In line with these findings, primary cardiomyocytes isolated from Pcsk9 KO treated with VLDL showed reduced mitochondrial mass compared to WT and the same was true for primary cardiomyocytes isolated Pcsk9/Ldlr DKO (Supplementary material online, Figure S6G), thus further excluding a role for the LDLR in the phenotype observed.
Figure 5.

PCSK9 effect on cardiac function is not dependent on the LDLR. (A and B) Running endurance on exhaustion test of Ldlr KO and Pcsk9/Ldlr DKO mice is displayed as running distance (P = 0.01) and running time (P = 0.01). (C) Results from the forelimb grip test are displayed (P = 0.23). (D and E) Left ventricular posterior wall thickness during systole (P = 0.01) and ejection fraction (P = 0.33) are shown. (F and G) Adenosine triphosphate quantification (P = 0.02) and energy charge (P = 0.02) of the heart. Data are presented as mean ± SD. (H) Metabolites of the Krebs cycle are shown (citrate, P = 0.003; α-ketoglutarate, P = 0.29; succinyl-CoA, P = 0.02; fumarate, P = 0.05; malate, P = 0.03; oxaloacetate, P = 0.0002) as fold of change ± SD. (I) Plasma levels of lactate (P = 0.03) are shown as mean ± SD. n = 6 mice per group. Non-parametric t-test was used to compare each group (*P < 0.05 and ***<0.001).
Circulating PCSK9 does not impact heart metabolism
In both human and mice, circulating PCSK9 is largely contributed by the liver. Therefore, to separate the effect of circulating vs. locally produced PCSK9 on heart function, we profiled cardiac function and heart morphology in mice lacking PCSK9 production selectively in the liver (AlbCre+/Pcsk9LoxP/LoxP mice) and therefore deficient for PCSK9 only in the circulation.20 The running distance and the running time observed in the exhaustion test were similar in AlbCre+/Pcsk9LoxP/LoxP mice compared to their AlbCre−/Pcsk9LoxP/LoxP counterpart (Figure 6A and B) as was muscular performance (Figure 6C). Accordingly, no differences emerged for left ventricular thickness during systole (Figure 6D), for ejection fraction (Figure 6E) as well as for several cardiac parameters (Supplementary material online, Figure S7A–E). Also, ATP production (Figure 6F), ATP energy charge (Figure 6G), Krebs cycle metabolites (Figure 6H), and glycolysis intermediates (Supplementary material online, Figure S7F) were not different in the heart of these mice. Circulating lactate levels (Figure 6J) and triglycerides (Supplementary material online, Figure S7G) were not affected in AlbCre+/Pcsk9LoxP/LoxP mice compared to their AlbCre−/Pcsk9LoxP/LoxP counterpart while cholesterol was significantly reduced in liver selective KO (Supplementary material online, Figure S7H). These data excluded a role for the deficiency of liver-produced (i.e. circulating) PCSK9 in the phenotype observed, rather pointing to locally produced PCSK9 deficiency as a driver of heart dysfunction.
Figure 6.

Circulating PCSK9 does not impact cardiac metabolism and heart structure. (A and B) Running endurance on exhaustion test of AlbCre−/Pcsk9LoxP/LoxP and AlbCre+/Pcsk9LoxP/LoxP mice is displayed as running distance (P = 0.68) and running time (P = 0.63). (C) Results from the forelimb grip test are displayed (P = 0.98). (D and E) Left ventricular posterior wall thickness during systole (P = 0.83) and ejection fraction (P = 0.98) are shown. (F and G) Adenosine triphosphate quantification (P = 0.27) and energy charge (P = 0.627) of the heart are presented. (H) Metabolites of the Krebs cycle (citrate, P = 0.26; α-ketoglutarate, P = 0.91; succinyl-CoA, P = 0.33; fumarate, P = 0.57; malate, P = 0.63; oxaloacetate, P = 0.009) are shown as fold of change ± SD. (I) Plasma levels of lactate (P = 0.50) are shown. Data are shown as mean ± SD; n = 5 mice per group. Non-parametric t-test was used to compare each group (*P < 0.05).
Genetic PCSK9 loss of function is associated with altered cardiac phenotype in humans
To further translate our findings in humans, we evaluated the impact of a PCSK9 loss-of-function variant (R46L) on echocardiographic-based markers of cardiac functionality in a cohort of 2606 subjects from the general population (PLIC study). A pilot analysis of cardiac profile in 12 heterozygous R46L carriers (Supplementary material online, Table S2) showed that these subjects displayed significant increase in left ventricular mass index (Figure 7A) but similar ejection fraction (Figure 7B) compared to age- and sex-matched WT subjects. Interestingly, leg and arm skeletal muscle mass were comparable between heterozygous subjects and WT carriers (Figure 7C).
Figure 7.

PCSK9 46L LOF individuals present heart failure with preserved ejection fraction. (A and B) Left ventricular mass index (g/m2) (P = 0.01) and ejection fraction (P = 0.10) of PCSK9 R46 and 46L carriers are shown. (C) Leg and arm skeletal muscle mass (quantified by Dual X-Rays Absorbimetry, following Hansen’s formula) (P = 0.26) is shown (R46, n = 516; 46L, n = 12). Non-parametric t-test was used to compare each group. (*P < 0.05).
Discussion
In this work, we demonstrate that PCSK9 plays a key role in controlling heart metabolism and function. When PCSK9 is absent, the expression of key receptors involved in lipid and lipoprotein uptake is increased and results in heart cholesterol accumulation, impaired beta-oxidation and mitochondrial activity, thus affecting cardiac metabolism and function (Graphical abstract).
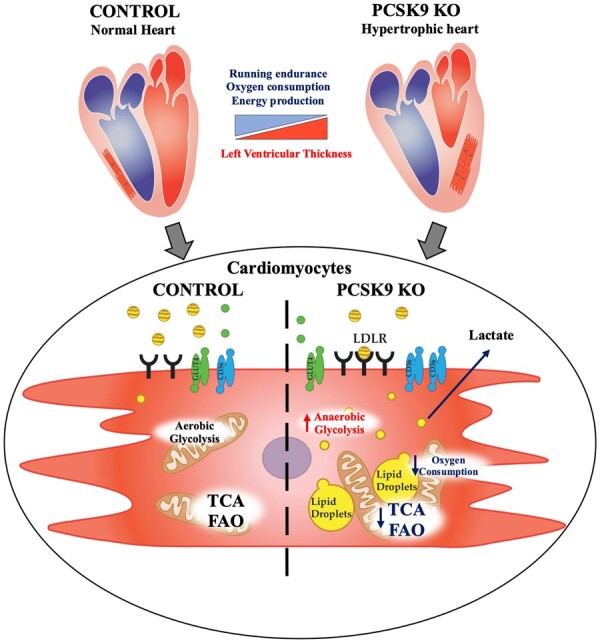
Impact of Pcsk9 deficiency on cardiac function and mitochondrial metabolism. Pcsk9 deficiency is associated with increased heart left ventricular thickness and reduced running performance independently of skeletal muscle alterations. Electron microscopy analysis showed increased cardiac accumulation of lipid droplets associated with a reduced density of mitochondrial cristae; this proffunctionally translated into impaired oxidative phosphorylation and mitochondrial metabolism in PCSK9 KO hearts.
Unlike the liver, the heart cannot synthesize large amounts of FAs and therefore lipid demand is primarily fulfilled with the uptake from the circulation, which however needs to be properly controlled to limit an excessive uptake which may result in lipid accumulation and cellular lipotoxicity. Under these circumstances, cardiomyocytes switch their metabolism towards anaerobic glycolysis, which however does not fully compensate the elevated energetic demand. Physiologically, this shift triggers a series of morphological adaptations in the heart including increased left ventricular wall thickness to maintain ejection fraction. This profile is typical of HFpEF where cardiomyocytes switch their metabolism from FA oxidation into mitochondria, to glycolysis and ketone body utilization24 as described in patients with hypertension25 and obesity,26 as well as with diabetes.27
Here we show that a similar profile is observed in PCSK9-deficient conditions in experimental models. A detailed profiling of cardiac metabolic signature revealed that oxygen consumption rate and ATP levels are reduced as a consequence of impaired mitochondrial function as suggested also by the changes in ETC protein complexes and activity. Previous observations highlighted that heart lipid accumulation drives lipotoxicity and contributes to heart metabolic switch; thus, the observation that PCSK9 deficiency results in increased cholesterol accumulation in the heart coupled with metabolic shift towards anaerobic glycolysis supports the hypothesis that PCSK9 plays a physiologically role in maintaining a proper balance of factors involved in lipid uptake by cardiomyocytes.
The obvious candidates for this activity are lipoprotein receptors, which, although expressed at a lower extent in the heart compared to other tissues, such as the liver, still play a critical role in controlling heart lipoprotein uptake and lipid metabolism.28 The observation that the expression of both LDLR and CD36 two PCSK9 targets, is increased in the heart of Pcsk9 KO mice could represent a matter of concern for patients treated with PCSK9 inhibitors, as the increase in LDLR recycling is responsible for increased lipoproteins uptake in liver and plasma cholesterol lowering, but might also influence heart lipid accumulation. To further elucidate this aspect, cardiac profile and running endurance were investigated in Pcsk9/Ldlr DKO mice and in Ldlr KO mice. Of note Pcsk9/Ldlr DKO mice still present heart dysfunction and the metabolic sequalae observed in Pcsk9 KO mice, while Ldlr KO had a significantly lower left ventricular mass/weight and heart LVPW thickness in systole compared to WT, thus excluding a role for PCSK9/LDLR axis in the cardiac phenotype observed. Moreover, this finding suggests that eventually LDLR-mediated lipoprotein uptake, at least in the heart, does not result in lipid accumulation and cardiac metabolism impairment. Could pharmacological inhibition of PCSK9 result in increased cardiac lipid uptake and heart dysfunction? Data from large interventional trials with PCSK9 inhibitors did not report an increased incidence of heart failure.29
Current therapies targeting PCSK9 include monoclonal antibodies which act by sequestering circulating PCSK9,30 and a gene silencing approach, which acts by selectively silencing PCSK9 mRNA expression in the liver.31 Given that the liver contributes to circulating PCSK9 levels, both approaches reduce circulating PCSK9 but do not affect local PCSK9 production, except for the liver. To explore whether a complete lack of circulating PCSK9 could impact heart metabolism, cardiac function was evaluated in an experimental model lacking PCSK9 expression selectively in the liver, thus presenting undetectable circulating PCSK9 levels but unaltered expression in other tissues20; a phenotype which mimics that observed following PCSK9 inhibitors therapy. In this experimental model, exercise performance, heart morphology, metabolic signature, and oxygen consumption were similar to those of WT mice. This observation is seminal to exclude any possible impact of circulating PCSK9 deficiency on heart metabolism and is in line with the data on cardiac function observed in clinical trials with anti-PCSK9 therapies.
Which cells are then providing PCSK9 that is critical for the maintaineance of cardiomyocyte homeostasis and lipid balance?
Under basal conditions, PCSK9 is expressed at very low levels in the heart (Supplementary material online, Figure S7I) but is induced in vivo under ischaemic conditions32 and in vitro following incubation with oxidized LDL.33 It has been hypothesized that PCSK9 induction immediately after ischemia/reperfusion injury might protect the heart in the acute phase by stimulating autophagic process and the removals of damaged mitochondria,32 an effect that could became deleterious over time as might lead to increased cell deterioration and cardiomyocyte death. It is also possible that PCSK9 produced locally by epicardial adipose tissue34 contributes to this pathophysiological change, a finding supported by the observation that the R46L variant is associated with increased epicardial fat accumulation in humans,23 independently of obesity or diabetes. However, we did not find any correlation between epicardial fat thickness and left ventricular mass (data not shown), supporting the hypothesis of an effect of PCSK9 on epicardial adipose tissue inflammation largely related to heart failure with reduced ejection fraction.35 Future studies in larger cohorts should address whether PCSK9 loss-of-function variants are associated with an altered cardiac phenotype independently of the lower cardiovascular risk provided by the long-life reduction in LDL-cholesterol levels observed under these circumstances. We have to acknowledge that we profiled heart function in full Pcsk9 KO mice and in liver selective PCSK9 KO models. While our findings excluded a role for circulating PCSK9 on the phenotype observed, a final confirmation for a selective role for PCSK9 in the heart should be determined in heart selective KO models. Similarly, the characterization of Pcsk9/CD36 DKO will be of help to investigate whether PCSK9 production following cardiac stress might represent a feedback mechanism contributing to maintain a proper balance between heart lipid uptake and lipid accumulation, thus limiting potential side effects of heart lipotoxicity.
Supplementary material
Supplementary material is available at European Heart Journal online.
Supplementary Material
Acknowledgments
Part of this work was carried out at NOLIMITS, an advanced imaging facility established by the Università degli Studi di Milano. We would thank Dr Vincenzo Conte and Dr Elena Vezzoli (Electron Microscopy Laboratory, Department of Biomedical Sciences for Health) for their technical help in electron microscopy work. Proteomics part of this work was performed at Unitech OMICs platform of the Università degli Studi di Milano. For their help in processing MS samples, we would like to thank Dr Fiorenza Faré, Dr Giulia Garrone, and Prof. Giangiacomo Beretta. The Genotype–Tissue Expression (GTEx) portal was used to evaluate PCSK9 expression in RNA-seq level within multiple tissues (http://www.gtexportal.org/home/gene/PCSK9).
Funding
The work of the authors is supported by Fondazione Cariplo 2016-0852 (G.D.N.) and 2019-1560 (F.B.); Telethon Foundation (GGP19146) (G.D.N.); WI218287 Cardiovascular Grant, PCSK9 Competitive Grant Program (PCSK9006) (G.D.N.); European Foundation for the Study of Diabetes (EFSD)/Lilly European Diabetes Research Programme (G.D.N.); Progetti di Rilevante Interesse Nazionale PRIN 2017 K55HLC (G.D.N.) and PRIN 2017 H5F943 (A.L.C.); “Cibo, Microbiota, Salute”, “Vini di Batasiolo S.p.A”, “Accademia di Medicina di Torino” AL_RIC19ABARA_01, The Peanut Institute Foundation “Research Award 2021” (A.B.); and “Post-Doctoral Fellowship 2020” by “Fondazione Umberto Veronesi” 2020-3318 (A.B.), 2021-4442 (F.B.). Department of excellence of Pharmacological and Biomolecular Sciences, Universita degli Studi di Milano (L.D.D.).
Conflict of interest: The authors have received research funding and/or honoraria for advisory boards, consultancy or speaker bureau from Aegerion (A.L.C.), Alnylam (G.D.N.), Amgen (A.L.C., G.D.N) AstraZeneca (A.L.C.), Eli Lilly (A.L.C.), Kowa (A.L.C.), Mediolanum (A.L.C.), Menarini (A.L.C.), Merck or MSD (A.L.C.), Novartis (G.D.N.), Pfizer (A.L.C., G.D.N.), Recordati (A.L.C.), Sanofi (A.L.C., G.D.N.), and Regeneron (A.L.C.). All other authors declared no conflict of interest.
Contributor Information
Lorenzo Da Dalt, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy.
Laura Castiglioni, Department of Pharmaceutical Sciences, Università degli Studi di Milano, Via Mangiagalli, 25, 20133 Milan, Italy.
Andrea Baragetti, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy; IRCCS Multimedica Hospital, Via Milanese, 300, 20099 Sesto San Giovanni, Italy.
Matteo Audano, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy.
Monika Svecla, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy.
Fabrizia Bonacina, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy.
Silvia Pedretti, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy.
Patrizia Uboldi, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy.
Patrizia Benzoni, Department of Biosciences, Università degli Studi di Milano, Via Celoria, 26, 20133 Milan, Italy.
Federica Giannetti, Department of Biosciences, Università degli Studi di Milano, Via Celoria, 26, 20133 Milan, Italy.
Andrea Barbuti, Department of Biosciences, Università degli Studi di Milano, Via Celoria, 26, 20133 Milan, Italy.
Fabio Pellegatta, Centro SISA per lo studio dell’Aterosclerosi, Ospedale Bassini, Via Massimo Gorki, 50, 20092 Cinisello Balsamo, Italy.
Serena Indino, Department of Biomedical Science for Health, Università degli Studi di Milano, Via Mangiagalli, 31, 20133 Milan, Italy.
Elena Donetti, Department of Biomedical Science for Health, Università degli Studi di Milano, Via Mangiagalli, 31, 20133 Milan, Italy.
Luigi Sironi, Department of Pharmaceutical Sciences, Università degli Studi di Milano, Via Mangiagalli, 25, 20133 Milan, Italy.
Nico Mitro, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy.
Alberico Luigi Catapano, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy; IRCCS Multimedica Hospital, Via Milanese, 300, 20099 Sesto San Giovanni, Italy.
Giuseppe Danilo Norata, Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Via balzaretti, 9, 20133 Milan, Italy; Centro SISA per lo studio dell’Aterosclerosi, Ospedale Bassini, Via Massimo Gorki, 50, 20092 Cinisello Balsamo, Italy.
References
- 1. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 2005;85:1093–1129. [DOI] [PubMed] [Google Scholar]
- 2. Pulinilkunnil T, Rodrigues B. Cardiac lipoprotein lipase: metabolic basis for diabetic heart disease. Cardiovasc Res 2006;69:329–340. [DOI] [PubMed] [Google Scholar]
- 3. van der Vusse GJ, van Bilsen M, Glatz JF. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res 2000;45:279–293. [DOI] [PubMed] [Google Scholar]
- 4. Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, Shulman GI, Blaner WS, Goldberg IJ. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem 2010;285:37976–37986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab 2012;15:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 2004;18:1692–1700. [DOI] [PubMed] [Google Scholar]
- 7. Tuunanen H, Ukkonen H, Knuuti J. Myocardial fatty acid metabolism and cardiac performance in heart failure. Curr Cardiol Rep 2008;10:142–148. [DOI] [PubMed] [Google Scholar]
- 8. Dei Cas A, Khan SS, Butler J, Mentz RJ, Bonow RO, Avogaro A, Tschoepe D, Doehner W, Greene SJ, Senni M, Gheorghiade M, Fonarow GC. Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC Heart Fail 2015;3:136–145. [DOI] [PubMed] [Google Scholar]
- 9. Haemmerle G, Moustafa T, Woelkart G, Büttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rülicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med 2011;17:1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Son NH, Yu S, Tuinei J, Arai K, Hamai H, Homma S, Shulman GI, Abel ED, Goldberg IJ. PPARγ-induced cardiolipotoxicity in mice is ameliorated by PPARα deficiency despite increases in fatty acid oxidation. J Clin Invest 2010;120:3443–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest 2003;111:419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perman JC, Boström P, Lindbom M, Lidberg U, StÅhlman M, Hägg D, Lindskog H, Scharin Täng M, Omerovic E, Mattsson Hultén L, Jeppsson A, Petursson P, Herlitz J, Olivecrona G, Strickland DK, Ekroos K, Olofsson SO, Borén J. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J Clin Invest 2011;121:2625–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Norata GD, Tavori H, Pirillo A, Fazio S, Catapano AL. Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc Res 2016;112:429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res 2014;114:1022–1036. [DOI] [PubMed] [Google Scholar]
- 15. Takahashi S, Sakai J, Fujino T, Hattori H, Zenimaru Y, Suzuki J, Miyamori I, Yamamoto TT. The very low-density lipoprotein (VLDL) receptor: characterization and functions as a peripheral lipoprotein receptor. J Atheroscler Thromb 2004;11:200–208. [DOI] [PubMed] [Google Scholar]
- 16. Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 2008;283:2363–2372. [DOI] [PubMed] [Google Scholar]
- 17. Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 2013;8:e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Demers A, Samami S, Lauzier B, Des Rosiers C, Ngo Sock ET, Ong H, Mayer G. PCSK9 induces CD36 degradation and affects long-chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. Arterioscler Thromb Vasc Biol 2015;35:2517–2525. [DOI] [PubMed] [Google Scholar]
- 19. Perego C, Da Dalt L, Pirillo A, Galli A, Catapano AL, Norata GD. Cholesterol metabolism, pancreatic β-cell function and diabetes. Biochim Biophys Acta Mol Basis Dis 2019;1865:2149–2156. [DOI] [PubMed] [Google Scholar]
- 20. Da Dalt L, Ruscica M, Bonacina F, Balzarotti G, Dhyani A, Di Cairano E, Baragetti A, Arnaboldi L, De Metrio S, Pellegatta F, Grigore L, Botta M, Macchi C, Uboldi P, Perego C, Catapano AL, Norata GD. PCSK9 deficiency reduces insulin secretion and promotes glucose intolerance: the role of the low-density lipoprotein receptor. Eur Heart J 2019;40:357–368. [DOI] [PubMed] [Google Scholar]
- 21. Lotta LA, Sharp SJ, Burgess S, Perry JRB, Stewart ID, Willems SM, Luan J, Ardanaz E, Arriola L, Balkau B, Boeing H, Deloukas P, Forouhi NG, Franks PW, Grioni S, Kaaks R, Key TJ, Navarro C, Nilsson PM, Overvad K, Palli D, Panico S, Quirós J-R, Riboli E, Rolandsson O, Sacerdote C, Salamanca-Fernandez E, Slimani N, Spijkerman AMW, Tjonneland A, Tumino R, van der A DL, van der Schouw YT, McCarthy MI, Barroso I, O’Rahilly S, Savage DB, Sattar N, Langenberg C, Scott RA, Wareham NJ. Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes: a meta-analysis. JAMA 2016; 316:1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, Voros S, Giugliano RP, Davey Smith G, Fazio S, Sabatine MS. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 2016; 375:2144–2153. [DOI] [PubMed] [Google Scholar]
- 23. Baragetti A, Balzarotti G, Grigore L, Pellegatta F, Guerrini U, Pisano G, Fracanzani AL, Fargion S, Norata GD, Catapano AL. PCSK9 deficiency results in increased ectopic fat accumulation in experimental models and in humans. Eur J Prev Cardiol 2017;24:1870–1877. [DOI] [PubMed] [Google Scholar]
- 24. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 2013;113:709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodeheffer RJ. Hypertension and heart failure: the ALLHAT imperative. Circulation 2011;124:1803–1805. [DOI] [PubMed] [Google Scholar]
- 26. Alpert MA, Lavie CJ, Agrawal H, Aggarwal KB, Kumar SA. Obesity and heart failure: epidemiology, pathophysiology, clinical manifestations, and management. Transl Res 2014;164:345–356. [DOI] [PubMed] [Google Scholar]
- 27. Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitUs. Circ Res 2019;124:121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res 2016;118:1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Santos RD, Stein EA, Hovingh GK, Blom DJ, Soran H, Watts GF, López JAG, Bray S, Kurtz CE, Hamer AW, Raal FJ. Long-term evolocumab in patients with familial hypercholesterolemia. J Am Coll Cardiol 2020; 75:565–574. [DOI] [PubMed] [Google Scholar]
- 30. Catapano AL, Pirillo A, Norata GD. New pharmacological approaches to target PCSK9. Curr Atheroscler Rep 2020; 22:24. [DOI] [PubMed] [Google Scholar]
- 31. Seidah NG, Prat A, Pirillo A, Catapano AL, Norata GD. Novel strategies to target proprotein convertase subtilisin kexin 9: beyond monoclonal antibodies. Cardiovasc Res 2019;115:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding Z, Wang X, Liu S, Shahanawaz J, Theus S, Fan Y, Deng X, Zhou S, Mehta JL. PCSK9 expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc Res 2018;114:1738–1751. [DOI] [PubMed] [Google Scholar]
- 33. Wolf A, Kutsche HS, Schreckenberg R, Weber M, Li L, Rohrbach S, Schulz R, Schlüter KD. Autocrine effects of PCSK9 on cardiomyocytes. Basic Res Cardiol 2020; 115:1093–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dozio E, Ruscica M, Vianello E, Macchi C, Sitzia C, Schmitz G, Tacchini L, Corsi Romanelli MM. PCSK9 expression in epicardial adipose tissue: molecular association with local tissue inflammation. Mediators Inflamm 2020;2020:1348913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J. Am. Coll. Cardiol 2018; 71:2360–2372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.