

SUMMARY
Dysregulated pre-mRNA splicing is an emerging Achilles’ heel of cancers and myelodysplasias. To expand the currently limited portfolio of small molecule drug leads, we screened for chemical modulators of the U2AF complex, which nucleates spliceosome assembly and is mutated in myelodysplasias. A hit compound specifically enhances RNA binding by a U2AF2 subunit. Remarkably, the compound inhibits splicing of representative substrates and stalls spliceosome assembly at the stage of U2AF function. Computational docking, together with structure-guided mutagenesis, indicates that the compound bridges the tandem U2AF2 RNA recognition motifs via hydrophobic and electrostatic moieties. Cells expressing a cancer-associated U2AF1 mutant are preferentially killed by treatment with the compound. Altogether, our results highlight the potential of trapping early spliceosome assembly as an effective pharmacological means to manipulate pre-mRNA splicing. By extension, we suggest that stabilizing assembly intermediates may offer a useful approach for small molecule inhibition of macromolecular machines.
Keywords: Spliceosome inhibition, ribonucleoprotein targeting, myelodysplastic syndrome, splicing factor mutation, therapeutic strategy, U2AF65, U2AF1, U2AF35, S34F-mutant
Graphical Abstract
eTOC Blurb
Many cancers and myelodysplasias are associated with pre-mRNA splicing defects. Chatrikhi et al describe a hit compound that stalls pre-mRNA splicing at the initial stages of spliceosome assembly by binding to U2AF2 and promoting U2AF2-RNA association. Treatment with the molecule alters splicing and preferentially kills cells expressing a cancer-relevant U2AF1-mutant.
INTRODUCTION
Alternative splicing of human pre-mRNAs is an essential step of gene expression and generates a multitude of tissue-specific isoforms from a much smaller number of genes (Pan et al., 2008; Wang et al., 2008). Genomes of hematologic malignances acquire recurrent mutations among a subset of pre-mRNA splicing factors (U2AF1, SF3B1, SRSF2, ZRSR2) (reviewed in (Dvinge et al., 2016)). The mutational hotspots typically cluster at key interfaces involved in the early stages of spliceosome assembly (Jenkins and Kielkopf, 2017). Dysregulated pre-mRNA splicing resulting from these mutations combined with other transcriptional abnormalities renders hematologic malignancies and cancers more sensitive to spliceosome inhibitors, relative to their “normal” counterparts (Hsu et al., 2015; Larrayoz et al., 2016; Lee et al., 2016; Obeng et al., 2016; Paolella et al., 2017; Seiler et al., 2018; Shirai et al., 2017; Xargay-Torrent et al., 2015). As such, defective pre-mRNA splicing offers a promising target for potential chemotherapies to treat cancers (reviewed in (Effenberger et al., 2017)). Although splicing modulators are not yet available for cancer treatment, a strong precedent for the success of pharmaceuticals targeting the pre-mRNA splicing pathway has been set by the recent FDA approvals of SMN2-directed drugs to treat spinal muscular atrophy (SMA) (reviewed in (Singh et al., 2020)). One major focus of pre-mRNA splicing inhibitors as potential chemotherapies for cancers has been small molecules targeting a druggable site of SF3B1 adjacent a conserved pre-mRNA adenosine (branchpoint) (Eskens et al., 2013; Hong et al., 2014; Seiler et al., 2018). The SF3B1-targeted compounds show promising abilities to selectively arrest the growth of cancer cells in a variety of models and patient samples (Hsu et al., 2015; Lagisetti et al., 2013; Larrayoz et al., 2016; Lee et al., 2016; Obeng et al., 2016; Paolella et al., 2017; Seiler et al., 2018; Shirai et al., 2017; Xargay-Torrent et al., 2015).
In view of the large uncharted pharmacological space created by the complexity of the spliceosome, there is rich promise for compounds that target any of the more than a hundred splicing factors beyond SF3B1. One promising splicing factor, for which the potential as a therapeutic target has yet to be explored, is the U2 small nuclear ribonucleoprotein auxiliary factor (U2AF). A heterodimer of U2AF2 and U2AF1 subunits recognizes and nucleates spliceosome assembly at the polypyrimidine (Py) tract (Singh et al., 1995) and AG-dinucleotide (Guth et al., 2001; Wu et al., 1999) consensus signals of the 3′ splice site. U2AF initially binds as a ternary complex with a third splicing factor, SF1, which subsequently is displaced by the SF3B1 subunit of the U2 particle. U2AF2 supports splicing at most sites, but depends on assistance from the U2AF1 subunit when the Py tract signal is degenerate (Guth et al., 2001; Wu et al., 1999). Notably, the U2AF1 subunit frequently carries an S34F/Y mutation, or less frequently Q157R/P, in approximately 12% of patients with myelodysplastic syndromes (MDS) (Graubert et al., 2012; Yoshida et al., 2011) or 3% with lung adenocarcinomas (Imielinski et al., 2012). Interestingly, U2AF1 is a paralogue of ZRSR2, another recurrently mutated splicing factor in MDS and chronic myelomonocytic leukemia, which functions as part of the minor U12 rather than U2-type spliceosome machinery (Madan et al., 2015; Shen et al., 2010). Albeit at lower frequencies, the U2AF2 subunit also acquires mutations in cancers and leukemias (Glasser et al., 2017; Kralovicova et al., 2020; Maji et al., 2020). Since U2AF2 recruits the SF3B1 subunit, the pre-clinical success of SF3B1 inhibitors suggests that targeting U2AF2 likewise could produce small molecule tools for investigating spliceosome assembly in normal cells and hematologic malignancies.
Here, we identify and characterize a small molecule modulator (NSC 194308) that increased, rather than reduced, association of the U2AF1–U2AF2–SF1–splice site RNA complex by binding a site between the U2AF2 RNA recognition motifs (RRM1 and RRM2). NSC 194308 inhibited pre-mRNA splicing by stalling spliceosome assembly at the point where U2AF helps recruit U2 snRNP to the branchpoint. These results set the stage for future optimization of a promising hit inhibitor of pre-mRNA splicing, and highlight a general strategy for inhibiting multi-stage processes by stabilizing assembly intermediates.
RESULTS
A High-Throughput Screen for U2AF–RNA Enhancers.
To screen for small molecule modulators of the U2AF-containing spliceosome, we focused on a complex among the U2AF1, U2AF2, and SF1 splicing factors (“U2AF”-complex) that recognizes the 3′ splice site in the initial stage of spliceosome assembly. Our U2AF protein complex included the relevant RNA binding domains (Figure 1A) and was expressed and purified as described (Fei et al., 2016; Okeyo-Owuor et al., 2015). We included the recurrent, MDS-associated S34F mutation of the U2AF1 subunit. Likewise, we chose an S34F-affected splice site of the DEK oncogene (Okeyo-Owuor et al., 2015) as the RNA binding site of the targeted U2AF complex in the screen. Fluorescence polarization (FP) of a 5′-fluorescein-labeled RNA oligonucleotide (FlRNA) was the principal readout of the screen (Figure 1B). Association of the ribonucleoprotein slows rotational diffusion of the FlRNA and hence retained more of the anisotropy of fluorophore emission following excitation with polarized light. In addition to comprising the relevant complex for early stage of 3′ splice site recognition, the inclusion of all three splicing factor subunits improved the signal of the screen by increasing the FP change following FlRNA binding compared to a single subunit alone. For sufficient dynamic range to monitor increases in RNA binding, we chose a baseline concentration of U2AF1S34F–U2AF2–SF1 protein complex (80 nM) within the initial phase of the DEK FlRNA binding curve (Figure 1C).
Figure 1.
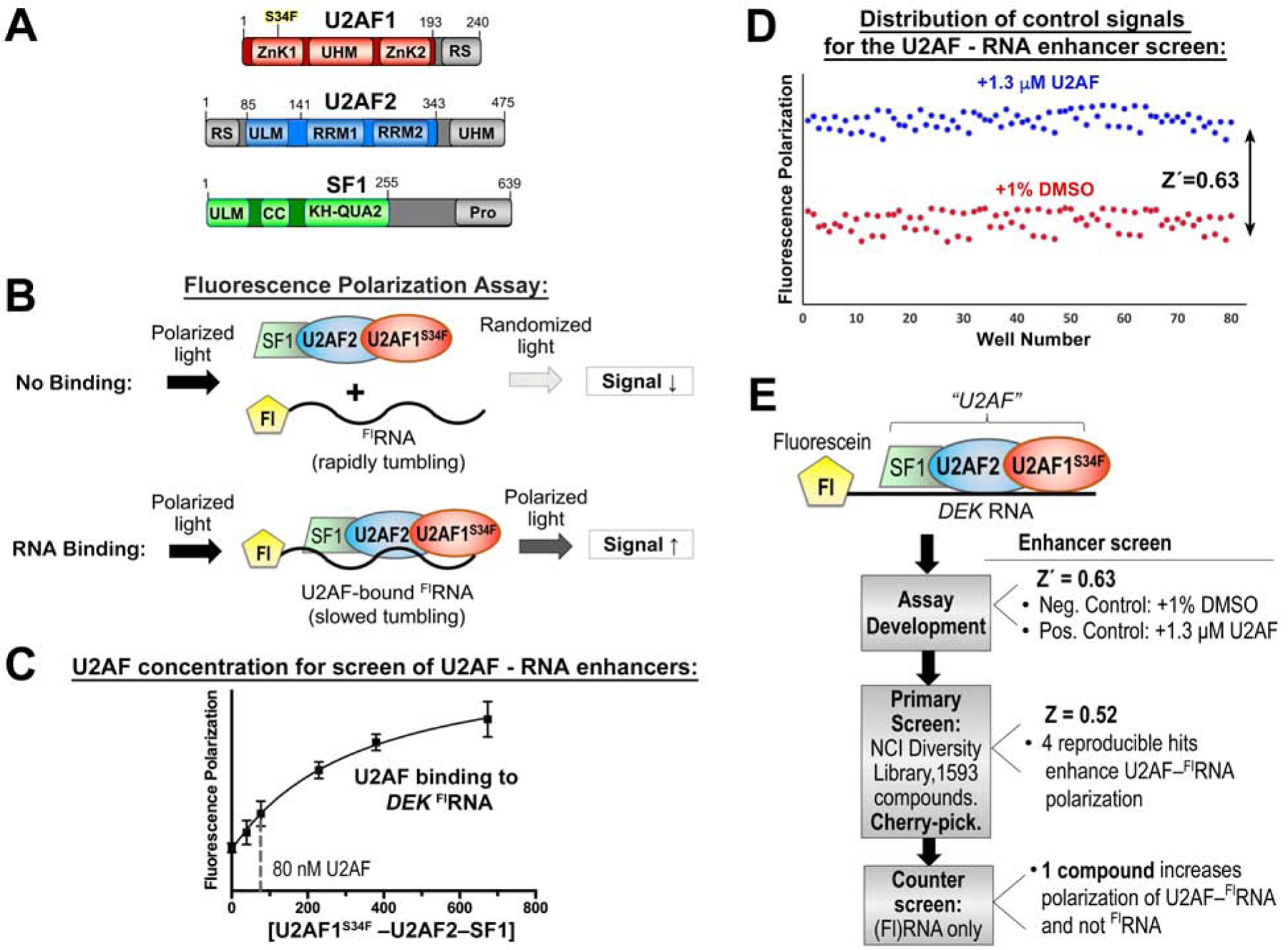
A high-throughput screen for modulators of U2AF–RNA complexes. (A) Domains of U2AF1, U2AF2, and SF1 splicing factors. Gray, region excluded from the screened expression constructs. (B) Schematic diagram of the fluorescence polarization RNA binding assay. Fl, 5′ fluorescein. (C) Fluorescence polarization curve for U2AF1S34F–U2AF2–SF1 binding into fluorescein DEK FLRNA. The nonlinear fit (solid curve) is overlaid on the mean data points ± SD of three replicates. A dashed line marks the protein concentration used for the enhancer screen. (D) Distribution of positive (blue) and negative (red) control signals used for the calculation of the Z′-factors of the assay. (E) Overview of the screen. Results are summarized to the right and detailed in Table S1.
Before screening chemical libraries, we first assessed the quality of the assay by measuring the FP of a plate comprising alternating rows of positive and negative control samples. We added excess U2AF1S34F–U2AF2–SF1 protein in a buffer including DMSO (1% v/v) to match the compound-containing samples as a positive control for enhancers of protein–RNA binding. The effective solvent of the compounds (1% v/v DMSO) served as a negative control. We reproducibly found that addition of unlabeled RNA reduced the FP, whereas excess U2AF protein increased the FP and the trivial amount of DMSO solvent had no detectable effect on the binding reaction. From these distributions, we calculated a statistical Z′-factor to evaluate the quality of the assay (Figure 1D). The Z′-factor (Zhang et al., 1999) of 0.63 for the set-up of a screen for enhancers of U2AF – RNA binding convincingly showed that the FP assay could measure gain of U2AF–RNA binding and set the stage to proceed with a chemical library screen.
We screened the 1593-compound Diversity Set V of the National Cancer Institute (NCI) Developmental Therapeutics Program, for which the modest size was suitable considering the yields of our protein preparation and provided a strong starting point of diverse, pharmacologically-desirable features for subsequent optimization of initial hits. An initial search for inhibitors of U2AF – RNA binding resulted in hit compounds that nonspecifically targeted the RNA alone (data not shown), which we set at a lower priority for further characterization. We next explored the possibility that small molecules could increase binding of the U2AF1S34F–U2AF2–SF1 complex to the relatively low affinity DEK splice site. The Z-factor of the screen was 0.52 (Figure 1E), indicating that hits could be expected under the conditions of this screen (Zhang et al., 1999). An initial pool of seven compounds (0.4% of the starting library) increased FP by at least 75% compared to a positive control signal (excess U2AF1S34F–U2AF2–SF1 protein). Replicates showed that four of these compounds (0.25% of the starting library) reproducibly increased the FP of the U2AF–RNA complex by >75% compared to the positive control. A single compound (NSC 194308, Figure 2A) specifically increased the FP of the U2AF1S34F–U2AF2–SF1–DEK FlRNA ribonucleoprotein without affecting the DEK FlRNA alone. The concentration of half-maximal enhancement (EC50 ~100 μM) was estimated from the increased FP of the U2AF1S34F–U2AF2–SF1–DEK FlRNA complex during titration with the hit enhancer (Figure 2A). Although the potency of the compound was low in this assay with the purified protein complex, we viewed the specific, ribonucleoprotein-directed activity of NSC 194308 as a promising start point for further investigation and optimization, particularly considering that small perturbations of the essential U2AF complex could have potentially significant consequences for living cells.
Figure 2.
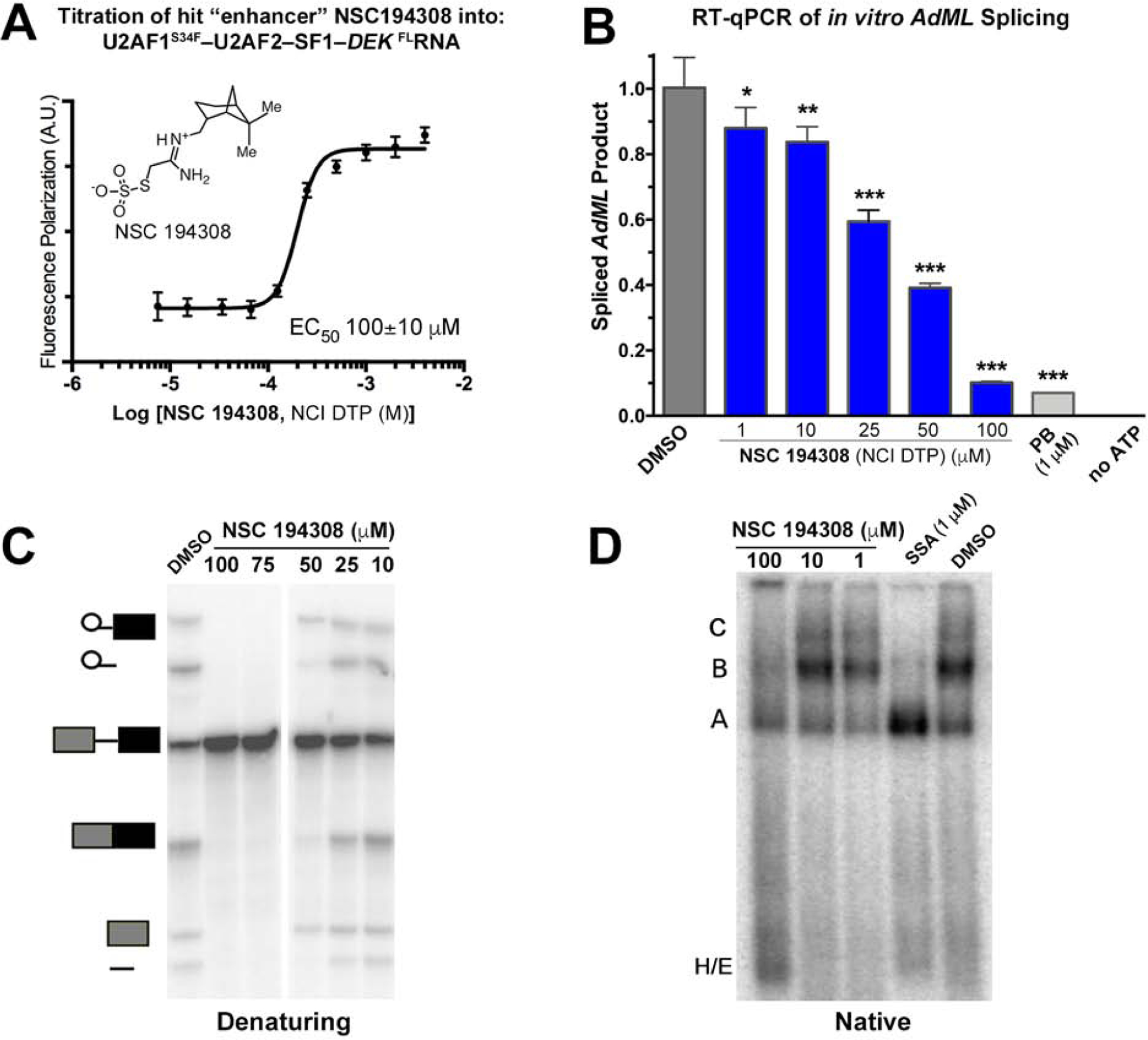
The hit enhancer of the U2AF2 – RNA complex stalls in vitro pre-mRNA splicing at the H/E-stage. (A) The fluorescence polarization dose-response of U2AF1S34F–U2AF2–SF1–DEK FLRNA titrated with hit enhancer (NSC 194308, chemical structure inset). (B) qRT-PCR of spliced AdML products. The relative amounts of spliced product were normalized to a mock-treated negative control (1% v/v DMSO, gray). The positive control is an SF3B1 inhibitor (PB, 1 µM pladienolide-B). Two-tailed, unpaired t-tests with Welch’s correction compared the indicated NSC 194308 concentration with the mock-treated control: *, p<0.05; **, p<0.005; ***, p<0.0005. (C) Denaturing gel analysis of the radiolabeled pre-mRNA substrate and spliced products from reactions treated with the indicated concentrations of NSC 194308 or a mock-treated control. Spliced products are schematically diagrammed to the left. (D) Native gel analysis of spliceosome assembly at 30 minutes in the presence of the indicated concentrations of NSC 194308, positive control (SSA, 1 µM spliceostatin-A), or mock-treated control. The identities of spliceosome complexes are indicated (left), with assembly occurring in the following order: H/E → A → B → C. NCI DTP, NCI Developmental Therapeutics Program. Data represented in A and B are mean ± SD of three replicates. Data represented in C and D are representative of three replicates. See also Figure S1.
Chemical synthesis of NSC 194308 hit compound.
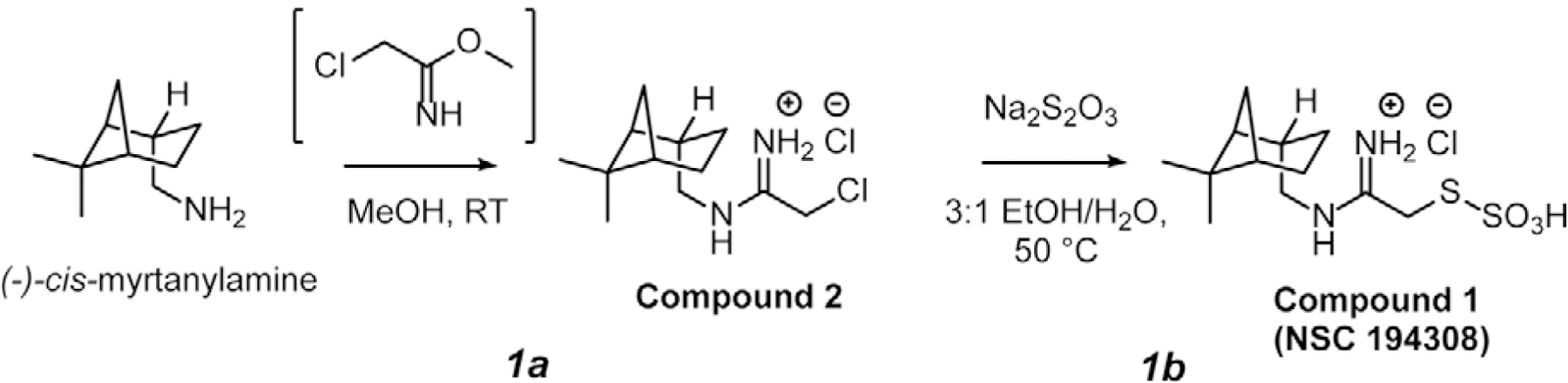
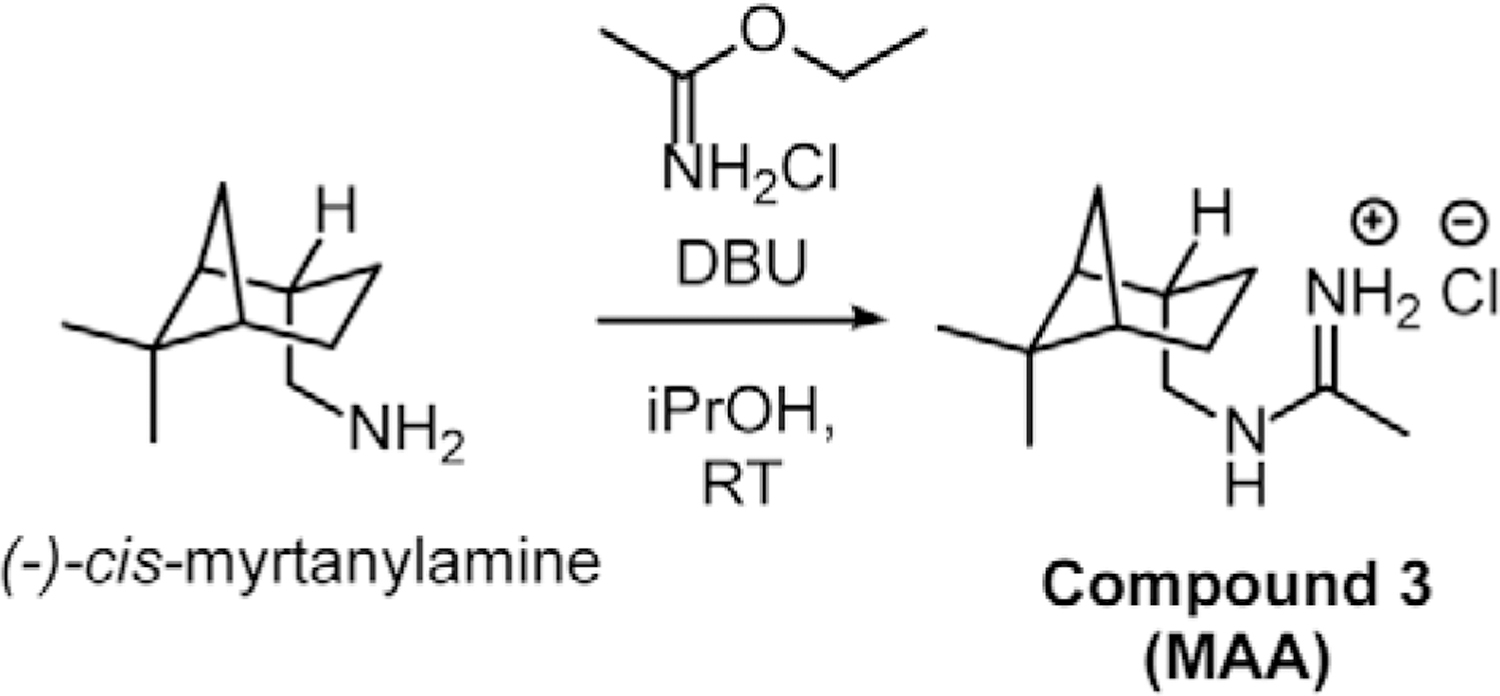
To obtain sufficient, pure NSC 194308 for the investigation of its functional activities, we synthesized the compound starting from (−)-cis-myrtanylamine. Following sequential reactions with chloroacetonitrile under strongly basic conditions followed by displacement of the chloride with sodium thiosulfate (Scheme 1), the compound was precipitated by acidification and its identity confirmed by NMR and X-ray crystallography of the intermediate (Compound 2 in Scheme 1, Star Methods), or NMR and mass spectrometry of the final product (Compound 1 in Scheme 1, Star Methods) (Figure S1A–C). The product showed similar potency for inhibition of in vitro pre-mRNA splicing as the compound obtained from the NCI repository (Figure S1D compared to Figure 2B). We also synthesized a (−)-cis-myrtanyl acetamidinium (MAA) variant (Compound 3 in Scheme 2) to probe structure-activity relationships (described below) and confirmed its identity by X-ray crystallography, NMR, and mass spectrometry (Figure S2A–C). Crystal structures of the NSC 194308 intermediate (Figure S1A) and the MAA variant (Figure S2A) established the absolute stereochemistry of the hit compound. We synthesized NSC 194308 de novo for use in subsequent experiments that required large amounts of material.
Scheme 1.
Scheme 2.
Hit U2AF–RNA “Enhancer” NSC 194308 blocks the pre-mRNA splicing process.
To test pre-mRNA splicing activities without potential complications from the intracellular metabolism or transport of the hit compound, we first examined the efficiencies of in vitro splicing reactions in HeLa nuclear extracts treated with increasing concentrations of NSC 194308 (Figure 2B–C). We used quantitative real-time reverse transcription (qRT)-PCR to detect splicing of a prototypical strong splice site from the Adenovirus Major Late promoter transcript (AdML). We compared a known SF3B1 inhibitor (pladienolide B) alongside mock-treatment with an equivalent amount of solvent (1% v/v DMSO) as positive and negative controls. Contrary to our expectation that an enhancer of U2AF–RNA interaction would stimulate pre-mRNA splicing, the NSC 194308 hit reduced splicing of the AdML substrate in the nuclear extract. We confirmed this result by denaturing gel electrophoresis to visualize the intermediates and products resulting from in vitro splicing of a radiolabeled pre-mRNA substrate (an AdML variant, Figure 2C). The pre-mRNA substrate appeared intact at the highest concentrations of NSC 194308, which ruled out nonspecific RNA degradation due to the compound. We concluded that the apparent concentration of half-maximal inhibition of in vitro splicing by NSC 194308 (IC50 ~35 µM, Figure 2B–C, Figure S1D) was similar to its apparent EC50 for promoting the purified U2AF – RNA complex (Figure 2A and 3E).
Figure 3.
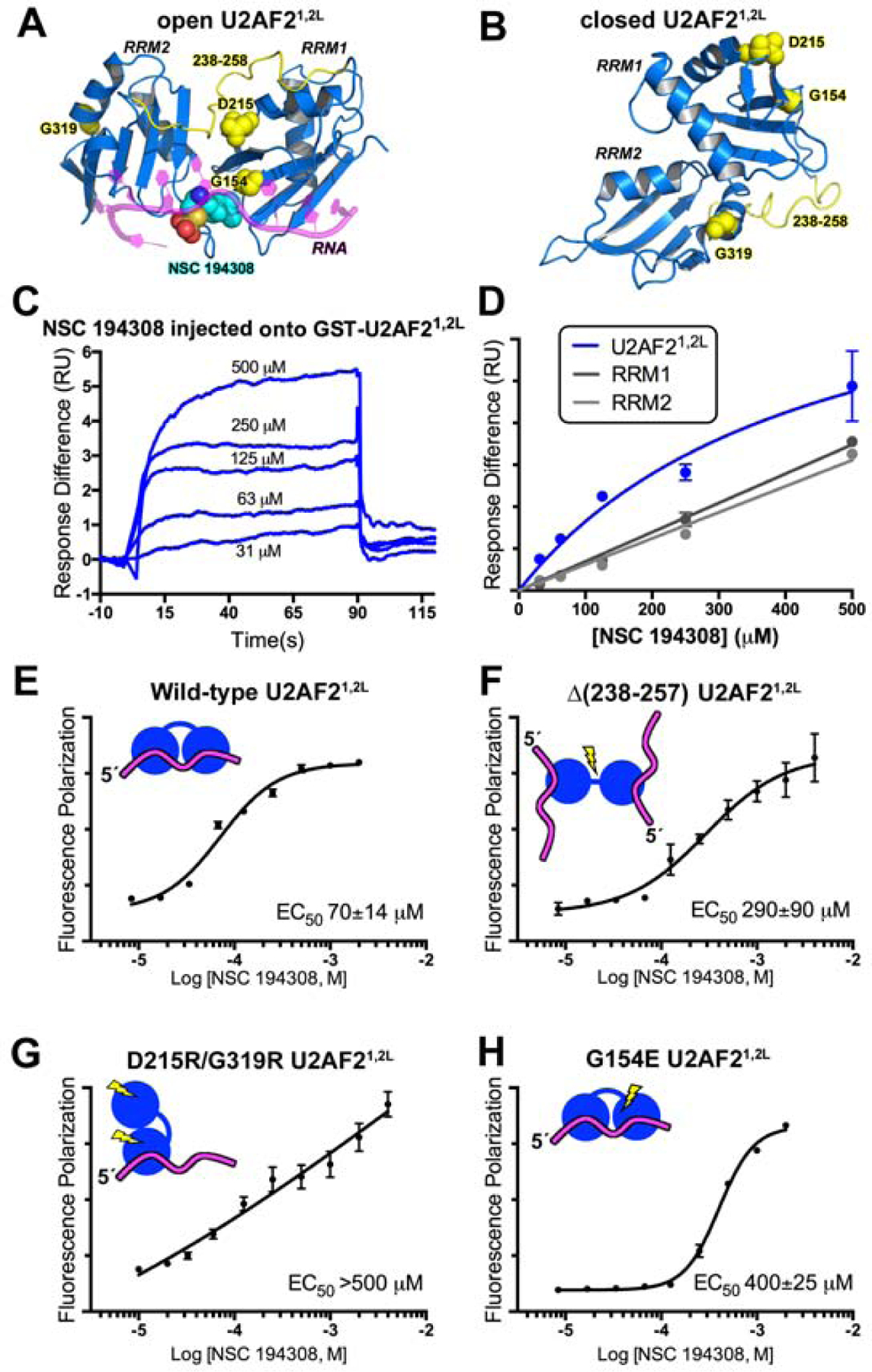
The hit enhancer NSC 194308 binds U2AF2 RRM1/RRM2 (U2AF21,2L). (A) The favorable predicted binding site for NSC 194308 is located between RRM1 and RRM2 (blue) of the open U2AF21,2L conformation (PDB ID 5EV4). Candock scores are listed in Table S2. The bound oligonucleotide (magenta) of the structure is overlaid for reference. Structure-guided mutants are yellow, and for single-site mutations, shown as space-filling CPKs; NSC 194308 is a CPK representation colored by atom: carbon, cyan; oxygen, red; sulfur, orange; nitrogen, blue. (B) Locations of the mutated-regions (yellow) shown on the NMR model (blue) of closed U2AF21,2L (PDB ID 2YH0). (C) Representative sensorgram showing the aligned responses of NSC 194308 injected at the indicated concentrations over an immobilized GST-U2AF21,2L surface. (D) Plot of the average responses (two replicates) from the saturated regions of the sensorgrams fit to a nonlinear steady-state model. NSC 194308 binding to GST-U2AF21,2L, blue; separate GST-RRM1, dark gray; GST-RRM2, light gray. (E-H) Fluorescence polarization dose-responses of the hit enhancer titrated into DEK FLRNA bound to U2AF21,2L variants, including (E) wild-type, (F) inter-RRM linker deletion, (G) D215R/G319R, or (H) G154E. The mean ± SD of three replicates are shown in E - H. Schematic diagrams of the expected conformations of the U2AF2 variants are inset and colored as in A.
To determine the mechanism of action for NSC 194308 inhibition of splicing, we proceeded to analyze the intermediates of spliceosome assembly in the presence of increasing concentrations of the compound using native gel electrophoresis (Figure 2D). The spliceosome assembles through a series of conformational and compositional changes that sequentially form the H-, E-, A-, B- and C- complexes (reviewed in (Wahl et al., 2009)). The U2AF and SF1 splicing factors bind the 3′ splice site in the H- and E-complexes and subsequently stabilize addition of SF3B1 to form the A-complex. With increasing amounts of NSC 194308, the C-complexes are lost and B complexes are drastically reduced. Complexes with similar mobility as the A-complex still form, but do not accumulate to the same levels as induced by an SF3B1-directed inhibitor of the spliceosome (spliceostatin-A) (Corrionero et al., 2011; Roybal and Jurica, 2010). More pre-mRNA migrates in the region of the gel associated with E-complexes, which is the point that U2AF joins the intron, along with the H-complexes that form in a splicing-independent manner. Because E- and A-complexes precede U2AF dissociation (Agafonov et al., 2011; Das et al., 2000), we concluded that NSC 194308 inhibits the pre-mRNA splicing process by stalling spliceosome assembly at a U2AF-dependent checkpoint for the Py tract, which occurs prior to tri-snRNP recruitment and catalytic activation.
NSC 194308 targets an inter-RRM cleft of the U2AF2 subunit.
Because we identified NSC 194308 as a hit compound in a screen using the ternary U2AF1S34F–U2AF2–SF1–DEK FlRNA ribonucleoprotein, the exact target of the compound was uncertain. As a first step towards identifying the binding site of NSC 194308, we used CANDOCK, a fragment-based docking software that leverages a knowledge-based forcefield for pose scoring (Fine et al., 2020). Although a structure of the intact, three protein complex was unavailable, we compared the scores of NSC 194308 docking on the piecewise structures of the human U2AF1, SF1, and two conformations of the U2AF2 subunit, an open RRM1/RRM2 optimally positioned to recognize uridine-rich RNA (Agrawal et al., 2016; Mackereth et al., 2011) or a closed RRM1/RRM2 arrangement that predominates in the absence of RNA (Mackereth et al., 2011) (Figure 3A–B, Table S2). The most favorable score for binding the hit enhancer was obtained for the open U2AF2 structure in comparison with closed U2AF2 or other components of the complex. The top ranked binding site wedged the NSC 194308 molecule between the tandem RRMs of the open U2AF2 conformation and near the bound oligonucleotide, suggesting a structural mechanism of action that stabilizes this major RNA-associated conformation.
To experimentally test the putative binding site of NSC 194308 between the U2AF2 RRMs, we monitored surface plasmon resonance (SPR) using a BIAcore T200 (Figure 3). The RRM-containing region of U2AF2 (U2AF21,2L, residues 141–343) was tethered to a sensor chip slide via an N-terminal GST tag then serial dilutions of NSC 194308 were injected over the surface. The RNA site was omitted due to its rapid dissociation and large response compared to the compound. The response following compound injection onto the GST-U2AF21,2L protein was corrected by subtracting the response of a GST-immobilized control cell. Although the rapid association and dissociation kinetics exceeded the reliable limit of the BIAcore instrument, the sensorgrams demonstrated reversible binding of the compound to the tethered U2AF2 protein. The one-site binding fit of the steady-state responses from a series of NSC 194308 concentrations suggested an apparent equilibrium dissociation constant (KD) of approximately 150 µM for the compound binding U2AF21,2L (Figure 3C–D). Although this value is a lower estimate due to the highest NSC 194308 concentration (~500 µM) that could be achieved without appreciable surface interaction with the compound, it is consistent with the EC50 of NSC 194308 for enhancing association of U2AF1S34F–U2AF2–SF1 with DEK FlRNA (Figure 2A). To further investigate the nature of the binding site, we compared NSC 194308 binding to the separate GST-tagged RRM1 or RRM2 domains of U2AF2 (Figure 3D). The steady-state responses of NSC 194308 injected over individual RRM domains were less than those observed for the intact RRM-containing region at equivalent concentrations. Moreover, a concentration-dependent plot of the steady-state response differences remained linear even at the highest concentrations of NSC 194308 (estimated KD >>>1 M), which typically indicates nonspecific interactions. Altogether, these SPR results demonstrated that NSC 194308 reversibly bound to the U2AF2 subunit in a manner that depended on the integrity of the tandem RRMs.
To further probe the location of the NSC 194308 binding site using a more quantitative assay, we compared the EC50s of structure-guided U2AF2 mutants in FP assays with the DEK FlRNA. Consistent with the docking and SPR results, the apparent potency of the hit compound for enhancing association of U2AF21,2L with DEK FlRNA was similar to the ternary U2AF1S34F–U2AF2–SF1 complex (Figure 3E). We compared the effects of three different structure-guided U2AF2 variants on the ability of NSC 194308 to enhance U2AF21,2L association with the DEK FlRNA. An internal deletion of the inter-RRM linker (residues 258–257) promotes separation of the two U2AF2 RRMs (Mackereth et al., 2011; Sickmier et al., 2006) with little reduction in the RNA binding affinity of a construct with otherwise identical boundaries (residues 141–342) (Agrawal et al., 2016). The linker deletion reduced the NSC 194308 sensitivity of the U2AF2 variant binding to RNA (EC50 290 µM) (Figure 3F). In a complementary investigation of the influence of the inter-RRM conformation on the responsiveness of U2AF2 to NSC 194308, we next prepared a double mutation (D215R/G319R) that is known to destabilize the open conformation while strengthening the closed conformation of the RRMs (Mackereth et al., 2011). The D215R/G319R U2AF2 binds RNA by isothermal titration calorimetry (Mackereth et al., 2011) and single molecule fluorescence approaches (Warnasooriya et al., 2020). Notably, the presence of the D215R/G319R mutation severely decreased the ability of NSC 194308 to enhance U2AF21,2L – RNA binding (Figure 3G). Thirdly, we introduced an electronegative G154E mutation that is expected to repel NSC 194308 from the docked site of U2AF2 (Figure 3H). We found that the G154E mutation reduced the responsiveness of U2AF21,2L – RNA binding to NSC 194308, consistent with the binding site of the docked model. Although the FP assay does not directly measure compound binding to protein, these results support the importance of the open U2AF2 RRM1/RRM2 interface for NSC 194308-mediated enhancement of U2AF2 – RNA binding, in agreement with the results of in silico docking and SPR sensorgrams.
Charged and hydrophobic groups of NSC 194308 contribute to U2AF2-targeted activity.
With the knowledge in-hand that NSC 194308 targets the U2AF2 subunit, we proceeded to investigate the importance of the distinct chemical groups of the compound for inhibition of pre-mRNA splicing in nuclear extract (Figure 4, Figure S2). We first synthesized a variant that omitted the anionic thiosulfate ((−)-cis-myrtanyl acetamidine) (Star Methods, Figure S2A–B). Removal of this functional group nearly abolished the anti-splicing activity of the compound. Next, we compared variations in the hydrophobic group by testing different compounds obtained from the NCI repository (Figure 4A). Reducing the hydrophobic portion of the compound to three carbons (NSC 187511) or to the minimal hexane scaffold (NSC 194285) significantly reduced the activity of the compound, whereas altering the shape by replacement with a globular adamantane (NSC 187514) had little effect (Figure 4, Figure S2F). Conversely, either extending the hexane group by a four-carbon linker from the charged group or adding additional hydrophobic methyl groups restored similar potency as the NSC 194308 parent. We concluded that both the anionic thiosulfate and hydrophobic contacts contributed to the anti-splicing activity of the NSC 194308 hit compound.
Figure 4.
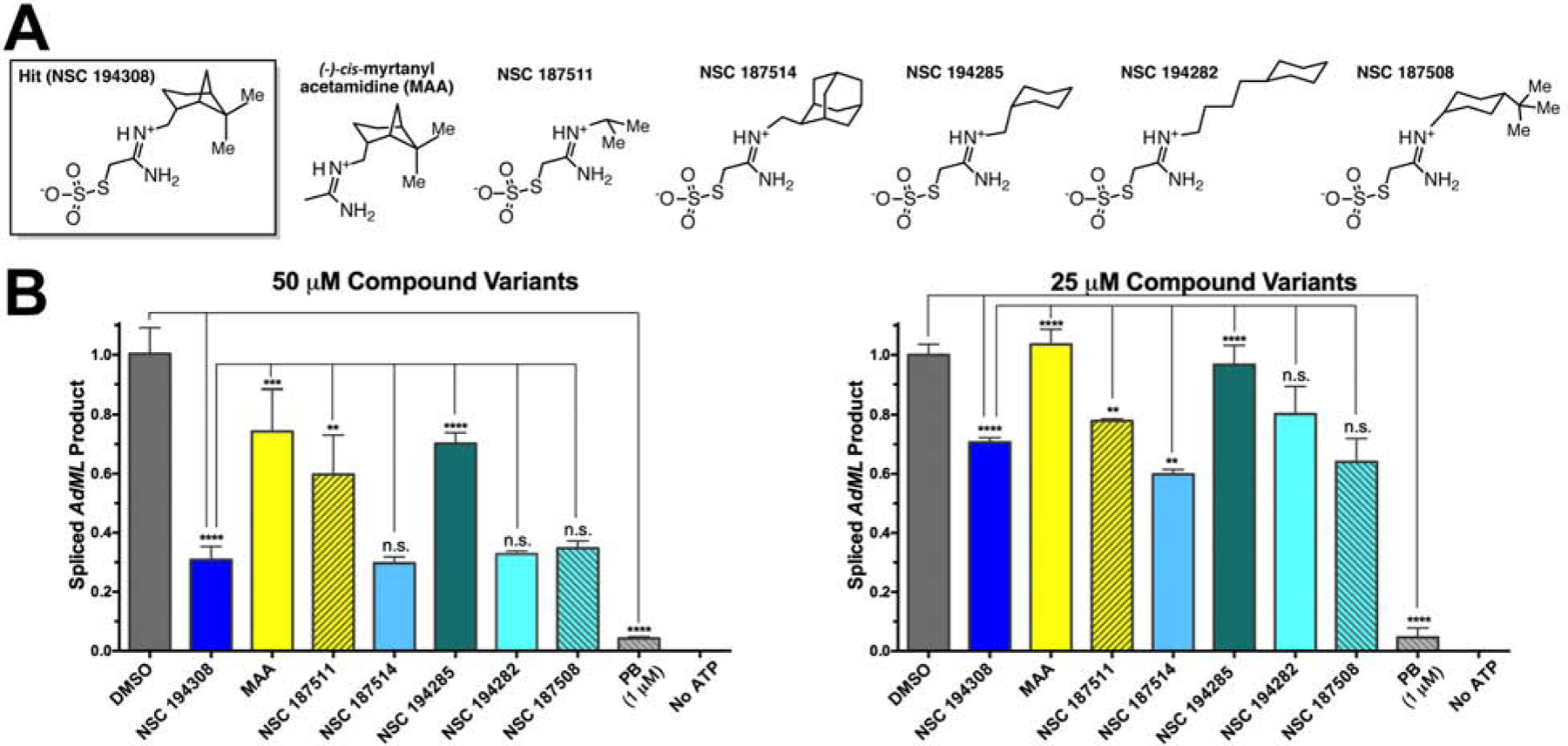
Comparison of NSC 194308 variants shows that anti-splicing activity depends on the thiosulfate and is sensitive to variations of the hydrophobic group. (A) Chemical structures of hit compound variants. The (−)-cis-myrtanyl acetamidine was synthesized de novo (Compound 3, Star Methods and Figure S2) and other compounds were obtained from the NCI DTP. Me, Methyl. (B) Comparison of the spliced AdML product from HeLa nuclear extract treated with either 50 μM (left) or 25 μM (right) compound variants. The products were detected by qRT-PCR, normalized to a mock-treated negative control (1% v/v DMSO), and compared to a pladienolide B (PB) positive control. The mean ± SD of three reactions are graphed. Two-tailed, unpaired t-tests with Welch’s correction: *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.00005. Titrations are shown in Figure S2.
NSC 194308 blocks U2AF2-sensitive splicing in cells.
Since NSC 194308 targets the U2AF2 subunit, we asked whether treatment with the hit compound would modulate U2AF2-dependent splicing in cells (Figure 5). We treated HEK 293T cells with increasing concentrations of NSC 194308 and isolated RNA after five hours of treatment, a timepoint at which nearly all cells remained viable. To compare the effects of reduced U2AF2 levels, we transfected HEK 293T cells with U2AF2-targeted siRNA and isolated RNAs either two- or three-days following transfection, which provided sufficient time for U2AF2 levels to decrease. First, we confirmed that U2AF2 levels were reduced by siRNA and remained constant after NSC 194308 treatment under the assay conditions (Figure S3). Then, we used qRT-PCR to analyze the NSC 194308-associated changes in the levels of three well-characterized transcripts (CCND1, DUSP11, and HTATSF1) that are subject to regulation by SF3B1 inhibitors and U2AF2-family proteins (Corrionero et al., 2011; Loerch et al., 2019; Seiler et al., 2018) (Figure 5A–C). The results shown in Figure 5 represent multiple biological replicates and are averages of three technical replicates normalized to GAPDH. The relative abundance of CCND1, DUSP11, and HTATSF1 was reduced by treatment with NSC 194308 to a similar extent as by knockdown of U2AF2. The levels of CCND1 were most sensitive to the compound (IC50 ~10 μM) compared to the other transcripts (IC50 ~75 μM). These apparent potencies of the compound for modulating transcript levels in cells agreed with the values for inhibiting pre-mRNA splicing in vitro.
Figure 5.
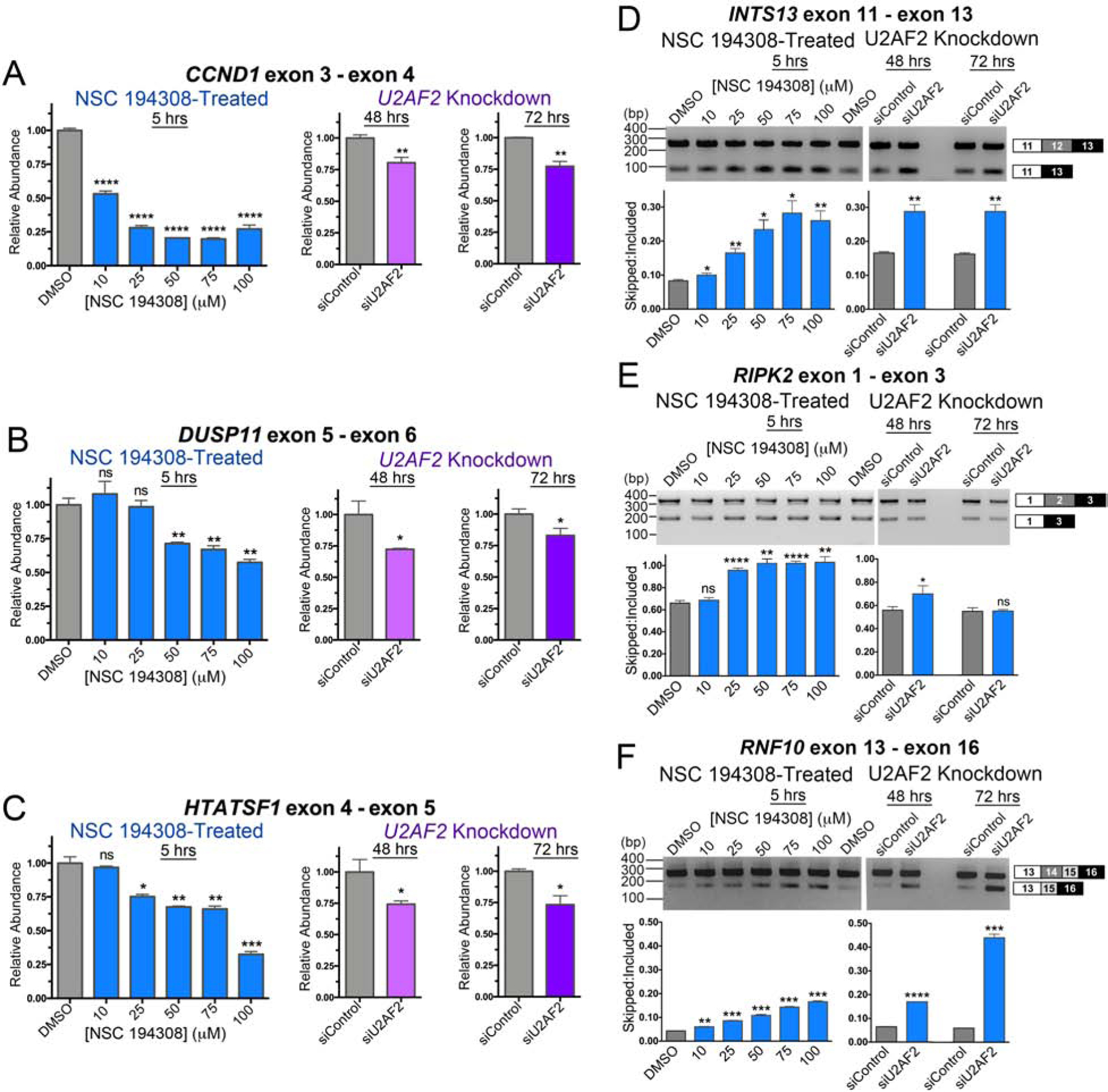
NSC 194308 alters splicing of U2AF2-sensitive transcripts in HEK 293T cells. (A - C) qRT-PCR of gene transcripts with known responsiveness to U2AF2 family members. NSC 194308-treated samples (blue, left) are compared with samples treated with U2AF2-targeted Stealth™ siRNAs (or control siRNA) for either two days (magenta, center) or three days (purple, right). The products of the indicated gene transcripts were normalized to GAPDH controls. The qRT-PCR results are mean ± SD of three reactions. (D - F) RT-PCR of the indicated U2AF2-regulated gene transcripts following either treatment with NSC 194308 (left) or U2AF2 knockdown (right). A mocked-treated control (0.1% v/v DMSO) matches the NSC 194308 solvent. The mean ± SD exon-skipped:included ratios of background-corrected bands from three reactions (six for DMSO control) are plotted below representative ethidium-bromide-stained agarose gels. The expected PCR products are schematized (right). Two-tailed, unpaired t-tests with Welch’s correction: *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.00005. Immunoblots are shown in Figure S3.
Although detected at the majority of 3′ splice sites (Shao et al., 2014; Zarnack et al., 2013), U2AF2 extensively regulates alterative splicing (e.g., (Cho et al., 2015; Ortuno-Pineda et al., 2012; Shao et al., 2014)). To investigate whether NSC 194308 specifically modulated U2AF2-dependent alternative splice sites, we next used reverse transcription (RT)-PCR to examine the effects of the compound on three representative U2AF2-dependent exon-skipping events (Figure 5D–F). Following knockdown of U2AF2 in HEK 293T cells, we noted robust increases in the skipping of INTS13 (also called ASUN) and RNF10 exons, whereas RIPK2 changes were subtle, in agreement with previous observations using HeLa cells (Shao et al., 2014). Remarkably, addition of NSC 194308 increased skipping of the U2AF2-sensitive exons in the spliced products. The similar effects of U2AF2 knockdown and NSC 194308 treatment were consistent with the ability of the compound to stall in vitro spliceosome assembly at the stages involving U2AF2 (Figure 2).
Cells expressing mutant U2AF1 have increased sensitivity to NSC 194308.
Considering the premise that human cells expressing mutant splicing factors are sensitized to splicing modulators, we asked whether NSC 194308 treatment would selectively decrease the survival of K562 leukemia cells expressing the MDS-associated S34F mutant of U2AF1. First, we tested the NSC 194308-sensitivity of a cell line with a stably integrated doxycycline-inducible, FLAG-tagged S34F-mutant U2AF1 compared to a FLAG-tagged wild-type U2AF1 control for overexpression (Figure S4). Treatment with NSC 194308 significantly reduced the survival and lowered the IC50 (p-value 0.0047 in unpaired t-test with Welch’s correction) of S34F mutant-induced K562 cells compared to the wild-type induced control cells (Figure 6A). Notably, the apparent IC50 of the K562 cells overexpressing the MDS-associated S34F mutation occurred at much lower NSC 194308 concentrations than the purified ribonucleoprotein complex (approximately 5 μM compared to 100 μM), suggesting that slight perturbations of the essential U2AF2 protein could have a larger functional impact in the context of prior insults to the pre-mRNA splicing pathway. Next, to reflect the levels of untagged U2AF1 expression expected in cancer cells, we edited either the S34F U2AF1 mutation or a wild-type control into a K562 cell line using CRISPR methods. Successful introduction of the mutation was verified by genomic sequencing and supported by S34F-associated changes in splicing (Figure 6B–G). Following NSC 194308 treatment, the survival and IC50 of the edited cell line expressing physiological levels of S34F-mutant U2AF1 also were significantly reduced (p-value 0.0065) compared to the wild-type counterpart (Figure 6A). The selective killing of S34F U2AF1-expressing cells by NSC 194308 was similar in magnitude to previously observed effects on analogous cell lines of an SF3B1 inhibitor, sudemycin D1 (approximately 2–4-fold selection) (Shirai et al., 2017).
Figure 6.
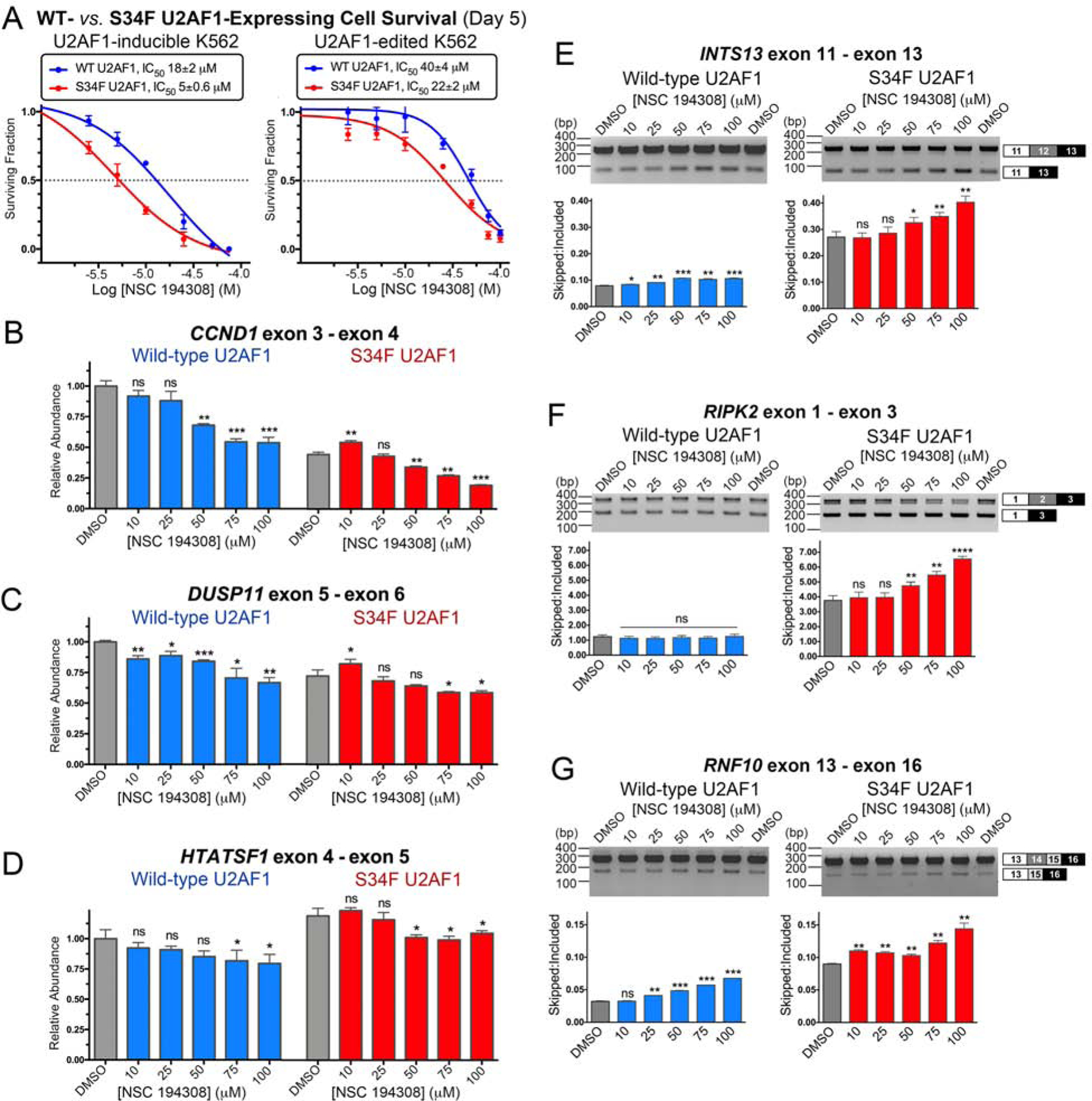
NSC 194308 preferentially kills K562 leukemia cells expressing MDS-associated S34F-mutant U2AF1 and alters splicing in these cells. (A) The fraction of viable cells (detected by trypan blue staining) normalized to a mock-treated sample (0.1% v/v DMSO, matching solvent) are plotted versus the log10 concentration of NSC 194308. The mean ± SD of three replicates are shown. K562 cells with stably-integrated, doxycycline-induced WT or S34F-mutant U2AF1 or CRISPR-edited endogenous U2AF1 gene were treated with the indicated concentrations of NSC 194308 for five days. The inhibitory concentrations (IC50) at half-maximal cell survival are inset above. (B - D) qRT-PCR (normalized to the GAPDH control of the WT sample) or (E - G) RT-PCR of the indicated gene transcripts from U2AF1-edited cells treated for seven hours with NSC 194308. The qRT-PCR results are mean ± SD of three reactions. The mean ± SD exon-skipped:included ratios of background-corrected bands from three reactions (six for DMSO control) are plotted below representative ethidium-bromide-stained agarose gels. Wild-type (WT), blue; S34F-mutant, red. Two-tailed, unpaired t-tests with Welch’s correction compare the indicated NSC 194308-treated sample with the matching WT or S34F mutant DMSO control: *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.00005. Immunoblots are shown in Figure S4.
With the knowledge that NSC 194308 inhibits U2AF2-dependent splicing and that U2AF1 is a heterodimeric partner of U2AF2, we thought that dysregulated splicing was likely to contribute to the sensitivity of S34F U2AF1-expressing cells to the compound. To explore this possibility, we examined representative transcripts isolated from edited K562 cell lines following seven-hour treatments with increasing concentrations of NSC 194308 (Figure 6). In general, the changes in transcripts from the NSC 194308-treated K562 leukemia cells were similar to those of RNAs from HEK 293T cells treated with NSC 194308 or U2AF2-targeted siRNA shown in Figure 5 and described in reference (Shao et al., 2014). In some cases, the presence of the S34F mutation of U2AF1 appeared to exacerbate or alter the NSC 194308-induced changes in gene expression. In particular, treatment with the compound further decreased the levels of CCND1 transcript or the exon-included RIPK2 splice form, which were already expressed at lower levels in the S34F-mutated cells than in the wild-type counterpart. We concluded that aggravation of splicing defects by NSC 194308 is a likely mechanism of action for its preferential killing of cells expressing mutant U2AF1.
DISCUSSION
In this study, we demonstrated that the U2AF2 splicing factor and its RNA binding properties can be targeted de novo by a small molecule. The success of our proof-of-concept screen implies that expansion of such an approach would identify additional U2AF2 modulators. We identified the binding site of NSC 194308 between the U2AF2 RRMs by a combination of docking, mutagenesis, and binding assays. This result is significant because the current repertoire of splicing factors targeted by small molecules is limited. Although the FDA-approved small molecule risdiplam (Evrysdi™) modulates pre-mRNA splicing, it binds a specific, AG-rich RNA motif of SMN2 for treatment of SMA rather than cancers (Wang et al., 2018). Antisense oligonucleotides show promise as modulators of specific splice sites, yet the ambiguity of the exact drivers of cancers, coupled with the difficulty of oligonucleotide delivery, remain on-going challenges confronting clinical applications to treat cancers. Despite a plethora of recurrent mutations in the early components of the pre-mRNA splicing machinery across multiple cancer types, a major class of clinically advanced spliceosome inhibitors universally target the same site of the splicing factor SF3B1 (reviewed in (Effenberger et al., 2017)). Screens for inhibitors of pre-mRNA splicing in nuclear extracts or cells are complex due to >100 participants in the splicing reaction, which complicates identifying the target for optimization. Considering the widespread role of U2AF2 in spliceosome assembly, small molecule modulators of U2AF2 have the potential to exploit the vulnerability of dysregulated pre-mRNA splicing in cancers. By analogy, a successful precedent has been set by SF3B1 inhibitors, which preferentially kill spliceosome-dysfunctional cells regardless of the source of the pre-mRNA splicing defects (Hsu et al., 2015; Larrayoz et al., 2016; Lee et al., 2016; Seiler et al., 2018; Shirai et al., 2017; Xargay-Torrent et al., 2015). Here, we demonstrated that a U2AF2-targeted small molecule can preferentially kill leukemia cell lines expressing the S34F-mutant U2AF1 that recurs in MDS (Figure 6), a disease currently without a chemotherapeutic cure.
An equally significant outcome of this project is the power of stabilizing stepwise intermediates of spliceosome assembly as a means to alter the splicing process. Notably, all three families of SF3B1-targeted splicing inhibitors stall spliceosome assembly at the A-stage near the time of U2AF2 release (Corrionero et al., 2011; Effenberger et al., 2014; Folco et al., 2011; Roybal and Jurica, 2010), although such inhibitors also can interfere with later stages of splicing (Effenberger et al., 2016). Certain quinone derivatives have been found to block the second catalytic step of splicing by stalling an intermediate similar to the C-complex (Berg et al., 2012). A distinct small molecule, identified in a screen for stage-specific inhibitors of spliceosome assembly, acts to block the conversion of B-stage spliceosomes to the activated BACT configuration (Sidarovich et al., 2017). At the earliest, E-stage of splicing, the SMN-C class of compounds interacts with the 5′ splice site of SMN exon 7 and promotes U1 small nuclear ribonucleoprotein (snRNP) association (Campagne et al., 2019; Sivaramakrishnan et al., 2017). By analogy with our findings for the NSC 194308 enhancer of U2AF2 – RNA complexes, we wonder if spliceosome assembly with, and hence splicing of, the SMN2 exon 7 would be stalled by elevated concentrations of the SMN-C modulator.
Recently, a chemical library screen identified phenothiazines as general inhibitors of protein-protein interactions with domains called U2AF homology motifs (UHM), among which U2AF2 carries a prototype (Jagtap et al., 2020). Optimized phenothiazines blocked the H/E-to-A complex transition of spliceosome assembly, presumably by inhibiting UHM – ligand interactions mediated by early stage splicing factors such as U2AF2. Here, we adopt a distinct strategy of enhancing, rather than inhibiting, U2AF2 action in the initial steps of splicing. Spliceosomes treated with our NSC 194308 hit compound are expected to accumulate at an E-like, U2AF2-containing stage, whereas phenothiazine-based inhibition of UHMs could stall even earlier (i.e., at an H-like stage). Since H- and E- complexes co-migrate by the standard native gel methods used by our colleagues (Jagtap et al., 2020) and in this work, detailing the mechanisms of action remains an important future direction for advancement of these compounds as meaningful tools to study the structural and functional consequences of spliceosome assembly. A second notable question would be whether phenothiazine-related compounds can perturb splicing in cells and (presumably thereby) preferentially kill cell lines expressing mutant splicing factors, as shown here for NSC 194308.
In addition to the spliceosome, many of the macromolecular machines of biology are assembled in a series of tightly regulated steps. This work illustrates the feasibility of blocking specific pathways by using small molecules to stabilize assembly intermediates, highlighting a potentially broad strategy for the development of new molecular tools and therapeutics.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for additional information and reagents should be directed to the Lead Contact, Clara L. Kielkopf (clara_kielkopf@urmc.rochester.edu).
Materials availability
Data and materials are available from the authors by reasonable request.
Data and code availability
The coordinates and structure factors are deposited in the Cambridge Crystallographic Data Centre (CCDC) with access codes CCDC 1981586 (Compound 2) and CCDC 1981585 (Compound 3). This study did not generate any code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and growth conditions
The human embryonic kidney (HEK) 293T (female) cell lines were obtained directly from ATCCR (CRL-3216™). HEK 293T cells were cultured in Dulbecco’s modified Eagle media (Gibco™), 10% fetal calf serum (R&D Systems Inc.), 2 mM L-glutamine and penicillin-streptomycin (pen-strep, Gibco™). As needed, cells were split with Trypsin-EDTA (Gibco™). The K562 (female) cell lines expressing doxycycline-inducible 3xFLAG wild-type or S34F U2AF1 have been described (Smith et al., 2019). Separately, the S34F mutation or matching wild-type control were introduced into a K562 background using CRISPR methods. K562 cells from ATCCR (CCL-243™), resuspended in Buffer R (Thermo Fisher Sci.), were gently mixed with the freshly prepared Hifi2-Cas9 (MCLAB) – gRNA (Synthego) ribonucleoprotein complex, single stranded donor oligodeoxynucleotide (ssODN) and electroporated using the Neon Electroporation System (Thermo Fisher Sci.). The clonal lines were expanded by limiting dilution and confirmed to harbor the desired substitution and zygosity by amplicon-based next-generation sequencing. The K562 cell lines were cultured in either RPMI 1640 (Gibco™) for the U2AF1-inducible cells or IMDM (Gibco™) for the U2AF1-edited cells, supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine and pen-strep. All cells were maintained at 37 °C in a humidified chamber containing 5% (v/v) CO2.
METHOD DETAILS
Preparation of proteins and RNAs for binding assays
The ternary protein complex of human U2AF1S34F (wild-type or S34F mutant, residues 1–193 of NCBI RefSeq NP_006749), U2AF2 (residues 85–471 of NP_001012496), and SF1 (residues 1–255 of NP_004621) was prepared as described (Fei et al., 2016). In brief, the U2AF1 and U2AF2 subunits were co-expressed in Escherichia coli and the tags were removed. The SF1 subunit was mixed with the heterodimer and the ternary complex was isolated by size exclusion chromatography in a buffer containing 25 mM HEPES pH 6.8, 150 mM NaCl, 3% (v/v) glycerol, 20 μM ZnCl2, 3 mM βME and 0.5 mM TCEP. The RNA recognition motif region of the U2AF2 subunit (residues 141–342) was prepared as described (Agrawal et al., 2016). The combined yield of protein prepared from approximately 12 liters of E. coli host cells (6 L co-expressing the human heterodimer plus 6 L expressing SF1) was used for each screen (for inhibitors or enhancers).
The RNA oligonucleotides were synthesized and HPLC purified by Dharmacon™ (Horizon Discovery). The DEK RNA oligonucleotide sequence is 5′-UAAGAAAUACUAAAUUAAUUUCUAG AAAAGAGUCU. Where indicated, a fluorescein (Fl) was tethered via a 6-methylene carbon linker to the 5′ terminus of the oligonucleotide.
Fluorescence polarization (FP) screens for U2AF–RNA modulators
The screening data are summarized in Table S1. Positive and negative control samples were plated in 24 replicates in 384-well flat bottom clear polystyrene plates (Corning, USA). A concentration of protein (80 nM) was chosen preceding the exponential phase of the binding curve of the low affinity DEK FlRNA site (20 nM). Excess U2AF1S34F–U2AF2–SF1 protein (1.3 μM) served as the positive control for enrichment of the complex and matching solvent (1% v/v DMSO) as the negative control. The samples were incubated at room temperature for 60 min, an empirically determined timepoint to equilibrate the reading. The FP was measured at 520 nm using an EnVision plate reader following excitation at 490 nm.
The NCI Diversity Set V library (1,593 compounds) in 96-well format (10 mM stock in DMSO) was transferred to 384-well plates and diluted to 1 mM in DMSO using a JANUS Varispan Automated Workstation (PerkinElmer). The U2AF1S34F–U2AF2–SF1 protein and FlDEK variant oligonucleotide were plated at a total 20 μL reaction volume per well. The JANUS pintool was used to transfer 100 nL aliquots from the library plate to the sample wells and achieve a 5 μM final concentration of each compound per well. The unlabeled RNA, excess protein, or DMSO were added to the appropriate positive and negative control wells as described above. The sample plates were incubated at room temperature for 60 min, the timepoint at which the Z′-factor had plateaued. Then, FP was measured using the EnVision plate reader. The Z′-factor is a statistical characteristic of the assay quality that is independent of the library and assessed from the mean values and standard deviations of the positive and negative control samples (μC+, μC−, σC+ and σC−):
The quality of the HTS was assessed using the statistical Z-score (Zhang et al., 1999), for which μs and μC− are the respective means, and σs and σC− are the standard deviations among of the samples and negative control signals:
The compounds that increased the FP of the sample wells by >75% compared to the signal of the positive controls were considered “hits”. To test the reproducibility of each hit compound’s effect compared to appropriate positive and negative controls, these initial hits were cherry-picked using the JANUS workstation and the FP measurements were replicated in triplicate. The FP dose-response curves of reproducible U2AF1S34F–U2AF2–SF1–FlDEK RNA modulators were measured using a Fluoromax-3 fluorimeter. The compound was titrated into a mixture of protein and RNA in the cuvette. The added volume of DMSO solvent was maintained at <2% (v/v) of the total sample volume. The sample was excited at 490 nm and the anisotropy measured at 520 nm. The data were fit with a sigmoidal dose response model using Prism (GraphPad Software).
Synthesis of hit compound NSC 194308 and variant
All reactions were carried out under an argon atmosphere with magnetic stirring, unless otherwise noted. Reagents were used as obtained from commercial suppliers without further purification. Dry solvents were dried for at least 24 hours over activated 3 Å molecular sieves before use in any reactions. Reactions were monitored by thin layer chromatography (TLC) separation on pre-coated silica gel 60 F254 glass-supported plates (Millipore-Sigma). The TLC plates were visualized under UV light then stained using p-anisaldehyde/sulfuric acid solution and gentle heating. NSC 194308 was dissolved in DMSO solvent at 10 mM stock concentration, stored at −80° C, and serial diluted in DMSO or matching buffers as needed immediately prior to each experiment. The 1H NMR spectra were recorded at room temperature on a 500 MHz Bruker Avance spectrometer. Chemical shifts are given in parts per million (ppm) referenced to solvent residual proton resonance (δ = 3.31 ppm for d4-MeOD or δ = 2.50 ppm for d6-DMSO). NMR data are reported as: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, dq = doublet of quartets, br = broad), coupling constants (J) given in Hz, and integration. The 13C NMR spectra were recorded at room temperature on a 125 MHz Bruker Avance spectrometer, with proton decoupling. Chemical shifts are given in parts per million (ppm) from referenced to solvent carbon resonance (δ = 49.00 for d4-MeOD).
Synthesis of Compound 2 from (−)-cis-myrtanylamine (Scheme 1a).
Metallic Na (23 mg, 1 mmol) was added to a flame-dried round bottom flask containing dry MeOH (5 ml), and an exothermic reaction took place with evolution of H2 gas. This solution was stirred for 1 h at room temperature, after which chloroacetonitrile (226 mg, 2.5 mmol) was added in a drop-wise fashion. This new solution was allowed to react at room temperature for 2 hours. Then, (−)-cis-myrtanylamine (306 mg, 1 mmol) was added dropwise, and the pH adjusted to roughly 1 with 3.3 M ethanolic HCl (circa 1 ml). The solution was allowed to react for 16 h at room temperature. The reaction mixture was concentrated, and isopropyl alcohol (iPrOH) (30 ml) was added, and the solids were triturated and filtered. The filtrate was concentrated to roughly half the initial volume (approximately 15 ml) and diethyl ether (15 ml) was added and the mixture was left to stand for 15 min. The product Compound 2 (2-chloro-N-(−)-cis-myrtanyl acetamidinium hydrochloride) was filtered off as a pink powder, and dried under vacuum (371 mg, 70% yield). The crystal structure of Compound 2 was determined as described below and is shown in Figure S1A. 1H NMR (500 MHz, d6-DMSO) δ 10.07 (br s, 1H), 9.54 (br s, 1H), 9.12 (br s, 1H), 4.41 (s, 2H), 3.22 (t, J = 6.9 Hz, 1H), 2.35 – 2.31 (m, 2H), 1.95 – 1.89 (m, 5H), 1.46 – 1.37 (m, 1H), 1.18 (s, 3H), 1.01 (s, 3H), 0.87 (d, J = 9.4 Hz, 1H) ppm. High-resolution mass spectrometry (HRMS) (electrospray ionization, ESI) (m/z): Calculated for C12H22ClN2 ([M+H]+): 229.1466; Observed: 229.1470.
Synthesis of Compound 1 from Compound 2 (Scheme 1b).
To a stirred solution of Compound 2 (66 mg, 0.25 mmol) in ethyl (Et) alcohol (0.75 ml) in an Erlenmeyer flask was added a solution of Na2S2O3 (40 mg, 0.25 mmol) in H2O (0.25 ml). The combined solution was heated to 50 °C for 1 h. Upon full consumption of the starting material by TLC monitoring, the solution was allowed to cool to room temperature. The solution was kept acidic by addition of 1 drop of concentrated hydrochloric acid (37% w/w) while stirring, and afterwards the mixture was allowed to stand at room temperature for 15 min without stirring. The product Compound 1 (NSC 194308) was filtered off as a white powder, and dried under vacuum (57 mg, 67% yield). 1H NMR (500 MHz, MeOD) δ 9.25 (br s, 1H), 8.94 (br s, 1H), 8.71 (br s, 1H), 3.98 (s, 2H), 3.30 (s, 2H), 2.47 – 2.40 (m, 2H), 2.12 – 1.90 (m, 5H), 1.54 (dt, J = 20.5, 8.5 Hz, 1H), 1.24 (s, 3H), 1.07 (s, 3H), 0.98 (d, J = 9.8 Hz, 1H) ppm. 13C NMR (125 MHz, d4-MeOD) δ 167.5, 49.8, 44.9, 42.5, 40.5, 39.6, 35.8, 33.9, 28.3, 26.9, 23.5, 20.7 ppm (Figure S1B–C). HRMS (ESI) (m/z): Calculated for C12H23N2O3S2 ([M+H]+): 307.1145; Observed: 307.1141.
Synthesis of Compound 3 from (−)-cis-myrtanylamine (Scheme 2).
1,8-Diazabicyclo[5.4.0]undec-7-ene (304 mg, 2 mmol) was added to a stirred solution containing dry iPrOH (2 ml), ethyl acetimidate hydrochloride salt (248 mg, 2 mmol) and (−)-cis-myrtanylamine (153 mg, 1 mmol) and the solution was allowed to stir at room temperature for 20 h. Water (20 ml) was added along with Et2O (20 ml) and the reaction was transferred to a separating funnel. The aqueous layer was extracted twice more with Et2O (20 ml each) and the combined organic layers were dried over MgSO4 and concentrated. The crude oil was dissolved in iPrOH (20 ml) and acidified with 3.3 M HCl in EtOH until a pH of roughly 1 was obtained. The turbid mixture was concentrated and the solid imidinium salt was triturated with EtOAc (20 ml) and filtered to yield white flakes of Compound 3 (N-(−)-cis-myrtanyl acetamidinium hydrochloride, MAA). The compound was then recrystallized from 19:1 acetone/iPrOH to give lustrous white needles (109 mg, 51% yield). The crystal structure of Compound 3 was determined as described below and is shown in Figure S2A. 1H NMR (500 MHz, d4-MeOD) δ 3.28 – 3.16 (m, 2H), 2.46 (dtd, J = 11.8, 6.1, 1.8 Hz, 1H), 2.42 – 2.32 (m, 1H), 2.22 (s, 3H), 2.11 – 1.90 (m, 5H), 1.59 – 1.43 (m, 1H), 1.24 (s, 3H), 1.07 (s, 3H), 0.98 (d, J = 9.8 Hz, 1H) ppm. 13C NMR (125 MHz, d4-MeOD) δ 166.1, 49.9, 44.9, 42.5, 40.5, 39.6, 34.0, 28.3, 26.9, 23.5, 20.7, 18.8 ppm (Figure S2B–C).
Crystal Structure Determinations of Compound 2 and Compound 3.
Compounds 2 and 3 were each crystallized from 19:1 acetone/iPrOH. Data sets were collected at 100 K using a Rigaku XtaLAB Synergy-S Dualflex diffractometer equipped with a HyPix-6000HE HPC area detector and PhotonJet (Cu) X-ray source. Structures were determined using ShelXT (Sheldrick, 2015b), refined using ShelXL (Sheldrick, 2015a), and converged to Rfactors of 0.118 and 0.039 for I>2σ(I). Structures were manipulated and figures generated using Olex2 (Bourhis et al., 2015) (Figures S1A and S2A).
In vitro splicing assays using nuclear extract
The DNA templates for AdML substrate pre-mRNAs have been described (Jurica et al., 2002; Reichert et al., 2002). For qRT-PCR splicing analysis, the pre-mRNAs were prepared using the mMESSAGE mMACHINE™ T7 transcription kit (Thermo Fisher Sci.) and purified using the MEGAclear™ transcription clean-up kit (Thermo Fisher Sci.). The in vitro splicing reaction used HeLa nuclear extract (ProteinOne) with a similar protocol as described (Effenberger et al., 2013), except that the reactions were incubated at 30 °C for one hour, heat-inactivated at 60 °C for 10 min, diluted two-fold, and finally stored at −80 °C. The spliced products were diluted to 1/40th of the total PCR reaction volume and quantified by Taqman qRT-PCR as described (Effenberger et al., 2013).
For denaturing gel splicing analysis, G(5′)ppp(5′)G-capped and 32P-UTP body-labeled pre-mRNA was created using standard T7 run-off transcription and gel purified. Nuclear extract was prepared from HeLa cells cultured in MEM/F12 1:1 and 5% (v/v) newborn calf serum and stored in 20 mM Tris pH 7.9, 0.1 M KCl, 0.2 mM EDTA, 20% (v/v) glycerol, 0.5 mM DTT (Dignam et al., 1983). The in vitro splicing reactions contained 10 nM pre-mRNA, 60 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg/mL tRNA, 40% (v/v) HeLa nuclear extract and increasing amounts of drug solved in 1% (v/v) DMSO, and were incubated at 30°C for 60 min. Following phenol:chloroform extraction and ethanol precipitation, RNAs were separated by electrophoresis in a 15% (v/v) polyacrylamide denaturing gel with 7 M urea in 1X TBE (45 mM Tris-borate, 1 mM EDTA). Gels were run at 35 W for 2 hours and visualized by phosphorimaging with a Typhoon Scanner (Molecular Dynamics).
For native gel analysis of spliceosome assembly, aliquots of the same splicing reactions were mixed with an equal volume of native loading buffer (20 mM Trizma base, 20 mM glycine, 25% (v/v) glycerol, 0.05% (w/v) cyan blue, 0.05% (w/v) bromophenol blue, 1 mg/mL heparin sulfate. Splicing complexes were separated by electrophoresis in a 2.1% (w/v) low-melt agarose native gel in 20 mM Tris, 20 mM glycine. Gels were run at 72 V for 3 h 50 min, vacuum-dried onto Whatman paper for 45 min at 65 °C, and visualized by phosphorimaging as described above.
Parameterization of docking software and preparation of structures
NSC 194308 was docked to the separate structures of the U2AF1, U2AF2, and SF1 components using CANDOCK, a fragment-based docking protocol that implements knowledge-based statistical functions for pose minimization and scoring (Fine et al., 2020). Unique chains of interest were extracted from the Protein Data Bank (PDB) structure coordinate files in preparation for docking, including PDB ID’s 5EV4 (Agrawal et al., 2016), 1JMT (Kielkopf et al., 2001), 1K1G (Selenko et al., 2003), 2YH0 (Mackereth et al., 2011), 4FXW (Wang et al., 2013), and 4YH8 (Yoshida et al., 2015). The Gen3D module of the Open Babel package (O’Boyle et al., 2011) was used to generate and optimize the geometry of a 3D model for the NSC 194308 compound. We used COFACTOR to predict candidate ligand binding sites for each protein structure based on homology to templates available in the wwPDB (Roy et al., 2012; Zhang et al., 2017). Finally, we used CANDOCK (Fine et al., 2020) to assess the fit of the NSC 194308 model coordinates for the candidate binding sites of the protein structures, with otherwise default values. The top minimized poses of docked NSC 194308 for each protein were scored relative to the knowledge-based forcefield as described (Bernard and Samudrala, 2009; Fine et al., 2020) (Table S2).
Biomolecular interaction by surface plasmon resonance
We used a Biacore™ T200 with CM5 sensor chips (GE Healthcare Inc.). We immobilized approximately 11,000 RU of anti-GST in a buffer containing 10 mM HEPES pH 6.8, 150 mM NaCl, 0.2 mM TCEP, 0.005% (v/v) P20 (HBS buffer). We then immobilized 900 – 1500 RU of GST on the reference surface and a similar amount of each GST-U2AF2 protein on the active surface of the sensor chip. The affinity experiments used matched injection and running buffers of HBS plus 1% (v/v) DMSO that were carefully and reproducibly matched. Each injection of NSC 194308 was repeated in duplicate at a flow rate of 30 µl −1 min and double-referenced to injections of the same buffer. The injection time was 90 s followed by 60 s dissociation. The surface was regenerated by application of 1 M NaCl for 60 s between injections. Data was analyzed using the BIAcore T200 Evaluation software (v3.2, GE Healthcare Inc.). The GST-U2AF21,2L responses were corrected for a control cell on which GST was immobilized. The responses at equilibrium (Req) were calculated by averaging 5 s of double-referenced data immediately before the end of the injection. The Req were plotted as a function of the corresponding NSC 194308 concentrations and fit to a one-site binding equation using Prism (GraphPad Software) to obtain the apparent equilibrium dissociation constant.
Transfections
U2AF2 levels were reduced by transfection of HEK 293T cells with Stealth™ siRNA HSS117616 (Thermo Fisher Sci.) and compared to a “lo GC” negative control Stealth™ siRNA using JetPrimeR Polyplus-transfection as instructed by the manufacturer. Cells were split after 24 hrs and harvested at two or three days post-transfection.
Viability Assays of K562 Cells
Inducible K562 cells were plated at 2.5×105 cells mL−1 in RPMI 1640 containing 0.25 μg mL−1 doxycycline (dox) (Takara Bio USA Inc.) in 10 cm plates and incubated at 95% humidity, 5% CO2, 37° C. After 48 hrs, cells were diluted in an equal volume of fresh media with dox and plated into 24 well dishes at 1×105 cells mL−1. Compound at the indicated concentration, or comparable DMSO control (0.1% v/v), was then added. Live cell numbers were measured after five days by trypan blue exclusion assays as suggested by the manufacturer (Invitrogen). CRISPR-edited K562 cells in IMDM without dox otherwise were compound-treated in an identical manner.
NSC 194308 treatments for RNA isolation
HEK293T cells were plated in 24 well plates at 1.2×105 cells/mL and incubated overnight. CRISPR-edited K562 were plated in 6 well plates at 2.5×105 cells mL−1 prior to treatment with compound. Cells were treated with the indicated amounts of NSC 194308 compound (from 1000x stocks in DMSO) or 0.1% (v/v) DMSO as a negative control. To observe the primary effects of the compound, HEK 293T cells or K562 cells were harvested after five or seven hours of exposure, respectively.
RT-PCR and qRT-PCR of RNAs from cell lines
Total RNA was isolated from harvested cells and DNase I-treated using the RNeasy Kit (Qiagen). The cDNAs were synthesized using Moloney murine leukemia virus RT with random primers (Invitrogen, Thermo Fisher Sci.). RNA levels of representative transcripts were analyzed in triplicate by quantitative PCR with SYBR™ Green using a Bio-Rad CFX thermal cycler. The qRT-PCR products were quantified by the relative standard curve method and normalized to the level of GAPDH RNA. Changes in the alternative splicing were investigated by PCR reactions. The bands were separated by gel electrophoresis on a 2% (w/v) agarose gel in TBE buffer, visualized by ethidium bromide stain, and imaged using a Bio-Rad Gel Doc XR+. Products of the RT-PCR reactions were quantified using FIJI (ImageJ) densitometry analysis. No products were detected in negative control reactions. The primer sequences are provided in Table S3.
Immunoblot analysis
Cells were lysed in a buffer containing 50 mM Tris pH 8.0, 10 mM EDTA, 1% (w/v) SDS, 1 mM DTT, with phosphatase and protease inhibitors. Protein was separated by SDS-PAGE, transferred onto PVDF membranes (Millipore-Sigma), then blocked with 5% (w/v) nonfat dry milk in TBS-T. Primary antibodies specific for GAPDH (Cell Signaling Technology Inc., Cat. No. 2118), U2AF2 (Millipore-Sigma, Cat. No. U4758), H2B (Cell Signaling Technology Inc., Cat. No. 12364), and U2AF1 (Abcam, Cat. No. ab86305) were diluted in blocking solution at 1:1000 (v/v) ratio. Anti-DYKDDDDK was diluted at 1:2000 (v/v) ratio. Anti-mouse IgG horseradish-peroxidase (Cat. No. NA931) and Anti-rabbit IgG horseradish-peroxidase (Cat. No. NA934), from GE Healthcare Inc., were diluted in blocking buffer at 1:5000 (v/v) ratio. Chemiluminescent signal, using Clarity Western ECL substrate (Bio-Rad), was detected on a Chemidoc™ Touch Imaging System (Bio-Rad).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are reported as mean ± SD, unless noted otherwise. The number of replicates for each experiment is indicated in the figure legends and was always at least n = 3. To assess statistical significance, two-tailed unpaired t-tests with Welch’s correction (which does not assume equal variance) were calculated in GraphPad Prism 6: *, p < 0.05; **, p < 0.005; ***, p < 0.0005; ****, p < 0.00005.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat#2118; RRID:AB_561053 |
| Rabbit monoclonal anti-Histone H2B | Cell Signaling Technology | Cat#12364; RRID:AB_2714167 |
| Rabbit polyclonal anti-DYKDDDDK Tag | Invitrogen™ / Thermo Fisher Inc. |
Cat#740001; RRID:AB_2610628 |
| Rabbit polyclonal anti-U2AF1 | Abcam | Cat# ab86305; RRID:AB_1925546 |
| Mouse monoclonal anti-U2AF2 | Sigma-Aldrich | Cat#U4758; RRID:AB_262122 |
| Anti-rabbit IgG-HRP | GE Healthcare | Cat#NA934 |
| Anti-mouse IgG-HRP | GE Healthcare | Cat#NA931 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HeLa Cell Nuclear Extract for in-vitro pre-mRNA Splicing | ProteinOne | Cat#P0002–2 |
| Pladienolide B | Santa Cruz Biotech. | Cat#sc-391691 |
| Doxycycline | Takara Bio USA, Inc. | Cat#631311 |
| NSC 194308 | NCI DTP | N/A |
| Diversity Set V library | NCI DTP | N/A |
| (−)-cis-myrtanylamine | Sigma-Aldrich | Cat#180807–1G |
| Critical Commercial Assays | ||
| iTaq™ Universal Probes One-Step Kit | Bio-Rad | Cat#1725141 |
| mMessage mMachine™ T7 Transcription Kit | Invitrogen™ / Thermo Fisher Inc. |
Cat#AM1344 |
| MEGAclear™ Transcription Clean-Up Kit | Invitrogen™ / Thermo Fisher Inc. |
Cat#AM1908 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat#27106 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| M-MLV Reverse Transcriptase | Invitrogen™ / Thermo Fisher Inc. |
Cat#28025013 |
| iQ SYBR Green Supermix | Bio-Rad | Cat#1708880 |
| Deposited Data | ||
| CCDC ID for Compound 2 | This paper | CCDC 1981586 |
| CCDC ID for Compound 3 | This paper | CCDC 1981585 |
| Experimental Models: Cell Lines | ||
| Human: 293T cells | ATCC® | CRL-3216 |
| Human: K562 cells expressing doxycycline-inducible 3X.Flag.U2AF1.WT | (Smith et al., 2019) | N/A |
| Human: K562 cells expressing doxycycline-inducible 3X.Flag.U2AF1.S34F | (Smith et al., 2019) | N/A |
| Human: K562 cells | ATCC® | CCL-243 |
| Human: K562 cells expressing CRISPR-edited U2AF1.WT | This paper | N/A |
| Human: K562 cells expressing CRISPR-edited U2AF1.S34F | This paper | N/A |
| Oligonucleotides | ||
| 5′-Fl DEK(−3U/−3C) RNA | Dharmacon | N/A |
| Stealth™ siRNA targeting U2AF2 | Invitrogen™ / Thermo Fisher Inc. |
Cat#10620319 |
| Stealth™ siRNA Negative Control Lo GC | Invitrogen™ / Thermo Fisher Inc. |
Cat#12935200 |
| Primers (Table S3) | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| U2AF heterodimer and SF1 constructs | (Okeyo-Owuor et al., 2015) | N/A |
| U2AF2 RNA binding domain construct | (Agrawal et al., 2016) | N/A |
| In vitro splicing substrate | (Jurica et al., 2002) | N/A |
| Software and Algorithms | ||
| GraphPad Prism v6.0 | GraphPad Software | www.graphpad.com |
| Biacore T200 Evaluation Software v3.2 | GE Healthcare Bio-Sciences Corp. | N/A |
| Open Babel v2.3.1 | (O’Boyle et al., 2011) | N/A |
| CANDOCK | (Fine et al., 2020) | N/A |
| ShelXT v2018/2 | (Sheldrick, 2015b) | N/A |
| ShelXL v2018/3 | (Sheldrick, 2015a) | N/A |
| Olex2 v1.2-ac3 | OlexSys, Ltd. | N/A |
SIGNIFICANCE.
Patients with myelodysplastic syndromes (MDS) and other cancers acquire defects in the molecular machinery for splicing of pre-mRNA transcripts into legible messages. Currently, MDS lacks a curative treatment aside from a bone marrow transplant. Small molecules are useful tools to investigate and target the defective pre-mRNA splicing machinery of MDS cells. Yet, the current arsenal of molecular inhibitors of RNA splicing is limited. In a proof-of-principle screen, we here discovered an anti-splicing compound (NSC 194308) that targets a key splicing factor, U2AF2. Normally, U2AF2 identifies the pre-mRNA splice site at the early stage of spliceosome assembly. U2AF2 participates in a critical network of splicing factors that often acquire MDS-associated mutations, including the well-studied U2AF1 and SF3B1 partners. By modulating U2AF2, we target an Achilles’ heel in the dysfunctional RNA splicing process of MDS cells. NSC 194308 stalls pre-mRNA splicing at this early-stage intermediate by binding an inter-RRM interface and enhancing U2AF2 association with the splice site RNA. Treatment with NSC 194308 exacerbates pre-mRNA splicing defects and preferentially kills human cells carrying a common MDS-mutation. By analogy with the pre-clinical success of SF3B1 inhibitors, optimized modulators of U2AF2 could serve as useful tools to dissect pre-mRNA splicing events and a step towards personalized treatments of MDS and spliceosome-dysregulated malignancies. Often, pharmaceutical approaches seek to block disease-relevant pathways by inhibiting key chemical reactions or macromolecular interactions. Yet, stabilizers of an interaction can be an avenue to block a pathway, especially in a multi-step process like spliceosome assembly and function. Here, we demonstrate the efficacy of a small molecule that promotes, rather than inhibits, an early-stage intermediate of spliceosome assembly. Such a strategy of stabilizing a pre-catalytic intermediate could be applied to stall other disease-relevant macromolecular assemblies for therapeutic intervention.
Highlights.
A small molecule binds to U2AF2 and promotes its association with splice site RNA
Promoting the U2AF2-RNA complex stalls the initial stages of spliceosome assembly
The small molecule exacerbates pre-mRNA splicing defects of U2AF1-mutant cells
Cancer-relevant U2AF1-mutant cells are preferentially killed by the small molecule
Acknowledgments:
We are grateful to Prof. A. V. Smrcka (U. Michigan) for advice designing the screen. This work was supported by grants from the National Institutes of Health (NIH R01 GM070503 to C.L.K. and R01 GM122279 to M.S.J.), the National Science Foundation (NSF CHE-1900050 to A.F.), UR Ventures (to C.L.K.), and the Edward P. Evans Foundation (to C.L.K., M.J.W., and T.A.G.). Work by R.S. was supported by an NIH Director’s Pioneer Award (1DP1OD006779), Clinical and Translational Sciences (NCATS) Award (UL1TR001412), and NCATS ASPIRE Design Challenge Award. A.M. was supported by NIH training grant T32 GM08646. Z.F. was supported by the National Library of Medicine (T15 LM012495). An NSF Major Research Instrumentation grant (CHE-1725028) supported the X-ray diffractometer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
Inclusion and Diversity: One or more of the authors of this paper received support from a program designed to increase minority representation in science.
REFERENCES
- Agafonov DE, Deckert J, Wolf E, Odenwalder P, Bessonov S, Will CL, Urlaub H, and Luhrmann R (2011). Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31, 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal AA, Salsi E, Chatrikhi R, Henderson S, Jenkins JL, Green MR, Ermolenko DN, and Kielkopf CL (2016). An extended U2AF65-RNA-binding domain recognizes the 3’ splice site signal. Nat Commun 7, 10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MG, Wan L, Younis I, Diem MD, Soo M, Wang C, and Dreyfuss G (2012). A quantitative high-throughput in vitro splicing assay identifies inhibitors of spliceosome catalysis. Mol Cell Biol 32, 1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard B, and Samudrala R (2009). A generalized knowledge-based discriminatory function for biomolecular interactions. Proteins 76, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourhis LJ, Dolomanov OV, Gildea RJ, Howard JA, and Puschmann H (2015). The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment - Olex2 dissected. Acta Crystallogr A Found Adv 71, 59–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagne S, Boigner S, Rudisser S, Moursy A, Gillioz L, Knorlein A, Hall J, Ratni H, Clery A, and Allain FH (2019). Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat Chem Biol 15, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Moon H, Loh TJ, Jang HN, Liu Y, Zhou J, Ohn T, Zheng X, and Shen H (2015). Splicing inhibition of U2AF65 leads to alternative exon skipping. Proc Natl Acad Sci U S A 112, 9926–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Minana B, and Valcarcel J (2011). Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Zhou Z, and Reed R (2000). Functional association of U2 snRNP with the ATP-independent spliceosomal complex E. Mol Cell 5, 779–787. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, and Roeder RG (1983). Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Anderson DD, Bray WM, Prichard BE, Ma N, Adams MS, Ghosh AK, and Jurica MS (2014). Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J Biol Chem 289, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Perriman RJ, Bray WM, Lokey RS, Ares M Jr., and Jurica MS (2013). A high-throughput splicing assay identifies new classes of inhibitors of human and yeast spliceosomes. J Biomol Screen 18, 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Urabe VK, and Jurica MS (2017). Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA 8, 10.1002/wrna.1381 . doi: 10.1002/wrna.138110.1002/wrna.1381. doi: 10.1002/wrna.1381 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, and Jurica MS (2016). Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA 22, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskens FA, Ramos FJ, Burger H, O’Brien JP, Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ, Baselga J, et al. (2013). Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res 19, 6296–6304. [DOI] [PubMed] [Google Scholar]
- Fei DL, Motowski H, Chatrikhi R, Prasad S, Yu J, Gao S, Kielkopf CL, Bradley RK, and Varmus H (2016). Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and Is required for cell survival. PLoS Genet 12, e1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine J, Konc J, Samudrala R, and Chopra G (2020). CANDOCK: Chemical Atomic Network-Based Hierarchical Flexible Docking Algorithm Using Generalized Statistical Potentials. J Chem Inf Model 60, 1509–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folco EG, Coil KE, and Reed R (2011). The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev 25, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser E, Agrawal AA, Jenkins JL, and Kielkopf CL (2017). Cancer-associated mutations mapped on high-resolution structures of the U2AF2 RNA recognition motifs. Biochemistry 56, 4757–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. (2012). Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth S, Tange TO, Kellenberger E, and Valcarcel J (2001). Dual function for U2AF35 in AG-dependent pre-mRNA splicing. Mol Cell Biol 21, 7673–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O’Brien J, and LoRusso P (2014). A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs 32, 436–444. [DOI] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. (2015). The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap PKA, Kubelka T, Soni K, Will CL, Garg D, Sippel C, Kapp TG, Potukuchi HK, Schorpp K, Hadian K, et al. (2020). Identification of phenothiazine derivatives as UHM-binding inhibitors of early spliceosome assembly. Nat Commun 11, 5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins JL, and Kielkopf CL (2017). Splicing factor mutations in myelodysplasias: Insights from spliceosome structures. Trends Genet 33, 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, and Moore MJ (2002). Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 8, 426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielkopf CL, Rodionova NA, Green MR, and Burley SK (2001). A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell 106, 595–605. [DOI] [PubMed] [Google Scholar]
- Kralovicova J, Borovska I, Kubickova M, Lukavsky PJ, and Vorechovsky I (2020). Cancer-Associated Substitutions in RNA Recognition Motifs of PUF60 and U2AF65 Reveal Residues Required for Correct Folding and 3’ Splice-Site Selection. Cancers (Basel) 12, 1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagisetti C, Palacios G, Goronga T, Freeman B, Caufield W, and Webb TR (2013). Optimization of antitumor modulators of pre-mRNA splicing. J Med Chem 56, 10033–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrayoz M, Blakemore SJ, Dobson RC, Blunt MD, Rose-Zerilli MJ, Walewska R, Duncombe A, Oscier D, Koide K, Forconi F, et al. (2016). The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia 30, 351–360. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. (2016). Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerch S, Leach JR, Horner SW, Maji D, Jenkins JL, Pulvino MJ, and Kielkopf CL (2019). The pre-mRNA splicing and transcription factor Tat-SF1 is a functional partner of the spliceosome SF3b1 subunit via a U2AF homology motif interface. J Biol Chem 294, 2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackereth CD, Madl T, Bonnal S, Simon B, Zanier K, Gasch A, Rybin V, Valcarcel J, and Sattler M (2011). Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature 475, 408–411. [DOI] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. (2015). Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6, 6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji D, Glasser E, Henderson S, Galardi J, Pulvino MJ, Jenkins JL, and Kielkopf CL (2020). Representative cancer-associated U2AF2 mutations alter RNA interactions and splicing. J Biol Chem 295, 17148–17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, and Hutchison GR (2011). Open Babel: An open chemical toolbox. J Cheminform 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. (2016). Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 30, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. (2015). U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortuno-Pineda C, Galindo-Rosales JM, Calderon-Salinas JV, Villegas-Sepulveda N, Saucedo-Cardenas O, De Nova-Ocampo M, and Valdes J (2012). Binding of hnRNP H and U2AF65 to respective G-codes and a poly-uridine tract collaborate in the N50–5’ss selection of the REST N exon in H69 cells. PLoS One 7, e40315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, and Blencowe BJ (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, Nichols CA, Agarwalla PK, Brown MS, Lamothe R, et al. (2017). Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elife 6, e23268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert VL, Le Hir H, Jurica MS, and Moore MJ (2002). 5’ exon interactions within the human spliceosome establish a framework for exon junction complex structure and assembly. Genes Dev 16, 2778–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Yang J, and Zhang Y (2012). COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 40, W471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal GA, and Jurica MS (2010). Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res 38, 6664–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P, Gregorovic G, Sprangers R, Stier G, Rhani Z, Kramer A, and Sattler M (2003). Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol Cell 11, 965–976. [DOI] [PubMed] [Google Scholar]
- Shao C, Yang B, Wu T, Huang J, Tang P, Zhou Y, Zhou J, Qiu J, Jiang L, Li H, et al. (2014). Mechanisms for U2AF to define 3’ splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol 21, 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM (2015a). Crystal structure refinement with SHELXL. Acta Crystallogr C Struct Chem 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM (2015b). SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr A Found Adv 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Zheng X, Luecke S, and Green MR (2010). The U2AF35-related protein Urp contacts the 3’ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev 24, 2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, White BS, Tripathi M, Tapia R, Ley JN, Ndonwi M, Kim S, Shao J, Carver A, Saez B, et al. (2017). Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat Commun 8, 14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmier EA, Frato KE, Shen H, Paranawithana SR, Green MR, and Kielkopf CL (2006). Structural basis of polypyrimidine tract recognition by the essential pre-mRNA splicing factor, U2AF65. Mol Cell 23, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidarovich A, Will CL, Anokhina MM, Ceballos J, Sievers S, Agafonov DE, Samatov T, Bao P, Kastner B, Urlaub H, et al. (2017). Identification of a small molecule inhibitor that stalls splicing at an early step of spliceosome activation. Elife 6, e23533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Valcarcel J, and Green MR (1995). Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science 268, 1173–1176. [DOI] [PubMed] [Google Scholar]
- Singh RN, Ottesen EW, and Singh NN (2020). The first orally deliverable small molecule for the treatment of spinal muscular atrophy. Neurosci Insights 15, 2633105520973985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaramakrishnan M, McCarthy KD, Campagne S, Huber S, Meier S, Augustin A, Heckel T, Meistermann H, Hug MN, Birrer P, et al. (2017). Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun 8, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Choudhary GS, Pellagatti A, Choi K, Bolanos LC, Bhagat TD, Gordon-Mitchell S, Von Ahrens D, Pradhan K, Steeples V, et al. (2019). U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat Cell Biol 21, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, and Luhrmann R (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, and Burge CB (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Schultz PG, and Johnson KA (2018). Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc Natl Acad Sci U S A 115, E4604–E4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Maucuer A, Gupta A, Manceau V, Thickman KR, Bauer WJ, Kennedy SD, Wedekind JE, Green MR, and Kielkopf CL (2013). Structure of phosphorylated SF1 bound to U2AF65 in an essential splicing factor complex. Structure 21, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnasooriya C, Feeney CF, Laird KM, Ermolenko DN, and Kielkopf CL (2020). A splice site-sensing conformational switch in U2AF2 is modulated by U2AF1 and its recurrent myelodysplasia-associated mutation. Nucleic Acids Res 48, 5695–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Romfo CM, Nilsen TW, and Green MR (1999). Functional recognition of the 3’ splice site AG by the splicing factor U2AF35. Nature 402, 832–835. [DOI] [PubMed] [Google Scholar]
- Xargay-Torrent S, Lopez-Guerra M, Rosich L, Montraveta A, Roldan J, Rodriguez V, Villamor N, Aymerich M, Lagisetti C, Webb TR, et al. (2015). The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget 6, 22734–22749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Park SY, Oda T, Akiyoshi T, Sato M, Shirouzu M, Tsuda K, Kuwasako K, Unzai S, Muto Y, et al. (2015). A novel 3’ splice site recognition by the two zinc fingers in the U2AF small subunit. Genes Dev 29, 1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Zarnack K, Konig J, Tajnik M, Martincorena I, Eustermann S, Stevant I, Reyes A, Anders S, Luscombe NM, and Ule J (2013). Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 152, 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Freddolino PL, and Zhang Y (2017). COFACTOR: improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res 45, W291–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, and Oldenburg KR (1999). A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors are deposited in the Cambridge Crystallographic Data Centre (CCDC) with access codes CCDC 1981586 (Compound 2) and CCDC 1981585 (Compound 3). This study did not generate any code.