

Abstract
Background & Aims:
Nonalcoholic Fatty Liver Disease (NAFLD), the hepatic correlate of the metabolic syndrome, is a major risk factor for hepatobiliary cancer (HBC). Although chronic inflammation is thought to be the root cause of all these diseases, the mechanism whereby it promotes HBC in NAFLD remains poorly understood. Here we aim to evaluate the hypothesis that inflammation-related dysregulation of the ESRP2-NF2-YAP/TAZ axis promotes HB carcinogenesis.
Methods:
We use murine NAFLD models, liver biopsies from NAFLD patients, human liver cancer registry data, and studies in liver cancer cell lines.
Results:
Our results confirm this hypothesis and support a model whereby chronic inflammation suppresses hepatocyte expression of ESRP2, an RNA splicing factor that directly targets and activates NF2, a tumor suppressor that is necessary to constrain YAP/TAZ activation.
Conclusions:
The resultant loss of NF2 function permits sustained YAP/TAZ activity that drives hepatocyte proliferation and de-differentiation, advantaging growth of cells with mutations that enable them to survive chronic oncogenic stress.
Keywords: epithelial splicing regulatory protein-2 (ESRP2), neurofibromatosis-2 (NF2), hippo kinase, alternative RNA splicing, nonalcoholic fatty liver disease (NAFLD), hepatocellular carcinoma (HCC), liver cancer, yes-associated protein (YAP)
Lay summary
Nonalcoholic fatty liver disease (NAFLD) increases the risk for hepatobiliary carcinogenesis. However, the underlying mechanism remains unknown. Our study demonstrates that chronic inflammation suppresses hepatocyte expression of ESRP2, an adult RNA splicing factor that activates NF2. Thus, inactive (fetal) NF2 loses the function to activate Hippo kinases and downstream YAP/TAZ activities increase, promoting hepatobiliary carcinogenesis in chronically injured liver, such as NAFLD. Liver cancer patients with more fetal NF2 mRNA have poor survival probability.
Graphical Abstract
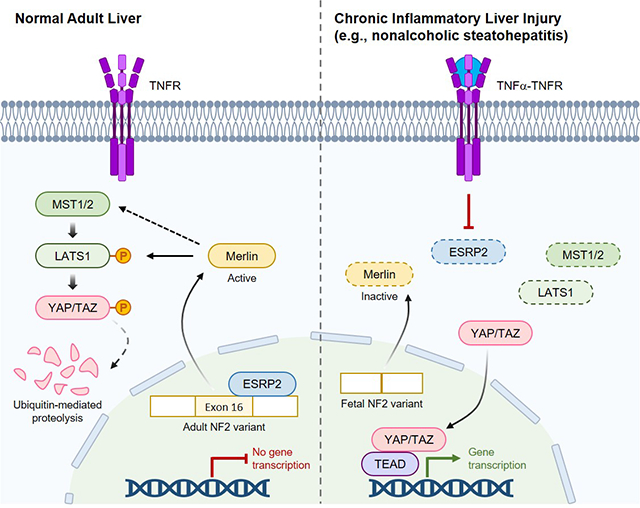
Introduction
Liver cancers related to nonalcoholic fatty liver disease (NAFLD) are typically incurable and predicted to progressively increase in incidence and prevalence due to the obesity pandemic which is fueling NAFLD pathogenesis.1 NAFLD increases the odds for all four types of liver cancer (i.e., hepatocellular carcinoma (HCC), intra- and extra- hepatic cholangiocarcinoma (CCa), and mixed HCC-CCa)1–3 and unlike most chronic liver diseases which mainly increase the risk for HCC, NAFLD often leads to cancer in non-cirrhotic liver.4 Because the mechanisms that enhance vulnerability to hepatobiliary carcinogenesis in NAFLD are poorly understood, developing biomarkers and interventions to improve prevention, diagnosis and treatment of NAFLD-related liver cancers has languished.
Chronic inflammation (CI) is believed to be a key driver of cancers in livers with NAFLD since NAFLD strongly associates with obesity and the metabolic syndrome (MetS),5 chronic inflammatory states that promote malignancies in general6 and which potentiate evolution of liver cancer in other liver diseases.7 In NAFLD, CI induces lipotoxicity and oxidative stress that eventually cause unrepairable DNA damage and malignant transformation, permitting cancerous liver cells to bypass replication check points and accumulate.7 This hypothesis seems plausible for the pathogenesis of NAFLD-related HCC since hepatocytes are proven targets for obesity-related lipotoxicity, but does not easily explain why NAFLD also increases the odds for CCa given that ductal cells in NAFLD livers do not accumulate excessive lipid.8 The fact that cancers of ductal cells associate with NAFLD suggests that normal regenerative responses may become corrupted by CI in NAFLD livers even before malignant transformation occurs.
Regeneration of both hepatocytes and biliary cells depends upon appropriate regulation of factors that control liver growth. Neurofibromatosis-2 (NF2) is a critical negative regulator of liver regeneration as evidenced by progressive accumulation of bipotent liver epithelial progenitors, hepatomegaly, HCC and bile duct hamartomas in Nf2-knockout mice.9–11 Conversely, Yes-associated protein (YAP) and its partner TAZ, are critical positive regulators of liver growth as proven by progressive hepatocyte apoptosis and bile duct loss in Yap-knockout mice.12 Importantly, NF2 and YAP/TAZ interact to control liver growth because inhibiting YAP/TAZ rescues the hepatocyte- and cholangiocyte- overgrowth phenotypes of Nf2-knockout mice.10 Further, YAP/TAZ induce NF2 to suppress their own activities.13 Together, these findings demonstrate that NF2 function is pivotal for regulating hepatocyte and ductal cell YAP/TAZ activity and in turn, indicate the importance of defining mechanisms that regulate NF2 activity since sustained activation of YAP promotes both hepatocyte and cholangiocyte neoplasia.12, 14
NF2 function in hepatocytes is critically controlled by epithelial splicing regulatory protein-2 (ESRP2), an epithelial cell-specific RNA splicing factor that is induced in hepatocytes as liver development ends.15 ESRP2 splices exon 16 into NF2 mRNA and this mature “adult” NF2 splice variant encodes a larger protein with increased function and thus, greater ability to suppress YAP/TAZ activity.16 Sustained activation of YAP/TAZ in adult mouse hepatocytes stimulates their proliferation and progressive de-differentiation into immature hepatocytes, ductal cells and hepatoblasts (i.e., bipotent progenitors of hepatocytes and ductal cells) and eventually results in liver cancers that resemble mixed HCC-CCa in humans.17 Loss of Esrp2 promotes activation of YAP/TAZ and hepatocytes in healthy Esrp2-KO mice are less mature and more proliferative than normal hepatocytes, proving that ESRP2 has an important role in constraining YAP/TAZ activity in adult liver.16, 18 We have shown that hepatocyte expression of Esrp2 is suppressed by TNFα and IL1β,18 pro-inflammatory cytokines that increase in obesity and promote both the MetS and NAFLD.19
It is unknown if the pro-inflammatory milieu fostered by obesity promotes suppression of Esrp2, reduced function of NF2 and/or increased YAP/TAZ activation in the liver. However, this possibility merits consideration since TNFα treatment stimulates healthy adult hepatocytes to de-differentiate into proliferative stem-like cells that self-assemble into tumor-like spheroids that re-differentiate into mature hepatocytes when TNFα is withdrawn.20 Further, this adult-to-fetal state change is accompanied by suppression of Esrp2 and can be reversed either by enforcing Esrp2 expression18 or inhibiting YAP/TAZ activity.21 Although decreased ESRP2 function and increased YAP/TAZ activity normalize when inflammation subsides, CI prolongs YAP/TAZ activation and thus, increases the time interval when p21 and other cell cycle inhibitors are subject to YAP/TAZ-mediated suppression.22 The resultant relaxation of normal mechanisms that check progression of DNA damaged cells through the cell cycle promotes cancer growth.23 Therefore, we hypothesized that NAFLD livers will exhibit reduced expression of Esrp2, relative depletion of adult NF2 splice variants and enrichment with less functional fetal NF2 splice variants, increased YAP/TAZ activity and induction of fetal liver cell markers before liver cancers develop, and persistence of this fetal-like phenotype in the cancers themselves.
Materials and methods
Materials and methods can be found in the supplementary methods and CTAT table.
Results
Mice with NASH exhibit reduced hepatocyte expression of adult splicing factor ESRP2
To determine whether ESRP2 and its targets are altered in NASH we fed mice the choline-deficient, L-amino acid defined high-fat diet (CDAHFD) for 22 weeks. Compared to chow-fed controls, CDAHFD-mice exhibited increased hepatic expression of Tnfα (Fig. 1A), liver injury (Fig. 1B), systemic glucose intolerance (Fig. 1C), suppression of hepatic Esrp2 mRNA (Fig. 1D) and protein (Fig. 1E), and reduced localization of ESRP2 in hepatocyte nuclei (Fig. 1F).
Fig. 1. ESRP2 expression is inversely related to severity of liver inflammation, liver injury and systemic glucose intolerance in mice with NASH.
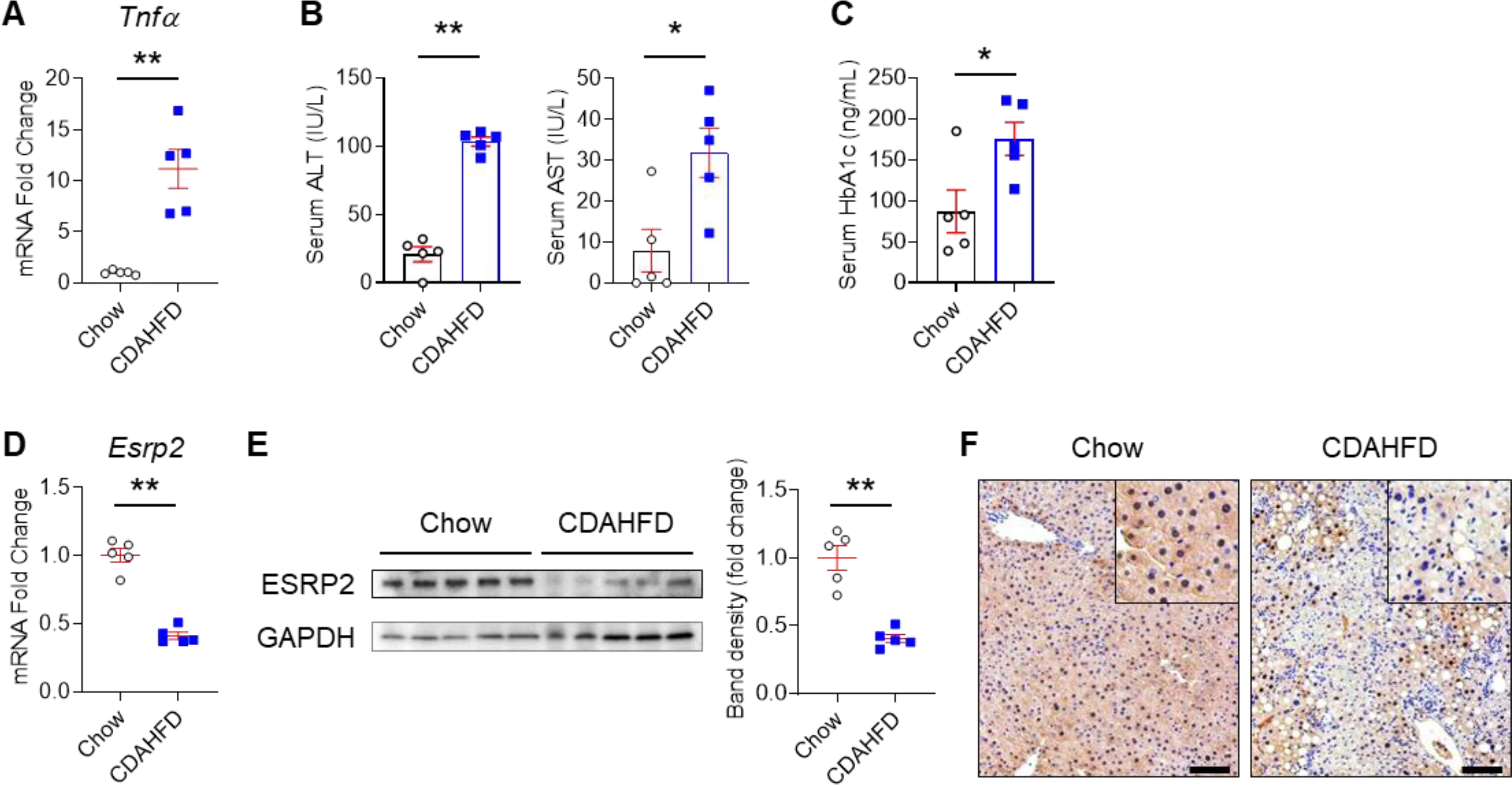
Mice were fed chow or choline-deficient, L-amino acid defined, high-fat diet (CDAHFD) for 22 weeks to induce nonalcoholic steatohepatitis (NASH). (A) qPCR for Tnfα in liver; (B) serum alanine transaminase (ALT), aspartate transaminase (AST); (C) hemoglobin A1c (HbA1c); (D) qPCR for Esrp2 in liver; (E) Immunoblots for ESRP2 in liver lysates normalized to GAPDH; (F) Liver immunohistochemistry (IHC) for ESRP2 (Scale bars = 100 μm). Results are graphed as dot plots (Chow: white circle, CDAHFD: blue square) with mean±s.e.m. (red bars); student t test was used for statistics (n=5 mice/group). *p < 0.05, **p < 0.005.
Enrichment with fetal NF2 splice variant, YAP/TAZ activation, and induction of fetal gene expression program in CDAHFD mice
ESRP2 generates the larger adult mRNA splicing variant of NF2 transcripts by splicing exon 16 into the smaller fetal NF2 mRNA splicing variant. Thus, larger adult NF2 splicing variants are less abundant when ESRP2 activity is low.16 The percent spliced in (PSI) of NF2 exon 16 (Chr22: Start:30079009 – End:30079053, GRCh37.p13) was significantly reduced in livers of CDAHFD mice (Fig. 2A). NF2 encodes an upstream Hippo kinase (HK) and loss of adult NF2 splice variants reduces activity of the HK cascade. This inhibits phosphorylation of YAP/TAZ, stabilizing them in their non-phosphorylated (active) states and increasing expression of YAP/TAZ-inducible genes.24 Compared to chow-fed controls, CDAHFD mice demonstrated reduced expression of the HK, MST1/2, decreased p-YAP/total YAP (i.e., YAP activation), TAZ accumulation (Fig. 2B), and induction of YAP/TAZ target-genes (e.g., Areg, Birc5) (Fig. 2C). YAP also regulates gene expression via post-transcriptional mechanisms and its activation suppresses Let-7 microRNA (miR)21, 25 and thereby, enhances accumulation of its direct mRNA target Igf2bp3,26 a stemness-associated RNA binding protein. Higher YAP activity in CDAHFD mice was accompanied by reduced expression of Let-7 miR and accumulation of Igf2bp3 mRNA (Fig. 2C). Igf2bp3 is generally down-regulated as fetal development ends but often re-induced in adult cancer where it marks cancer stem cells.27 Igf2bp3 is also induced in subpopulations of nonmalignant liver cells that exhibit stem-like growth capacity in regenerating livers.21 Various markers of liver progenitors were increased in livers of CDAHFD mice, including Krt7, Sox9 and Gli2 (Fig. 2C&D) while expression of C/ebpα (an anti-proliferative gene that is up-regulated as liver development ends28) was repressed (Fig. 2C&D). Immunostaining confirmed accumulation of KRT7-expressing hepatocytes (Fig. 2E) in CDAHFD-mice. The aggregate data suggest that CDAHFD-induced NASH suppresses an ESRP2-dependent adult RNA splicing program and promotes accumulation of less mature, more fetal-like hepatocytes.
Fig. 2. Fetal Nf2 variants accumulate, activating YAP/TAZ and promoting adult-to-fetal reprogramming in mice with NASH.
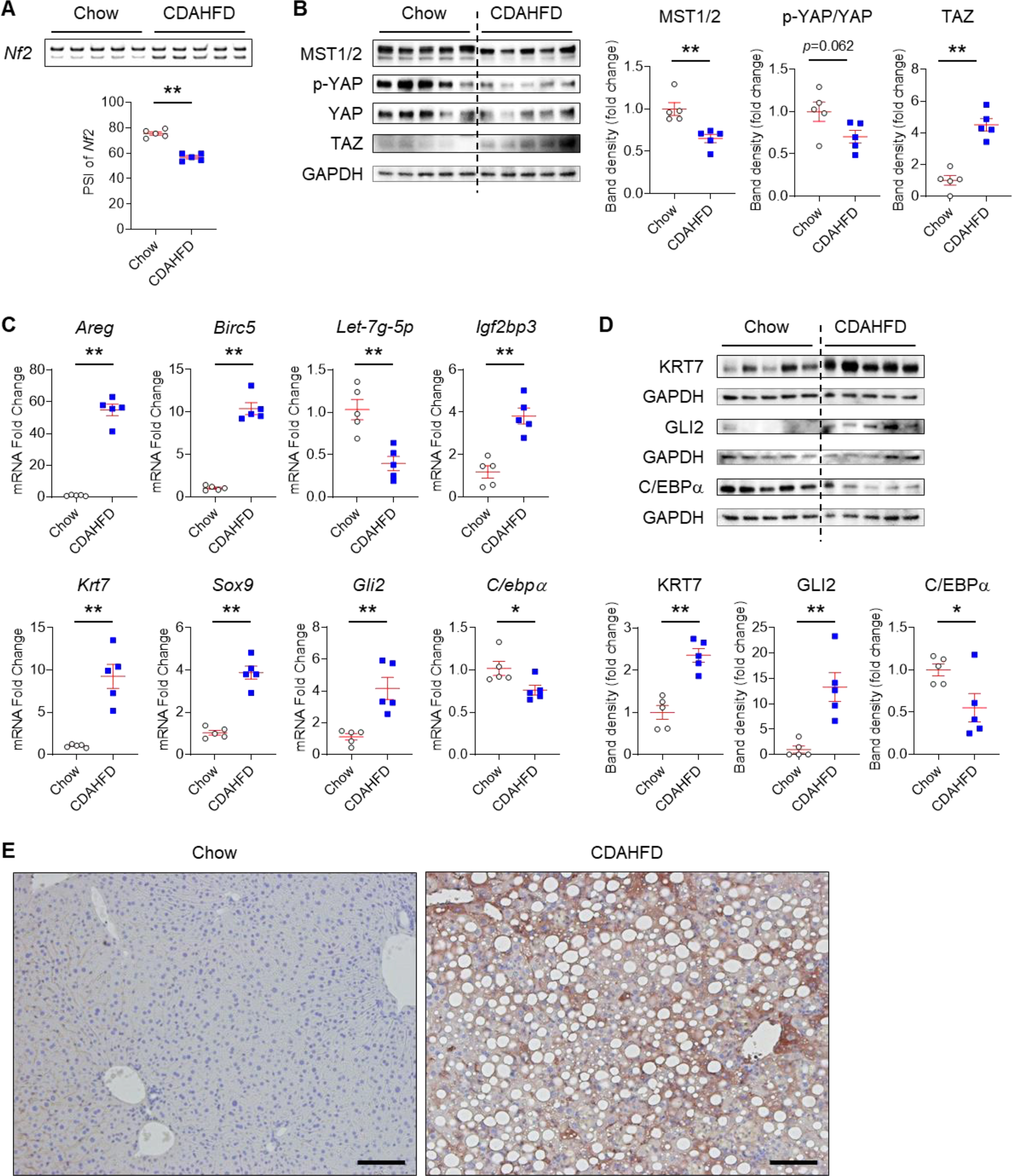
(A) Alternative splicing of Nf2 in livers of chow- and CDAHFD-fed mice. The larger (upper) band includes exon 16 (45 nucleotides in length), while the smaller (lower) band lacks this exon. Differences in percent spliced in (PSI) values are shown. (B) Immunoblots for MST1/2, phosphorylated YAP (p-YAP), total YAP and TAZ normalized to GAPDH in liver lysates. (C) qPCR for YAP/TAZ targets (Areg and Birc5), a microRNA suppressed by YAP (Let-7g-5p), an oncofetal gene that is regulated by Let-7 (Igf2bp3), stem/progenitor cell markers (Krt7, Sox9, Gli2), and a mature hepatocyte marker (C/ebpα); (D) Immunoblots for KRT7, GLI2 and C/EBPα normalized to GAPDH. Results are shown as dot plots (Chow: white circle, CDAHFD: blue square) with mean±s.e.m. (red bars); student t test was used for statistics (n=5 mice/group). *p < 0.05, **p < 0.005. (E) Liver IHC for KRT7 (Scale bars = 100 μm).
Fetal NF2 variants are enriched in precancerous livers and liver cancers in mouse models
To determine the generalizability and potential implications of these findings, we screened two additional mouse models of NASH for changes in NF2 splicing variants. Mice fed methionine choline-depleted, ethionine-supplemented (MCDE) diets for 3 weeks develop severe NASH but recover normal liver histology by 3 weeks after the diet is discontinued.29 In this model, changes in NF2 splice variants tightly paralleled changes in NASH activity: adult NF2 splice variants decreased (and fetal splice variant accumulated) when NASH was present and recovered to baseline levels when NASH resolved (Fig. S1A). DIAMOND™ mice provide another mouse model of NASH that models progressive NASH and evolution of NASH-related liver cancer in humans.30 In DIAMOND™ mice, adult NF2 splice variants became relatively depleted after 24 weeks of feeding NASH-inducing diets, declined further when feeding was extended to 32 weeks, and remained at this low level at 52 weeks, by which time 92% of male DIAMOND™ mice developed liver cancers30 (Fig. S1B).
Although hepatocarcinogenesis is particularly penetrant in DIAMOND™ mice, the other two murine models of NASH that exhibit enrichment with fetal NF2 variants also evoke a premalignant state and increase liver cancer incidence.29, 31 NF2 gene single nucleotide polymorphisms and genetic mutations have been associated with hepatic tumorigenesis in humans.32 Therefore, we examined livers of Mdr2-KO mice (a model of hepatocarcinogenesis induced by chronic bile acid toxicity33) for evidence of adult NF2 depletion and fetal NF2 enrichment. Compared to livers of littermate controls, non-malignant livers of Mdr2-KO mice exhibited decreased adult NF2 splicing variants and enrichment with the fetal NF2 splicing variant (Fig. S1C). Adult NF2 splicing variants were further depleted (and fetal NF2 splicing variants increased) in the primary liver cancers that developed in the Mdr2-depleted strain (Fig. S1C). Hence, the aggregate data suggest that NASH and other types of chronic inflammatory liver injury suppress an adult RNA splicing program, permitting accumulation of a fetal variant of NF2 that inhibits HK signaling that would otherwise inactivate YAP/TAZ in hepatocytes. The resultant YAP/TAZ activation promotes a fetal gene expression program and progressive accumulation of fetal-like liver cells before overt liver cancers develop and persist in the cancers themselves, suggesting that the process is favorable for hepatocarcinogenesis.
Human NASH and liver cancer show ESRP2 suppression, enrichment with fetal NF2 splice variants, and YAP/TAZ activation
NASH increases the risk for both HCC and cancers of the biliary tree (CCa).1–3 Similar to mouse models of NASH that increase the odds for liver cancer, humans with NASH exhibit hepatic induction of developmental signaling pathways,34 activation of YAP/TAZ,35 accumulation of YAP(+) ductal cells (a.k.a., the ductular reaction)36, and liver enrichment for pathways associated with cancer.37, 38 To determine if humans with NAFLD also exhibit reduced expression of ESRP2 we analyzed publicly-available liver RNA seq data.38 ESRP2 mRNAs were significantly lower in NAFLD cohorts with severe NAS score than respective controls without significant liver disease or NAFLD with mild NAS score (Fig. 3A). To determine if adult splicing variants of NF2 were similarly suppressed in NAFLD, we isolated and assayed RNA from archived liver samples from another 47 NAFLD patients, 14 patients with no or mild nonspecific liver abnormalities, 10 patients with HCC, and 1 patient with NASH cirrhosis and CCa (Table 1). Compared to non-NAFLD controls, NAFLD livers exhibited significantly lower NF2 exon 16 PSI, confirming that both the adult splicing factor, ESRP2, and the specific NF2 splicing variant that it generates are reduced in human NAFLD, permitting relative enrichment with fetal NF2 variants (Fig. 3B). We were unable to demonstrate correlations between fetal NF2 accumulation and severity of histologic steatosis, NAS activity, hepatocyte ballooning or fibrosis in this relatively small cohort (Fig. S2). However, we noted that NF2 PSI was significantly lower in the HCC samples than in either non-NAFLD control or NAFLD livers (Fig. 3B), suggesting that human liver cancers are particularly enriched with the fetal NF2 splice variant.
Fig. 3. ESRP2 suppression, fetal NF2 variant enrichment, and YAP/TAZ activation in human NASH and liver cancer.
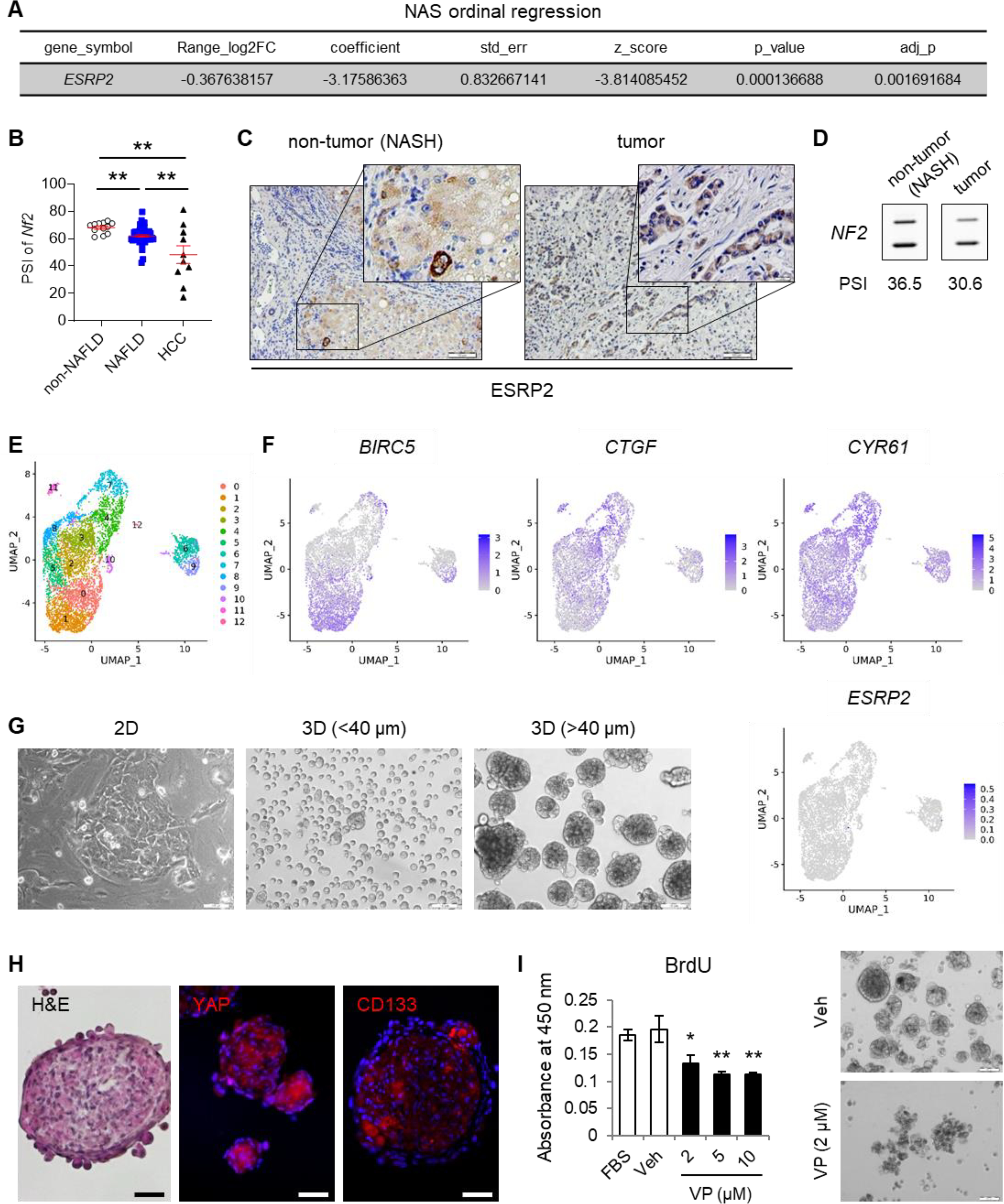
(A) Ordinal regression analysis of published data38 shows that ESRP2 gene expression profiles vary with NAFLD activity score (NAS) in humans. (B) Alternative splicing of NF2 in livers from individuals with normal livers (n=12), NAFLD (n=47), or HCC (n=10). PSI values are shown as mean±s.e.m; student t test was used. *p < 0.05, **p < 0.005. (C) IHC for ESRP2 in liver from patient with NASH-cirrhosis and NASH-driven CCa. Scale bars = 100 μm. (D) Alternative splicing of NF2 in human NASH cirrhosis (non-tumor) and NASH-CCa (tumor). PSI values are beneath blots. (E) UMAP plot of NASH-CCa isolate. (F) Distribution of YAP/TAZ targets (BIRC5, CTGF, CYR61) and ESRP2 among the 13 NASH-CCa clusters analyzed by scRNA-seq. (G) Representative images of cancer cells isolated from NASH-CCa and cultured on standard tissue culture plates (2D) or ultra-low attachment plates for anchorage-independent spheroid formation (3D). Tumor spheroids were sorted by size (smaller or larger than 40 μm diameter); scale bars = 100 μm. (H) Representative H&E and immunofluorescent staining of tumor spheroids for YAP, CD133, and DAPI; scale bars = 50 μm. (I) Bromodeoxyuridine (BrdU) incorporation in NASH-CCa tumor spheroids before and after treatment with verteporfin (VP), an inhibitor of YAP/TAZ. Either fetal bovine serum (FBS) or vehicle (Veh, 0.1% DMSO) were used as controls. Representative images of tumor spheroids before (Veh) and after VP (2 μM) treatment are shown; scale bars = 100 μm. UMAP, uniform manifold approximation and projection; DAPI, 4’,6-diamidino-2-phenylindole.
In our preclinical models, accumulation of fetal NF2 splice variants and evidence of YAP/TAZ activation occurred in injured livers before liver cancers emerged and persisted in the cancers. To determine if a similar process might occur in human NASH-related cancer, we analyzed NF2 splicing variants in a patient with NASH cirrhosis who died from complications related to locally invasive hilar CCa. Both liver tissues from NASH cirrhosis and NASH-CCa showed little expression of nuclear ESRP2 and only a few cells with cytosolic ESRP2 (Fig. 3C). In this patient with NASH-related cirrhosis and CCa, NF2 exon 16 PSI was lower in the cirrhotic non-tumor liver (Fig. 3D) than in our other NAFLD cohort (Fig. 3B) and even lower in the CCa than in adjacent cirrhotic non-tumor liver (Fig. 3D). Single-cell RNA sequencing (scRNA-seq) analysis of the CCa (Fig. 3E) showed that YAP-target genes (e.g., BIRC5, CTGF, and CYR61) were strongly expressed in subpopulations of cells in the cancer, while cells expressing ESRP2 were extremely rare (Fig. 3F). Subpopulations of cancer cells co-expressed YAP or TAZ and NUAK2, an essential effector of YAP-driven hepatomegaly and hepatobiliary carcinogenesis39 and/or other markers of proliferation, such as PCNA, Ki67 and various cyclins (Fig. S3). We confirmed markedly low expression of ESRP2, accumulation of fetal NF2, and strong expression of YAP-target genes in other human CCa (Fig. S4&S5). Further, the CCa cells from our patient propagated when placed in 2D culture and readily generated tumor spheroids when cultured in conditions that favor anchorage independent growth (Fig. 3G). YAP and the liver cancer stem cell marker CD133 were strongly expressed in these tumor spheroids (Fig. 3H) and treating with a YAP inhibitor significantly reduced their proliferative activity (Fig. 3I). These findings support the concept that YAP activation is an important determinant of NASH-related CCa growth, as has been reported in HCC17, 39 and non-NASH-related CCa40 and together, suggest that YAP activation may be enabled by fetal NF2 splice variants that accumulate when ESRP2 is absent.
ESRP2 and its target, NF2, control YAP/TAZ activity and growth of malignant liver cells
To more directly examine the relationship between YAP activation, NF2 RNA splicing, ESRP2 expression and cancer cell phenotypes, we manipulated ESRP2 expression in Huh-7 cells, a human HCC line. Compared to Huh-7 cells treated with control adenoviral vectors (AdGFP), Huh-7 cells treated with adenoviral vectors carrying ESRP2 (AdESRP2) expressed significantly higher levels of ESRP2 mRNA (Fig. 4A) and protein (Fig. 4B), but significantly lower levels of the fetal NF2 splicing variant that lacks exon 16 and thus, are relatively enriched with the larger adult NF2 splicing variant that harbors exon 16 (Fig. 4C). Importantly, increasing ESRP2 expression and up-regulating levels of the adult NF2 splice variant significantly reduced activation of YAP/TAZ in Huh-7 cells (Fig. 4D&E). Furthermore, AdESRP2-treated Huh-7 cells formed smaller tumor spheroids compared to AdGFP-treated control Huh-7 cells (Fig. 4F). To more directly determine the role of NF2 RNA splicing for these outcomes, we used a CRISPR-Cas9 approach to delete exon 16 from NF2 in other Huh-7 cells (Fig. S6) and generated 2 clones with differing levels of fetal NF2 enrichment. Only about 14% of the NF2 mRNA in clone 2 remained the adult NF2 splice variant, whereas ~33% of NF2 in the parental Huh-7 cells was the larger, adult NF2 splicing variant (Fig. 5A). The relatively modest enrichment with fetal NF2 observed in clone 2 was sufficient to: i) reduce phosphorylation of HKs (MST1/2 and LATS1), and their targets (YAP/TAZ) (Fig. 5C), ii) increase YAP/TAZ activity as evidenced by increased expression of YAP/TAZ target genes (e.g., AREG, CTGF, CYR61, PTGS2), iii) induce expression of a stem-progenitor marker (e.g., SOX4), and iv) suppress expression of the mature hepatocyte marker C/EBPα (Fig. 5B). Most importantly, compared to parental Huh-7 cells, Huh-7 ΔE16 cells that were enriched with the fetal NF2 splice variant generated both more and larger tumor spheroids (Fig. 5D). Together, these results provide direct evidence that the adult RNA splicing factor ESRP2, regulates splicing of NF2 mRNAs to control the HK cascade and resultant activation/deactivation of its downstream targets, YAP/TAZ, in liver cancer cells, supporting the tenet that the NASH-related suppression of ESRP2 and resultant enrichment with fetal NF2 splicing variants enables liver carcinogenesis.
Fig. 4. Fetal NF2 variant enrichment promotes neoplastic growth of liver cancer cells.
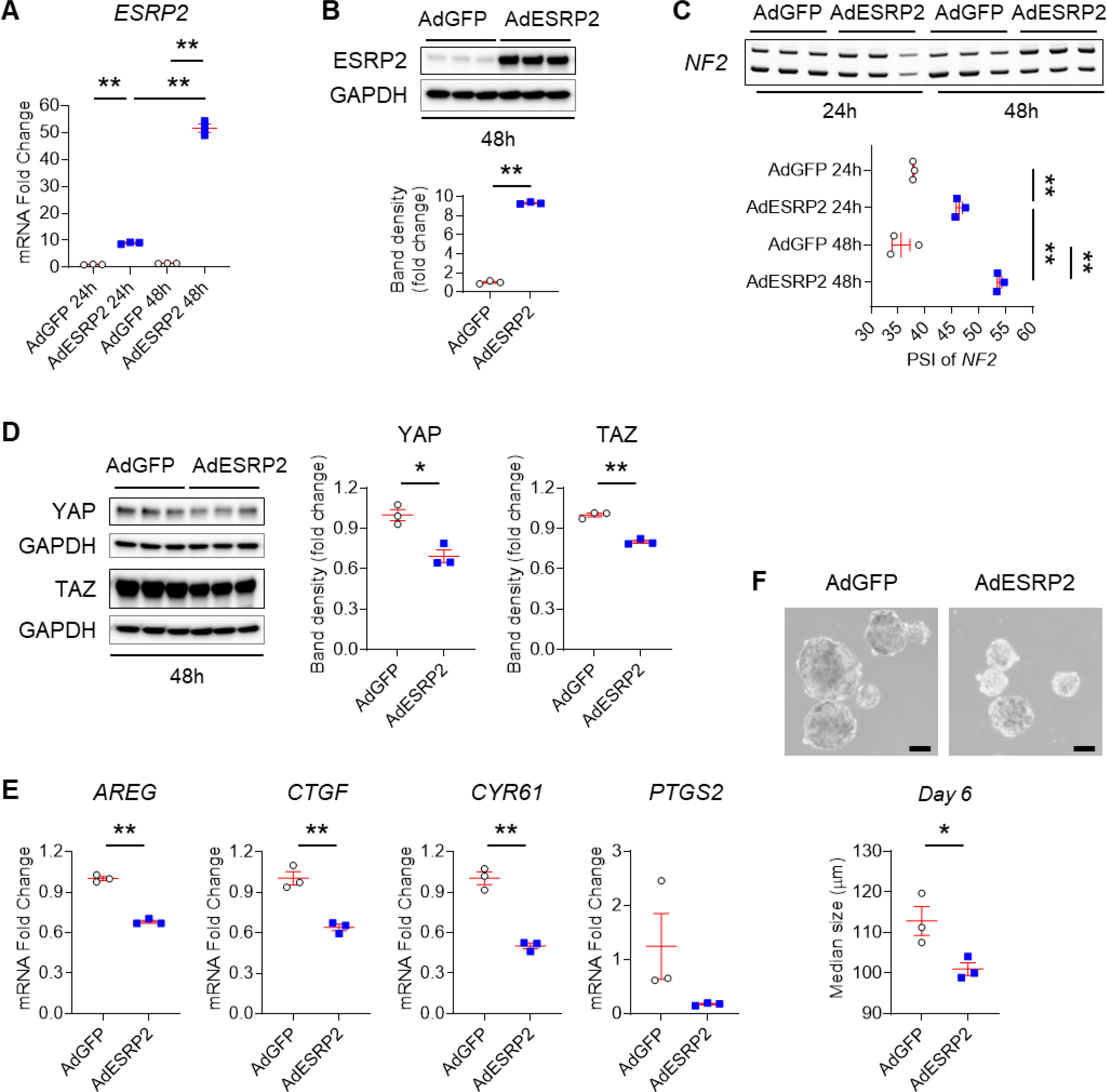
Huh-7 cells were transduced with GFP-tagged (AdGFP) or ESRP2-overexpressing (AdESRP2) adenoviruses for 24 or 48 hours (n=3 repeats/group/time). (A) qPCR for ESRP2; (B) Immunoblots for ESRP2 normalized to GAPDH in cells harvested after 48 hours; (C) Alternative splicing of NF2; (D) Immunoblots for YAP and TAZ normalized to GAPDH; (E) qPCR for YAP/TAZ targets (AREG, CTGF, CYR61, PTGS2). (F) Representative images of AdGFP and AdESRP2 transduced Huh-7 cell tumorspheres on culture day 6. Scale bars = 100μm. Size of tumor spheroids was assessed using Image J 1.8.0 software. Results are graphed as dot plots (AdGFP: white circle, AdESRP2: blue square) with mean±s.e.m. (red bars), and statistical analyses were performed by one-way ANOVA with Tukey’s corrections or using student t test. *p < 0.05, **p < 0.005.
Fig. 5. Fetal NF2 variant enrichment promotes neoplastic growth of liver cancer cells.
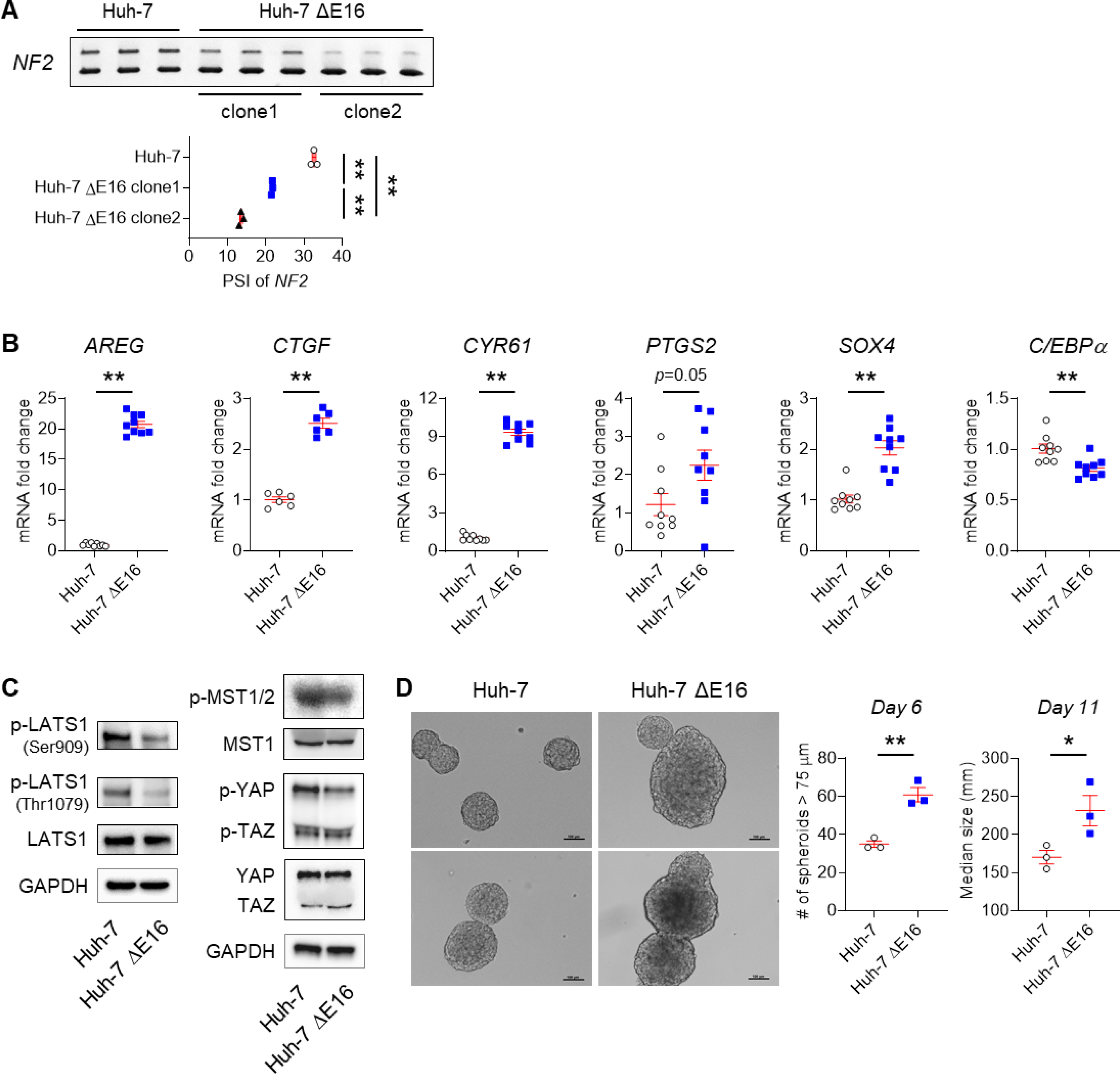
(A-D) CRISPR-Cas9 was used to remove exon 16 from NF2 in Huh-7 cells (ΔE16). Results from three experimental replicates are shown. (A) Alternative splicing of NF2 in parental Huh-7 cells and two different ΔE16 clones; (B) qPCR for YAP/TAZ targets (AREG, CTGF, CYR61, PTGS2), a stem cell marker (SOX4) and a mature hepatocyte marker (C/EBPα) in parental WT Huh-7 cells and the second clone of Huh-7 ΔE16 cells. (C) Immunoblots for p-LATS1 on either Ser909 or Thr1079 residue, total LATS1, p-MST1 on Thr183/ p-MST2 on Thr180, total MST1, p-YAP on Ser127, p-TAZ on Ser89 and total YAP/TAZ normalized to GAPDH in WT and ΔE16 Huh-7 cells cultured in anchorage-independent conditions for 11 days. (D) Representative images of WT and ΔE16 Huh-7 cell tumorspheres on culture day 11. Scale bars = 100μm. Tumor spheroids (>75 μm) were counted on culture day 6 (n=3/group); size was assessed on culture day 11 using Image J 1.8.0 software. Results are graphed as dot plots (Parental Huh-7: white circle, Huh-7 ΔE16: blue square) with mean±s.e.m. (red bars), and statistical analyses were performed by one-way ANOVA with Tukey’s corrections or using student t test. *p < 0.05, **p < 0.005.
TCGA data show worse outcomes in human liver cancers with less ESRP2 and more fetal NF2 variants
Pro-inflammatory cytokines that accumulate in patients with severe alcohol-related steatohepatitis (e.g., TNFα, IL1β) suppress ESRP2 expression in hepatocytes.18 However, whether or not suppressing ESRP2 influences the evolution of human liver cancer has never been reported. In liver sections from 5 NASH patients with incidentally identified HCC, we found that cancer cells were essentially devoid of ESRP2 (Fig. 6A). To determine if suppression of ESRP2 and/or accumulation of the fetal NF2 splice variant associate with HCC or CCa outcomes in humans, we evaluated The Cancer Genome Atlas (TCGA) databases using either the Gene Expression Profiling Interactive Analysis (GEPIA) 241, a web resource for gene expression analysis, or Wanderer42, a web tool visualizing gene expression profiles. In the TCGA HCC cohort, ESRP2 expression in the tumor tended to be lower than in non-tumorous liver, although this difference did not achieve statistical significance (Fig. 6B). On the other hand, HCC recurrence was significantly more likely in tumors with lower expression of ESRP2 (Fig. 6C). More importantly, low tumor expression of ESRP2 also significantly reduced both overall survival and progression-free survival in HCC patients (Fig. 6D). Suppression of the adult splicing factor, ESRP2, promotes accumulation of fetal NF2 splice variants that lack exon 16. The TCGA SpliceSeq43, a web-based resource for alternative mRNA splicing in cancer, showed that PSI of NF2 exon 16 is lower in HCC tumor samples (64.5%) than in non-tumor samples (83.1%), indicating that the exon 16 of NF2 mRNA is largely skipped in HCC (Fig. 6E). In addition, compared to non-tumor livers, HCC in the TCGA dataset were significantly enriched with a variant of NF2 mRNAs (ENST00000338641.8), a transcript that excludes exon 16 to encode the fetal NF2 splice variant (Fig. 6F). The exclusion of NF2 exon 16 (i.e. accumulation of fetal NF2 with low PSI) was significantly correlated with lower ESRP2 gene expression (Fig. 6G). Further, patients whose tumors had high fetal NF2 had significantly worse survival probability than patients with low HCC levels of fetal NF2 (Fig. 6H). The aggregate results identify ESRP2 suppression as a novel inflammation-associated mechanism that contributes to liver cancer growth in people.
Fig. 6. Fetal NF2 variant enrichment and ESRP2 reduction correlate with poor survival in HCC patients.

(A) Representative image of IHC for ESRP2 in liver sections from patient with NASH-associated HCC. Scale bars = 100 μm. (B) Analysis of ESRP2 expression in HCC samples (n=369) of The Cancer Genome Atlas (TCGA) dataset compared to non-tumor samples (n=160) from TCGA and GTEx datasets. Expression is shown as log2(TPM+1) with median; values > |log2FC| = 1 and p-value <0.05 are considered to be differentially expressed. (C) ESRP2 gene expression in HCC tumor samples from the TGCA data set grouped by recurrence (No: n=51, Yes: n=20). Data is shown as log2(x+1) RPKM with median, student t test was used for statistics. *p < 0.05. (D) Kaplan-Meier plots demonstrate the effect of ESRP2 gene expression on survival. Plots were generated using KM-plotter (https://kmplot.com). (E) PSI values of NF2 exon 16 in HCCs (n=417) from TCGA SpliceSeq dataset compared to the respective adjacent non-tumor livers. (F) Expression of an NF2 variant excluding exon 16 (ENST00000338641.8) in HCC (n=369) tumor samples from TCGA dataset compared to non-tumor samples (n=160) from TCGA and GTEx datasets. Data are shown as log2(TPM+1) with median; genes with > |log2FC| = 1 and p-value <0.05 are considered to be differentially expressed. (G) Correlation between the level of adult NF2 splicing variant and ESRP2 level in TCGA HCC samples. Point plot illustrating the correlation between NF2 exon 16 splicing (PSI) and ESRP2 expression. Pearson correlation (r) and p-value (p) are shown. The X-axis shows the log2 aggregate expression level of ESRP2 as quantified by normalized read counts. The Y-axis shows the NF2 exon 16 PSI (linear). (H) Overall and disease-free survival analyses of HCC patients in the TCGA dataset based on mRNA expression level of the fetal variant of NF2 (ENST00000338641.8). TPM, transcripts per million; RPKM, reads per kilobase per million mapped reads; HR, hazard ratio.
Discussion
Our studies identify a role for the epithelial cell splicing factor, ESRP2, and its direct target NF2, in hepatobiliary carcinogenesis. ESRP2 is induced in hepatocytes as liver development ends and regulates splicing in ~20% of hepatocyte mRNAs.15 NF2, a tumor suppressor and core component of the HK cascade,16 is one of the hepatocyte mRNAs that are directly regulated by ESRP2. ESRP2 splices exon 16 into fetal NF2 mRNA to generate an adult transcript that is translated into a protein with enhanced NF2 activity and this promotes greater inactivation/destabilization of YAP/TAZ.16 Reducing YAP/TAZ activity suppresses further liver growth.10 Suppressing YAP also facilitates liver maturation since sustained activation of YAP in hepatocytes causes them to de-differentiate into bi-potent progenitors that can become either hepatocytes or cholangiocytes,14 and hepatobiliary cancers develop in mice if NF2 inhibition9–11 or YAP activation12, 14 persist.
Chronic hepatocyte and/or biliary injury are risk factors for hepatobiliary cancer in both mice and humans.33, 44 Hence, NAFLD, one of the most common causes of chronic liver disease,1 has also become a leading cause of primary hepatobiliary malignancy.1–3 NAFLD-related liver cancers are often attributed to CI and consequent insulin resistance/hyperinsulinemia.3 Proinflammatory cytokines also suppress hepatocyte expression of ESRP2 and promote accumulation of fetal NF2 variants.18 Here, we demonstrate that livers of mice and humans with NAFLD exhibit significant suppression of ESRP2, loss of adult NF2 variants, and accumulation of fetal NF2 variants. These new findings provide a plausible mechanism for earlier reports by ourselves36 and others35 which noted accumulation of YAP/TAZ in nuclei of hepatocytes and ductular cells in NAFLD patients and mouse models of NAFLD. In addition, we have now correlated these responses with evidence of increased YAP activity and markers of de-differentiated liver epithelial cells in NAFLD livers from both species, and directly manipulated NF2 in a human liver cancer cell line to prove that it is the dysregulation of the ESRP2-NF2 axis that causes increased YAP activity and hepatocyte de-differentiation. Importantly, defective ESRP2 regulation of HK signaling is not simply pertinent for NAFLD because here we also show that fetal NF2 variants accumulate in the pre-malignant livers of mice with chronic cholestatic liver injury caused by disruption of the Mdr2 gene, and we previously reported that livers of humans and mice with alcohol-related liver injury demonstrate suppression of ESRP2 and accumulation of fetal NF2 variants.18 Given that loss of NF2 function can cause liver cancer,9–11 we propose that inflammation-related dysregulation of the ESRP2-NF2 axis facilitates hepatobiliary carcinogenesis in many inflammatory liver diseases.
The significance of defective ESRP2-NF2 regulation in liver cancer pathogenesis is further supported by our analysis of TCGA data which reveals significantly worse liver cancer outcomes in patients whose tumors are relatively depleted of ESRP2 and enriched with fetal NF2 variants. These human data complement and extend our findings in several mouse models of chronic liver disease that are known to promote liver cancer. Those results show that dysregulation of the ESRP2-NF2-YAP/TAZ axis occurs before cancer develops and remains dysregulated in the cancer itself. Our analysis of a cholangiocarcinoma that developed in a patient with NAFLD cirrhosis confirmed that fetal NF2 variants are high in cirrhotic non-tumor liver and even more enriched in the cancer. Importantly, scRNA-seq analysis of this NAFLD-related cancer demonstrated that many proliferative cells in the cancer co-express YAP/TAZ. Further, cancer maintenance depends on YAP/TAZ activity because treating spheroids derived from this malignancy with a YAP inhibitor suppressed growth dramatically.
Since a recent study has shown that transcription of ESRP2 is androgen-regulated in prostate cancer,45 we reviewed TCGA data for sex-related differences in ESRP2 mRNA in HCC. However, we were unable to detect differences (data not shown) maybe because TCGA data lacks critical information about sex hormone status and the later may be influenced by age and co-morbidities, including underlying liver disease. Future research will be necessary to evaluate this issue more directly in liver cancer given that liver cancer is more predominant in males and post-menopausal women.46 While our studies reveal a previously unsuspected role for ESRP2-mediated post-transcriptional regulation of NF2 in liver carcinogenesis, more research will be necessary to delineate the mechanisms involved and determine whether or not these mechanisms can be targeted therapeutically. Work by others has proven that NF2 deletion causes liver cancer in otherwise healthy mice by enhancing YAP/TAZ activity.9–11 Activated YAP/TAZ normally induce NF2, providing a negative feedback mechanism to terminate further YAP/TAZ activation.13 Our results suggest that the fetal NF2 variants that accumulate in injured livers are less able to implement this feedback inhibition of YAP/TAZ than adult NF2 variants. Further, a relatively small shift in the relative abundance of fetal versus adult NF2 variants in Huh-7 cells was sufficient to phenocopy findings in Nf2-knockout mice, significantly potentiating YAP/TAZ activation and malignant traits. Thus, it is conceivable that biologically relevant suppression of YAP/TAZ might be achieved during chronic liver injury by developing approaches to limit fetal NF2 accumulation. Future studies in mouse models are needed to address this definitively. The importance of ESRP2 for controlling the abundance of fetal NF2 variants (and thus, YAP/TAZ activity) in liver cells also remains to be determined. Such information will dictate the utility of targeting ESRP2 to optimize NF2 tumor suppressor activity and interrupt hepatobiliary carcinogenesis. It has been suggested that ESRP2 expression is restricted to hepatocytes in adult livers15 and our data indicate that suppression of ESRP2 both permits hepatocytes to become more proliferative and to acquire cholangiocyte markers, suggesting that the ESRP2-NF2-YAP/TAZ axis may regulate the ductular reaction. Affirmation of this hypothesis would have broad clinical implications as the intensity of the ductular reaction correlates with the severity of liver inflammation, risk for fibrosis progression, and development of liver cancer in many liver diseases, including NAFLD.
In summary, we present a novel model to explain why NAFLD (and other types of chronic inflammatory liver injury) promote liver cancer. Inflammatory cytokines that increase during the metabolic syndrome (and other causes of liver injury) suppress ESRP2, an RNA splicing factor that generates the adult variant of NF2. This reduces the relative abundance of adult NF2 and enriches hepatocytes with fetal NF2. Because fetal NF2 encodes a HK that is much less effective for de-activating YAP/TAZ, YAP/TAZ activity is increased in hepatocytes of injured livers and sustained loss of NF2 tumor suppressor function drives hepatocytes to become more proliferative and de-differentiated (i.e., cholangiocyte-like). These hepatocyte-derived, reactive ductal-type cells release paracrine factors that stimulate neighboring stromal cells to become fibrogenic and that recruit immune cells which perpetuate local liver inflammation. The latter re-enforces ESRP2 suppression and maintains predominance of fetal NF2, limiting YAP/TAZ deactivation, and imposing a selection pressure that advantages liver cells with mutations that enable survival during chronic oncogenic stress caused by sustained YAP/TAZ activation.
Supplementary Material
Highlights.
Hepatocyte expression of ESRP2 is suppressed and fetal NF2 accumulates in chronically injured livers including NASH.
Fetal NF2 loses the function to activate Hippo kinases and thus, YAP/TAZ activities increase.
Aberrant expression of ESRP2-NF2-YAP/TAZ axis promotes carcinogenesis in chronically injured liver.
Liver cancer patients with low ESRP2 and high fetal NF2 mRNA have high recurrence and poor survival probability.
Acknowledgements
We very thank the NAFLD Clinical-Translational Research Program and the patients who generously donated samples for biomedical research.
Financial support
This work was supported by NIH grants DK106633, AA010154, DK077794 and the Florence McAlister Professorship of Medicine to AMD, and a postdoc fellowship from the Duke Regeneration Next Initiative and NRF of Korea 2021R1C1C1003904 to JH.
This work was partially supported by a DoD Prostate Cancer Research Program Health Disparity Research Award PC131972 to SRP PI and JAF Co-I, a NIH Feasibility Studies to Build Collaborative Partnerships in Cancer Research P20 Award 1P20-CA202925–01A1 to SRP Overall PI and JAF PI of Pilot Project One, a NIH Basic Research in Cancer Health Disparities R01 Award R01CA220314 to SRP PI and JAF Co-I, a Prostate Cancer Foundation Movember Challenge Award #18CHAL04 to SRP and JAF Multi-PI and MA YI, a DoD Prostate Cancer Research Program Health Disparity Research Award PC180980 to JAF and SRP Co-I, and a DoD Lung Cancer Research Program Idea Development Award – New Investigator LC190367 to JAF PI.
Abbreviations
- ALT
alanine transaminase
- AST
aspartate transaminase
- CCa
cholangiocarcinoma
- CDAHFD
choline-deficient, L-amino acid defined high-fat diet (60% kcal from fat)
- CI
chronic inflammation
- DIAMOND
Diet Induced Animal Model Of Nonalcoholic fatty liver Disease
- ESRP2
epithelial splicing regulatory protein-2
- GEPIA
Gene Expression Profiling Interactive Analysis
- HbA1c
hemoglobin A1c
- HCC
hepatocellular carcinoma
- HK
hippo kinase
- MCDE
methionine choline-depleted, ethionine-supplemented
- MetS
metabolic syndrome
- miR
microRNA
- NAFLD
nonalcoholic fatty liver disease
- NF2
neurofibromatosis-2
- PSI
percent spliced in
- scRNA-seq
single-cell RNA sequencing
- TCGA
The Cancer Genome Atlas
- YAP
yes-associated protein
Footnotes
Conflict of interest
The authors have declared that no conflict of interest exists.
Data availability
The data supporting the findings of this study are available within the article and/or supplementary materials.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Said A, Ghufran A. Epidemic of non-alcoholic fatty liver disease and hepatocellular carcinoma. World J Clin Oncol 2017;8:429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wongjarupong N, Assavapongpaiboon B, Susantitaphong P, Cheungpasitporn W, Treeprasertsuk S, Rerknimitr R, et al. Non-alcoholic fatty liver disease as a risk factor for cholangiocarcinoma: a systematic review and meta-analysis. BMC Gastroenterol 2017;17:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol 2012;56:1384–1391. [DOI] [PubMed] [Google Scholar]
- [4].Alexander J, Torbenson M, Wu TT, Yeh MM. Non-alcoholic fatty liver disease contributes to hepatocarcinogenesis in non-cirrhotic liver: A clinical and pathological study. J Gastroenterol Hepatol 2013;28:848–854. [DOI] [PubMed] [Google Scholar]
- [5].Lonardo A, Ballestri S, Marchesini G, Angulo P, Loria P. Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig Liver Dis 2015;47:181–190. [DOI] [PubMed] [Google Scholar]
- [6].Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 2019;51:27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang YM, Kim SY, Seki E. Inflammation and liver cancer: Molecular mechanisms and therapeutic targets. Semin Liver Dis 2019;39:26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 2016;150:1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, et al. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev 1998;12:1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 2010;19:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Benhamouche S, Curto M, Saotome I, Gladden AB, Liu C-H, Giovannini M, et al. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev 2010;24:1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 2007;17:2054–2060. [DOI] [PubMed] [Google Scholar]
- [13].Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 2015;29:1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bhate A, Parker DJ, Bebee TW, Ahn J, Arif W, Rashan EH, et al. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat Commun 2015;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bangru S, Arif W, Seimetz J, Bhate A, Chen J, Rashan EH, et al. Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat Struct Mol Biol 2018;25:928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nishio M, Sugimachi K, Goto H, Wang J, Morikawa T, Miyachi Y, et al. Dysregulated YAP1/TAZ and TGF-β signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc Natl Acad Sci U S A 2016;113:E71–E80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hyun J, Sun Z, Ahmadi AR, Bangru S, Chembazhi UV, Du K, et al. Epithelial splicing regulatory protein 2–mediated alternative splicing reprograms hepatocytes in severe alcoholic hepatitis. J Clin Invest 2020;130:2129–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mirea A-M, Tack CJ, Chavakis T, Joosten LA, Toonen EJ. IL-1 family cytokine pathways underlying NAFLD: towards new treatment strategies. Trends Mol Med 2018;24:458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Peng WC, Logan CY, Fish M, Anbarchian T, Aguisanda F, Álvarez-Varela A, et al. Inflammatory cytokine TNFα promotes the long-term expansion of primary hepatocytes in 3D culture. Cell 2018;175:1607–1619. e1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hyun J, Oh S-H, Premont RT, Guy CD, Berg CL, Diehl AM. Dysregulated activation of fetal liver programme in acute liver failure. Gut 2019;68:1076–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Muramatsu T, Imoto I, Matsui T, Kozaki K-i, Haruki S, Sudol M, et al. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis 2011;32:389–398. [DOI] [PubMed] [Google Scholar]
- [23].Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 2017;17:93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Moya IM, Halder G. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol 2019;20:211–226. [DOI] [PubMed] [Google Scholar]
- [25].Chaulk SG, Lattanzi VJ, Hiemer SE, Fahlman RP, Varelas X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J Biol Chem 2014;289:1886–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kugel S, Sebastián C, Fitamant J, Ross KN, Saha SK, Jain E, et al. SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 2016;165:1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen C-L, Tsukamoto H, Liu J-C, Kashiwabara C, Feldman D, Sher L, et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest 2013;123:2832–2849. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [28].Shiojiri N, Takeshita K, Yamasaki H, Iwata T. Suppression of C/EBP α expression in biliary cell differentiation from hepatoblasts during mouse liver development. J Hepatol 2004;41:790–798. [DOI] [PubMed] [Google Scholar]
- [29].Syn WK, Jung Y, Omenetti A, Abdelmalek M, Guy CD, Yang L, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 2009;137:1478–1488. e1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 2016;65:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ikawa-Yoshida A, Matsuo S, Kato A, Ohmori Y, Higashida A, Kaneko E, et al. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int J Exp Pathol 2017;98:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang N, Zhao Z, Long J, Li H, Zhang B, Chen G, et al. Molecular alterations of the NF2 gene in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Oncol Rep 2017;38:3650–3658. [DOI] [PubMed] [Google Scholar]
- [33].Philips GM, Chan IS, Swiderska M, Schroder VT, Guy C, Karaca GF, et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS One 2011;6:e23943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012;55:1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 2016;24:848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, et al. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J Hepatol 2015;63:962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, et al. Gene expression predicts histological severity and reveals distinct molecular profiles of nonalcoholic fatty liver disease. Sci Rep 2019;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yuan W-C, Pepe-Mooney B, Galli GG, Dill MT, Huang H-T, Hao M, et al. NUAK2 is a critical YAP target in liver cancer. Nat Commun 2018;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015;6:17206–17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for largescale expression profiling and interactive analysis. Nucleic Acids Res 2019;47:W556–W560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Díez-Villanueva A, Mallona I, Peinado MA. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin 2015;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ryan M, Wong WC, Brown R, Akbani R, Su X, Broom B, et al. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res 2016;44:D1018–D1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chan IS, Guy CD, Chen Y, Lu J, Swiderska-Syn M, Michelotti GA, et al. Paracrine Hedgehog signaling drives metabolic changes in hepatocellular carcinoma. Cancer Res 2012;72:6344–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Munkley J, Li L, Krishnan SG, Hysenaj G, Scott E, Dalgliesh C, et al. Androgen-regulated transcription of ESRP2 drives alternative splicing patterns in prostate cancer. Elife 2019;8:e47678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wands J Hepatocellular carcinoma and sex. N Engl J Med 2007;357:1974–1976. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.