

Summary
Defects in translation lead to changes in the expression of proteins that can serve as drivers of cancer formation. Here, we show that cytosolic NAD+ synthesis plays an essential role in ovarian cancer by regulating translation and maintaining protein homeostasis. Expression of NMNAT-2, a cytosolic NAD+ synthase, is highly upregulated in ovarian cancers. NMNAT-2 supports the catalytic activity of the mono(ADP-ribosyl) transferase (MART) PARP-16, which mono(ADP-ribosyl)ates (MARylates) ribosomal proteins. Depletion of NMNAT-2 or PARP-16 leads to inhibition of MARylation, increased polysome association and enhanced translation of specific mRNAs, aggregation of their translated protein products, and reduced growth of ovarian cancer cells. Furthermore, MARylation of the ribosomal proteins, such as RPL24 and RPS6, inhibits polysome assembly by stabilizing eIF6 binding to ribosomes. Collectively, our results demonstrate that ribosome MARylation promotes protein homeostasis in cancers by fine-tuning the levels of protein synthesis and preventing toxic protein aggregation.
Keywords: ADP-ribosylation, Cancer, Mono(ADP-ribose), Mono(ADP-ribosyl)ation (MARylation), mRNA translation, NAD+, NAD+ synthesis, NAD+ sensor, NMNAT-2, Ovarian cancer, PARP-16, Protein aggregation, Protein synthesis, Ribosomes, Translation
Graphical Abstract
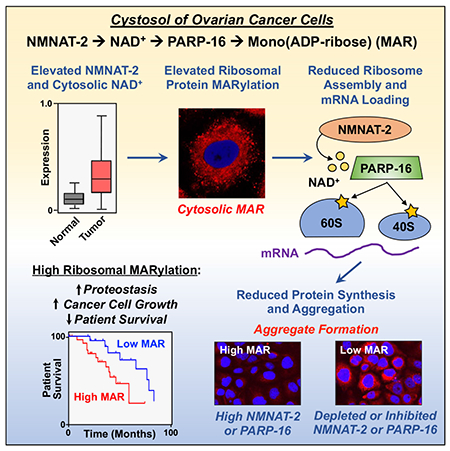
In brief:
Ribosome MARylation and proteostasis in cancer
Introduction
NAD+ is an important metabolite that regulates diverse cellular pathways by acting as a cofactor for redox reactions, as well as a substrate for enzymes such as poly(ADP-ribosyl) transferases (PARPs) and sirtuins (Chiarugi et al., 2012). Unlike in redox reactions where NAD+ is converted to its reduced form (NADH), PARPs consume NAD+ by cleaving the ADP-ribose (ADPR) moiety and covalently attaching it to amino acids in specific substrate proteins. PARPs are the major consumers of NAD+ in the cell and their activity is dependent on the cell’s ability to replenish NAD+. NAD+ can be re-synthesized from nicotinamide (NAM), a byproduct of ADP-ribosylation (ADPRylation) reactions, through a salvage pathway. The final step in the synthesis of NAD+ in this pathway is mediated by nicotinamide mononucleotide adenylyl transferases (NMNATs) that have distinct subcellular localizations, expression levels, and functions (Mori et al., 2014; Orsomando et al., 2012; Sorci et al., 2007). NMNAT-1 is nuclear, while NMNAT-2 is associated with the golgi and acts in the cytoplasm (Berger et al., 2005; Lau et al., 2010; Mayer et al., 2010). NMNAT-3 can localize to the mitochondria or cytoplasm depending on cell-type (Berger et al., 2005; Hikosaka et al., 2014; Yamamoto et al., 2016). We recently demonstrated that NAD+ synthesis and function is compartmentalized in cells (e.g., with NMNAT-1 supporting nuclear PARP-1 activity during adipogenesis) (Ryu et al., 2018), but NAD+ compartmentalization and its functional significance in cancer has yet to be investigated.
The PARP family of enzymes contains 17 members, each of which has distinct subcellular localizations, enzymatic activities (PARP versus MART), and protein substrates (Vyas et al., 2013). As such, the PARP family of enzymes collectively regulates a wide range of molecular mechanisms and cellular functions, including transcription, mRNA processing and stability, and DNA damage responses (Kim et al., 2020). While the majority research to date has been focused on understanding the biological importance of poly(ADP-ribosyl)ation (PARylation), predominantly mediated by the nuclear PARPs (PARP-1 and PARP-2), little is known about the biological importance of MARylation and the enzymes that catalyze these reactions. Most PARP family are monoenzymes that mediate substrate MARylation, and most of these enzymes are localized to the cytosol (Daugherty et al., 2014; Vyas et al., 2013). Therefore, we hypothesized that compartmentalized NAD+ synthesis regulates the activity of PARP monoenzymes that are involved in cytosolic processes dysregulated in cancer.
Fast dividing cells, such as cancer cells, must maintain optimum protein levels. Ribosomes are a hub for this regulation: they are the molecular machines that synthesize proteins, and they also recruit a plethora of proteins to assist in protein clearance. Dysregulation of ribosome function is causative for several disorders, such as neurodegeneration and cancer (Klinge and Woolford, 2019). Recent studies have shown that the central components of the ribosome, including the repertoire of ribosomal proteins, can be regulated and diversified to control protein translation (Sauert et al., 2015). This regulation is mediated, in part, by post-translational modifications (PTMs) of ribosomal proteins, including phosphorylation, acetylation, and ubiquitylation, among others (Simsek and Barna, 2017). Recent studies have begun to link ribosome biogenesis, ribosome function, and translation to cellular outcomes in cancers (Brighenti et al., 2015; Bustelo and Dosil, 2017; Dai and Lu, 2008; van Sluis and McStay, 2014). Interestingly, ADP-ribosylation of the translation machinery is a well-characterized outcome of intoxication by several human bacterial pathogens (e.g., Corynebacterium diphtheria, Pseudomonas aeruginosa, and Vibrio cholerae), whose toxins (diphtheria toxin, exotoxin A, and cholix toxin, respectively) ADP-ribosylate elongation factor-2 (eEF2), an essential component of the protein translation machinery, on a unique diphthamide residue in domain IV (Deng and Barbieri, 2008; Jorgensen et al., 2008). ADP-ribosylation of eEF2 halts protein synthesis and causes cell death (Challa et al., 2021; Deng and Barbieri, 2008; Mateyak and Kinzy, 2013; Simon et al., 2014). Here, we explore the relationships between cytosolic NAD+ synthesis, ribosome MARylation, protein synthesis, proteostasis in ovarian cancer.
Results
NMNAT-2 controls cytoplasmic NAD+ levels and ribosomal protein MARylation
Ovarian cancers have a unique expression pattern of the NMNATs; the levels of NMNAT2 mRNA are upregulated and NMNAT3 mRNA levels are downregulated in ovarian cancers when compared to noncancerous ovarian tissue (Figure 1A). We previously showed that compartmentalized NMNAT-1 and NMNAT-2 compete for their shared substrate, nicotinamide mononucleotide (NMN), during adipocyte differentiation (Ryu et al., 2018). To determine if compartmentalization of NAD+ synthesis occurs in ovarian cancer cells, we generated human OVCAR3 ovarian cancer cells with shRNA-mediated knockdown of NMNAT1 or NMNAT2. Knockdown of either had no observable effect on the total cellular NAD+ level (Figure S1A). To analyze the effects of NMNAT1 or NMNAT2 knockdown on the subcellular levels of NAD+, we used genetically-encoded, nuclear- or cytosol-specific NAD+ sensors and live-cell imaging (Cambronne et al., 2016; Ryu et al., 2018). NMNAT2 knockdown reduced the cytosolic NAD+ levels and elevated the nuclear NAD+ levels in OVCAR3 cells, while NMNAT1 knockdown had the opposite effect (Figure 1B, 1C and S1B. S1C). Ectopic expression of wild-type (Wt) mouse NMNAT-2 or a catalytically dead mutant (H24D) (Ali et al., 2016; Ryu et al., 2018; Yalowitz et al., 2004) in OVCAR3 cells whose endogenous NMNAT-2 was depleted, revealed a dependence on NMNAT-2 catalytic activity for the cytosolic NAD+ signal; expression of wild-type NMNAT-2 increased cytosolic NAD+ levels, but inhibited nuclear NAD+ levels. In contrast, ectopic expression of NMNAT-1 reduced cytosolic NAD+ levels, but enhanced nuclear NAD+ levels (Figure S1D). Together, these data indicate that NMNAT-2 is required for cytosolic NAD+ synthesis and affects NAD+ homeostasis in ovarian cancer cells.
Figure 1. NAD+ synthesis and ADPRylation are compartmentalized in ovarian cancer cells.
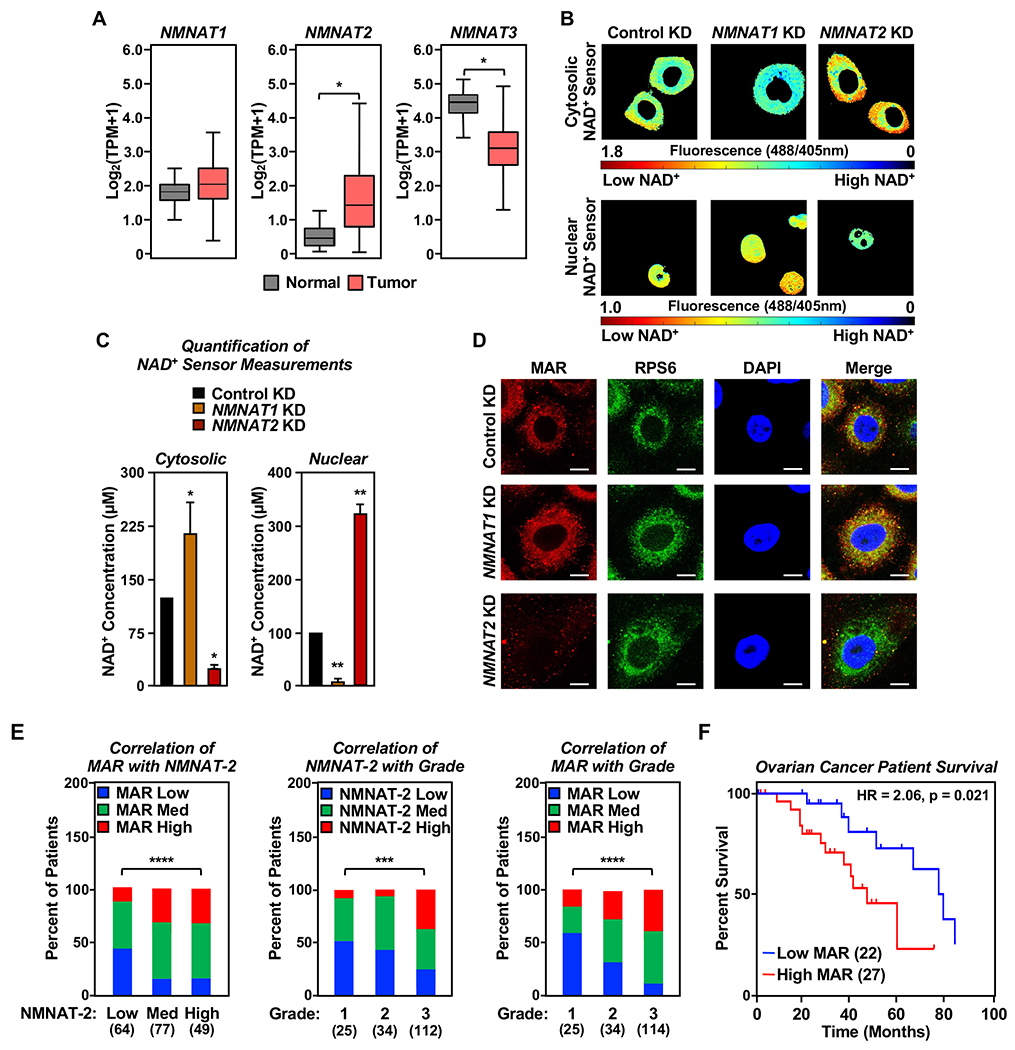
(A) RNA-seq expression data for NMNAT1, NMNAT2, and NMNAT3 mRNAs in ovarian cancer tissues, expressed as transcripts per million (TPM). (TCGA ovarian cancer samples, n = 426) compared to normal ovarian tissues (GTEx data, n = 88) (* p < 0.05).
(B and C) NMNAT-1 and NMNAT-2 regulate compartment-specific NAD+ levels in OVCAR3 cells with NMNAT1 or NMNAT2 knockdown (KD). The fluorescence images in (B) were generated using cytosolic and nuclear NAD+ sensors. The scale bar shows the inverse relationship between fluorescence and NAD+ level. Each bar in the graph in (C) represents the mean ± SEM of the NAD+ concentrations calculated using sensor(488/405 nm)/control(488/405 nm) fluorescence ratios determined by live cell imaging using a standard curve. (n = 3, ANOVA, * p < 0.05, ** p < 0.001).
(D) Co-localization of MAR and RPS6, a ribosomal protein, in OVCAR3 cells as determined by immunofluorescent staining. Representative images are shown. Scale bar = 10 μm.
(E) MAR levels positively correlate with NMNAT-2 expression in ovarian cancer patient samples, and high grade ovarian cancers have higher levels of NMNAT-2 and MAR. IHC analysis for MAR and NMNAT-2 using ovarian cancer tissue microarrays. The number of patients in each group are indicated below the graphs (Chi-square test, *** p<0.001, **** p < 0.0001).
(F) High MAR levels by IHC are a predictor of poor survival in ovarian cancer patients. Analysis of progression free survival using the immunohistochemistry staining for MAR in high grade serous ovarian cancer tissues (n = 49). HR: Hazard ratio.
See also Figure S1.
NMNAT-1 co-localizes with PARP-1 on chromatin in the nucleus and regulates PARP-1 activity by providing NAD+ for localized consumption (Zhang et al., 2012). Since NMNAT-2 regulates cytosolic NAD+ levels, we hypothesized that NMNAT-2 supports the activity of cytosolic PARPs/MARTs in ovarian cancer cells. To test this, we monitored cellular MAR and PAR levels by immunofluorescent staining after NMNAT1 or NMNAT2 knockdown using MAR and PAR detection reagents that we developed previously (Gibson et al., 2017). The results from these assays demonstrate that: (1) MAR localizes primarily to the cytosol and PAR localizes primarily to the nucleus, (2) NMNAT-2 depletion reduces cytosolic MAR levels, but does not affect nuclear PAR levels, and (3) NMNAT-1 depletion reduces nuclear PAR levels, but does not affect cytosolic MAR levels (Figure 1D and Figure S1E). Surprisingly, the majority of the MAR signal co-localized with a ribosomal protein, RPS6 (Figure 1D). These results suggest that NAD+ synthesized by NMNAT-2 regulates MARylation in ovarian cancer cells.
To explore this in more detail in a cancer context, we performed immunohistochemistry (IHC) analysis of ovarian cancer tissue microarrays to evaluate NMNAT-2 and MAR levels in ovarian cancer patient samples. We observed a positive correlation between NMNAT-2 and MAR levels, with high-grade ovarian cancers having the highest levels of NMNAT-2 and MAR (Figure 1E and Figure S1F). Of note, MAR staining in 56 high grade serous ovarian cancer patient samples from UT Southwestern Medical Center revealed that patients with high MAR levels have poor progression-free survival outcomes (Figure 1F and Figure S1G).
Following up on our observation that a majority of MAR signal co-localized with the ribosomal protein RPS6, we isolated ribosomes from OVCAR3 cells and blotted for MAR and PAR. We found that ribosomal proteins are highly MARylated, but not PARylated (Figure 2A). Depletion of NAD+ using an NAD+ synthesis inhibitor (i.e., FK866, which inhibits NAMPT) (Figure S1A) demonstrated that decreased NAD+ levels can abrogate ribosomal protein MARylation (Figure 2B). Indeed, NMNAT-2 depletion in OVCAR3 cells reduced ribosomal protein MARylation, whereas NMNAT-1 depletion had little effect (Figure 2C). Moreover, ectopic expression of an RNAi-resistant wild-type mouse NMNAT-2 (Nmnat2), but not the catalytically dead mutant, enhanced ribosomal protein MARylation in OVCAR3 cells subjected to NMNAT2 knockdown (Figure 2D), indicating that NMNAT-2 catalytic activity is required for ribosomal protein MARylation. Interestingly, NMNAT1 knockdown enhanced ribosome MARylation, and re-expression of an RNAi-resistant wild-type mouse NMNAT-1 (Nmnat1), but not a catalytically dead mutant, inhibited ribosomal protein MARylation (Figure S1H). Collectively, these results suggest that cytosolic NAD+ synthesis by NMNAT-2 is required for ribosomal protein MARylation.
Figure 2. NMNAT-2-dependent MARylation of ribosomal proteins inhibits protein synthesis.
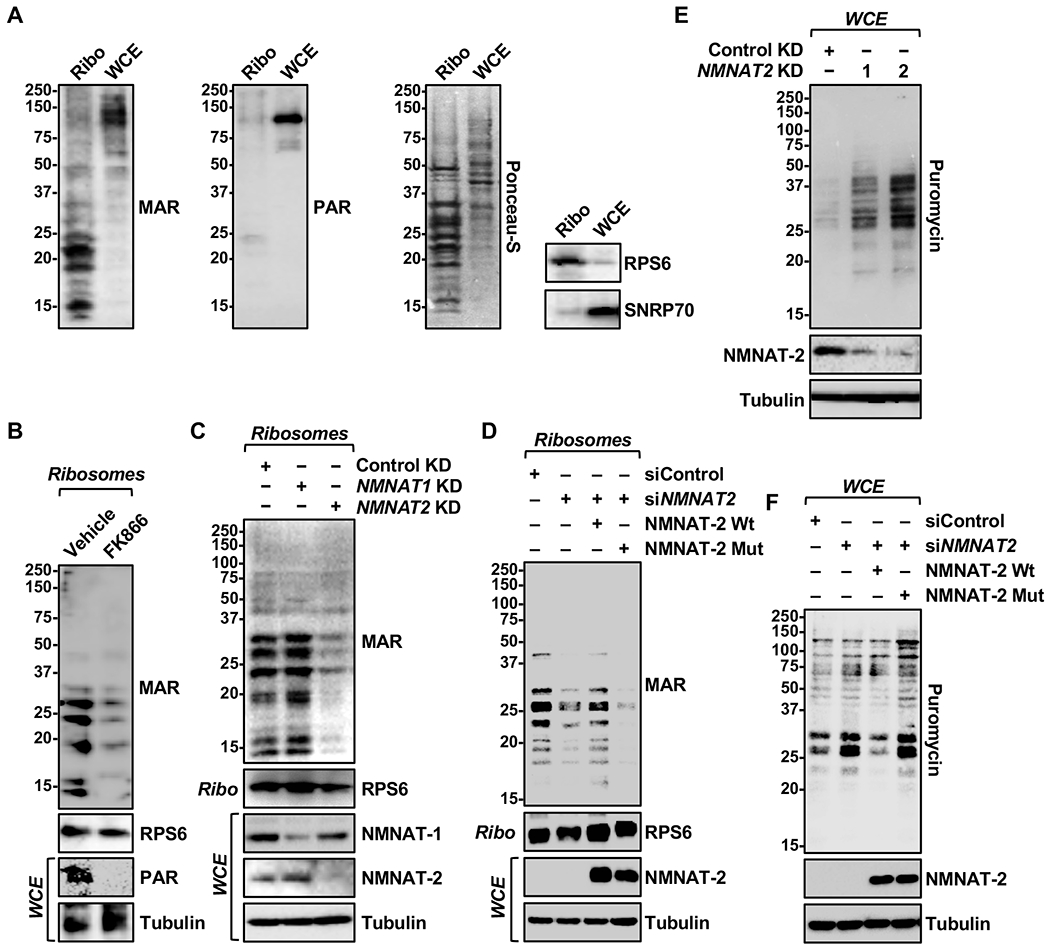
(A) MARylation, but not PARylation, is detected in ribosomal fractions. Western blot analysis for MAR and PAR of ribosomal fractions or whole cell extracts prepared from OVCAR3 cells. RPS6 and SNRP70 were used as the markers for ribosomal and nuclear fractions, respectively.
(B) FK866 treatment reduces both MARylation and PARylation in OVCAR3 cells. Western blot analysis of ribosomal fractions or whole cell extracts isolated from OVCAR3 cells treated with 20 nM FK866 for 48 hours.
(C) NMNAT-2 depletion inhibits ribosomal protein MARylation. Western blot analysis of ribosomal fractions or whole cell extracts isolated from OVCAR3 cells subjected to NMNAT2 knockdown.
(D) NMNAT-2 catalytic activity is required for ribosomal protein MARylation. Western blot analysis of ribosomal fractions or whole cell extracts prepared from OVCAR3 cells subjected to Dox-induced expression of wild-type (Wt) or catalytically dead (H24D) mouse NMNAT-2 (Nmnat2; siRNA-resistant) followed by siRNA-mediated knockdown of NMNAT2.
(E) NMNAT-2 depletion enhances protein synthesis in OVCAR3 cells. Western blot analysis of puromycin incorporation assays from OVCAR3 cells subjected to NMNAT1 or NMNAT2 knockdown.
(F) Ectopic expression of NMNAT-2 overexpression inhibits protein synthesis. Western blot analysis of puromycin incorporation assays prepared from OVCAR3 cells subjected to Dox-induced expression of wild-type (Wt) or catalytically dead (H24D) mouse NMNAT-2 (Nmnat2; siRNA-resistant) followed by siRNA-mediated knockdown of NMNAT2.
See also Figure S1.
Ribosomal protein MARylation inhibits protein synthesis
To examine the effects of NMNAT-2-regulated ribosomal MARylation on mRNA translation, we used puromycin incorporation as a measure of protein synthesis levels in OVCAR3 cells with NMNAT-2 depletion or 293T cells with NMNAT-2 ectopic expression. Surprisingly, we observed that puromycin incorporation, as determined by Western blotting, was higher in cells with NMNAT2 knockdown (Figure 2E). Furthermore, puromycin incorporation was inhibited by the ectopic expression of wild-type NMNAT-2, but not by the catalytically dead mutant (Figure 2F). In contrast, NMNAT-1 enhanced protein synthesis in a catalytic-dependent manner (Figure S1I). Together, these data indicate that NMNAT-2, which supports ribosomal protein MARylation, acts to inhibit protein synthesis in a manner that depends on its catalytic activity.
To identify the MART(s) required for NMNAT-2-regulated ribosomal protein MARylation, we used an siRNA knockdown screen targeting cytosolic MARTs expressed in OVCAR3 cells (Figure S2, A and B). The effects of knockdown were assayed by immunofluorescent staining (Figure 3A and Figure S2C and S2E) and Western blotting of ribosomal fractions (Figure 3B and Figure S2D) for MAR and RPS6. This screen identified that PARP-16 as the only MART whose knockdown had significant effect on bulk cytosolic, as well as ribosomal protein, MARylation. These results were confirmed by shRNA-mediated depletion of PARP-16 in OVCAR3 cells. Similar to NMNAT-2 depletion, PARP-16 depletion dramatically reduced ribosomal protein MARylation (Figure 3C) and stimulated protein synthesis (Figure 3D). PARP-16 is a tail-anchored, ER-resident protein that regulates the ER stress response by modifying the key enzymes in the pathway, such as PERK (Jwa and Chang, 2012). It also regulates the nuclear translocation machinery by MARylation of KAPß1 (Di Paola et al., 2012). We found that PARP-16 is highly enriched in ribosomal fractions from OVCAR3 cells (Figure 3E). Importantly, NMNAT-2 and PARP-16 interact in cells in a manner that is independent of NMNAT-2 catalytic activity, but only wild-type and not catalytically dead mutant NMNAT-2 (W92G) enhances PARP-16 auto-MARylation (Figure S3A and 3F), providing a direct link between NMNAT-2 catalytic activity and PARP-16 catalytic activity through NAD+ production.
Figure 3. PARP-16 and NMNAT-2 regulates ribosomal protein MARylation-dependent protein homeostasis.
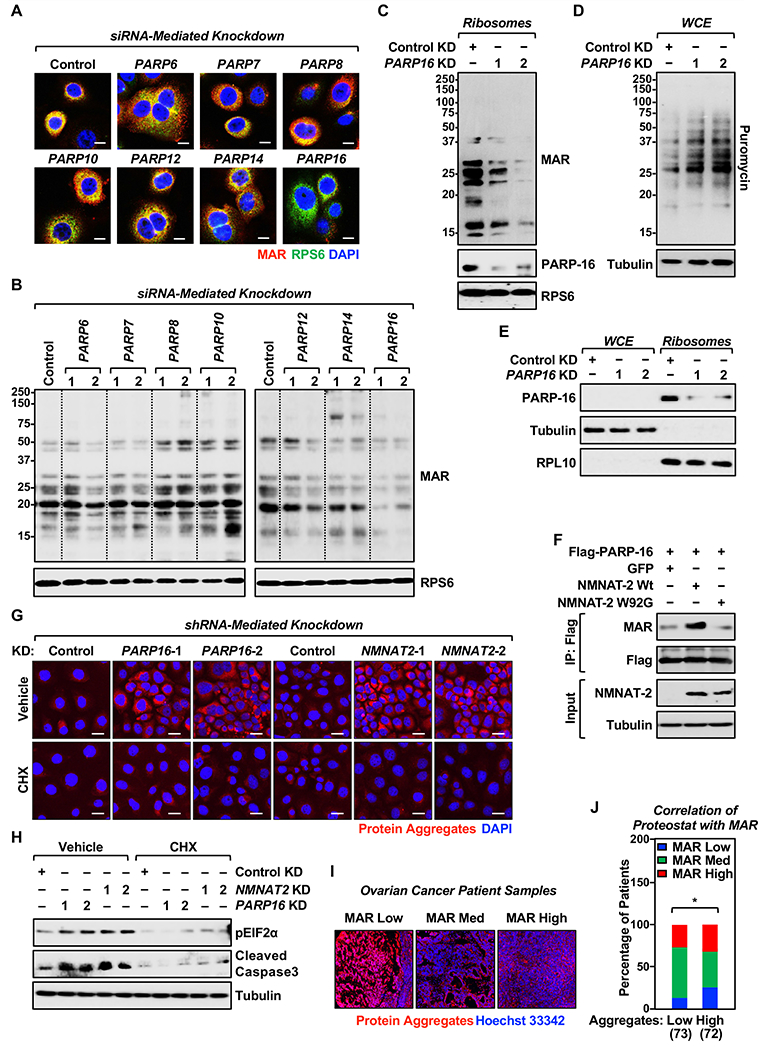
(A and B) PARP-16 mediates ribosomal protein MARylation. OVCAR3 cells were subjected to knockdown with two different siRNAs targeting each of the expressed cytosolic PARP monoenzymes. (A) Representative images from immunofluorescent staining for MAR, RPS6, and DNA (DAPI). The results from siRNA #2 targeting each PARP monoenzymes are shown. Scale bar = 10 μm. (B) Western blot analysis of ribosomal fractions from cells treated as described in (A).
(C) PARP-16 knockdown reduces ribosomal protein MARylation. Western blot analysis for MAR and PARP-16 of ribosomal fractions prepared from OVCAR3 cells subjected to shRNA-mediated knockdown of PARP16. RPS6 was used as the marker for ribosomal fractions.
(D) PARP-16 depletion enhances protein synthesis in OVCAR3 cells. Western blot analysis of puromycin incorporation assays from OVCAR3 cells subjected to PARP16 knockdown.
(E) PARP-16 associates with ribosomes. Cell fractionation and Western blot analysis of PARP-16 in whole cell extracts and ribosomal fractions prepared from OVCAR3 cells. RPL10 and tubulin were used as markers/loading controls for the ribosomal fractions and whole cell extracts, respectively.
(F) NMNAT-2 regulates PARP-16 activity. PARP-16 was immunoprecipitated from 293T cells ectopically expressing Flag-tagged wild-type (Wt) or catalytically dead (W92G) NMNAT-2 and subjected to Western blotting for MAR and Flag.
(G) Depletion of PARP-16 or NMNAT-2 promotes the accumulation of protein aggregates. Staining of protein aggregates using Proteostat aggresome detection reagent in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Treatment with a low dose of cycloheximide (10 μg/mL) for 16 hours inhibits the accumulation of the aggregates. Scale bar = 25 μm.
(H) Depletion of PARP-16 or NMNAT-2 causes proteotoxicity. OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown were assayed for eIF2α phosphorylation and cleaved caspase-3 by Western blotting. Inhibition of translation by cycloheximide blocks the phosphorylation of eIF2α and caspase-3 cleavage.
(I and J) MAR levels negatively correlate with protein aggregation in ovarian cancer patient samples. (I) Representative images of IHC analysis for Proteostat aggresome detection reagent staining using ovarian cancer tissue microarrays. (J) IHC analysis for MAR and Proteostat aggresome detection reagent staining using ovarian cancer tissue microarrays. The number of patients in each group are indicated below the graphs (Chi-square test, * p<0.05).
See also Figures S2, S3, and S4.
Ribosomal protein MARylation inhibits protein aggregation
Proper control of protein synthesis is required to support the growth of cancer cells. Cancer cells require high levels of protein synthesis to support their anabolic processes, but high levels of protein production can cause ER stress and lead to the formation of toxic protein aggregates (Han et al., 2013). Given the prior findings that PARP-16 regulates ER biology (Di Paola et al., 2012; Jwa and Chang, 2012) and our current results showing that PARP-16 depletion enhances protein synthesis, we postulated that loss of PARP-16 induces accumulation of protein aggregates. Indeed, OVCAR3 cells with depletion of PARP-16 or NMNAT-2 have high levels of protein aggregate formation in the cytoplasm (Figure 3G and Figure S3B). As a consequence, the cells exhibit elevated levels of phospho-eIF2α and caspase-3 cleavage, indicating high levels of proteotoxicity-mediated apoptosis (Figure 3H). The accumulation of the protein aggregates, induction of eIF2α phosphorylation, and cleavage of caspase-3 were alleviated by inhibition of protein synthesis using a low dose of cycloheximide (CHX; Figure 3, G and H, and Figure S3B). In contrast, induction of ER stress using thapsigargin or inhibition of the integrated stress response using ISRIB (Sidrauski et al., 2013) had little effect on PARP-16- and NMNAT-2-dependent increases in protein synthesis (Figure S3, C and D). Thus, the effects of PARP-16 or NMNAT-2 depletion on protein synthesis is independent of their effects on ER biology. Rather, dysregulated protein synthesis due to PARP-16 or NMNAT-2 depletion causes ER stress.
Importantly, a similar decrease in ribosomal protein MARylation, as well as increases in protein synthesis and protein aggregation, after PARP-16 or NMNAT-2 depletion were observed in multiple ovarian cancer cell lines, OVCAR4, SKOV3, and HCC5044 (Figure S4, A–I). Similar results were observed in the neuroblastoma cell line, SH-SY5Y, which also has elevated levels of NMNAT2 expression (Ryu et al., 2018); NMNAT-2 depletion caused a decrease in ribosomal protein MARylation, as well as increases in protein synthesis and protein aggregation (Figure S4, J–L). Collectively, these results indicate that reducing ribosomal protein MARylation by depleting PARP-16 or NMNAT-2 promotes protein synthesis and protein aggregation. IHC analysis of ovarian cancer tissue microarrays revealed a negative correlation between protein aggregation and MAR levels in patient samples (Figure 3, I and J), as expected based on our other observations.
NMNAT-2 and PARP-16 regulate ovarian cancer growth
Given the observed effects of PARP-16 or NMNAT-2 depletion on mRNA translation in ovarian cancer cells, we examined their role in ovarian cancer phenotypes. Depletion of PARP-16 or NMNAT-2 inhibited the growth of OVCAR3 cells in culture (Figure S5A). Inhibition of protein synthesis using a low dose of cycloheximide abrogated the inhibition of cell growth mediated by PARP-16 or NMNAT-2 depletion (Figure S5A). Depletion of PARP-16 or NMNAT-2 had a more profound effect on anchorage-independent growth of OVCAR3 cells (Figure 4A) and reduced OVCAR3 xenograft tumor growth in mice (Figure 4B and C, and Figure S5B). With respect to the latter, PARP-16 or NMNAT-2 depletion in the OVCAR3 xenograft tumors promoted enhanced protein synthesis in vivo as determined by puromycin incorporation assays (Figure 4D and Figure S5C), protein aggregation, and apoptosis (Figure 4E). IHC analysis of ovarian cancer tissue microarrays revealed a negative correlation between cleaved caspase 3 staining and MAR levels in patient samples (Figure S5D).
Figure 4. NMNAT-2 and PARP-16 support ovarian cancer cell growth through ribosomal protein MARylation.
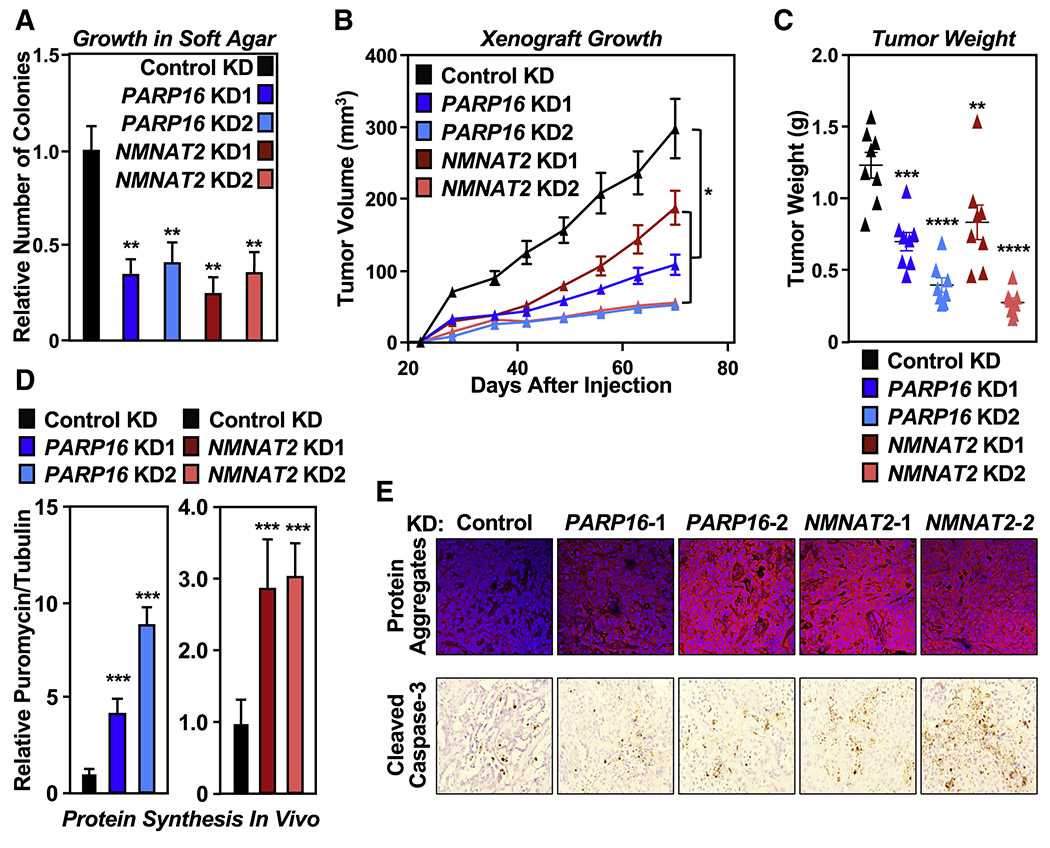
(A) Depletion of PARP-16 or NMNAT-2 inhibits the anchorage-independent growth of OVCAR3 cells. Soft agar assay of OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Each bar in the graph represents the mean ± SEM of the relative number of colonies (n = 3, one-way ANOVA, ** p < 0.01).
(B) Depletion of PARP-16 or NMNAT-2 inhibits the in vivo growth of xenograft tumors formed from OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown (n = 8 per group, ANOVA, * p<0.05).
(C) Weights of tumors formed from OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown (n = 8, one-way ANOVA, ** p<0.01, *** p<0.001, **** p<0.0001).
(D) Depletion of PARP-16 or NMNAT-2 enhances protein synthesis and protein aggregation in vivo. Each bar in the graph in (D) represents the mean ± SEM of the relative ratios of Western blot signals of puromycin to tubulin (n = 3, t-test with Holm-Sidak correction, *** p<0.001).
(E) Depletion of PARP-16 or NMNAT-2 causes proteotoxicity in vivo. Analysis of xenograft tumors described in (B) with Proteostat aggresome detection reagent staining and IHC using an antibody that recognizes cleaved caspase-3.
See also Figure S5.
A stem-loop element in the 3’UTRs of mRNAs directs ribosome loading upon NMNAT-2 or PARP-16 depletion
Next, we aimed to determine how ribosomal protein MARylation affects ribosome function and suppresses protein synthesis. Polysome profiling in OVCAR3 cells revealed the enrichment of ribosomal protein MARylation in fractions containing assembled monosomes and, to a lesser extent, polysomes, but not in fractions containing free ribosomal subunits (Figure 5A and Figure S6A). This suggests that ribosomal protein MARylation may affect assembly or function of polysomes. To test this, we performed polysome RNA-sequencing (RNA-seq) assays and observed altered loading of >2,000 mRNAs on the polysomes when PARP-16 or NMNAT-2 were depleted, with two-thirds showing increased loading (Figure 5B). The transcripts showing increased loading encode proteins in pathways that are important in cancer, such as gene transcription, G-protein-coupled receptor (GPCR) signaling, and chromatin organization (Figure 5C).
Figure 5. Ribosomal protein MARylation regulates polysome function through 3’ UTR stem-loop structures in mRNAs.
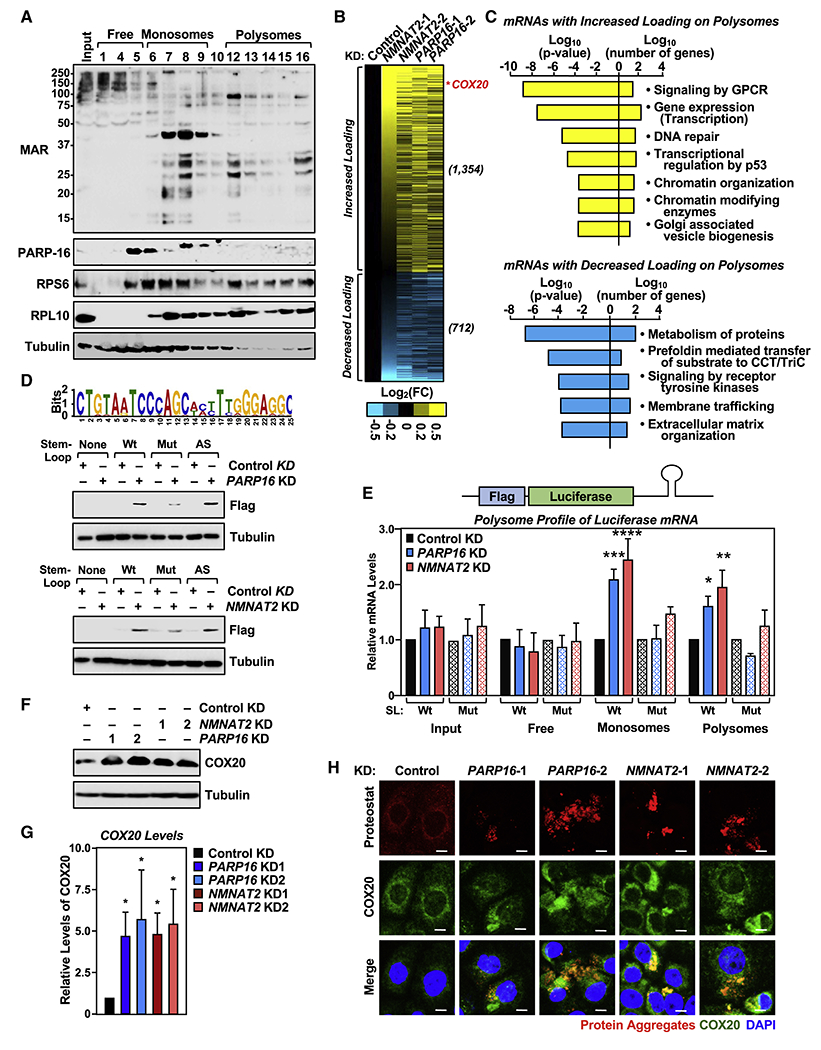
(A) Ribosomal protein MARylation is enriched in the monosome and polysome fractions of OVCAR3 cells. Western blot analysis for MAR and PARP-16 of the sucrose density gradient fractions prepared from OVCAR3 cells. RPS6 and RPL10 were used as markers for the small and large ribosomal subunits, respectively.
(B and C) Depletion of NMNAT-2 or PARP-16 alters mRNA loading on polysomes. RNA-sequencing assay of mRNAs associated with polysomes isolated from OVCAR3 cells subjected to NMNAT2 or PARP16 knockdown. (B) Heatmap representation of mRNAs that exhibited altered loading on the polysomes when NMNAT-2 or PARP-16 were depleted. (C) Gene GO analysis of these mRNAs.
(D) Identification of a transferable stem-loop motif in the 3’UTRs of mRNAs enriched on polysomes after NMNAT2 or PARP16 knockdown. (Top panel) Sequence of the motif with the highest score. (Bottom panels) The stem-loop motif in the 3’UTR of Flag-luciferase mRNA is required for translational regulation by PARP-16 and NMNAT-2. Western blot analysis for Flag-luciferase of lysates from PARP16 or NMNAT2 knockdown OVCAR3 cells that were transfected with the indicated Flag-luciferase constructs.
(E) Addition of the stem-loop motif to the 3’UTR regulates polysome loading of Flag-luciferase mRNA. RT-qPCR analysis of Flag-luciferase mRNA isolated from the density gradient fractions corresponding to free ribosomal subunits, monosomes, and polysomes from PARP-16 or NMNAT-2 depleted OVCAR3 cells. Each bar in the graph represents the mean ± SEM of the relative Flag-luciferase mRNA levels (n = 3, two-way ANOVA, * p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001).
(F and G) Depletion of PARP-16 or NMNAT-2 enhances COX20 protein levels. (F) Western blot analysis for COX20 in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Each bar in the graph in (G) represents the mean ± SEM of the ratio of the levels of COX20 to tubulin (n = 3, two-way ANOVA, * p < 0.05).
(H) Depletion of PARP-16 or NMNAT-2 promotes the accumulation of COX20 protein aggregates in OVCAR3 cells. Co-staining of protein aggregates using Proteostat aggresome detection reagent and COX20 in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Scale bar = 25 μm.
See also Figure S6.
Since regulatory elements in the untranslated regions (UTRs) of mRNAs can regulate mRNA translation (Gallie et al., 1996; Kuersten and Goodwin, 2003; Mazumder et al., 2003), we performed a sequence analysis of the UTRs from the mRNAs whose loading was altered when PARP-16 or NMNAT-2 were depleted. This revealed a set of sequences with likely stem-loop secondary structures in the 3’UTRs of genes with increased loading onto polysomes (Figure S6B). We generated a reporter construct that contained the top motif hit from the sequence analysis in the 3’UTR of a cDNA encoding Flag-luciferase in a mammalian expression vector (SL Wt). We also generated Flag-luciferase constructs with the stem-loop sequence mutated to either disrupt the stem-loop structure (SL Mut) or to form a stem-loop structure with an antisense sequence (SL AS) (Figure S6C). We then monitored the levels of Flag-luciferase protein produced from these constructs in OVCAR3 cells by Western blotting. We found that depletion of PARP-16 or NMNAT-2 increased Flag-luciferase protein levels when the wild-type (SL Wt) or antisense (SL AS) stem-loops were present in the construct, but not when the stem-loop structure was disrupted (SL Mut) (Figure 5D). While the steady-state levels of the Flag-luciferase mRNA was the same for all three constructs (Figure S6D), we observed a higher monosome and polysome loading of Flag-luciferase mRNA containing the wild-type, but not the mutant, stem-loop sequence when PARP-16 or NMNAT-2 was depleted (Figure 5E). An unrelated mRNA, RPL19, did not exhibit altered monosome or polysome loading under the same conditions (Figure S6E). These results identify a 3’ UTR element that is responsive to cytosolic NAD+ synthesis mediated by NMNAT-2 and ribosomal protein MARylation mediated by PARP-16.
To determine if increased loading of mRNAs on polysomes upon inhibition of ribosomal protein MARylation results in increased synthesis of the cognate proteins and subsequent aggregation, we performed co-localization assays of Flag-luciferase protein and the protein aggregates shown in Figure 3E. We observed a clear overlap between Flag-luciferase protein and the protein aggregates upon depletion of PARP-16 or NMNAT-2 (Figure S6F). Interestingly, COX20, whose mRNA contains the top hit stem-loop structure in its 3’UTR and exhibited increased polysome loading upon PARP-16 or NMNAT-2 knockdown (Figure 5B), also demonstrated enhanced protein expression in PARP-16 or NMNAT-2 depleted cells (Figs. 5F and 5G). Consistent with these results, immunofluorescent staining confirmed that COX20 protein expression is elevated upon PARP-16 and NMNAT-2 depletion and COX20 localizes to the protein aggregates (Figure 5H). In a similar assay, we did not observe colocalization of RPS6 with the protein aggregates (Figure S6G). COX20 is a cytochrome c oxidase assembly factor that regulates mitochondrial respiration (Bourens and Barrientos, 2017; Bourens et al., 2014; Elliott et al., 2012), but the effects of COX20 aggregation on its function remain to be studied. Nevertheless, the results from these experiments indicate that loss of ribosomal protein MARylation due to depletion of PARP-16 or NMNAT-2 results in altered polysome loading of mRNAs that contain specific functional motifs in their 3’UTRs, resulting in enhanced protein synthesis and aggregation. Consistent with these observations, expression of wild-type, but not catalytically dead NMNAT-2, inhibited expression of the Flag-luciferase reporter with wild-type stem-loop (Figure S6H and S6I).
Ectopic expression of NMNAT-2 enhances protein synthesis in normal fallopian tube cells
Thus far our results indicate an important role for NMNAT-2 and PARP-16 in the regulation of translation in cancer cells. A remaining question is if this pathway is specific for cancers. To answer this, we used normal fallopian tube cells FT194 and FT282, since high grade serous ovarian cancers are thought to arise from cells originating in the fallopian tubes (Labidi-Galy et al., 2017; Medeiros et al., 2006). The expression levels of NMNAT-2 and PARP-16 are low in these cells compared to ovarian cancer cells (Figure 6A). Surprisingly, in contrast to what we observed in the cancer cells, NMNAT2 knockdown in the normal fallopian tube cells reduced protein synthesis (Figure 6B), while ectopic expression of NMNAT-2 enhanced protein synthesis in a catalytic-dependent manner (Figure 6C). Ectopic expression of PARP-16 reduced protein synthesis in these cells and blocked the increase in protein synthesis mediated by NMNAT-2 (Figure 6D). These data suggest that NMNAT-2 stimulates protein synthesis in normal cells with low PARP-16 levels, perhaps through another cytosolic PARP or by shuttling the NAD+ into metabolic regulation, but works through PARP-16 in cancer cells to suppress protein synthesis. Consistent with this, RNA-sequencing of polysomes isolated from fallopian tube cells, as well as OVCAR3 cells whose endogenous NMNAT-2 was depleted, revealed that ectopic expression of NMNAT-2 suppressed polysome loading of the set of mRNAs whose loading was upregulated by NMNAT2 and PARP16 knockdown in OVCAR3 cells (Figure 6E–6G). Importantly, the same sets of genes showed the same responses to NMNAT-2 expression (and opposite responses to NMNAT-2 depletion) across the three cells lines tested (Figure 6G).
Figure 6. Role of NMNAT-2 and PARP-16 in the regulation of translation in normal fallopian tube cells.
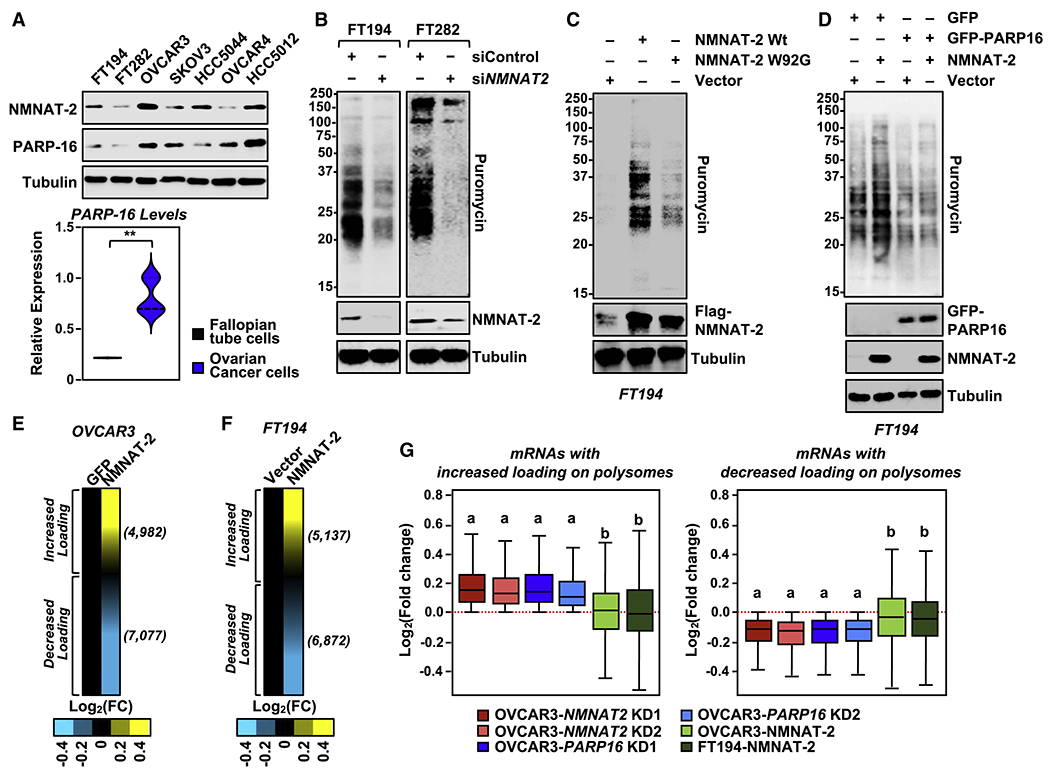
(A) NMNAT-2 and PARP-16 levels are higher in ovarian cancer cells. (Top panel) Representative images of Western blot analysis of lysates prepared from a panel of fallopian tube cells and ovarian cancer cells. (Bottom panel) Violin plot of average expression of PARP-16 obtained from three independent biological replicates (t-test, ** p < 0.01).
(B) Depletion of NMNAT-2 suppresses protein synthesis in fallopian tube cells. Western blot analysis of puromycin incorporation assays from FT194 and FT282 cells subjected to siRNA-mediated NMNAT2 knockdown.
(C) NMNAT-2 expression enhances protein synthesis in FT194 cells. Western blot analysis of puromycin incorporation assays from FT194 cells subjected to Dox-induced expression of NMNAT-2.
(D) Ectopic expression of PARP-16 inhibits protein synthesis in FT194 cells. Western blot analysis of puromycin incorporation assays from FT194 cells transfected with GFP-epitope tagged PARP-16 and Dox-induced expression of NMNAT-2.
(E) Ectopic expression of NMNAT-2 alters mRNA loading on polysomes. Heatmaps showing the results of RNA-sequencing assay of mRNAs associated with polysomes isolated from OVCAR3 cells subjected to Dox-induced expression of wild-type mouse NMNAT-2 (Nmnat2) followed by siRNA-mediated knockdown of NMNAT2.
(F) Ectopic expression of NMNAT-2 alters mRNA loading on polysomes. Heatmaps showing the results of RNA-sequencing assay of mRNAs associated with polysomes isolated from FT194 cells subjected to Dox-induced expression of wild-type NMNAT-2.
(G) Ectopic expression of NMNAT-2 partially reverses the loading of mRNA onto polysomes of genes whose polysome loading is altered with depletion of NMNAT-2 and PARP-16 (Fig. 5B) with ectopic expression of NMNAT-2 in OVCAR3 and FT194 cells (p-value < 2.2e-16).
Site-specific MARylation of RPL24 and RPS6 inhibits polysome formation
Next, we identified the sites of MARylation on ribosomes. We observed that ribosomal proteins in OVCAR3 cells are primarily MARylated at Glu and Asp residues (Figure S7A). Using mass spectrometry we identified MARylation sites in about a dozen ribosomal proteins (Figure S7, B and C). Five of these sites are located at the interface between the 40S and 60S subunits in proteins that are known to regulate 80S monosome formation, such as RPL24 (Figure 7A). To identify the mechanisms by which site-specific ribosomal protein MARylation might impact protein synthesis, we further characterized the MARylation of RPL24. We first validated that RPL24 MARylation is regulated by PARP-16 (Figure S7, D and E) and that Glu4 is indeed a site of MARylation (Figure 7B). Loss of RPL24 MARylation by mutation of Glu4 to Gln phenocopies NMNAT-2 or PARP-16 depletion; it enhances protein synthesis, protein aggregation, COX20 expression (Figure 7, C–E and Figure S7, F–H), and expression of the Flag-luciferase with wild-type stem-loop (Figure S7P). Moreover, expression of the MAR-deficient mutant RPL24-E4Q blocked PARP-16-mediated translational regulation (Figure S7, J and K). This demonstrates that the effect of PARP-16 loss on translation is through its regulation of RPL24 MARylation at Glu4. Similar to PARP-16 depletion, expression of RPL24-E4Q induces apoptosis, which can be relieved by inhibition of protein synthesis using cycloheximide (Figure 7F and Figure S7I).
Figure 7. Site-specific MARylation of RPL24 at Glu4 inhibits polysome formation.
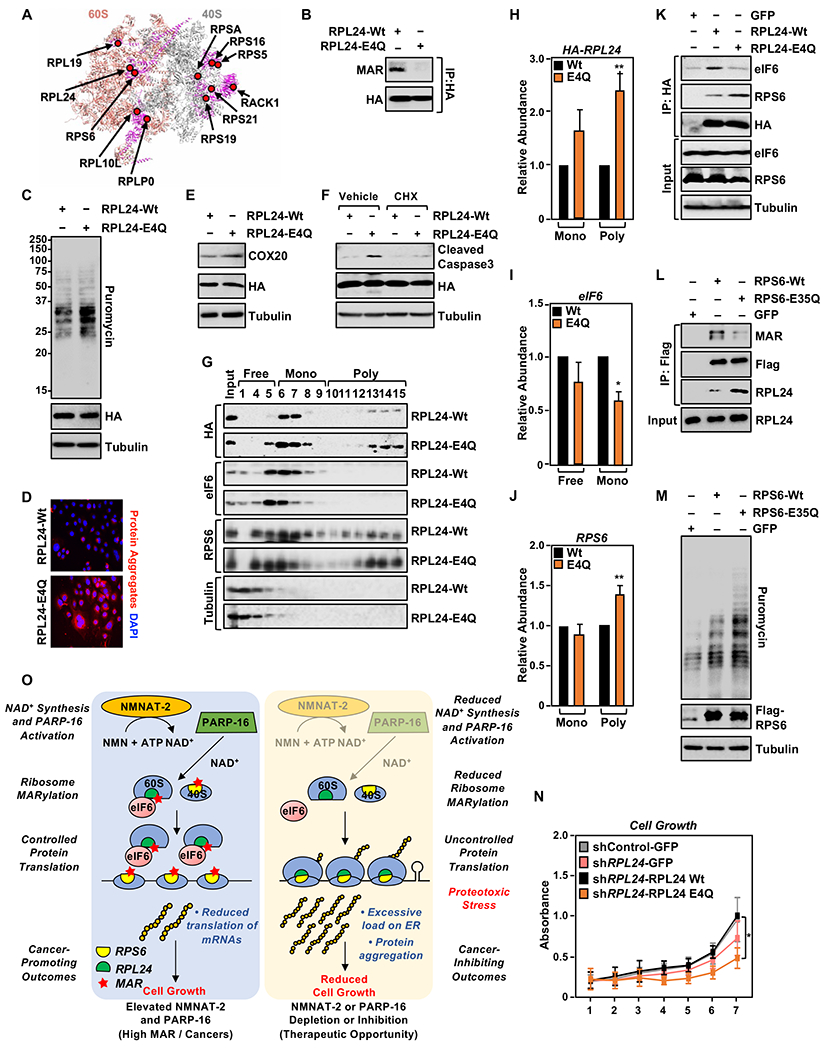
(A) Spatial distribution of the proteins modified by MARylation in the 80S ribosome (PDB ID: 4V6X). RPL24, which is located at the 60S-40S interface, is MARylated.
(B) RPL24 is MARylated at Glu 4. HA-tagged RPL24 was immunoprecipitated from OVCAR3 cells ectopically expressing wild-type (Wt) or MARylation deficient (E4Q) RPL24 and subjected to Western blotting for MAR and HA.
(C) RPL24-E4Q expression enhances protein synthesis in OVCAR3 cells. Western blot analysis of puromycin incorporation assays from OVCAR3 cells subjected to Dox-induced expression of RPL24.
(D) RPL24-E4Q expression promotes the accumulation of protein aggregates. Staining of protein aggregates using Proteostat aggresome detection reagent in OVCAR3 cells subjected to Dox-induced expression of RPL24.
(E) RPL24-E4Q expression enhances COX20 protein levels. Western blot analysis for COX20 in OVCAR3 cells subjected to Dox-induced expression of RPL24.
(F) Loss of RPL24 MARylation induces apoptosis. OVCAR3 cells subjected to Dox-induced expression of RPL24 were assayed for caspase 3 cleavage by Western blotting. Inhibition of translation by cycloheximide blocks the cleavage of caspase 3.
(G - J) Loss of RPL24 MARylation induces polysome formation. (G) Western blot analysis for HA-tagged RPL24, eIF6 and RPS6 of the sucrose density gradient fractions prepared from OVCAR3 cells subjected to Dox-induced expression of RPL24. Each bar in the graph in (H) represents the mean ± SEM of the relative abundance of RPL24, eIF6, and RPS6 in monosomes or polysomes (n = 4, Student’s t-test, * p < 0.05 and ** p<0.01).
(K) Loss of Glu4 MARylation inhibits RPL24 interaction with eIF6. HA-tagged RPL24 was immunoprecipitated from OVCAR3 cells with Dox-induced expression of RPL24 and subjected to Western blotting for eIF6, RPS6, and HA.
(L) MARylation of RPS6 at Glu 35 inhibits binding to RPL24. Flag-tagged RPS6 was immunoprecipitated from OVCAR3 cells subjected to Dox-induced expression of wild-type (Wt) or MARylation deficient (E35Q) RPS6 and subjected to Western blotting for MAR, RPL24 and Flag.
(M) RPS6-E35Q expression enhances protein synthesis in OVCAR3 cells. Western blot analysis of puromycin incorporation assays from OVCAR3 cells subjected to Dox-induced expression of RPS6.
(N) RPL24-E4Q expression inhibits cell growth. OVCAR3 cells subjected to Dox-induced knockdown and re-expression of RPL24 for 7 days and crystal violet staining was performed, (n = 4, one-way ANOVA, * p < 0.01).
(O) Schematic of the mechanisms by which NMNAT-2/NAD+ and PARP-16/MAR regulate protein homeostasis and ovarian cancer growth. Additional details are provided in the text.
Post-translational modifications such as acetylation of RPL24 affect its interaction with eIF6, a negative regulator of subunit joining (Lebreton et al., 2006; Wilson-Edell et al., 2014). In comparison to wild-type RPL24 (RPL24-Wt), RPL24-E4Q is more highly enriched in polysome fractions (Figure 7, G and H, and Figure S7L). RPL24-E4Q-expressing cells have greater enrichment of RPS6 in polysomes and reduced enrichment of eIF6 in ribosomes (Figure 7, G to J, and Figure S7L). Results from co-immunoprecipitation assays suggest that loss of RPL24 MARylation inhibits the interaction of RPL24 with eIF6, while enhancing its interaction with RPS6 and other proteins located in the 40S subunit (Figure 7K and Figure S7M). These data suggest that MARylation of RPL24 at Glu4 inhibits 80S monosome formation in-part by modulating the eIF6-60S complex. Similarly, we observed that mutation of a MARylation site in RPS6 (RPS6-E35Q) enhanced binding to RPL24 (Figure 7L), polysome formation (Figure S7O) and increased protein synthesis (Figure 7M).
In summary, we showed that loss of MARylation of RPL24 or RPS6 enhances protein synthesis which is driven by increased polysome assembly (Figure 7O). This pathway plays an essential role in control of cell growth as OVCAR3 cells that are subjected to knockdown and re-expression of RPL24-E4Q suppresses cell growth (Figure 7N and Figure S7N). These data highlight the significance of site-specific ribosomal protein MARylation in polysome assembly by modulating protein-protein interactions. Collectively, the results from this study shed light on a novel role for ribosomal protein MARylation in the regulation of protein homeostasis and the growth of ovarian cancer cells.
Discussion
Our results from this study connect cytosolic NAD+ synthesis by NMNAT-2 and cytosolic protein MARylation by PARP-16 in a molecular pathway that integrates cellular metabolism, ribosome function, and protein homeostasis (Figure 7O). This pathway is naturally amplified in ovarian cancers through the over-expression of NMNAT-2, providing a means for the cancer cells to maintain proteostasis during accelerated cell growth. Recent studies have shed light on proteotoxic stress as a key blockade for cancer cell transformation, and activation of proteins that can help alleviate this stress are required for optimal cell survival and growth (Chui et al., 2019; Guang et al., 2019). For example, enhanced translation of ATF4 mRNA downstream of c-Myc expression is required to overcome Myc-induced cell death. Once translated, ATF4 serves as a transcription factor that promotes the expression of genes involved in antioxidant response, autophagy, amino acid biosynthesis, and transport (Pathria et al., 2019; Tameire et al., 2019). Thus, understanding the underlying molecular mechanisms that support cancer growth by reducing proteotoxic stress are essential.
In our study, we observed that the NMNAT-2/PARP-16-dependent cytosolic MARylation pathway mediates ribosomal protein MARylation and helps to maintain protein homeostasis by generally inhibiting protein synthesis, but also directing the polysome loading and translation of a select set of mRNAs (Figure 7O). NAD+ produced by NMNAT-2 directly supports the catalytic activity of PARP-16. Consistent with this model, depletion of NMNAT-2 or PARP-16 relieves the inhibition of protein synthesis, leading to uncontrolled protein synthesis and the accumulation of protein aggregates. The control of mRNA translation by ribosomal MARylation is dependent, in part, on stem-loop regulatory elements present in the 3’UTRs of the mRNAs that exhibit enhanced polysome loading upon NMNAT-2 or PARP-16 depletion. Although the precise mechanism of how these stem-loop structures directs translational regulation has not been elucidated, the elements are transferable and can enhance the polysome loading of a heterologous mRNA upon NMNAT-2 or PARP-16 depletion. Structural elements in the 3’UTR of mRNAs are key for translational regulation (Mazumder et al., 2003). One possible mechanism by which the stem-loop structures could be directing translational regulation of ribosomal MARylation is by enhancing 5’-3’ looping of the mRNAs to increase ribosome recycling and translation initiation (Bai et al., 2013; Mangus et al., 2003; Wells et al., 1998). Or, the stem-loop structure could assist in loading of the mRNAs to polysomes, likely through RNA binding proteins (Matoulkova et al., 2012). Higher levels of polysome formation when ribosome MARylation is reduced might synergize with the 3’UTR-mediated loading of mRNAs to polysomes. The molecular details of this pathway provide a unique view of how cells can integrate production of metabolites, post-translational modification of proteins, and nucleic acid-based regulatory elements to control essential macromolecular processes.
We have also identified several ribosomal proteins that are modified by MARylation in ovarian cancer cells and characterized the mechanistic role of site-specific MARylation of RPL24 in the control of mRNA translation. We observed that expression of a MARylation deficient mutant, RPL24-E4Q, enhances protein synthesis and proteotoxic stress by promoting polysome assembly. Our data suggest that MARylation of RPL24 supports the interaction of eIF6 to the ribosomes and loss of this interaction by mutating Glu4 leads to higher polysome assembly. Release of eIF6 from the ribosomes is an essential step in ribosome assembly in the cytosol, and errors in this pathway are implicated in cancers and cancer-susceptibility syndromes, such as Shwachman–Bodian–Diamond syndrome (Gandin et al., 2008; Menne et al., 2007; Miluzio et al., 2011; Pressato et al., 2012). In addition to RPL24, a number of the MARylated proteins are localized to the 60S-40S interface; these MARylation events may also affect monosome assembly and protein synthesis similar to RPL24 MARylation. Moreover, other ribosomal proteins with roles in eIF6 binding, such as RPL10 (Bussiere et al., 2012; Ceci et al., 2003; Zhou et al., 2019), are also MARylated and may affect eIF6 binding to the ribosome and polysome assembly.
We have found that cancer cells can exploit this pathway to balance their need to maintain proteostasis as they accelerate growth, which in many cases relies on increased ribosome biogenesis and enhanced protein synthesis (Bhat et al., 2015; Truitt and Ruggero, 2016). Many ovarian cancers have elevated levels of NMNAT-2 expression, which correlate with enhanced MARylation levels. Dependence on NMNAT-2 in this pathway, however, creates a vulnerability in the cancer cells that can be exploited to inhibit the growth of the cells, (e.g., NMNAT-2 depletion). Importantly, high NMNAT-2 expression correlates with tumor grade, and high MARylation levels are a significant indicator of poor progression-free survival in ovarian cancers. Together, our data indicate that ovarian cancer cells depend on NMNAT-2 for the maintenance of elevated levels of cytoplasmic NAD+, which promotes protein homeostasis through PARP-16-dependent MARylation of ribosomal proteins, thereby regulating ribosome function and regulating protein synthesis. The NMNAT-2/NAD+ and PARP-16/MAR axis acts as a ‘guardrail’ that prevents fast-growing cancer cells with elevated protein synthesis from ‘falling off the cliff into toxic protein aggregation. Speculatively, such mechanisms may play a role in other diseases of protein aggregation, such as those that occur in the neurons. These and other possibilities will be explored in future studies.
Limitations of the Study
Recent studies have identified tyrosine residues on ribosomal proteins as acceptors of ADPR. The strategy used in our study for mass spectrometric identification of MARylation sites is limited to Asp/Glu residues, hence we cannot rule out additional sites of modifications and their functional importance. While we observed that MAR is highly enriched in ribosomes using biochemical and cell-based assays, we mainly used macrodomains 2 and 3 from PARP-14 to detect MAR. Future studies are required to test the specificity of this reagent towards specific modified residues. While we provide data from several independent assays in multiple cancer cell lines and patient samples to evaluate the molecular, cellular, and biological effects of reduced ribosome MARylation, additional studies are required to generalize our findings and model to human cancers in situ. Lastly, the NAD+ sensors used in this study have a low dynamic range. While we used standard curves from NAD+-permeabilized cells to determine the NAD+ levels in intact cells reliably, use of newer NAD+ detection strategies might improve the sensitivity.
STAR★Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, W. Lee Kraus, Ph.D. (Lee.Kraus@utsouthwestern.edu).
Materials availability
All cell lines and DNA constructs are available by request from W. Lee Kraus. The mono(ADP-ribose) detection reagent is available for purchase from EMD Millipore. The cpVenus-based NAD+ sensors were obtained under a material transfer agreement from Dr. Michael Cohen, Oregon Health and Sciences University.
Data and code availability
The RNA-seq and data sets generated specifically for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) using the superseries accession number GSE146458. The new mass spec data sets generated for these studies are available as supplemental data provided with this manuscript. The original data generated in this study can be accessed from the Mendeley dataset (https://data.mendeley.com/datasets/whw4z4cng3/draft?a=e2e1a38a-19e0-4078-9d5b-1bbd2251f34d)
Experimental Models and Subject Details
Cell culture
OVCAR3, OVCAR4, 293T, FT194, FT282, SKOV3, and SH-SY5Y cells were purchased from the American Type Cell Culture (ATCC). HCC5044 and HCC5012 cells were obtained from Dr. Adi Gazdar (Thu et al., 2017). The ovarian cancer cells were maintained in RPMI (Sigma-Aldrich, R8758) supplemented with 10% fetal bovine serum, 1% GlutaMax (Thermo Fisher Scientific, 35050061) and 1% penicillin/streptomycin. 293T cells were cultured in DMEM (Sigma-Aldrich, D5796) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The fallopian tube cells were cultured in DMEM-F12 (VWR, 45000-346) supplemented with 10% fetal bovine serum. SH-SY5Y cells were cultured in DMEM (Sigma-Aldrich, D5796) supplemented with 20% fetal bovine serum and 1% penicillin/streptomycin. Fresh cell stocks were replenished every three months. All cell lines were tested for mycoplasma every 6 months.
Generation of cell lines with stable knockdown or ectopic expression
Cells were transfected with either lentiviruses or retroviruses for stable knockdown or ectopic expression, respectively. We generated lentiviruses by transfection of the pLKO.1 constructs described above, together with an expression vector for the VSV-G envelope protein (pCMV-VSV-G, Addgene plasmid no. 8454), an expression vector for GAG-Pol-Rev (psPAX2, Addgene plasmid no. 12260), and a vector to aid with translation initiation (pAdVAntage, Promega) into 293T cells using GeneJuice transfection reagent (Novagen, 70967) according to the manufacturer’s protocol. The resulting viruses were used to infect the ovarian cancer cells in the presence of 7.5 μg/mL polybrene 24 hours and 48 hours, respectively, after initial 293T transfection. Stably transduced cells were selected with puromycin (Sigma, P9620; 1 μg/mL).
Retroviruses were generated by transfection of the pQXCIH NMNAT2 constructs described above, together with an expression vector for the VSV-G envelope protein (pCMV-VSV-G), into Phoenix Ampho cells using GeneJuice transfection reagent (Novagen, 70967) according to the manufacturer’s protocol. The resulting viruses were used to infect 293T cells in the presence of 7.5 μg/mL polybrene 24 hours and 48 hours, respectively, after initial 293T transfection. Stably transduced cells were selected with hygromycin (50 μg/mL).
Inducible ectopic protein expression in OVCAR3 cells
Recombinant lentiviruses were generated by transfecting the pINDUCER20 vectors into 293T cells as described above. OVCAR3 cells were infected with the lentiviruses with subsequent selection using 500 μg/mL G418 sulfate (Sigma, A1720). For inducible expression of RPL24, RPS6, or NMNAT-2, the cells were treated with 1 μg/mL Doxycycline (Dox) for 48 hours.
Inducible ectopic protein expression in FT 194 cells
Recombinant lentiviruses were generated by transfecting the pINDUCER20 vectors into 293T cells as described above. FT194 cells were infected with the lentiviruses with subsequent selection using 150 μg/mL G418 sulfate (Sigma, A1720). For inducible expression of NMNAT-2 cells were treated with 1 μg/mL Dox for 48 hours.
Generation of cell lines with inducible knockdown
Recombinant lentiviruses were generated by transfecting the pTRIPZ vectors into 293T cells as described above. Dox-inducible NMNAT2 knockdown in SH-SY5Y cells was described previously (Ryu et al., 2018). Briefly, after the stable expression of shNMNAT2 was obtained, knockdown was induced by treating the cells with 1 μg/mL doxycycline for 72 hours. OVCAR3 cells with ectopic expression of RPL24 as described above were infected with the lentiviruses, with subsequent selection using 500 μg/mL G418 sulfate, and then were treated with 1 μg/mL Dox for 48 hours.
siRNA-mediated knockdown and ectopic protein expression in OVCAR3 cells
For siRNA-mediated knockdown of endogenous NMNAT2 and re-expression of mouse Nmnat2 (resistant to the human-targeted RNAi sequence), the above described OVCAR3 cells expressing pInducer-Nmnat2 were treated with 1 μg/mL Dox at the time of plating. Twenty-four hours later, siRNAs were transfected using Lipofectamine RNAiMAX reagent (Invitrogen, 13778150) according to the manufacturer’s instructions. The cells were treated with 0.5 μg/mL Dox for 48 hours.
Mice used for in vivo experiments
All mouse xenograft experiments were performed in compliance with the Institutional Animal Care and Use Committee (IACUC) at the UT Southwestern Medical Center. Female NOD/SCID/gamma (NSG) mice at 6-8 weeks of age were used.
Method Details
Cell treatments
Ovarian cancer cells were treated with various inhibitors as described herein. For NAD+ depletion, cells were treated with a NAMPT inhibitor, FK866 (20 nM; Sigma, F8557) for 48 hours. For inhibition or activation of ER stress, the cells were treated with ISRIB (1 μM; Sigma, SML0843) and thapsigargin (250 nM; Tocris, 1138), respectively, for 3 hours. For inhibiting protein synthesis in the protein aggregation assays, the cells were treated with cycloheximide (10 μg/mL; Sigma, C7698) for 16 hours.
Antibodies
The custom rabbit polyclonal antisera against NMNAT-1 was made in-house as described previously (Ryu et al., 2018). The custom rabbit polyclonal antisera against NMNAT-2 was raised against bacterially-expressed, purified recombinant human NMNAT-2 (Pocono Rabbit Farm and Laboratory) and screened in-house against a range of NMNAT-2 antigens. The custom rabbit polyclonal antiserum against PARP-1 was generated in-house by using purified recombinant amino-terminal half of PARP-1 as an antigen (now available Active Motif; cat. no. 39559). The custom recombinant antibody-like anti-poly(ADP-ribose) binding reagent (anti-PAR) and anti-mono(ADP-ribose) binding reagent (anti-MAR) were generated and purified in-house (now available from Millipore Sigma, MABE1031 and MABE1076, respectively) (Gibson et al., 2017). The other antibodies used were as follows: rabbit polyclonal against PARP-16 (Abcam, ab84641 and GeneTex, GTX123450), mouse monoclonal against NMNAT-2 (Abcam, ab56980), ß-tubulin (Abcam, ab6046), SNRP70 (Abcam, ab83306), RPS6 (Cell Signaling Technologies, 2317S), RPL10 (Biorad, VPA00362), RPS5 (Santa Cruz, sc-390935), RPS19 (Santa Cruz, sc-100836), Puromycin (Millipore, MABE343), Flag (Sigma-Aldrich, F3165), COX20 (Proteintech, 25752-1-AP and Sigma, HPA045490), phospho-eIF2α (Cell signaling, 9721), eIF2α (Cell signaling, 9722), RPL24 (Proteintech, 17082-1-AP), eIF6 (Cell signaling, 3833T), rabbit monoclonal against RPS6 (Cell signaling, 2217S), cleaved caspase-3 (Cell signaling, 9661S), mouse monoclonal against HA (Sigma-Aldrich, H3663), rabbit polyclonal against HA (Abcam, ab9110), rabbit polyclonal against Flag (Invitrogen, PA1-984B), rabbit IgG (Invitrogen, 10500C), goat anti-rabbit HRP-conjugated IgG (Pierce, 31460), and goat anti-mouse HRP-conjugated IgG (Pierce, 31430).
siRNAs-mediated knockdown
The siRNA oligos used to knockdown the PARP enzymes were as follows:
PARP6 (siRNA1: SASI_Hs02_0035 3150, siRNA2: SASI_Hs01_00036196)
PARP7 (siRNA1: SASI_Hs01_00202925, siRNA2: SASI_Hs01_00202926)
PARP8 (siRNA1: SASI_Hs01_00139643, siRNA2: SASI_Hs01_00139644)
PARP10 (siRNA1: SASI_Hs01_00042392, siRNA2: SASI_Hs02_00360347)
PARP12 (siRNA1: SASI_Hs01_00151522, siRNA2: SASI_Hs01_00151523)
PARP14 (siRNA1: SASI_Hs02_00350199, siRNA2: SASI_Hs01_00178227)
PARP16 (siRNA1: SASI_Hs01_00071323, siRNA2: SASI_Hs01_00071325)
The siRNAs for the PARPs and NMNAT2 (SASI_Hs02_00346871) and the control siRNA (SIC001) were purchased from Sigma. All the siRNA oligos were transfected at a final concentration of 30 nM using Lipofectamine RNAiMAX reagent (Invitrogen, 13778150) according to the manufacturer’s instructions. The cells were used for various assays 48 hours after siRNA transfection.
Vectors for ectopic expression and knockdown
The vectors described below were generated using the oligonucleotide primers described in the next section. All constructs were verified by sequencing.
shRNAs targeting NMNAT2, NMNAT, and PARP16.
pLKO vectors expressing shRNAs targeting the mRNA sequences of human NMNAT2 (shRNA1: TRCN0000035439, shRNA2: TRCN0000035440), NMNAT1 (TRCN0000111436), PARP16 (shRNA1: TRCN0000433598, shRNA2: TRCN0000053169), and control shRNA (SHC002) were purchased from Sigma.
Dox-inducible shRNA knockdown of NMNAT2 in SH-SY5Y cells were described previously (Ryu et al., 2018). The pTRIPZ vectors encoding shRNAs targeting human NMNAT2 were purchased from Dharmacon (shRNA1: V3THS400730, shRNA2: V3THS_400733) and the control pTRIPZ vector was used as described previously (Ryu et al., 2018). The pTRIPZ vector encoding shRNAs targeting human RPL24 was purchased from Horizon Discovery (RHS4696-200748120).
Mammalian expression vectors.
Retroviral NMNAT-2 expression vectors were generated to express NMNAT2 mRNA that is resistant to targeting by shRNA2. The cDNA insert was amplified by PCR from pCMV-NMNAT2 and cloned into the pQXCIH and pInducer20 vectors. Mutations in the shRNA targeting region were incorporated using the protocol adapted from the Quickchange site-directed mutagenesis kit (Agilent). The plasmid for expressing the GFP epitope-tagged PARP-16 was obtained from Dr. Michael Cohen. The plasmids for Dox-inducible expression of wild-type and catalytically dead mouse NMNAT-1 and mouse NMNAT-2 were described previously (Ryu et al., 2018).
A sequence corresponding to the stem-loop structure (SL) enriched in the 3’UTRs of mRNAs with increased polysome loading upon NMNAT2 or PARP16 knockdown was cloned downstream of Flag-luciferase cDNA in the pcDNA3 vector (Addgene, 58792). In addition to the wild-type sequences, sequences with mutations in the stem-loop structure that either destroy the stem-loop or reverse the orientation (antisense) of the stem-loop were also cloned into the pCDNA3 Flag-luciferase vector.
Expression vectors for cpVenus-based nuclear and cytoplasmic NAD+ sensors and their corresponding cpVenus-only controls were kindly provided by Dr. Michael Cohen and Dr. Richard Goodman (Cambronne et al., 2016).
The plasmid for Dox-inducible expression of C-terminal HA epitope-tagged RPL24 was generated using a cDNA for RPL24 that was amplified from the pCMV3 cDNA clone obtained from Sino biologicals (HG20845-G) and subcloned into the pInducer20 vector. The E4Q mutation was introduced into the pCMV3-RPL24 plasmid using the protocol adapted from the Quickchange site-directed mutagenesis kit (Agilent).
The plasmid for Dox-inducible expression of C-terminal Flag epitope-tagged RPS6 was generated using a cDNA for RPS6 that was amplified from the pcDNA3.1+/C-(K)DYK cDNA clone obtained from GenScript (OHu02539) and subcloned into the pInducer20 vector. The E35Q mutation was introduced into the pcDNA3.1+/C-(K)DYK-RPS6 plasmid using the protocol adapted from the Quickchange site-directed mutagenesis kit (Agilent).
List of oligonucleotide primers used for cloning
Cloning primers for pQXCIH NMNAT2
Forward: 5’- ACCTGCAGGAATTGATCCGCATGGACTACAAGGATGACG -3’
Reverse: 5’- AATTAAGCGTACGAGGCCTACTAGCCGGAGGCATTGATG -3’
Primers for generating RNAi-resistant NMNAT2
Forward 1: 5’- GTTTGAAAGAGCAAGAGATTATCTGCAC -3’
Reverse 1: 5’- GTGCAGATAATCTCTTGCTCTTTCAAAC -3’
Forward 2: 5’- AAAGAGCAAGAGACTACCTGCACAAAAC -3’
Reverse 2: 5’- GTTTTGTGCAGGTAGTCTCTTGCTCTTT -3’
Primers for generating catalytically inactive NMNAT2 (W92G)
Forward: 5’- ACCAGGACACCGGGCAGACGACCTGCAG -3’
Reverse: 5’- CTGCAGGTCGTCTGCCCGGTGTCCTGGT -3’
Primers for cloning pInducer NMNAT2
Forward: 5’- TCCGCGGCCCCGAACTAGTGATGGACTACAAGGATGACG -3’
Reverse: 5’- GTTTAATTAATCATTACTACCTAGCCGGAGGCATTGATG -3’
Primers for cloning pCDNA3.1(+) PARP16
Forward: 5’-GCGAAGCTTATGGACTACAAAGACGATGACGACAAGCAGCCCTC AGGCTGG-3’
Reverse: 5’-GCGGGATCCTTATCTTTTCGCACGATTCCAAAAG-3’
Primers for cloning stem-loops into pCDNA3 Flag-luciferase
Wildtype SL Forward: 5’-GCGGAAAGTCCAATTGTAAACACCTGTAATCCCAGCACTTTGGGAGGCCGAGGTG-3’
Wildtype SL Reverse: 5’-TAGGGCCCTCTAGATGCATGCACCTCGGCCTCCCAAAGTGCTGGGATTACAGGTGT-3’
Mutant SL Forward: 5’-GCGGAAAGTCCAATTGTAAACACCTGTAATCCCAGCACTTTCCCTCGCCGAGGTG-3’
Mutant SL Reverse: 5’-TAGGGCCCTCTAGATGCATGCACCTCGGCGAGGGAAAGTGCTGGGATTACAGGTGT-3’
Antisense SL Forward: 5’-GCGGAAAGTCCAATTGTAAGTGGAGCCGGAGGGTTTCACGACCCTAATGTCCACA-3’
Antisense SL Reverse: 5’-TAGGGCCCTCTAGATGCATGTGTGGACATTAGGGTCGTGAAACCCTCCGGCTCCAC-3’
Primers for generating RPL24-E4Q
Forward: 5’-ATGAAGGTCCAGCTGTGCAGTTTTAG-3’
Reverse: 5’-CTAAAACTGCACAGCTGGACCTTCAT-3’
Primers for cloning pInducer-RPL24
RPL24 Wt Forward: 5’-TCCGCGGCCCCGAACTAGTGATGAAGGTCGAGCTGTGC-3’
RPL24 E4Q Forward: 5’-TCCGCGGCCCCGAACTAGTGATGAAGGTCCAGCTGTGC-3’
RPL24 Reverse: 5’-GTTTAATTAATCATTACTACTTAGGCGTAGTCAGGCAC-3’
Primers for cloning p!nducer-RPS6
Forward: 5’- TCCGCGGCCCCGAACTAGTGATGAAGCTGAACATCTCC -3’
Reverse: 5’- GTTTAATTAATCATTACTACTTATCACTTATCGTCGTCATC -3’
Primers for generating RPS6-E35Q
Forward: 5’- CGTATGGCCACACAAGTTGCTGCTGACGCT -3’
Reverse: 5’- AGCGTCAGCAGCAACTTGTGTGGCCATACG -3’
Preparation of cell lysates
Cells were cultured and treated as described above before the preparation of cell extracts.
Whole cell lysates.
At the conclusion of the treatments, the cells were washed twice with ice-cold PBS and resuspended in Lysis Buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing 1 mM DTT, 250 nM ADP-HPD, 10 μM PJ-34, 1x complete protease inhibitor cocktail (Roche, 11697498001) and phosphatase inhibitors (10 mM sodium fluoride, 2 mM sodium orthovanadate, and 10 mM ß-glycerophosphate). The cells were vortexed for 30 seconds in the Lysis Buffer and then centrifuged at full speed for 15 minutes at 4°C in a microcentrifuge to remove the cell debris.
Nuclear and cytoplasmic fractionation.
For the nuclear and cytoplasmic fractions, the cells were washed twice with ice-cold PBS and collected by scraping in Isotonic Buffer (10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 0.3 M sucrose, 1 mM DTT) containing 1x complete protease inhibitor cocktail, 250 nM ADP-HPD, 10 μM PJ-34, and phosphatase inhibitors (10 mM sodium fluoride, 2 mM sodium orthovanadate, and 10 mM ß-glycerophosphate). The cells were incubated on ice in the Isotonic Buffer for 15 minutes to allow the cells to swell and then lysed by the addition of 0.6% IGEPAL CA-630 with vortexing for 10 seconds. The nuclei from the lysed cells were pelleted by centrifugation at 10,000 x g for 30 seconds in a table top centrifuge, and the supernatant was collected as the cytoplasmic fraction. The pelleted nuclei were washed twice with ice-cold PBS and resuspended in Nuclear Extraction Buffer (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 1 mM EDTA, 1% IGEPAL CA-630, 1 mM DTT) containing 1x complete protease inhibitor cocktail, 250 nM ADP-HPD, 10 μM PJ-34, and phosphatase inhibitors (10 mM sodium fluoride, 2 mM sodium orthovanadate, and 10 mM ß-glycerophosphate). The nuclei were incubated on ice for 30 minutes for nuclear protein extraction. All the samples were centrifuged at full speed for 5 minutes at 4°C in a microcentrifuge to clarify and the supernatants were collected for immunoblotting.
Ribosome fractionation.
Ribosomal fractions were isolated from the cells as described previously (Kim et al., 2019). Briefly, the cells were plated into 150 cm diameter dishes at 90% confluence one day prior to the assay. The cells were then washed three times with ice-cold PBS and were scraped gently into 1.5 mL Buffer A (50 mM Tris-HCl, pH 7.5, 250 mM sucrose, 250 mM KCl, 5 mM MgCl2) supplemented with protease inhibitors, phosphatase inhibitors, ADP-HPD, and PJ-34 as described above. IGEPAL-CA-630 was then added to a final concentration of 0.7% (v/v) and the cells were incubated on ice for 15 minutes with frequent mixing. Five percent of each lysate was removed and stored for input or whole cell extract. The remaining portion of each lysate was centrifuged at 750 x g for 10 minutes at 4°C in a microcentrifuge, and the supernatants were centrifuged again at full speed for 10 minutes at 4°C in a microcentrifuge to remove nuclear proteins and transferred to a new tube. The concentration of KCl in the lysates was adjusted to 500 mM using a 3 M KCl stock and the lysates were loaded onto a 2.5 mL sucrose cushion (50 mM Tris-HCl pH 7.5, 1 M sucrose, 0.5 M KCl, 5 mM MgCl2,) in polypropylene tubes (Beckman Coulter, 328874). The samples were centrifuged for 4 hours at 210,000 x g in a Beckman coulter Optima L-80 XP ultracentrifuge using a SW60Ti rotor. After the centrifugation, the supernatant and sucrose cushion in each tube were discarded, and the ribosomal pellet was resuspended in Buffer C (50 mM Tris-HCl pH 7.5, 25 mM KCl, 5 mM MgCl2) supplemented with protease, phosphatase, and PARG (i.e., ADP-HPD) inhibitors.
Isolation of polysomes.
To isolate polysomes, 5 million cells were plated in 15 cm diameter dishes 24 hours prior to the assay. Polysomes were isolated from the cells using a previously described protocol (Morita, 2013) with some modifications. Briefly, the cells were treated with 100 μg/mL cycloheximide for 10 minutes, then washed three times with ice-cold PBS containing 100 μg/mL cycloheximide. The cells were collected by gentle scraping in 500 μL Polysome Lysis Buffer (15 mM Tris HCl pH 7.4, 15 mM MgCl2, 250 mM NaCl, 1% Triton X-100 in DEPC water) supplemented with 1 mM DTT, 100 μg/mL cycloheximide, and 400 U/mL RNase inhibitor (Promega; N2611), as well as the protease, phosphatase, PARG (ADP-HPD), and PARP (PJ-34) inhibitors noted above. The resuspended cells were vortexed for 30 seconds and centrifuged at full speed for 15 minutes at 4°C in a microcentrifuge. Five percent of the lysate was aliquoted to be used as input for measuring the steady state mRNA or protein levels. RNA content was measured by reading the absorbance at 260 nm, and equal amounts of RNA were loaded onto 10-50% sucrose gradients. The gradients were centrifuged at 125,000 x g for 2 hours at 4°C in a Beckman coulter Optima L-80 XP ultracentrifuge using a SW60Ti rotor. The gradient was collected as 250 μL fractions in 2 mL microfuge tubes. The RNA content in these fractions were measured by reading the absorbance at 260 nm and the peaks corresponding to monosomes and polysomes were noted.
The proteins were precipitated from these fractions using methanol-chloroform. Briefly, 900 μL of methanol were added to each 250 μL fraction with mixing by inversion, then 225 μL of chloroform were added with mixing by vortexing. Finally, 675 μL of ddH2O were added to the tubes, followed by vortexing until a precipitate was observed. The samples were centrifuged at full speed for 5 minutes at 4°C in a microcentrifuge. The upper phase was removed by aspiration and the protein pellet was washed by adding 750 μL methanol with gentle mixing. The protein pellet was re-collected by centrifugation at full speed for 5 minutes at 4°C in a microcentrifuge. After the protein pellets were allowed to air dry briefly, they were dissolved in 1x SDS-PAGE loading solution, heated at 50°C for 10 minutes, and boiled at 100°C for SDS-PAGE and subsequent immunoblotting.
Immunoblotting
Protein concentrations of the cell lysates were determined using a Bio-Rad Protein Assay Dye Reagent (Bio-Rad, 5000006). Equal volumes of lysates containing the same concentrations of protein were boiled at 100°C for 5 minutes after addition of 1/4 volume of 4x SDS-PAGE Loading Solution (250 mM Tris, pH 6.8, 40% glycerol, 0.04% Bromophenol Blue, 4% SDS), run on polyacrylamide-SDS gels, and transferred to nitrocellulose membranes. After blocking with 5% nonfat milk in TBST, the membranes were incubated with the primary antibodies described above in TBST with 0.02% sodium azide, followed by anti-rabbit HRP-conjugated IgG (1:5000) or anti-mouse HRP-conjugated IgG (1:5000). Immunoblot signals were detected using an ECL detection reagent (Thermo Fisher Scientific, 34577, 34095).
Measurement of total intracellular NAD+ levels
For NAD+ measurements, 1.5 million cells were lysed with 0.5 M perchloric acid and neutralized with 0.55 M K2CO3. The samples were then centrifuged and the supernatants were collected for NAD+ measurements. Total intracellular NAD+ levels were measured using a NAD+/NADH colorimetric assay kit (Cyclex, CY-1253) following the manufacturer’s instructions. Three replicates were performed and statistical differences between control and experimental samples were determined using one-way ANOVA analysis.
Puromycin incorporation assays
Protein synthesis was determined using puromycin incorporation assays as previously described (Schmidt et al., 2009). Briefly, cells were plated at 80% confluence in 6-well plates. The following day, the cells were treated with 10 μg/mL puromycin for 15 minutes at 37°C. Whole cell extracts were prepared from these cells and puromycin incorporation was visualized by immunoblotting using an antibody against puromycin.
GTEx and TCGA tissue expression analyses
The expression of NMNAT1, NMNAT2, and NMNAT3 in normal and cancer tissues was determined based on RPKM values using GEPIA (Tang et al., 2017).
RNA isolation and reverse transcription-quantitative real-time PCR (RT-qPCR)
OVCAR3 cells were transfected with different siRNAs and total RNA was isolated using the Qiagen RNAeasy Plus Mini kit (Qiagen, 74136) according to the manufacturer’s protocol. Total RNA was reverse transcribed using oligo(dT) primers and MMLV reverse transcriptase (Promega, PR-M1705) to generate cDNA. For the experiments with the luciferase/stem-loop constructs, RNA was reverse transcribed using random hexamer primers (Millipore, 11034731001) and MMLV reverse transcriptase. The cDNA samples were subjected to RT-qPCR using gene-specific primers, as described below. Target gene expression was normalized to the expression of RPL19 mRNA. All experiments were performed a minimum of three times with independent biological replicates to ensure reproducibility and a statistical significance of at least p < 0.05. Statistical differences between control and experimental samples were determined using the Student’s t-test.
RT-qPCR primers
RPL19 forward: 5’- ACATCCACAAGCTGAAGGCA-3’
RPL19 reverse: 5’- TGCGTGCTTCCTTGGTCTTA -3’
PARP6 forward: 5’- AGTTCTGGAATGATGACGACTCG -3’
PARP6 reverse: 5’- GTGGGTGTCGATACAGGTCAG -3’
PARP7 forward: 5’- CCAAAACCAGTTTCTTTGGGAG -3’
PARP7 reverse: 5’- CAGATTCCATCTACCACATCC -3’
PARP8 forward: 5’- TGTGCTAGTTACTACAGAGCCA -3’
PARP8 reverse: 5’- CCCCATCATAGTTCACCTGCC -3’
PARP10 forward: 5’- TACGGGAAGGGCGTGTATTTC -3’
PARP10 reverse: 5’- GCCACGAACACCGCCTTAT -3’
PARP12 forward: 5’- ATCTGCCAGCAGAACTTTGA -3’
PARP12 reverse: 5’- AACATCGTGTGGGTCTGCGTGT -3’
PARP14 forward: 5’- CTATGGATGCCAAGAATGGC -3’
PARP14 reverse: 5’- CTGGTCTGGAGTACGTATCA -3’
PARP16 forward: 5’- ATGGTAGCCGCCTAGAAAACT -3’
PARP16 reverse: 5’- CCCTCTCCGAACAAGGATGTC -3’
Luciferase forward: 5’- GAAGCGAAGGTTGTGGATCT -3 ’
Luciferase reverse: 5’- TGTAGCCATCCATCCTTGTC -3’
Immunofluorescent staining and confocal microscopy of cultured cells
The following microscopy-based protocols for cultured cells were used to determined cellular MAR and PAR localization and amounts, nuclear and cytoplasmic NAD+ levels, and protein aggregation levels in cells.
Immunostaining for MAR and PAR.
OVCAR3 cells were seeded on 8-well chambered slides (Thermo Fisher, 154534) one day prior to the experiment. The cells were washed twice with PBS, fixed in 4% paraformaldehyde for 15 minutes at room temperature, and washed three times with PBS. The cells were permeabilized for 5 minutes using Permeabilization Buffer (PBS containing 0.01% Triton X-100), washed three times with PBS, and incubated for 1 hour at room temperature in Blocking Solution (PBS containing 1% BSA, 10% FBS, 0.3M Glycine and 0.1% Tween-20). The fixed cells were incubated with a mixture of RPS6 antibody at a 1:200 dilution and the MAR or PAR detection reagents at a concentration of 20 μg/mL in PBS overnight at 4°C, followed by three washes with PBS. The cells were then incubated with a mixture of Alexa Fluor 594 donkey anti-rabbit IgG (ThermoFisher, A-21207) and Alexa Fluor 488 goat anti-mouse IgG (ThermoFisher, A-11001) each at a 1:500 dilution in PBS for 1 hour at room temperature. After incubation, the cells were washed three times with PBS. Finally, coverslips were placed on cells coated with VectaShield Antifade Mounting Medium with DAPI (Vector Laboratories, H-1200) and images were acquired using an inverted Zeiss LSM 780 confocal microscope.
Protein aggregation assay.
The levels of protein aggregation in cells were measured using Proteostat Protein Aggregation Kit (Enzo; ENZ-51035) according to the manufacturer’s protocol. Briefly, the cells were plated in 8-well chambered slides and treated ± cycloheximide (10 μg/mL) in normal growth media for 16 hours. The cells were then fixed in 4% paraformaldehyde for 30 minutes, permeabilized in Permeabilization Buffer (1x assay buffer containing 0.5% Triton X-100, 3 mM EDTA pH 8.0) for 30 minutes at 4°C. The cells were then treated with the Proteostat aggresome detection reagent for 30 minutes at room temperature. After washing with PBS, the coverslips were fixed with VectaShield containing DAPI, and confocal imaging was performed using a Zeiss LSM 780 microscope.
For quantifying the levels of protein aggregation, the cells were plated in a 96-well plate and treated ± 10 μg/mL cycloheximide for 16 hours. The cells were then fixed and permeabilized as described above, and then were stained with the Proteostat aggresome detection reagent and Hoechst 33342 for 30 minutes at room temperature. The cells were then washed twice with PBS and the fluorescence intensities at ex/em 500/600 nm (for Proteostat aggresome dye) and ex/em of 350/461 nm (for Hoechst 33342) were measured. Fluorescence intensity from a well not containing any cells was subtracted as background from the other measured intensities. The data were represented as a ratio of the mean florescence intensities relative to control knockdown cells. Four independent biological replicates were performed for each condition.
Image analysis.
The fluorescence intensities captured by the confocal imaging were analyzed by Fiji ImageJ software (Schindelin et al., 2012). The intensity and contrast of the images were further adjusted in Microsoft Powerpoint and same changes were applied to all of the samples.
Determination of nuclear and cytoplasmic NAD+ levels using cpVenus-based sensors
OVCAR3 cells expressing nuclear or cytoplasmic NAD+ sensors and their corresponding cpVenus-only controls (Cambronne et al., 2016) were used to measure changes in subcellular NAD+ levels as previously described (Ryu et al., 2018). The cells were transfected with the respective plasmids for cpVenus or NAD+ sensors in pCDNA3 expression vectors using GeneJuice. Twenty-four hours later, the cells were trypsinized and plated in 8-well glass bottom chamber slides (Thermo Fisher, 15411) in cell culture medium. Before microscopy, the medium was replaced with Fluorobrite medium (Thermo Fisher, A1896701) supplemented with 10% fetal bovine serum, and live-cell imaging was performed. Representative images were taken at 63x magnification on a Zeiss LSM 880 confocal microscope as previously described (Ryu et al., 2018).
For quantifying the relative NAD+ levels, the cells were treated as described above and the confocal images were acquired at 10x magnification. The ratios of fluorescence intensities at 405 nm to 488 nm of sensor and cpVenus controls were determined. Three independent biological replicates were performed for each condition.
Image analysis.
For NAD+ measurements, we used Fiji software to process the images to obtain TIFF files. The images were then processed in MATLAB using a custom script as described previously (Ryu et al., 2018) to obtain pixel-by-pixel ratiometric images of intensities at the 488 nm and 405 nm wavelengths. For quantification of NAD+ levels, ratios of fluorescence measurements at 488 nm and 405 nm of both sensor and cpVenus controls were obtained using Fiji software.
Permeabilization of cells to NAD+.
OVCAR3 cells were cultured as described above and the cells were then permeabilized with 0.002% digitonin and simultaneously treated with the indicated amounts of NAD+ with incubation at room temperature for 15 min. The samples were then subjected to live cell imaging and analyzed as described above. All of the ratiometric values were plotted relative to the values obtained using 10 mM NAD+.
Quantification of intracellular NAD+ levels.
To determine the intracellular NAD+ levels, the standard curve generated from the NAD+ permeabilization assays described above were fitted to a sigmoidal regression model using GraphPad Prism 9. The ratiometric values relative to 10 μM NAD+ were entered into the equation as a y value to calculate the intracellular (nuclear or cytoplasmic) NAD+ levels. To measure the changes in NAD+ levels under different experimental conditions, the ratiometric values of fluorescence measurements were obtained by live cell imaging analysis, as described above, and normalized to the samples with lowest values (e.g., NMNAT2 knockdown for cytosol). The NAD+ concentrations were then determined from the standard curve using the relative ratiometric values.
siRNA screen to identify PARPs that mediate MARylation of ribosomes
OVCAR3 cells were plated into 6-well plates and transfected with 30 nM each of the PARP mRNA-targeting siRNAs, as described before. Two different siRNAs per PARP mRNA were used. Twenty-four hours after transfection, the cells were trypsinized and collected. A portion of the cells was plated in 8-well chambered slides for immunofluorescent staining and the remaining cells were plated in 6-well plates for RNA-isolation. Knockdown efficiency of the siRNAs was measured by RT-qPCR, and MARylation levels were determined using immunofluorescent staining, as described above.
A separate set of cells was collected in parallel for immunoblotting of ribosome fractions with MAR detection reagent and RPS6 antibody. The intensity of the MAR and RPS6 signals from the immunoblots were determined using ImageJ software (Schneider et al., 2012) and the intensity of the MAR signals were normalized to RPS6 levels. Three independent biological replicates were performed for each condition and statistical differences versus the control were determined using Student’s t-test.
Determination of auto-activation of PARP-16
293T cells with stable expression of GFP, NMNAT-2 (WT), and NMNAT-2 (W92G) were transfected with a pCDNA3.1 vector containing a cDNA encoding Flag-epitope tagged PARP-16 using GeneJuice transfection reagent. Forty-eight hours after transfection, the cells were harvested in ice-cold PBS and then lysed in 250 mM Lysis Buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl, 1 mM EDTA, 1% IGEPAL CA-630, 10% glycerol, and 1 mM DTT) containing 1x complete protease inhibitor cocktail, phosphatase inhibitors, PARG inhibitor, and PARP inhibitor as described above. Equal volumes of lysate containing same amounts of total protein were used to immunoprecipitate PARP-16 by incubating with anti-Flag M2 affinity resin (Sigma, A2220-5mL) on a nutator at 4°C. After incubation overnight, the beads were washed three times for 5 minutes each at 4°C with High Salt Wash Buffer (50 mM Tris-HCl pH 7.5, 375 mM NaCl, 1 mM EDTA, 1% IGEPAL CA-630, 10% glycerol, and 1 mM DTT) containing 1x complete protease inhibitor cocktail, phosphatase inhibitors, PARG inhibitor, and PARP inhibitor as described above. The beads were boiled in 1x SDS-PAGE loading buffer. The samples were run on an 8% SDS-PAGE gel and transferred to a nitrocellulose membrane for immunoblotting. Autoactivation of PARP-16 was determined by immunoblotting with MAR detection reagent as described above.
Co-Immunoprecipitation of NMNAT-2 with PARP-16
293T cells with stable expression of GFP, NMNAT-2 (WT), and NMNAT-2 (W92G) were transfected with a pEGFP vector containing a cDNA encoding GFP-epitope tagged PARP-16 using GeneJuice transfection reagent. Forty-eight hours after transfection, the cells were harvested in ice-cold PBS and then lysed in IP Lysis Buffer (50 mM Tris-HCl pH7.5, 0.15 M NaCl, 1.0 mM EDTA, 1% NP-40 and 10% glycerol, supplemented with fresh 1 mM DTT, 250 nM ADP-HPD, 10 μM PJ34, 1x complete protease inhibitor cocktail). Equal volumes of lysate containing same amounts of total protein were used to immunoprecipitate PARP-16 by incubating with anti-GFP and protein G beads on a nutator at 4°C. After incubation overnight, the beads were washed five times for 5 minutes each at 4°C with IP Lysis Buffer. The beads were boiled in 1x SDS-PAGE loading buffer. The samples were run on an 10% SDS-PAGE gel and transferred to a nitrocellulose membrane for immunoblotting.
Co-Immunoprecipitation of RPL24 interacting proteins
OVCAR3 cells cultured in 15 cm diameter plates were treated with 1 μg/mL of Dox for 48 hours to induce the expression of HA-tagged RPL24. The cells were washed twice with ice-cold PBS, and collected in IP Lysis Buffer (50 mM Tris-HCl pH7.5, 0.15 M NaCl, 1.0 mM EDTA, 1% NP-40 and 10% glycerol, supplemented with fresh 1 mM DTT, 250 nM ADP-HPD, 10 μM PJ34, 1x complete protease inhibitor cocktail). The cell lysates were vortexed for 30 seconds and cell debris was cleared by centrifugation for 10 minutes at 4°C at 15,000 g. The protein concentrations in the supernatants were measured using a Bradford assays.
An equal amount of protein was used for each IP condition. The cell lysates were incubated with 2 μg of mouse monoclonal antibody against the HA tag and protein G agarose beads overnight at 4°C with gentle rotation. The beads were then washed five times with IP Lysis Buffer for 5 minutes each at 4°C with gentle mixing. The beads were then heated at 100°C for 5 minutes in 1x SDS-PAGE loading buffer. The immunoprecipitated proteins were run on a 12% SDS-PAGE gel and probed with the indicated antibodies.
Co-immunoprecipitation of RPS6-interacting proteins
OVCAR3 cells cultured in 15 cm diameter plates were treated with 1 μg/mL of Dox for 48 hours to induce the expression of HA-tagged RPL24. The cells were washed twice with ice-cold PBS, and collected in high salt IP Lysis Buffer (50 mM Tris-HCl pH7.5, 0.50 M NaCl, 1.0 mM EDTA, 1% NP-40 and 10% glycerol, supplemented with fresh 1 mM DTT, 250 nM ADP-HPD, 10 μM PJ34, 1x complete protease inhibitor cocktail). The cell lysates were vortexed for 30 seconds and cell debris was cleared by centrifugation for 10 minutes at 4°C at 15,000 g. The protein concentrations in the supernatants were measured using a Bradford assays.
An equal amount of protein was used for each IP condition. The cell lysates were incubated with anti-FLAG M2 agarose resin (Sigma, A2220) overnight at 4°C with gentle rotation. The beads were then washed five times with high salt IP Lysis Buffer for 5 minutes each at 4°C with gentle mixing. The beads were then heated at 100°C for 5 minutes in 1x SDS-PAGE loading buffer. The immunoprecipitated proteins were run on a 12% SDS-PAGE gel and probed with the indicated antibodies.
Polysome RNA-sequencing
Generation of RNA-seq libraries.
For performing polysome RNA-seq, two biological replicates of total RNA isolated from polysome fractions and corresponding input lysates were used. The total RNA was then enriched for polyA+ RNA using Dynabeads Oligo(dT)25 (Invitrogen). The polyA+ RNA was then used to generate strand-specific RNA-seq libraries as described previously (Zhong et al., 2011). The RNA-seq libraries were subjected to QC analyses (final library yield, and the size distribution of the final library DNA fragments) and sequenced using an Illumina HiSeq 2000.
Analysis of RNA-seq data.
The raw data were subjected to QC analyses using the FastQC tool (Andrews, 2010). The reads were then mapped to the human genome (hg38) using the spliced read aligner TopHat, version.2.0.13 (Kim et al., 2013). Transcriptome assembly was performed using cufflinks v.2.2.1 (Trapnell et al., 2010) with default parameters. The transcripts were merged into two distinct, non-overlapping sets using cuffmerge, followed by cuffdiff (Trapnell et al., 2010) to call the differentially regulated transcripts.
Normalizing enrichment in polysomes.
The FPKM expression values from the cuffdiff analysis above were used to determine the changes in gene expression levels. All genes with FPKM below one in each of the inputs were discarded. The genes whose levels of mRNAs in polysomes normalized to the levels in input are different in the NMNAT2 knockdown and PARP16 knockdown samples compared to the control samples were integrated to find the commonly regulated gene set. These values were used in the heatmap and subsequent analysis.
Transcriptome data analysis.
The differentially expressed genes from the analysis described above were used for subsequent analyses. Gene ontology analyses were determined using the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources website for gene ontology analysis (Huang da et al., 2009) for genes specifically altered in the NMNAT2 knockdown and PARP16 knockdown polysome-bound RNA compared to the control samples. Heat maps were generated using Java TreeView (Saldanha, 2004) for the genes significantly enriched or depleted in all of the indicated conditions. Box plot representations were used to compare the log2(fold change) for genes in the different experimental conditions compared to matched controls. Box plots were generated using custom scripts in R. Wilcoxon rank sum tests were performed to determine the statistical significance of all comparisons.
MEME analysis.
The 5’UTR and 3’UTR regions for genes the enriched genes and depleted genes were collected (hg38, Gencode v. 29). The sequences were then formatted to run a MEME analysis (Bailey et al., 2009) using a custom awk script, and sequences less than 8 nucleotides were removed using seqkit. MEME was then used to retrieve motifs using standard parameters and a maximum width size of 25 and RNAfold web server was used to predict the secondary structures of these motifs.
Functional analysis of 3’UTR stem-loop (SL) sequences
Flag-luciferase immunoblotting assays.
Expression of Flag-luciferase was used to evaluate 3’UTR sequence-dependent translation regulation by NMNAT-2 and PARP-16. OVCAR3 cells with control, NMNAT2, or PARP16 knockdown were plated into 6-well plates and the cells were transfected with 0.5 μg of the Flag-luciferase-SL constructs. Twenty-four hours after transfection, whole cell lysates were prepared by lysing the cells in 1x cell lysis buffer from the Promega luciferase assay kit (Promega, E1500) for immunoblotting, or total RNA was isolated as described above.
Polysome profiling of the luciferase mRNA.
For polysome profiling of the luciferase mRNA, OVCAR3 cells with control, NMNAT2, or PARP16 knockdown were plated into 15 cm diameter cell culture dishes and the cells were transfected with 4 μg of the wild-type stem-loop or mutant stem-loop Flag-luciferase-SL constructs. Twenty four hours after transfection, the cells were lysed and subjected to gradient centrifugation to isolate polysome fractions as described above. RNA was isolated from the fractions corresponding to free proteins (fractions 1-4), monosomes (fractions 7-10), and polysomes (fractions 12-16). Equal amounts of RNA were used for reverse transcription to generate cDNAs for RT-qPCR analysis. Three independent biological replicates were performed for each condition and two-way ANOVA was used for determining the statistical significance.
Analysis of Flag-luciferase in the protein aggregates.
For analyzing the co-localization of Flag-luciferase with protein aggregates, OVCAR3 cells were plated in 8-well chambered slides and, 24 hours later, they were transfected with the wild-type or mutant stem-loop Flag-luciferase-SL constructs. Forty-eight hours after transfection, the cells were fixed and permeabilized as described above for the protein aggregation assays. After permeabilization, the cells were blocked with 3% BSA in PBS for 1 hour and then incubated with Flag antibody overnight at 4°C. The cells were then washed with PBS and incubated with Alexa 488-conjugated mouse secondary antibody for 1 hour at room temperature. The cells were then washed with PBS and incubated with the Proteostat aggresome detection reagent in assay buffer for 30 minutes at room temperature. The cells were then washed with PBS and mounted with VectaShield containing DAPI. Confocal imaging was performed using a Zeiss LSM 780 microscope.
Identification and validation of COX20 as a target by immunoblot analysis.
To validate stem-loop-dependent translational regulation by MARylation with an endogenous protein, we first identified the targets using the polysome profiling data and MEME analysis as described in the previous section. COX20 was selected for further validation because the COX20 mRNA contains the identified stem-loop structure in its 3’UTR and it has increased polysome association upon NMNAT-2 or PARP-16 depletion. The expression of COX20 protein was determined by Western blot analysis as described above. Briefly, the OVCAR3 cells were plated into 6-well plates and 24 hours later the cells were lysed using the whole cell lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM DTT, 250 nM ADP-HPD, 10 μM PJ-34 supplemented with protease and phosphatase inhibitors). The lysates were run on a 10% SDS PAGE gel and transferred to a nitrocellulose membrane as described above. The membrane was probed first with COX20 antibody and then with ß-tubulin antibody. The intensities of COX20 and ß-tubulin signals from the immunoblots were determined using ImageJ software and the intensity of the COX20 signals were normalized to tubulin levels. Three independent biological replicates were performed for each condition and statistical differences versus the control were determined using Student’s t-test.
Analysis of COX20 and RPS6 in the protein aggregates.
Localization of COX20 or RPS6 to protein aggregates was determined using immunofluorescence assays. Briefly, the indicated cells were fixed, permeabilized and blocked as described above for the analysis of Flag-luciferase. The cells were then incubated with COX20 or RPS6 antibody at a dilution of 1:200 in PBS overnight at 4°C. The cells were then washed with PBS, incubated first with Alexa 488-conjugated rabbit secondary antibody or mouse secondary antibody for COX20 or RPS6 respectively for 1 hour and then with the Proteostat aggresome detection reagent for 30 minutes at room temperature. The cells were then washed with PBS, mounted on a coverslip and confocal imaging was performed. Three independent biological replicates were performed for each condition and the representative images were shown.
Cell growth assays
Cell growth assays with cycloheximide treatment.
OVCAR3 cells were plated at a density of 3,000 cells per well in a 96-well plate. The cells were then treated with either 1 pg/mL cycloheximide or with the vehicle control (ethanol). The cells were fixed with 4% paraformaldehyde on 0 and 7 days after treatment and washed with water. The fixed cells were then stained with crystal violet (0.5% crystal violet in 20% methanol) for 30 minutes with gentle shaking at room temperature. The stained cells were washed with water and air dried. The crystal violet was then dissolved in 10% acetic acid and the absorbance at 570 nm was measured using a spectrophotometer. The absorbance of a blank well was subtracted from the samples and the values were normalized to the values at day 0. Three independent biological replicates were performed to ensure reproducibility. Statistical differences were determined using one-way ANOVA.
Cell growth assays for OVCAR3 cells with ectopic expression of RPL24.
OVCAR3 cells with Dox-inducible knockdown and re-expression of RPL24 were plated at a density of 10,000 cells per well in a 24-well plate. The cells were maintained in growth medium containing 0.5 μg/mL puromycin, 200 μg/mL G418, and 1 μg/mL Dox for the indicated amount of time.
Anchorage independent growth assays
Soft agar assays were used to determine the changes in anchorage independent growth of the OVCAR3 cells. Briefly, the bottom agar layer was made by adding 1 mL of 6% agar (Thermo Scientific, J10907-22) in complete growth medium in a 12-well plate. The agar layer was allowed to solidify at 37°C for 30 minutes. The top layer, 1 mL of 3% agar containing 10,000 cells, was added gently over the bottom layer and the cells were cultured for 3 weeks. The cells were replenished with 300 μL fresh medium once per week. After 3 weeks, the colonies were stained with 0.5% crystal violet dissolved in 20% methanol. Excess crystal violet stain was washed with water and the plates were imaged. The number of colonies per well were manually counted. Three independent biological replicates were performed and statistical differences were determined using one-way ANOVA.
Xenograft experiments
To establish ovarian cancer xenografts, 2 × 106 OVCAR3 cells with control, NMNAT2, or PARP16 knockdown were injected subcutaneously in 100 μL into the flank of the mice in a 1:1 ratio of PBS and Matrigel (Fisher, CB 40230). The weight of the mice was monitored once per week and tumor growth measured using electronic calipers approximately once a week. Tumor volumes were calculated using a modified ellipsoid formula: Tumor volume = ½ (length × width2). At the end of the experiment, the mice were treated with puromycin (40 nmol/g of body weight) for 30 minutes and the mice were euthanized to collect the xenograft tissue. The tissue was cut into several small pieces and separate portions were either snap-frozen in liquid nitrogen or fixed using 4% paraformaldehyde. The frozen tissues were pulverized using a tissue mill and lysed in Whole Cell Lysis Buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM DTT, 250 nM ADP-HPD, 10 μM PJ-34 supplemented with protease and phosphatase inhibitors) for extraction of protein. The protein samples were analyzed by immunoblotting as described above.
Immunohistochemistry of ovarian cancer samples
Ovarian cancer tissue microarrays were purchased from US Biomax (Cat. no. OV2001a). Paraffin sections were deparaffinized in xylene and rehydrated sequentially in 100%, 95%, 80%, and 70% ethanol prepared in ddH2O. Antigen retrieval was performed by boiling the sections in 10 mM citrate, pH 6.0, for 10 minutes. After cooling to room temperature for 20 minutes, the sections were incubated in 3% H2O2 in methanol for 15 minutes. After washing with PBS, the sections were blocked with 5% normal goat serum (Thermo Fisher Scientific, 50062Z) in PBS at room temperature for 1 hour and then incubated with primary antibodies (1:500 for MAR and NMNAT-2, 1:300 for cleaved caspase 3) overnight at 4°C in PBS containing 1% BSA. After washing with PBS, the sections were incubated with biotin-conjugated horse anti-rabbit secondary antibody at room temperature for 1 hour. The sections were then incubated with Vectastain ABC reagent (Vector Laboratories, PK-7200-NB) for 30 minutes. The peroxidase was then developed using DAB reagent (Abcam, ab64238). After counterstaining with hematoxylin solution (Sigma, HHS16), the sections were dehydrated by sequential washing with 70%, 80%, 95%, and 100% ethanol and mounted with Permount Mounting Medium (Fisher Scientific, SP15).
Immunohistochemical staining for MAR was performed on a Dako Autostainer Link 48 system. Briefly, 5 μm paraffin sections were baked for 20 minutes at 60°C, then deparaffinized and hydrated before the antigen retrieval step. Heat-induced antigen retrieval was performed at pH 6 for 20 minutes in a Dako PT Link. The tissue was incubated with a peroxidase blocker and then MAR detection reagent (1:400) for 20 minutes. The staining was visualized using the EnVision FLEX visualization system. The intensity of staining for each antibody was scored on a scale of 0-3, where 3 was the highest intensity (expression) by a pathologist blinded to the experimental conditions.
Detection of protein aggregates in ovarian cancer samples
The ovarian cancer tissue microarray was baked at 65°C for 30 minutes, followed by deparaffinization, rehydration, antigen retrieval and peroxide blocking as described above. The tissues were then blocked and permeabilized in Permeabilization Buffer (1x assay buffer containing 0.5% Triton X-100, 3 mM EDTA pH 8.0) for 30 minutes at 4°C. The tissues were then incubated with the Proteostat aggresome detection reagent for 30 minutes at room temperature. After washing with PBS, the tissues were incubated with 1 μg/mL Hoescht 33342 nuclear stain for 5 minutes at room temperature. The slide was washed with PBS and coverslips were fixed with ProLong Diamond antifade mountant (Fisher Scientific, P36970) and stored in dark at room temperature for 24 hours. The slide was then scanned using a Zeiss Axioscan microscope.
Dot blotting to determine the residues on which ribosomal proteins are MARylated
Ribosomal fractions or whole cell extracts were prepared from OVCAR3 cells as described above. The lysates were incubated overnight with 100 mM HEPES pH 8.8 and 0.75 M hydroxylamine (Sigma, 438227). Four microliters of each lysate was spotted onto a nitrocellulose membrane. The membranes were air dried for 10 minutes, blocked with 5% nonfat milk in TBST and Western blotting was performed using the antibodies indicated.
Identification of ADP-ribosylation sites on ribosomal proteins
Isolation of ribosomes for mass spectrometric analysis.
To identify the sites of ADP-ribosylation on ribosomal proteins, ribosomes were isolated from OVCAR3 cells as described above. OVCAR3 cells were seeded into twelve 15 cm diameter plates at a density of 90% one day prior to ribosome isolation. The isolated ribosomes were resuspended in Buffer C (50 mM Tris-HCl pH 7.5, 25 mM KC1, 5 mM MgCl2) supplemented with protease, phosphatase, and PARG (i.e., ADP-HPD) inhibitors. EDTA was added to a final concentration of 5 mM and the sample was incubated at 4°C for 30 minutes to dissociate the ribosomal complexes. An equal volume of 200 mM HEPES pH 8.8 and 0.75 M hydroxylamine (Sigma, 438227) were added to the ribosome fractions, followed by incubated at 4°C for 16 hours. The fractions were concentrated using centrifugal filter units (Millipore, UFC501024), boiled in 4x SDS loading solution, followed by SDS-PAGE. The bands were excised from the gel and processed for mass spectrometric analysis. Three independent biological replicates were performed.
LC-MS/MS analysis.
Gel slices were digested overnight with trypsin (Promega) following reduction and alkylation with DTT and iodoacetamide, respectively (Sigma–Aldrich). The samples were subjected to solid-phase extraction cleanup with an Oasis HLB plate (Waters) and the resulting samples were injected onto an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex). The samples were injected onto a 75 μm i.d., 75-cm long EasySpray column (Thermo) and eluted with a gradient from 0-28% Buffer B over 90 min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water and Buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer was operated in positive ion mode with an ion transfer tube temperature of 275°C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2-7. Dynamic exclusion was set for 25 s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v2.2 (Thermo), with peptide identification performed using Sequest HT searching against the human protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, and oxidation of Met and hydroxamic acid modification of Asp and Glu were set as variable modifications. The false-discovery rate (FDR) cutoff was 1% for all peptides.
Determination of RPL24 MARylation
The following protocol was used to determine the MARylation levels of RPL24. Cells were transfected with a pCMV3 vector containing a cDNA encoding HA-epitope tagged RPL24 using GeneJuice transfection reagent. Forty-eight hours after transfection, the cells were harvested in ice-cold PBS and then lysed in 500 mM Lysis Buffer (50 mM Tris-HCl pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% IGEPAL CA-630, 10% glycerol, and 1 mM DTT) containing 1x complete protease inhibitor cocktail, phosphatase inhibitors, PARG inhibitor, and PARP inhibitor as described above. Equal volumes of lysate containing same amounts of total protein were used to immunoprecipitate RPL24 by incubating with mouse monoclonal antibody against HA and protein G agarose beads (Thermo Fisher, 15920010) at 4°C with gentle shaking. After incubation overnight, the beads were washed three times for 5 minutes each at 4°C with the 500 mM Lysis Buffer. The beads were boiled in 1x SDS-PAGE loading buffer. The samples were run on a 12% SDS-PAGE gel and transferred to a nitrocellulose membrane for immunoblotting with MAR detection reagent as described above.
• Quantification and Statistical Analysis
All sequencing-based genomic experiments were performed a minimum of two times with independent biological samples. Statistical analyses for the genomic experiments were performed using standard genomic statistical tests as described above. All gene specific qPCR-based experiments were performed a minimum of three times with independent biological samples. All Western blotting experiments with quantification were performed a minimum of three times with independent biological samples and analyzed by Image Lab 6.0. Statistical analyses were performed using GraphPad Prism 9. All tests and p-values are provided in the corresponding figures or figure legends.
Supplementary Material
Table S1. MARylated ribosomal proteins in OVCAR3 cells identified using mass spectrometry, Related to Figure 7 and Figure S7.
Ribosomal fractions isolated from OVCAR3 cells were treated with hydroxylamine (NH2OH) for site identification. The trypsin digested (Protein ID) hydroxylamine treated (Site ID) samples were subjected to LC-MS/MS analysis. The “Column Heading” key provides annotation information describing the metrics of each of the LC-MS/MS-identified peptides. The “Tab” key provides details about all of the other worksheets contained within this spreadsheet.
Figure S1. NMNATs mediate compartmentalized NAD+ synthesis, Related to Figures 1 and 2.
(A) Depletion of NMNAT-1 or NMNAT-2 does not affect total cellular NAD+ levels in OVCAR3 cells. Total NAD+ measurements in OVCAR3 cells subjected to shRNA-mediated NMNAT1 or NMNAT2 knockdown. As a control for the assay, OVCAR3 cells expressing control shRNA were treated with 20 nM FK866 for 48 hours. Each bar in the graph represents the mean ± SEM of the relative levels of NAD+ (n = 3, one-way ANOVA, ***p < 0.001).
(B) Depletion of NMNAT-1 or NMNAT-2 does not affect fluorescence intensities of the cpVenus control sensors. Fluorescence images of cytosolic or nuclear cpVenus control sensors in NMNAT1 or NMNAT2 knockdown OVCAR3 cells.
(C) Dose-response curves of sensors and controls upon NAD+ permeabilization. The ratiometric values of fluorescence ratios (488/405 nm) of NAD+ sensor and cpVenus controls were measured by live cell image analysis and the values were plotted relative to 10 mM NAD+. Each point represents the mean ± SEM, n = 3.
(D) NMNAT-2 catalytic activity is required for cytosolic NAD+ synthesis. (Left two panels) OVCAR3 cells, which were subjected to Dox-induced expression of mouse NMNAT-2 and siRNA-mediated knockdown of NMNAT2, were used for NAD+ sensor measurements. (Right two panels) OVCAR3 cells, which were subjected to Dox-induced expression of mouse NMNAT-1 and siRNA-mediated knockdown of NMNAT1, were used for NAD+ sensor measurements. Relative changes in NAD+ levels were calculated using sensor(488/405 nm)/control(488/405 nm) fluorescence ratios using a standard curve. (n = 3, t-test, * p < 0.05, ** p < 0.01, **** p < 0.0001).
(E) Nuclear PARylation is sensitive to NMNAT-1 depletion, but not NMNAT-2 depletion. Localization of PAR and RPS6, a ribosomal protein, in OVCAR3 cells subjected to shRNA-mediated NMNAT1 or NMNAT2 knockdown as determined by immunofluorescent staining. Representative images are shown. Scale bar = 10 μm.
(F) Representative images of immunohistochemical staining for NMNAT-2 and MAR using an ovarian cancer tissue microarray.
(G) Representative images of immunohistochemical staining for MAR in ovarian cancer tissues.
(H) Expression of NMNAT-1 inhibits ribosomal protein MARylation. Western blot analysis of ribosomal fractions or whole cell extracts prepared from OVCAR3 cells subjected to Dox-induced expression of wild-type (Wt) or catalytically dead (W170A) mouse NMNAT-1 (Nmnat1) followed by siRNA-mediated knockdown of NMNAT1.
(I) Ectopic expression of NMNAT-1 enhances protein synthesis. Western blot analysis of puromycin incorporation assays prepared from OVCAR3 cells subjected to Dox-induced expression of wild-type (Wt) or catalytically dead (W170A) mouse NMNAT-1 (Nmnat1) followed by siRNA-mediated knockdown of NMNAT1.
Figure S2. Small scale siRNA screen to identify the MART(s) required for ribosomal protein MARylation, Related to Figure 3.
(A) RNA-seq analysis of the expression of members of the PARP family of enzymes in OVCAR3 cells. Highlighted in red are the cytosolic MARTs.
(B) RT-qPCR analysis in OVCAR3 cells of the mRNAs encoding MARTs that were subjected to knockdown. Each MART was targeted by two different siRNAs. Each bar in the graph represents the mean ± SEM of the mRNA levels of the indicated PARPs, normalized to the levels of RPL19 (n = 3, t-test with the Holm-Sidak correction, ** p < 0.001).
(C) Representative images of OVCAR3 cells subjected to immunofluorescent staining for MAR, RPS6, and DNA (DAPI). The results from siRNA #1 targeting each PARP monoenzymes are shown. Scale bar = 10 μm.
(D) Quantification of the Western blots shown in Figure 3B. Each bar in the graph represents mean ± SEM of the relative expression of MAR to RPS6 ratio (n = 3, t-test with the Holm-Sidak correction, ** p < 0.01).
(E) Uncropped images of OVCAR3 cells subjected to immunofluorescent staining for MAR, RPS6, and DNA (DAPI). The results from siRNA #1 (Figure 3A) and siRNA #2 (Figure S2C) targeting each PARP monoenzymes are shown.
Figure S3. MARylation-dependent regulation of protein synthesis is unaltered by the ER-stress pathway, Related to Figure 3.
(A) NMNAT-2 interacts with PARP-16. GFP-epitope tagged PARP-16 was immunoprecipitated from 293T cells ectopically expressing Flag-tagged wild-type (Wt) or catalytically dead (W92G) NMNAT-2 and subjected to Western blotting for NMNAT-2 and GFP.
(B) Quantitative analysis of protein aggregation in OVCAR3 cells. Fluorescence measurements of protein aggregates using Proteostat aggresome detection reagent and Hoechst 3342 dye in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Treatment with a low dose of cycloheximide (10 μg/mL) for 16 hours inhibits the accumulation of the aggregates. Each bar in the graph represents the mean ± SEM of the ratio of fluorescence intensities of the Proteostat and Hoechst 3342 signals (n = 4, t-test, * p < 0.05, ** p < 0.01).
(C and D) Altering the ER stress levels does not affect the regulation of protein synthesis by PARP-16 or NMNAT-2. Western blot analysis of puromycin incorporation and phosphorylation of eIF2α in OVCAR3 cells subjected to PARP16 knockdown (C) or NMNAT2 knockdown (D) and treated with ISRIB (1 μM) or thapsigargin (250 nM) for 3 hours.
Figure S4. NMNAT-2 and PARP-16 regulate protein synthesis in other types of cancer cells, Related to Figure 3.
(A) Depletion of NMNAT-2 or PARP-16 reduces ribosomal protein MARylation in OVCAR4 cells. Western blot analysis of ribosomal fractions from OVCAR4 cells subjected to shRNA-mediated NMNAT2 or PARP16 knockdown.
(B and C) Depletion of NMNAT-2 or PARP-16 induces protein synthesis and protein aggregation in OVCAR4 cells. Analysis of puromycin incorporation as assessed by Western blotting (B) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (C) in OVCAR4 cells subjected to NMNAT2 or PARP16 knockdown.
(D) Depletion of NMNAT-2 or PARP-16 reduces ribosomal protein MARylation in SKOV3 cells. Western blot analysis of ribosomal fractions from SKOV3 cells subjected to shRNA-mediated NMNAT2 or PARP16 knockdown. (E and F) Depletion of NMNAT-2 or PARP-16 induces protein synthesis and protein aggregation in SKOV3 cells. Analysis of puromycin incorporation as assessed by Western blotting (E) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (F) in SKOV3 cells subjected to NMNAT2 or PARP16 knockdown.
(G) Depletion of NMNAT-2 or PARP-16 reduces ribosomal protein MARylation in HCC5044 cells. Western blot analysis of ribosomal fractions from HCC5044 cells subjected to shRNA-mediated NMNAT2 or PARP16 knockdown.
(H and I) Depletion of NMNAT-2 or PARP-16 induces protein synthesis and protein aggregation in HCC5044 cells. Analysis of puromycin incorporation as assessed by Western blotting (H) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (I) in HCC5044 cells subjected to NMNAT2 or PARP16 knockdown.
(J) Depletion of NMNAT-2 reduces ribosomal protein MARylation in SH-SY5Y neuroblastoma cells. Western blot analysis of ribosomal fractions from SH-SY5Y cells subjected to doxycycline (Dox) treatment for 72 hours to induce shRNA-mediated knockdown of NMNAT2.
(K and L) Depletion of NMNAT-2 enhances protein synthesis and protein aggregation in SH-SY5Y cells. Analysis of puromycin incorporation as assessed by Western blotting (E) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (F) in SH-SY5Y cells subjected to doxycycline (Dox) treatment for 72 hours to induce shRNA-mediated knockdown of NMNAT2.
Figure S5. NMNAT-2 and MARylation or ribosomal proteins are required for the growth of ovarian cancers, Related to Figure 4.
(A) Blocking mRNA translation restores cell growth in PARP-16- or NMNAT-2-depleted OVCAR3 cells. OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown were cultured in the presence of vehicle or cycloheximide (1 μg/mL) for 7 days and crystal violet staining was performed. Each bar in the graph represents the mean ± SEM of the relative levels of cell growth (n = 3, one-way ANOVA, * p < 0.05).
(B) Images of xenograft tumors formed from PARP16 or NMNAT2 knockdown OVCAR3 cells at the end of the experiment.
(C) Depletion of PARP-16 or NMNAT-2 enhances protein synthesis in vivo. Western blot analysis of puromycin incorporation in the OVCAR3 xenograft tumors described in (C).
(D) IHC analysis for correlation of MAR and NMNAT-2 with cleaved caspase-3 using ovarian cancer tissue microarrays. The number of patients in each group is indicated below the graphs (Chi-square test, * p<0.05, **** p < 0.0001).
Figure S6. Depletion of PARP-16 or NMNAT-2 promotes the accumulation of protein aggregates with a Flag-luciferase-3’ UTR stem-loop reporter, Related to Figure 6.
(A) mRNA distribution in the gradient centrifugation fractions for the samples used in Fig. 5A.
(B) Summary of the top five high complexity (i.e., non-repetitive) motifs in the 3’UTR of mRNAs that have higher polysome loading upon depletion of PARP-16 or NMNAT-2.
(C) Predicted secondary structures of the top motif hit with a predicted stem-loop structure (SL WT), mutated motif with predicted disrupted stem-loop structure (SL Mut), and antisense sequence with a predicted stem-loop structure (SL AS).
(D) The stem-loop motif does not affect the steady state levels of Flag-luciferase mRNA in a fusion construct. RT-qPCR analysis of Flag-luciferase mRNA in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown (Student’s t-test, none of the values are statistically significant).
(E) Depletion of PARP-16 or NMNAT-2 does not alter polysome loading of RPL19. RT-qPCR analysis of RPL19 mRNA isolated from the density gradient fractions corresponding to free ribosomal subunits, monosomes, and polysomes from PARP-16 or NMNAT-2 depleted OVCAR3 cells. Each bar in the graph represents the mean ± SEM of the relative RPL19 mRNA levels (n = 3, Two-way ANOVA, none of the values reached statistical significance).
(F) The stem-loop motif controls aggregation of Flag-luciferase when PARP-16 or NMNAT-2 are depleted. Co-staining of protein aggregates using Proteostat aggresome detection reagent and Flag-luciferase using Flag antibody in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Scale bar = 25 μm.
(G) RPS6 does not localize to protein aggregates in OVCAR3 cells. Costaining of protein aggregates using Proteostat aggresome detection reagent and RPS6 in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Scale bar = 25 μm.
(H and I) Ectopic expression of NMNAT-2 inhibits expression of the stem-loop containing Flag-luciferase. Quantification (H) and representative images (I) of Western blot analysis for Flag-luciferase of lysates from OVCAR3 cells that were subjected to Dox-induced expression of mouse NMNAT-2 (Nmnat2) followed by siRNA-mediated knockdown of NMNAT2 and transfection with the indicated Flag-luciferase constructs.
Figure S7. Identification of MARylation sites in ribosomal proteins, Related to Figure 7.
(A) Ribosomal proteins are modified at Glu and Asp residues. Dot blots of OVCAR3 cell ribosomal fractions (for MARylation) and whole cell extracts (for PARylation) subjected to treatment with NaCl or hydroxylamine, the latter to remove ADPR from the proteins.
(B) Venn diagram depicting the overlap of three replicates of the ribosomal proteins modified by MARylation in OVCAR3 cells as determined by mass spectrometry after hydroxylamine treatment.
(C) Ribosomal protein MARylation sites identified in three replicates of mass spectrometry.
(D and E) PARP-16 regulates RPL24 MARylation. HA-tagged RPL24 was immunoprecipitated from 293T cells ectopically expressing PARP-16 in (D) and OVCAR3 cells subjected to siRNA-mediated knockdown of PARP16 in (E) and assayed for MAR and HA by Western blotting.
(F) Quantification of the results from Figure 6C showing puromycin incorporation levels in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the ratio of puromycin to tubulin levels (n = 3, Student’s t-test, * p < 0.05).
(G) Quantitative analysis of protein aggregation in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the ratio of fluorescence intensities of the Proteostat and Hoechst 33342 signals (n = 4, Student’s t-test, ** p < 0.01).
(H) Quantification of the results from Figure 6E showing COX20 levels in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the relative expression of COX20 to tubulin ratio (n = 3, Student’s t-test; * p < 0.05).
(I) Quantification of the results from Figure 6F showing cleaved caspase-3 levels in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the relative levels of cleaved caspase-3 to tubulin ratio (n = 3, Student’s t-test, * p < 0.05).
(J and K) RPL24-E4Q blocks PARP-16 mediated increase in protein synthesis. (J) Western blot analysis of puromycin incorporation in OVCAR3 cells expressing Dox-induced wild-type (Wt) or mutant (E4Q) RPL24 subjected to siRNA-mediated PARP16 knockdown. Each bar in the graph in (K) represents the mean ± SEM of the relative ratios of Western blot signals of puromycin to tubulin (n = 3, Student’s t-test, ** p < 0.001).
(L) Quantitative representation of the Western blots in Figure 6G showing the simultaneous release of eIF6 and association of RPS6 from monosomal and polysomal fractions, respectively, when RPL24-E4Q is expressed.
(M) Loss of Glu4 MARylation inhibits RPL24 interaction with multiple proteins in the 40S subunit. HA-tagged RPL24 was immunoprecipitated from OVCAR3 cells with Dox-induced expression of RPL24 and subjected to Western blotting for RPS19, RPSA, and HA.
(N) Expression of RPL24 in OVCAR3 cells subjected to Dox-induced knockdown and re-expression of RPL24 that were used in the growth assays in Figure 7N.
(O) Loss of RPS6 MARylation induces polysome formation. Western blot analysis for Flag-tagged RPS6 and RPL24 in the sucrose density gradient fractions prepared from OVCAR3 cells subjected to Dox-induced expression of RPS6.
(P) Western blot analysis for Flag-luciferase in lysates from OVCAR3 cells that were subjected to Dox-induced expression of RPL24 and transfected with the indicated Flag-luciferase constructs.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PARP-1 | Active Motif | Cat. No. 39559; RRID: AB_2793257 |
| Anti-mono-ADP-ribose binding reagent | Millipore | Cat. No. MABE1076; RRID: AB_2665469 |
| Anti-poly-ADP-ribose binding reagent | Millipore | Cat. No. MABE1031; RRID: AB_2665467 |
| PARP-16 | Abcam | Cat. No. ab84641; RRID:AB_1925296 |
| RPS6 | Cell Signaling | Cat. No. 2317S; RRID:AB_2238583 |
| NMNAT-2 | This study | N/A |
| NMNAT-1 | Ryu et al., 2018 | N/A |
| RPL10 | Biorad | Cat. No. VPA00362 |
| FLAG | Sigma-Aldrich | Cat. No. F3165; RRID:AB_259529 |
| β-tubulin | Abcam | Cat. No. ab6046; RRID:AB_2210370 |
| Puromycin | Millipore | Cat. No. MABE343; RRID:AB_2566826 |
| COX20 | Proteintech | Cat. No. 25752-1-AP |
| Phospho-eIF2α | Cell Signaling | Cat. No. 9721; RRID:AB_330951 |
| eIF2α | Cell Signaling | Cat. No. 9722; RRID:AB_2230924 |
| SNRP70 | Abcam | Cat. No. ab83306; RRID:AB_10673827 |
| RPL24 | Proteintech | Cat. No. 17082-1-AP; RRID:AB_2181728 |
| eIF6 | Cell Signaling | Cat. No. 3833T; RRID:AB_2096520 |
| Rabbit monoclonal against RPS6 | Cell Signaling | Cat. No. 2217S; RRID:AB_331355 |
| Cleaved caspase-3 | Cell Signaling | Cat. No. 9661S; RRID:AB_2341188 |
| Mouse monoclonal against HA | Sigma-Aldrich | Cat. No. H3663; RRID:AB_262051 |
| Rabbit polyclonal against HA | Abcam | Cat. No. ab9110; RRID:AB_307019 |
| Rabbit polyclonal against Flag | Invitrogen | Cat. No. PA1-984B; RRID: AB_347227 |
| PARP-16 | GeneTex | Cat. No. GTX123450; RRID: AB_11171112 |
| RPS5 | Santa Cruz | Cat. No. sc-390935; RRID:AB_2713966 |
| RPS19 | Santa Cruz | Cat. No. sc-100836; RRID: AB_1129199 |
| COX20 | Sigma | Cat. No. HPA045490; RRID: AB_10962869 |
| Alexa Fluor 594 donkey anti-rabbit IgG | ThermoFisher | Cat. No. A-21207; RRID:AB_141637 |
| Alexa Fluor 488 goat anti-mouse IgG | ThermoFisher | Cat. No. A-11001; RRID:AB_2534069 |
| Goat anti-rabbit HRP-conjugated IgG | ThermoFisher | Cat. No. 31460; RRID: AB_228341 |
| Goat anti-mouse HRP-conjugated IgG | ThermoFisher | Cat. No. 31430; RRID: AB_10960845 |
| Rabbit IgG | ThermoFisher | Cat. No. 10500C; RRID: AB_2532981 |
| Biological samples | ||
| Ovarian cancer TMA | US Biomax | Cat. No. OV2001a |
| Ovarian cancer tissues | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| FK866 | Sigma | F8557; CAS: 658084-64-1 |
| ISRIB | Sigma | SML0843; CAS: 1597403-47-8 |
| Thapsigargin | Tocris | 1138; CAS: 67526-95-8 |
| Cycloheximide | Sigma | C7698; CAS: 66-81-9 |
| Critical commercial assays | ||
| RNeasy kit | QIAGEN | Cat. No. 74134 |
| NAD+/NADH colorimetric assay kit | Cyclex | Cat. No. CY-1253 |
| Proteostat Protein Aggregation Kit | Enzo | Cat. No. ENZ-51035 |
| Deposited data | ||
| RNA-seq | This study | GEO: GSE146458 |
| Original data files | This study | Mendeley dataset: http://data.mendeley.com/login?redirectPath=/datasets/whw4z4cng3/draft?a=e2e1a38a-19e0-4078-9d5b-1bbd2251f34d |
| Experimental models: cell lines | ||
| OVCAR3 | ATCC | RRID: CVCL_0465 |
| OVCAR4 | ATCC | RRID: CVCL_1627 |
| SH-SY5Y | ATCC | RRID: CVCL_0019 |
| 293T | ATCC | Cat. No. CRL-3216 |
| Phoenix | ATCC | RRID: SCR_003163 |
| SKOV3 | ATCC | RRID: CVCL_0532 |
| HCC5044 | Thu et al., 2016 | N/A |
| HCC5012 | Thu et al., 2016 | N/A |
| FT 194 | ATCC | RRID: CVCL_A4AW |
| FT 282 | ATCC | RRID: CVCL_A4AX |
| Experimental models: organisms/strains | ||
| Mouse: NOD-SCID Gamma | Mouse Breeding Core at UT Southwestern | N/A |
| Recombinant DNA | ||
| pINDUCER20-RPL24-HA wild-type and mutant | This study | N/A |
| pCMV3-RPL24-HA | Sino Biologicals | HG20845-G |
| pcDNA3.1+/C-(K)DYK-RPS6 | GenScript | OHu02539 |
| pINDUCER20-RPS6-Flag wild-type and mutant | This study | N/A |
| pQXCIH-Flag-NMNAT2, wild-type and mutant | This study | N/A |
| pInducer20-Flag-NMNAT2, wild-type and mutant | This study | N/A |
| pCDNA3.1+-Flag-PARP16 | This study | N/A |
| pCDNA3-Flag-Luciferase-SL | This study | N/A |
| pCDNA3-NAD+ sensors and cpVenus controls | Cambronne et al., 2016 | N/A |
| pLKO.1-NMNAT2 | Sigma | Cat. No. TRCN0000035439, TRCN0000035440 |
| pLKO.1-PARP16 | Sigma | Cat. No. TRCN0000433598, TRCN0000053169 |
| pLKO.1-NMNAT1 | Sigma | Cat. No. TRCN0000111436 |
| pTRIPZ-NMNAT2 | Dharmacon | Cat. No. V3THS400730, V3THS_400733 |
| pInducer20-Nmnat2, wild-type and mutant | Ryu et al., 2018 | N/A |
| pInducer20-Nmnat1, wild-type and mutant | Ryu et al., 2018 | N/A |
| pTRIPZ-RPL24 | Horizon Discovery | Cat. No. RHS4696-200748120 |
| Control shRNA | Sigma | SHC002 |
| pLKO.1 | Addgene | Plasmid No. 10878 |
| pINDUCER20 | Addgene | Plasmid No. 44012 |
| pCMV-VSV-G | Addgene | Plasmid No. 8454 |
| pAdVAntage | Promega | Cat. No. TB207 |
| psPAX2 | Addgene | Plasmid No. 12260 |
| Sequence-based reagents | ||
| Primers for molecular cloning | See STAR Methods | N/A |
| Software and algorithms | ||
| FastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Tophat | (Kim et al., 2013) | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | (Trapnell et al., 2010) | http://cole-trapnell-lab.github.io/cufflinks/ |
| DAVID Bioinformatics Resources | LHRI | https://david.ncifcrf.gov/content.jsp?file=citation.htm |
| Java TreeView | Saldanha, 2004 | http://jtreeview.sourceforge.net/ |
| MEME | Bailey et al., 2009 | https://meme-suite.org/ |
| RNAfold | N/A | http://ma.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi |
Highlights.
The cytosolic NAD+ synthase NMNAT-2 is highly upregulated in ovarian cancers.
Ribosome MARylation requires NAD+ from NMNAT-2, as well as the MART PARP-16.
Site-specific MARylation of ribosomes inhibits polysome assembly and mRNA loading.
It also inhibits translation, maintains proteostasis, and supports cancer cell growth.
Acknowledgements
We thank Dr. Andrew Kelleher and Dr. Rebecca Gupte for critical comments on this manuscript. We also thank Dr. Xiaolu Cambronne and Dr. Michael Cohen for the cpVenus-based NAD+ sensors and PARP-16 constructs. We acknowledge and thank the following UT Southwestern core facilities: Live Cell Imaging Core for microscopy support (Dr. Katherine Luby-Phelps), Next Generation Sequencing Core for deep sequencing services (Vanessa Schmid), Proteomics Core Facility for identification of sites of MARylation (Dr. Andrew Lemoff), Histopathology Core for histology services, Tissue Management Shared Resource for providing human tissues and immunohistochemical support, and the Whole Brain Microscopy Facility for imaging TMA slides (Dr. Denise Ramirez).
Funding
This work was supported by a grant from the NIH/NIDDK (R01 DK069710) to W.L.K., funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment to W.L.K., and a postdoctoral fellowship from the Ovarian Cancer Research Alliance (GAA202103-0003) to S.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
W.L.K. is a founder and consultant for Ribon Therapeutics, Inc and ARase Therapeutics, Inc. He is also coholder of U.S. Patent 9,599,606 covering the ADP-ribose detection reagent used herein, which has been licensed to and is sold by EMD Millipore.
References
- Ali YO, Allen HM, Yu L, Li-Kroeger D, Bakhshizadehmahmoudi D, Hatcher A, McCabe C, Xu J, Bjorklund N, Taglialatela G, et al. (2016). NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLoS Biol 14, e1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FASTQC. A quality control tool for high throughput sequence data. [Google Scholar]
- Bai Y, Zhou K, and Doudna JA (2013). Hepatitis C virus 3′UTR regulates viral translation through direct interactions with the host translation machinery. Nucleic Acids Res 41, 7861–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, and Ziegler M (2005). Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280, 36334–36341. [DOI] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, and Topisirovic I (2015). Targeting the translation machinery in cancer. Nat Rev Drug Discov 14, 261–278. [DOI] [PubMed] [Google Scholar]
- Bourens M, and Barrientos A (2017). Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J Biol Chem 292, 7774–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourens M, Boulet A, Leary SC, and Barrientos A (2014). Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum Mol Genet 23, 2901–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighenti E, Trere D, and Derenzini M (2015). Targeted cancer therapy with ribosome biogenesis inhibitors: a real possibility? Oncotarget 6, 38617–38627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere C, Hashem Y, Arora S, Frank J, and Johnson AW (2012). Integrity of the P-site is probed during maturation of the 60S ribosomal subunit. J Cell Biol 197, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR, and Dosil M (2017). Ribosome biogenesis and cancer: basic and translational challenges. Curr Opin Genet Dev 48, 22–29. [DOI] [PubMed] [Google Scholar]
- Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, and Goodman RH (2016). Biosensor reveals multiple sources for mitochondrial NAD(+). Science 352, 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceci M, Gaviraghi C, Gorrini C, Sala LA, Offenhauser N, Marchisio PC, and Biffo S (2003). Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature 426, 579–584. [DOI] [PubMed] [Google Scholar]
- Challa S, Stokes MS, and Kraus WL (2021). MARTs and MARylation in the Cytosol: Biological Functions, Mechanisms of Action, and Therapeutic Potential. Cells 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, Dolle C, Felici R, and Ziegler M (2012). The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer 12, 741–752. [DOI] [PubMed] [Google Scholar]
- Chui MH, Doodnauth SA, Erdmann N, Tiedemann RE, Sircoulomb F, Drapkin R, Shaw P, and Rottapel R (2019). Chromosomal Instability and mTORC1 Activation through PTEN Loss Contribute to Proteotoxic Stress in Ovarian Carcinoma. Cancer Res 79, 5536–5549. [DOI] [PubMed] [Google Scholar]
- Dai MS, and Lu H (2008). Crosstalk between c-Myc and ribosome in ribosomal biogenesis and cancer. J Cell Biochem 105, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Young JM, Kerns JA, and Malik HS (2014). Rapid evolution of PARP genes suggests a broad role for ADP-ribosylation in host-virus conflicts. PLoS Genet 10, e1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, and Barbieri JT (2008). Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu Rev Microbiol 62, 271–288. [DOI] [PubMed] [Google Scholar]
- Di Paola S, Micaroni M, Di Tullio G, Buccione R, and Di Girolamo M (2012). PARP16/ARTD15 is a novel endoplasmic-reticulum-associated mono-ADP-ribosyltransferase that interacts with, and modifies karyopherin-ss1. PLoS One 7, e37352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott LE, Saracco SA, and Fox TD (2012). Multiple roles of the Cox20 chaperone in assembly of Saccharomyces cerevisiae cytochrome c oxidase. Genetics 190, 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallie DR, Lewis NJ, and Marzluff WF (1996). The histone 3′-terminal stem-loop is necessary for translation in Chinese hamster ovary cells. Nucleic Acids Res 24, 1954–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V, Miluzio A, Barbieri AM, Beugnet A, Kiyokawa H, Marchisio PC, and Biffo S (2008). Eukaryotic initiation factor 6 is rate-limiting in translation, growth and transformation. Nature 455, 684–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Conrad LB, Huang D, and Kraus WL (2017). Generation and Characterization of Recombinant Antibody-Like ADP-Ribose Binding Proteins. Biochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang MHZ, Kavanagh EL, Dunne LP, Dowling P, Zhang L, Lindsay S, Bazou D, Goh CY, Hanley C, Bianchi G, et al. (2019). Targeting proteotoxic stress in cancer: A review of the role that protein quality control pathways play in oncogenesis. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka K, Ikutani M, Shito M, Kazuma K, Gulshan M, Nagai Y, Takatsu K, Konno K, Tobe K, Kanno H, et al. (2014). Deficiency of nicotinamide mononucleotide adenylyltransferase 3 (Nmnat3) causes hemolytic anemia by atering the glycolytic flow in mature erythrocytes. Journal of Biological Chemistry 289, 14796–14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jorgensen R, Purdy AE, Fieldhouse RJ, Kimber MS, Bartlett DH, and Merrill AR (2008). Cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. J Biol Chem 283, 10671–10678. [DOI] [PubMed] [Google Scholar]
- Jwa M, and Chang P (2012). PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1alpha-mediated unfolded protein response. Nat Cell Biol 14, 1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, and Kraus WL (2019). Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Challa S, Jones A, and Kraus WL (2020). PARPs and ADP-ribosylation in RNA biology: from RNA expression and processing to protein translation and proteostasis. Genes Dev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge S, and Woolford JL Jr. (2019). Ribosome assembly coming into focus. Nature reviews Molecular cell biology 20, 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuersten S, and Goodwin EB (2003). The power of the 3′ UTR: translational control and development. Nat Rev Genet 4, 626–637. [DOI] [PubMed] [Google Scholar]
- Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S, Phallen J, et al. (2017). High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun 8, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Dolle C, Gossmann TI, Agledal L, Niere M, and Ziegler M (2010). Isoform-specific targeting and interaction domains in human nicotinamide mononucleotide adenylyltransferases. J Biol Chem 285, 18868–18876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton A, Saveanu C, Decourty L, Rain JC, Jacquier A, and Fromont-Racine M (2006). A functional network involved in the recycling of nucleocytoplasmic pre-60S factors. J Cell Biol 173, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangus DA, Evans MC, and Jacobson A (2003). Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol 4, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateyak MK, and Kinzy TG (2013). ADP-ribosylation of translation elongation factor 2 by diphtheria toxin in yeast inhibits translation and cell separation. J Biol Chem 288, 24647–24655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoulkova E, Michalova E, Vojtesek B, and Hrstka R (2012). The role of the 3′ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol 9, 563–576. [DOI] [PubMed] [Google Scholar]
- Mayer PR, Huang N, Dewey CM, Dries DR, Zhang H, and Yu G (2010). Expression, localization, and biochemical characterization of nicotinamide mononucleotide adenylyltransferase 2. J Biol Chem 285, 40387–40396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder B, Seshadri V, and Fox PL (2003). Translational control by the 3′-UTR: the ends specify the means. Trends Biochem Sci 28, 91–98. [DOI] [PubMed] [Google Scholar]
- Medeiros F, Muto MG, Lee Y, Elvin JA, Callahan MJ, Feltmate C, Garber JE, Cramer DW, and Crum CP (2006). The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol 30, 230–236. [DOI] [PubMed] [Google Scholar]
- Menne TF, Goyenechea B, Sanchez-Puig N, Wong CC, Tonkin LM, Ancliff PJ, Brost RL, Costanzo M, Boone C, and Warren AJ (2007). The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet 39, 486–495. [DOI] [PubMed] [Google Scholar]
- Miluzio A, Beugnet A, Grosso S, Brina D, Mancino M, Campaner S, Amati B, de Marco A, and Biffo S (2011). Impairment of cytoplasmic eIF6 activity restricts lymphomagenesis and tumor progression without affecting normal growth. Cancer Cell 19, 765–775. [DOI] [PubMed] [Google Scholar]
- Mori V, Amici A, Mazzola F, Di Stefano M, Conforti L, Magni G, Ruggieri S, Raffaelli N, and Orsomando G (2014). Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 9, e113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Alain T, Topisirovic I and Sonenberg N (2013). Polysome profiling analysis. Bio-protocol 3. [Google Scholar]
- Orsomando G, Cialabrini L, Amici A, Mazzola F, Ruggieri S, Conforti L, Janeckova L, Coleman MP, and Magni G (2012). Simultaneous single-sample determination of NMNAT isozyme activities in mouse tissues. PLoS ONE 7, e53271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Lee JS, Hasnis E, Tandoc K, Scott DA, Verma S, Feng Y, Larue L, Sahu AD, Topisirovic I, et al. (2019). Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat Cell Biol 21, 1590–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressato B, Valli R, Marletta C, Mare L, Montalbano G, Lo Curto F, Pasquali F, and Maserati E (2012). Deletion of chromosome 20 in bone marrow of patients with Shwachman-Diamond syndrome, loss of the EIF6 gene and benign prognosis. Br J Haematol 157, 503–505. [DOI] [PubMed] [Google Scholar]
- Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, and Kraus WL (2018). Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Sauert M, Temmel H, and Moll I (2015). Heterogeneity of the translational machinery: Variations on a common theme. Biochimie 114, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6, 275–277. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon NC, Aktories K, and Barbieri JT (2014). Novel bacterial ADP-ribosylating toxins: structure and function. Nat Rev Microbiol 12, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, and Barna M (2017). An emerging role for the ribosome as a nexus for post-translational modifications. Curr Opin Cell Biol 45, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, Orsomando G, and Magni G (2007). Initial-rate kinetics of human NMN-adenylyltransferases: Substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry 46, 4912–4922. [DOI] [PubMed] [Google Scholar]
- Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, Salas Salinas C, Chinga F, Monroy AM, Fu W, et al. (2019). ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol 21, 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, and Zhang Z (2017). GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45, W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu KL, Papari-Zareei M, Stastny V, Song K, Peyton M, Martinez VD, Zhang YA, Castro IB, Varella-Garcia M, Liang H, et al. (2017). A comprehensively characterized cell line panel highly representative of clinical ovarian high-grade serous carcinomas. Oncotarget 8, 50489–50499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt ML, and Ruggero D (2016). New frontiers in translational control of the cancer genome. Nat Rev Cancer 16, 288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Sluis M, and McStay B (2014). Ribosome biogenesis: Achilles heel of cancer? Genes Cancer 5, 152–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Chesarone-Cataldo M, Todorova T, Huang YH, and Chang P (2013). A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun 4, 2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SE, Hillner PE, Vale RD, and Sachs AB (1998). Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2, 135–140. [DOI] [PubMed] [Google Scholar]
- Wilson-Edell KA, Kehasse A, Scott GK, Yau C, Rothschild DE, Schilling B, Gabriel BS, Yevtushenko MA, Hanson IM, Held JM, et al. (2014). RPL24: a potential therapeutic target whose depletion or acetylation inhibits polysome assembly and cancer cell growth. Oncotarget 5, 5165–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalowitz JA, Xiao S, Biju MP, Antony AC, Cummings OW, Deeg MA, and Jayaram HN (2004). Characterization of human brain nicotinamide 5′-mononucleotide adenylyltransferase-2 and expression in human pancreas. Biochem J 377, 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Hikosaka K, Mahmood A, Tobe K, Shojaku H, Inohara H, and Nakagawa T (2016). Nmnat3 Is dispensable in mitochondrial NAD level maintenance in vivo. PLOS ONE 11, e0147037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, Ryu KW, Gamble MJ, and Kraus WL (2012). Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. J Biol Chem 287, 12405–12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, Xiang JZ, Fei Z, and Giovannoni JJ (2011). High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc 2011, 940–949. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Musalgaonkar S, Johnson AW, and Taylor DW (2019). Tightly-orchestrated rearrangements govern catalytic center assembly of the ribosome. Nat Commun 10, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. MARylated ribosomal proteins in OVCAR3 cells identified using mass spectrometry, Related to Figure 7 and Figure S7.
Ribosomal fractions isolated from OVCAR3 cells were treated with hydroxylamine (NH2OH) for site identification. The trypsin digested (Protein ID) hydroxylamine treated (Site ID) samples were subjected to LC-MS/MS analysis. The “Column Heading” key provides annotation information describing the metrics of each of the LC-MS/MS-identified peptides. The “Tab” key provides details about all of the other worksheets contained within this spreadsheet.
Figure S1. NMNATs mediate compartmentalized NAD+ synthesis, Related to Figures 1 and 2.
(A) Depletion of NMNAT-1 or NMNAT-2 does not affect total cellular NAD+ levels in OVCAR3 cells. Total NAD+ measurements in OVCAR3 cells subjected to shRNA-mediated NMNAT1 or NMNAT2 knockdown. As a control for the assay, OVCAR3 cells expressing control shRNA were treated with 20 nM FK866 for 48 hours. Each bar in the graph represents the mean ± SEM of the relative levels of NAD+ (n = 3, one-way ANOVA, ***p < 0.001).
(B) Depletion of NMNAT-1 or NMNAT-2 does not affect fluorescence intensities of the cpVenus control sensors. Fluorescence images of cytosolic or nuclear cpVenus control sensors in NMNAT1 or NMNAT2 knockdown OVCAR3 cells.
(C) Dose-response curves of sensors and controls upon NAD+ permeabilization. The ratiometric values of fluorescence ratios (488/405 nm) of NAD+ sensor and cpVenus controls were measured by live cell image analysis and the values were plotted relative to 10 mM NAD+. Each point represents the mean ± SEM, n = 3.
(D) NMNAT-2 catalytic activity is required for cytosolic NAD+ synthesis. (Left two panels) OVCAR3 cells, which were subjected to Dox-induced expression of mouse NMNAT-2 and siRNA-mediated knockdown of NMNAT2, were used for NAD+ sensor measurements. (Right two panels) OVCAR3 cells, which were subjected to Dox-induced expression of mouse NMNAT-1 and siRNA-mediated knockdown of NMNAT1, were used for NAD+ sensor measurements. Relative changes in NAD+ levels were calculated using sensor(488/405 nm)/control(488/405 nm) fluorescence ratios using a standard curve. (n = 3, t-test, * p < 0.05, ** p < 0.01, **** p < 0.0001).
(E) Nuclear PARylation is sensitive to NMNAT-1 depletion, but not NMNAT-2 depletion. Localization of PAR and RPS6, a ribosomal protein, in OVCAR3 cells subjected to shRNA-mediated NMNAT1 or NMNAT2 knockdown as determined by immunofluorescent staining. Representative images are shown. Scale bar = 10 μm.
(F) Representative images of immunohistochemical staining for NMNAT-2 and MAR using an ovarian cancer tissue microarray.
(G) Representative images of immunohistochemical staining for MAR in ovarian cancer tissues.
(H) Expression of NMNAT-1 inhibits ribosomal protein MARylation. Western blot analysis of ribosomal fractions or whole cell extracts prepared from OVCAR3 cells subjected to Dox-induced expression of wild-type (Wt) or catalytically dead (W170A) mouse NMNAT-1 (Nmnat1) followed by siRNA-mediated knockdown of NMNAT1.
(I) Ectopic expression of NMNAT-1 enhances protein synthesis. Western blot analysis of puromycin incorporation assays prepared from OVCAR3 cells subjected to Dox-induced expression of wild-type (Wt) or catalytically dead (W170A) mouse NMNAT-1 (Nmnat1) followed by siRNA-mediated knockdown of NMNAT1.
Figure S2. Small scale siRNA screen to identify the MART(s) required for ribosomal protein MARylation, Related to Figure 3.
(A) RNA-seq analysis of the expression of members of the PARP family of enzymes in OVCAR3 cells. Highlighted in red are the cytosolic MARTs.
(B) RT-qPCR analysis in OVCAR3 cells of the mRNAs encoding MARTs that were subjected to knockdown. Each MART was targeted by two different siRNAs. Each bar in the graph represents the mean ± SEM of the mRNA levels of the indicated PARPs, normalized to the levels of RPL19 (n = 3, t-test with the Holm-Sidak correction, ** p < 0.001).
(C) Representative images of OVCAR3 cells subjected to immunofluorescent staining for MAR, RPS6, and DNA (DAPI). The results from siRNA #1 targeting each PARP monoenzymes are shown. Scale bar = 10 μm.
(D) Quantification of the Western blots shown in Figure 3B. Each bar in the graph represents mean ± SEM of the relative expression of MAR to RPS6 ratio (n = 3, t-test with the Holm-Sidak correction, ** p < 0.01).
(E) Uncropped images of OVCAR3 cells subjected to immunofluorescent staining for MAR, RPS6, and DNA (DAPI). The results from siRNA #1 (Figure 3A) and siRNA #2 (Figure S2C) targeting each PARP monoenzymes are shown.
Figure S3. MARylation-dependent regulation of protein synthesis is unaltered by the ER-stress pathway, Related to Figure 3.
(A) NMNAT-2 interacts with PARP-16. GFP-epitope tagged PARP-16 was immunoprecipitated from 293T cells ectopically expressing Flag-tagged wild-type (Wt) or catalytically dead (W92G) NMNAT-2 and subjected to Western blotting for NMNAT-2 and GFP.
(B) Quantitative analysis of protein aggregation in OVCAR3 cells. Fluorescence measurements of protein aggregates using Proteostat aggresome detection reagent and Hoechst 3342 dye in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Treatment with a low dose of cycloheximide (10 μg/mL) for 16 hours inhibits the accumulation of the aggregates. Each bar in the graph represents the mean ± SEM of the ratio of fluorescence intensities of the Proteostat and Hoechst 3342 signals (n = 4, t-test, * p < 0.05, ** p < 0.01).
(C and D) Altering the ER stress levels does not affect the regulation of protein synthesis by PARP-16 or NMNAT-2. Western blot analysis of puromycin incorporation and phosphorylation of eIF2α in OVCAR3 cells subjected to PARP16 knockdown (C) or NMNAT2 knockdown (D) and treated with ISRIB (1 μM) or thapsigargin (250 nM) for 3 hours.
Figure S4. NMNAT-2 and PARP-16 regulate protein synthesis in other types of cancer cells, Related to Figure 3.
(A) Depletion of NMNAT-2 or PARP-16 reduces ribosomal protein MARylation in OVCAR4 cells. Western blot analysis of ribosomal fractions from OVCAR4 cells subjected to shRNA-mediated NMNAT2 or PARP16 knockdown.
(B and C) Depletion of NMNAT-2 or PARP-16 induces protein synthesis and protein aggregation in OVCAR4 cells. Analysis of puromycin incorporation as assessed by Western blotting (B) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (C) in OVCAR4 cells subjected to NMNAT2 or PARP16 knockdown.
(D) Depletion of NMNAT-2 or PARP-16 reduces ribosomal protein MARylation in SKOV3 cells. Western blot analysis of ribosomal fractions from SKOV3 cells subjected to shRNA-mediated NMNAT2 or PARP16 knockdown. (E and F) Depletion of NMNAT-2 or PARP-16 induces protein synthesis and protein aggregation in SKOV3 cells. Analysis of puromycin incorporation as assessed by Western blotting (E) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (F) in SKOV3 cells subjected to NMNAT2 or PARP16 knockdown.
(G) Depletion of NMNAT-2 or PARP-16 reduces ribosomal protein MARylation in HCC5044 cells. Western blot analysis of ribosomal fractions from HCC5044 cells subjected to shRNA-mediated NMNAT2 or PARP16 knockdown.
(H and I) Depletion of NMNAT-2 or PARP-16 induces protein synthesis and protein aggregation in HCC5044 cells. Analysis of puromycin incorporation as assessed by Western blotting (H) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (I) in HCC5044 cells subjected to NMNAT2 or PARP16 knockdown.
(J) Depletion of NMNAT-2 reduces ribosomal protein MARylation in SH-SY5Y neuroblastoma cells. Western blot analysis of ribosomal fractions from SH-SY5Y cells subjected to doxycycline (Dox) treatment for 72 hours to induce shRNA-mediated knockdown of NMNAT2.
(K and L) Depletion of NMNAT-2 enhances protein synthesis and protein aggregation in SH-SY5Y cells. Analysis of puromycin incorporation as assessed by Western blotting (E) and protein aggregation as assessed by staining using Proteostat protein aggregation detection reagent (F) in SH-SY5Y cells subjected to doxycycline (Dox) treatment for 72 hours to induce shRNA-mediated knockdown of NMNAT2.
Figure S5. NMNAT-2 and MARylation or ribosomal proteins are required for the growth of ovarian cancers, Related to Figure 4.
(A) Blocking mRNA translation restores cell growth in PARP-16- or NMNAT-2-depleted OVCAR3 cells. OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown were cultured in the presence of vehicle or cycloheximide (1 μg/mL) for 7 days and crystal violet staining was performed. Each bar in the graph represents the mean ± SEM of the relative levels of cell growth (n = 3, one-way ANOVA, * p < 0.05).
(B) Images of xenograft tumors formed from PARP16 or NMNAT2 knockdown OVCAR3 cells at the end of the experiment.
(C) Depletion of PARP-16 or NMNAT-2 enhances protein synthesis in vivo. Western blot analysis of puromycin incorporation in the OVCAR3 xenograft tumors described in (C).
(D) IHC analysis for correlation of MAR and NMNAT-2 with cleaved caspase-3 using ovarian cancer tissue microarrays. The number of patients in each group is indicated below the graphs (Chi-square test, * p<0.05, **** p < 0.0001).
Figure S6. Depletion of PARP-16 or NMNAT-2 promotes the accumulation of protein aggregates with a Flag-luciferase-3’ UTR stem-loop reporter, Related to Figure 6.
(A) mRNA distribution in the gradient centrifugation fractions for the samples used in Fig. 5A.
(B) Summary of the top five high complexity (i.e., non-repetitive) motifs in the 3’UTR of mRNAs that have higher polysome loading upon depletion of PARP-16 or NMNAT-2.
(C) Predicted secondary structures of the top motif hit with a predicted stem-loop structure (SL WT), mutated motif with predicted disrupted stem-loop structure (SL Mut), and antisense sequence with a predicted stem-loop structure (SL AS).
(D) The stem-loop motif does not affect the steady state levels of Flag-luciferase mRNA in a fusion construct. RT-qPCR analysis of Flag-luciferase mRNA in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown (Student’s t-test, none of the values are statistically significant).
(E) Depletion of PARP-16 or NMNAT-2 does not alter polysome loading of RPL19. RT-qPCR analysis of RPL19 mRNA isolated from the density gradient fractions corresponding to free ribosomal subunits, monosomes, and polysomes from PARP-16 or NMNAT-2 depleted OVCAR3 cells. Each bar in the graph represents the mean ± SEM of the relative RPL19 mRNA levels (n = 3, Two-way ANOVA, none of the values reached statistical significance).
(F) The stem-loop motif controls aggregation of Flag-luciferase when PARP-16 or NMNAT-2 are depleted. Co-staining of protein aggregates using Proteostat aggresome detection reagent and Flag-luciferase using Flag antibody in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Scale bar = 25 μm.
(G) RPS6 does not localize to protein aggregates in OVCAR3 cells. Costaining of protein aggregates using Proteostat aggresome detection reagent and RPS6 in OVCAR3 cells subjected to PARP16 or NMNAT2 knockdown. Scale bar = 25 μm.
(H and I) Ectopic expression of NMNAT-2 inhibits expression of the stem-loop containing Flag-luciferase. Quantification (H) and representative images (I) of Western blot analysis for Flag-luciferase of lysates from OVCAR3 cells that were subjected to Dox-induced expression of mouse NMNAT-2 (Nmnat2) followed by siRNA-mediated knockdown of NMNAT2 and transfection with the indicated Flag-luciferase constructs.
Figure S7. Identification of MARylation sites in ribosomal proteins, Related to Figure 7.
(A) Ribosomal proteins are modified at Glu and Asp residues. Dot blots of OVCAR3 cell ribosomal fractions (for MARylation) and whole cell extracts (for PARylation) subjected to treatment with NaCl or hydroxylamine, the latter to remove ADPR from the proteins.
(B) Venn diagram depicting the overlap of three replicates of the ribosomal proteins modified by MARylation in OVCAR3 cells as determined by mass spectrometry after hydroxylamine treatment.
(C) Ribosomal protein MARylation sites identified in three replicates of mass spectrometry.
(D and E) PARP-16 regulates RPL24 MARylation. HA-tagged RPL24 was immunoprecipitated from 293T cells ectopically expressing PARP-16 in (D) and OVCAR3 cells subjected to siRNA-mediated knockdown of PARP16 in (E) and assayed for MAR and HA by Western blotting.
(F) Quantification of the results from Figure 6C showing puromycin incorporation levels in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the ratio of puromycin to tubulin levels (n = 3, Student’s t-test, * p < 0.05).
(G) Quantitative analysis of protein aggregation in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the ratio of fluorescence intensities of the Proteostat and Hoechst 33342 signals (n = 4, Student’s t-test, ** p < 0.01).
(H) Quantification of the results from Figure 6E showing COX20 levels in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the relative expression of COX20 to tubulin ratio (n = 3, Student’s t-test; * p < 0.05).
(I) Quantification of the results from Figure 6F showing cleaved caspase-3 levels in OVCAR3 cells subjected to Dox-induced RPL24 expression. Each bar in the graph represents the mean ± SEM of the relative levels of cleaved caspase-3 to tubulin ratio (n = 3, Student’s t-test, * p < 0.05).
(J and K) RPL24-E4Q blocks PARP-16 mediated increase in protein synthesis. (J) Western blot analysis of puromycin incorporation in OVCAR3 cells expressing Dox-induced wild-type (Wt) or mutant (E4Q) RPL24 subjected to siRNA-mediated PARP16 knockdown. Each bar in the graph in (K) represents the mean ± SEM of the relative ratios of Western blot signals of puromycin to tubulin (n = 3, Student’s t-test, ** p < 0.001).
(L) Quantitative representation of the Western blots in Figure 6G showing the simultaneous release of eIF6 and association of RPS6 from monosomal and polysomal fractions, respectively, when RPL24-E4Q is expressed.
(M) Loss of Glu4 MARylation inhibits RPL24 interaction with multiple proteins in the 40S subunit. HA-tagged RPL24 was immunoprecipitated from OVCAR3 cells with Dox-induced expression of RPL24 and subjected to Western blotting for RPS19, RPSA, and HA.
(N) Expression of RPL24 in OVCAR3 cells subjected to Dox-induced knockdown and re-expression of RPL24 that were used in the growth assays in Figure 7N.
(O) Loss of RPS6 MARylation induces polysome formation. Western blot analysis for Flag-tagged RPS6 and RPL24 in the sucrose density gradient fractions prepared from OVCAR3 cells subjected to Dox-induced expression of RPS6.
(P) Western blot analysis for Flag-luciferase in lysates from OVCAR3 cells that were subjected to Dox-induced expression of RPL24 and transfected with the indicated Flag-luciferase constructs.
Data Availability Statement
The RNA-seq and data sets generated specifically for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) using the superseries accession number GSE146458. The new mass spec data sets generated for these studies are available as supplemental data provided with this manuscript. The original data generated in this study can be accessed from the Mendeley dataset (https://data.mendeley.com/datasets/whw4z4cng3/draft?a=e2e1a38a-19e0-4078-9d5b-1bbd2251f34d)