

Abstract
The correct targeting and insertion of tail-anchored (TA) integral membrane proteins is critical for cellular homeostasis. TA proteins are defined by a hydrophobic transmembrane domain (TMD) at their C-terminus and are targeted to either the ER or mitochondria. Derived from experimental measurements of a few TA proteins, there has been little examination of the TMD features that determine localization. As a result, the localization of many TA proteins are misclassified by the simple heuristic of overall hydrophobicity. Because ER-directed TMDs favor arrangement of hydrophobic residues to one side, we sought to explore the role of geometric hydrophobic properties. By curating TA proteins with experimentally determined localizations and assessing hypotheses for recognition, we bioinformatically and experimentally verify that a hydrophobic face is the most accurate singular metric for separating ER and mitochondria-destined yeast TA proteins. A metric focusing on an 11 residue segment of the TMD performs well when classifying human TA proteins. The most inclusive predictor uses both hydrophobicity and C-terminal charge in tandem. This work provides context for previous observations and opens the door for more detailed mechanistic experiments to determine the molecular factors driving this recognition.
Keywords: co-chaperones, EMC, GET pathway, protein targeting, SND pathway, tail-anchored proteins
1 |. INTRODUCTION
Biogenesis of membrane proteins is an essential yet complicated process necessary for maintaining cellular homeostasis. Synthesized by ribosomes in the cytosol, membrane proteins account for approximately a third of the proteome and must be targeted to specified membranes (reviewed in References 1–3). A hydrophobic alpha-helical stretch, often a transmembrane domain (TMD), encodes this information and its position within an open reading frame dictates the cellular machinery responsible for its recognition and targeting.3 While computational methods have refined the ability to detect and predict cellular localization of these integral membrane proteins over time,4 the precise molecular signals continue to be elusive. Historically, decoding known signals into detailed rules has proven difficult given their great variation and the lack of sequence motifs–thus these signals are often discussed at a high level, for example, hydrophobic alpha-helical stretches. Despite the inability to define these rules, cellular chaperones accurately recognize the various signals to sort substrates into their distinct cellular destinations.
Here, we attempt to address one class of membrane proteins, tail-anchored (TA) proteins, found across cellular compartments and involved in a variety of roles including vesicle trafficking, protein translocation, quality control and apoptosis (reviewed in References 2,5–7). TA proteins are marked by a single TMD near their C-terminus and account for approximately 2% of the genome.6,8–10 Because of the position of their signals, TA proteins are translated by the ribosome and then post-translationally targeted primarily to the endoplasmic reticulum (ER) or outer mitochondrial membrane. The TMD and C-terminal residues following have been demonstrated to be necessary and sufficient for correct targeting in many experimental contexts.11,12 Thus, it is suggested that the information recognized by TA protein targeting pathways is contained within the TMD and neighboring residues.
The recent identification of a new route for TA proteins to the ER membrane has challenged how we previously differentiated between mitochondria and ER-bound TA proteins.3,10,13 To date, while the cellular components involved in mitochondrial TA protein targeting remain unclear, multiple overlapping pathways have been identified for TA protein targeting to the ER membrane.2,6,14–17 The first identified and most studied pathway is the Guided Entry of TA protein (GET) pathway.14,15 Consisting of six proteins, Sgt2 and Get1–5, the GET pathway is responsible for targeting ER-bound TA proteins (“ER TA proteins” for simplicity) with more hydrophobic TMDs. In yeast, the co-chaperone Sgt2 first captures TA proteins from Ssa1 and, with the aid of Get4 and Get5, transfers the client to the ATPase Get3 that acts as the central targeting factor of the pathway.3,18–21 An ER membrane bound Get1/2 complex facilitates disassociation of the Get3/TA complex and insertion of the TA protein into the membrane. Recently, Guna and colleagues demonstrated that human Get3 (HsGet3) fails to bind to TA proteins with relatively low hydrophobicity within their TMDs. These proteins instead are inserted into the ER membrane by the ER Membrane Complex (EMC).17 A 10-subunit complex, the EMC inserts TA proteins delivered by calmodulin. For TA proteins with moderately hydrophobic TMDs, both the GET pathway and EMC can facilitate insertion. A third dedicated pathway capable of targeting TA proteins into the ER membrane is the SRP-independent (SND) pathway.16 Snd1, the first component of the SND pathway, interacts with the ribosome and possibly the nascent chain while the membrane bound Snd2 and Snd3 interact with the translocon complex. In the absence of the GET pathway, the SND pathway is capable of targeting ER TA proteins with TMDs further away from their C-termini. These overlapping pathways, dependent on either hydrophobicity or signal positions, highlight the diversity in these proteins and the difficulty in identifying a common characteristic of ER-destined TMDs.16
General patterns have been observed based on exploration of targeting information within the TMD and the C-terminal residues of TA proteins. ER TA proteins tend to have more hydrophobic TMDs10,13,17,22 while some mitochondria TA proteins are amphipathic.10 By modifying the positive charge following their TMDs with an example TMD, studies have shown how insertion by the GET pathway into the ER membrane can be impaired.13,23 Distinction between peroxisomal and mitochondria TA proteins have been made based on the charge of their C-terminal tails, whereas mitochondria and ER TA proteins in mammals are differentiated by a combination of TMD hydrophobicity and C-terminal charge.24 A charged tail was overcome by increasing the hydrophobicity of the TMD, directing the mitochondrial TA protein to the ER. Guna and colleagues determine a threshold in total hydrophobicity by modifying a model TMD to delineate substrates that are inserted either via the GET or EMC pathways.17 Throughout these previous works, the ability of these rules to separate ER vs mitochondrial TA proteins at-large has not been systematically assessed, so their broader applicability is still unclear.
With multiple pathways with overlapping substrates, understanding the factors within substrates recognized for targeting is critical. Here we show that formalizing previously suggested criteria, while adequate, are not sufficient for classifying ER TA proteins with moderately hydrophobic TMDs suggested to be substrates of the EMC insertase. We demonstrate through computational and experimental methods that classifying TA proteins by the presence of a hydrophobic face in their TMD is more inclusive, properly capturing both ER TMDs with low hydrophobicity and mitochondrial TA proteins in both yeast and humans.
2 |. RESULTS
2.1 |. Curating TA proteins with experimentally determined localizations
In order to screen TA proteins to identify a concise criterium for localization, we first curated a comprehensive set of TA proteins from the yeast proteome pulling together localizations across public repositories and publication-associated datasets. We screened the reference yeast genome from UniProt25 for putative TA proteins and filtered for unique genes longer than 50 residues (Figure 1A). Uniprot and TOPCONS226 were used to identify proteins with a single TMD within 30 amino acids of the C-terminus5 that lacked a predicted signal peptide (as determined by SignalP 4.127). While this set encompasses proteins previously predicted as TA proteins,28,29 it is larger (95 vs 55 or 56) and we believe a more accurate representation of the repertoire of TA proteins (Figure 1B). Based on their UniProt-annotated and Gene Ontology Cellular Components (GO CC) localizations,30,31 TA proteins were subcategorized as ER-bound (encompassing labels including cell membrane, Golgi apparatus, nucleus, lysosome and vacuole membrane and referred to as ER TA proteins), mitochondrial (inner and outer mitochondria membrane [IMM & OMM]), peroxisomal, and unknown (Figure 1C). This set is readily available for future analyses (Table S1). The majority of proteins have no annotated cellular localization. Several previously identified TA proteins are not identified by our pipeline and excluded from this new set. These proteins include OTOA (otoancorin) that contains a predicted signal peptide, FDFT1 (squalene synthase or SQS) with two predicted hydrophobic helices by this method, and YDL012C which has a TMD with very low hydrophobicity.17,28 This analysis was also applied to the human genome and a list of 573 putative TA proteins was compiled and annotated based on published localizations (Figure 1A-C). Like with the yeast list, the human list is larger than previous reports (573 vs 411), and the majority of the proteins have no annotated localization.
FIGURE 1.
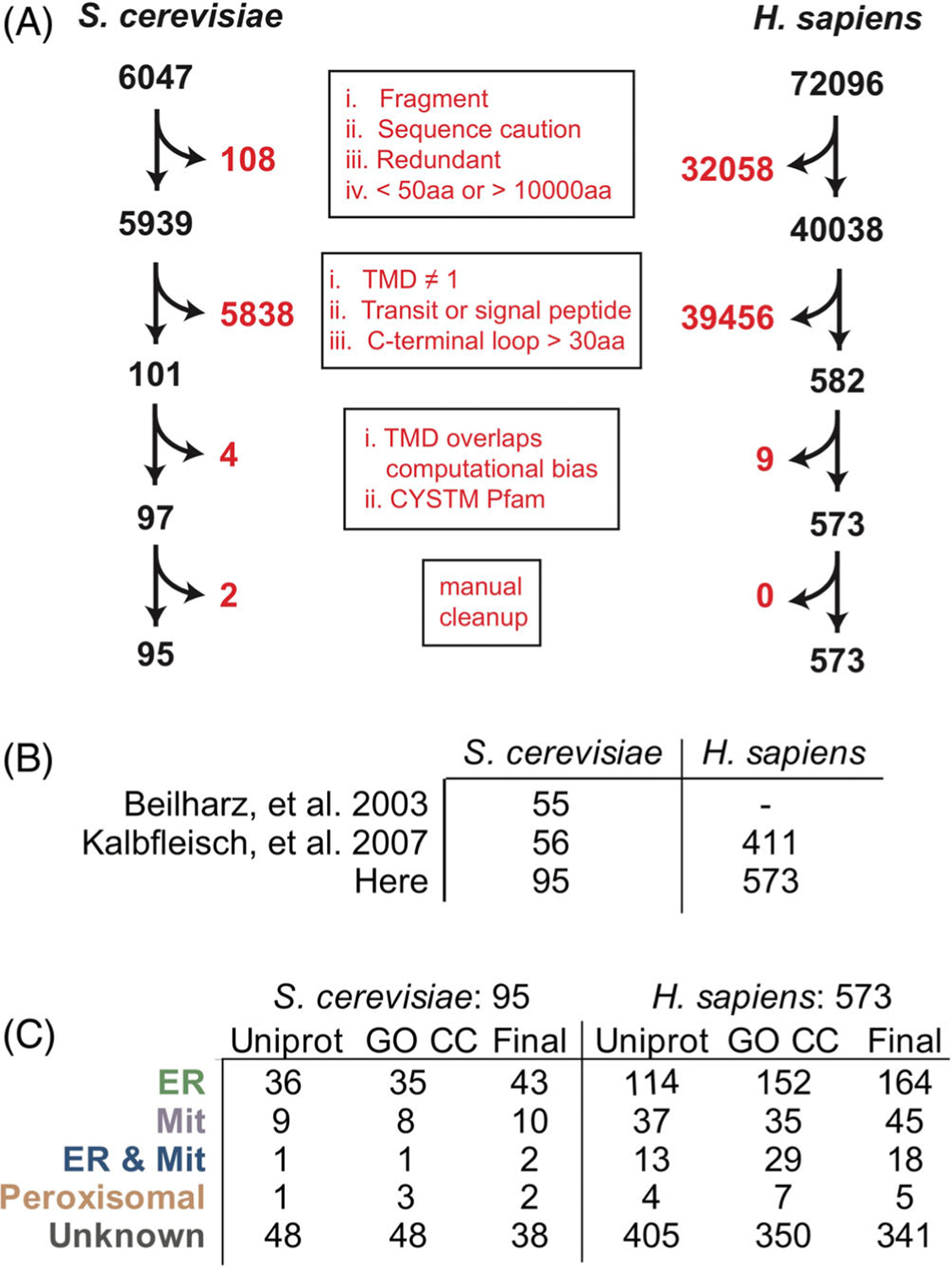
Compiling a list of TA proteins from the human and yeast genomes. (A) A schematic of the pipeline used to gather TA proteins by filtering the Human and Yeast proteomes for TA proteins. (B) A comparison of the TA proteins collected for the analyses here vs previous datasets. (C) Localizations gathered from Uniprot entry Subcellular Localizations (CC) and Gene Ontology Cellular Compartment (GO) annotations. Those with conflicts were resolved by manually parsing the literature to build the final set
2.2 |. Assessing current metrics for TA classification
To identify factors encoded within TA proteins that ensure correct localization, we began by considering several posited properties including the charge following the TMD, TMD length and TMD hydrophobicity. Previous reports suggest that the presence of positively charged residues following the TMD of mitochondria-bound TA proteins prevents insertion into the ER membrane.13,23 The number of positively charged C-terminal residues for all 95 yeast proteins was calculated, avoiding issues associated with defining the extent of TMDs by counting any charge from the center of the predicted TMD to the C-terminus. No clear separation is observed when plotting TA proteins with known localizations by number of positively charged residues (Figure 2A). As a metric this does a poor job distinguishing between the two; six ER-annotated proteins have a C-terminal positive charge of three or more and one out of the eight mitochondria-annotated proteins has no C-terminal positive charge. Furthermore, neither negative nor net charge of the C-terminal loop separates ER from mitochondrial TA proteins (Figure 2B,C). While modulating the C-terminal positive charge affects localization,13 cells do not solely use this signal to specify protein localization. Considering the difference in lipid compositions of the ER and mitochondrial membranes, a signal might be encoded in the TMD lengths, but this metric also fails to separate the two sets (Figure 2D).
FIGURE 2.
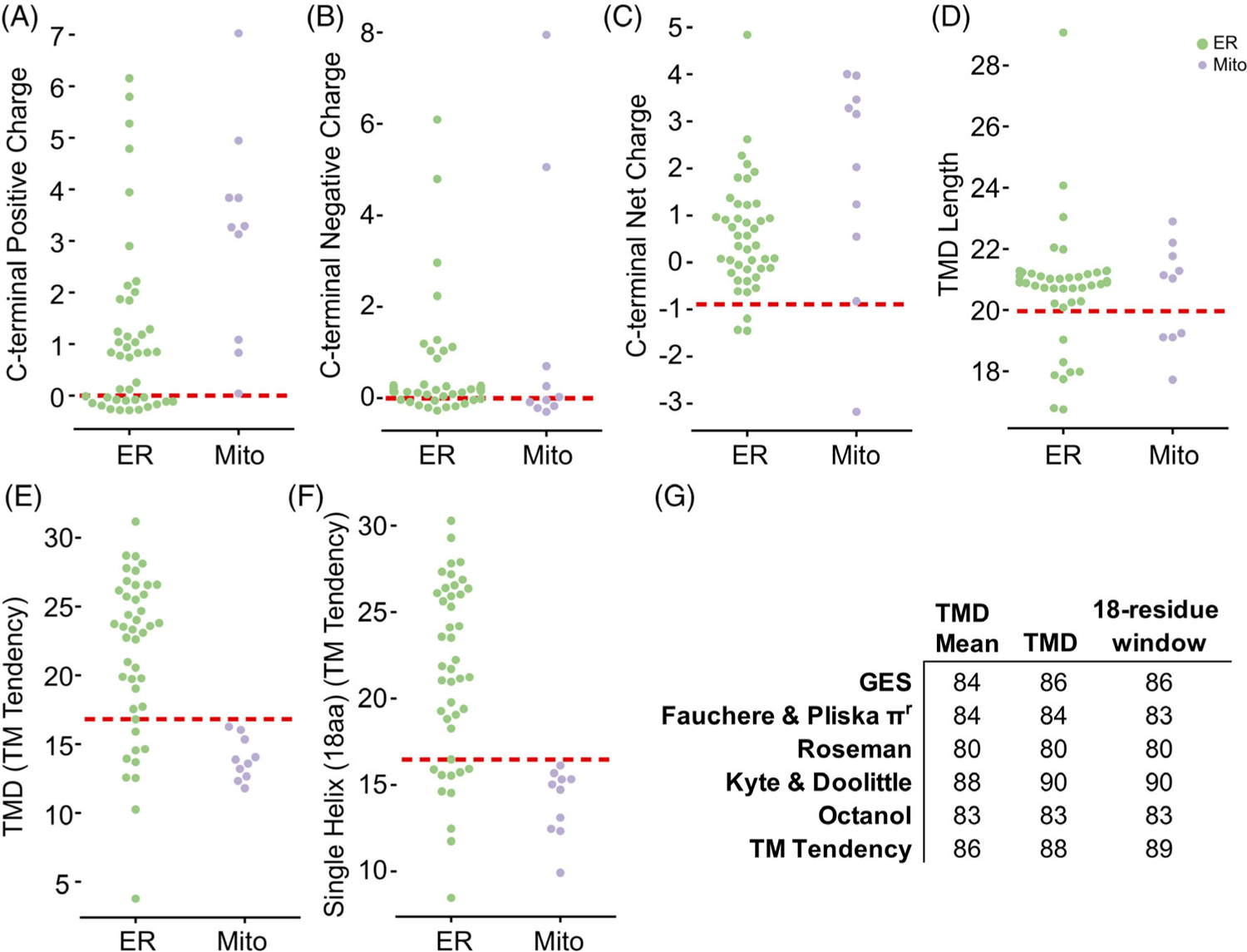
Investigating properties encoded in the C-terminal residues of TA proteins. For A-F, Jitter plots of property distribution for predicted TA proteins identified as ER (green) or mitochondria (purple) with the best predictive threshold indicated by a dashed red line. Properties visualized are for the C-terminal number of (A) positive residues, (B) negative residues, and (C) net charge and then for (D) TMD length, (E) TMD hydrophobicity, and (F) maximum hydrophobicity of an 18-residue stretch. (G) The AUROC across various hydrophobicity scales for the mean, total, and 18-residue windows of the predicted TMDs
TMD hydrophobicity is the proposed localization-determining feature of TA proteins in studies thus far.3,10,13 The TM tendency scale, used here and in past studies with TA targeting,3,17 is a statistical hydrophobicity scale that incorporates both hydrophobicity and helical propensity into a single value assigned to each of the 23 amino acids by using amino acid propensities in TMDs known at the time of its creation32 (Figure 2E). The total hydrophobicity (sum of each residue’s hydrophobicity value) of a TMD sufficiently splits ER and mitochondrial proteins but places a significant number of ER TA proteins among mitochondrial TA proteins. In other words, the total hydrophobicity can classify GET pathway substrates as ER-bound but fails to identify substrates of the EMC insertase that are also ER TA proteins.17 For example, the TMD of squalene synthase, a bona fide EMC substrate,17 has a lower hydrophobicity than that of model mitochondrial TA protein, Fis1 (Total TM Tendency = 12.5 vs 18.78, respectively). Limiting the hydrophobicity to a single helix stretch, that is, 18aa, sees no improvement in classification (Figure 2F).
To examine this inability to correctly classify lower hydrophobic ER TA proteins, we comprehensively assess hydrophobicity across a variety of established scales32–36 (Figure 2G) and then quantitatively assess predictive power using the receiver operating characteristic (ROC) framework (for a primer, see Reference 37). An ROC curve captures how well a numerical score separates two categories, here ER vs mitochondria, and whose figure of merit is the area under the curve (AUROC). This is a more accurate representation of prediction than simpler numbers like accuracy and precision, which require setting a specific threshold in a numerical score because it accounts for sensitivity and selectivity. A perfect separation gives an AUROC of 100 whereas a random separation results in an AUROC of 50. No matter the hydrophobicity scale used, the total hydrophobicity captures the ER vs mitochondria split to varying extents. In each case, the mean hydrophobicity performs more poorly, yet considering the most hydrophobic 18-residue single-helix stretch results in a slight improvement in predictive ability suggesting that a subset of the helix can explain recognition (Figure 2G).
2.3 |. TMD residue organization better classifies TA protein localization
We wondered if TA protein classification could be improved by carefully assessing the hydrophobicity of the TMDs. Data showing that Sgt2 (a co-chaperone in the GET pathway) binds a TMD of a minimal length of 11 residues suggests only a subset of each helix may be necessary to classify localization.12 Indeed, the maximum hydrophobicity of segments, specified by the number residues selected, better classifies ER vs mitochondrial TA proteins across hydrophobicity scales (Figure 3A,B).
FIGURE 3.
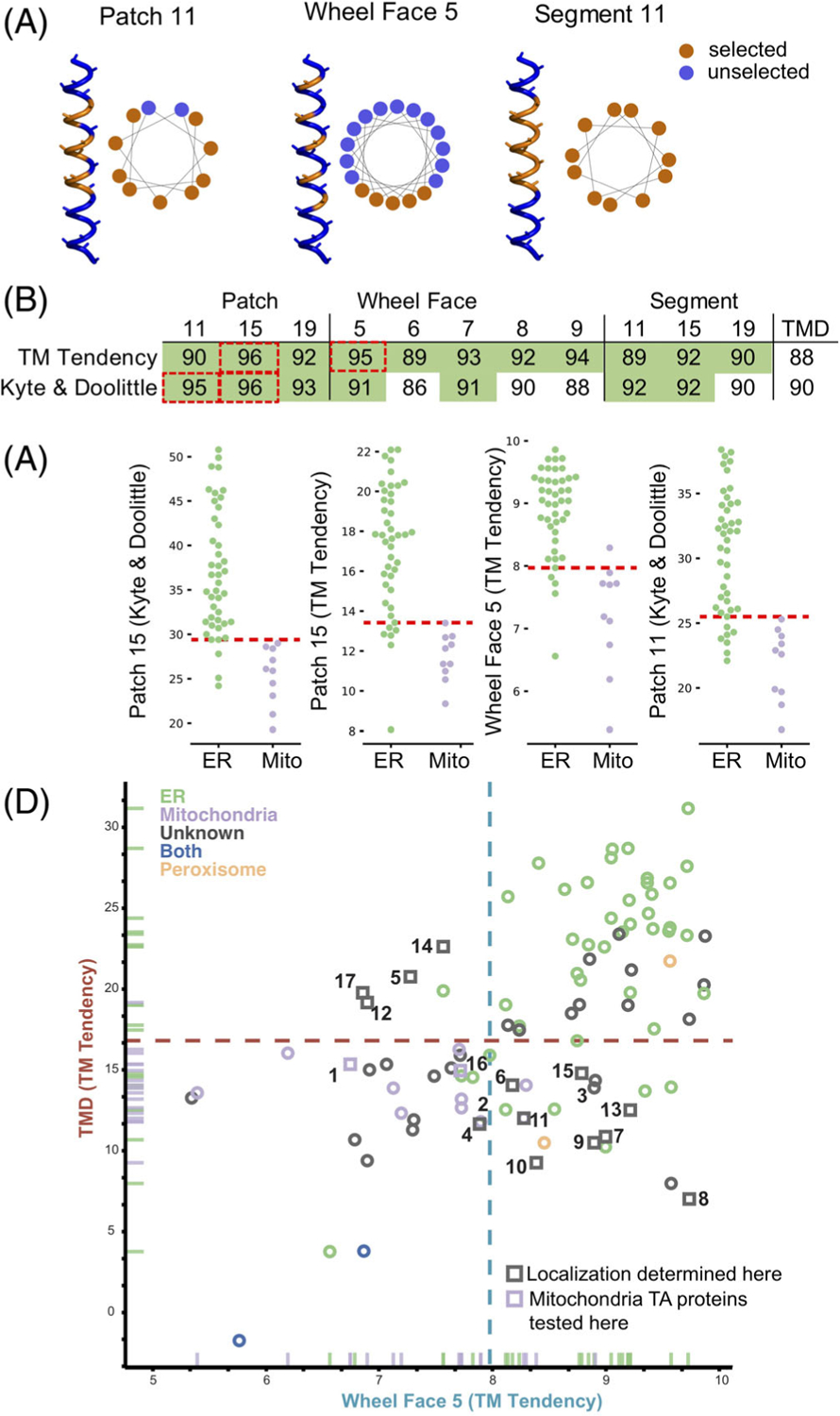
Analyzing different geometries of hydrophobic residues in TMDs to improve classification. (A) Alpha-helices and helical wheel plots illustrating the residues selected (orange) for each metric tested, patch, wheel face and segment, showing residues selected and not selected (blue) in each analysis. (B) AUROC values for the metrics illustrated in (A) and total hydrophobicity. (C) Jitter plots as in Figure 2 for the top four hydrophobic metrics: Patch 15 (Kyte & Doolittle scale), Patch 15 (TM Tendency scale), Wheel Face 5 (TM Tendency scale) and Patch 11 (Kyte & Doolittle scale). Red dashed line indicates the best predictive threshold. (D) 2D comparison plot of total hydrophobicity (y-axis) and a Wheel Face 5 (TM Tendency scale) (x-axis). TA proteins are colored by localization, ER ( green), mitochondria (purple), Unknown ( gray), both mitochondria and ER (blue), and peroxisome (orange). TA proteins selected for experimental determination of localizations are marked squares. Dashed lines indicate best predictive threshold
Furthermore, it was also reported that TMDs where the most hydrophobic residues cluster to one side of a helical wheel plot,38 a 2D representation of an alpha-helix, bind more efficiently to Sgt2.12 We sought to examine if this clustering is a feature of ER TA proteins and absent in mitochondria TA proteins. This clustering we define as a helical wheel face (Wheel Face) and specify a length by the number of residues selected (Figure 3A,B). We also extend the face along the sides of the helix, defining a Patch, selecting three of the four residues in a single turn of a helix. Patch geometries are specified by length of the segment considered, that is, Patch 11 is confined in a 11 segment residues with 9 residues selected (Figure 3A,B). Improvements in classification over the total hydrophobicity metric are seen in several cases (Figure 3B, green, Figure S1B, green, Table S2). The metrics with the best classification capability are Patch 15 (Kyte & Doolittle and TM Tendency), Wheel Face 5 (TM Tendency scale) and Patch 11 (Kyte & Doolittle scale; Figure 3B, dashed red box). These metrics have an improved AUROC value of 96, 96, 95 and 95, respectively, compared to the TMD hydrophobicity score of 90 (Kyte & Doolittle) and 88 (TM Tendency; Figure 3B). At the best threshold of the ROC curve, these metrics correspond to five, seven, six and eight miscategorized proteins, respectively. A scatter plot illustrates how these metrics translate to improved separation of ER and mitochondrial TA proteins (Figure 3C).
Other hydrophobic geometries were also explored as potential competing hypotheses: residues in a line (every fourth residue), rectangle (one residue plus two residues two away on either side) or star (two adjacent residues and one residue two away on either side; Figure S1A,B). As with the Patch geometries, these geometries are specified by the length of the TMD considered. Again, improvements are seen in geometries that present hydrophobic patches, that is, Rectangle 9 and Star 8, where line geometries rarely improved classification regardless of scale used (Figure S1B). Given the relative dearth of experimental data and the substantial number of hypotheses being tested (geometries and hydrophobicity scales), it is difficult to definitively say if one geometry is the sole deciding factor for localization based only on bioinformatics. Regardless of the hydrophobicity scale used, it is clear that the organization of hydrophobic residues within a TMD is important for targeting TA proteins to their intended membranes.
2.4 |. Testing the localization of unknown TA proteins
We then tested if either face (Wheel Face or Patch), Segment, or TMD hydrophobicity metrics enabled us to predict the localization of unknown TA proteins. To do this we selected a subset of unknown TA proteins, whose localization would be predicted differently by TMD and Wheel Face 5 metrics using the TM Tendency scale (Figure 3D, numbered gray points, Table S3). This selection was made because of the strong AUROC and biochemical data suggesting TA protein containing a helical wheel face bind more efficiently to Sgt2. Several in this group have a hydrophobicity less than the previously suggested cut-off for EMC substrates17 (Figure 3D, lower right quadrant). Our experimental setup based on that from Rao et al–GFP is fused N-terminally to the TMD and C-terminal residues of the unknown TA protein (Figure 4A yellow panel). Localization is determined by overlap with either a BFP-tagged mitochondria presequence that marks the mitochondria (Figure 4A cyan panel) and a tdTomato-tagged Sec63 acting as an ER marker (Figure 4A magenta panel).13 Overlap was determined computationally using two algorithms we developed: one to segment individual cells in brightfield and another to determine which fluorescence probe the GFP overlapped with on a per cell basis (Table S3).
FIGURE 4.
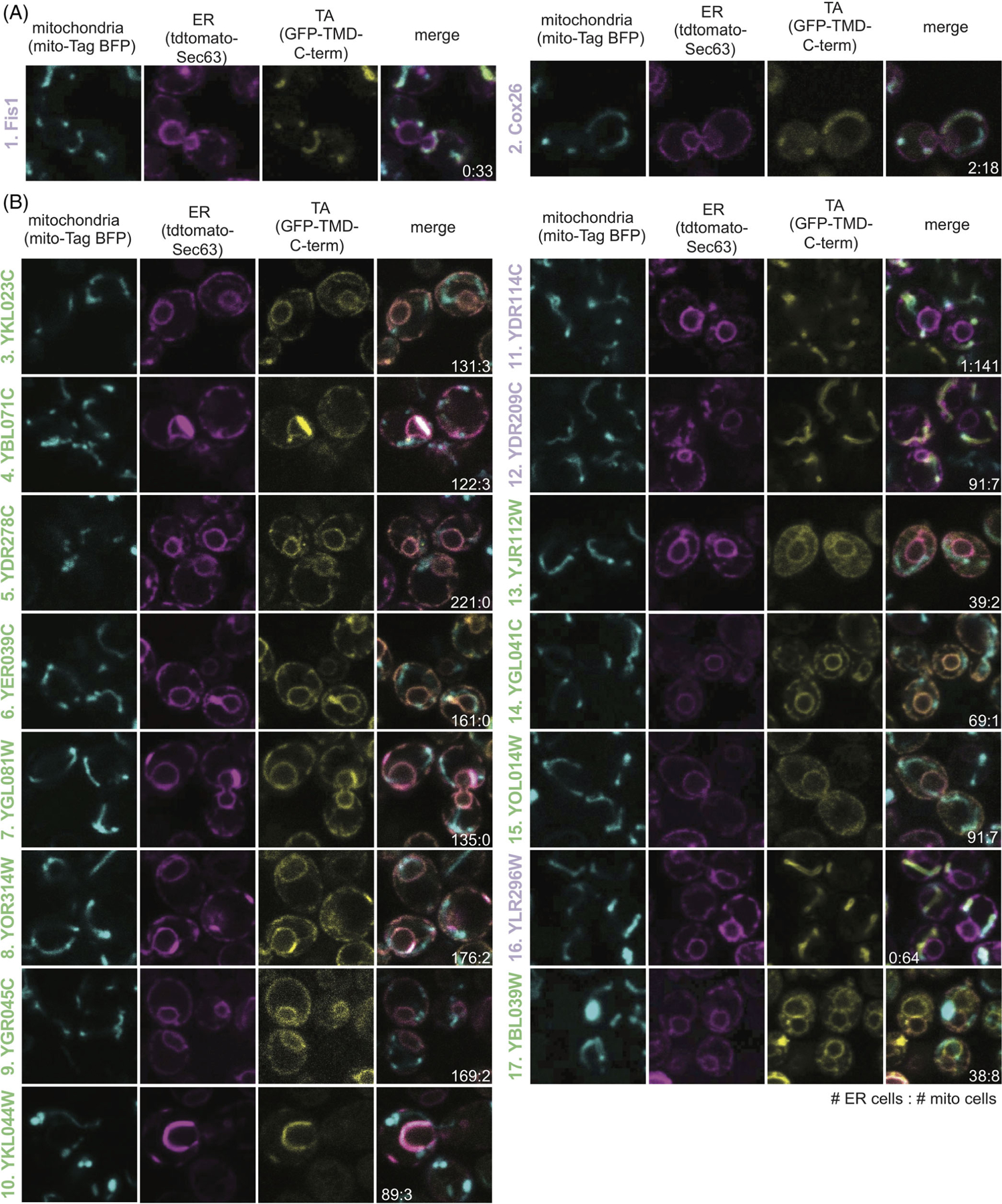
Localization of unknown yeast TA proteins. The ER (magenta panel) and mitochondria (cyan panel) were labeled with tdTomato and BFP, respectively. TA protein localization was visualized by GFP (yellow panel) and colocalization was determined by overlap (merge panel). The ratio of the number of cells with the TA protein localizing to the ER vs the mitochondria are noted in the merge image. Numbered as in Figure 3D with labels colored based on their determined localizations: ER (green) and mitochondria (purple). TA proteins include (A) two mitochondrial TA proteins with known localizations and (B) 15 with unknown localizations
This experimental setup and computational analysis were first applied to the known mitochondria proteins Fis1 and Cox26.13,39,40 The analysis correctly determines these proteins to colocalize with BFP, thus correctly classifying them as mitochondria TA proteins (Figure 4A). We then experimentally tested the 15 Unknown TA proteins where 11 localize to the ER, three to the mitochondria, and one to another cellular compartment (Figure 4B, Table 1). The localization of this latter TA protein cannot be determined by our experimental setup except to say it does not clearly colocalize with the ER or mitochondria markers visually or through our computational analysis (Table S3). The shape of the organelle is consistent with localization to the ER-derived vacuole (Figure 4B, #17).41 In total, we report the first localization of 10 previously Unknown TA proteins.
TABLE 1.
Best performing hydrophobicity metrics when combined with charge are those restricted to shorter segments of a helix in humans
| Metric | Scale | Organism | Correct (%) | Misclassified mito | Misclassified ER | Miclassified total |
|---|---|---|---|---|---|---|
| Patch 11 | TM Tendency | H. sapiens | 82% | 26 | 12 | 38 |
| TMD | Kyte & Doolittle | H. sapiens | 82% | 33 | 5 | 38 |
| Segment 19 | Kyte & Doolittle | H. sapiens | 81% | 29 | 9 | 38 |
| Segment 11 | TM Tendency | H. sapiens | 81% | 27 | 12 | 39 |
| Segment 15 | TM Tendency | H. sapiens | 81% | 25 | 14 | 39 |
| Wheel face 9 | TM Tendency | H. sapiens | 81% | 26 | 13 | 39 |
| Wheel face 7 | TM Tendency | H. sapiens | 81% | 27 | 13 | 40 |
| Segment 11 | Kyte & Doolittle | H. sapiens | 81% | 28 | 12 | 40 |
| Segment 15 | Kyte & Doolittle | H. sapiens | 81% | 30 | 10 | 40 |
| Patch 19 | TM Tendency | H. sapiens | 80% | 27 | 14 | 41 |
| Patch 15 | TM Tendency | H. sapiens | 80% | 30 | 11 | 41 |
| Segment 19 | TM Tendency | H. sapiens | 80% | 26 | 15 | 41 |
| Patch 19 | Kyte & Doolittle | H. sapiens | 80% | 30 | 12 | 42 |
| Patch 11 | Kyte & Doolittle | H. sapiens | 79% | 28 | 15 | 43 |
| Patch 15 | Kyte & Doolittle | H. sapiens | 78% | 28 | 17 | 45 |
| TMD | TM Tendency | H. sapiens | 78% | 31 | 14 | 45 |
| Wheel face 5 | TM Tendency | H. sapiens | 77% | 28 | 19 | 47 |
| Patch 15 | TM Tendency | S. cerevisiae | 88% | 7 | 2 | 9 |
| Wheel face 5 | TM Tendency | S. cerevisiae | 87% | 6 | 4 | 10 |
| Wheel face 7 | TM Tendency | S. cerevisiae | 87% | 6 | 4 | 10 |
| Patch 11 | TM Tendency | S. cerevisiae | 87% | 6 | 4 | 10 |
| TMD | TM Tendency | S. cerevisiae | 87% | 6 | 4 | 10 |
| Segment 15 | TM Tendency | S. cerevisiae | 86% | 6 | 5 | 11 |
| Segment 19 | TM Tendency | S. cerevisiae | 86% | 6 | 5 | 11 |
| Patch 19 | TM Tendency | S. cerevisiae | 84% | 7 | 5 | 12 |
| Segment 11 | TM Tendency | S. cerevisiae | 83% | 6 | 7 | 13 |
Note: A ranked comparison of the best performing hydrophobicity metric when combined with a C-terminal charge cut-off for both human and yeast TA proteins.
Several datasets report protein localizations in yeast but are not yet, or partially, integrated into bioinformatics databases like Uniprot. One in particular was of use for this study, reporting the localizations assigned by qualitatively accessing the pattern of protein expression in images of 17 TA proteins in the Unknown category42(Table S4). Coincidentally, a few of these proteins were included in our experimental test set, for a combined 27 new TA proteins with previously unknown localizations (Tables S3 and S4). Of the TA proteins identified by Weill and colleagues, all but one, YKL044W, was confirmed (Table S3). Given the ability to mark ER and mitochondria and quantitate colocalization on a per-cell basis, we use the localization determined here throughout our analysis, that is, YKL044W localizes to the ER. Collectively, we have compiled a list of 27 TA proteins and their localizations that have yet to be integrated into protein databases or reported: 20 ER, six mitochondrial, and one peroxisomal.
2.5 |. Reassessing classification metrics using newly determined localizations
The newly determined localizations were compared to the predicted localizations of the best performing hydrophobicity metrics. Total hydrophobicity metrics across all scales only correctly predict 9 or 14 of the 26 ER and mitochondria TA proteins. Experimental localizations from this work and the Schuldiner Lab42 result in a putative yeast TA protein list with 88% having known localizations (Figure 5A). With most localizations known, comparing metrics based on AUROC values is a good representation of the overall dataset (Table S5). The best performing metrics were Wheel Face 7 (TM Tendency) and Wheel Face 5 (TM Tendency), with scores of 89 and 88, respectively vs the TMD hydrophobicity AUROC score of 76 (Table S5). These metrics correctly predicted the localization of 19 out of 26 and 17 out of 26, respectively, of the subset of our test set that localized to the ER or mitochondria (Figure 5A,B). A Patch geometry using the Fauchere & Pliska scale performs well when predicting new localizations–correctly predicting 18 of 26 localizations (Figure 5A). Segment metrics performed similarly when predicting new localizations and their AUROC values improved with the inclusion of the new localizations (Figure 5A). In all, metrics focused on the organization of hydrophobic residues within the TMD of TA proteins better predict TA protein localization–the best consider just a five or seven residue face or a fraction of the TMD.
FIGURE 5.

A hydrophobic Wheel Face metric of 5 or 7 residues best separates ER and mitochondria TA proteins. (A) A ranking of the five best performing hydrophobicity metrics compared to the TMD hydrophobicity metrics of the appropriate hydrophobicity scales (TM Tendency, Fauchere & Pliska and Kyte and Doolittle). The number of correctly predicted localizations as well as the final AUROC scores are used to assess the effectiveness of each metric. The total number of correctly classified yeast TA proteins is also noted. The two metrics directly compared in the 2D comparison plot in (B) are highlighted in blue (TM Tendency, Wheel Face 5, x-axis) and red (TM Tendency, TMD, y-axis). Hydrophobicities are plotted and TA proteins are colored as they were in Figure 3D. Newly determined localizations from Figure 4 (black outlined) and Weill et al (squares) are filled in with the appropriate colors, ER (green), mitochondria (purple) and peroxisome (orange)
2.6 |. Expanding this metric to human TA proteins
We next applied this analysis to the human genome. Using our compiled list of 573 putative human TA proteins, we sought to identify a more inclusive set of criteria for ER- vs mitochondria-bound TA proteins. The best performing hydrophobicity scales in the yeast dataset were TM tendency and Kyte & Doolittle, so the other scales were not further considered with the human dataset. While TMD hydrophobicity metrics correctly capture mitochondria TA proteins, they fail to capture many ER TA proteins (Figure 6A, Table S6). Quantitatively assessing all metrics, we see slight improvements in classification with metrics using patches or segments compared to total hydrophobicity (Figure 6A,B, Table S6). The metric with the highest AUROC score is Patch 11 (Kyte and Doolittle). Many proteins in our dataset have a single report of their localizations in databases. There is potential for changes to these localizations as seen with many Bcl-2 family members (Figure 6B filled blue points) where there exist multiple reports of these proteins localizing to the ER and/or to the mitochondria. While this may be unique to these TA proteins, as their function to regulating apoptosis is tied in with their transport between the two membranes, some reported localizations may be the product of over-expression. Future work verifying and determining localizations of human TA proteins will likely result in improvements in classification by a metric derived from hydrophobic geometries.
FIGURE 6.
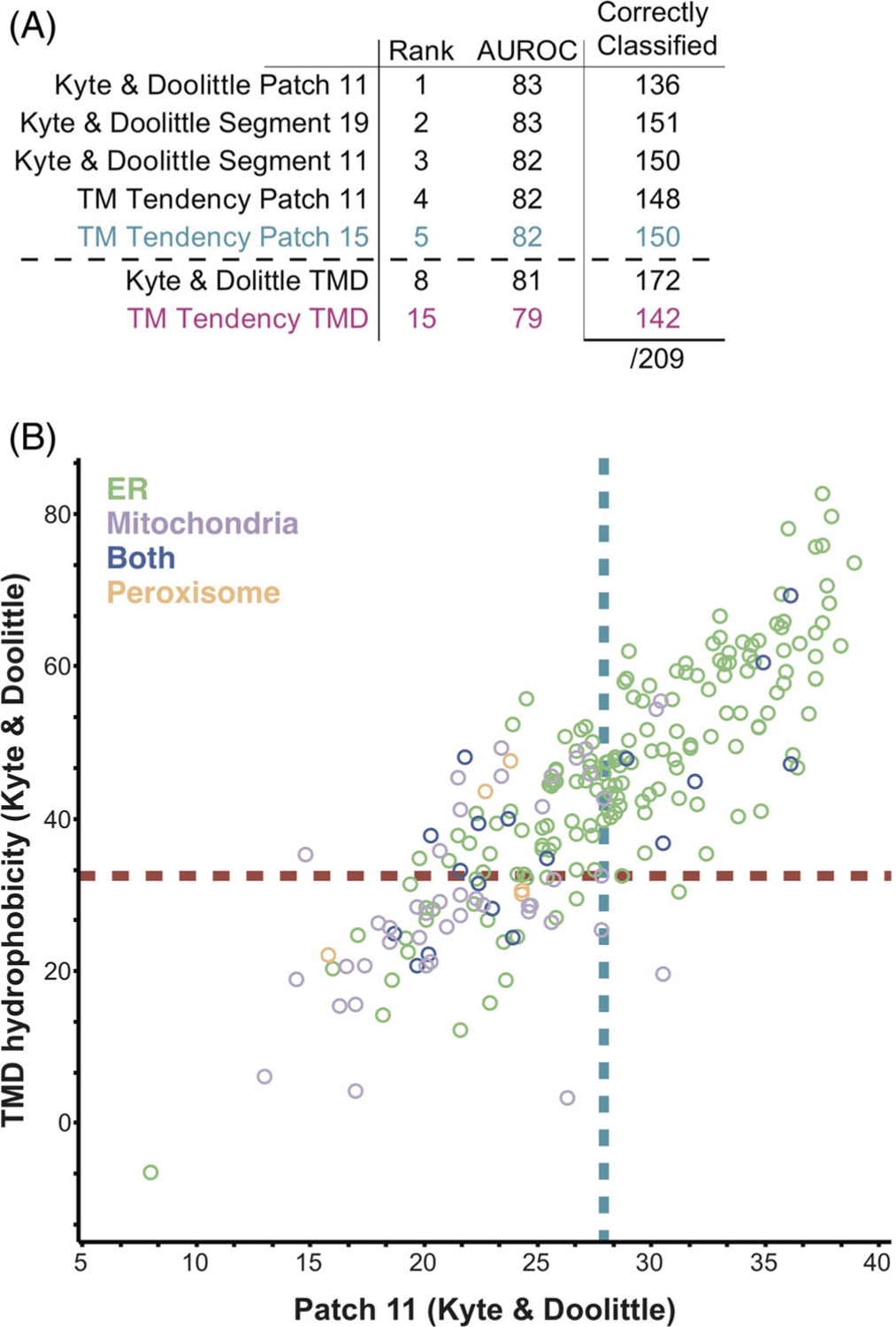
Human ER and mitochondrial TA proteins can be separated by the most hydrophobic 11 residues segment. (A) A table of the with the AUROC values of the best performing hydrophobicity metrics and the overall TMD hydrophobicity, along with their ranking. The number of total misclassified proteins are separated by ER and mitochondria TA proteins. (B) 2D comparison for the human dataset of TMD hydrophobicity and Patch 11 metrics using the Kyte and Doolittle scale. Hydrophobicities are plotted and TA proteins are colored as in Figure 3D. Unknown TA proteins are not plotted
2.7 |. Determining a two-step criterion for localization determination
We then tested if combining a hydrophobicity geometry with a C-terminal charge metric resulted in more accurate classification of TA proteins. Costello and colleagues demonstrated in mammals, distinctions between ER, mitochondria and peroxisomal TA proteins can be made using a combination of charge and TMD hydrophobicity cut-offs.24 They suggest mitochondria TA proteins have tails that are less charged than peroxisomal TA proteins, but more charged than ER TA proteins, which are generally more hydrophobic than mitochondria TA proteins. Previous reports demonstrated the GET pathway fails to insert TA proteins with a sufficiently charged C-terminus.13 This selectivity filter was seen at the membrane and cytosolic components were unaffected by the presence of a charge. Perhaps this rejection of TA proteins with a C-terminal charge is seen across all ER targeting pathways in both yeast and humans. To further explore this, we determined anything to be above the hydrophobicity cut-off to be classified as ER-bound and anything below the cut-off to be passed through a charge filter. When analyzing the number of C-terminal positive residues following the TMD of TA proteins that fall below the hydrophobicity cut-off, we find that a benchmark of three positive residues best separates ER and mitochondria TA proteins–mitochondria TA proteins generally contain at least three charged residues. We applied this secondary filter to our best performing yeast metrics (Wheel Face 5 and Wheel Face 7 residues) and the TMD hydrophobicity (Table 1). In these cases, the three metrics perform the same, misclassifying 10 TA proteins. Intriguingly, a Patch 15 metric does best, correctly classifying 88% of all yeast TA proteins. A metric utilizing both a helical wheel face and C-terminal charge does slightly better than that using TMD hydrophobicity and charge, but the significance of that improvement is difficult to determine based on this small dataset.
The human dataset is larger, and we sought to apply this tandem metric application to our list of putative TA proteins (Figure 7). Similar to what was observed in the yeast dataset, improvements in classification are seen (Table 1). Interestingly, applying a C-terminal charge sequentially to hydrophobic metrics constrained to a fragment of ~11 residues, either a Patch (TM tendency) or the entire segment (Kyte & Doolittle), and the TMD hydrophobicity metric (Kyte & Doolittle), perform equally well, each misclassifying 38 TA proteins. Most hydrophobicity metrics performed similarly with either scale, suggesting a subset of the TMD is required for correct targeting (Table 1). It is clear that in both human and yeast, a combination of hydrophobicity and C-terminal charge filters are necessary for correct classification as was demonstrated in the context of the GET pathway. The hydrophobicity window can be limited to a fraction of the TMD and still perform as well as the entire TMD.
FIGURE 7.
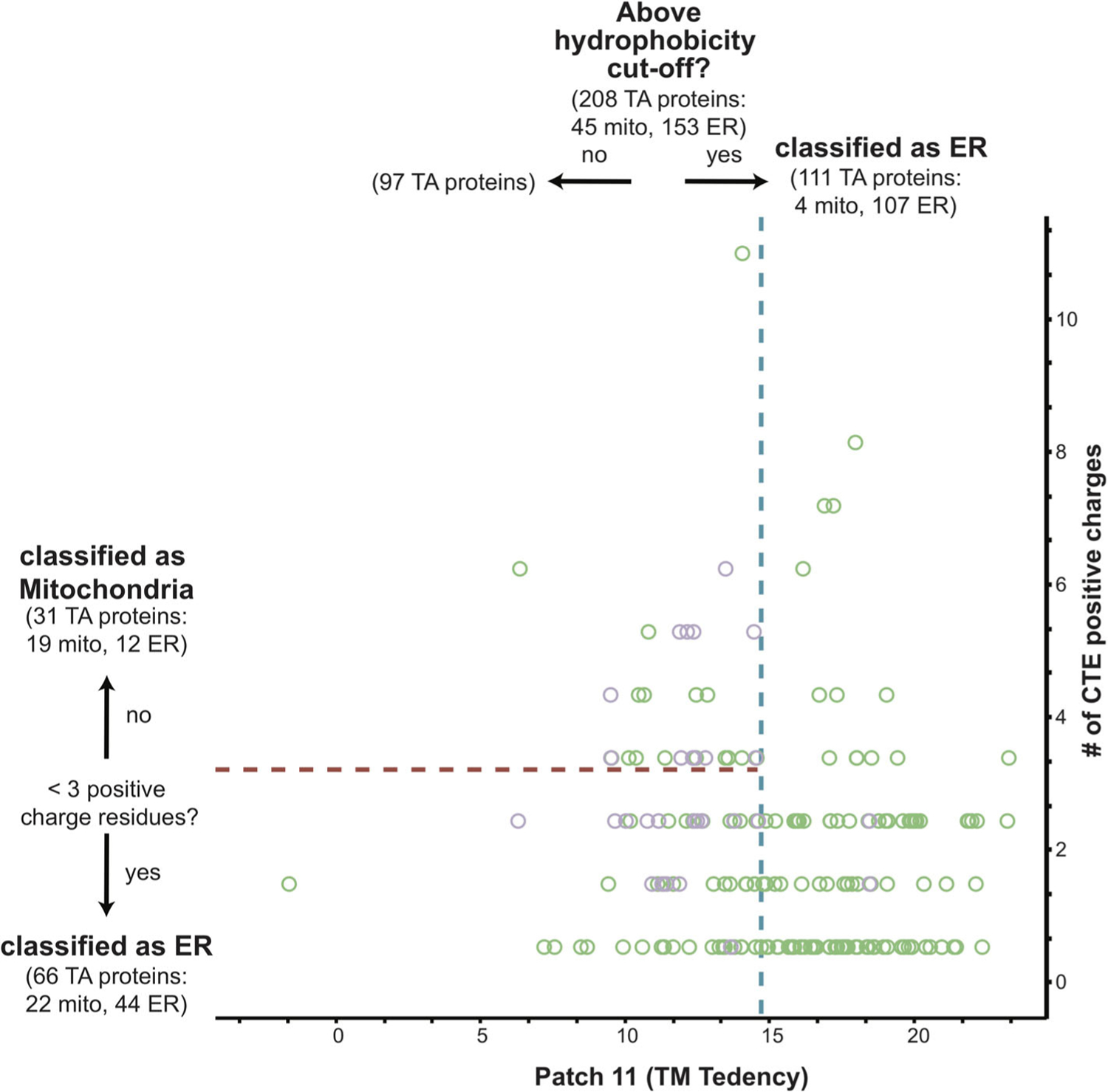
Combining a hydrophobicity and C-terminal charge metric results in a more effective predictor. The most hydrophobic 11 amino acid segment of all human TA protein TMDs with known localizations to either the ER (green) or mitochondria (purple) was calculated using the Kyte and Doolittle scale and plotted along the x-axis. The number of positive charge residues was counted and plotted along the y-axis. The best fit cut-off for the hydrophobicity metric (blue dotted line) and charge metric (red dotted line) are marked. The number of ER and mitochondria TA proteins captured in each step is denoted in the corresponding quadrant
3 |. DISCUSSION
Decoding the signaling information in membrane proteins responsible for their correct targeting to cellular membranes is still a mystery. For the class of membrane proteins with a single TMD and no signal peptide, TA proteins, some observations have been made to distinguish between those destined for the ER and those destined for the mitochondria. This report provides an extensive analysis of yeast and human TA proteins to identify a set of criteria to distinguish between ER- and mitochondria-bound TA proteins. This study also includes an expansion of putative TA proteins in both humans and yeast as well as newly determined experimental localization of several yeast TA proteins.
An initial separation by hydrophobicity can be applied to TA proteins, relegating TMDs with high hydrophobicities as ER proteins. A secondary filter can be applied to those below the cut-off classifying TA proteins with at least three charged residues following their TMDs as mitochondria-bound and the rest as ER-bound (Figure 7). This sequential selectivity was noted in the yeast GET pathway.13 In this case, it was demonstrated that the cytosolic targeting factors Sgt2 and Get3 bind to optimal TMDs based on a combination of high hydrophobicity and helical propensity. Regardless of hydrophobicity, TA proteins containing a charged C-termini were not inserted into ER microsomes. The analysis here demonstrates that generally ER TA proteins, not just GET substrates, lack charges in their C-terminus.
When determining the effectiveness of a hydrophobicity metric alone, metrics that focus on a hydrophobic geometry, a hydrophobic face in yeast and a hydrophobic segment restricted to 11 to 19 residues in humans, perform better than the hydrophobicity of the entire TMD. Applying the charge filter reveals that total hydrophobicity is as effective as hydrophobic face or segment metrics. Differences in the best performing hydrophobicity metrics between the yeast and human dataset could be explained by the observation that SGTA is more permissive to client binding than Sgt2.12 Collectively, these datasets demonstrate that a fraction of the TMD is necessary and sufficient for correct localization. Interestingly, in the human dataset, some of the best performing metrics are limited to an 11-residue window, concurring with reports that SGTA recognizes TMDs of at least 11 amino acids.12
While biochemical data suggested that clustering hydrophobic residues to one side of a helix increased binding to Sgt2, a co-chaperone in an ER TA protein targeting pathway, a cellular role of this hydrophobic face remained unclear.12 From the bioinformatic analysis and experimental localization data presented here, we demonstrate most yeast ER TA proteins contain a hydrophobic face–made of five to seven adjacent residues along a helical wheel plot. The two components of the GET pathways that directly bind to TA proteins, Sgt2 and Get3, both have binding sites composed of a hydrophobic groove. One could imagine the hydrophobic face in clients buried in the hydrophobic groove of Sgt2 and Get3, enhancing the hydrophobic binding interactions. Perhaps cellular factors involved in targeting TA proteins to the ER recognize this face and future identified ER TA protein binding partners will also feature a helical hand for client binding.
In this work, we provide a comprehensive bioinformatics analysis of naturally occurring TA proteins in the yeast and human genomes. While comprehensive, subtle differences in each metric’s geometries and hydrophobic scales cannot easily be differentiated analyzing just wild-type proteins. Similar work has helped disentangle the positional dependence of hydrophobicity in the insertion of integral membrane proteins.43 Likewise, future work could better define the geometry and hydrophobic scale needed for TA targeting by larger scale mutational analyses, perhaps even transforming the question of TA targeting into that of sequence selection/enrichment.44
The targeting of TA proteins presents an intriguing and enigmatic problem for understanding the biogenesis of this important class of proteins. How subtle differences in clients modulate the interplay of hand-offs that direct these proteins to the correct membrane remains to be understood. Through in vivo imaging of yeast cells and computational analysis, we provide more clarity to client discrimination. A major outcome of this is the clear preference for a hydrophobic face on ER TA proteins of low hydrophobicity. In yeast, this alone is sufficient to predict the destination of a TA protein. In mammals, and likely more broadly in metazoans, while clearly an important component, alone the hydrophobic face cannot fully discriminate targets. For a full understanding, we expect other factors to contribute, reflective of the increased complexity of higher eukaryotes, perhaps involving more players.16
4 |. METHODS
4.1 |. Assembling a database of putative TA proteins and their TMDs
Proteins identified from UniProt25 containing a single TMD within 30 residues of the C-terminus were separated into groups based on their localization reported in UniProt. The topology of all proteins with 3 TMs or fewer was further analyzed using TOPCONS26 to avoid missed single-pass TM proteins. Proteins with a predicted signal peptide,27 an annotated transit peptide, problematic cautions, or with a length less than 50 or greater than 1000 residues were excluded. Proteins localized to the ER, golgi apparatus, nucleus, endosome, lysosome and cell membrane were classified as ER-bound, those localized to the outer mitochondrial membrane were classified as mitochondria-bound, those localized to the peroxisome were classified as peroxisomal proteins, and those with unknown localization were classified as unknown. Proteins with a compositional bias overlapping with the predicted TMD were also excluded. A handful of proteins and their inferred localizations were manually corrected or removed (see notebook and Table S1).
4.2 |. Assessing the predictive power of various hydrophobicity metrics
We thoroughly examined the metrics relating hydrophobicity, both published and by our own exploration, to better understand their relationship to protein localization. Notably, we recognized that a TMD’s hydrophobic moment (μH)33 was a poor predictor of localization, for example, although a Leu18 helix is extremely hydrophobic, it has (μH) = 0 since opposing hydrophobic residues are penalized in this metric. To address this, we define a metric that capture the presence of a hydrophobic face of the TMD: the maximally hydrophobic cluster on the face. For this metric, we sum the hydrophobicity of residues that orient sequentially on one side of a helix when visualized in a helical wheel diagram. While a range of hydrophobicity scales were predictive using this metric, we selected the TM Tendency scale32 to characterize the TMDs of putative TA proteins and determined the most predictive window by assessing a range of lengths from 4 to 12 (this would vary from three turns of a helix to six).
By considering sequences with inferred ER or mitochondrial localizations, we calculated the Area Under the Curve of a Receiver Operating Characteristic (AUROC) to assess predictive power. As we are comparing a real-valued metric (hydrophobicity) to a 2-class prediction, the AUROC is better suited for this analysis over others like accuracy or precision (a primer37). Because of many fewer mitochondrial proteins (ie, a class imbalance), we also confirmed that ordering hydrophobicity metrics by AUROC was consistent with the ordering produced by the more robust, but less common, Average Precision (see notebook).
4.3 |. Constructing plasmids for live cell imaging
A p416ADH-GFP-Fis1 plasmid and a mt-TagBFP described in Rao et al were gifted to us from the Walter lab, UCSF13,45 and a Sec63-tdtomato was a gift from Sebastian Schuck, ZMVH, Universitat Heidelberg. TMDs sequences were ordered from Twist Biosciences (San Francisco, CA) with flanking HindIII and XhoI sites. GFP-TMD constructs were made by restriction enzyme digestion (New England Biolabs, USA) of the p416ADH-GFP-Fis1 plasmid and the genes ordered from Twist Biosciences followed by T4 DNA (New England Biolabs, USA) ligation of the template and TMD fragments.
4.4 |. Live cell imaging
The yeast strain used are those described in Rao et al, also a gift from the Walter Lab, UCSF. Strains containing each GFP fused TMD were grown in appropriate selection media. Coverslips were prepped by coating with 0.1 mg/mL concavalin A (Sigma, USA) in 0.9% NaCl solution. Cells were immobilized on coverslips at a concentration of 5000 cells/mm2 (plates at 1.8 cm2, thus 9 × 108 cells/well) and imaged using a Nikon LSM800 (Nikon, Japan). Images were collected at wavelengths 488, 514 and 581 nm and were processed with ImageJ46 and two in-house image processing algorithms.
4.5 |. Image processing to determine localization
Yeast cells were segmented using deep learning-based tools. The variable pattern of DIC images with mixed low and high contrasts for back-grounds and cell bodies (signal variance of each whole image ranging from 67.4 to 2706.3, a ×40 difference–average, median and SD of signal variance for all images were, respectively 645.6, 563.8 and 419.1) prevented using classical gradient based methods to successfully segment cells. We adopted and compared two contemporary tools, YeastSpotter, a Mask-RCNN method dedicated to yeast cells,47 and Cellpose, a generalist method trained on a large pool of cell images.48 Note that, the former was not trained on yeast cell images but used a model pretrained on a larger set of other cell images to build a friendly tool for yeast cell segmentation. Cellpose is a more sophisticated tool whose pretrained models have learned to segment well based on a myriad of intensity gradient values and image styles. It has shown to achieve high quality segmentation on an extended variety of cell images, including in our yeast cells images, producing superior results when compared to YeastSpotter with the advantage of running faster on GPUs (tested on Nvidia RTX 2080 Ti). We thus exclusively used Cellpose with its cyto pretrained model to segment yeast cells in all our DIC images. We used maximum intensity projections of up to two or three slices per image stack but mostly a single slice was sufficient to create a single representative image for segmentation. Spurious, tiny, segmented regions whose size were shown to be outliers were automatically removed using an area opening morphological operation.
Individual cells were isolated by applying the mask to the corresponding florescent images of each of the three wavelengths. Masks less than 7.5 μm2 corresponded to incorrectly identified, incomplete, or out-of-plane cells and were omitted from analysis. Masks were applied to each florescence channel. An empirical threshold was applied to each channel to identify true florescence from background, and the percentage of each cell with co-localized GFP and BFP or GFP and tdTomato was then calculated. Localization was then determined identifying which pair of channels (GFP&BFP vs GFP&tdTomato) had greater overlap, that is, OverlapGFP&BFP > OverlapGFP&tdTomato resulted in a mitochondria annotation. The number of individual cells in each category were counted. Outputs from this algorithm were verified by manually inspecting individual images.
Supplementary Material
{kind=link}
ACKNOWLEDGMENTS
The authors thank members of the Clemons lab for support and discussion. The authors would also like to thank the Caltech Biological Imaging center for the use of their microscopes for our live cell imaging as well as the Caltech Center for Advanced Methods in Biological Image Analysis for aiding in the isolation of individual cells in our images. This work was supported by National Institutes of Health (NIH) Grant R01GM097572 (to William M. Clemons), NIH/National Research Service Award Training Grant 5T32GM07616 (to Shyam M. Saladi and Michelle Y. Fry) and a National Science Foundation Graduate Research fellowship under Grant 1144469 (to Shyam M. Saladi).
Funding information
National Science Foundation Graduate Research, Grant/Award Number: 1144469; NIH/National Research Service Award Training, Grant/Award Number: 5T32GM07616; National Institutes of Health, Grant/Award Number: R01GM097572
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/tra.12809.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
All code employed is available openly at github.com/clemlab/sgt2a-modeling with analysis done in Jupyter Lab/Notebooks using Python 3.6 enabled by Numpy, Pandas, Scikit-Learn, BioPython, bebi103,49 and Bokeh as well as in Rstudio/Rmarkdown Notebooks enabled by packages within the Tidyverse ecosystem.
REFERENCES
- 1.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Cohen J Mol Biol. 2001;305:567–580. [DOI] [PubMed] [Google Scholar]
- 2.Fry MY, Clemons WM. Complexity in targeting membrane proteins. Science. 2018;359:390–391. [DOI] [PubMed] [Google Scholar]
- 3.Guna A, Hegde RS. Transmembrane domain recognition during membrane protein biogenesis and quality control. Curr Biol. 2018;28: R498–R511. [DOI] [PubMed] [Google Scholar]
- 4.Almagro Armenteros JJ, Sønderby CK, Sønderby SK, Nielsen H, Winther O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017;33:3387–3395. [DOI] [PubMed] [Google Scholar]
- 5.Borgese N, Colombo S, Pedrazzini E. The tale of tail-anchored proteins. J Cell Biol. 2003;161:1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chartron JW, Clemons WM, Suloway CJM. The complex process of GETting tail-anchored membrane proteins to the ER. Curr Opin Struct Biol. 2012;22:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabu C, Schmid V, Schwappach B, High S. Biogenesis of tail-anchored proteins: the beginning for the end? J Cell Sci. 2009;122:3605–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kutay U, Hartmann E, Rapoport TA. A class of membrane proteins with a C-terminal anchor. Trends Cell Biol. 1993;3:72–75. [DOI] [PubMed] [Google Scholar]
- 9.Denic V. A portrait of the GET pathway as a surprisingly complicated young man. Trends Biochem Sci. 2012;37:411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wattenberg B, Lithgow T. Targeting of C-terminal (tail)-anchored proteins: understanding how cytoplasmic activities are anchored to intracellular membranes. Traffic. 2001;2:66–71. [DOI] [PubMed] [Google Scholar]
- 11.Wang F, Brown EC, Mak G, Zhuang J, Denic V. A chaperone cascade sorts proteins for posttranslational membrane insertion into the endoplasmic reticulum. Mol Cell. 2010;40:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin K-F, Fry MY, Saladi SM, William M, Clemons J. Molecular basis of tail-anchored integral membrane protein recognition by the cochaperone Sgt2. J Biol Chem. 2021;296:100441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao M, Okreglak V, Chio US, Cho H, Walter P, Shan S-O. Multiple selection filters ensure accurate tail-anchored membrane protein targeting. Elife. 2016;5:e21301–e21324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuldiner M, Metz J, Schmid V, et al. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell. 2008; 134:634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stefanovic S, Hegde RS. Identification of a targeting factor for post-translational membrane protein insertion into the ER. Cell. 2007;128: 1147–1159. [DOI] [PubMed] [Google Scholar]
- 16.Aviram N, Costa EA, Arakel EC, et al. The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature. 2016;540:134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guna A, Volkmar N, Christianson JC, Hegde RS. The ER membrane protein complex is a transmembrane domain insertase. Science. 2018; 359:470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chio US, Cho H, Shan S-O. Mechanisms of tail-anchored membrane protein targeting and insertion. Annu Rev Cell Dev Biol. 2017;33: 417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao S, Hegde RS. Membrane protein insertion at the endoplasmic reticulum. Annu Rev Cell Dev Biol. 2011;27:25–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao S, Hegde RS. A calmodulin-dependent translocation pathway for small secretory proteins. Cell. 2011;147:1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho H, Shan S-O. Substrate relay in an Hsp70-cochaperone cascade safeguards tail-anchored membrane protein targeting. EMBO J. 2018; 37:e99264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chitwood PJ, Juszkiewicz S, Guna A, Shao S, Hegde RS. EMC is required to initiate accurate membrane protein topogenesis. Cell. 2018;175:1507, e16–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Figueiredo Costa B, Cassella P, Colombo SF, Borgese N. Discrimination between the endoplasmic reticulum and mitochondria by spontaneously inserting tail-anchored proteins. Traffic. 2018;19:182–197. [DOI] [PubMed] [Google Scholar]
- 24.Costello JL, Castro IG, Camões F, et al. Predicting the targeting of tail-anchored proteins to subcellular compartments in mammalian cells. J Cell Sci. 2017;130:1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. 2020; 1–10. [DOI] [PMC free article] [PubMed]
- 26.Tsirigos KD, Peters C, Shu N, Käll L, Elofsson A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015;43:W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen H. Predicting secretory proteins with signalP. Protein Function Prediction. New York, NY: Springer; 2017:59–73. [DOI] [PubMed] [Google Scholar]
- 28.Beilharz T, Egan B, Silver PA, Hofmann K, Lithgow T. Bipartite signals mediate subcellular targeting of tail-anchored membrane proteins in Saccharomyces cerevisiae. J Biol Chem. 2003;278:8219–8223. [DOI] [PubMed] [Google Scholar]
- 29.Kalbfleisch T, Cambon A, Wattenberg BW. A bioinformatics approach to identifying tail-anchored proteins in the human genome. Traffic. 2007;8:1687–1694. [DOI] [PubMed] [Google Scholar]
- 30.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gene Ontology Consortium. The gene ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao G, London E. An amino acid “transmembrane tendency” scale that approaches the theoretical limit to accuracy for prediction of transmembrane helices: relationship to biological hydrophobicity. Prot Sci. 2006;15:1987–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrohobic moment plot. J Mol Biol. 1984;179:125–142. [DOI] [PubMed] [Google Scholar]
- 34.Fauchere J-L, Pliska V. Hydrophobic parameters π of amino acid side chains from partitioning of N-acetyl amino acid amides. Eur J Med Chem. 1983;18:369–375. [Google Scholar]
- 35.Roseman MA. Hydrophilicity of polar amino acid side-chains is markedly reduced by flanking peptide bonds. J Mol Biol. 1988;200:513–522. [DOI] [PubMed] [Google Scholar]
- 36.Wimley WC, Creamer TP, White SH. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 1996;35:5109–5124. [DOI] [PubMed] [Google Scholar]
- 37.Swets J, Dawes R, Monahan J. Better decisions through science. Sci Am. 2000;283:82–87. [DOI] [PubMed] [Google Scholar]
- 38.Schiffer M, Edmundson AB. Use of helical wheels to represent the structures of proteins and to identify segments with helical potential. Biophys J. 1967;7:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levchenko M, Wuttke J-M, Römpler K, et al. Cox26 is a novel stoichiometric subunit of the yeast cytochrome c oxidase. BBA – Mol Cell Res. 1863;2016:1624–1632. [DOI] [PubMed] [Google Scholar]
- 40.Hartley AM, Lukoyanova N, Zhang Y, et al. Structure of yeast cytochrome c oxidase in a supercomplex with cytochrome bc1. Nat Struct Mol Biol. 2019;26(1):78–83. 10.1038/s41594-018-0172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128: 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weill U, Yofe I, Sass E, et al. Genome-wide SWAp-tag yeast libraries for proteome exploration. Nat Methods. 2018;15:617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hessa T, Meindl-Beinker NM, Bernsel A, et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. [DOI] [PubMed] [Google Scholar]
- 44.Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okreglak V, Walter P. The conserved AAA-ATPase Msp1 confers organelle specificity to tail-anchored proteins. Proc Natl Acad Sci USA. 2014;111:8019–8024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu AX, Zarin T, Hsu IS, Moses AM. YeastSpotter: accurate and parameter-free web segmentation for microscopy images of yeast cells. Bioinformatics. 2019;35:4525–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stringer C, Wang T, Michaelos M, Pachitariu M. Cellpose: a generalist algorithm for cellular segmentation. Nat Methods. 2021;18:100–106. [DOI] [PubMed] [Google Scholar]
- 49.Bois JA. justinbois/bebi103: Version 0.1.0 [Internet]. 10.22002/D1.1615 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code employed is available openly at github.com/clemlab/sgt2a-modeling with analysis done in Jupyter Lab/Notebooks using Python 3.6 enabled by Numpy, Pandas, Scikit-Learn, BioPython, bebi103,49 and Bokeh as well as in Rstudio/Rmarkdown Notebooks enabled by packages within the Tidyverse ecosystem.