

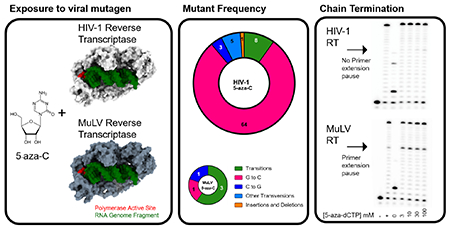
Abstract
5-aza-cytidine (5-aza-C) has been shown to be a potent human immunodeficiency virus type 1 (HIV-1) mutagen that induces G-to-C hypermutagenesis by incorporation of the reduced form (i.e., 5-aza-dC, 5-aza-dCTP). Evidence to date suggests that this lethal mutagenesis is the primary antiretroviral mechanism for 5-aza-C. To investigate the breadth of application of 5-aza-C as an antiretroviral mutagen, we have conducted a comparative, parallel analysis of the antiviral mechanism of 5-aza-C between HIV-1 and gammaretroviruses – i.e., murine leukemia virus (MuLV) and feline leukemia virus (FeLV). Intriguingly, in contrast to the hallmark G-to-C hypermutagenesis observed with HIV-1, MuLV and FeLV did not reveal the presence of a significant increase in mutational burden, particularly that of G-to-C transversion mutations. The effect of 5-aza-dCTP on DNA synthesis revealed that while HIV-1 RT was not inhibited by 5-aza-dCTP even at 100 μM, 5-aza-dCTP was incorporated and significantly inhibited MuLV RT, generating pause sites and reducing the fully extended product. 5-aza-dCTP was found to be incorporated into DNA by MuLV RT or HIV-1 RT, but only acted as a non-obligate chain terminator for MuLV RT. This biochemical data provides an independent line of experimental evidence in support of the conclusion that HIV-1 and MuLV have distinct primary mechanisms of antiretroviral action with 5-aza-C. Taken together, our data provides striking evidence that an antiretroviral mutagen can have strong potency via distinct mechanisms of action among closely related viruses, unlinking antiviral activity from antiviral mechanism of action.
Keywords: retrovirus, cytosine analog, mutagenesis, lentivirus, gammaretrovirus
Graphical Abstract
Introduction
Antiviral development has a shorter history than vaccine development, given that idoxuridine, the first antiviral, received approval for use in 1963 [1, 2]. Since that time, over ninety antiviral drugs from thirteen different functional groups have been approved for the treatment of nine human viral diseases – HIV-1, hepatitis B virus, hepatitis C virus, herpesvirus, human cytomegalovirus, varicella-zoster virus, respiratory syncytial virus, influenza virus, and human papilloma virus [1]. For HIV-1, the antiretroviral drugs include nucleoside reverse transcriptase (RT) inhibitors, nonnucleoside RT inhibitors, acyclic nucleoside phosphonate analogues, protease inhibitors, integrase inhibitors, and entry inhibitors.
Antiviral drugs are widely recognized as having a primary antiviral mechanism of action. For example, antiviral effects are typically caused by particularly structural characteristics of compounds, which is not a property of nucleoside analogues [3, 4]. The intense interest in antiviral drug repurposing/repositioning has demonstrated that antiviral drug that can be used as an antiviral for another virus typically affects the same target using the same antiviral mechanism of action [5]. Furthermore, the discovery of new activities to treat various diseases, including viral diseases remains a priority [6, 7]. The COVID-19 pandemic attracted strong interest in the use of drug repurposing though this has not been as fruitful as was hoped. When successful, drug repurposing can discover new indications for current medications by 1) reducing the risk of failure in drug development, 2) reducing the drug development timeline, and 3) improving the overall cost effectiveness of drug development. These advantages make drug repurposing less risky with a potentially more rapid return on investment in drug development. Importantly, repurposed drugs can reveal new targets and pathways that can be further exploited.
HIV infects approximately 36.7 million individuals worldwide and remains a global pandemic [8].The high rate of HIV-1 mutagenesis results in a viral quaspecies that can be observed both within infected individuals as well as in populations. HIV-1 quasispecies are readily capable of rapidly evolving antiviral drug resistance, and have served as a major barrier in the successful development of an AIDS vaccine(s). The high mutation rate of HIV-1 represents a key biological property that has important implications to immune evasion, drug resistance, and alternations in cellular tropism.
Lethal mutagenesis is a relatively novel antiviral approach in which viral infection is extinguished in the presence of a viral mutagen, which elevates the virus mutation rate [9]. Lethal mutagens investigated thus far have included nucleobase or nucleoside analogs that base-pair promiscuously because of properties including conformational flexibility, tautomerization, structural rearrangement, or ionization [10]. For HIV-1, several small molecules have been identified, including 5-aza-cytidine (5-aza-C), 5-aza-2′-deoxycytidine (5-aza-dC), 5-hydroxy-2’-deoxycytidine, and KP-1212 [11–14]. Notably, their effects have been demonstrated in cell culture under specific conditions and multiplicity of infection, and broad applicability in a variety of conditions is limited.
A previous report has indicated that 5-aza-C has antiviral activity during both the early and late phases of the HIV-1 life cycle, implicating its incorporation during reverse transcription as well as synthesis of the viral genomic RNA by RNA polymerase II, respectively [15]. During the late phase of HIV-1 replication (i.e., viral genome RNA synthesis), 5-aza-C primarily induces C-to-G transversions in HIV-1, while during the early phase of replication (i.e., reverse transcription), 5-aza-C primarily induces G-to-C transversions [15]. Previous studies have proposed that the G-to-C transversions are the result of incorporation of 5-aza-C into minus-strand viral DNA, and is followed by 5-aza-C hydrolysis and deformylation into ring-opened remnants [16]. The ring-opened remnants are thought to mispair with deoxycytidine during plus-strand viral DNA synthesis, which results in mutation fixation of G-to-C transversion mutations in the proviral DNA. Alternatively, 5-aza-C hydrolysis products could be directly incorporated by HIV-1 RT during viral DNA synthesis.
It is notable that 5-aza-C is closely related to 5-aza-dC, which can induce HIV-1 lethal mutagenesis [11, 17, 18]; and is a more potent lethal mutagen [15]. 5-aza-dC has anti-HIV-1 activity (i.e., EC50 ~ 200 nM) in the absence of significant cytotoxicity, and has been shown to act in synergy with gemcitabine and resveratrol [11, 19]. The combination of 5-aza-dC with gemcitabine has been demonstrated to exhibit antiviral activity in a murine leukemia virus (MuLV)-based mouse model for AIDS [20]. Another study described a 5-aza-dC divalerate prodrug that was more stable and maintained anti-HIV-1 activity [21]. HIV-1 replication in the presence of 5-aza-dC has demonstrated an increase in virus mutagenesis and a specific elevation of G-to-C transversions, which is an uncommon mutation observed in HIV-1 populations [11]. The determinants for 5-aza-dC incorporation were investigated by use of next-generation sequencing with five amplicons prepared from HIV-1 proviral DNA that was recovered from untreated or 5-aza-dC-treated cells [18]. Sequence analysis revealed an increase in the HIV-1 mutation frequency, with 5-aza-dC inducing a ~ 4.1-fold increase in the overall mutation frequency (i.e., all mutation types) and a ~ 155-fold increase in the G-to-C transversion mutation frequency [18]. Interestingly, G-to-C frequencies significantly varied between different sequence positions (up to ~ 80-fold), yet mutational hotspots (i.e., upper outliers within the distribution of mutation frequencies) were not reported. All guanine positions that were analyzed, and most cytosine position, were found to be susceptible targets of 5-aza-dC. These findings demonstrated that 5-aza-dC-mediated HIV-1 mutagenesis was promiscuous and without hallmark mutational hotspots – clearly distinct from that of APOBEC3G-mediated HIV-1 hypermutagenesis.
To more broadly investigate this potent antiretroviral mutagen, we have conducted comparative, parallel analyses of the antiviral mechanism of 5-aza-C among different retroviruses, specifically between HIV-1 and gammaretroviruses – i.e., murine leukemia virus (MuLV) and feline leukemia virus (FeLV). In these parallel studies, MuLV and HIV-1 vectors of comparable design were constructed and contained a reporter gene cassette expressing the mCherry and GFP fluorescence proteins. A FeLV vector that expresses the GFP gene was used to analyze another gammaretrovirus. Vector viruses of both HIV-1 and MuLV were produced and used to infect target cells +/− 5-aza-C at concentrations that were ~ EC75 for each virus. Mutant frequencies were determined by flow cytometry, and total DNA from infected cells was purified, the GFP gene was amplified and sequenced for virus mutation spectra analyses. Quite surprisingly, in contrast to the hallmark G-to-C hypermutagenesis observed with HIV-1, the presence of a significant increase in mutational burden was not observed with MuLV, particularly G-to-C transversion mutations. The effect of 5-aza-dCTP on DNA synthesis was tested with MuLV and HIV-1 RT in vitro. While HIV-1 RT was not inhibited by 5-aza-dCTP even at 100 μM, 5-aza-dCTP was incorporated by and significantly inhibited MuLV RT, generating pause sites and reducing the fully extended product. These biochemical observations provide an independent line of experimental evidence in support of the conclusion that HIV-1 and MuLV have a distinct mechanism of antiretroviral action with 5-aza-C. The data obtained with FeLV was generally consistent to that from MuLV. Taken together, our data indicate that a viral mutagen can have distinct mechanisms of action among closely related viruses.
Materials and methods
Cell lines, plasmids, and chemicals.
The HEK 293T cell line (a human embryonic kidney 293 cell line containing the SV40 T-antigen) was obtained from the American Type Culture Collection (Manassas, VA) and the Platinum-GP cell line was from Cell Biolabs, Inc. (San Diego, CA). The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: U373-MAGI-CXCR4CEM (MAGI; a human glioblastoma cell line expressing a modified CEM-derived CXCR4 gene linked to a puromycin resistance gene) cells contributed by Dr. Michael Emerman [22, 23], U87MG cells (a glioma cell line) contributed by Dr. Bruce Chesebro, raltegravir (catalog # 11680) and zidovudine (AZT; catalog # 3485). The plasmids pNL4-3 MIG and FeLV-GFP have been previously described [17, 24]. The MuLV reporter plasmid pLN-MIG was created by subcloning the mCherry-IRES-GFP from pNL4-3 MIG into pLN [25] at BclI/RsrII. pHCMV-VSVG was provided by J. Burns (University of California, San Diego). RT resistant mutants (M184I, M184V, V208M (FeLV)) were created as previously described [26–28]. Virus was produced by co-transfecting 293T cells with pHCMV-VSV-G and pNL4-3 MIG or FeLV-GFP using linear polyethylenemine (PEI, Polysciences, Inc.) as previously described [29]. Platinum-GP cells were transfected with pLN-MIG using GenJet™ In Vitro DNA Transfection Reagent (SignaGen Laboratories). Virus was collected at 48- or 72-h post-transfection, concentrated in VivaSpin® 20, 100,000 MWCO columns (Sartorius, Germany), DNaseI-treated, and frozen in aliquots at − 80 °C.
5-Aza-C (5-Azacytidine) was purchased from Sigma-Aldrich (St. Louis, MO), while 5-aza-dC divalerate (referred to below simply as 5-aza-dC), a pro-drug form of 5-aza-dC, was synthesized by the Center for Drug Design at the University of Minnesota [30]. The 5-aza-dCTP used in these studies was purchased from Jena Bioscience (Jena, Germany). All drugs were dissolved in the appropriate solvent and were stored in aliquots at − 20 °C.
Vector virus infectivity analysis.
MAGI or U87MG cells were infected as described previously [17]. Cells were pre-treated with 5-aza-C (Sigma-Aldrich), AZT, raltegravir, or DMSO (vehicle) for 2 h prior to infection. Cells were harvested at 48 h to 72 h after infection, and analyzed on an LSR II flow cytometer (BD Biosciences). Data analysis was performed using FlowJo® and GraphPad Prism. Infectivity and mutation frequency were determined as previously described [17].
DNA sequencing analysis.
MAGI or U87MG cells were treated with drug or vehicle for two hours before infection with reporter virus. Drug and virus were removed 24 h post-infection, and cells were harvested 72 h after infection. A fraction of the cells was analyzed for infectivity. Vehicle controls had between 40% and 80% infection, while 5-aza-C treated cells had between 8% and 15% infection. Remaining cells were washed with PBS and DNA was extracted with the HighPure PCR Template Kit (Roche). DNA was amplified with Phusion Hot Start II High Fidelity DNA Polymerase and primers specific to EGFP. The forward primer used was 5’- TCAAGCGTATTCAACAAGG-3’ and the reverse primer was 5’-CATTGTTAGCTGCTGTATTGC-3’. Purified amplification products were ligated to the pGEM-T vector (Promega) and transformed into the DH5α strain of E. coli. Plasmid DNA was purified and subjected to Sanger DNA sequencing analysis. Three independent experiments were performed for each virus, from independently produced vector virus stocks. Mutation analysis was performed using the SeqMan program of the Lasergene 7 software package (DNAStar).
Quantitative PCR analysis of reverse transcription products.
U87MG cells were treated with drug or vehicle for two hours before infection with virus. A heat inactivated control (95°C for 30 minutes) was included for each virus. Cells were harvested 18 hours after infection. A portion of cells were immediately re-plated and harvested 48-72 hours after infection for infectivity determination. Vehicle controls had between 20 and 30% infection. Remaining cells were washed with PBS, and DNA was extracted with the HighPure PCR Template Kit. Quantitative PCR (qPCR) was performed as previously described, using primers specific to a late reverse transcription product for HIV-1 [11], MuLV [31] and FeLV (Forward 5’ - CTGACTCGTGGTCTCGGTGTTC ; Reverse 5’ - GGCCAGCTTACCTCCTGATG ). Samples were normalized to 18S rRNA, and are described as a percentage of the vehicle control for each experiment. The heat inactivated controls were analyzed to determine plasmid carryover – all samples had less than 0.5% carryover.
Cell-free 5-aza-dCTP incorporation assay by purified reverse transcriptase.
The single nucleotide incorporation assay used has been previously described [27]. Briefly, this assay uses a 5′ P32 radio-labelled 23-mer primer (P) annealed to a 24-mer template (T) with a single G overhang. Extension from an 23-mer to 24-mer indicates that 5-aza-dCTP has been incorporated by RT. 20 μL reactions contained 200 fmol template/primer, 2 μL 5-aza-dCTP at concentrations indicated or 50 μM of dNTPs for the positive control, 4 μL of purified RT (HIV-1 NL4-3 or MuLV, normalized by titrating enzyme activity), 25 mM Tris–HCI, pH 8.0, 2 mM dithiothreitol, 100 mM KCl, 5 mM MgCl2, and 10 μM oligo(dT). Reactions were incubated at 37 °C for 5 min and then quenched with 10 μL of 40 mM EDTA and 99 % (vol/vol) formamide at 95 °C for 2 min. The reactions were resolved on a 20 % urea-PAGE gel (American Bio Sequel NE reagent) and imaged using Pharos FX molecular imager (Bio-Rad). HIV-1 RT (HXB2) was purified as previously described (Kim B: Genetic selection in Escherichia coli for active human immunodeficiency virus reverse transcriptase mutants. Methods 1997, 12:318 324). MuLV RT was obtained from New England Biolabs (Beverly, MA).
Primer extension assay.
An RT primer extension assay was performed as previously described [27], A 5′ 32P-labeled 17-mer DNA primer (5′-CGCGCCGAATTCCCGCT-3′) was annealed to a 40-mer RNA template (5’-AAGCUUGGCUGCAGAAUAUUGCUAGCGGGAAUUCGGCGCG-3’) in the presence of 100 mM NaCl, 10 mM Tris–HCl (pH 8.0) and 1 mM EDTA. 20 μL reaction mixtures contained 10 nM template-primer, 4 μL of purified HIV-1 RT or MuLV RT (normalized by titrating enzyme activity), 5 μM of dATP, dGTP and TTP (ThermoScientific), 2 μL 5-aza-dCTP at concentrations indicated, 12.5 mM Tris–HCl (pH 7.5), 12.5 mM NaCl, 2.5 mM MgCl2 and 20 μM oligo(dT). Reactions were initiated upon addition of RT and incubated at 37 °C for 5 min. Reactions were terminated with 10 μL of 40 mM EDTA and 99 % formamide. The products were resolved on a 14 % urea-PAGE gel (American Bio Sequel NE reagent), imaged using Pharos FX molecular imager (Biorad) and analyzed using ImageLab software.
Cell cytotoxicity assay analysis.
U373-MAGI (5000 cells/well) were plated in a 96-well plate 24 hours prior to drug treatment. Cells were treated for five days, and proliferation was quantified using the CellTiter-Glo kit from Promega according to the manufacturer’s instructions. Cells treated with DMSO were used as a no-drug control. The luminescence was recorded using an Orion microplate luminometer from Berthold Detection Systems (Huntsville, AL). The data were normalized to relative cell count by setting the value for no-drug cells at 100% for each experiment and then dividing the data for the other samples by the number used to convert the no-drug cells to 100%. This conversion was normalized for differences in luciferase activity between experiments.
Statistical analysis.
All figures were created in Microsoft Office for Mac 2011, version 14.5.2 (Redmond, WA), or GraphPad Prism, version 5.0 (GraphPad Software, Inc., La Jolla, CA). To determine the half maximal inhibitory concentration (IC50) of 5-aza-C of each virus, infectivity data were normalized to the data for the vehicle control, plotted against log-transformed drug concentrations, and subjected to nonlinear regression in GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). Data were analyzed by calculating the mean ± standard deviation (sd). Differences between groups were analyzed by either one-way ANOVA test (infectivity and qPCR experiments) or “N-1” Chi-squared test (mutation frequency). A P-value of <0.05 was considered statistically significant.
Results
5-aza-C reduces MuLV, FeLV and HIV-1 infectivity, but only increases HIV-1 mutagenesis
Previous studies have demonstrated that 5-azacytidine (5-aza-C) and its reduced form 5-aza-2’-deoxycytidine (5-aza-dC) inhibited the infectivity of HIV-1 and FeLV [11, 12, 24, 32]. We hypothesized that 5-aza-C inhibits all retroviruses, including gamma-retrovirus MuLV, through the same antiviral mechanisms. To test this hypothesis, three viral vectors were assessed, including HIV-MIG (mCherry-IRES-GFP), MuLV-MIG and FeLV-GFP (Figure 1). MAGI cells were treated with 5-aza-C (Figure 2A) or vehicle 2 h before infection and infectivity, defined by GFP+ and mCherry+ cells, was determined by flow cytometry. As expected, HIV-1-MIG and FeLV-GFP were inhibited by 5-aza-C, as was the MuLV-MIG (Figure 2B). The IC50 of 5-aza-C was 13 μM (95% confidence interval of 9.5 μM-18 μM) for FeLV,, 49 μM (95% confidence interval of 43 μM-58 μM) for MuLV, and 83 μM (95% confidence interval of 76 μM-91 μM) for HIV-1, respectively. Another study showed that 5-aza-C has an IC50 of 112 μM with HIV, a difference that may be attributed to the different viral vectors used [12].
Figure 1. Retroviral vectors for monitoring virus infectivity and mutagenesis.
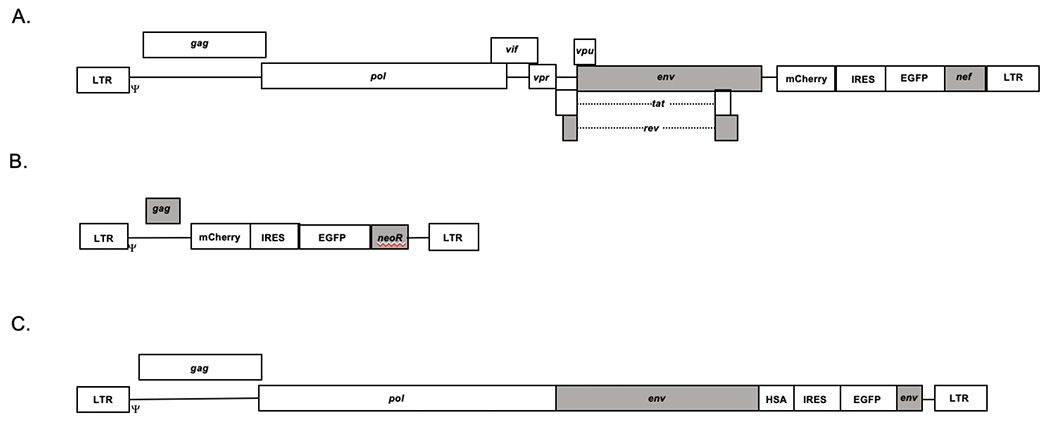
(A) Human immunodeficiency virus type 1 (HIV-1) vector. The HIV-1 vector HIV-1 MIG has been previously described [12], and contains a mCherry, internal ribosomal entry site (IRES), EGFP gene expression cassette. (B) Murine leukemia virus (MuLV)-based retroviral vector. The pLN vector was used to introduce a gene expression cassette including the mCherry gene, an IRES and the EGFP gene (see Materials and Methods). (C) Feline leukemia virus vector. A gene expression cassette containing the heat stable antigen (HSA), IRES, and the EGFP gene was introduced into the envelope gene region [24]. Each of these vectors was pseudotyped with the VSV-G protein to produce vector virus to infect target cells (Materials and Methods). The grey boxes represent genes containing mutations, which eliminated their functionality.
Figure 2. Effect of 5-aza-C treatment on infectivity and mutant frequency of MuLV and HIV-1.
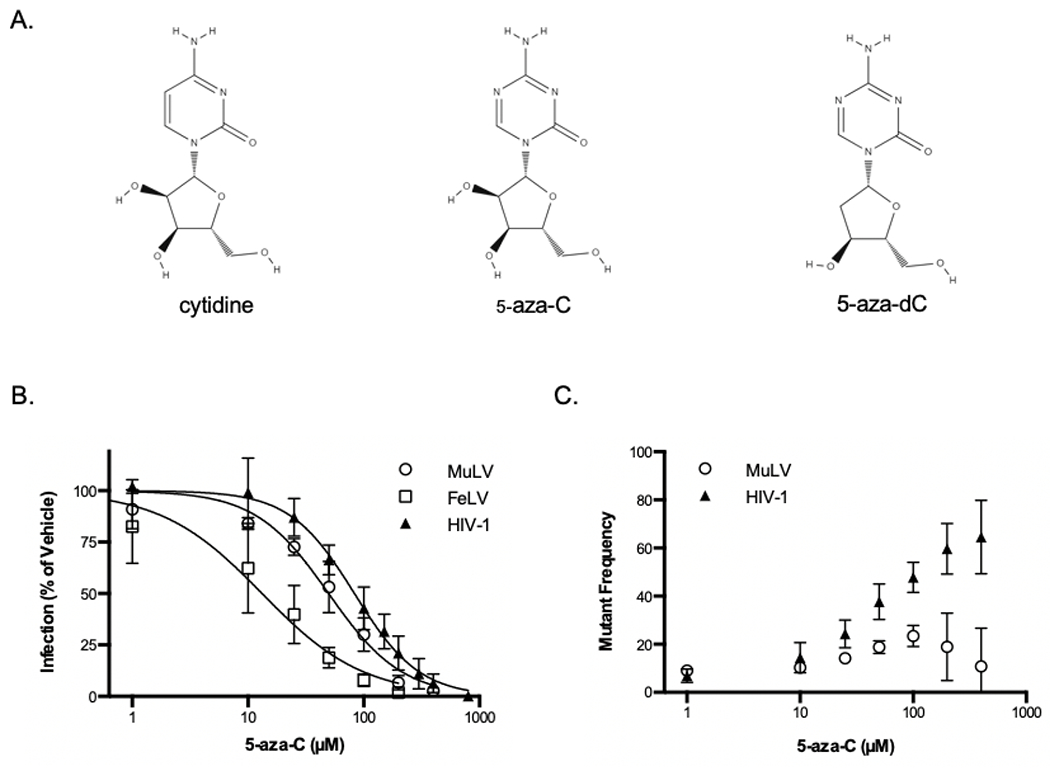
(A) Structures of cytidine, 5-aza-C, and its active derivative 5-aza-2’-deoxycytidine. (B and C) U373-MAGI cells were treated with increasing concentrations of 5-aza-C (i.e., 1, 10, 25, 50, 100, 200, and 400 μM) for two hours prior to infection with MuLV, FeLV, or HIV-1 vectors. Cells were harvested at 48 hours after infection for determination of infection (B) and mutant frequency (C) by flow cytometry. Mutant frequency is calculated by the ratio of single positive populations (mCherry+ only and GFP+ only). Data represent the mean ± SD of at least four independent experiments, normalized to the vehicle control.
5-aza-C has been reported to induce G-to-C hypermutation in the HIV-1 genome and thus increasing the mutational load [12, 18]. As shown in Figure 1, both MuLV-MIG and HIV-MIG contain two reporter genes, GFP and mCherry. If a mutation occurs in either reporter gene causing it to lose fluorescent capability, we will observe mutants that express only GFP or mCherry. Therefore, the mutant frequency can be quantified by the ratio of single positive populations (mCherry+ only and GFP+ only). With the data from flow cytometry, we calculated the relative mutant frequency of MuLV and HIV-1 genome, and observed that HIV-1 had a higher mutant frequency than that of MuLV with the treatment of 5-aza-C (Figure 2C).
To further investigate this observation, we conducted Sanger sequencing of the GFP gene in the proviral DNA. U373-MAGI cells were treated with the relative EC75 concentrations of 5-aza-C and then infected with MuLV, FeLV or HIV-1. DNA was extracted from infected cells at 72 hours after infection, and a portion of the GFP gene was amplified and subjected to Sanger sequencing. Surprisingly, our results showed that the MuLV GFP had no significant increase in mutation frequency when treated with 5-aza-C compared to vehicle control (Figure 3A; Table 1). However, treatment with 5-aza-C increased the mutation frequency of HIV-1 and FeLV genome significantly. Although, without considering the one hypermutated sequence, the mutation frequency of FeLV genome in the presence of 5-aza-C was not significantly increased compared to vehicle group control as well (Figure 3B). Nevertheless, 5-aza-C induced a significantly higher mutation frequency in HIV-1 than MuLV or FeLV whether the hypermutated sequences are considered or not (Figure 3A, 3B; Table 1). Furthermore, we found that the mutational load induced by 5-aza-C varied. MuLV GFP had the lowest mutational load with only one mutation in five mutants that were sequenced (Figure 3C). Three FeLV mutants had one, two or ten mutations. HIV-1 had the highest mutational load, ranging from one to eleven mutations per mutant sequenced. We observed one G-to-A hypermutant in both FeLV-GFP and HIV-1-EGFP mutants, which has 10 or 11 mutations, respectively. Additionally, mutational spectra analysis showed that treatment with 5-aza-C most commonly induced G-to-C mutations to HIV-1-GFP (Figure 4). Among the overall five mutations detected in 5-aza-C-treated MuLV-EGFP, only one was G-to-C and the other one was C-to-G. Similarly, three of the five mutations detected in 5-aza-C-treated FeLV-EGFP turned out to be G-to-C. In short, our results demonstrate that 5-aza-C didn’t significantly increase the mutation frequency and cause G-to-C hypermutation of MuLV and FeLV, as it does with HIV-1. Differences in the mechanism of action are likely due to structural differences between the HIV-1 RT and the gammaretroviral RTs. The general trend of the locations of mutations in the MuLV and FeLV proviral DNA suggested that they were distinct to those characterized for HIV-1 (Figure 5). Previous use of reporter genes for analysis of mutation spectra and mutation frequency has demonstrated a strong coincidence in comparable analyses observed with viral sequences in the proviral genome [11, 12, 15, 18].
Figure 3. Effect of 5-aza-C on mutation frequency.
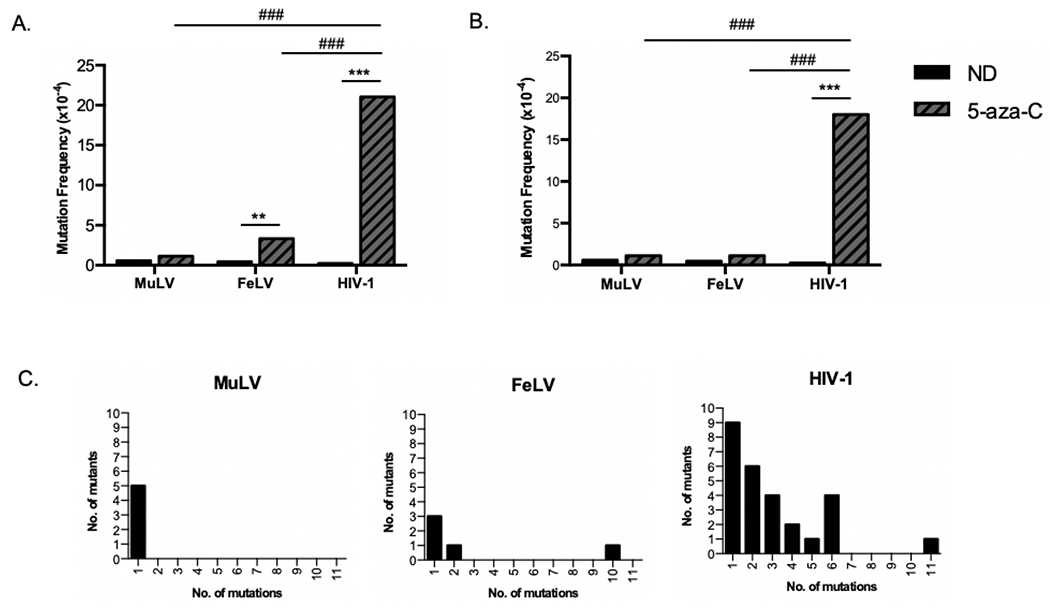
U373-MAGI cells were treated with ~EC75 concentration of 5-aza-C (i.e., 200 μM for MLV; 50 or 75 μM for FeLV; 400 μM for HIV-1) for two hours prior to infection with MuLV, FeLV, or HIV-1. DNA was harvested at 72 hours after infection, and a portion of the EGFP gene was amplified and subjected to Sanger sequencing. (A) Mutation frequency was determined by the number of mutations detected per 10,000 base pairs sequenced. (B) Mutation frequency after removing hypermutated sequences (>7 mutations per 615bp amplicon). (C) Mutational load of each amplicon sequenced. Results are combined from three independent experiments. ** = p < 0.01, *** = p < 0.001 compared to no drug (ND); ŧŧŧ = p < 0.001 compared to 5-aza-C treated HIV-1.
Table 1.
Effect of 5-aza-C on mutation frequency in MuLV, FeLV, and HIV-1.
| Virus╲ | 5-aza-C | No. of clones sequenced | No. of clones with mutations | No. of mutations | No. of base pairs sequenced | Mutation frequency (D/bp) | Fold increase |
|---|---|---|---|---|---|---|---|
| MuLV | − | 57 | 2 | 2 | 34,904 | 5.7x10−5 | / |
| MuLV | + | 72 | 5 | 5 | 44,033 | 1.1x10−4 | 2.0 |
| FeLV | − | 71 | 2 | 2 | 43,298 | 4.6x10−5 | / |
| FeLV a | + | 74 (75) |
4 (5) |
5 (15) |
45,389 (46,004) |
1.1x10−4 (3.3x10−4) |
2.4 (7.1) |
| HIV-1 | − | 72 | 0 | 0 | 44160 | <2.3x10−5 | / |
| HIV-1 a | + | 73 (74) |
26 (27) |
81 (92) |
43843 (44458) |
1.8x10−3 (2.1x10−3) |
>82 (>90) |
Numbers in parentheses include hypermutants (> 7 mutations / 615 bp amplicon).
Figure 4. Mutation spectra for 5-aza-C- or vehicle-treated viruses.
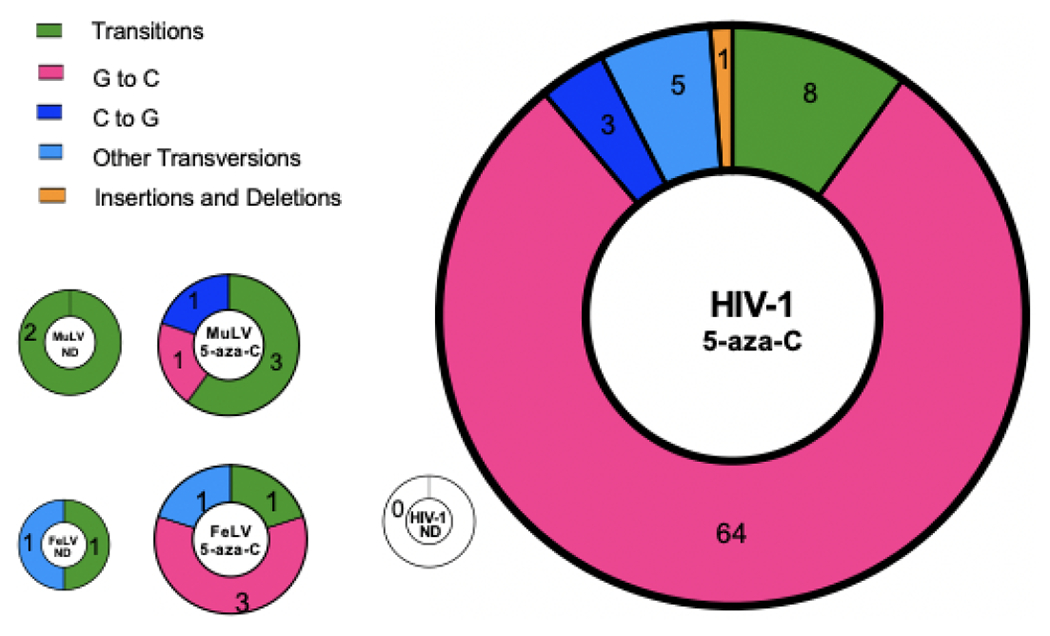
The frequency of each type of mutation is expressed as a percentage of total mutations. The area of each pie chart is sized according to total mutation frequency; HIV-1 ND pie chart is sized to the upper limit of its mutation frequency. Total numbers of each mutation type are indicated in each slice.
Figure 5.
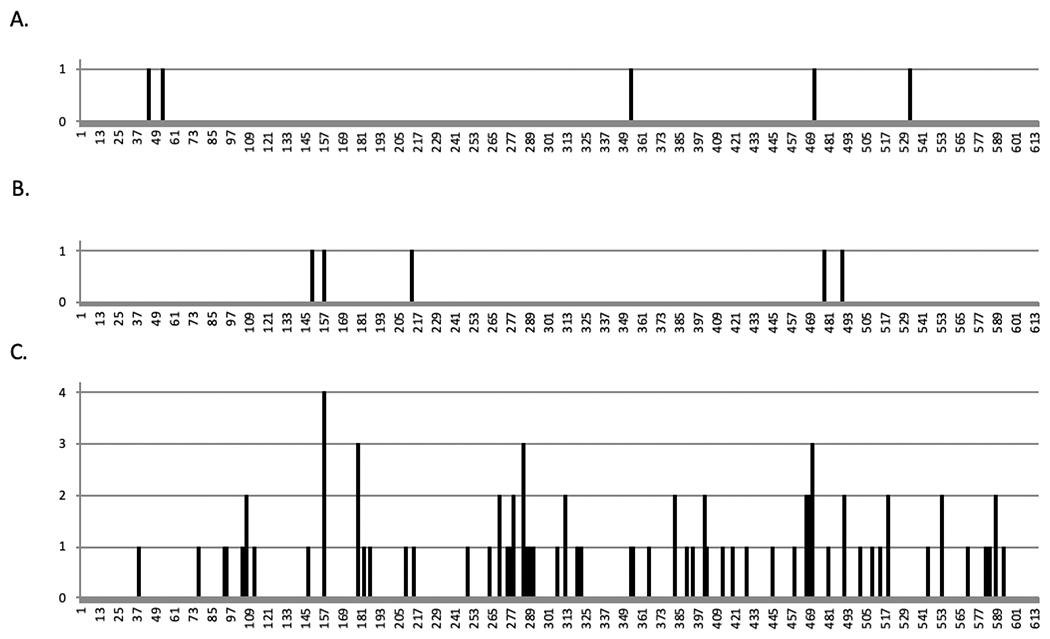
Localization of 5-aza-C-induced mutations. Location on EGFP amplicon is shown for each mutation in MuLV (A), and FeLV (B), and HIV-1 (C). The y-axis indicates the number of amplicons containing a mutation at that location.
To investigate whether mutation in the highly conserved YMDD motif (which can confer high levels of resistance to cytosine analogs) of RT would impact the susceptibility of FeLV (YVDD) or HIV-1 (YMDD) to 5-aza-C, experiments were conducted in which virus infection was analyzed between the wt parent and the YMDD mutant. Following infection of 5-aza-C treated target cells with wt or YMDD mutant virus, we observed that mutation of the YMDD motif did not impact the relative susceptibility to 5-aza-C (Figure 6). Additionally, there was no difference in mutant frequency between wt HIV-1 and the YMDD derivatives in the presence of 5-aza-C (Figure 6). Differences in nucleoside insertion fidelity of the HIV-1 M184V RT variant compared to wt RT observed in in cell-free studies [33] were not inferred in this virological assay.
Figure 6. Effect of mutations in the reverse transcriptase YMDD motif on 5-aza-C susceptibility and viral mutant frequency.
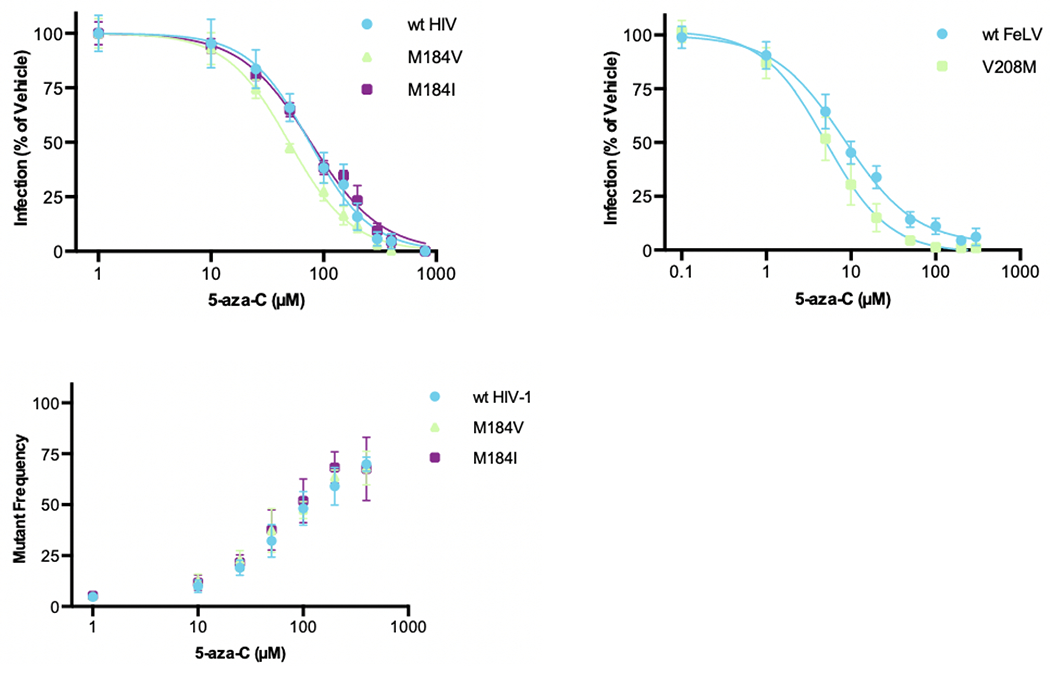
U373-MAGI cells were treated with increasing concentrations of 5-aza-C (i.e., 1, 10, 25, 50, 100, 150, 200, 300, 400 and 800 μM for wt HIV-1 and RT variants; 0.1, 1, 5, 10, 20, 50, 10, 200, 300 μM for FeLV and RT variant) for two hours prior to infection with HIV-1 or FeLV vectors. Cells were harvested at 48 hours after infection for determination of changes in the percent of infected cells and the viral mutant frequency as determined by flow cytometry. Mutant frequency analysis was not done with FeLV given that the vector contained only a GFP marker and not both the mCherry and GFP markers. Data represent the mean +/− SD of at least three independent experiments, and were normalized to the vehicle control (ND).
5-aza-C reduced the synthesis of late RT products for MuLV, FeLV and HIV-1
The incorporation of 5-aza-C, or its metabolites, during reverse transcription can result in either lethal mutagenesis of virus or chain termination of the DNA/RNA strand. Since 5-aza-C did not cause lethal mutagenesis of MuLV and FeLV, we examined the generation of the reverse transcription products in the presence of 5-aza-C. MAGI cells were treated with 5-aza-C two hours before infection. Cells were harvested at 18 hours after infection for quantification of a late reverse transcription product by qPCR. AZT, a well-known RT inhibitor, was used as a positive control, and raltegravir, an integrase inhibitor, was used as a negative control. Infection of cells in the presence of 5-aza-C treatment was monitored along with monitoring the synthesis of late RT products (Figure 7). Raltegravir was found to not impact on late RT product accumulation. In contrast, AZT decreased the synthesis of late RT products. Intriguingly, 5-aza-C reduced the synthesis of late RT products for HIV-1 as well as MuLV and FeLV, indicating that diminution of late RT products occurs with all viruses. Since mutation accumulation was only observed with HIV-1 and not the gammaretroviruses (Figure 3), MuLV RT may act as a non-obligate chain terminator (see below). For HIV-1, the reduction of late RT products may be due to pausing and reduced polymerase extension following 5-aza-C incorporation. The phenotype observed with 5-aza-C and HIV-1 is similar to that observed with favipiravir and some RNA viruses [34–38]. Taken together, these observations suggest that 5-aza-C can impact both viral mutagenesis as well as viral DNA synthesis for HIV-1 and only viral DNA synthesis for MuLV and FeLV.
Figure 7. 5-aza-C treatment inhibits reverse transcription of MuLV and HIV-1.
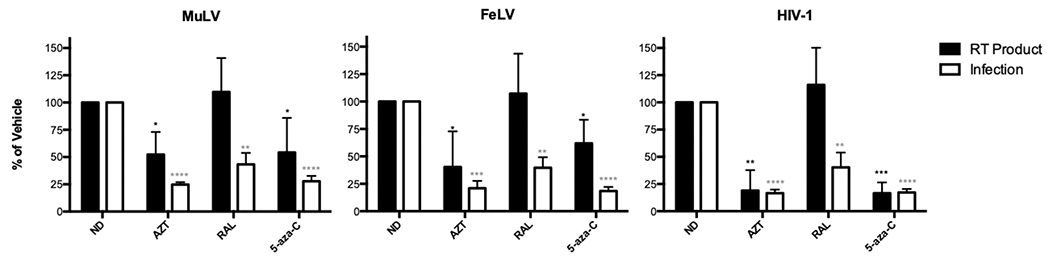
U373-MAGI cells were treated with AZT, raltegravir (RAL), or 5-aza-C for two hours prior infection. Cells were harvested at 18 hours after infection for quantification of a late reverse transcription product by qPCR. The AZT concentrations used were 4-5 nM for MuLV, 2-3 nM for FeLV and 5-10 nM for HIV-1. RAL concentrations used were 5-100 nM for MuLV, 5-50 nM for FeLV and 100 nM for HIV-1. 5-aza-C concentrations used were 150 μM for MuLV, 80 μM for FeLV and 400 μM for HIV-1. The data shown represent the mean +/− SD of three independent experiments, and were normalized to the vehicle control (ND). *** = P < 0.001, ** = P < 0.01, * = P < 0.05 compared to RT product of no drug (ND); **** = P < 0.0001, *** = P < 0.001, ** = P < 0.01 compared to infection of no drug (ND).
5-aza-dCTP can be incorporated into DNA by MuLV RT or HIV-1 RT, but only acts as a non-obligate chain terminator for MuLV RT
To further confirm our assumption that 5-aza-CTP is incorporated into viral genomes during reverse transcription, an in vitro incorporation assay was conducted using a 5’ 32P-labeled 23-mer DNA primer annealed to a 24-mer DNA template with a single G overhang incubated with MuLV or HIV-1 RT and increasing concentrations of 5-aza-dCTP. The reactions were later resolved on a 20 % urea-PAGE gel. As shown in Figure 8A, the 5-aza-dCTP with a concentration of over 0.5 mM was shown to be incorporated into the end of the 32P-labeled 23-mer DNA primer with either MuLV or HIV-1 RT. The lower band intensity observed at higher concentrations of 5-aza-dCTP in reactions with MuLV RT could be due to variation in loading (such variation is internally controlled by determination of the incorporation kinetics by plotting the ratios between extended vs. unextended primers).
Figure 8. Biochemical analysis of 5-aza-dC-triphosphate.
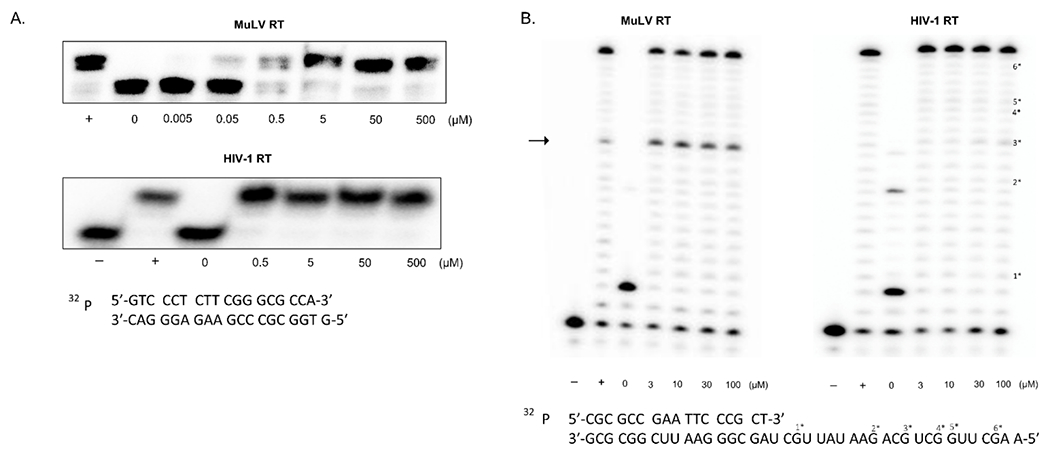
(A) Incorporation of 5-aza-dC-TP by MuLV or HIV-1 reverse transcriptase. A 5’ 32P-labeled 23-mer DNA primer annealed to a 24-mer DNA template with a single G overhang was incubated with MuLV or HIV-1 RT and increasing concentrations of 5-aza-dC-TP. The minus (−) and plus (+) refer to absence or addition of dCTP. Increasing concentrations of 5-aza-dC-TP (i.e., 0, 0.5, 5, 50, 500 μM) is indicated. (B) Extension of MuLV or HIV-1 RT beyond an incorporated 5-aza-dC-MP. A 5’ 32P-labeled 17-mer DNA primer annealed to a 40–mer RNA template was extended by MuLV or HIV-1 RT with fixed dATP, dGTP, and TTP concentrations. Increasing concentrations of 5-aza-dC-TP (i.e., 0, 3, 10, 30, 100 μM) was added to the reaction. indicates that no RT is added; while “+” indicates that plus dCTP is added. Template/primer sequences are shown in the bottom. “#*” indicates the location of each “G” in the template and its related extension product on the gel if extension pauses at that location. The arrow indicates the primer extension paused at the third 5-aza-CTP incorporation site.
An in vitro primer extension assay was also conducted to assess whether 5-aza-CTP can act as a chain terminator. For this assay, a 5′ 32P-labeled 17-mer DNA primer was annealed to a 40-mer RNA template. Reactions were initiated upon addition of RT and resolved on a 14 % urea-PAGE gel after termination. In Figure 8B, there was an intermediate product shown on the gel for MuLV RT. It was observed that the 5’ 32P-labeled 23-mer DNA primer was extended with MuLV RT, but that the primer extension paused at the third 5-aza-CTP incorporation site (see arrow). However, only final full-length extension product was seen for HIV-1 RT without significant pausing. One explanation for these observations is that 5-aza-CMP incorporation may alter local template/primer (T/P) structure after incorporation, which may cause the MuLV RT to fall off from the T/P, generating stronger pause sites. In contrast, this T/P structure change may not be large enough to cause the HIV-1 RT to fall off from the T/P. Taken together, our data from these in vitro biochemical experiments show that 5-aza-dCTP can be incorporated into the DNA strand with both MuLV and HIV-1 RTs, but that strong termination of the DNA strand extension only occurs with the MuLV RT, and only weak pausing that did not affect the accumulation of full length product was observed with HIV-1 RT.
Discussion
We previously characterized 5-aza-cytidine (5-aza-C) as a potent HIV-1 mutagen that induces G-to-C hypermutagenesis by incorporation of the reduced form (i.e., 5-aza-dC, 5-aza-dCTP) [12, 15]. Evidence to date suggests that this lethal mutagenesis is the primary antiretroviral mechanism for 5-aza-C [15]. In this study, we sought to more broadly investigate this potent antiretroviral mutagen by conducting comparative, parallel studies of the antiviral mechanism of 5-aza-C between HIV-1 and gammaretroviruses, i.e., MuLV and FeLV. Surprisingly, in contrast to the hallmark G-to-C hypermutagenesis that was observed with HIV-1, MuLV and FeLV did not reveal the presence of a significant increase in virus mutational burden, particularly that of G-to-C transversion mutations. This observation suggested a distinct antiviral mechanism of action between HIV-1 and gammaretroviruses. Our analysis of the effect of 5-aza-dCTP on DNA synthesis by MuLV and HIV-1 RT revealed that HIV-1 RT was found to not be inhibited by 5-aza-dCTP - even at concentrations of 100 μM. In contrast, 5-aza-dCTP was incorporated and significantly inhibited MuLV RT, generating pause sites and reducing the fully extended product. These intriguing biochemical observations support the conclusion that HIV-1 and MuLV have distinct mechanisms of antiretroviral action with 5-aza-dC. This notable observation argues that a HIV-1 mutagen can have a distinct mechanism of action compared to the closely related gammaretroviruses (i.e., MuLV as well as FeLV). This implies that potent antiviral activity can be manifested by distinct mechanisms among closely related viruses.
A previous comparative analysis conducted a parallel study of novel antiretroviral drugs between HIV-1 and HIV-2 [39]. HIV-2 infects about two million people worldwide and has fewer treatment options than HIV-1. It was noted that 5-aza-C was more potent against HIV-2 than HIV-1, which highlights the importance of minor differences between viruses that can impact antiviral potency. For example, AZT is active against both HIV-1 and HIV-2, but the antiviral pathways are distinct [40]. This is likely due to structural differences among the HIV-1 and HIV-2 RTs in regards to the ATP binding sites. Non-nucleoside RT inhibitors that are quite potent at inhibiting HIV-1 replication, possess no activity against HIV-2, and have been found to be inactive against feline immunodeficiency virus, which is a feline lentivirus [41]. HIV-2 encodes for the Vpx protein, which is involved in the degradation of the dNTP pool regulator SAMHD1 for the successful infection of macrophages and other myeloid cells which are known to have low dNTP pools [42, 43]. This could potentially influence antiviral activity of some agents. While closely related viruses may possess varying susceptibilities against an antiviral agent, each virus may utilize different pathways towards the development of antiviral drug resistance. Interestingly, favipirivir has been shown to have a mechanism of action that can act as a chain terminator and/or cause lethal mutagenesis, depending upon the RNA virus [34–38].
There have been two previous models proposed to explain the antiretroviral activity of 5-aza-C during the early phase of HIV-1 replication. In the first model, 5-aza-C is incorporated during reverse transcription as a deoxyribonucleotide (i.e., 5-aza-dCTP) [12]. The incorporation of 5-aza-dCTP would require the activity of cellular enzyme ribonucleotide reductase (RNR), which would convert the diphosphate form of 5-aza-C to 5-aza-dC, and this would then be phosphorylated to create 5-aza-dCTP [12]. Previous studies have reported that roughly 10-20% of 5-aza-C is reduced to 5-aza-dC by the action of RNR [15], which would allow 5-aza-dCTP to be incorporated during reverse transcription. The incorporation of 5-aza-dCTP is likely favored, given that HIV-1 RT residue Y115 acts as a steric gate, and can prevent ribonucleotides from efficient incorporation [44]. However, it remains a distinct possibility that 5-aza-C could be incorporated as 5-aza-CTP during HIV-1 reverse transcription. In this regard, HIV-1 RT has been observed to incorporate endogenous ribonucleotides at significant levels when deoxyribonucleotides levels are depleted – causing high nucleoside triphosphate [NTP]/deoxynucleoside triphosphate [dNTP] ratios – as found in non-dividing cells such as in macrophages [45–47]. High concentrations of 5-aza-C have been previously found to result in potent antiviral activity in cell culture [12, 15], which could result in significant incorporation of 5-aza-CTP.
The data presented indicate that in the cell culture conditions studied, 5-aza-C treatment resulted in an increase in the virus mutation frequency – specifically high levels of G-to-C mutagenesis – with HIV-1, but not with MuLV or FeLV (Figure 3, 4). Intriguingly, analysis of late RT products at these 5-aza-C concentrations resulted in similar reductions of viral DNA products for HIV-1, MuLV and FeLV (Figure 7). This observation indicates that the increase of viral mutagenesis observed with HIV-1 coincides with a reduction in the efficiency in viral DNA synthesis. This is somewhat reminiscent to that observed with favipiravir and some RNA viruses. Both purified HIV-1 and MuLV RTs were shown to incorporate 5-aza- CTP, but only MuLV RT was observed to result in pausing using the selected primer/template. One possible explanation for this apparent discrepancy between the cell culture results versus that obtained with the purified RTs is that the reaction conditions used did not reveal pausing for HIV-1 RT on the selected primer/template. Another possible explanation is that an analysis of a diverse number of templates would reveal sequences where HIV-1 RT may also be prone to pausing, as would likely be predicted from the results observed in Figure 7. Future analyses will be helpful in further delineating the differences seen between these observations.
While an antiviral drug may possess a widely recognized antiviral mechanism of action, it is likely that this is a primary antiviral mechanism, and that other secondary antiviral mechanisms can contribute to the over susceptibility of a virus to an antiviral agent. Differences in the mechanism of action are likely impacted by structural differences between the HIV-1 and MuLV RTs. Figure 9 shows an alignment of the RT active sites of MuLV, FeLV and HIV-1, as well as a ribbon and space-filling diagram of the amino acid residues in the RT active sites. Analysis of the MuLV and HIV-1 conserved active sites suggests that there are amino acid substitutions that impact the electrostatic charge potential and that could impact the interactions with 5-aza-dC (Figure 9). Key residues in the active site can impact enzyme fidelity. The relative cell-free base substitution error rates of HIV-1, MuLV and FeLV using the lacZα peptide gene in M13mp2, i.e., 2 x 10 −4, 3.3 x 10 −5, and 5.8 x 10 −6 mutations per target base pair, respectively, have been previously analyzed and provide a point of reference in differences between these enzymes in regards to fidelity [26, 48, 49]. Future studies that analyze subtle structural differences in the catalytic domains of these RTs will likely provide important insights for advancing these studies to more precisely define the molecular basis for antiviral mechanism(s) of action. The comparative investigation of a HIV-1 mutagen by analysis with another member of the Retroviridae provides further insights into the breadth of antiretroviral activity associated with an agent possessing anti-HIV-1 activity. Our studies help to emphasize that against the same general viral target (i.e., RT), there can be more than one antiviral mechanism of activity at play, and that the dominant antiviral mechanism of action can be different among viruses – even closely related viruses where amino acid and/or structural differences can result in differences in molecular interactions that manifest different antiviral outcomes.
Figure 9. Comparison of the MuLV and HIV-1 reverse transcriptase active sites.
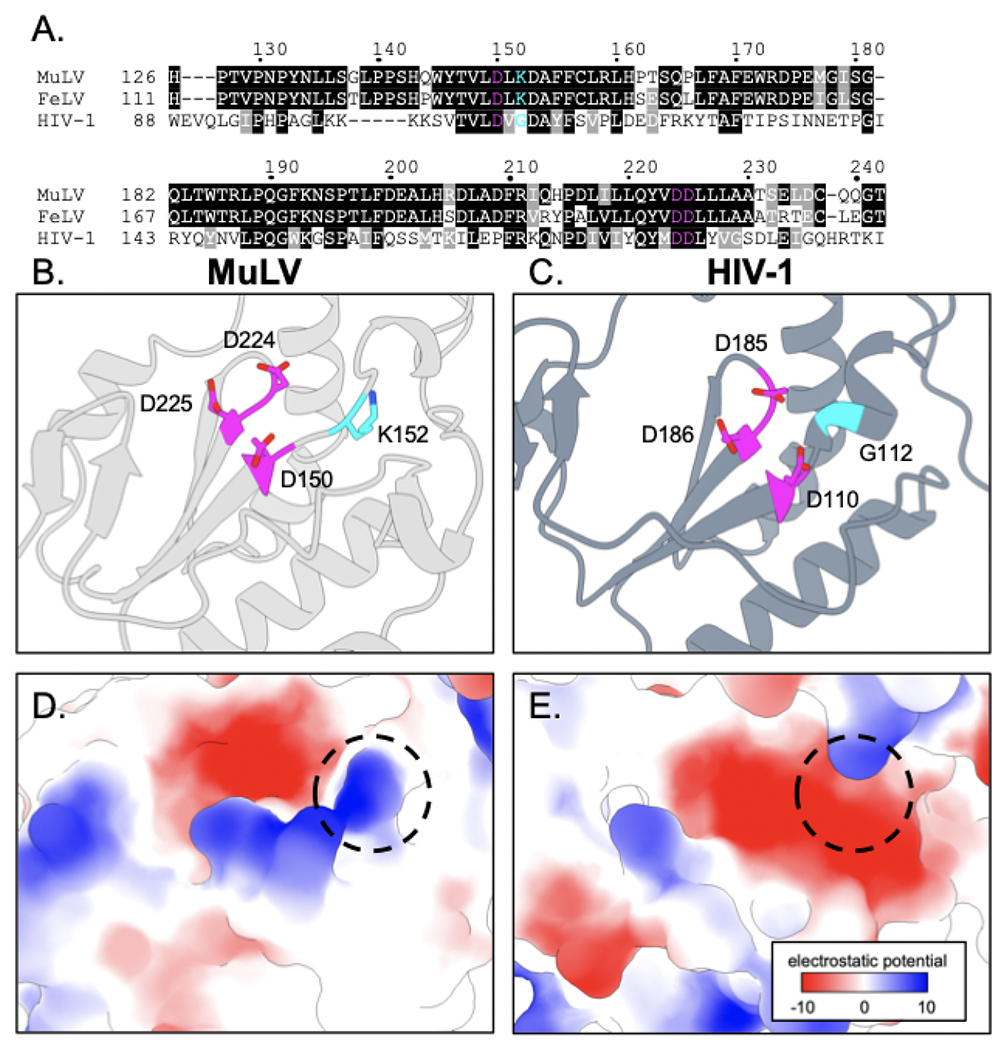
(A) Multiple sequence alignment of reverse transcriptase (RT) amino acid residues spanning the active sites of MuLV, FeLV, and HIV-1 (GenBank accession nos. AAA66622.1, M18247.1, and AAK08484.2 respectively) [50]. Identical amino acid residues (black highlighted), similar amino acid residues (grey highlighted), and positions with variable amino acid residues (no highlights) are indicated. (B and C) Conserved catalytic active site aspartate residues (magenta) for MuLV RT (grey ribbons – PDB ID: 1MML) and HIV-1 RT (slate blue ribbons – PDB ID: 4IFV). MuLV RT K152 and HIV-1 RT G112 (cyan) indicate the variable amino acid residues adjacent to the active site that are highlighted on the corresponding structures [51, 52]. (D and E) Coulombic electrostatic potential for the variable RT active sites. Variable residues K152 and G112 from MuLV and HIV-1, respectively, are indicated (dashed circle). The RT renderings were generated with ChimeraX [53, 54].
Research Highlights.
5-aza-C has previously been shown to be a HIV-1 mutagen
Comparative analysis of 5-aza-C was done between HIV-1 and MuLV
5-aza-C did not significantly increase the mutational burden in MuLV
A viral mutagen can have potent activity against related viruses via distinct mechanisms
Acknowledgements
This research was supported by NIH grants R01 GM56615. Y.M. was supported by T32 AI83196 (Institute for Molecular Virology Training Program); N.T. was supported by NIH T32 DA007097 and F32 AI150351; W.G. was supported by NIH T32 OD010993. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH R01 GM129325 and the Office of Cyber Infrastructure and Computational Biology, NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Literature Cited
- [1].De Clercq E, Li G. Approved Antiviral Drugs over the Past 50 Years . Clin Microbiol Rev. 2016;29:695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Prusoff WH. Synthesis and biological activities of iododeoxyuridine, an analog of thymidine. Biochim Biophys Acta. 1959;32:295–6. [DOI] [PubMed] [Google Scholar]
- [3].Seley-Radtke KL, Yates MK. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res. 2018;154:66–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yates MK, Seley-Radtke KL. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antiviral Res. 2019;162:5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mercorelli B, Palu G, Loregian A. Drug Repurposing for Viral Infectious Diseases: How Far Are We? Trends Microbiol. 2018;26:865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58. [DOI] [PubMed] [Google Scholar]
- [7].Farha MA, Brown ED. Drug repurposing for antimicrobial discovery. Nat Microbiol. 2019;4:565–77. [DOI] [PubMed] [Google Scholar]
- [8].Organization WH. Global AIDS Update 2016. 2016.
- [9].Perales C, Gallego I, de Avila AI, Soria ME, Gregori J, Quer J, et al. The increasing impact of lethal mutagenesis of viruses. Future Med Chem. 2019;11:1645–57. [DOI] [PubMed] [Google Scholar]
- [10].Bonnac LF, Mansky LM, Patterson SE. Structure-activity relationships and design of viral mutagens and application to lethal mutagenesis. J Med Chem. 2013;56:9403–14. [DOI] [PubMed] [Google Scholar]
- [11].Clouser CL, Patterson SE, Mansky LM. Exploiting drug repositioning for discovery of a novel HIV combination therapy. Journal of virology. 2010;84:9301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dapp MJ, Clouser CL, Patterson S, Mansky LM. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. Journal of virology. 2009;83:11950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Harris KS, Brabant W, Styrchak S, Gall A, Daifuku R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 2005;67:1–9. [DOI] [PubMed] [Google Scholar]
- [14].Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:1492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rawson JM, Daly MB, Xie J, Clouser CL, Landman SR, Reilly CS, et al. 5-Azacytidine Enhances the Mutagenesis of HIV-1 by Reduction to 5-Aza-2’-Deoxycytidine. Antimicrob Agents Chemother. 2016;60:2318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jackson-Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R. Mutagenicity of 5-aza-2’-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:4681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rawson JM, Heineman RH, Beach LB, Martin JL, Schnettler EK, Dapp MJ, et al. 5,6-Dihydro-5-aza-2’-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorg Med Chem. 2013;21:7222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rawson JM, Landman SR, Reilly CS, Bonnac L, Patterson SE, Mansky LM. Lack of mutational hot spots during decitabine-mediated HIV-1 mutagenesis. Antimicrob Agents Chemother. 2015;59:6834–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Clouser CL, Chauhan J, Bess MA, van Oploo JL, Zhou D, Dimick-Gray S, et al. Anti-HIV-1 activity of resveratrol derivatives and synergistic inhibition of HIV-1 by the combination of resveratrol and decitabine. Bioorganic & medicinal chemistry letters. 2012;22:6642–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Clouser CL, Holtz CM, Mullett M, Crankshaw DL, Briggs JE, O’Sullivan MG, et al. Activity of a novel combined antiretroviral therapy of gemcitabine and decitabine in a mouse model for HIV-1. Antimicrob Agents Chemother. 2012;56:1942–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clouser CL, Bonnac L, Mansky LM, Patterson SE. Characterization of permeability, stability and anti-HIV-1 activity of decitabine and gemcitabine divalerate prodrugs. Antiviral chemistry & chemotherapy. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Harrington RD, Geballe AP. Cofactor requirement for human immunodeficiency virus type 1 entry into a CD4-expressing human cell line. Journal of virology. 1993;67:5939–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vodicka MA, Goh WC, Wu LI, Rogel ME, Bartz SR, Schweickart VL, et al. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology. 1997;233:193–8. [DOI] [PubMed] [Google Scholar]
- [24].Greggs WM 3rd, Clouser CL, Patterson SE, Mansky LM. Discovery of drugs that possess activity against feline leukemia virus. J Gen Virol. 2012;93:900–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980–2, 4–6, 9–90. [PMC free article] [PubMed] [Google Scholar]
- [26].Operario DJ, Reynolds HM, Kim B. Comparison of DNA polymerase activities between recombinant feline immunodeficiency and leukemia virus reverse transcriptases. Virology. 2005;335:106–21. [DOI] [PubMed] [Google Scholar]
- [27].Daly MB, Roth ME, Bonnac L, Maldonado JO, Xie J, Clouser CL, et al. Dual anti-HIV mechanism of clofarabine. Retrovirology. 2016;13:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dapp MJ, Heineman RH, Mansky LM. Interrelationship between HIV-1 fitness and mutation rate. J Mol Biol. 2013;425:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Clouser CL, Bonnac L, Mansky LM, Patterson SE. Characterization of permeability, stability and anti-HIV-1 activity of decitabine and gemcitabine divalerate prodrugs. Antiviral chemistry & chemotherapy. 2014;23:223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schneider WM, Wu DT, Amin V, Aiyer S, Roth MJ. MuLV IN mutants responsive to HDAC inhibitors enhance transcription from unintegrated retroviral DNA. Virology. 2012;426:188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Greggs WM 3rd, Clouser CL, Patterson SE, Mansky LM. Broadening the use of antiretroviral therapy: the case for feline leukemia virus. Ther Clin Risk Manag. 2011;7:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wainberg MA, Drosopoulos WC, Salomon H, Hsu M, Borkow G, Parniak M, et al. Enhanced fidelity of 3TC-selected mutant HIV-1 reverse transcriptase. Science. 1996;271:1282–5. [DOI] [PubMed] [Google Scholar]
- [34].Shannon A, Selisko B, Le NT, Huchting J, Touret F, Piorkowski G, et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat Commun. 2020;11:4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].de Avila AI, Gallego I, Soria ME, Gregori J, Quer J, Esteban JI, et al. Lethal Mutagenesis of Hepatitis C Virus Induced by Favipiravir. PLoS One. 2016;11:e0164691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].de Avila AI, Moreno E, Perales C, Domingo E. Favipiravir can evoke lethal mutagenesis and extinction of foot-and-mouth disease virus. Virus Res. 2017;233:105–12. [DOI] [PubMed] [Google Scholar]
- [37].Escribano-Romero E, Jimenez de Oya N, Domingo E, Saiz JC. Extinction of West Nile Virus by Favipiravir through Lethal Mutagenesis. Antimicrob Agents Chemother. 2017;61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Baranovich T, Wong SS, Armstrong J, Marjuki H, Webby RJ, Webster rG, et al. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. Journal of virology. 2013;87:3741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Beach LB, Rawson JM, Kim B, Patterson SE, Mansky LM. Novel inhibitors of human immunodeficiency virus type 2 infectivity. J Gen Virol. 2014;95:2778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Alvarez M, Nevot M, Mendieta J, Martinez MA, Menendez-Arias L. Amino acid residues in HIV-2 reverse transcriptase that restrict the development of nucleoside analogue resistance through the excision pathway. J Biol Chem. 2018;293:2247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Galilee M, Alian A. The structure of FIV reverse transcriptase and its implications for non-nucleoside inhibitor resistance. PLoS Pathog. 2018;14:e1006849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004;279:51545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J Biol Chem. 2012;287:21570–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nguyen LA, Domaoal RA, Kennedy EM, Kim DH, Schinazi RF, Kim B. Pre-steady state kinetic analysis of HIV-1 reverse transcriptase for non-canonical ribonucleoside triphosphate incorporation and DNA synthesis from ribonucleoside-containing DNA template. Antiviral Res. 2015;115:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kennedy EM, Gavegnano C, Nguyen L, Slater R, Lucas A, Fromentin E, et al. Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J Biol Chem. 2010;285:39380–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kennedy EM, Amie SM, Bambara RA, Kim B. Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J Biol Chem. 2012;287:14280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Daddacha W, Noble E, Nguyen LA, Kennedy EM, Kim B. Effect of ribonucleotides embedded in a DNA template on HIV-1 reverse transcription kinetics and fidelity. J Biol Chem. 2013;288:12522–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Boyer JC, Bebenek K, Kunkel TA. Unequal human immunodeficiency virus type 1 reverse transcriptase error rates with RNA and DNA templates. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:6919–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roberts JD, Preston BD, Johnston LA, Soni A, Loeb LA, Kunkel TA. Fidelity of two retroviral reverse transcriptases during DNA-dependent DNA synthesis in vitro. Mol Cell Biol. 1989;9:469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Notredame C, Higgins dG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17. [DOI] [PubMed] [Google Scholar]
- [51].Georgiadis MM, Jessen SM, Ogata CM, Telesnitsky A, Goff SP, Hendrickson WA. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure. 1995;3:879–92. [DOI] [PubMed] [Google Scholar]
- [52].Bauman JD, Patel D, Dharia C, Fromer MW, Ahmed S, Frenkel Y, et al. Detecting allosteric sites of HIV-1 reverse transcriptase by X-ray crystallographic fragment screening. J Med Chem. 2013;56:2738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pettersen EF, Goddard TD, Huang CC, Meng eC, Couch GS, Croll TI, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]