

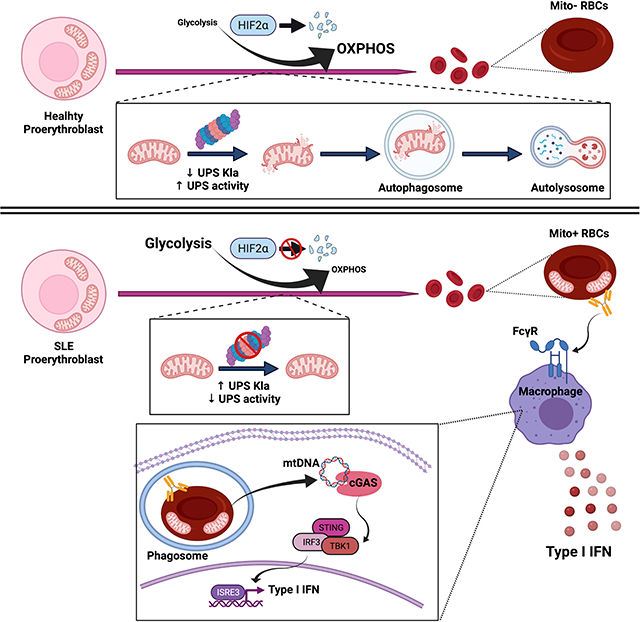
SUMMARY
Emerging evidence supports that mitochondrial dysfunction contributes to Systemic Lupus Erythematosus (SLE) pathogenesis. Here we show that programmed mitochondrial removal, a hallmark of mammalian erythropoiesis, is defective in SLE. Specifically, we demonstrate that during human erythroid cell maturation, a hypoxia-inducible factor (HIF)-mediated metabolic switch is responsible for the activation of the ubiquiting proteasome system (UPS), which precedes and its necessary for the autophagic removal of mitochondria. A defect in this pathway leads to accumulation of red blood cells (RBCs) carrying mitochondria (Mito+ RBCs) in SLE patients and in correlation with disease activity. Antibody-mediated internalization of Mito+ RBCs induces Type I interferon (IFN) production through activation of cGAS in macrophages (M⏀). Accordingly, SLE patients carrying both Mito+ RBCs and opsonizing antibodies display the highest levels of blood Interferon Stimulated Gene (ISG) signatures, a distinctive feature of SLE.
In Brief:
A subgroup of SLE patients fail to engage HIF regulated metabolic and proteasomal pathways causing the accumulation of mitochondria-containing red blood cells. These cells, when engulfed by macrophages activate cGAS/STING dependent inflammation.
Graphical Abstract
INTRODUCTION
SLE is considered a prototype ”Type I interferonopathy”. Thus, recombinant IFN induces a lupus-like syndrome indistinguishable from spontaneous SLE (Ronnblom et al., 1991; Stewart, 2003). In addition, an ISG transcriptional signature is present in the blood of a large fraction of patients (Banchereau et al., 2017). Furthermore, genome wide association studies (GWAS) highlighted the contribution to SLE susceptibility of genes encoding molecules involved in nucleic acid (NA) degradation and sensing as well as Type I IFN signaling pathways (Langefeld et al., 2017).
Mitochondrial dysfunction has been linked to SLE pathogenesis. Thus, SLE neutrophils extrude oxidized mitochondrial DNA (ox mtDNA), a potent interferogenic stimulus (Caielli et al., 2016; Lood et al., 2016). Moreover, accumulation of succinate-driven mitochondrial reactive oxygen species (mtROS) is a feature of a unique CD4+ T cell helper subopulation expanded in children with SLE (Caielli et al., 2019).
To determine whether mitochondrial dysfunction affects other cell lineages in SLE, we studied the life-cycle of RBCs, as mitochondrial clearance is a feature of their maturation (An et al., 2015). Using both small molecule inhibitors as well as cells from patients with genetically defined defects in UPS function (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated Temperature Syndrome – CANDLE) (Brehm et al., 2015), we demonstrate that a HIF-mediated metabolic switch regulates the activation of the UPS, which is upstream of mitophagy, during human erythroid cell maturation. Furthermore, we show that a defect in this pathway prevents the activation of the UPS and subsequently blocks mitophagy, resulting in the accumulation of interferogenic Mito+ RBCs, in SLE patients.
RESULTS
RBCs from patients with active SLE retain mitochondria
Programmed mitochondrial removal takes place during normal development in both lens epithelial and the erythroid lineage (Zhang and Ney, 2010). Erythroid cell differentiation evolves through morphologically distinct nucleated precursor stages prior to enucleation and final maturation of reticulocytes into RBCs, when mitochondria are completely removed. Surprisingly, using Mitotracker Deep Red (MTDR), we observed up to 37.2% of mature (CD235a+ CD71−) RBCs containing mitochondria (Mito+ RBCs) in a fraction (69.2%, n=26) of SLE patients (Figures 1A). These cells were not present in either healthy donors (HD, n=8) or patients with Juvenile Dermatomyositis (JDM, n=8) (Figure 1A). Mitotracker signal was abolished by carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), a mitochondrial uncoupling agent, supporting the specify of the staining (Figure 1A). Accordingly, both immunohistochemistry for the mitochondrial marker COXIV and transmission electron microscopy (TEM) analyses confirmed the presence of mitochondria-like organelles in a fraction of mature SLE RBCs (Figures 1B and 1C).
Figure 1. RBCs from patients with active SLE retain mitochondria.
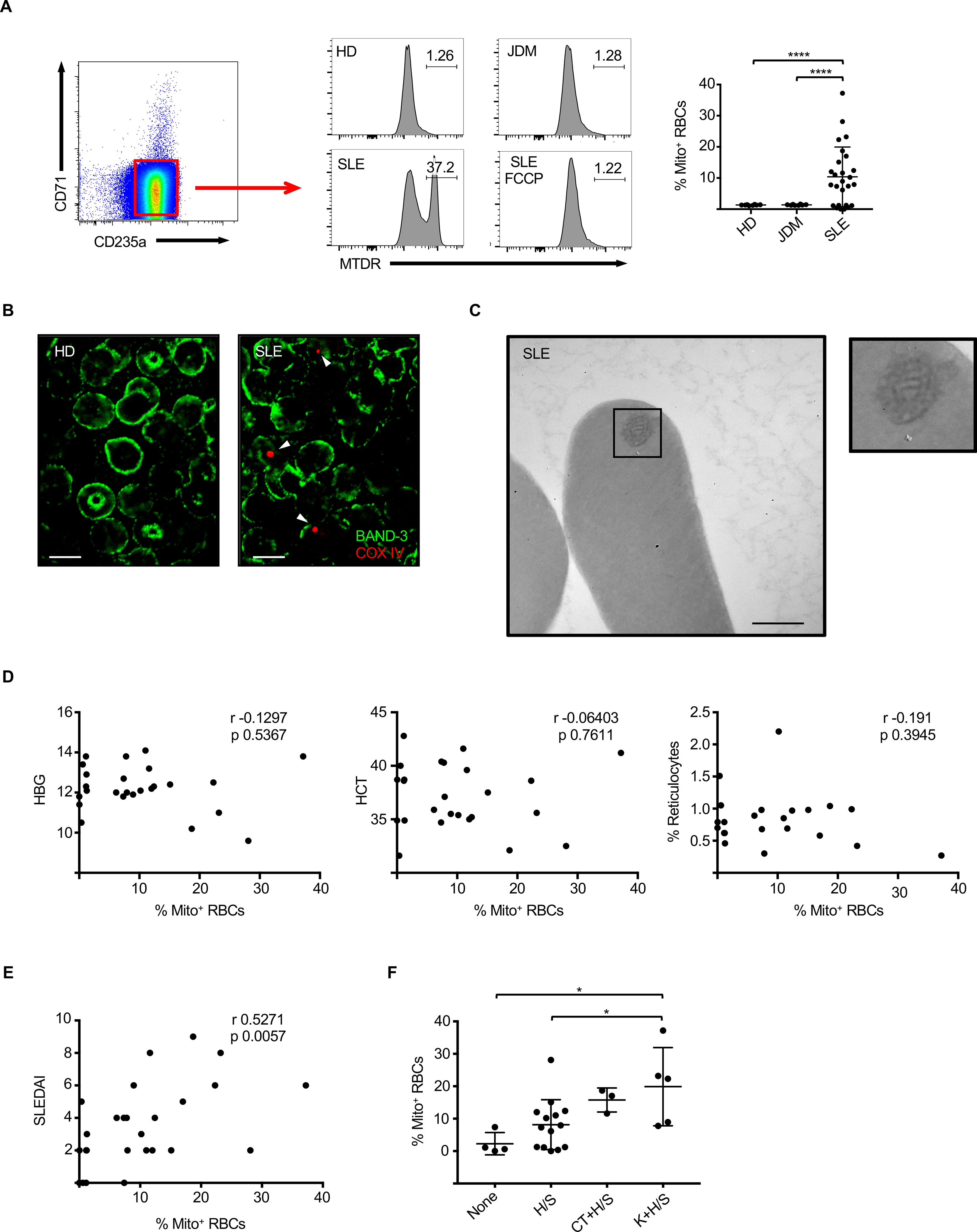
(A) Representative FACS plot and quantification of mature RBCs (CD235a+ CD71− cells) that contain mitochondria (Mito+ RBCs) in HD (n=8), JDM (n=8) and SLE (n=26) patients. (MTDR - MitoTracker Deep Red; FCCP - p-trifluoromethoxyphenylhydrazone; HD – Healthy donor). (B) RBCs Mature RBCs were isolated from HD and SLE and immunostained for the mitochondrial marker COXIV and the mature RBCs marker Band-3. Scale bar = 5 μm. (C) Transmission electron microscopy (TEM) analysis shown the presence of mitochondria-like organelles in mature SLE RBCs. Scale bar = 500 nm. (D) Lack of correlation between the percentage of circulating Mito+ RBCs and blood paramethers associated with anemia such as hemoglobin content (HBG; n=25), hematocrit (HCT; n=25), % of circulating reticulocytes (n=21). (E) Correlation between the percentage of circulating Mito+ RBCs and disease activity assessed by the SLEDAI score. (n=26). (F) Percentage of circulating Mito+ RBCs in SLE patients stratified based on alterations of selected SLEDAI components. None: no alterations in any SLEDAI components (n=4); H/S: alterations in serological/hematological parameters only (n=14); CT+H/S: connective tissue involvement + serological/hematological parameters (n=3); K+H/S: kidney involvement + serological/hematological parameters (n=5). In (B and C) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A) and (F); Pearson correlation test in (D) and (E)].
The percentage of circulating Mito+ RBCs did not correlate with either anemia, a condition that could drive immature erythroid precursors into the circulation (Kim and Nemeth, 2015), ethnicity or treatment (Figure 1D and Table S1). Importantly, it correlated with disease activity as measured by the SLE Disease Activity Index (SLEDAI) (Gladman et al., 2002) (Figure 1E). Because this index is a composite score evaluating multiple parameters, we further assessed whether the presence of Mito+ RBCs correlated with a particular SLEDAI component. Thus, patients were classified into four groups according to the following components: no SLEDAI parameters (“none”), serological/hematological parameters only (“H/S”), connective tissue involvement and serological/hematological parameters (“CT+H/S”) or kidney involvement and serological/hematological parameters (“K+H/S”), reflecting increasing levels of disease severity (Banchereau et al., 2016). Interestingly, Mito+ RBCs were especially enriched in the latter group, supporting their association with disease activity (Figure 1F).
PBMCs-derived RBCs as a model to study human erythropoiesis
Adult erythropoiesis takes place in the bone marrow via three stages: early erythropoiesis, terminal erythroid differentiation and reticulocyte maturation. Early erythropoiesis involves the differentiation of hematopoietic stem and progenitor cells (HSPCs) into proerythroblasts. Terminal erythroid differentiation consists of a stepwise transition from proerythroblasts to enucleated reticulocytes. Finally, reticulocytes mature into RBCs upon removing organelle remnants (An et al., 2015).
To study the mechanism of mitochondrial removal during human terminal erythroid differentiation, we generated proerythroblasts starting from PBMCs (van den Akker et al., 2010). As previously described, at the end of the expansion phase (0 h) most cells in these cultures displayed a proerythroblast phenotype characterized by high CD36, CD71 and CD49d expression and low CD235a and CD233 expression (Figures 2A, 2B and 2C). Proerythroblasts were then allowed to mature in the presence of high dose erythropoietin, which induced progressive loss of CD71, CD36 and CD49d coupled with gradual increased expression of CD235a and CD233 (Figures 2B and 2C). As maturation progressed, cells accumulated RBCs specific membrane proteins (GYPA, EBP42, EBP41, SLC4A1, SLC2A1), enzymes (CA2, PRDXs, BLVRB, SOD1, CA1, BPGM) as well as different hemoglobins (HB) (Figures 2D and 2E). Furthermore, we observed an increase in expression of erythroid-associated transcription factors (GATA1 and KLF1) and a concomitant decrease in expression of HSPCs-associated transcription factors (GATA2 and ETV6) (Figure 2F) (Love et al., 2014). Overall, our data support that this in vitro system accurately recapitulates the human erythroid maturation process.
Figure 2. PBMC-derived RBCs as a model to study human erythropoiesis.
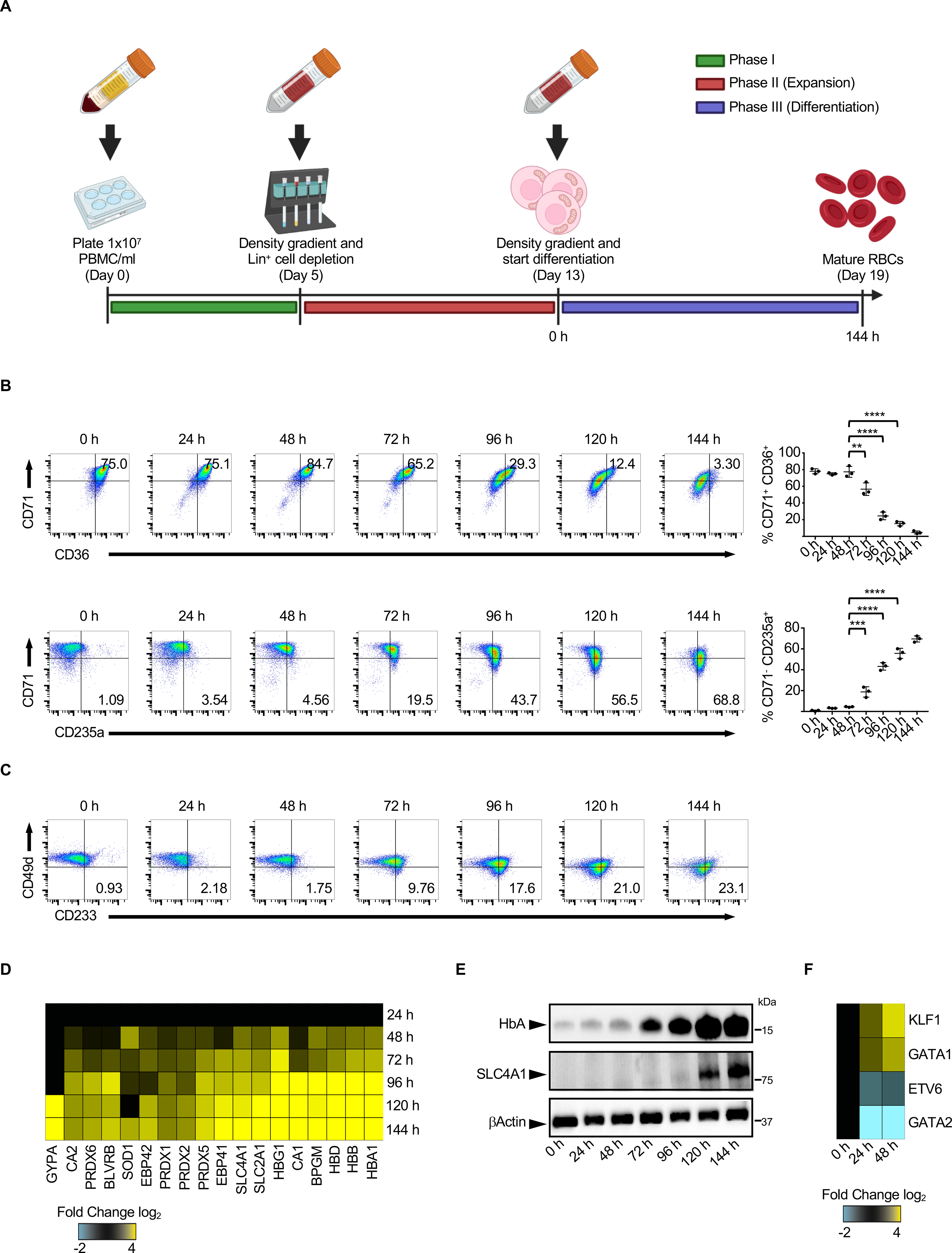
(A) Schematic representation of the culture protocol used to generate RBCs from PBMCs. Expression of CD71, CD36 and CD235a (B) or CD233 and CD49d (C) on the cell surface of proerythroblasts during the differentiation phase in a typical culture. (n=3). Proteomic (D) and Western blot (E) analysis of RBC-specific proteins expressed by proerythroblasts during the differentiation phase. (F) Changes in the expression of representative erythroid-specific transcription factors during proerythroblast maturation. In (C, D, E and F) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (B)].
Activation of the UPS is critical for progression of mitophagy during human erythropoiesis
Mitophagy is involved in the removal of mitochondria during the final stage of murine reticulocyte maturation (Sandoval et al., 2008; Schweers et al., 2007). However, its role in human terminal erythroid differentiation has not been completely elucidated (Betin et al., 2013).
Using immunoblotting analysis, we monitored the levels of mitochondrial proteins during the final stage of human proerythroblast maturation. We expected to observe the synchronized degradation of all mitochondrial proteins, since the content of autophagosomes is non-selectively degraded via acidic lysosomal hydrolases during mitophagy (Pickles et al., 2018). Surprisingly, our results showed that mitochondrial proteins fell into two major groups according to their degradation rate. Thus, while a group of proteins were rapidly removed during the differentiation process (rapidly removed mitochondrial proteins; RRMPs), another group remained unaffected until later time points (slowly removed mitochondrial proteins; SRMPs; Figure 3A). This suggests that mitophagy might not be the only mechanism that regulates mitochondrial degradation during human erythropoiesis.
Figure 3. Activation of the UPS is critical for mitophagy during human erythropoiesis.

(A) Representative Western blot analysis and quantification of ribosomal proteins (RPL29), rapidly removed mitochondrial proteins (RRMPs) and slowly removed mitochondrial proteins (SRMPs) in proerythroblasts collected at different stages of terminal erythroid maturation. (n=3). (B) Representative Western blot analysis of RPL29, RRMPs and SRMPs in proerythroblasts differentiated in the presence of UPS (MG132 or bortezomib) or autophagy (Bafilomycin A1 or E-64d/Pepstatin) inhibitors. (C) Representative immunofluorescence images showing colocalization of the lysosomal maker LAMP1 with COXIV (RRMP) or ATP5A (SRMP) in proerythroblasts collected at different stages of terminal erythroid maturation. White arrowheads indicate colocalization. Scale bar = 2 μm. (D) Relative chymotrypsin-like UPS activity over time during proerythroblast maturation. (n=5). (E) Autophagy flux over time during proerythroblast maturation. (n=3). (F) Western blot analysis and quantification of COXIV and ATP5A levels in HD and CANDLE proerythroblasts 24 h and 144 h post-differentiation. (HD – Healthy donor). (G) Representative FACS plot of Mitotracker Deep Red (MTDR) staining on mature RBCs (gated on CD235a+ CD71− cells) isolated from CANDLE3 patient. (H) Western blot analysis of selected mitochondrial proteins in proerythroblast transfected with short hairpin RNAs (shRNAs) specific for 15- Lipoxygenase (ALOX15). In (B, C and H) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A), (D) and (E)].
A growing body of evidence shows that selected mitochondrial proteins are rapidly degraded by the UPS before mitochondrial remnants are removed through mitophagy (Wei et al., 2017; Yoshii et al., 2011). Consistently, the degradation of RRMPs was sensitive to UPS inhibitors (MG132 or bortezomib) but not to autophagy inhibitors (Bafilomycin A1 or E-64d/Pepstatin) (Figures 3B and S1A). In contrast, autophagy blockade significantly reduced the elimination of SRMPs (Figures 3B and S1A). Indeed, while the RRMP COXIV never associated with lysosomes, the SRMP ATP5A co-localized with LAMP1+ compartments before being degraded (Figures 3C and S1B). In addition, RRMPs shared the degradation pattern with ribosomes, which have been shown to be exclusively removed by the UPS (Nguyen et al., 2017) (Figures 3A, 3B and S1A). All togheter, these results suggest that RRMPs are degraded in a UPS-dependent autophagy-independent manner, while SRMPs are removed by mitophagy. In support of being activated upstream of mitophagy, the UPS activity reached maximum values during the earlier stages of proerythroblast differentiation, while autophagic flux increased only at later time points (Figures 3D, 3E and S1C). Consistenlty, the appearance of swollen mitochondria, reminiscent of depolarized organelles degraded by the UPS (Yoshii et al., 2011), preceded the formation of autophagosomes/autolysosomes containing mitochondrial remnants (Figure S2A).
Degradation of selected mitochondrial proteins by the UPS has been reported not only to precede, but also to be necessary for mitophagy in non-erythroid cells (Chan et al., 2011; Tanaka et al., 2010; Wei et al., 2017). Indeed, although UPS inhibition did not affect the number of autophagic vacuoles (Figure S2B), it significantly decreased the incorporation of mitochondria into autophagosomes (Figure S2C) and blocked the removal of SRMPs by autophagy (Figures 3B and S1A). These results indicate that proteasomal degradation of selected mitochondrial proteins is critical to trigger mitophagy in human proerythroblasts. Of note, mtDNA is removed by autophagy as its degradation i) follows the kinetic of SRMPs and ii) is sensitive to both UPS and autophagy inhibitors (Figures S2D and S2E).
To further confirm these results, we obtained blood samples from three patients with CANDLE syndrome carrying different additive loss-of-function mutations in UPS subunits (Brehm et al., 2015). Similar to MG132-treated cells, proerythroblasts generated from CANDLE patient’s PBMCs displayed defective mitochondrial removal in the absence of a concomitant maturation delay and in correlation with in vitro reduced UPS activity (Figures 3F, S2F and S2G). Interestingly, a significant fraction (55%) of Mito+ RBCs were detected in the blood of the patient whose proerythroblasts showed the lowest UPS activity in vitro (Figure 3G).
Studies in rabbit reticulocytes highlighted that intramitochondrial proteins (i.e., IMM and matrix proteins) can be degraded by the cytosolic UPS through the effect of 15-Lipoxygenase (encoded by ALOX15 gene). This enzyme permeabilizes mitochondrial membranes and permits the cytosolic proteolytic machinery to access the matrix compartment (Grullich et al., 2001; van Leyen et al., 1998). Accordingly, shRNA-mediated ALOX15 knockdown abrogated the degradation of both luminal RRMPs (COXIV) and SRMPs (HSP60) without affecting the removal of RRMPs facing the cytosol (TOMM40) (Figures 3H and S2H). Similar results were obtained with the ALOX15 inhibitor eicosatetraynoic acid (ETYA) (Figure S2I). These data support a role of ALOX15 in granting the cytosolic UPS access to luminal proteins, a necessary step to complete mitophagy.
HIF-2α-mediated metabolic reprogramming is upstream of UPS activation during human erythropoiesis
Pluripotent stem cells (PSCs) switch from glycolysis to mitochondrial oxidative phosphorylation (OXPHOS) as they differentiate (Ito and Suda, 2014). However, little is known about the regulation of glucose and mitochondrial metabolism during proerythroblast differentiation. Hints that UPS activity might be regulated by cell metabolism have recently emerged (Jagoe et al., 2002; Meul et al., 2020; Zhang et al., 2003). We then explored whether proerythroblasts, similar to PSCs, undergo a metabolic switch from glycolysis toward OXPHOS as they differentiate, and if this process is required to increase UPS activity.
Consistent with a preference for anaerobic respiration, extracellular acidification rate (ECAR), both basal and after oligomycin treatment (GR; glycolytic reserve), as well as lactate production were highest in proerythroblasts (0 h) and decreased upon differentiation (24 h) (Figures 4A and 4B). Accordingly, ECAR in differentiating proerythroblasts responded minimally to glucose (Figure S3A). To assess mitochondrial respiration, we exposed the cells to oligomycin and FCCP; this reveals the maximal respiratory capacity (MRC). Differentiating proerythroblasts showed high MRC and spare respiratory capacity (SRC), measured as the difference between basal and maximal oxygen consumption rate (OCR) after FCCP, as well as OCR/ECAR ratio, all indicative of increased commitment to OXPHOS (Figures 4C and 4D).
Figure 4. HIF-2α-mediated metabolic reprogramming is upstream of UPS activation during human erythropoiesis.

(A) Quantification of ECAR (n=5) and glycolytic reserve (n=3) in human proerythroblasts before (0 h) and after (24 h) differentiation. (B) Relative lactate levels in the medium of human proerythroblasts before (0 h) and after (24 h) differentiation. (n=4). (C) Quantification of maximal and spare respiratory capacity in human proerythroblasts before (0 h) and after (24 h) differentiation. (n=4). (D) OCR/ECAR ratio in human proerythroblasts before (0 h) and after (24 h) differentiation. (n=4). (E) OCR response to UK5099, oligomycin (Oligo), FCCP and antimycin A + rotenone (R/A) in human proerythroblasts 24 h post-differentiation. F) Relative chymotrypsin-like UPS activity in proerythroblasts differentiated in the presence or absence of UK5099. (n=3). Representative Western blot analysis (G) and quantification (H) of selected mitochondrial proteins in proerythroblasts differentiated in the presence or absence of UK5099 or DMOG. (I) Representative native gel analysis of UPS complexes from native cell lysates of proerythroblasts before (0 h) and after 24 h or 48 h post differentiation. The resolution of double-capped (30S) and single-capped (26S) UPSs is indicated together with the 20S UPS complexes. (J) Relative chymotrypsin-like UPS activity in proerythroblasts differentiated in the presence or absence of sodium L-lactate. (n=3). (K) Kla levels on immunoprecipitated UPS from proerythroblasts before (0 h) and after differentiation (24 h) in the presence or absence of sodium L-lactate. (L) Representative Western blot analysis of the expression kinetics of HIF-2α and BNIP3 in proerythroblasts differentiated in the presence or absence of DMOG. # denotes non specific bands. (M) Relative chymotrypsin-like UPS activity in proerythroblasts differentiated in the presence or absence of DMOG. (n=3). In (E, G, I, K and L) results are representative of at least three independent experiments. Data are means ± SEM. [Two-tailed unpaired Student t test].
To confirm these observations, we perfomed unbiased intracellular metabolomic profiling of proerythroblasts before and after differentiation. Undifferentiated proerythroblasts (0 h) transformed significant segments of the metabolome in opposite direction compared to their differentiating counterparts (24 h). In the glycolysis pathway, differentiating proerythroblasts significantly increased D-glucose, suggesting lower hexokinase (HK) flux and decreased input flux into glycolysis. This was also evident from the lower levels of downstream glycolytic intermediates. At the final step of glycolysis, differentiating cells significantly decreased lactate and increased acetyl-CoA, suggesting a reduced lactate dehydrogenase (LDHA) flux and an enhanced pyruvate dehydrogenase (PDH) flux (Figure S3B).
Consistent with a metabolic switch away from glycolysis, levels of glycolytic enzymes (HK2, GAPDH, PGAM1, PKLR, PKM, LDHA, LDHB) and glucose transporters (SLC2A3) were significantly downregulated during the differentiation process (Figures S3C and S3D). Taken together, these results support that an energetic demand increase during terminal erythroid differentiation requires a switch from glycolysis toward OXPHOS.
To establish a link between mitochondrial activity and UPS activation, we differentiated proerythroblasts in the presence of the pyruvate carrier (MPC) inhibitor UK5099 as pyruvate is the main substrate oxidized by mitochondria in proerythroblasts (Figure S3E). As previously reported (Zhong et al., 2015), UK5099 dampened the metabolic switch by increasing glycolysis and attenuating OXPHOS (Figures 4E and S3F). Furthermore, this treatment significantly reduced UPS activation and mitochondrial removal (Figures 4F, 4G and 4H), thus supporting that UPS activity, in proerythroblasts, is under metabolic control. Similar results were obtained when mitochondrial respiration was disrupted with FCCP or with the ATP synthase inhibitor oligomycin (Figures S3G and S3H).
To further investigate the underlying mechanism required for metabolic activation of the UPS, we first analyzed whether the metabolic switch affects the expression of UPS subunits and/or Nrf2, a transcription factor that upregulates UPS activity (Chapple et al., 2012). However, their overall expression was not significantly altered (Figure S3I).
In addition to increasing UPS abundance, cells adjust UPS-mediated degradation by regulating UPS assembly. We then used native gels with immunoblotting to resolve the different active UPS complexes in the cells. This method allows the discrimination into free 20S UPSs, single- (26S) and double-capped (30S) UPS complexes (Rousseau and Bertolotti, 2018). Differentiating proerythroblasts showed increased amount of assembled single- and double-capped UPS complexes, whereas the level of free 20S UPSs was slightly but not significantly increased (Figure 4I).
Post-translational modifications (PTM) are recognized to be an important underlying mechanism for the regulation of UPS assembly (Kors et al., 2019). Metabolites such as acetate, succinate and malonate are involved in PTM of lysine residues (Hirschey and Zhao, 2015). Exposure of purfied UPS to each of these metabolites, however, did not significantly affect the chymotrypsin-like activity (Figure S3J). More recently, lactate-derived lysine lactylation (Kla) has been identified as a new metabolite-induced PTM (Zhang et al., 2019). To analyze the impact of this glycolytic end-product on UPS activity, we exposed purified UPS to different concentrations of sodium L-lactate or L-lactic acid. Interestingly, the chymotrypsin-like activity was significantly reduced upon treatment with both of these metabolites (Figure S3J). Indeed, proerythroblasts cultured in the presence of sodium L-lactate showed a reduction of UPS activation (Figure 4J). To investigate whether changes in Kla were involved in the regulation of UPS activity by lactate, we examined the dynamics of this PTM in both total proteins and immunoprecipitated UPS. Following the decrease in intracellular lactate (Figure S3B), both total protein as well as UPS Kla levels were reduced during proerythroblast differentiation and this was prevented when sodium L-lactate was added to the culture (Figures 4K and S3K). Thus, lactate levels regulate the metabolic switch-mediated UPS activation by modulating UPS Kla levels.
When oxygen is limited, cells tend to favor anaerobic glycolysis over OXPHOS. This metabolic switch is mediated through the oxygen-sensitive transcription factors HIFs. HIFs promote an oxidative metabolism to glycolysis switch through transcriptional activation of genes that regulate glucose uptake and glycolysis. They also promote the expression of PDKs, which prevent pyruvate from entering the TCA cycle, thus blocking OXPHOS (Lee et al., 2020a). Consistent with the possibility that HIFs regulate the erythroblast metabolic switch, HIF-2α as well as its transcriptional target BNIP3 and a set of well-defined HIFs target genes were all downregulated during proerythroblast differentiation (Figures 4L and S3L). Indeed, the hypoxic mimetic drug dimethyloxalylglycine (DMOG), which prevents HIF- 2α degradation, blunted glycolysis to OXPHOS conversion (Figures 4L and S3M). In accordance with an impaired metabolic switch, DMOG-treated cells showed both decreased UPS activation and mitochondrial removal (Figures 4G, 4H and 4M). These findings support that HIF- 2α degradation regulates the metabolic switch away from glycolysis and towards OXPHOS during proerythroblast differentiation.
Impaired HIF-2α-mediated metabolic switch and UPS activation in SLE erythroblasts
To uncover the mechanism underlying defective mitochondrial removal in SLE, we generated RBCs from patient’s PBMCs. Similar to our ex vivo findings, two groups of SLE patients emerged according to whether their proerythroblasts removed (hereinafter referred to as Remover – R) or not (hereinafter referred to as Non Remover – NR) mitochondria during in vitro differentiation (Figures 5A, 5B and S4A). Importantly, the NR phenotype is associated with high SLEDAI score but not with delayed maturation, a feature of Type I IFN signaling (Means and Krantz, 1993), or with reduced expression of erythroid-associated transcription factors (Figures S4B, S4C and S4D). Furthermore, both mitochondrial mass (measured by mitochondrial DNA copy number) and biogenesis, assessed by the expression of i) PGC1α (Puigserver et al., 1998) or ii) transcripts involved in mtDNA replication (Falkenberg, 2018), were similar between SLE R and NR proerythroblasts (Figures S4E, S4F and S4G).
Figure 5. Impaired HIF-2α-mediated metabolic switch and UPS activation in SLE erythroblasts.
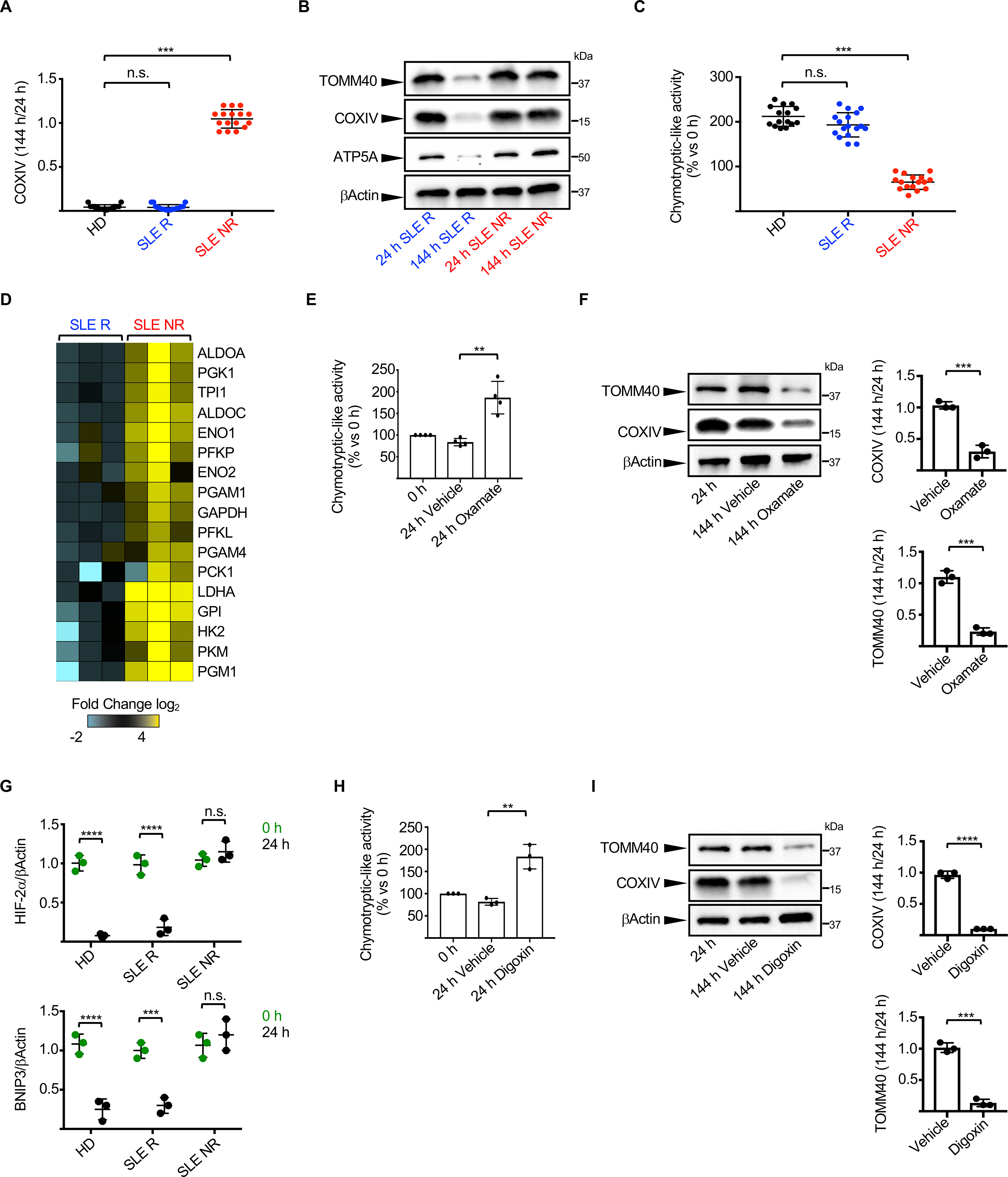
(A) Ratio of COXIV levels in HD (n=15), SLE Removers (R; n=17) and SLE Non-Removers (NR; n=16) proerythroblasts 144 h and 24 h post-differentiation. (HD – Healthy donor). (B) Representative Western blot analysis of selected mitochondrial proteins in proerythroblasts from one representative SLE R and one representative NR 24 h and 144 h post-differentiation. (C) Relative chymotrypsin-like UPS activity in HD (n=15), SLE R (n=17) and SLE NR (n=16) proerythroblasts 24 h post-differentiation. (D) Differential gene expression analysis between SLE R (n=3) and NR (n=3) of selected genes involved in the glycolytic and gluconeogenesis pathways. Data are normalized to SLE R. Relative chymotrypsin-like UPS activity (E, n=4) and levels of selected mitochondrial proteins (F, n=3) in SLE NR proerythroblasts differentiated in the presence or absence of Sodium Oxamate. (G) Quantification of HIF-2α and BNIP3 protein levels in SLE R and NR proerythroblasts. Data are normalized to βActin. (n=3). Relative chymotrypsin-like UPS activity (H) and levels of selected mitochondrial proteins (I) in SLE NR proerythroblasts differentiated in the presence or absence of Digoxin. (n=3). In (B, F and I) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A) and (C); Two-tailed unpaired Student t test in (E), (F), (G), (H) and (I)].
As we have linked UPS activation with mitochondrial removal, we next explored whether impaired activation of the UPS was a feature of NR SLE proerythroblasts. Indeed, proerythroblasts from these patients failed to increase UPS activity upon differentiation (Figure 5C).
Decreased in UPS subunits or lysine-48 linked polyubiquitin conjugates (K48-Poly Ub), a well-established signal for UPS-mediated protein degradation, are among the most common mechanisms associated with reduced UPS activity (Collins and Goldberg, 2017). However, levels of both UPS subunits and total K48-Poly Ub, which mirrors mitochondrial polyubiquitin conjugates (Lavie et al., 2018), were similar in NR and R proerythroblasts (Figures S4H and S4I). Thus, in accordance with previous findings (Levin et al., 2018; Lokireddy et al., 2015; Vilchez et al., 2012), reduced UPS activity in SLE NR is not associated with defective formation of K48-Poly Ub, as the ubiquitination machinery is properly activated in this group of patients.
In line with our observation that a switch from glycolysis to OXPHOS is required to trigger UPS activation in proerythroblasts, both the gene expression and bioenergetic profiles of NR showed decreased glycolytic to OXHPOS conversion 24 h post-differentiation compared to R proerythroblasts (Figures 5D and S4J). Indeed, differentiation of NR proerythroblasts in the presence of sodium oxamate, a drug that specifically inhibits glycolysis and promotes OXPHOS (Ait-Ali et al., 2015; Haschemi et al., 2012; Lee et al., 2020b; Son et al., 2013; Valvona and Fillmore, 2018) restored UPS activation and mitochondrial removal (Figures 5E and 5F).
Recently, reduced UPS activation has been linked to defective glycolysis to OXPHOS conversion due to Complex-I (C-I) downregulation (Meul et al., 2020). However, the levels of C-I subunits were comparable between R and NR proerythroblasts (Figure S4K). Futhermore, aspartate supplementation, which restored UPS activity in case of C-I dysfunction (Meul et al., 2020), did not rescue UPS activation in the latter group of patients (Figure S4L).
We then examined whether defective degradation of HIF-2α may be upstream of the impaired metabolic switch observed in NR proerythroblasts. Accordingly, HIF-2α, BNIP3 and a set of HIFs target genes were not down-regulated during NR proerythroblast differentiation (Figures 5G and S4M). Furthermore, UPS activation and mitochondrial removal were restored in NR proerythroblasts in the presence of the HIF inhibitor digoxin (Zhang et al., 2008) (Figures 5H and 5I). These data confirm the role of HIF-2α-mediated metabolic switch in UPS activation and mitochondrial removal during human erythropoiesis and link a defect in this pathway with the presence of Mito+ RBCs in SLE.
Mito+ RBCs and opsonizing anti-RBC antibodies drive Type I IFN production in vitro and correlate with the IFN signature in SLE patients
Reticuloendothelial M⏀ are involved in the silent clearance of aged, oxidized and IgG-opsonized RBCs (Klei et al., 2017). Phagocytosis of cells carrying excessive amount of danger associated molecular patterns (DAMPs) has been shown, however, to stimulate Type I IFN production in trans (Ahn et al., 2018). As mtDNA is a powerful DAMP (Grazioli and Pugin, 2018), we surmised that ingestion of Mito+ RBCs by M⏀ might contribute to the IFN response observed in SLE.
To address this possibility, we first co-cultured in vitro generated M⏀ with RBCs obtained under three different conditions: aged, oxidized and IgG- opsonized. While all three conditions significantly increased RBCs phagocytosis, opsonization was both the most efficient and the most relevant to an autoantibody-mediated disease like SLE (Figure S5A). We, therefore, selected it for the next set of studies.
Compared to opsonized Mito− RBCs, opsonized Mito+ RBCs triggered significantly higher production of TFNα as well as IP-10, a chemokine downstream of IFN signaling (Figure 6A). Importantly, both the kinetics of RBC uptake and degradation as well as the levels of phagolysosomal acidification were comparable in M⏀ that engulfed opsonized Mito− or Mito+ RBCs (Figures S5B, S5C, S5D and S5E), suggesting that these processes are not involved in the pro-inflammatory phenotype induced by the latter (Martinez et al., 2011).
Figure 6. Mito+ RBCs and opsonizing anti-RBC antibodies drive Type I IFN production in vitro and correlate with the SLE IFN signature.
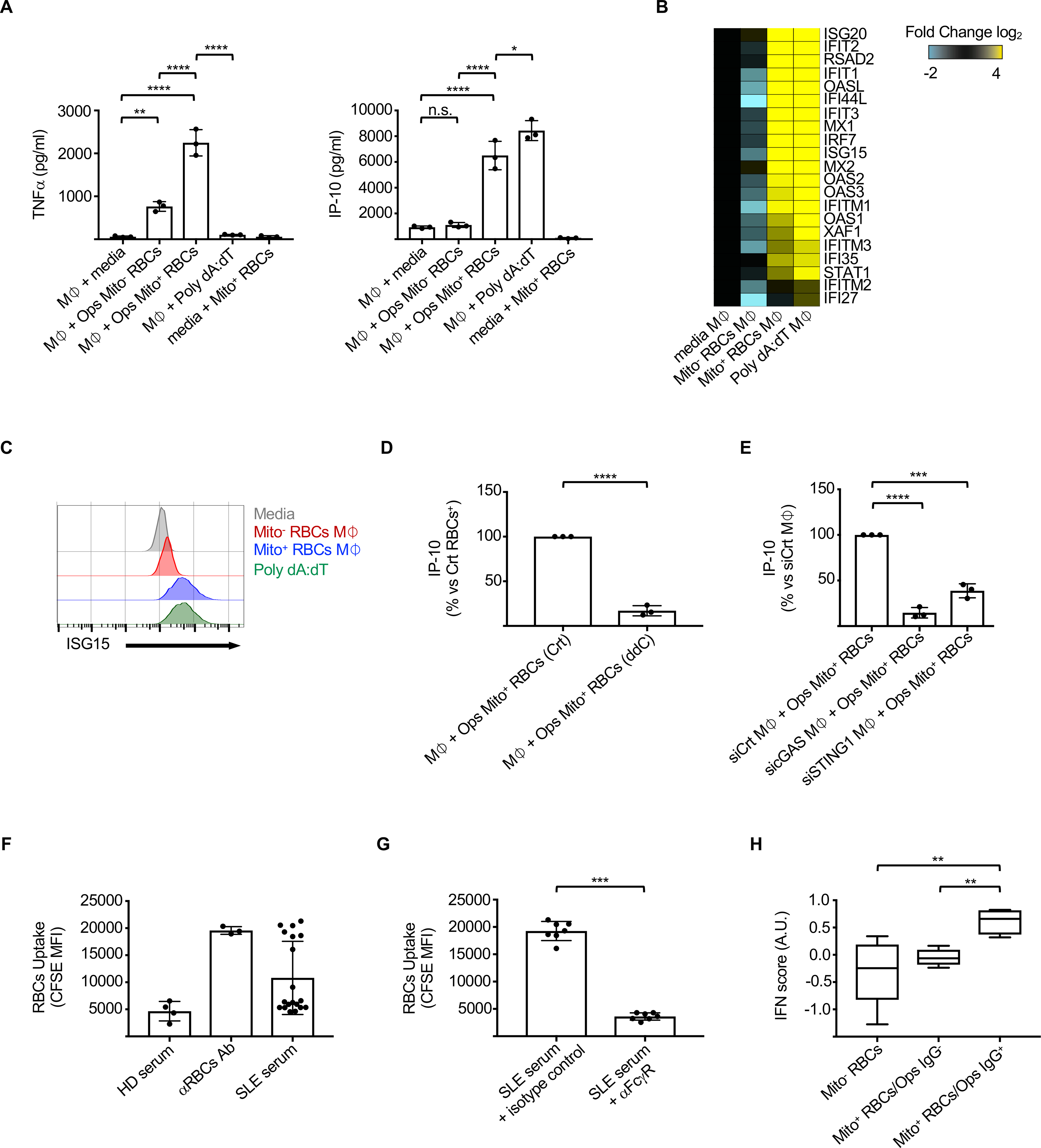
(A) Levels of TNFα and IP-10 in the supernatants of M⏀ cultured with medium, opsonized (Ops) Mito− or Mito+ RBCs or Poly dA:dT. (n=3). Heat map of differentially expressed interferon-stimulated genes (ISGs) and (B) intracellular ISG15 protein levels (C) in M⏀ phagocytized opsonized Mito− or Mito+ RBCs (Mito− or Mito+ RBCs M⏀). Poly dA:dT is positive control. (D) Normalized IP-10 levels in the supernatants of M⏀ cultured with opsonized Mito+ RBCs generated in the presence or absence or 2′3′-dideoxycytidine (ddC). (n=3). (E) Normalized IP-10 levels in the supernatants of M⏀ transfected with the indicated siRNA and then cultured with opsonized Mito+ RBCs. (n=3). (F) Phagocytosis of CFSE-labeled HD RBCs, treated as indicated, by M⏀. (n=5 HD serum; n= 19 SLE serum). (HD – Healthy donor). (G) Phagocytosis of CFSE-labeled HD RBCs, pretreated with SLE serum, in the presence of isotype control or anti-FcγR antibody. (n=7). (H) IFN score in SLE patients with Mito− RBCs (n=4) or with Mito+ RBCs that carry (Ops IgG+; n=4) or not (Ops IgG−; n=6) opsonizing antibodies in their sera. In (C) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A), (E) and (H); Two-tailed unpaired Student t test in (D) and (G)].
Consistent with the secreted cytokine profile, M⏀ that had ingested opsonized Mito+ RBCs (hereinafter Mito+ RBCs M⏀), but not those that ingested opsonized Mito− RBCs (hereinafter Mito− RBCs M⏀), upregulated the expression of ISGs (Figures 6B, 6C, S5F and S5G). ISG expression was downstream of Type I IFNs, as conditioned supernatant from Mito+ RBCs M⏀ failed to induce ISGs expression upon Type I IFN receptor blockade (Figure S5H).
The role of Mito+ RBC-derived mtDNA in M⏀ activation was next explored by generating Mito+ RBCs in the presence of 2’ 3’-dideoxycytidine (ddC), which inhibits mtDNA replication (Dalakas et al., 2001) (Figure S5I). Treatment with ddC drastically decreased IP-10 production by M (Figure 6D). MtDNA can engage TLR9 or cGAS to trigger Type I IFN (Caielli et al., 2016; West et al., 2015). As human M⏀ do not express TLR9 (Jarrossay et al., 2001), we focused on the role of cGAS (Motwani et al., 2019). M⏀ knockdown of cGAS, STING1 or IRF3 largely abrogated IP-10 production (Figures 6E, S5J and S5K). These data indicate that Mito+ RBC-derived mtDNA is a potent stimulus for Type I IFN production by M⏀ in trans in a cGAS-dependent manner.
To address the in vivo relevance of our findings, we first classified SLE patients according to the presence of Mito+ RBCs in whole blood and the expression of an ISG transcriptional signature in their ex-vivo PBMCs (Banchereau et al., 2016). In addition, we assessed the opsonizing capacity of the patient sera through the ability to enhance HD RBC phagocytosis in vitro (Fendel et al., 2007). As expected, a significantly higher phagocytic rate was observed when HD RBCs were pre-treated with sera from a fraction (36.8%) of SLE patients (Figures 6F and S5L). This effect was FcγR-dependent, supporting the role of opsonizing antibodies in this process (Figure 6G). Importantly, higher ISG transcriptional activity was detected in patients harboring the combination of Mito+ RBCs and opsonizing serum (Figures 6H and S5M), thus supporting the contribution of this pathway in the induction and/or amplification of Type I IFN production in a distinct group of SLE patients.
DISCUSSION
Emerging evidence suggests that mitochondrial and metabolic abnormalities play important roles in SLE pathogenesis. Thus, activation of mTORC1 and increased mitochondrial metabolic rate lead to abnormal CD4+ T cell activation in SLE patients (Morel, 2017; Oaks et al., 2016). Furthermore, succinate-driven mtROS accumulation is a feature of Th10 cells, an extra-follicular CD4+ T cell helper subopulation expanded in SLE (Caielli et al., 2019). Finally, dysregulation of mitochondrial quality control in SLE neutrophils induces the extrusion of pro-inflammatory ox mtDNA (Caielli et al., 2016; Lood et al., 2016).
Mitochondrial clearance is a feature of RBC maturation (An et al., 2015). Previous mechanistic studies on how mitochondria are removed during murine erythropoiesis focused on mitophagy without considering the role of the UPS (Schweers et al., 2007; Zhang and Ney, 2010). Here, we provide evidence that the UPS and mitophagy jointly play an essential role in mitochondrial removal during human erythropoiesis, with UPS activation being both upstream and necessary for mitophagy to proceed. These results are in agreement with previous studies in non-erythroid cells showing that UPS-dependent degradation of selected mitochondrial proteins is required for mitophagy (Chan et al., 2011; Tanaka et al., 2010; Wei et al., 2017). As a proof of concept, proerythroblasts either treated with UPS inhibitors or generated from the PBMCs of CANDLE syndrome patients, fail to remove their mitochondria. Importantly, we show that Type I IFN is not responsible for mitochondrial retention in SLE RBCs, but rather delays proerythroblast maturation, as reported (Means and Krantz, 1993).
Metabolic control of UPS activity has been described before (Jagoe et al., 2002; Zhang et al., 2003). Recently, a direct connection between metabolites and UPS assembly through PTM has emerged (Kors et al., 2019; Meul et al., 2020). Thus, addition of O-GlcNAc to the UPS ATPase subunit Rpt2 increased the stability of proteins by reducing their UPS-dependent degradation (Zhang et al., 2003). Furthermore, histone deacetylase (HDAC) inhibitors increased the acetylation of cardiac UPS subunits and led to enhanced proteolytic capacity (Wang et al., 2013). Here we show that lactate-induced PTM of UPS subunits, in the form of lysine lactylation, reduces UPS activity by affecting its assembly. Emerging evidence shows that lactate has regulatory functions in both innate and adaptive immune cells and induces changes in gene expression by regulating histone Kla levels (Haas et al., 2016; Zhang et al., 2019). A role of lactate-induced Kla in the regulation of UPS activity has, however, never been described before.
Mito+ RBCs have been reported in patients with Pearson’s and Rett syndrome but their potential pathogenic role was not addressed (Ahlqvist et al., 2015; Sbardella et al., 2017).
We here demonstrate that the concomitant presence of Mito+ RBCs and opsonizing antibodies in a significant group of SLE patients correlates with an enhaced blood ISG transcriptional signature. Opsonizing antibodies might directly recognize surface determinants on RBCs, or be associated with complement fixing immune complexes (ICs) through complement receptor type 1 (CR1), also on the RBC surface. Interestingly, while the direct binding of antibodies to RBCs is a hallmark of autoimmune hemolytic anemia (AHA), IC binding to CR1 is not associated with hemolysis (Hanaoka et al., 2018). Indeed, patients in our cohort displaying both Mito+ RBCs and opsonizing antibodies did not suffer from overt hemolysis, supporting the latter scenario. Patients with CANDLE syndrome also harbor an exuberant ISG signature of unknown origin (Brehm et al., 2015). The contribution of Mito+ RBCs to this signature and/or to the overall CANDLE inflammatory phenotype will require further studies.
Both genetic dissection of monogenic lupus and longitudinal immune-monitoring of SLE patients with sporadic disease support a broad activation of the IFN system (Banchereau et al., 2016; Omarjee et al., 2019). Yet, therapeutic blockade of Type I IFN or its receptor yielded negative or only modest beneficial results, respectively, in phase III clinical trials (Goulden and Isenberg, 2020; Kalunian, 2016; Morand et al., 2020). There is therefore an urgent need to classify patients according to dysregulated pathways upstream of IFN production, which could lead to improved molecular stratification and ultimately therapeutic success. Extracellular nucleic acids (NAs) have been extensively associated with Type I IFN production in SLE. Thus, NA-containing ICs are internalized via FcγRs and engage endosomal sensors in pDCs (Guiducci et al., 2010; Means et al., 2005; Vallin et al., 1999). Furthermore, circulating microparticle-bound chromatin DNA induces Type I IFN production by pDCs and expansion of DNA-reactive B cells (Sisirak et al., 2016; Soni et al., 2020). Finally, cell-free NAs, including neutrophil-derived ox mtDNA, are abundant in SLE fluids (Duvvuri and Lood, 2019; Lee et al., 2014; Truszewska et al., 2020) and upon internalization into pDCs, trigger Type I IFN production in a TLR9 dependent manner (Caielli et al., 2016). Recently, cytosolic leakage of mtDNA through VDAC pores was shown to induce cell-intrinsic cGAS activation in lupus (Kim et al., 2019). However, mtDNA is unlikely to induce cell-intrinsic cGAS activation in Mito+RBCs, as these cells do not express DNA sensors (Bryk and Wisniewski, 2017). Rather, the pathway that we here uncover relies on cytosolic sensing within macrophages of cell-associated mtDNA that fails to be removed from mature RBCs. This pathway is enhanced by the presence of anti-RBC antibodies, a feature of SLE (Hair et al., 2020), and it might be amenable to a variety of therapeutic approaches, including metabolic intervention.
Limitations of study
There are several limitations to the current study that will require further investigation. First, the inteferogenic capacity of Mito+ RBCs is limited to in vitro-based experiments. In fact, the role of Type I IFN in murine models of SLE remains controversial (Moore and Putterman, 2020), and whether the UPS plays a role in mitochondrial removal during murine erythropoiesis has not been addressed (Sandoval et al., 2008). Importantly, the mechanism(s) controlling HIF-2α degradation/stabilization during human erythropoiesis need to be resolved. Finally, the nature and origin of opsonizing anti-RBCs autoantibodies in pediatric SLE patients remain to be investigated.
STAR METHODS TEXT
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Virginia M. Pascual (vip2021@med.cornell.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA-seq data generated during this study are available in the GEO database under the accession number GSE163073. Other data generated in the current study are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Children and adolescents with SLE and JDM were enrolled from the Pediatric Rheumatology clinics at Texas Scottish Rite Hospital for Children and Children’s Medical Center in Dallas (TX). Study procedures were followed in accordance with protocols approved by the Institutional Review Boards at the University of Texas Southwestern Medical Center (092010–167), at the Baylor Scott and White Research Institute (011–200 and 007–221) and Weill Cornell Medical College (1711018757). Assent was obtained from patients between 7 and 17 years of age. Patients were evaluated by a standardized protocol during routine clinic visits every three months and more frequently if clinical symptoms warranted evaluation. Blood was collected in ACD tubes (Beckton Dickson) and laboratory measurements were recorded. Clinical disease activity was assessed using the SLE Disease Activity Index (SLEDAI-2K). Relevant clinical information is depicted in Table S1. CANDLE subjects were enrolled as part of the National Institutes of Health institutional review board-approved natural history studies and were genetically defined as previously reported (Brehm et al., 2015). Relevant information pertaining to these patients is depicted in Table S2. Healthy donor Leukopaks were obtained from the New York Blood Center.
METHOD DETAILS
Proerythroblast expansion and differentiation from human PBMCs.
Proerythroblasts were expanded and differentiated starting with Peripheral Blood Mononuclear cells (PBMCs). PBMCs were isolated by density purification using Lymphoprep™ (Stem Cell Technologies) and cells were cultured as described previously (van den Akker et al., 2010) but with modifications. Briefly, PBMCs were seeded at 1×107 cells/mL in expansion medium (phase 1), consisting of StemSpan™ Serum-Free Expansion Medium (SFEM, Stem Cell Technologies) supplemented with SCF (100 ng/mL; Stem Cell Technologies), PROCRIT® (2 U/mL; Janssen), dexamethasone (1 μM; Sigma–Aldrich), IGF-1 (40 ng/mL; Stem Cell Technologies), cholesterol-rich lipids (40 μg/mL; Sigma–Aldrich) and IL-3 (10 ng/mL; Stem Cell Technologies). After 5 days, erythroblasts were purified by density purification using Lymphoprep™ and a lineage depletion step (Lineage Cell Depletion Kit; Miltenyi Biotech) was performed to ensure complete removal of lineage positive cells at this stage. Cells were reseeded in expansion medium without IL-3 (expansion phase - phase 2) and kept between 1.5 and 2×106/mL by daily counting and medium changes. After 8 days of expansion cells were washed with phosphate-buffered saline (PBS) and reseeded in differentiation medium consisting of SFEM medium (differentiation phase - phase 3) supplemented with PROCRIT® (10U/mL; Janssen), insulin (10 μg/mL; Santa Cruz Biotech), Human Peripheral Blood Plasma (3%; Stem Cell Technologies), 3,3′,5-Triiodo-L-thyronine (1 μM; Sigma–Aldrich), IGF-1 (40ng/mL; Stem Cell Technologies) and holo-transferrin (0.5 mg/mL; Santa Cruz Biotech). Cells were collected every 24 h for phenotypic characterization and kept between 2–4×106 cells/mL by daily counting and medium changes. UK5099 (2 μM; Santa Cruz Biotech), Dimethyloxalylglycine (DMOG, 100 μM; Santa Cruz Biotech), Sodium Oxamate (25 mM; Santa Cruz Biotech), Digoxin (100 nM; Sigma–Aldrich), MG132 (5 μM; Santa Cruz Biotech), Bortezomib (5 μM; Santa Cruz Biotech), Bafilomycin A1 (100 nM; Sigma–Aldrich), E-64d/Pepstatin (1 μg/mL – 5 μM; Sigma–Aldrich), Eicosatetraynoic acid (ETYA, 100 μM; Sigma–Aldrich), Sodium L-lactate (5 mM; Sigma–Aldrich), FCCP (1 μM; Santa Cruz Biotech), Oligomycin A (1 μM; Santa Cruz Biotech), L-Aspartic acid (10 mM; Sigma–Aldrich) or Human Recombinant Interferon α2β (IFNα2β 1000 U/mL; Schering Corp.) were added to the culture during phase 3.
Proerythroblast expansion and differentiation from human CD34+ cells.
Frozen human bone marrow CD34+ hematopoietic stem and progenitor cells were obtained from Stem Cell Technologies. These cells were differentiated into mature erythroid cells utilizing a three-phase culture protocol. In phase 1 (day 1 – 5), cells were cultured at a density of 105/mL in Iscove’s Modified Dulbecco’s Media (IMDM; Thermo-Fisher) supplemented with 3% Human Peripheral Blood Plasma (Stem Cell Technologies), 3% human serum (Sigma–Aldrich), 3 IU/mL heparin (Sigma–Aldrich), 10 μg/mL insulin (Sigma–Aldrich), 200 μg/mL holo-transferrin (Santa Cruz Biotech), 2 U/mL PROCRIT® (Jenssen), 10 ng/mL SCF (Stem Cell Technologies) and 10 ng/mL IL-3 (Stem Cell Technologies). In phase 2 (day 5 – 10), IL-3 was omitted from the medium. In phase 3 (day 10 – 16), cells were cultured at a density of 2×106/mL, with both IL-3 and SCF omitted from the medium, the holotransferrin concentration was increased to 1 mg/mL and the PROCRIT® concentration was increased to 10 U/mL. Cells were cultured at 37°C and 5% CO2.
shRNA transfection of human CD34+ cells.
shRNA transfection of CD34+ cells was performed using the Nucleofector™ 2b Device (Lonza) according to the protocol provided by the manufacturer, with some modifications. Briefly, after 7 days of expansion, 4×106 CD34+ cells were gently resuspended in 100 μl of Human CD34+ Nucleofector™ Solution (Lonza), mixed with 1 μg of shALOX15 plasmid (Santa Cruz Biotech) and pulsed with the program U- 01. As a control a nontargeting shRNA control plasmid (shCrt, Santa Cruz Biotech) was used. Immediately after nucleofection, cells were transferred into prewarmed phase 2 medium for 48 h. Cells stably expressing shRNA were selected with puromycin (1 μg/mL; Santa Cruz Biotech) for an additional 48 h before starting the differentiation process in phase 3 medium. Transfection and puromycin selection efficiency were validated by using 1 μg of copGFP Control Plasmid (Santa Cruz Biotech).
Monocyte enrichment and differentiation into macrophages (M⏀).
PBMCs were isolated from healthy donor Leukopaks with Lymphoprep™ gradient centrifugation. Monocytes were then isolated with EasySep™ Human Monocyte Enrichment Kit (Stem Cell Technologies) following the manufacturer protocol and then differentiated into M⏀ in macrophages-SFM medium (Thermo-Fisher) supplemented with GM-CSF (50 ng/mL; Sargramostim, Partner Therapeutics). At day 3, non-adherent cells were removed by replacing the culture media. Cells were used for co-culture experiments at day 5.
Forward transfection of primary human M⏀.
Forward transfection of primary human M⏀ was performed as described previously (Troegeler et al., 2014). Briefly, at day 3 of M⏀ culture, the culture media was aspirated gently to ensure the removal of floating cells. Fresh complete Roswell Park Memorial Institute medium (cRPMI) was added to the attached cells to a final volume of 250 μl per well in 24- well plates. The ON- TARGETplus SMARTpool siRNA targeting cGAS, STING1 and IRF3 were purchased from Horizon. For the formation of the lipid- siRNA complexes, 110.25 μl of warm RPMI medium (without FBS) were mixed with 11 μl of HiPerFect tranfection reagent (Qiagen). siRNA was added at the final concentration of 200 nm, and the lipid- siRNA mix incubated for 20 min at room temperature during which the mixture was inverted gently a few times to enhance the formation of complexes. At the end of the incubation period, 125 μl of the lipid- siRNA complexes were added drop- wise onto the media of adherent M⏀ accompanied by a gentle rocking of the plate. The plates were then placed at 37°C and 5% CO2. After 6 h, cRPMI supplemented with GM- CSF (final concentration of 50 ng/ mL) was added 500 μl. After 48 h, cells were assessed for protein suppression efficiency by immunoblotting and used for co-culture experiments.
In vitro phagocytosis assay.
Mito+ and Mito− RBCs were generated from HD PBMCs in the presence or absence of MG132 (5 μM) respectively. To deplete mtDNA, 2′,3′-Dideoxycytidine (ddC, 100 μM; Sigma–Aldrich) was added during the culture together with MG132. RBCs were then opsonized with anti-human RBC antibody (diluted 1:250; Rockland), stained with 2 μM CFSE (Thermo-Fisher) or with 100 ng/mL pHrodo™ Red SE (Thermo-Fisher) and then co-cultured with macrophages at a 1:2 (M⏀ to RBCs ratio) for 18 h in cRPMI medium with 10% FBS. For some experiments, RBCs were oxidized with Copper (II) sulfate (0.5 mM; Santa Cruz Biotech) and Sodium L-ascorbate (100 mM; Santa Cruz Biotech) or aged with Calcium Chloride (2.5 mM; Santa Cruz Biotech) and A23187 (0.5 μM; Sigma–Aldrich) for 30 min at 37°C. The supernatants were collected and frozen for the quantification of cytokine levels with the CBA assay (BD Bioscience). Non-engulfed RBCs were removed by three washes with cold PBS, phagocytes were collected using trypsin and EDTA and assessed for phagocytosis by flow cytometry or immunofluorescence microscopy upon counterstaining the actin filaments with AlexaFluor 647 Phalloidin (Thermo-Fisher) and nuclear DNA with Hoechst 33342 (Thermo-Fisher). Pre-complexed Poly dA:dT/ LyoVec™ (referred as Poly dA:dT. 1 μg/mL; Invivogen) was used as a positive control.
M⏀ culture with conditioned supernatants.
M⏀ were co-cultured with opsonized Mito+ RBCs at 1:2 M⏀ to RBCs ratio for 18 h in cRPMI medium with 10% FBS. Supernatants were harvested, filtered (0.1 μm) to remove any contaminating debris and then added to untreated M⏀ for 18 h. IFN neutralization was achieved by pre-treating the M⏀ with anti-human IFNAR2 neutralizing antibody (20 μg/mL; PBL assay science) for 30 min at 37°C.
Flow cytometry.
For PBMC-derived erythroid cell characterization, 3×105 cells were collected every 24 h during the phase 3 of the differentiation process and analyzed by flow cytometry for CD36 (Biolegend), CD235a (Biolegend), CD71 (Biolegend), CD49d (Biolegend), and CD233 (Miltenyi Biotec) expression. For mitochondrial content in mature RBCs, PBMCs were removed from peripheral blood by density centrifugation using Lymphoprep™ (Stem Cell Technologies). The cell pellet was washed four times (3000 rpm, 8 min, 4°C) with PBS supplemented with 2 mM EDTA and 10 mM Glucose. The cell pellet was then re-suspended in PBS supplemented with 10 mM Glucose and MitoTracker Deep Red (MTDR, 200 nM; Thermo-Fisher) and incubated for 30 min at 37°C. The cell pellet was then washed four times with PBS, treated with Fc Block (BD Bioscience) and stained with the antibodies. Where described, FCCP (Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; 500 nM) was added during the incubation with MTDR. The fluorescence intensity of MTDR was measured, in the mature CD71− CD235a+ RBCs population, by flow cytometry. For the detection of intracellular ISG15, 5×105 cells were resuspended in 4% paraformaldehyde. After 20 min incubation on ice, cells were washed with FACS buffer (0.5% FBS and 0.09% sodium azide in PBS). Cells were then stained with anti-human ISG15/UCRP PE-conjugated antibody (R&D systems) in Cytofix/Cytoperm Fixation and Permeabilization Solution (BD Biosciences) following the manufacturer’s instructions. After 30 min of incubation on ice, cells were washed with ice cold Cytofix/Cytoperm buffer, resuspended in FACS buffer and analyzed. For TMRM staining, cells were cultured with media or FCCP (1 μM) for 24 h and then resuspended in PBS containing TMRM (10 μM) and incubated at 37°C in the dark for 30 min. Cells were washed with PBS and analyzed. Samples were acquired using FACS Canto II™ (BD Biosciences) or FACS Melody™ (BD Biosciences). Data were analyzed using Flowjo software.
Immunofluorescent microscopy (IF).
For blood RBC staining, PBMCs were removed from peripheral blood by density centrifugation using Lymphoprep™ (Stem Cell Technologies). After removal of contaminating reticulocytes and neutrophils with CD71 and CD15 microbeads (Miltenyi Biotech) respectively, RBCs were pelleted, washed twice in PBS containing 5 mM glucose and then fixed for 5 min in 100% methanol. Cells were rinsed three times then permeabilized in PBS containing 0.1 M glycine (rinsing buffer) plus 0.1% Triton X-100 for 5 min. To ensure complete neutralization of unreacted aldehydes, cells were then incubated in rinsing buffer at room temperature for 30 min. After incubation, all nonspecific binding was blocked by incubation again for 60 min in blocking buffer (PBS containing 0.05 mM glycine, 0.2% fish skin gelatin and 0.05% sodium azide). Staining was performed by using anti-COXIV (1 μg/mL; Abcam) and anti-SLC4A1/Band 3 (1 μg/mL; Abcam) antibodies diluted in blocking buffer. Isotype specific anti–mouse or anti–rabbit Alexa Fluor 488 or Alexa Fluor 568 (Thermo-Fisher) were used as secondary antibodies. After labeling, resuspended RBCs were allowed to attach to cover slips coated with poly-L-lysine, and the cover slips were mounted by using ProLong™ Gold Antifade Mountant (Thermo-Fisher). For in vitro generated erythroblasts staining, cells were settled on poly-L-lysine coated glass coverslips and then fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were permeabilized with 0.05% Triton X-100 in PBS for 5 min at room temperature and then treated with blocking buffer for 30 min at room temperature. Staining was performed by using anti-COXIV (1 μg/mL; Abcam), anti-ATP5A (1 μg/mL; Abcam) and anti-LAMP1 (1 μg/mL; Abcam) antibodies diluted in blocking buffer. Isotype specific anti–mouse or anti–rabbit Alexa Fluor 488 or Alexa Fluor 568 (Thermo-Fisher) were used as secondary antibodies. The samples were examined with a Leica TCS SP5 confocal laser-scanning microscope equipped with a 63x/1.4 oil objective. Fiji/ImageJ software was used for analysis. The percentage of co-localization was calculated from the Manders’ Overlap Coefficient using the Fiji/ImageJ Coloc2 plugin.
Transmission electron microscopy (TEM).
Cells were fixed by immersion in 2.5% glutaraldehyde (Sigma–Aldrich), 4% paraformaldehyde (Sigma–Aldrich), 0.02% picric acid (Sigma–Aldrich) in 0.1 M sodium cacodylate buffer (pH 7.3) and fixation was continued overnight at 4°C. Cells were washed and post-fixed with 1% Osmium tetroxide (Sigma–Aldrich) and 1.5% Potassium ferricyanide (III) (Sigma–Aldrich) aqueous solution for 60 min. After washing, cells were stained with 1.5% aqueous Uranyl acetate (Sigma–Aldrich) solution for 30 min and then dehydrated through graded ethanol series, followed by acetonitrile (15 minutes per step). Samples were then embedded in Embed 812 resin (Electron Microscopy Sciences) and sections were cut at 55–60 nm (silver-gold) using a Diatome diamond knife (Diatome) on a Leica Ultracut S ultramicrotome (Leica). Sections were then contrasted with lead citrate and viewed on a JSM 1400 electron microscope (JEOL) operated at 100 kV. Digital images were recorded using a Veleta 2K x2K camera (EMSIS).
Determination of lactate concentration.
The lactate concentration was measured using a Lactate Assay Kit (Sigma–Aldrich) according to the manufacturer’s instructions.
Quantification of mtDNA abundance/ mtDNA copy number.
The abundance of mtDNA was assessed by Real-Time PCR as described previously (Caielli et al., 2016). Briefly, total DNA was extracted with DNAzol™ Reagent (Thermo-Fisher). DNA concentration was assessed with Quant-iT™ PicoGreen™ dsDNA Assay Kit (Thermo-Fisher). Real-Time PCR was performed with Power SYBR™ Green PCR Master Mix (Thermo-Fisher) with 3 ng of isolated DNA and 0.5 μM of the following primers: mtDNA encoded NADH dehydrogenase subunit 1 (ND1; 5′-GCATTCCTAATGCTTACCGAAC-3′ and 5′-AAGGGTGGAGAGGTTAAAGGAG-3′); genomic DNA encoded Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 5′-AGGCAACTAGGATGGTGTGG-3′ and 5′-TTGATTTTGGAGGGATCTCG-3′).
RNA preparation and sequencing library preparation.
RNA preparation and sequencing were performed at the Baylor Research Institute (BRI) Genomic Core Facility. Total RNA was isolated from cell lysates using the RNeasy Micro Kit (Qiagen), including on-column deoxyribonuclease digestion, and was analyzed for quality using the RNA 6000 Pico Kit (Agilent Technologies). Poly(A)-enriched next-generation sequencing library construction was performed using the KAPA mRNA Hyper Prep Kit (KAPA Biosystems) with 100 ng of input total RNA and 15 amplification cycles according to the manufacturer’s protocol. The quality and quantity of the individual libraries was assessed using the High Sensitivity DNA Kit (Agilent). The libraries were equimolar pooled and an additional 1X bead clean-up was used to remove adapter dimer. The pooled libraries were quantitated using the KAPA Library Quantification Kit, Universal (KAPA Biosystems) and sequenced on an Illumina NextSeq 500 with paired-end 75-base-pair (bp) read lengths.
RNA sequencing data processing and analysis.
Quality control of raw reads was performed with FastQC. Reads were aligned to the reference human genome (GRCh38) using HISAT2 after quality and adapter trimming by Cutadapt. After sorting binary alignment map files by name using samtools, the HTSeq-count program was used to quantify total numbers of read counts mapped to the genome. The RNA sequencing data analysis was performed in the R programming language. The DESeq2 R package was used for size factor and dispersion estimation calculation and differential gene expression analysis.
Modular analysis and INF score.
Modular analysis was performed on RNA sequencing data obtained from SLE PBMCs using the second generation of a modular framework analysis as previously described (Chaussabel et al., 2008). IFN score was extrapolated from the values of the three “Interferon Modules” (M1.2, M3.4, and M5.12). Module transcript content and annotations are available online at http://websle.com/.
SDS-PAGE and Western blot.
Cells were washed in PBS, and then lysed with RIPA Lysis and Extraction Buffer (Thermo-Fisher) in the presence of Halt Protease and Phosphates Inhibitor Cocktail (Thermo-Fisher). Samples were incubated on ice for 30 min and then centrifuged (13,000 g for 10 min at 4°C) to remove cellular debris. The supernatants, containing the protein fraction, were collected and stored at −80°C until further analysis. Protein concentration was estimated using the BCA Protein Assay Kit (Thermo-Fisher) following the manufacturer’s instructions. 10–30 μg of cell lysate was resuspended in 5× Lane Marker Reducing Sample Buffer (Thermo-Fisher), boiled for 5 min at 100°C, and then subjected to electrophoresis with Mini-PROTEAN TGX precast electrophoresis gels (Bio-Rad Laboratories). The proteins were then transferred to PVDF membranes with Trans-Blot Turbo Transfer System (Bio-Rad Laboratories), blocked for 1 h in 5% nonfat dry milk in TBST (Tris Buffered Saline containing 0.1% Tween-20), and incubated overnight at 4°C with the primary antibodies. After washing in TBST, the membranes were incubated for 1 h at room temperature with Poly HRP-conjugated anti–rabbit or anti–mouse IgG (Thermo-Fisher). ECL™ Western Blotting Detection Reagent (Sigma–Aldrich) were used for detection. Digital images were acquired with ChemiDoc MP System equipped with Image Lab Software (Bio-Rad Laboratories). To measure autophagic flux cells were treated, 3 h before harvesting, with the lysosomotropic agents bafilomycin A1 (100 nM; Sigma–Aldrich) and then assessed for LC3B-II levels by Western blot.
Bioenergetics measurements.
3×105 cells (erythroblasts) were seeded on a Seahorse XF Mini Analyzers plates (Agilent Technologies) coated with Corning® Cell-Tak™ Cell and Tissue Adhesive (Corning). For oxidative profiling, the assay medium was (Agilent Technologies) containing 25 mM glucose, 2 mM glutamine and 1 mM sodium pyruvate and manually adjusted to pH 7.4 with NaOH 0.1 N. To measure mitochondrial respiration, 1 μM Oligomycin A (to block ATP synthesis), 2 μM FCCP (to uncouple mitochondria proton pumping) and 1 μM antimycin A + rotenone (to block the electron transport chain) were injected during measurement of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in a Seahorse XF Mini Analyzers (Agilent Technologies). For mitochondrial fuel test assay, the mitochondrial pyruvate carrier inhibitor UK5099 (2 μM), the glutaminase inhibitor BPTES (3 μM) or the carnitine palmitoyl-transferase 1A inhibitor Etomoxir (4 μM) were injected before oligomycin. For glycolytic profiling, the assay medium was Seahorse XF Base Medium (Agilent Technologies) containing 2 mM glutamine and manually adjusted to pH 7.4 with NaOH 0.1 N. Glycolysis, monitored as the ECAR, was measured after the addition of 10 mM D(+)Glucose.
UPS activity assay.
UPS activity was measured as previously described (Brehm et al., 2015). Briefly, erythroblasts were gently lysed in TSDG buffer (10 mM Tris-HCl, 1.1 mM MgCl2, 10 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 2 mM ATP, 10% (v/v) glycerol, pH 7.0) with 3 freeze-thaw cycles to prevent UPS denaturation. 10 μg cell of lysates were assayed for protease activity in assay buffer (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 2 mM ATP) with either 100 μM Suc-Leu-Leu-Val-Tyr-aminomethyl-coumarin (Suc-Leu-Leu-Val-Tyr-AMC, Enzo Life Sciences) for chymotryptic-like activity, BZ-Val-Gly-Arg-AMC (Enzo Life Sciences) for tryptic-like activity, or Z-Leu-Leu-Glu-AMC (Enzo Life Sciences) for caspase-like activity. UPS activity was determined by measuring, over time, the fluorescence of AMC liberated from peptides substrate (Ex/Em: 380nm/460nm). For purified UPS activity, 2 μg of Human 26S Proteasome Protein (R&D Systems) were incubated, in TSDG buffer, with the indicated concentrations of sodium acetate, sodium succinate, sodium malonate, sodium L-lactate, L-lactic acid or with epoxomicin (10 μM; Sigma–Aldrich) for 30 min at room temperature. The chymotryptic-like activity was then measured as described above.
Native gel electrophoresis.
15 μg protein of TSDG lysates were mixed with 5x native loading buffer (250 mM Tris, 50% Glycerol, 0.01% Bromphenol blue) and loaded onto commercially available 3%–8% gradient NuPAGE Novex Tris-acetate gels (Thermo-Fisher). Native UPS complexes were separated at 150 V for 4 h at 4°C. After native gel electrophoresis, the gels were incubated for 15 min in a solubilization buffer (66 mM Na2CO3, 2% SDS, 1.5% β-mercaptoethanol) at room temperature in order to facilitate the subsequent blotting of the proteins using the previously described Western blot technique.
Purification of UPS complexes.
Intact UPS complexes were isolated from TSDG lysates (15 μg) with the Proteasome Purification Kit (Enzo) according to the protocol provided by the manufacturer. The immunoprecipiated UPS complexes were then subjected to SDS-PAGE and immunoblotting using the previously described Western blot technique. To avoid interference of heavy and light antibody chains, Clean-Blot™ IP Detection Reagent (Thermo-Fisher) was used instead of Poly HRP-conjugated secondary antibody.
Detection of opsonizing antibodies in SLE sera.
The presence of opsonizing antibodies in SLE sera was assessed by using a modified version of an in vitro based erythrophagocytosis assay (Fendel et al., 2007). Briefly, HD EDTA- blood was washed twice with 1 mL PBS and 50 μl of blood pellet were incubated in 950 μl PBS containing 2 nM CFSE (Thermo-Fisher) for 10 min at room temperature. The staining was stopped by adding 1 mL FCS, followed by three washes with PBS. Subsequently, 10 μl of blood pellet were washed and incubated for 1 h in HD or SLE sera diluted in PBS (25% v/v). After incubation, samples were washed extensively in PBS and 2 μl blood pellet (corresponding to 2×107 RBCs) were used for incubation with monocyte-derived M⏀. RBCs and M⏀ were co-incubated, at a 1:10 M⏀ to RBCs ratio, for 18 h at 37°C and 5% CO2. Were described, M⏀ were pre-treated with anti-FcγR neutralizing antibody (20 μg/mL; Biolegend). Non-ingested RBCs were lysed in 0.9 mL ice- cold distilled water for 1 min. Thereafter, 0.1 mL 10× PBS was added to restore isotonicity. Cell debris and hemoglobin was removed by four washes in PBS and cells were analyzed by flow cytometry.
Proteomic analysis.
Proteomic analysis was performed in the Proteomics Resource Center (PRC) at The Rockefeller University. 10 ug of protein lysates (prepared in PBS with 0.5 % Triton X100) were added to 20 uL of 50 mM ammonia-carbonate (ABC) buffer and 120 uL of ice-cold acetone. Proteins were then precipitated o/n at −20°C. Precipitates were dissolved in 30 uL 8 M Urea/10 mM DTT/50 mM ABC buffer and sonicated in an ultrasound bath for 45 min at RT. For proteins alkylation, 5 uL of 190 mM 2-iodoacetamide (Sigma–Aldrich) was added and samples were incubated for 90 min in the dark. Samples were then treated with 0.5 ug of Endoproteinase Lys-C (New England Biolabs) and 0.8 ug of trypsin (Sigma–Aldrich) and digested for 5 h. Samples were solid phase extracted and analyzed using a 50 min nano flow gradient on Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer (Thermo-Fisher). Data were analyzed using Proteome Discoverer™ Software (Thermo-Fisher) and MaxQuant Software (MaxQuant).
Polar metabolomics analysis.
Polar metabolomic analysis was performed at the Weill Cornell Medicine (WCM) Meyer Cancer Center Proteomics & Metabolomics Core Facility. Metabolites were extracted from 2×106 cells using 80% methanol. Pre-cooled 80% methanol (1 mL) was added to each sample and kept in −80°C overnight. Samples were then centrifuged at 4°C for 15 minutes at 14,000 rpm. The supernatants were extracted. Targeted LC/MS analyses were performed on a Q Exactive Orbitrap mass spectrometer (Thermo-Fisher) coupled to a Vanquish UPLC system (Thermo-Fisher). The Q Exactive operated in polarity-switching mode. A Sequant ZIC-HILIC column (2.1 mm i.d. × 150 mm, Merck) was used for separation of metabolites. Flow rate was set at 150 μL/min. Buffers consisted of 100% acetonitrile for mobile A, and 0.1% NH4OH - 20 mM CH3COONH4 in water for mobile B. Gradient ran from 85% to 30% A in 20 min followed by a wash with 30% A and re-equilibration at 85% A. Metabolites were identified on the basis of exact mass within 5 ppm and standard retention times. Relative metabolite quantitation was performed based on peak area for each metabolite. All data analysis was done using in-house written scripts.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as means ± SEM. Statistical analyses were performed using GraphPad Prism (version 9, GraphPad Software). Comparison of two parameters was performed using two-tailed unpaired Student t test. Multiparameter analyses of cell-based studies were performed with two-way analysis of variance (ANOVA) followed by Tukey post hoc test for multiple comparisons. Statistical significance was defined as ****p<0.0001; ***p<0.001; **p<0.005; *p<0.05; n.s.=not significant.
Supplementary Material
Figure S1. Activation of the UPS is critical for mitophagy during human erythropoiesis, Related toFigure 3. (A) Quantification of RPL29, COXIV and ATP5A levels in proerythroblasts cultured in the presence of UPS (MG132 or bortezomib) or autophagy (Bafilomycin A1 or E-64d/Pepstatin) inhibitors. (n=3). (B) Representative immunofluorescence images and quantification (n=15 cells/time point) showing colocalization of the lysosomal maker LAMP1 with COXIV (RRMP) or ATP5A (SRMP) in proerythroblasts collected at different stages of terminal erythroid maturation. White arrowheads indicate colocalization. Scale bar = 2 μm. (C) Relative tryptic- and Caspase-like proteasome activities over time during proerythroblast maturation. (n=4). In (B) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A) and (D); Two-tailed unpaired Student t test in (C)].
Figure S2. Activation of the UPS is critical for mitophagy during human erythropoiesis, Related toFigure 3. Representative electron micrograph of proerythroblasts 24/48 h (left) 72/96 h (middle) and 120/144 h (right) post-differentiation. Star indicates swollen mitochondria with ruptured membranes and loss of cristae structure. Black arrowheads indicate autophagosome. M=mitochondria. Scale bar = 500 nm. (B) Number of autophagosomes per cell in proerythroblasts differentiated for 120/144 h with or without MG132. (n=5). (C) Representative electron micrograph of proerythroblasts differentiated for 120/144 h with or without MG132. Black arrowhead indicates autophagosome with mitochondrial cargo. White arrowhead indicates empty autophagosome. M=mitochondria. Percentage of autophagosomes containing mitochondrial cargo is also shown. (n=5). Scale bar = 200 nm. (D) Relative mtDNA abundance in proerythroblasts collected at different stages of terminal erythroid maturation. (n=3). (E) Relative mtDNA abundance in proerythroblasts cultured in the presence of UPS (MG132 or bortezomib) or autophagy (Bafilomycin A1 or E-64d/Pepstatin) inhibitors. (n=3). (F) Relative chymotrypsin-like proteasome activity in three CANDLE patient proerythroblasts 24 h post-differentiation. (HD – Healthy donor). (G) Percentage of CD71+/CD36+ cells in HD and three CANDLE patient proerythroblasts 24 h and 144 h post-differentiation. (H) ALOX15 protein expression demonstrating shRNA-mediated knockdown efficiency. (I) Levels of selected mitochondrial proteins in proerythroblasts differentiated in the presence or absence of eicosatetraynoic acid (ETYA). (n=3). In (A) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (D) and (E); Two-tailed unpaired Student t test in (B), (C) and (I)].
Figure S3. HIF-2α-mediated metabolic reprogramming is upstream of proteasome activation during human erythropoiesis, Related toFigure 4. (A) ECAR response to glucose in human proerythroblasts before (0 h) and after (24 h) differentiation. (n=3). (B) Heat map analysis of intracellular metabolites in human proerythroblasts before (0 h) and after (24 h) differentiation. (C) Heat map analysis showing genes involved in glycolysis and gluconeogenesis in proerythroblasts 0, 24 and 48 h post-differentiation. (D) Western blot analysis of selected glycolytic enzymes in proerythroblasts 0, 24 and 48 h post-differentiation. # denotes non specific bands. (E) Normalized spare respiratory capacity values, of human proerythroblasts 24 h post-differentiation, in the presence of UK5099, Etomoxir or BPTES. (n=3). (F) ECAR response to UK5099 in human proerythroblasts 24 h post-differentiation. (G) Tetramethylrhodamine (TMRM) staining on proerythroblasts treated with vehicle of FCCP for 24 h. (H) Levels of selected mitochondrial proteins in proerythroblasts differentiated in the presence or absence of FCCP or Oligomycin. (I) Western blot analysis of Nrf2 and selected proteasome subunits in proerythroblasts 0, 24 and 48 h post-differentiation. (J) Relative chymotrypsin-like proteasome activity of purified proteasome treated as described. (n=3). (K) Western blot analysis of Kla levels in proerythroblasts whole cell lysate (WCL) before (0 h) and after (24 h) differentiation. (n=3). (L) Heat map analysis of RNA-seq data showing a set of HIFs target genes in proerythroblasts 0, 24 and 48 h post-differentiation. (M) ECAR and OCR responses of human proerythroblasts differentiated for 24 h in the presence or absence of DMOG. In (D), (F), (G), (H), (I) and (M) results are representative of at least three independent experiments. Data are means ± SEM. [Two-tailed unpaired Student t test in (A) and (K); One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (E) and (J)].
Figure S4. Impaired HIF-2α-mediated metabolic switch and proteasome activation in SLE erythroblasts, Related toFigure 5. (A) Ratio of TOMM40 protein levels in HD (n=5), SLE R (n=5) and SLE NR (n=5) proerythroblasts 144 h and 24 h post-differentiation. (HD – Healthy donor). (B) SLEDAI values of SLE R (n=6) and SLE NR (n=7). (C) Representative FACS plot and quantification of cell surface expression of CD71 and CD36 in SLE R, SLE NR and SLE R differentiated in the presence of IFNα2β. (n=3). (D) Relative expression (R.E.) of erythroid-associated transcription factors in SLE R and NR proerythroblasts 24 h post-differentiation. (n=3). (E) MtDNA copy number in in SLE R and NR proerythroblasts 0 and 24 h post-differentiation. (n=3). (F) Representative Western blot analysis of PGC1α levels in differentiating SLE R and NR proerythroblasts. G) R.E. of proteins associated with mtDNA replication in SLE R and NR proerythroblasts 24 h post-differentiation. (n=3). (H) Gene expression analysis, between SLE R (n=3) and NR (n=3), of selected proteasome subunits. (I) Representative Western blot analysis of lysine-48 linked polyubiquitin conjugates (K48-Poly Ub) in WCL from HD, SLE R and SLE NR proerythroblasts 24 h post-differentiation. (J) ECAR and OCR response, in SLE R and NR proerythroblasts 24 h post-differentiation, to oligomycin (Oligo), FCCP and antimycin A + rotenone (R/A). (K) R.E. of proteasome subunits in SLE R and NR proerythroblasts 24 h post-differentiation. (n=3). (L) Relative chymotrypsin-like UPS activity in SLE NR proerythroblasts differentiated in the presence or absence of Aspartate. (M) Differential gene expression analysis, between SLE R (n=3) and NR (n=3), of selected HIFs target genes. In (F), (I) and (J) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A); Two-tailed unpaired Student t test in (B), (C), (D), (E) and (G)].
Figure S5. Mito+ RBCs and opsonizing anti-RBC antibodies drive Type I IFN production in vitro and correlate with the SLE IFN signature, Related toFigure 6. (A) Quantification of M⏀ phagocytized CFSE-labeled Mito+ RBCs that were treated as indicated. (Mito+ RBCs = untreated RBCs; Ops Mito+ RBCs = RBCs opsonized with anti-RBCs antibodies; Aged Mito+ RBCs = A23187/CaCl2 treated RBCs; Oxidized Mito+ RBCs = sodium ascorbate/CuSO4 treated RBCs). (n=3) (B) Quantification of M⏀ phagocytized opsonized CFSE-labeled Mito− or Mito+ RBCs at various time points at 1:2 M⏀:RBCs ratio or at various ratios for 18 h. (n=3). (C) Quantification of M⏀ with intracellular fragmented CFSE fluorescence upon phagocytosis of opsonized CFSE-labeled Mito− or Mito+ RBCs. (n=5 fields). Scale bar = 10 μm. (D) Quantification of residual CFSE fluorescence in M⏀ incubated with opsonized CFSE-labeled Mito− or Mito+ RBCs for 24 h, then chased for the indicated time points. (n=3). (E) Quantification of pHrodo fluorescence in M⏀ incubated with opsonized pHrodo-labeled Mito− or Mito+ RBCs. (n=3). (F) FACS gating strategies of M⏀ engulfing opsonized CFSE-labeled Mito− or Mito+ RBCs. (G) Western blot analysis for selected ISGs in M⏀ phagocytized opsonized Mito− or Mito+ RBCs (Mito− or Mito+ RBCs M⏀). Poly dA:dT is positive control. # denotes non specific bands. (H) Western blot analysis on M⏀ treated with the supernatant from M⏀ phagocytized opsonized Mito+ RBCs in the presence of anti-IFNAR2 antibody. # denotes non specific bands. (I) Relative mtDNA abundance in Mito+ RBCs generated with or without 2′,3′-dideoxycytidine (ddC). (n=3). (J) cGAS, STING1 and IRF3 protein expression demonstrating shRNA-mediated knockdown efficiency. (K) Normalized IP-10 levels in the supernatants of M⏀ transfected with IRF3 siRNA and then cultured with opsonized Mito+ RBCs. (n=3). (L) Representative histogram plot for phagocytosis of CFSE-labeled HD RBCs, treated as indicated, by M⏀. (M) Expression levels of three IFN-regulated modules in SLE patients with Mito− RBCs (n=4) or with Mito+ RBCs that carry (Ops IgG+; n=4) or not (Ops IgG−; n=6) opsonizing antibodies in their sera. In (G) and (H) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A) and (M); Two-tailed unpaired Student t test in (B), (C), (D), (E), (I) and (K)].
HIF2α degradation promotes UPS activation and mitophagy during human erythropoiesis.
Defective HIF2α degradation leads to accumulation of Mito+ RBCs in SLE patients.
Uptake of opsonized Mito+ RBCs by macrophages induces IFN production via cGAS/STING.
Highest ISG scores define SLE patients with Mito+ RBCs and opsonizing antibodies.
ACKNOWLEDGMENTS
We are grateful to our SLE and JDM patients, their families, the healthy individuals who participated in our study and the members of the Pediatric Rheumatology Clinics at Texas Scottish Rite Hospital for Children and the Children’s Medical Center in Dallas,TX. We thank Dr. A. Darehshouri (UTSW medical center – Dallas) and Dr. L. Cohen-Gould (WCMC – New York) for help with transmission electron microscopy studies, and the Genomics Core at the Baylor Scott and White Research Institute (Dallas, TX) for RNA sequencing. This work was supported by grants NIAMS CORT P50AR070594 Center for Lupus Research (to V.P. and J.F.B.), NIAID NIH U19 AI082715 (to V.P.) and funds from the Drukier Institute for Children’s Health at Weill Cornell Medicine.
DECLARATION OF INTERESTS
V.P. has received consulting honoraria from Sanofi, Astra-Zeneca and Moderna and is the recipient of a research grant from Sanofi and a contract from Astra Zeneca. J.F.B. is a member of the S.A.B. of Neovacs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahlqvist KJ, Leoncini S, Pecorelli A, Wortmann SB, Ahola S, Forsstrom S, Guerranti R, De Felice C, Smeitink J, Ciccoli L, et al. (2015). MtDNA mutagenesis impairs elimination of mitochondria during erythroid maturation leading to enhanced erythrocyte destruction. Nat Commun 6, 6494. [DOI] [PubMed] [Google Scholar]
- Ahn J, Xia T, Rabasa Capote A, Betancourt D, and Barber GN (2018). Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 33, 862–873 e865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Ali N, Fridlich R, Millet-Puel G, Clerin E, Delalande F, Jaillard C, Blond F, Perrocheau L, Reichman S, Byrne LC, et al. (2015). Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 161, 817–832. [DOI] [PubMed] [Google Scholar]
- An X, Schulz VP, Mohandas N, and Gallagher PG (2015). Human and murine erythropoiesis. Curr Opin Hematol 22, 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau R, Cepika AM, Banchereau J, and Pascual V (2017). Understanding Human Autoimmunity and Autoinflammation Through Transcriptomics. Annu Rev Immunol 35, 337–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, Cepika AM, Acs P, Turner J, Anguiano E, et al. (2016). Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 165, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betin VM, Singleton BK, Parsons SF, Anstee DJ, and Lane JD (2013). Autophagy facilitates organelle clearance during differentiation of human erythroblasts: evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy 9, 881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A, Reinhardt A, Almeida de Jesus A, et al. (2015). Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 125, 4196–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk AH, and Wisniewski JR (2017). Quantitative Analysis of Human Red Blood Cell Proteome. J Proteome Res 16, 2752–2761. [DOI] [PubMed] [Google Scholar]
- Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M, et al. (2016). Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213, 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, Banchereau R, Xu Z, Chandra M, Chung CH, et al. (2019). A CD4(+) T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med 25, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, and Chan DC (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple SJ, Siow RC, and Mann GE (2012). Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol 44, 1315–1320. [DOI] [PubMed] [Google Scholar]
- Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, Stichweh D, Blankenship D, Li L, Munagala I, et al. (2008). A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity 29, 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, and Goldberg AL (2017). The Logic of the 26S Proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas MC, Semino-Mora C, and Leon-Monzon M (2001). Mitochondrial alterations with mitochondrial DNA depletion in the nerves of AIDS patients with peripheral neuropathy induced by 2’3’-dideoxycytidine (ddC). Lab Invest 81, 1537–1544. [DOI] [PubMed] [Google Scholar]
- Duvvuri B, and Lood C (2019). Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front Immunol 10, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg M (2018). Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem 62, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendel R, Mordmuller B, Kreidenweiss A, Rudat A, Steur C, Ambrosch C, Kirstein M, Berdel WE, Kremsner PG, and Brandts C (2007). New method to quantify erythrophagocytosis by autologous monocytes. Cytometry A 71, 258–264. [DOI] [PubMed] [Google Scholar]
- Gladman DD, Ibanez D, and Urowitz MB (2002). Systemic lupus erythematosus disease activity index 2000. J Rheumatol 29, 288–291. [PubMed] [Google Scholar]
- Goulden B, and Isenberg D (2020). Anti-IFNalphaR Mabs for the treatment of systemic lupus erythematosus. Expert Opin Biol Ther, 1–10. [DOI] [PubMed] [Google Scholar]
- Grazioli S, and Pugin J (2018). Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front Immunol 9, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grullich C, Duvoisin RM, Wiedmann M, and van Leyen K (2001). Inhibition of 15-lipoxygenase leads to delayed organelle degradation in the reticulocyte. FEBS Lett 489, 51–54. [DOI] [PubMed] [Google Scholar]
- Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, Chan JH, Wright T, Punaro M, Bolland S, et al. (2010). TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465, 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, and Mauro C (2016). Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem Sci 41, 460–471. [DOI] [PubMed] [Google Scholar]
- Hair P, Goldman DW, Li J, Petri M, Krishna N, and Cunnion K (2020). Classical complement activation on human erythrocytes in subjects with systemic lupus erythematosus and a history of autoimmune hemolytic anemia. Lupus 29, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaoka H, Iida H, Kiyokawa T, Takakuwa Y, and Kawahata K (2018). A positive direct Coombs’ test in the absence of hemolytic anemia predicts high disease activity and poor renal response in systemic lupus erythematosus. Lupus 27, 2274–2278. [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al. (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab 15, 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, and Zhao Y (2015). Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol Cell Proteomics 14, 2308–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, and Suda T (2014). Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol 15, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagoe RT, Lecker SH, Gomes M, and Goldberg AL (2002). Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J 16, 1697–1712. [DOI] [PubMed] [Google Scholar]
- Jarrossay D, Napolitani G, Colonna M, Sallusto F, and Lanzavecchia A (2001). Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol 31, 3388–3393. [DOI] [PubMed] [Google Scholar]
- Kalunian KC (2016). Interferon-targeted therapy in systemic lupus erythematosus: Is this an alternative to targeting B and T cells? Lupus 25, 1097–1101. [DOI] [PubMed] [Google Scholar]
- Kim A, and Nemeth E (2015). New insights into iron regulation and erythropoiesis. Curr Opin Hematol 22, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, et al. (2019). VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klei TR, Meinderts SM, van den Berg TK, and van Bruggen R (2017). From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front Immunol 8, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kors S, Geijtenbeek K, Reits E, and Schipper-Krom S (2019). Regulation of Proteasome Activity by (Post-)transcriptional Mechanisms. Front Mol Biosci 6, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langefeld CD, Ainsworth HC, Cunninghame Graham DS, Kelly JA, Comeau ME, Marion MC, Howard TD, Ramos PS, Croker JA, Morris DL, et al. (2017). Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun 8, 16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie J, De Belvalet H, Sonon S, Ion AM, Dumon E, Melser S, Lacombe D, Dupuy JW, Lalou C, and Benard G (2018). Ubiquitin-Dependent Degradation of Mitochondrial Proteins Regulates Energy Metabolism. Cell Rep 23, 2852–2863. [DOI] [PubMed] [Google Scholar]
- Lee HT, Lin CS, Lee CS, Tsai CY, and Wei YH (2014). Increased 8-hydroxy-2’-deoxyguanosine in plasma and decreased mRNA expression of human 8-oxoguanine DNA glycosylase 1, anti-oxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leucocytes in patients with systemic lupus erythematosus. Clin Exp Immunol 176, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Chandel NS, and Simon MC (2020a). Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 21, 268–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Ji X, Nissim I, and Long F (2020b). Malic Enzyme Couples Mitochondria with Aerobic Glycolysis in Osteoblasts. Cell Rep 32, 108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A, Minis A, Lalazar G, Rodriguez J, and Steller H (2018). PSMD5 Inactivation Promotes 26S Proteasome Assembly during Colorectal Tumor Progression. Cancer Res 78, 3458–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokireddy S, Kukushkin NV, and Goldberg AL (2015). cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A 112, E7176–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, and Kaplan MJ (2016). Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love PE, Warzecha C, and Li L (2014). Ldb1 complexes: the new master regulators of erythroid gene transcription. Trends Genet 30, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, and Green DR (2011). Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 108, 17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Means RT Jr., and Krantz SB (1993). Inhibition of human erythroid colony-forming units by tumor necrosis factor requires beta interferon. J Clin Invest 91, 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, and Luster AD (2005). Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 115, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meul T, Berschneider K, Schmitt S, Mayr CH, Mattner LF, Schiller HB, Yazgili AS, Wang X, Lukas C, Schlesser C, et al. (2020). Mitochondrial Regulation of the 26S Proteasome. Cell Rep 32, 108059. [DOI] [PubMed] [Google Scholar]
- Moore E, and Putterman C (2020). Are lupus animal models useful for understanding and developing new therapies for human SLE? J Autoimmun 112, 102490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, et al. (2020). Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med 382, 211–221. [DOI] [PubMed] [Google Scholar]
- Morel L (2017). Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol 13, 280–290. [DOI] [PubMed] [Google Scholar]
- Motwani M, Pesiridis S, and Fitzgerald KA (2019). DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 20, 657–674. [DOI] [PubMed] [Google Scholar]
- Nguyen AT, Prado MA, Schmidt PJ, Sendamarai AK, Wilson-Grady JT, Min M, Campagna DR, Tian G, Shi Y, Dederer V, et al. (2017). UBE2O remodels the proteome during terminal erythroid differentiation. Science 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oaks Z, Winans T, Huang N, Banki K, and Perl A (2016). Activation of the Mechanistic Target of Rapamycin in SLE: Explosion of Evidence in the Last Five Years. Curr Rheumatol Rep 18, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omarjee O, Picard C, Frachette C, Moreews M, Rieux-Laucat F, Soulas-Sprauel P, Viel S, Lega JC, Bader-Meunier B, Walzer T, et al. (2019). Monogenic lupus: Dissecting heterogeneity. Autoimmun Rev 18, 102361. [DOI] [PubMed] [Google Scholar]
- Pickles S, Vigie P, and Youle RJ (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, and Spiegelman BM (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839. [DOI] [PubMed] [Google Scholar]
- Ronnblom LE, Alm GV, and Oberg KE (1991). Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann Intern Med 115, 178–183. [DOI] [PubMed] [Google Scholar]
- Rousseau A, and Bertolotti A (2018). Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol 19, 697–712. [DOI] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, and Wang J (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbardella D, Tundo GR, Campagnolo L, Valacchi G, Orlandi A, Curatolo P, Borsellino G, D’Esposito M, Ciaccio C, Cesare SD, et al. (2017). Retention of Mitochondria in Mature Human Red Blood Cells as the Result of Autophagy Impairment in Rett Syndrome. Sci Rep 7, 12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 104, 19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, Rashidfarrokhi A, Yeste A, Panea C, Chida AS, et al. (2016). Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 166, 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son MJ, Jeong BR, Kwon Y, and Cho YS (2013). Interference with the mitochondrial bioenergetics fuels reprogramming to pluripotency via facilitation of the glycolytic transition. Int J Biochem Cell Biol 45, 2512–2518. [DOI] [PubMed] [Google Scholar]
- Soni C, Perez OA, Voss WN, Pucella JN, Serpas L, Mehl J, Ching KL, Goike J, Georgiou G, Ippolito GC, et al. (2020). Plasmacytoid Dendritic Cells and Type I Interferon Promote Extrafollicular B Cell Responses to Extracellular Self-DNA. Immunity 52, 1022–1038 e1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TA (2003). Neutralizing interferon alpha as a therapeutic approach to autoimmune diseases. Cytokine Growth Factor Rev 14, 139–154. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, and Youle RJ (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191, 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troegeler A, Lastrucci C, Duval C, Tanne A, Cougoule C, Maridonneau-Parini I, Neyrolles O, and Lugo-Villarino G (2014). An efficient siRNA-mediated gene silencing in primary human monocytes, dendritic cells and macrophages. Immunol Cell Biol 92, 699–708. [DOI] [PubMed] [Google Scholar]
- Truszewska A, Wirkowska A, Gala K, Truszewski P, Krzemien-Ojak L, Perkowska-Ptasinska A, Mucha K, Paczek L, and Foroncewicz B (2020). Cell-free DNA profiling in patients with lupus nephritis. Lupus 29, 1759–1772. [DOI] [PubMed] [Google Scholar]
- Vallin H, Perers A, Alm GV, and Ronnblom L (1999). Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J Immunol 163, 6306–6313. [PubMed] [Google Scholar]
- Valvona CJ, and Fillmore HL (2018). Oxamate, but Not Selective Targeting of LDH-A, Inhibits Medulloblastoma Cell Glycolysis, Growth and Motility. Brain Sci 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Akker E, Satchwell TJ, Pellegrin S, Daniels G, and Toye AM (2010). The majority of the in vitro erythroid expansion potential resides in CD34(−) cells, outweighing the contribution of CD34(+) cells and significantly increasing the erythroblast yield from peripheral blood samples. Haematologica 95, 1594–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leyen K, Duvoisin RM, Engelhardt H, and Wiedmann M (1998). A function for lipoxygenase in programmed organelle degradation. Nature 395, 392–395. [DOI] [PubMed] [Google Scholar]
- Vilchez D, Boyer L, Morantte I, Lutz M, Merkwirth C, Joyce D, Spencer B, Page L, Masliah E, Berggren WT, et al. (2012). Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 489, 304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Fang C, Zong NC, Liem DA, Cadeiras M, Scruggs SB, Yu H, Kim AK, Yang P, Deng M, et al. (2013). Regulation of acetylation restores proteolytic function of diseased myocardium in mouse and human. Mol Cell Proteomics 12, 3793–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R Jr., Mishra P, and Levine B (2017). Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168, 224–238 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, and Mizushima N (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286, 19630–19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, and Kudlow JE (2003). O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115, 715–725. [DOI] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, et al. (2008). Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A 105, 19579–19586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Ney PA (2010). Reticulocyte mitophagy: monitoring mitochondrial clearance in a mammalian model. Autophagy 6, 405–408. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Li X, Yu D, Li X, Li Y, Long Y, Yuan Y, Ji Z, Zhang M, Wen JG, et al. (2015). Application of mitochondrial pyruvate carrier blocker UK5099 creates metabolic reprogram and greater stem-like properties in LnCap prostate cancer cells in vitro. Oncotarget 6, 37758–37769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Activation of the UPS is critical for mitophagy during human erythropoiesis, Related toFigure 3. (A) Quantification of RPL29, COXIV and ATP5A levels in proerythroblasts cultured in the presence of UPS (MG132 or bortezomib) or autophagy (Bafilomycin A1 or E-64d/Pepstatin) inhibitors. (n=3). (B) Representative immunofluorescence images and quantification (n=15 cells/time point) showing colocalization of the lysosomal maker LAMP1 with COXIV (RRMP) or ATP5A (SRMP) in proerythroblasts collected at different stages of terminal erythroid maturation. White arrowheads indicate colocalization. Scale bar = 2 μm. (C) Relative tryptic- and Caspase-like proteasome activities over time during proerythroblast maturation. (n=4). In (B) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A) and (D); Two-tailed unpaired Student t test in (C)].
Figure S2. Activation of the UPS is critical for mitophagy during human erythropoiesis, Related toFigure 3. Representative electron micrograph of proerythroblasts 24/48 h (left) 72/96 h (middle) and 120/144 h (right) post-differentiation. Star indicates swollen mitochondria with ruptured membranes and loss of cristae structure. Black arrowheads indicate autophagosome. M=mitochondria. Scale bar = 500 nm. (B) Number of autophagosomes per cell in proerythroblasts differentiated for 120/144 h with or without MG132. (n=5). (C) Representative electron micrograph of proerythroblasts differentiated for 120/144 h with or without MG132. Black arrowhead indicates autophagosome with mitochondrial cargo. White arrowhead indicates empty autophagosome. M=mitochondria. Percentage of autophagosomes containing mitochondrial cargo is also shown. (n=5). Scale bar = 200 nm. (D) Relative mtDNA abundance in proerythroblasts collected at different stages of terminal erythroid maturation. (n=3). (E) Relative mtDNA abundance in proerythroblasts cultured in the presence of UPS (MG132 or bortezomib) or autophagy (Bafilomycin A1 or E-64d/Pepstatin) inhibitors. (n=3). (F) Relative chymotrypsin-like proteasome activity in three CANDLE patient proerythroblasts 24 h post-differentiation. (HD – Healthy donor). (G) Percentage of CD71+/CD36+ cells in HD and three CANDLE patient proerythroblasts 24 h and 144 h post-differentiation. (H) ALOX15 protein expression demonstrating shRNA-mediated knockdown efficiency. (I) Levels of selected mitochondrial proteins in proerythroblasts differentiated in the presence or absence of eicosatetraynoic acid (ETYA). (n=3). In (A) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (D) and (E); Two-tailed unpaired Student t test in (B), (C) and (I)].
Figure S3. HIF-2α-mediated metabolic reprogramming is upstream of proteasome activation during human erythropoiesis, Related toFigure 4. (A) ECAR response to glucose in human proerythroblasts before (0 h) and after (24 h) differentiation. (n=3). (B) Heat map analysis of intracellular metabolites in human proerythroblasts before (0 h) and after (24 h) differentiation. (C) Heat map analysis showing genes involved in glycolysis and gluconeogenesis in proerythroblasts 0, 24 and 48 h post-differentiation. (D) Western blot analysis of selected glycolytic enzymes in proerythroblasts 0, 24 and 48 h post-differentiation. # denotes non specific bands. (E) Normalized spare respiratory capacity values, of human proerythroblasts 24 h post-differentiation, in the presence of UK5099, Etomoxir or BPTES. (n=3). (F) ECAR response to UK5099 in human proerythroblasts 24 h post-differentiation. (G) Tetramethylrhodamine (TMRM) staining on proerythroblasts treated with vehicle of FCCP for 24 h. (H) Levels of selected mitochondrial proteins in proerythroblasts differentiated in the presence or absence of FCCP or Oligomycin. (I) Western blot analysis of Nrf2 and selected proteasome subunits in proerythroblasts 0, 24 and 48 h post-differentiation. (J) Relative chymotrypsin-like proteasome activity of purified proteasome treated as described. (n=3). (K) Western blot analysis of Kla levels in proerythroblasts whole cell lysate (WCL) before (0 h) and after (24 h) differentiation. (n=3). (L) Heat map analysis of RNA-seq data showing a set of HIFs target genes in proerythroblasts 0, 24 and 48 h post-differentiation. (M) ECAR and OCR responses of human proerythroblasts differentiated for 24 h in the presence or absence of DMOG. In (D), (F), (G), (H), (I) and (M) results are representative of at least three independent experiments. Data are means ± SEM. [Two-tailed unpaired Student t test in (A) and (K); One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (E) and (J)].
Figure S4. Impaired HIF-2α-mediated metabolic switch and proteasome activation in SLE erythroblasts, Related toFigure 5. (A) Ratio of TOMM40 protein levels in HD (n=5), SLE R (n=5) and SLE NR (n=5) proerythroblasts 144 h and 24 h post-differentiation. (HD – Healthy donor). (B) SLEDAI values of SLE R (n=6) and SLE NR (n=7). (C) Representative FACS plot and quantification of cell surface expression of CD71 and CD36 in SLE R, SLE NR and SLE R differentiated in the presence of IFNα2β. (n=3). (D) Relative expression (R.E.) of erythroid-associated transcription factors in SLE R and NR proerythroblasts 24 h post-differentiation. (n=3). (E) MtDNA copy number in in SLE R and NR proerythroblasts 0 and 24 h post-differentiation. (n=3). (F) Representative Western blot analysis of PGC1α levels in differentiating SLE R and NR proerythroblasts. G) R.E. of proteins associated with mtDNA replication in SLE R and NR proerythroblasts 24 h post-differentiation. (n=3). (H) Gene expression analysis, between SLE R (n=3) and NR (n=3), of selected proteasome subunits. (I) Representative Western blot analysis of lysine-48 linked polyubiquitin conjugates (K48-Poly Ub) in WCL from HD, SLE R and SLE NR proerythroblasts 24 h post-differentiation. (J) ECAR and OCR response, in SLE R and NR proerythroblasts 24 h post-differentiation, to oligomycin (Oligo), FCCP and antimycin A + rotenone (R/A). (K) R.E. of proteasome subunits in SLE R and NR proerythroblasts 24 h post-differentiation. (n=3). (L) Relative chymotrypsin-like UPS activity in SLE NR proerythroblasts differentiated in the presence or absence of Aspartate. (M) Differential gene expression analysis, between SLE R (n=3) and NR (n=3), of selected HIFs target genes. In (F), (I) and (J) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A); Two-tailed unpaired Student t test in (B), (C), (D), (E) and (G)].
Figure S5. Mito+ RBCs and opsonizing anti-RBC antibodies drive Type I IFN production in vitro and correlate with the SLE IFN signature, Related toFigure 6. (A) Quantification of M⏀ phagocytized CFSE-labeled Mito+ RBCs that were treated as indicated. (Mito+ RBCs = untreated RBCs; Ops Mito+ RBCs = RBCs opsonized with anti-RBCs antibodies; Aged Mito+ RBCs = A23187/CaCl2 treated RBCs; Oxidized Mito+ RBCs = sodium ascorbate/CuSO4 treated RBCs). (n=3) (B) Quantification of M⏀ phagocytized opsonized CFSE-labeled Mito− or Mito+ RBCs at various time points at 1:2 M⏀:RBCs ratio or at various ratios for 18 h. (n=3). (C) Quantification of M⏀ with intracellular fragmented CFSE fluorescence upon phagocytosis of opsonized CFSE-labeled Mito− or Mito+ RBCs. (n=5 fields). Scale bar = 10 μm. (D) Quantification of residual CFSE fluorescence in M⏀ incubated with opsonized CFSE-labeled Mito− or Mito+ RBCs for 24 h, then chased for the indicated time points. (n=3). (E) Quantification of pHrodo fluorescence in M⏀ incubated with opsonized pHrodo-labeled Mito− or Mito+ RBCs. (n=3). (F) FACS gating strategies of M⏀ engulfing opsonized CFSE-labeled Mito− or Mito+ RBCs. (G) Western blot analysis for selected ISGs in M⏀ phagocytized opsonized Mito− or Mito+ RBCs (Mito− or Mito+ RBCs M⏀). Poly dA:dT is positive control. # denotes non specific bands. (H) Western blot analysis on M⏀ treated with the supernatant from M⏀ phagocytized opsonized Mito+ RBCs in the presence of anti-IFNAR2 antibody. # denotes non specific bands. (I) Relative mtDNA abundance in Mito+ RBCs generated with or without 2′,3′-dideoxycytidine (ddC). (n=3). (J) cGAS, STING1 and IRF3 protein expression demonstrating shRNA-mediated knockdown efficiency. (K) Normalized IP-10 levels in the supernatants of M⏀ transfected with IRF3 siRNA and then cultured with opsonized Mito+ RBCs. (n=3). (L) Representative histogram plot for phagocytosis of CFSE-labeled HD RBCs, treated as indicated, by M⏀. (M) Expression levels of three IFN-regulated modules in SLE patients with Mito− RBCs (n=4) or with Mito+ RBCs that carry (Ops IgG+; n=4) or not (Ops IgG−; n=6) opsonizing antibodies in their sera. In (G) and (H) results are representative of at least three independent experiments. Data are means ± SEM. [One-way analysis of variance (ANOVA) with Tukey post hoc test for multiple comparisons in (A) and (M); Two-tailed unpaired Student t test in (B), (C), (D), (E), (I) and (K)].
Data Availability Statement
The RNA-seq data generated during this study are available in the GEO database under the accession number GSE163073. Other data generated in the current study are available from the Lead Contact upon request.