

Abstract
Pancreatic cancer is a recalcitrant cancer with one of the lowest 5-year survival rates. A hallmark of pancreatic cancer is the prevalence of oncogenic mutation in the KRAS gene. The KRAS oncogene plays a critical role in the initiation and maintenance of pancreatic tumors and its signaling network represents a major target for therapeutic intervention. A number of inhibitors have been developed against kinase effectors in various Ras signaling pathways. Their clinical activity, however, has been disappointing thus far. More recently, covalent inhibitors targeting the KRASG12C oncoprotein have been developed. These inhibitors showed promising activity in KRASG12C mutant pancreatic cancer in early clinical trials. This review will present an updated summary of our understanding of mutant KRAS function in pancreatic cancer and discuss therapeutic strategies that target oncogenic KRAS signaling in this disease.
Keywords: KRAS, pancreatic cancer, target therapy, G12C, MAPK pathway
BACKGROUND
Pancreatic cancer is one of the most lethal forms of cancer. In 2021, approximately 60,430 patients are expected to be diagnosed with pancreatic cancer and approximately 48,220 patients are expected to die from the disease (1). Around 85% of pancreatic cancers are pancreatic ductal adenocarcinoma (PDAC) that arise from the malignant transformation of the ductal epithelial cells that form the capillary-like duct system that carries enzymes and other secretions away from the pancreas. Despite advances in diagnosis and treatment in the past decade, the 5-year survival rate of pancreatic cancer is approximately 9% (2). This poor prognosis is in part due to two factors. The first factor is a lack of good early diagnostic methods. Patients with early-stage pancreatic cancer often lack specific symptoms, and no reliable early biomarkers have been developed for non-invasive diagnosis of early-stage, resectable tumors. As a result, at the time of diagnosis patients often present with locally advanced or metastatic disease. The second factor is a lack of effective treatment. Current treatment of unresectable pancreatic cancer primarily involves chemotherapies that shrink tumors but are limited by the emergence of drug resistance (3). Targeted therapies have been of limited success. For example, olaparib has been approved for PDACs that bear mutations in homologous recombination genes but is indicated in only a small fraction of patients with limited survival benefit.
Genetically, PDAC is defined with a handful of recurring mutations in oncogene and tumor suppressor genes whose mutations are associated with disease progression (4). Amongst these are four canonical mutations – KRAS (~85%), TP53 (60–70%), CDKN2A (>50%), and SMAD4 (~50%). Genes involved in epigenetic regulation (ARID1A, ARID1B, SMARCA1, MLL2, MLL3, KDM6A) and the DNA damage response (ATM, BRCA2) are also found to be mutated, albeit at lower frequencies in PDAC (2, 5). Genetic analysis of clinical specimens indicates that KRAS mutation is an early event present in stage 1 pancreatic intraepithelial neoplasia (PanIN). Acquisition of mutations in CDKN2A, TP53 and SMAD4 are associated with PanIN progression and the development of invasive PDAC (Figure 1). The extremely high frequency of KRAS mutation in PDAC parallels that of BRAF mutation in melanoma. This suggests that the Ras signaling pathway as a key oncogenic driver of PDAC development is a prime target for inhibitor development. In addition to the above somatic mutations, pancreatic tumors can be further classified into several subtypes based on their histopathological features, gene expression profiles and their stromal and immune milieu (5). These different molecular subtypes are likely to influence the function of a driver oncogene such as KRAS and how tumor cells respond to targeted therapies.
Figure 1.

Common oncogenic mutations in the development of pancreatic cancer. PDAC, pancreatic ductal adenocarcinoma; PanIN, pancreatic intraepithelial neoplasia.
Current treatment of unresectable pancreatic cancer primarily relies on chemotherapies including gemcitabine and 5-FU. Median survival on various chemotherapies is poor and often does not exceed 12–18 months (3). Resistance to chemotherapy is attributable to multiple factors. Pancreatic cancer often presents a dense, fibrotic tumor microenvironment with low blood vessel density. This hampers drug delivery to tumor cells. Cancer-associated fibroblasts in an activated tumor stroma may provide pro-survival paracrine signaling to cancer cells, thus reducing the cytotoxic effect of chemotherapeutic agents. Pancreatic cancer often harbors an immunosuppressive tumor microenvironment (2). As a result, these tumors are refractory to immune checkpoint blockage using PD-1 and PD-L1 antibodies.
The dominance of KRAS mutations suggests targeted therapies against the Ras signaling network could present an effective treatment modality for PDAC. However, efforts in this direction thus far have yielded little clinical benefit (see below). Several targeted therapies have been approved for PDAC. The first approved, EGFR inhibitor erlotinib in combination with gemcitabine, gave a modest survival benefit compared to gemcitabine alone and was effectively abandoned by the community after negative data for EGFR-directed therapy in KRAS-mutant colorectal cancer emerged (6). More recently, tissue agnostic targeted therapy has included a small fraction of patients with PDAC. The TRK kinase inhibitors larotrectinib and entrectinib have been approved for solid tumors harboring a NTRK-fusion gene (7). NTRK-fusion occurs in only 0.5% of PDAC and the benefit of TRK inhibitors have not been systematically evaluated in PDAC beyond individual cases. Finally, in late-stage PDAC with germline mutation in BRCA1 or BRCA2 (~7.5% of the patients), the PARP inhibitor olaparib could extend progression free survival but not overall survival (8). Thus, identifying new therapeutic modalities for pancreatic cancer represents an urgent need.
Overview of the KRAS oncogene and its function
The KRAS gene encodes a member of the Ras family of small GTPases. KRAS and its two closely related paralogs, HRAS and NRAS, are frequently mutated in human cancer. KRAS mutation is found in ~85% of PDACs, ~45% of colorectal adenocarcinomas (CRC), ~30% of lung adenocarcinomas, and at lower frequencies in a wide array of tumor types (9). HRAS and NRAS mutations are also found in many tumor types, albeit at a lower frequency than KRAS. The vast majority of mutations in the Ras genes are mis-sense mutations at three hotspot residues G12, G13 and Q61. These mutations critically impair Ras GTPase activity and effectively lock the Ras protein in its GTP bound, active state. Collectively, mutations in the three Ras genes occur in over 10% of all human cancer. Currently, there are no effective, targeted therapies against most tumors with Ras mutation. In addition, Ras mutation is often a biomarker associated with poor prognosis and poor response to other targeted agents. Thus, solving the Ras problem is a high-priority area of cancer research (10).
The signaling network and molecular biology of Ras has been extensively reviewed recently (9, 11, 12) and a summary is provided here. KRAS, HRAS and NRAS are peripheral membrane proteins. They share a highly homologous G-domain which contains the GTPase catalytic site and two regions (switch I and II) that undergo conformational change upon GDP/GTP exchange. The C-terminus of these protein are more divergent, and KRAS can undergo alternative splicing to give rise to two isoforms KRAS4A and KRAS4B with distinct C-terminus sequences. Post-translational modification of Ras proteins at their C-terminus by a series of enzymes results in their farnesylation (HRAS, NRAS, KRAS4A and KRAS4B) and palmitoylation (HRAS, NRAS and KRAS4A). These lipid attachments enable Ras to be associated with the plasma membrane and other endomembrane compartments in the cell and are essential for Ras function.
Ras proteins are signal transduction molecules whose activity is controlled by cell surface growth factor, cytokine and hormone receptors. Growth factor receptor activation stimulates GDP-GTP exchange on Ras that is facilitated by a family of guanine nucleotide exchange factors (GEFs) that are recruited to adaptor proteins binding the receptor. The GTP-bound Ras undergoes a conformational change that enables its switch I and II regions to interact with and activate a number of downstream effector proteins. Subsequent GTP hydrolysis by Ras, a reaction that is greatly accelerated by its binding to GTPase-activating proteins (GAPs), returns Ras to the inactive, GDP-bound state. The aforementioned hotspot mutations in Ras either impair GAP binding (G12 and G13 mutations) or the intrinsic GTPase activity (Q61 mutations) of Ras. These mutations dramatically shift the equilibrium of Ras protein to exist mostly in the GTP-bound state and effectively uncouple Ras proteins from upstream receptor inputs and drive constitutive signaling to downstream Ras effector pathways (13).
A major reason for Ras mutations being frequently selected in cancer is that Ras signals to multiple effector pathways whose activation favors oncogenic transformation. These include the MAP-kinase (MAPK) pathway, the PI 3-kinase (PI3K)/AKT/mTOR pathway, the small GTPases Rho, Rac and Ral, and phospholipase C (Figure 2). Together, these pathways regulate cell proliferation, cell growth, cell survival, cell metabolism, cell motility and a host of gene transcriptional programs. By constitutively activating these pathways, the Ras oncoprotein confers growth, survival and metastatic advantages to the cancer cell (11).
Figure 2.
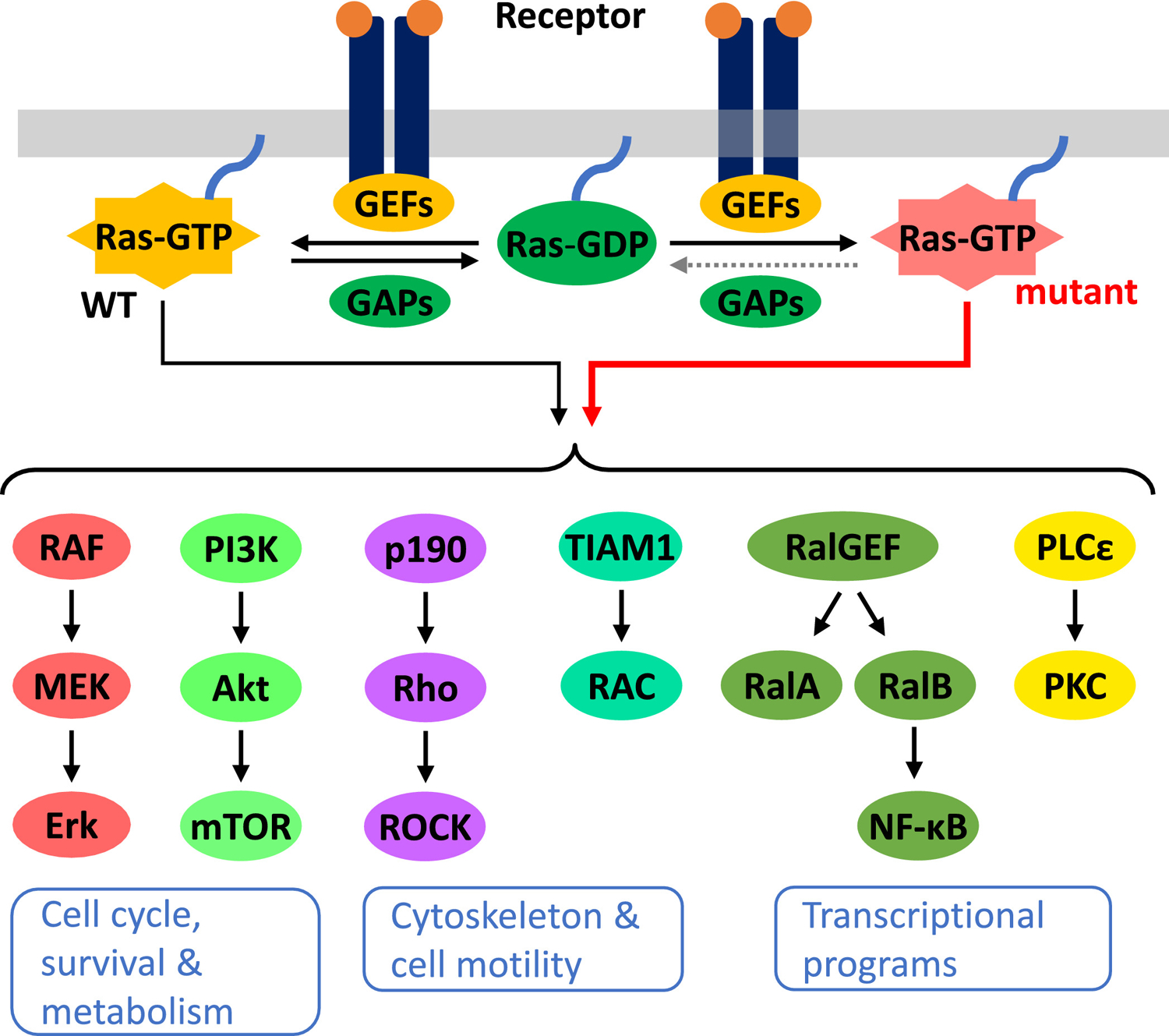
A simplified schematic of the Ras signaling network with major canonical effector pathways shown. Oncogenic mutation in Ras locks it in the GTP-bound, active form, therefore resulting in constitutive signaling to downstream pathways. GEFs, guanine nucleotide exchange factors; GAPs, GTPase-activating proteins; WT, wild type.
Currently, it is not fully understood why KRAS mutation is more prevalent than HRAS and NRAS mutation in adenocarcinomas. These genes are expressed at different levels in different epithelial compartments, and it is possible KRAS expression is optimal for driving cell proliferation and oncogenic transformation. It has been shown that mutant KRAS is more effective at promoting colonic epithelial cell proliferation in mice (14) and stemness properties in tumor cells through its ability to suppress non-canonical Wnt signaling (15). Thus, KRAS might possess distinct signaling properties that make it a more effective oncogene than HRAS and NRAS. Among KRAS mutant adenocarcinomas, a tissue-specific pattern of codon mutation is observed. For example, mutations at G13, K117 and A246 are more common in CRC than PDAC and lung cancer. Among the codon 12 mutations, the G12C mutation is prevalent in lung cancer (likely due to smoking-induced mutagenesis) but rare in PDAC and CRC (Figure 3). G12D and G12V are the most common in PDAC, which also shows enrichment for the G12R mutation that is rarely found in CRC lung and lung cancer (16). It is not clear why different KRAS codon mutations are selected in different tissues, although context-dependent signaling by a specific mutant could contribute to this bias (17).
Figure 3.
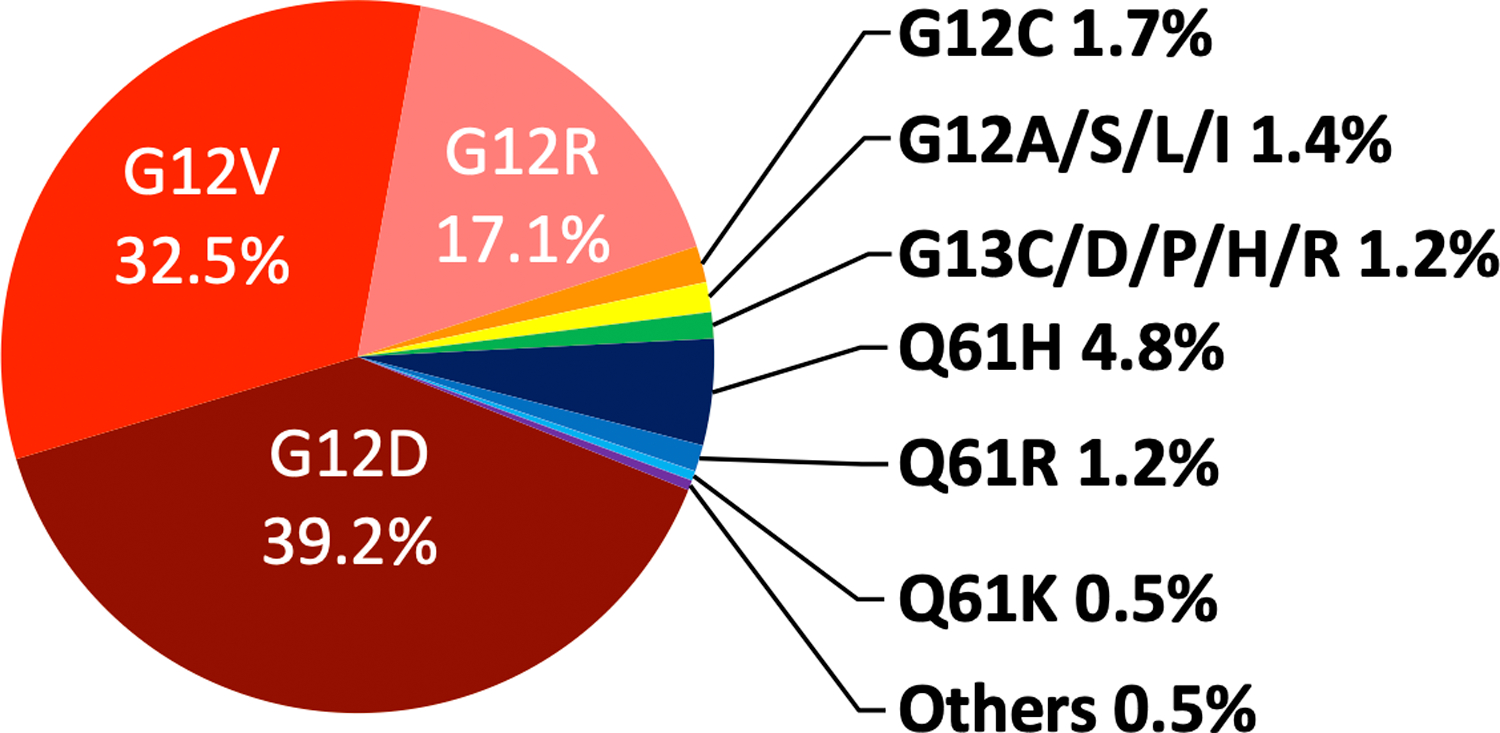
Distribution of KRAS mutations in pancreatic cancer. The analysis was done using publicly available data from the cBioPortal database (48, 49) that includes 665 KRAS mutant tumor samples from four large scale pancreatic cancer studies (50–53).
Therapeutic targeting of the Ras signaling network
Targeting the Ras oncoprotein
The Ras signaling network is a target-rich space that has receive intense focus for pharmacological intervention (9, 18). Early efforts at inhibiting Ras oncoprotein have not yielded effective Ras inhibitors due to the challenging biochemical properties of the Ras protein as a drug target. Inhibitors that block farnesyltransferase to prevent Ras membrane localization failed to gain traction in clinical studies due to alternative mechanisms of Ras membrane localization and concerns with these inhibitors’ pleiotropic effect on the membrane localization of other small GTPase. Thus, for two decades Ras has remained an “undruggable” target.
Recently, innovative chemical biology has led to the breakthrough discovery of inhibitors against the KRASG12C oncoprotein (19). The structural feature of these inhibitors consists of a moderately-reactive cysteine cross-linking group and a scaffold that binds to the G-domain of Ras. In this configuration, these compounds selectively react with the codon 12 cysteine on the mutant KRASG12C protein but are far less reactive towards cysteine residues on other proteins. Covalent cross-linking to codon 12 cysteine leads to an induced fit of the inhibitor in KRASG12C and trap the protein in its inactive, GDP-bound state (20–23). Consequently, these inhibitors effectively and specifically extinguish oncogenic signaling from mutant KRASG12C but do not interfere with the function of any WT Ras proteins or non-G12C mutant Ras proteins. These inhibitors represent a major breakthrough in our ability to directly inhibit Ras. In preclinical studies, KRASG12C inhibitors have demonstrated significant and selective in vitro toxicity in KRASG12C mutant cancer cells; and these inhibitors are effective at inhibiting the growth of KRASG12C mutant xenograft and PDX tumors in mice (21–23). In early clinical studies, they have demonstrated promising activity in patients whose tumors harbor KRASG12C mutation (23, 24).
Non-G12C KRAS oncoproteins remain a therapeutic challenge. Several alternative strategies have been developed to target mutant KRAS. These include the use of siRNAs against KRAS (25), the use of mutant KRAS peptide for anticancer vaccine (26, 27), and the development of adoptive T-cell therapy with T-cell receptors that specifically react to mutant KRAS peptides (28, 29). Although each of these approaches has its own set of translational challenges, their success could significantly broaden the spectrum of KRAS mutations that are targetable.
Inhibitors targeting Ras effector pathways
A number of highly specific and potent small molecule inhibitors have been developed to target major Ras effector pathways in an effort to block oncogenic Ras signaling. The characteristics of the targets and their respective inhibitors have been reviewed in detail recently (9, 12). The MAPK and PI3K pathways received most attention due to their critical role in mediating the proliferative, survival and metabolic effects of the KRAS oncogene. In the MAPK pathway, inhibitors of BRAF kinase can paradoxically activate this pathway in KRAS mutant cells due to their ability to promote the dimerization of BRAF and CRAF and the activation of CRAF. Inhibitors targeting MEK and ERK kinases further downstream can effectively block MAPK pathway signaling. Targeting the PI3K pathway, multiple inhibitors for class Ia PI3K, AKT, and mTOR have been developed to block this pathway. Despite promising anti-tumor effects in preclinical models of KRAS mutant cancer, MAPK and PI3K pathway inhibitors have not demonstrated significant clinical activity in patients with KRAS mutant tumors, including in pancreatic cancer (see below). Understanding the cause of this disconnect is key to improving the therapeutic efficacy of Ras effector pathway inhibitors. One potential explanation is that, giving the multitude of effector pathways that KRAS can engage, inhibiting one or two pathways is insufficient to block the oncogenic activity of KRAS. Another potential explanation is that inhibitors of KRAS effector pathways have a narrower therapeutic window than KRAS inhibitors, and their on-target toxicity in normal tissue prevents dosing to a higher level in patients. A third potential explanation is that effector pathway utilization by mutant KRAS is heterogenous and context-dependent in cancer cells, thus making KRAS mutant tumors – even those from the same tissue – heterogeneous in their sensitivity to the inhibition of a given Ras effector pathway (30, 31).
Inhibition of Ras synthetic lethal and collateral dependency partners
To expand the target space in Ras mutant cancer cells, RNAi and CRISPR-based genetic screens have been carried out to identify synthetic lethal partners of the KRAS oncogene whose suppression confers selective toxicity in KRAS mutant cells. A number of synthetic lethal genes have been identified using genome-wide RNAi and CRISPR screens, and many of these are druggable targets (32, 33). These studies have revealed two important themes. First, synthetic lethal interactions with KRAS tends to be context-dependent and they reflect the underlying genetic heterogeneity of cancer cell lines. A particular synthetic lethality interaction, therefore, is often observed only among a subset of KRAS mutant cancer cell lines. Second, synthetic lethal partners of KRAS often play a critical role in stress-response pathways – such as those involved in DNA damage response, genomic stability, cell survival, metabolic adaption, proteotoxic and oxidative stress – that serve to alleviate various forms of oncogenic stress associated with tumorigenesis (34). Inhibition of these pathways therefore exacerbates oncogenic stress in KRAS mutant cells and impairs their viability (32, 33). An example is the mitotic kinase PLK1. We originally identified PLK1 as a synthetic lethal partner of mutant KRAS in colorectal cancer cells through a genome-wide RNAi screen and showed that PLK1 inhibition leads to more pronounced genomic instability in KRAS mutant cells (35). Recently, the PLK1 inhibitor onvansertib has been evaluated in combination with chemotherapy and bevacizumab in KRAS mutant metastatic colorectal cancer in a phase II trial, and this combination seems to demonstrate favorable tolerability and clinical response (36).
To improve the efficacy of inhibitors, genetic and pharmacological screens have been carried out to identify collateral dependency partners of KRAS and MAPK pathway inhibitors that could enhance their lethality in KRAS mutant cells. These experiments have identified receptor tyrosine kinase (RTK) signaling to PI3K and mTOR (23, 37–40), as well as the autophagy pathway (30) as collateral dependencies with KRAS and MAPK pathway inhibitors. These insights could serve as a useful guide for the rational testing of drug combinations in KRAS mutant tumors in clinical trials.
Resistance mechanisms to KRAS and MAPK pathway inhibitors
Like other signal transduction pathways in the cell, the Ras/MAPK pathway has evolved to sense changes in receptor activity. In order to maintain the pathway’s dynamic range under different levels of receptor activity, a number of negative feedback mechanisms have evolved. For example, ERK can phosphorylate CRAF to inhibit its activity; MAPK pathway can induce the expression of DUSP phosphatases to dephosphorylate ERK and Sprouty and SPRED proteins to inhibit Ras activation (41). Prolonged activation of the Ras/MAPK pathway can also lead to the feedback inhibition of RTK signaling (42). In KRAS and BRAF mutant cancer cells, constitutive activation of the MAPK pathway strongly induces these negative feedback loops as a maladaptive attempt to constrain pathway activity. As a result, acute inhibition of KRAS or MAPK in KRAS and BRAF mutant cells often results in a bi-phasic response. Initially, the pathway is effectively shut down by the inhibitor. This, however, also diminishes the activity of the negative feedback loops. Consequently, the activity of the MAPK pathway gradually rebounds to a new steady state level despite the continuous presence of the inhibitor, and increased activation of RTKs and its downstream PI3K/AKT pathway is often evident in this second phase (41, 42). This inhibitor-induced loss of negative feedback therefore reduces the efficacy of KRAS and MEK inhibitors. Target combinations that can abrogate this adaptive resistance mechanism, including vertical drug combination within the MAPK pathway (43) and co-inhibition of parallel RTK/PI3K signaling (40, 44, 45), could result in a more sustained inhibition of cell viability in KRAS mutant cells.
KRAS mutant cells could also escape Ras/MAPK inhibitors by undergoing epithelial-mesenchymal transition (EMT). In these cells, MAPK and PI3K signaling are sustained by RTKs including FGFR and IGFR, rendering them less sensitive to KRAS or MEK inhibitors. Strategies that reverse the EMT transcriptional program, as well as co-targeting FGFR and IGFR signaling in EMT-induced cells could re-sensitize them towards KRAS and MEK inhibitors (46, 47)
KRAS mutation in the context of pancreatic cancer
Frequency and codon bias of KRAS mutations in pancreatic cancer
An analysis of the cBioPortal database (48, 49) using four large scale pancreatic cancer studies (50–53) indicated that KRAS mutation occurs in 85.8% (665/775) of tumor samples. Most are missense mutations that occur in the three hotspot residues G12, G13 and Q61 (Figure 3). KRASG12D and KRASG12V are most prevalent, at 39.2 and 32.5% of all KRAS mutations, respectively. As mentioned, the G12R mutation also occurs at high frequency in PDAC (17% of all KRAS mutations). The reason for this bias is unclear, although evidence suggests that the KRASG12R oncoprotein has distinct biochemical properties and is less effective at activating PI3K signaling and micropinocytosis (54). The KRASG12C mutant allele, whose protein product is inhibitable with the recently developed KRASG12C covalent inhibitors, occurs only in 1.7% of KRAS mutant PDACs.
KRAS mutation in the initiation, progression and prognosis of pancreatic cancer
KRAS mutation is an early oncogenic event in pancreatic cancer. KRAS mutation is readily detectable in 25% and 38% of PanIN-1A and PanIN-1B, respectively (55). These findings indicate that KRAS mutation is likely an early and initiating event in human pancreatic cancer. This is similar to lung adenocarcinomas where KRAS mutation is also detected in early lesions (56). In contrast, in colorectal cancer KRAS mutation is a progression event that occurs after the initiation event of mutation in the Wnt pathway (57). Mouse models also support the role of Kras mutation as an initiating event in pancreatic cancer.
Acquisition of mutations in tumor suppressor genes CDKN2A, TP53, and SMAD4 enables early stage PanIN to progress to late stage PanIN and, ultimately invasive PDAC (2). The higher frequency of KRAS mutation observed in late-stage PDAC compared to early-stage PanIN suggests that KRAS mutation might confer additional fitness benefits, and thus be further selected, during tumor progression. Studies in human cancer cell lines and in mouse models show that the majority of KRAS mutant PDAC cells are continuously addicted to the KRAS oncogene for their survival.
Because KRAS mutation is an early event, it is unclear whether, and how, the cellular role of the KRAS oncogene change during progression. As noted earlier, KRAS signaling can regulate multiple effector pathways (Figure 2). It is possible that mutant KRAS can play distinct roles in driving cell proliferation, cell survival and cell migration at different stages of PDAC as the tumor progresses, through the differential engagement of its downstream signaling pathways.
KRAS mutation in the prognosis of pancreatic cancer
Multiple studies have investigated KRAS mutation as a prognostic factor in patients with PDAC (58) and have arriving at mixed findings. For example, in localized and rescetable PDACs, KRAS mutation was found to be either negatively associated with or not associated with survival (59, 60). In locally advanced and metastatic PDACs, KRAS mutation was found to be either negatively associated with or not associated with survival with chemotherapy (61, 62). There seems to be a consensus that KRAS mutation negatively influences response to EGFR-targeted therapy (61, 63). Large cohort studies with more homogenous clinical and tumor profiles are needed to further clarify the prognostic value of KRAS mutation in PDAC. Given the high prevalence of KRAS mutation in pancreatic cancer, an alternative view might be to understand what therapies might confer better response in the minority of patients with KRAS WT pancreatic cancer.
Molecular Function of the KRAS oncogene in pancreatic cancer
Insights from human pancreatic cancer cell lines
A large body of work has established that cancer cells with KRAS mutation exhibit strong oncogene addiction to KRAS. RNAi mediated gene knockdown studies and CRISPR mediated gene knockout studies in hundreds of cancer cell lines have shown that KRAS mutant pancreatic, lung and colorectal adenocarcinoma cells are more sensitive to the loss of the KRAS oncogene than KRAS WT cancer cell lines (64, 65) (Figure 4). These findings demonstrate that KRAS mutant pancreatic cancer cells are dependent on the function of the KRAS oncogene for their survival. It is remarkable that some of these cell lines have been cultured for years in media containing high levels of growth factors, yet their addiction to the KRAS oncogene remained stable. The inability of growth factor signaling to abrogate KRAS addiction might be due to the activation of negative feedback loops in KRAS mutant cells that down-regulate receptor signaling.
Figure 4.
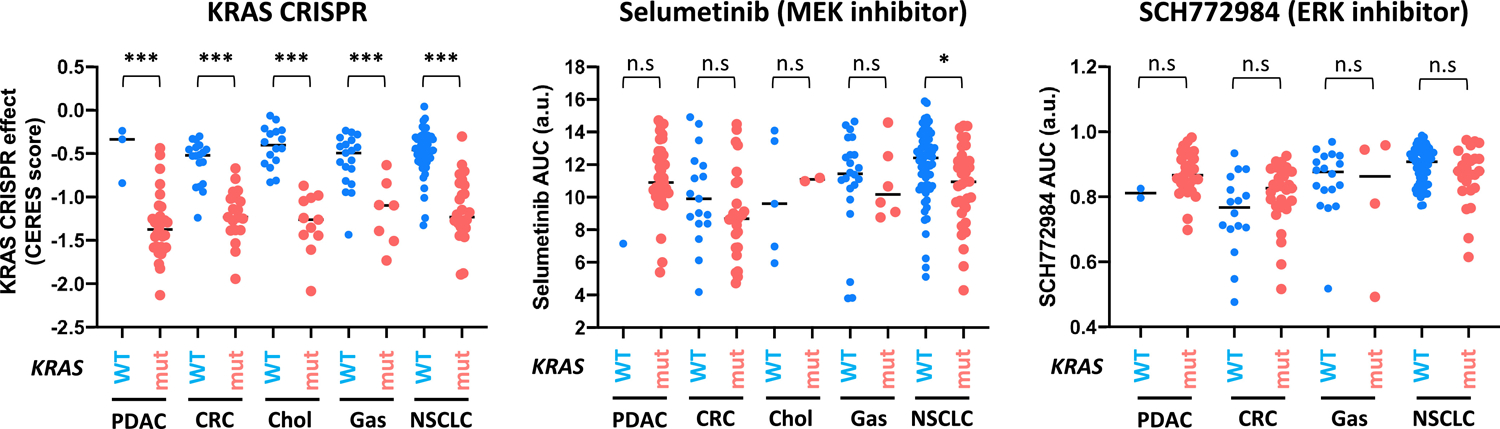
The sensitivity of KRAS mutant and WT cell lines to CRISPR-mediated KRAS knockout, to the MEK inhibitor selumetinib and to the ERK inhibitor SCH772984. The analysis was done using publicly available data from the DepMap database. For the KRAS CRISPR data, normalized sensitivity score (CERES) was used (65). Within each tumor type, KRAS mutant cells are more sensitive to KRAS knockout than KRAS WT cells (*** p < 0.001). For the drug sensitivity data, normalized area under the curve (AUC) data was used for both selumetinib (CDT2 dataset) and SCH772984 (Sanger GDSC2 dataset) (69, 70). Within each tumor type, KRAS mutant cells are not significantly (n.s.) more sensitive to the inhibitor than KRAS WT cells, with the exception of NSCLC cells to selumetinib (* p = 0.01). Each data point represents an individual cell line. PDAC, pancreatic ductal adenocarcinoma; CRC, colorectal adenocarcinoma; Chol, cholangiocarcinoma; Gas, gastric adenocarcinoma; NSCLC, non-small cell lung cancer; a.u. arbitrary units.
In KRAS mutant pancreatic cancer cells, acute silencing of KRAS via RNAi or CRISPR results in cell cycle arrest, apoptosis and suppression of anchorage-independent proliferation in vitro, and attenuated tumor growth and metastasis in xenograft models (66–68). Silencing mutant KRAS down-regulates the activity of multiple Ras effector pathways including the MAPK and PI3K pathways (67, 68). Inhibitors targeting these Ras effector pathways, therefore, should exhibit selectivity towards KRAS mutant pancreatic cancer cells. However, large-scale drug sensitivity studies using the MEK and ERK inhibitors did not reveal the same degree of selectivity in KRAS mutant cells (69, 70) (Figure 4). This is not entirely surprising because the MAPK pathway is critical for driving cell proliferation and cell survival in cells without KRAS mutation, whereas KRAS itself is not (likely due to redundant signaling from HRAS and NRAS). This may also mean that inhibition of KRAS itself will have a greater therapeutic window than targeting the MAPK pathway downstream.
Insights from animal models of pancreatic cancer driven by mutant KRAS
Genetically engineered mouse models (GEMMs) of Kras mutant pancreatic cancer have provided valuable insights on the molecular mechanism of this disease (71). Selective expression of the Kras oncogene in the mouse pancreas can be achieved in several ways, including the use of Cre-mediate activation of a latent KrasG12D mutant allele (LSL-Kras) (72) or a mutant KasG12D transgene under the control of a tetracyclin-inducible promoter (iKras) (73). These mouse models have validated the role of Kras mutations as a tumor initiating event in pancreatic cancer. Expression of mutant Kras alone from embryonic pancreatic cells results in PanIN with histological and molecular features that resemble PanIN in human PDACs (72). GEMMs with compound mutations that combine Kras mutation with the inactivation of tumor suppressor genes Cdkn2a and Tp53, or with the inactivation of the TGFβ-SMAD4 signaling pathway in the pancreas develop invasive and metastatic PDAC (74–76), thus validating the role of these tumor suppressor pathways in PDAC progression (4). Mouse PDAC tumors driven by mutant Kras also experience metabolic alteration and are dependent on glucose and glutamine (73, 77). Extinguishing the expression of mutant Kras in the pancreas led to rapid tumor regression, thus demonstrating a critical role of Kras in tumor maintenance (73, 78).
Mouse models also shed light on how pancreatic cancer develops. Pancreatic cancer models from Pdx1- and Ptf1a-promoter mediated Kras expression result in wide-spread Kras expression in most pancreas cells during early development. Expression of a mutant Kras transgene from the Krt19 promoter, which is active in ductal epithelial cells but not in other cell types in the pancreas, failed to induce tumor (79). On the other hand, activation of the LSL-Kras allele using elastase-driven Cre, which is expressed in acinar cells, results in PanIN development (80). Thus, acinar cells could also serve as the cell of origin for PDAC and acinar-ductal metaplasia could occur during PanIN development. Induction of mutant Kras expression in the adult pancreas was far less efficient at inducing tumor than it does in young mice. Tumor development in adult mice, however, can be greatly accelerated by chemically induced pancreatitis. Thus, inflammatory signaling is likely to play a critical role in pancreatic cancer development (80, 81). The mechanism by which this occurs is not fully understood, one explanation is that the inflammatory tumor microenvironment can suppress Kras oncogene-induced senescence during tumor initiation (81).
Recently, normal and tumor organoid models have been developed from both human and mouse pancreas (82, 83). Normal pancreatic organoids enable the rapid modeling of genetic interactions among candidate driver mutations in pancreatic cancer. Tumor organoid models preserve many of the histological and molecular features of pancreatic tumors and their spectrum of heterogeneity. Together, these organoid models could serve as a powerful system for new target discovery, for the identification of biomarkers that are associated with efficacy of existing therapeutics, and for the validation of novel therapeutic agents in pancreatic cancer (83).
Genetic mechanisms that bypass KRAS oncogene addiction in pancreatic cancer cells
Although pancreatic cancer cell lines exhibit strong addiction to the KRAS oncogene, extinguishing mutant KRAS expression can ultimately lead to adaptation in cancer cells that by-pass their KRAS dependency. In the context of pancreatic cancer, KRAS dependency can be by-passed by the elevated expression of the transcription factor YAP1, which promotes EMT and confers cell-intrinsic resistance to KRAS loss (84, 85). Overexpression of the histone deacetylase HDAC5 also enables bypass of KRAS extinguishment through cytokine-mediate remodeling of the tumor microenvironment (86). These findings indicate that KRAS mutant pancreatic tumors could ultimately escape targeted inhibition of the KRAS oncoprotein, paralleling the observation with BRAF and EGFR inhibitors in patients with BRAF mutant melanoma and EGFR mutant non-small cell lung cancer, respectively.
Clinical activity of targeted therapies against KRAS signaling pathways in pancreatic cancer
Ras effector pathway inhibitors
Pancreatic cancer has been a focus for the clinical development of targeted therapies against the Ras signaling network due to the high prevalence of KRAS mutation in this disease. A number of highly selective and potent inhibitors have been developed against kinases in the Ras signaling network particularly those in the MAPK and PI3K/mTOR pathways (9, 12). Despite promising preclinical activities, single agent inhibitors targeting the MAPK and PI3K/mTOR pathways have yielded poor clinical efficacy in phase II trials. Single agent phase II trials of the MEK inhibitors CI-1040 and selumetinib showed that these agents were well tolerated but did not give superior response compared to chemotherapy (87, 88). In a phase I/II trial, a combination of the MEK inhibitor pimasertib and gemcitabine did not give superior survival rate than gemcitabine alone (89). Inhibitors targeting the ERK kinases including ulixertinib, LY3214996, and MK-8353 are being evaluated in early-stage trials that include patients with pancreatic cancer; currently there are insufficient data reported to evaluate their efficacy. In single agent phase II trials of the mTOR inhibitors temsirolimus and everolimus, very little clinical activity was seen (90, 91). Combination of these inhibitors with gemcitabine-based chemotherapy also did not yield significant improvement in clinical activity (92, 93). These early trial data indicates that targeting a single component in the Ras signaling network either alone or in combination with chemotherapy is unlikely to yield significant survival benefits.
KRASG12C inhibitors
Two KRASG12C inhibitors, sotorasib (AMG510) and adagrasib (MRTX849) have entered clinical development and they have demonstrated promising activities in solid tumors with KRASG12C mutation, particularly among patients with KRASG12C non-small cell lung cancer (23, 24). In a phase II trial with sotorasib, 12 patients with KRASG12C pancreatic cancer were enrolled. Among these 12, 1 patient had a partial response, 8 patients had stable disease and 2 patients had progressive disease (24). One patient with PDAC enrolled on the Phase 1/2 study of MRTX849 (NCT03785249); it has been reported in abstract form that this patient has had a partial response (32nd EORTC-NCI-AACR Symposium, 2020). These exciting early data suggest that KRASG12C inhibitors will achieve good disease control and survival extension in patients with KRASG12C pancreatic cancer in phase III trials. Whether depth and duration of response will be comparable to that in lung cancer remains to be determined. Meanwhile, the G12D inhibitor MRTX1133 is advancing through pre-IND studies. This will be very important for the 39% of patients with KRASG12D-mutated PDAC.
Combination therapies
The challenges seen with single agent inhibitors targeting Ras effector pathways indicate that combination therapy is necessary to control oncogenic KRAS signaling and achieve better clinical response. KRASG12C inhibitors in combination with either chemotherapy or with Ras effector pathway inhibitors could further improve their clinical efficacy. Preclinical data indicates that KRASG12C inhibition leads to a pro-inflammatory tumor microenvironment and potentiates tumor response to immune checkpoint inhibitors (22). If this observation can be validated in clinical trials, it could potentially result in significant survival improvement for patients with KRASG12C pancreatic cancer.
For pancreatic cancer with non-G12C KRAS mutations, it is likely that combination therapy should include a MAPK pathway inhibitor given this pathway’s central role in driving cell proliferation and cell survival downstream of KRAS. Such combination would require careful consideration of the therapeutic window and on-target toxicity of the agents. In a phase II study combining the MEK inhibitor selumetinib with the AKT inhibitor MK-2206 in patients with metastatic pancreatic cancer, those receiving the drug combination actually did slightly worse than the control group receiving the mFOLFOX chemotherapy (94). This outcome is likely due, in part, to the toxicity of the drug combination as the combination arm had a higher fraction of patients that experienced grade 3 or higher toxicity and discontinued treatment (94). Thus, a combination therapy needs to demonstrate clear genotype-specific toxicity in KRAS mutant pancreatic cancer cells vs. KRAS WT cells in normal tissues in order to have a good therapeutic window. To do so, drug combinations could aim to either deepen inhibition of the MAPK pathway, exacerbate oncogenic stress, or exploit collateral dependencies in KRAS mutant tumors. Several drug combinations that test these principles are currently undergoing clinical trials in patients with pancreatic cancer, summarized in Table 1.
Table 1:
Selected clinical trials that evaluate combination of MEK inhibitors with other targeted agents in patients with pancreatic cancer. Trials were selected to represent different scientific rationales.
| Clinical Trial ID | Target 1 (agent) | Target 2 (agent) | Mechanistic Rationale |
|---|---|---|---|
| NCT04005690 (Phase I) | MEK (cobinetinib) | PARP (Olaparib) | Co-targeting DNA-repair vulnerability in KRAS mutant cells (98) |
| NCT04132505, NCT03825289 (Phase I) | MEK (Binimetinib, trametinib) | Autophagy/lysosome (hydroxychloroquine) | Autophagy is a collateral metabolic dependency in KRAS mutant cells (30, 95, 96) |
| NCT02428270 (Phase II) | MEK (trametinib) | FAK (GSK2256098) | FAK-mediated cell adhesion signaling supports KRAS-driven transformation (99) |
| NCT04390243 (Phase II) | MEK (benimetinib) | RAF (encorafenib) | Vertical combination for sustained MAPK pathway inhibition (43, 100) |
| NCT01222689 (phase II) | MEK (selumetinib) | EGFR (erlotinib) | Co-targeting upstream RTK to prevent feedback and parallel signaling (101) |
Recently, through combinatorial RNAi screens and hypothesis-driven approaches, we and others have identified the autophagy pathway as a collateral dependency partner with MAPK pathway inhibitors in pancreatic cancer cells with KRAS mutation (30, 95, 96). Autophagy is upregulated in KRAS mutant cancer cells and it serves as a cell survival mechanism when cancer cells experience metabolic stress (97). In KRAS mutant pancreatic cancer cells, inhibition of the MAPK pathway downregulates glucose uptake and forces the cell to become more dependent on autophagy for survival (95, 96). Co-inhibition of the MAPK pathway and the autophagy pathway therefore leads to exacerbated metabolic stress in the KRAS mutant context. Clinical trials testing this hypothesis are currently underway (Table 1).
Conclusions
KRAS mutation is a hallmark of pancreatic cancer. Basic and translational research over the past two decades has established a mechanistic picture of how the KRAS oncogene drives pancreatic cancer development. A growing number of highly selective inhibitors are now available to target KRAS and its signaling network. The recent discovery of KRASG12C inhibitors and their delivery to the clinic represents a breakthrough that could transform pancreatic cancer treatment. Ongoing clinical trials testing new drug combination could lead to the identification of new therapeutic strategies that improve patient survival in the near future.
Acknowledgement
This work was supported by the Intramural Research Program of the National Institutes of Health and the National Cancer Institute with a grant ZIA BC 011732 to Ji Luo. The content is solely the responsibility of the author and does not necessarily represent the official views or policies of the National Institutes of Health and the Department of Health and Human Services. The mention of trade names, commercial products or organizations does not imply endorsement from the US Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
No known conflict of interest for the author.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2020;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371(11):1039–49. [DOI] [PubMed] [Google Scholar]
- 3.Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15(6):333–48. [DOI] [PubMed] [Google Scholar]
- 4.Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156(6):1821–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2019;16(4):207–20. [DOI] [PubMed] [Google Scholar]
- 6.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–6. [DOI] [PubMed] [Google Scholar]
- 7.Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med. 2018;378(8):731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019;381(4):317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13(11):828–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25(3):272–81. [DOI] [PubMed] [Google Scholar]
- 11.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med. 2018;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170(1):17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40(5):600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang MT, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, et al. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell. 2015;163(5):1237–51. [DOI] [PubMed] [Google Scholar]
- 16.Haigis KM. KRAS Alleles: The Devil Is in the Detail. Trends Cancer. 2017;3(10):686–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, et al. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov. 2019;9(6):738–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCormick F Progress in targeting RAS with small molecule drugs. Biochem J. 2019;476(2):365–74. [DOI] [PubMed] [Google Scholar]
- 19.Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15(11):771–85. [DOI] [PubMed] [Google Scholar]
- 20.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172(3):578–89 e17. [DOI] [PubMed] [Google Scholar]
- 22.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–23. [DOI] [PubMed] [Google Scholar]
- 23.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C Inhibitor, MRTX849, Provides Insight Toward Therapeutic Susceptibility of KRAS Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2019. [DOI] [PMC free article] [PubMed]
- 24.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383(13):1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan TL, Fellmann C, Lee CS, Ritchie CD, Thapar V, Lee LC, et al. Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discov. 2014;4(10):1182–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaft JE, Litvak A, Arcila ME, Patel P, D’Angelo SP, Krug LM, et al. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin Lung Cancer. 2014;15(6):405–10. [DOI] [PubMed] [Google Scholar]
- 27.Kubuschok B, Pfreundschuh M, Breit R, Hartmann F, Sester M, Gartner B, et al. Mutated Ras-transfected, EBV-transformed lymphoblastoid cell lines as a model tumor vaccine for boosting T-cell responses against pancreatic cancer: a pilot trial. Hum Gene Ther. 2012;23(12):1224–36. [DOI] [PubMed] [Google Scholar]
- 28.Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375(23):2255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sim MJW, Lu J, Spencer M, Hopkins F, Tran E, Rosenberg SA, et al. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc Natl Acad Sci U S A. 2020;117(23):12826–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A. 2019;116(10):4508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan TL, Amzallag A, Bagni R, Yi M, Afghani S, Burgan W, et al. Differential Effector Engagement by Oncogenic KRAS. Cell Rep. 2018;22(7):1889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu B, Luo J. Synthetic lethal genetic screens in Ras mutant cancers. Enzymes. 2013;34 Pt. B:201–19. [DOI] [PubMed] [Google Scholar]
- 33.Aguirre AJ, Hahn WC. Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harb Perspect Med. 2018;8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahn DH, Erlander M, Ridinger M, Samuelsz E, Barzi A, Bekaii-Saab T, et al. Phase Ib/II study of the polo-like kinase 1 (PLK1) inhibitor, onvansertib, in combination with FOLFIRI and bevacizumab for second line treatment of KRAS-mutated metastatic colorectal cancer. Annals of Oncology. 2020;31:S427. [Google Scholar]
- 37.Sulahian R, Kwon JJ, Walsh KH, Pailler E, Bosse TL, Thaker M, et al. Synthetic Lethal Interaction of SHOC2 Depletion with MEK Inhibition in RAS-Driven Cancers. Cell Rep. 2019;29(1):118–34 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molina-Arcas M, Moore C, Rana S, van Maldegem F, Mugarza E, Romero-Clavijo P, et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci Transl Med. 2019;11(510). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lou K, Steri V, Ge AY, Hwang YC, Yogodzinski CH, Shkedi AR, et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal. 2019;12(583). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin Cancer Res. 2019;25(2):796–807. [DOI] [PubMed] [Google Scholar]
- 41.Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res. 2010;16(13):3329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kun E, Tsang YTM, Ng CW, Gershenson DM, Wong KK. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev. 2020;92:102137. [DOI] [PubMed] [Google Scholar]
- 43.Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res. 2020;26(7):1633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, et al. SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors. Cell Rep. 2019;26(1):65–78 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR blockade reverts resistance to KRAS G12C inhibition in colorectal cancer. Cancer Discov. 2020. [DOI] [PMC free article] [PubMed]
- 46.Peng DH, Kundu ST, Fradette JJ, Diao L, Tong P, Byers LA, et al. ZEB1 suppression sensitizes KRAS mutant cancers to MEK inhibition by an IL17RD-dependent mechanism. Sci Transl Med. 2019;11(483). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y, et al. Epithelial-to-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov. 2016;6(7):754–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52. [DOI] [PubMed] [Google Scholar]
- 52.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173(2):321–37 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, et al. Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020;10(1):104–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7(1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yokota J, Kohno T. Molecular footprints of human lung cancer progression. Cancer Sci. 2004;95(3):197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361(25):2449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17(3):153–68. [DOI] [PubMed] [Google Scholar]
- 59.Shin SH, Kim SC, Hong SM, Kim YH, Song KB, Park KM, et al. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas. 2013;42(2):216–22. [DOI] [PubMed] [Google Scholar]
- 60.Schultz NA, Roslind A, Christensen IJ, Horn T, Hogdall E, Pedersen LN, et al. Frequencies and prognostic role of KRAS and BRAF mutations in patients with localized pancreatic and ampullary adenocarcinomas. Pancreas. 2012;41(5):759–66. [DOI] [PubMed] [Google Scholar]
- 61.Kim ST, Lim DH, Jang KT, Lim T, Lee J, Choi YL, et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol Cancer Ther. 2011;10(10):1993–9. [DOI] [PubMed] [Google Scholar]
- 62.Bournet B, Muscari F, Buscail C, Assenat E, Barthet M, Hammel P, et al. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boeck S, Jung A, Laubender RP, Neumann J, Egg R, Goritschan C, et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol. 2013;48(4):544–8. [DOI] [PubMed] [Google Scholar]
- 64.McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell. 2017;170(3):577–92 e10. [DOI] [PubMed] [Google Scholar]
- 65.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell. 2017;170(3):564–76 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fleming JB, Shen GL, Holloway SE, Davis M, Brekken RA. Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: justification for K-ras-directed therapy. Mol Cancer Res. 2005;3(7):413–23. [DOI] [PubMed] [Google Scholar]
- 67.Zorde Khvalevsky E, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(51):20723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cancer Cell Line Encyclopedia C, Genomics of Drug Sensitivity in Cancer C. Pharmacogenomic agreement between two cancer cell line data sets. Nature. 2015;528(7580):84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perez-Mancera PA, Guerra C, Barbacid M, Tuveson DA. What we have learned about pancreatic cancer from mouse models. Gastroenterology. 2012;142(5):1079–92. [DOI] [PubMed] [Google Scholar]
- 72.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–50. [DOI] [PubMed] [Google Scholar]
- 73.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. [DOI] [PubMed] [Google Scholar]
- 76.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20(22):3147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122(2):639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63(9):2005–9. [PubMed] [Google Scholar]
- 80.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. [DOI] [PubMed] [Google Scholar]
- 81.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19(6):728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018;8(9):1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158(1):171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158(1):185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hou P, Kapoor A, Zhang Q, Li J, Wu CJ, Li J, et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020;10(7):1058–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–62. [DOI] [PubMed] [Google Scholar]
- 88.Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–23. [DOI] [PubMed] [Google Scholar]
- 89.Van Cutsem E, Hidalgo M, Canon JL, Macarulla T, Bazin I, Poddubskaya E, et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int J Cancer. 2018;143(8):2053–64. [DOI] [PubMed] [Google Scholar]
- 90.Javle MM, Shroff RT, Xiong H, Varadhachary GA, Fogelman D, Reddy SA, et al. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer. 2010;10:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27(2):193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karavasilis V, Samantas E, Koliou GA, Kalogera-Fountzila A, Pentheroudakis G, Varthalitis I, et al. Gemcitabine Combined with the mTOR Inhibitor Temsirolimus in Patients with Locally Advanced or Metastatic Pancreatic Cancer. A Hellenic Cooperative Oncology Group Phase I/II Study. Target Oncol. 2018;13(6):715–24. [DOI] [PubMed] [Google Scholar]
- 93.Kordes S, Klumpen HJ, Weterman MJ, Schellens JH, Richel DJ, Wilmink JW. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother Pharmacol. 2015;75(6):1135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients With Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017;3(4):516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25(4):620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell Metab. 2017;25(5):1037–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med. 2017;9(392). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Konstantinidou G, Ramadori G, Torti F, Kangasniemi K, Ramirez RE, Cai Y, et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 2013;3(4):444–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rebecca VW, Alicea GM, Paraiso KH, Lawrence H, Gibney GT, Smalley KS. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2014;27(6):1154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7(1):86–93. [DOI] [PubMed] [Google Scholar]