

Abstract
The neuropeptide Y (NPY) family is a peptide-activated G protein-coupled receptor system conserved across all bilaterians, and is involved in food intake, learning, and behavior. We hypothesized that comparing the NPY system in evolutionarily ancient organisms can reveal structural determinants of peptide recognition and receptor activation conserved in evolution. To test this hypothesis, we investigated the homologous FLP/NPR system of the protostome C. elegans. For three prototypic peptide–receptor complexes representing different ligand types, we integrate extensive functional data into structural models of the receptors. Common features include acidic patches in the extracellular loops (ECLs) of the receptors that cooperatively ‘draw’ the peptide into the binding pocket, which was functionally validated in vivo. A structurally conserved glutamate in the ECL2 anchors the peptides by a conserved salt bridge to the arginine of the RFamide motif. Beyond this conserved interaction, peptide binding show variability enabled by receptor-specific interactions. The family-conserved residue Q3.32 is a key player for peptide binding and receptor activation. Altered interaction patterns at Q3.32 may drastically increase the efficacy to activate the receptor.
Keywords: G protein-coupled receptor (GPCR), FLP neuropeptide, binding pocket, structure-activity relationship, C. elegans
Graphical Abstract
INTRODUCTION
A substantial portion of G protein-coupled receptors (GPCRs) is activated by endogenous peptide or protein ligands. These peptide and protein GPCRs are involved in many physiological processes and are important pharmacological targets. Understanding how peptides interact with their receptors is an essential step towards the development of more potent and selective probe molecules that can validate these receptors as targets and ultimately spur therapeutic development. Specifically, the recognition of the large and flexible peptides by the receptor and the mechanisms for receptor-subtype selectivity are intensively studied.
Reflecting their essential physiological functions, many peptide GPCR systems are conserved down to basic animals, which opens up avenues to study conserved aspects of peptide–receptor interactions, and a potential co-evolution of the binding pockets. Among several other systems, the neuropeptide Y (NPY) system has been present already in the common ancestor of protostomes and deuterostomes, the ‘ur-bilaterian’ [1,2], and is thus expected to have homologs in all bilaterians. In humans, the NPY family has an essential role in regulating food intake and is intensively studied as a potential target for modulating food consumption in the context of obesity [3–5]. Additional functions of the NPY system include the regulation of memory retention, mood and anxiety [6–9]. NPY and the related peptides PYY and PP activate four cognate GPCRs (Y1, Y2, Y4 and Y5) in humans. The peptides feature a C-terminal arginine-phenylalanine/tyrosine sequence with an amidated carboxyl-terminus (-RxRF/Yamide), which is essential for their activity. High-resolution structural insights of NPY binding to its cognate receptors are currently lacking, but structural models suggest that the arginine residues of this sequence contact a conserved D6.59 on the top of transmembrane helix 6 (TM6) of the receptors, and that the C terminus of the peptide accommodates a binding pocket in the TM bundle [10–12].
Recently, we showed that the FMRFamide-like peptide (FLP)/neuropeptide receptor-resemblence (NPR) system of the protostome Caenorhabditis elegans is, beyond sequence similarities, pharmacologically highly similar to the human NPY system [13]. Most notably, the peptide ligands feature cross-species activity, and human NPY receptors can functionally replace C. elegans npr-1 null mutants in an in vivo context [13]. The FLP/NPR system in C. elegans is expanded and comprises more than 70 FLP ligands (incl. isoforms originating from local genome duplications) and 40 NPR receptors [14,15] with apparently redundant activation profiles [13]. Interestingly, many of the short peptides only contain a minimal C-terminally conserved –ϕRFamide (ϕ=hydrophobic) sequence, and lack a second Arg/Lys residue at the −4 position which still suffices for receptor activation. To facilitate comparison to human NPY-like peptides, positions are counted from the C terminus. FLP-21 (GLGPRPLRFamide) activates several NPRs. It features nanomolar potencies for NPR-1 and NPR-11, and also activates the human Y2 and Y4 receptor with sub-micromolar potency [13]. This peptide carries a second arginine residue at the −5 position (one position displaced from the ‘classic’ −4 position of NPY). Recently, three FLPs with increased length and a ‘classic’ C-terminal -RxRF/Yamide have been functionally characterized (FLP-27, FLP-33, FLP-34), which act at NPR-11 and human NPY receptors [13], but have limited activity at other NPRs [13]. Isoform 1 of FLP-34 (FLP-34–1: ADISTFASAINNAGRLRYamide), activates NPR-11 with nanomolar potency.
Here, we illuminate how FLP-21 and FLP-34–1 bind NPR-1 and NPR-11 to resolve the contribution of the C-terminal -RFamide and –RxRYamide motif, respectively, for receptor selectivity and the activation process. For each of the three peptide–receptor combinations (FLP-21 – NPR-1; FLP-21 – NPR-11; FLP-34–1 – NPR-11), we integrate experimental restraints into a comparative model, and dock the peptides with full flexibility, which converged into well-defined binding poses. We demonstrate that binding of the FLP ligands is heavily dependent on the conserved C-terminal motifs, which involves a salt bridge of the arginine at the −2 position (RFamide) to a conserved acidic receptor residue in the ECL2. Additional acidic clusters aid the guidance of the peptides into the binding pocket. Moreover, the C-terminal Phe/Tyr-amide is required for both, affinity to the receptor and receptor activation. The shape of the binding pocket and the receptor residues involved in peptide binding are conserved to a high degree in the human NPY receptors, suggesting a similar mode of action. Mutation of the family-wide conserved Q3.32 in the transmembrane binding pocket of NPR-11 leads to an altered binding mode of FLP-21 at this receptor, which increases G protein signaling, thus identifying interactions for efficacious receptor activation.
RESULTS
The C-terminal residues of FLP-21 are critical for binding and activation of NPR-1.
One aim of this study was to characterize the binding and activation of two NPRs (NPR-1 and NPR-11) with two prototypic FLP ligands (FLP-21 and FLP-34–1 prototypic for C-terminal - RFamide and -RxRYamide motifs, respectively), and compare this with the human NPY receptors.
We first set out to identify the residues in the 9 aa FLP-21 that are important for binding and activation of NPR-1. We performed a structure-activity study with several peptide variants to assess the importance of the conserved C-terminal sequence in comparison to the N-terminal part of the peptide (Figures 1, 2) for activation of NPR-1, measured as decrease of cellular cAMP levels. R8 was very sensitive to exchanges. Removing the charge in a [R8Q]-FLP-21 rendered the peptide inactive up to 10 μM concentration, just like a [R8A] variant, and even the very mild exchange to homoarginine (R8hArg), keeping the charge but extending the residue length by one methylene group, resulted in > 900-fold loss of potency to activate the receptor (Figure 1b). At position F9, hydrophobic bulk, but also the aromaticity was important, as [F8L]-FLP-21 was almost inactive, and [F8Cha]-FLP-21 (Cha, cyclohexylalanine) increased the EC50 by > 600-fold. Adding a hydroxyl group to F9 (F9Y) was tolerated relatively well, and resulted in a mild 14-fold loss of receptor activation. Still, these data indicate that the binding pocket is relatively narrow (Figure 1b). We also investigated the C-terminal amidation, a conserved feature of FLPs as well as NPY peptides of chordates. Removing the amidation to generate a free acid (-CONH2 to -COOH), or the further deletion of the entire carboxyl group ([F9tyramide]-FLP-21) resulted in inactive peptides up to concentrations of 10 μM (Figure 1b). This suggests that the amidation is sterically required, but the negative charge of the free acid under physiological conditions interferes with binding.
Figure 1. The conserved C terminus of FLP-21 (-RFamide) is highly important for binding and activation of the C. elegans NPR-1 receptor.
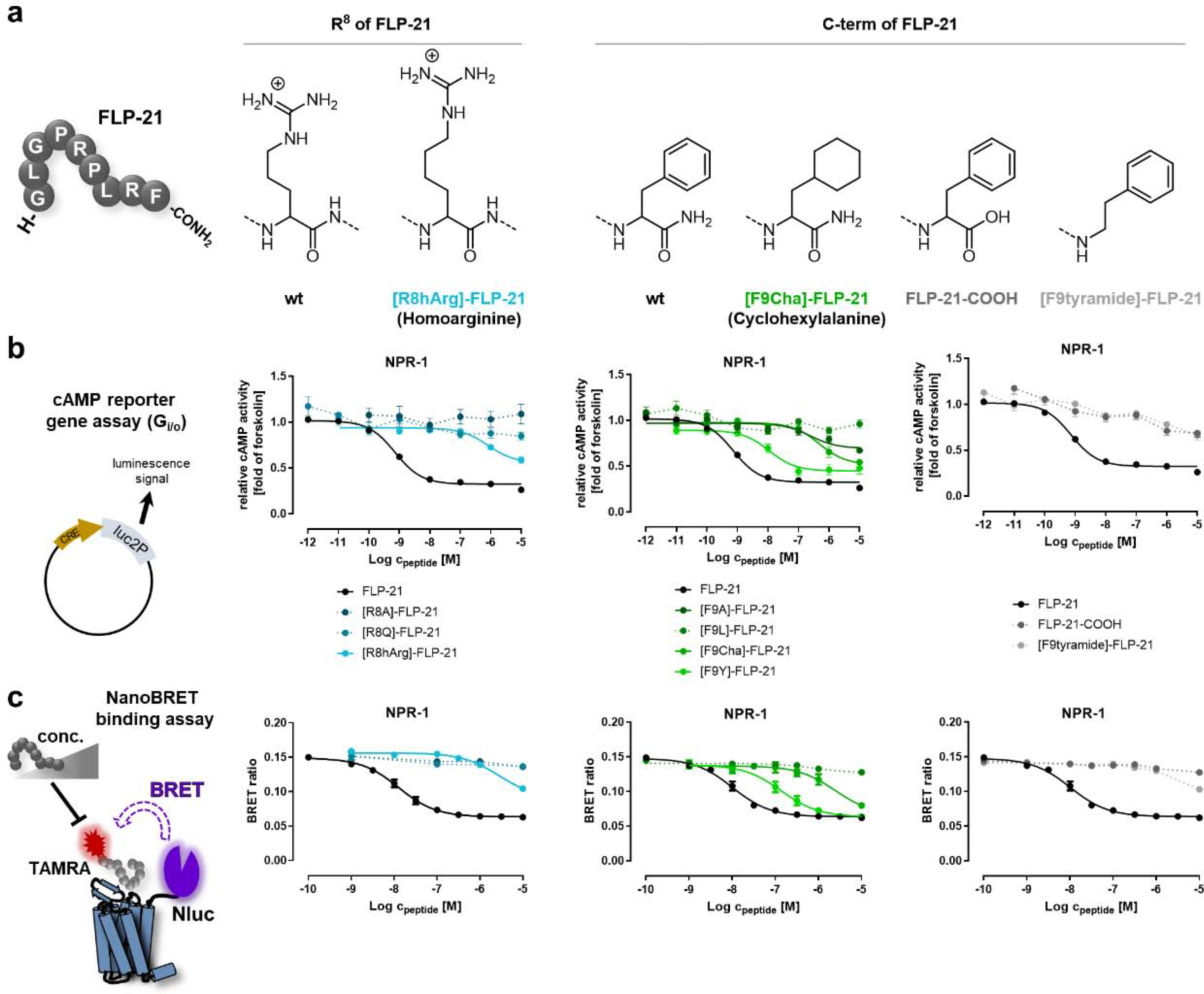
a) Amino acid sequence of FLP-21 and chemical structure of unnatural amino acid exchanges in comparison to the wild type peptide. R8 of FLP-21 was replaced with homoarginine (hArg), F9 was replaced with cyclohexylalanine (Cha). C-terminal amidation (wild type) was either deamidated (FLP-21-COOH) or the entire carboxygroup was removed ([F9tyramide]-FLP-21). b) Each position in the conserved C-terminal RFamide sequence is highly important for activating NPR-1 and even very weak modifications of these positions drastically impair the activity as measured by a cAMP reporter gene assay. NPR-1 couples to Gαi/o, thus, receptor activity was measured by the inhibition of cAMP production induced by 5 μM forskolin (set to 1.0), i.e. lower values correspond to high receptor activity. c) C-terminal variants of FLP-21 also display very weak affinity to the receptor, as seen in a NanoBRET displacement binding assays. For this purpose, the C. elegans receptors were fused at their N terminus with a nanoluciferase (Nluc), while peptides were modified with a tetramethylrhodamine (TAMRA) fluorophore at their N terminus. Addition of unlabeled peptide displaces the TAMRA-labeled peptide, visible as a decrease in BRET signal. All data represent mean ± SEM of n≥3 independent experiments conducted in technical triplicate.
Figure 2. C-terminal FLP-21 modifications lead to a drastic loss of NPR-1 receptor activation, while changes in the more N-terminal part are tolerated well.
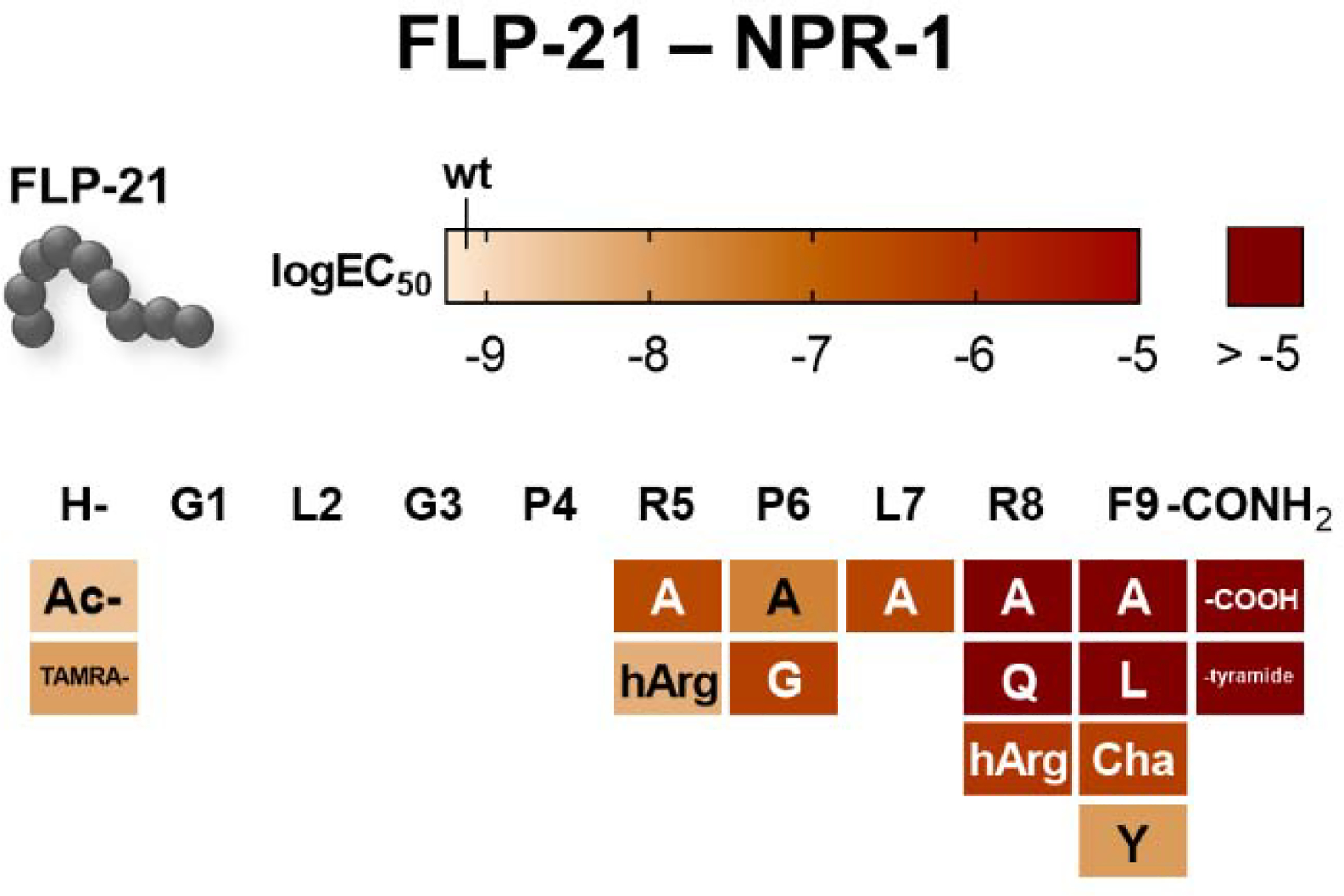
The activity was tested cAMP reporter gene assay (Gi/o). The activity of all synthesized FLP-21 variants is shown as a heatmap. LogEC50 values are encoded in a color gradient (light orange to dark red, high to low potency). FLP-21 wild type activity is indicated in the legend bar. Substitutions are in one-letter code (standard amino acids) or three-letter code (unnatural substitutions; hArg: homoargenine; Cha: cyclohexylalanine). N terminus (H-) was either acetylated (Ac-) or labeled with a tetramethylrhodamine fluorophore (TAMRA-). C-terminal amidation (indicated as -CONH2) was either deamidated (-COOH) or the entire carboxygroup was removed (-tyramide).
To resolve whether these variations of the C-terminal RFamide motif also cause a similar loss of binding or merely impair the activation process itself, we devised a displacement binding assay based on NanoBRET between a nanoluciferase (Nluc) that is genetically fused to the N terminus of the receptor and tetramethylrhodamine-labeled FLP-21 ([TAMRA]-FLP-21; Figure 1c). We verified the functionality of the Nluc-fused receptor and TAMRA-modified peptide variants (SI: Table S1, S2). The peptides containing variations of the RF-amide had severely impaired binding affinities (SI: Table S3), which basically mirrored the results of the signaling assay (SI: Table S2). For instance, the Ki of [F9Y]-FLP is shifted by 11-fold, matching the 14-fold shift of the EC50 in the cAMP assay relative to wild type FLP-21. This demonstrates that all of the chemical entities in the C terminus are required for receptor binding and activation (Figure 1c).
Next, we investigated residues in the N-terminal and central part of FLP-21, which are less conserved within FLPs (summarized in Figure 2, SI: Table S2, S3). One particularly interesting residue is a second arginine R5 (−5 position). Substitution of R5 of FLP-21 to alanine resulted in a 368-fold decreased EC50 in the cAMP assay, suggesting a high relevance for receptor activation, although the effect is not as drastic as for [R8A]-FLP-21 (Figure 2, SI: Table S2). Two hydrophobic residues, P6 and L7, space R5 and R8. This hydrophobicity significantly contributes to FLP-21 binding at NPR-1, as alanine substitutions decreased the activity by 34- and 497-fold for [P6A]-FLP-21 and [L7A]-FLP-21, respectively (Figure 2, SI: Table S2). The variant [P6G]-FLP-21 even had a > 500-fold shifted EC50 compared to the wild type peptide. Since proline and glycine can occupy similar backbone conformations, we attributed this effect to the lack of hydrophobicity rather than conformational restriction.
In contrast, modifications at the N terminus of FLP-21 did not severely affect receptor activation. Acetylation of the free N terminus of the peptide or even the addition of the bulky TAMRA fluorophore resulted in only 3-fold and 12-fold reduced potency in receptor activation, respectively (Figure 2, SI: Table S2), indicating that binding of FLP-21 to NPR-1 is mediated foremost through the C terminus, while the N terminus is solvent exposed.
In the next step, we investigated the binding pocket of NPR-1 (Figure 3a–e, SI: Table S1). The initial set of receptor residues we analyzed covered functionally important positions of the human NPY receptors, which are conserved in NPR-1. These were T2.61 and Q3.32, putatively serving as a polar anchor for the amidated C terminus and side chain of Q34 of NPY at the Y2R [11], the ‘toggle switch’ residue W6.48 [12,16], and two acidic residues, D/E6.59 and E5.27(ECL2) (E5.24 for NPY receptors), that are suggested to bind the arginines of the NPY peptides [10–12,17]. Even though the residue numbering seems to differ, the position of the E5.24/5.27 in the ECL2 is conserved, and is located two residues next to the conserved disulfide bridge in the ECL2 (equivalent to position 45.52 in the current GPCRdb numbering [18], but we chose to retain the numbering as used for the human NPY receptors for comparison). Moreover, we extended our selection by residues that point into the putative peptide binding pocket in a homology model of NPR-1 (see methods for details on homology modeling), and are possible complements to the important arginine and hydrophobic/aromatic residues of the peptide: W2.70, M4.57, Y4.60, F5.24, T5.39, I6.58, D6.61, D6.62, D7.26, D7.27, Y7.28, and Y7.32.
Figure 3. Identification of critical positions of NPR-1 for receptor activation and contact points to the ligand FLP-21.
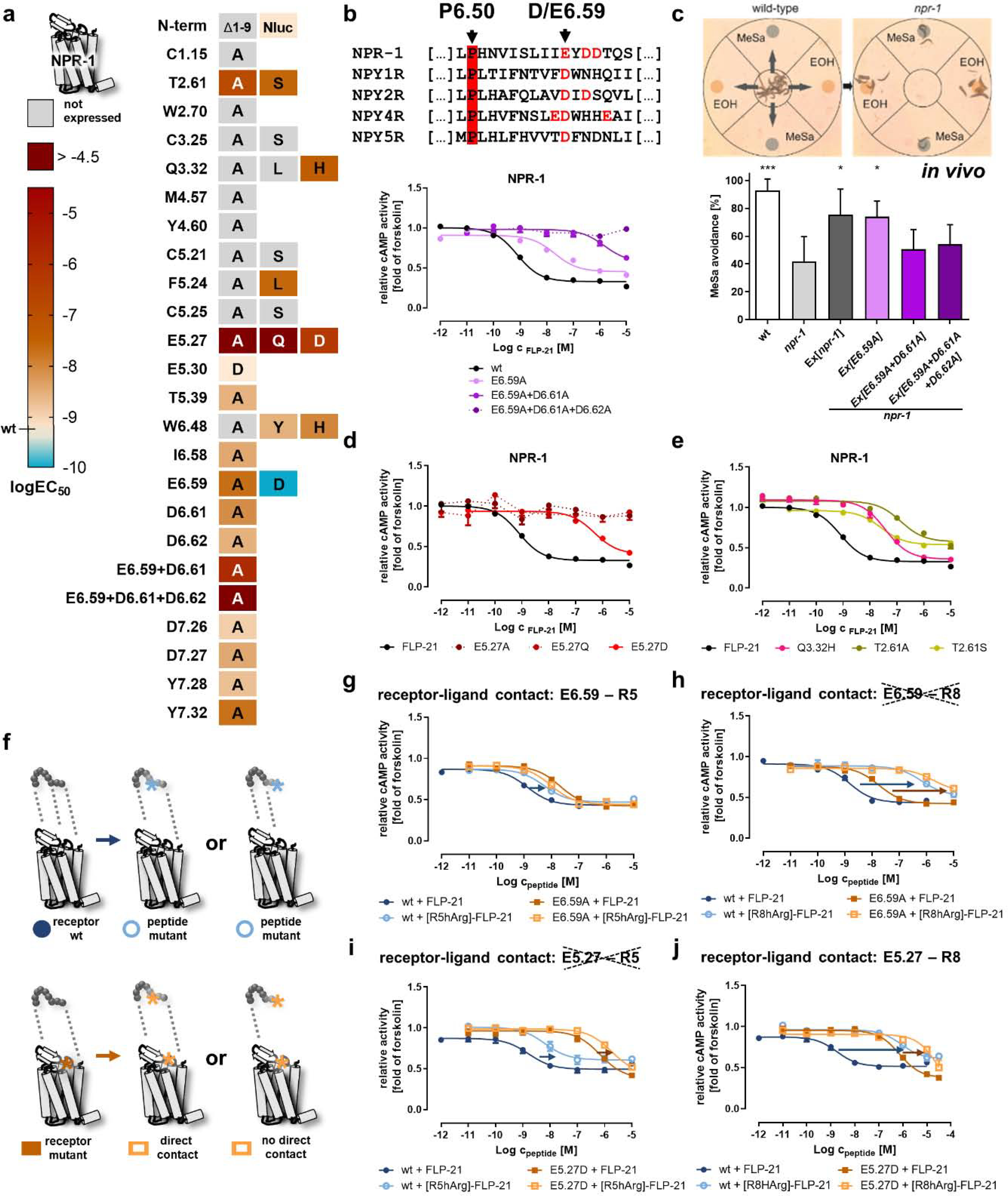
a) Activity of NPR-1 variants depicted as heatmap. The logEC50 values were determined in a cAMP reporter gene assay (Gi/o), and are color-coded from light orange (wild type, high potency) to dark red (logEC50 > −5). A gain of function is indicated by cyan color. The nomenclature of the receptor positions follows Ballesteros und Weinstein [55]. “Not expressed” indicates receptor variants that are not correctly folded and transported to the plasma membrane (cf. SI: Figure S1). Selected concentration-response curves are shown in panels b, d, e. b) Alignment of TM6/ECL3 residues of NPR-1 with the human NPY receptors. A cluster of acidic residues in NPR-1 is shown in red, and the conserved D/E6.59 is indicated by an arrow. Mutation of this acidic cluster gradually decreases receptor activity. c) The acidic cluster in TM6/ECL3 is also critical for receptor function in vivo in C. elegans. This is measured by phenotypic rescue of a npr-1 null mutant in a methyl salicylate (MeSa) avoidance assay (see methods for details). npr-1 deficient animals lose MeSa avoidance, which is rescued by transgenic expression of npr-1 driven by a npr-1 promoter (Ex[npr-1]). The single exchange of E6.59 to alanine in NPR-1 still gives sufficient receptor activity for a phenotypic rescue (Ex[E6.59A]), while the combination mutants Ex[E6.59A+D6.61A] and Ex[E6.59A+D6.61A+D6.62A] are indistinguishable from the npr-1 null mutant. Data are shown as mean ± SD of n ≥ 4 independent experiments (N ≥ 60 worms in each experiment). *p < 0.05; ***p < 0.001 compared to the npr-1 null mutant. d) The receptor position E5.27 in ECL2 is highly important for receptor activation and even a very mild mutation (E5.27D) leads to a drastic EC50 shift of > 400-fold, while exchanges to glutamine or alanine are expressed, but not activatable up to peptide concentrations of 10 μM. e) In addition to the two acidic clusters in TM6 and ECL2, mutation of two hydrophilic positions T2.61 and Q3.32 decreases receptor activation by > 30-fold. f) Double-cycle mutagenesis to find interacting residues between NPR-1 and FLP-21. Mutation of a functionally important ligand position eliminates one binding interaction (light blue) compared to the wild type situation (dark blue), which leads to a decreased receptor activity (first round; upper panel). In the second round (lower panel), the receptor is mutated (light orange). If the two tested positions interact, this will not further affect the receptor activity, as the corresponding interaction had been eliminated by the peptide mutation before (lower panel, middle). In contrast, if the receptor mutation does not interact with the tested ligand position, the receptor activity is further decreased (lower panel, right). g/h/i/j) Receptor positions E6.59 and E5.27 were tested against [R5/8]-FLP-21 mutants. EC50 shifts are indicated with arrows (blue/orange). A reduced shift and thus a direct interaction (smaller orange arrow compared to blue arrow) was found for [R5A]-FLP-21 to E6.59A and for [R8hArg]-FLP-21 to E5.27D, but not for the other combinations. b, d, e, g-j: Data represent x-fold of forskolin (mean ± SEM) of n ≥ 3 independent experiments.
All NPR-1 receptor mutants contained an enhanced yellow fluorescent fusion protein (eYFP) at their C terminus, and were first analyzed for correct folding and location to the plasma membrane in live cell fluorescence microscopy (SI: Figure S1). Correctly expressed NPR-1 mutants were tested in cAMP reporter gene assays with wild type FLP-21 (summarized in Figure 3a).
The most outstanding feature of the binding pocket in NPR-1 is the clustering of several acidic residues in the ECL3 that might serve as potential anchors for the arginine residues of the peptide ligand. The mutation of residues D7.26 and D7.27 (ECL3) to alanine did not severely affect receptor activation (EC50 shifts < 5-fold). In contrast, mutation of residues E6.59, D6.61 and D6.62 in the ECL3 towards TM6 gradually decreased receptor activation (Figure 3b). The effect of the single alanine mutants was 4 to 25-fold (SI: Table S1), but already the combination variant E6.59A+D6.61A displayed nearly 1900-fold increased EC50 values, and the triple mutant E6.59A+D6.61A+D6.62A was inactive up to peptide concentrations of 10 μM. The additional EC50 shifts of the double/triple mutants by far exceed the values that can be expected by simple additive effects (~250-fold for E6.59A+D6.61A, calculated from SI: Table S1), indicating these residues act in a cooperative manner. To verify that this is also the case in vivo, we used our previously established methyl salicylate (MeSa) avoidance rescue assay [13]. Loss of NPR-1 in C. elegans alters certain chemoattractance/~avoidance behaviors, and results in different responses to substances such as MeSa [19]. Wild type C. elegans (N2) avoid MeSa, a behavior which is greatly reduced in npr-1 null mutants (Figure 3c). This phenotype can be rescued by transgenic expression of npr-1 driven by the npr-1 promoter (Ex[npr-1], positive control). Strikingly, an npr-1 construct with the single E6.59A mutation still leads to a phenotypic rescue, but the double/triple combination of the mutation with D6.61A and D6.62A was inactive, underlining the cooperative function of this cluster in vivo.
The second highly conserved acidic residue in NPR-1 is located near the conserved disulfide bridge in the ECL2. Mutation of this structurally conserved residue E5.27 to alanine or glutamine results in no measurable receptor activation with peptide concentrations up to 10 μM, indicating that this residue is critical for receptor activation (Figure 3d). Even a very mild mutation of E5.27 to aspartate, thereby shortening the length of the side chain by one methylene group, results in a drastic > 600-fold decreased potency of FLP-21 to activate the receptor.
Having identified important residues at the peptide and at the receptor side, we focused on pinpointing the peptide–receptor interactions by double-cycle mutagenesis [20]. In the first cycle (mutagenesis at the peptide), a peptide mutant shows decreased activity when tested with the wild type receptor, indicating loss of a specific binding interaction. This peptide variant will display the same activity at a receptor that is mutated at the exact position that interacts with the mutated position of the peptide (second cycle, mutagenesis of the peptide combined with the receptor mutant). In contrast, if the peptide is mutated in a position that belongs to another site of interaction, additional effects will be observed (Figure 3f).
Using this strategy, we identified two salt bridges between of R5 and R8 of FLP-21 to E6.59 and E5.27 of NPR-1, respectively (Figure 3g–j; SI: Table S4). [R5hArg]-FLP-21 has a 7-fold shifted EC50 at the wild type receptor (EC50 = 5.2 nM versus 0.8 nM for wild type FLP-21, blue arrow in Figure 3g). The receptor variant E6.59A displayed a 25-fold shifted EC50 relative to the wild type NPR-1 when stimulated with FLP-21 (EC50 = 20 nM). Combining peptide and receptor mutations did not induce a further rightward-shift of the EC50 (EC50 = 9.4 nM, 0.5-fold of E6.59A–FLP-21; orange curves in Figure 3g), as expected when these positions directly interact. We also tested the NPR-1 mutant E6.59D. When stimulated with wild type FLP-21, this receptor mutant displayed a 6-fold improved EC50 compared to the wild type receptor (EC50 0.13 nM), indicating that a reduced distance between R5 and E6.59D even increases the activity of the complex. Stimulating this receptor mutant with [R5hArg]-FLP-21, thus re-introducing the ‘missing’ methylene group at the peptide side, then again led to a 5-fold rightward-shift of the EC50, and restored the wild type situation (SI: Table S4), underlining an interaction of R5 of FLP-21 to E6.59 of NPR-1. We conducted an analogous series of experiments using the mutants D6.61A and D6.62A. These residues, however, did not specifically interact with R5 of FLP-21, while the combination mutant E6.59A+D6.61A confirmed the data of the E6.59A single mutant (SI: Table S4). We further crosschecked if R8 of FLP-21 might simultaneously interact with position E6.59 of NPR-1 (Figure 3h, SI: Table S4). Stimulation of the E6.59A mutant of NPR-1 with [R8hArg]-FLP-21 resulted in 91-fold less potent receptor activation compared to wild type FLP-21 at this receptor mutant. This further loss of activity argues against a strong direct interaction for this combination. However, the additional loss in potency for this peptide variant was less than at the wild type receptor (91-fold versus 976-fold, orange versus blue arrow in Figure 3h, SI: Table S4), which may indicate a partial interaction.
The contact pattern of receptor position E5.27 to the arginine residues of FLP-21 is reversed. The receptor mutant E5.27D displays a > 600-fold shifted EC50 relative to the wild type receptor (EC50 = 495 nM) when stimulated with FLP-21. Stimulation of this NPR-1 mutant with [R5hArg]-FLP-21 resulted in an additional loss of potency, similar to the stimulation at the wild type receptor (blue/orange arrows, 6.6-fold to 3.7-fold shift, respectively, Figure 3i). On the contrary, a R8hArg exchange in FLP-21 was much less detrimental for the activation of the E5.27D mutant of NPR-1 compared to the wild type receptor (Figure 3j; orange/blue arrows, 34-fold to 976-fold shift, respectively), suggesting a direct interaction between these residues.
Taken together, binding of FLP-21 to NPR-1 is heavily dependent on the five C-terminal amino acids, in particular the conserved -RFamide. Two salt bridges between [R5]-FLP21 to E6.59, and [R8]-FLP-21 to E5.27 were identified by double-cycle mutagenesis and can be applied to guide the computational docking of the peptide into a homology model of NPR-1.
The C-terminal amino acids of FLP-21 are most important for binding and activation of NPR-11, in a similar way as to NPR-1.
The receptor NPR-11 is also activated by FLP-21, albeit with 64-fold reduced potency (EC50 51 nM) compared to FLP-21–NPR-1 (EC50 0.8 nM). There are a few specific differences in the binding pocket between NPR-1 and NPR-11 that might contribute to this difference in potency. NPR-11 sequence also contains the conserved acidic residue E5.23 (E5.27 in NPR-1, E5.24 in human NPY receptors) as well as an additional acidic cluster in ECL2 (E5.26, E5.29). However, the conserved acidic residue at the top of TM6 is not located at position 6.59 in NPR-11, but displaced to position E6.61, while there is a threonine at position 6.59. Further conserved residues of the NPY family, such as T2.61, Q3.32 or F7.35 are also present in NPR-11. In addition, we chose to mutate T2.64, M2.68, L4.51, I5.20, T5.39, Q5.46, and N6.58 based on their position in the receptor model (Figure 4a).
Figure 4. Identification of critical residues for activation of NPR-11 by FLP-21.
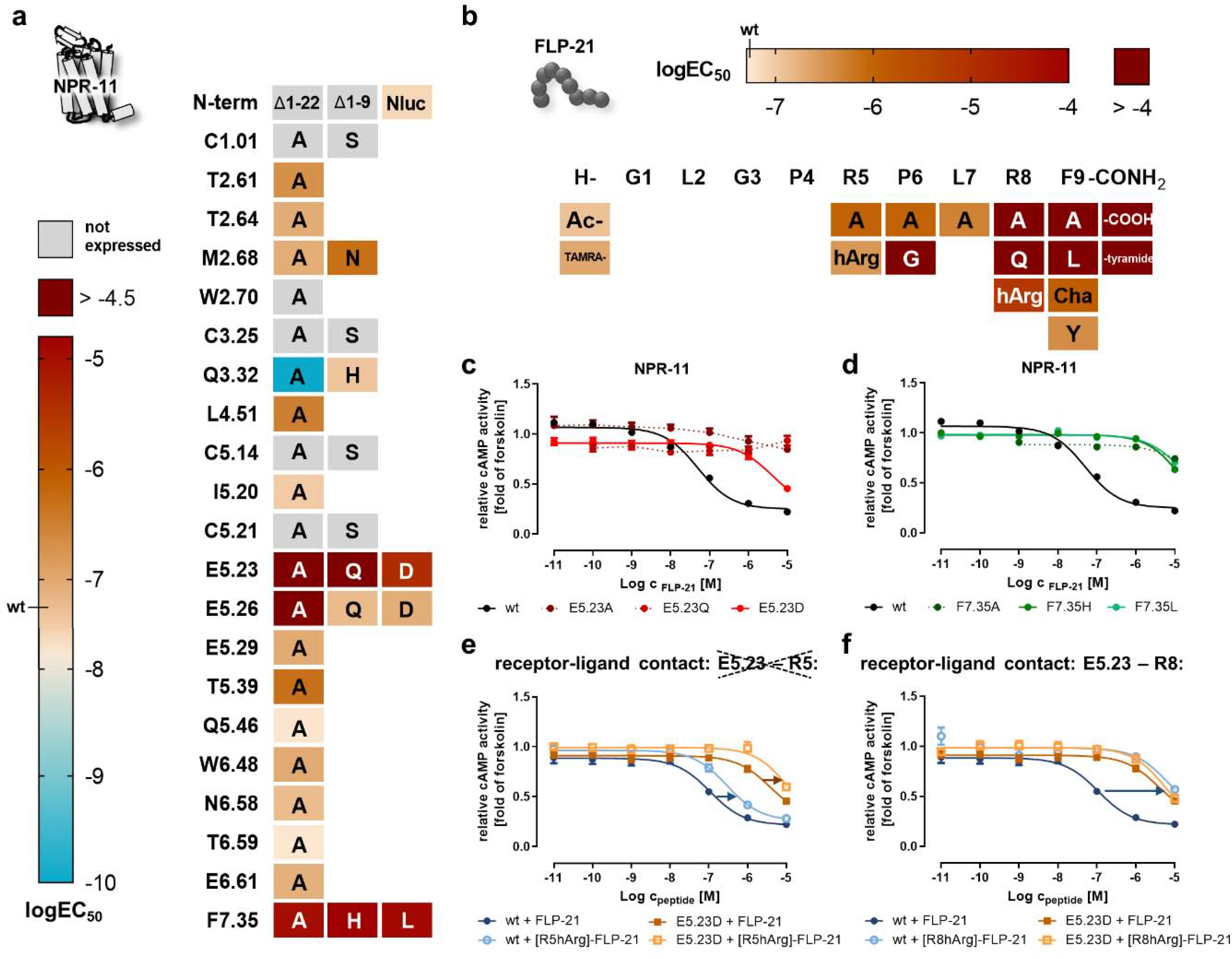
Peptide and receptor mutants were tested in a cAMP reporter gene assay (Gi/o) to derive logEC50 values. a) Heatmap of the activity of mutated NPR-11 positions. The logEC50 values are color-coded from light orange (wild type; high potency) to dark red (logEC50 > −4.5). A gain of function is indicated by cyan color. The nomenclature of the receptor positions follows Ballesteros and Weinstein [55]. “Not expressed” indicates receptor variants that are not correctly folded and transported to the plasma membrane (cf. SI: Figure S2). Selected concentration-response curves are shown in panels c and d. b) Heatmap showing the activity of FLP-21 ligand variants. The logEC50 values are color-coded from light orange (wild type) to dark red (logEC50 > −4). c) The receptor position E5.23 in ECL2 is highly important for receptor activation and even a very mild mutation (E5.23D) leads to a drastic EC50 shift of >70-fold, while exchanges to glutamine or alanine are expressed, but are not activated by FLP-21 up to a concentration of 10 μM. d) Mutation of receptor position F7.35 to alanine, histidine or leucine reduces receptor activation >250-fold. e/f) Receptor position E5.23 was tested against position R5 (e) or R8 (f) of FLP-21 to pinpoint the interacting residue by double-cycle mutagenesis (cf. Figure 3f). EC50 shifts are indicated by arrows (blue/orange). Stimulating of NPR-11 mutant E5.23D with [R8hArg]-FLP-21 does not further increase the EC50 compared to stimulation with wild type FLP-21 (f), indicating a direct interaction. In contrast, stimulation of NPR-11 mutant E5.23D with [R5hArg]-FLP-21 produces a further rightward shift of the EC50 compared to FLP-21, undermining a direct contact (e). c-f: Data represent x-fold of forskolin (mean ± SEM) of n ≥ 3 independent experiments.
On the peptide side, the effects of amino acid exchanges within the sequence of FLP-21 for binding and activation of NPR-11 were overall very similar to NPR-1 (Figure 4b). Changes to the N terminus of the peptide were tolerated well, while the five C-terminal amino acids are critical for binding and activating the receptor. [R8A]- or [R8Q]-FLP-21 were inactive up to concentrations of 10 μM, however, exchanging R8 to the longer hArg retained some activity and appeared less disruptive for activation of NPR-11 compared to NPR-1 (EC50 shift at NPR-11: 135-fold compared to 976-fold at NPR-1; SI: Table S2). At position F9, [F9A]- and [F9L]-as well as FLP-21-COOH or [F9tyramide]-FLP-21 were inactive. In contrast, [F9Y]-FLP-21 displayed wild type-like activity, and the non-aromatic [F9Cha]-FLP-21 peptide variant resulted in a moderate, 20-fold reduced EC50, suggesting that the binding pocket is large and does not absolutely require the aromaticity. The hydrophobicity at position P6 was similarly important at NPR-11 compared to NPR-1 (EC50 shift of [P6A]-FLP-21: 18-fold at NPR-11, compared to 34-fold at NPR-1), but mutation of L7 to alanine was tolerated relatively well with only 10-fold increased EC50, suggesting that this position is not essential for binding and activation of NPR-11. As for FLP-21 binding to NPR-1, changes in the potency of receptor activation and binding affinities occurred at the same scale (SI: Table S2, S3).
We investigated the peptide-binding pocket of NPR-11 analogously to the NPR-1. The correct folding and expression at the plasma membrane was verified for all NPR-11 variants by live cell fluorescence microscopy (SI: Figure S2), and then tested for activation by FLP-21 in cAMP reporter gene assays (logEC50 values are summarized in Figure 4a). The glutamate residue E5.23 in the ECL2 of NPR-11 was essential for the receptor activation by FLP-21, in a similar manner to E5.27 in NPR-1 receptor (Figure 4c). While mutation of E5.23 to alanine or glutamine showed no receptor activation with peptide concentrations up to 10 μM, the very mild mutation to aspartic acid, reducing the length of the side chain by one methylene group, invoked a >70-fold decreased potency of receptor activation. The nearby residue E5.26 also contributed to receptor activation as removing the charge at this position in the E5.26A mutant rendered the receptor inactive, while mutation to glutamine or aspartate, thus retaining a certain length and polarity, behaved like wild type.
Since E/D6.59 is not present in NPR-11, we hypothesized that E6.61 could take over its function. However, we found no significant difference between wild type NPR-11 (EC50 51 nM) and E6.61A mutant (EC50 81 nM) for receptor activation. Also residue T6.59, which replaces the otherwise conserved D/E6.59, and the adjacent N6.58 did not significantly contribute to the activation of NPR-11 by FLP-21, as the corresponding alanine mutants displayed wild type-like activities.
Mutation of T2.61 and Q3.32 affected NPR-11 much differently than NPR-1. A receptor variant carrying a T2.61A mutation only modestly decreased the EC50 by 4-fold. Mutation of Q3.32 to histidine did not affect activation of NPR-11 (in contrast to the same mutation at NPR-1). The variant Q3.32A (not expressed in NPR-1) surprisingly resulted in a dramatic gain of function (> 500-fold, EC50 0.002-fold of the wild type receptor), which was not readily intuitive and is discussed below. Furthermore, we identified F7.35 as an important bulky residue in NPR-11 (Figure 4d). The F7.35A mutant was inactive; introducing the smaller and more polar histidine, or the medium-sized hydrophobic leucine residue re-introduced some activity, but the EC50 was still shifted by > 200-fold.
Next, we aimed to identify the direct interaction partner of the highly important glutamate E5.23 of NPR-11, supposedly R5 or R8 of FLP-21, by double-cycle mutagenesis (Figure 4e–f, SI: Table S5). Replacing R5 of FLP-21 with homo-arginine leads to the same, relatively weak reductions in the potency of receptor activation at the wild type and E5.23D mutant of NPR-11 (blue/orange arrows in Figure 4e). This additional loss of function at the E5.23D mutant clearly suggests that E5.23 does not interact with R5 of FLP-21. On the other hand, a [R8hArg]-FLP-21 variant severely loses activity at the wild type receptor (135-fold impaired EC50, blue arrow in Figure 4f). However, such a loss of function of the mutated peptide compared to the wild type peptide does not occur at the E5.23D mutant (orange lines superimpose in Fig. 4f), because the underlying receptor-ligand interaction had been disturbed before by mutation of E5.23 and is not further weakened by changes at the peptide side. These data strongly indicate a direct interaction between R8 of FLP-21 and E5.23 of NPR-11. The interaction between R8 of FLP-21 and the conserved glutamate residue in the ECL2 is highly similar to the interaction between FLP-21 and NPR-1. In contrast to the binding pocket at NPR-1, R5 and L7 of FLP-21 only weakly affect binding and activation at NPR-11.
FLP-34–1 containing a C-terminal -RxRYamide motif activates NPR-11 more potently than FLP-21.
Out of all FLPs in C. elegans, FLP-34–1 is most similar to the human NPY peptide family. With 18 aa in length, it is a long FLP and the only one with a C-terminal tyrosine (all others carry a C-terminal phenylalanine). Moreover, FLP-34–1 contains the C-terminal RxRF/Yamide sequence with only one residue spacing the two C-terminal arginine residues, just as the chordate NPY family. FLP-34–1 activates NPR-11 with the highest potency (EC50 1.1 nM) out of all peptides tested [13] and is almost 50-fold more potent than FLP-21. Notwithstanding these differences in the potency of receptor activation, the binding affinity of wild type FLP-34–1 (Ki) at NPR-11 was 107 nM (SI: Table S3), which was about the same affinity as FLP-21 at this receptor (Ki = 79 nM, SI: Table S3). To understand the binding and activation of FLP-34–1 to NPR-11 (summarized in Figure 5), we first synthesized a series of peptide variants to assess the contribution of the single positions. As observed before, the changes in the potency of receptor activation induced by the mutated peptides were similar to the changes in the binding affinity (Figure 5B, Table S6). Substitution of either arginine in the C-terminal motif resulted in >900-fold decreased potency of receptor activation. Combination of both mutations (R15A+R17A) resulted in > 3500-fold decrease in receptor activation (SI: Table S6), which is far less than what would be expected from additive effects, thus suggesting both arginines are part of the same interaction network. The exact length of the R15 side chain appeared not so critical, as exchange to the longer homo-arginine was tolerated well (2-fold decrease of EC50), while mutation or R17 to hArg invoked a >70-fold decrease in receptor activation, suggesting a more tightly regulated interaction at R17 than at R15. L16 mutation to alanine of FLP-34–1 resulted in > 140-fold decrease in receptor activation, in analogy to the requirement of hydrophobicity at the −3/−4 positions in FLP-21. As expected, the very C-terminal Y18 residue is critical for the activity of the peptide and Y18A substitution rendered the peptide inactive up to 10 μM. In contrast, substituting the Y18 to phenylalanine did not change the activity of the peptide within the experimental error (EC50 1.2-fold of wild type FLP-34–1), indicating that the additional hydroxyl functionality does not take over major additional functionalities compared to the otherwise conserved C-terminal phenylalanine of FLPs at NPR-11. Moving more towards the N terminus of the peptide, mutation of G14 to alanine or glutamic acid, one position before the conserved C-terminal motif, was well tolerated (1.8-fold and 0.7-fold EC50 compared to FLP-34–1, respectively). Moreover, N-terminal modification of FLP-34–1 with a bulky TAMRA fluorophore did not affect receptor activation (EC50 0.4 nM, wild type FLP-34–1 1.1 nM), suggesting that the most C-terminal residues of the peptide predominantly determine the receptor interaction.
Figure 5. Identification of critical residues for activation of NPR-11 by FLP-34–1.
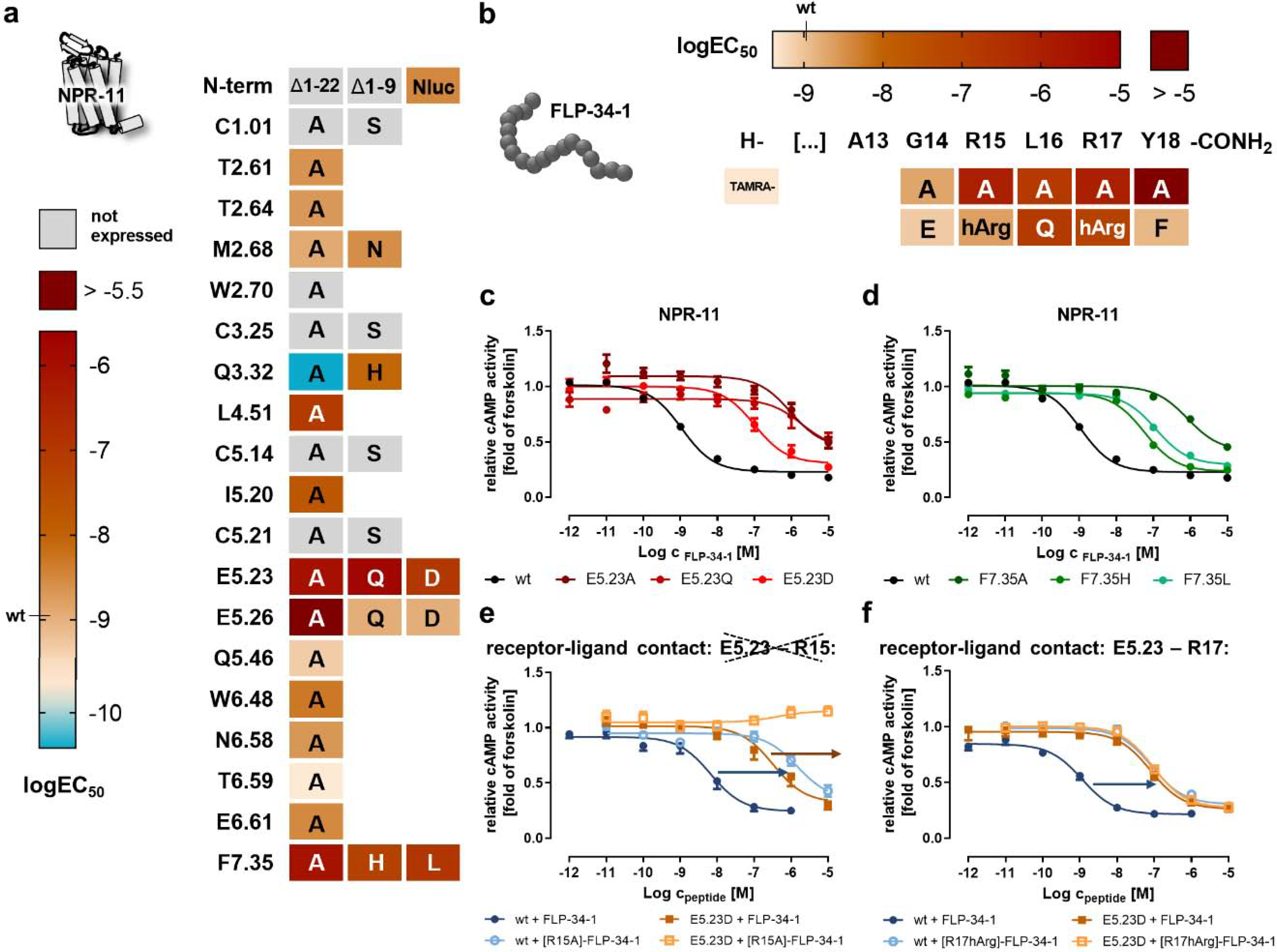
Peptide and receptor mutants were tested in a cAMP reporter gene assay (Gi/o) to derive logEC50 values. a) Heatmap of the activity of mutated NPR-11 positions. The logEC50 values are color-coded from light orange (wild type; high potency) to dark red (logEC50 > −5.5). A gain of function is indicated by cyan color. The nomenclature of the receptor positions follows Ballesteros and Weinstein [55]. “Not expressed” indicates receptor variants that are not correctly folded and transported to the plasma membrane (cf. SI Figure S2). Selected concentration-response curves are shown in panels c and d. b) Heatmap showing the activity of FLP-34–1 ligand variants. The logEC50 values are color-coded from light orange (wild type) to dark red (logEC50 > −5). c) The receptor position E5.23 in ECL2 is highly important for receptor activation and even a very mild mutation (E5.23D) leads to a drastic EC50 shift of >90-fold, while exchanges to glutamine or alanine are expressed, but are not activated by FLP-34–1 up to a concentration of 10 μM. d) Mutation of receptor position F7.35 to alanine, histidine or leucine reduces receptor activation > 50-fold. e/f) Receptor position E5.23 was tested against position R15 (e) and R17 (f) of FLP-34–1 to pinpoint the interacting residue by double-cycle mutagenesis (cf. Figure 3f). EC50 shifts are indicated by arrows (blue/orange). Stimulation of NPR-11 mutant E5.23D with [R17hArg]-FLP-34–1 does not further increase the EC50 compared to stimulation with wild type FLP-34–1 (f), indicating a direct interaction. In contrast, stimulation of NPR-11 mutant E5.23D with [R15hArg]-FLP-34–1 produces a further rightward shift of the EC50 compared to FLP-34–1, undermining a direct contact (e). c-f: Data represent x-fold of forskolin (mean ± SEM) of n ≥ 3 independent experiments.
On the receptor side, we tested the same NPR-11 receptor mutants with FLP-34–1 as before for FLP-21 (Figure 5a, SI: Table S7). Again, position E5.23 in the ECL2 appeared most critical for receptor activation by FLP-34-1, as removing the charge of the side chain resulted in almost 1000-fold reduced potency, and the receptor activity was also sensitive to E5.23D mutation (92-fold increased EC50, Figure 5c). At E5.26, the presence of a polar residue was required for receptor activation. In TM6 and TM7, N6.58, T6.59 and E6.61 did not significantly contribute to receptor activation, while the bulky and aromatic F7.35 was required, in a very similar manner to activation with FLP-21 (Figure 5d). As for the stimulation with FLP-21, mutation of Q3.32 to alanine resulted in a gain of function for the activation of NPR-11 by FLP-34–1, although by ‘only’ 20-fold (EC50 0.05-fold of wild type receptor). Interestingly, mutation of Q3.32 to histidine resulted in 8-fold decreased potency of receptor activation, suggesting that this position has a somewhat different role in the binding pocket of NPR-11 for FLP-34–1 compared to FLP-21. We observed additional differences at positions L4.51 and I5.20(ECL2). While these residues were not critical for activation of NPR-11 by FLP-21, mutation of these residues to alanine reduced the potency of receptor activation by FLP-34–1 by 76-fold and 15-fold, respectively.
Finally, we investigated the FLP-34–1 – NPR-11 complex for direct peptide–receptor interactions by double-cycle mutagenesis (Figure 5e–f, SI: Table S8). Since the effects of the acidic residues in the ECL2 for activation of NPR-11 by FLP-34–1 looked very similar to the situation with FLP-21, particularly at position E5.23, we reasoned that this residue might interact with R17 of FLP-34–1 (−2 position, analogous to R8 of FLP-21). This proved to be the case: The combination of E5.23D mutant of NPR-11 with [R17hArg]-FLP-34–1 did not further shift the EC50 of receptor activation compared to stimulation of this receptor variant with wild type FLP-34–1 (0.9-fold, orange curves in Figure 5f, compared to the 77-fold shift at the wild type receptor in blue), thus suggesting a direct interaction. In contrast, stimulating the E5.23D receptor mutant with [R15A]-FLP-34–1 induced a further rightward shift (orange arrow in Figure 5e), arguing against a direct interaction.
Taken together, the receptor activation and binding data highlight the critical role of the C-terminal RxRYamide motif of FLP-34–1. The position of the salt bridge is conserved between the −2 position of the peptide (R17 in FLP-34–1) and E5.23 in the ECL2, analogous to the interaction of FLP-21 at NPR-1 and NPR-11, while the exact positioning of R15 remained elusive. There was a strong overlap in the ligand-binding pocket with many of the receptor residues being important for activation by FLP-21 and FLP-34–1. However, residues L4.51 and I5.20(ECL2) have a unique function for mediating activation by FLP-34–1.
FLP-21 and FLP-34–1 docked into comparative models of NPR-1 and NPR-11 and their interfaces.
To get a better understanding of the peptide-binding pocket, we generated homology models of NPR-1 and NPR-11, and docked the ligands into these models using the generated functionality data as restraints [21]. The models were created based on three experimentally determined class A GPCR structures in a multi-template hybridization approach (Y1 (PDB code 5ZBQ); ETB (5GLI); K-OR-1 (4DJH)). These templates were selected based on the high sequence identity to NPR-1 and NPR-11, which was 29% and 35%, respectively (template alignment to NPR-1 and NPR-11 in SI: Table S9). NPR-1 and NPR-11 residues were threaded onto the coordinates of each template structure and hybridized into one comparative model for NPR-1 and NPR-11. Each homology model contains parts of the threaded structures complemented with de novo generated loops, in case of missing loop coordinates in the template structures. Homology modeling included an energy minimization step (see methods for details). NPR-1 and NPR-11 carry two cysteine residues in their ECL2. Based on the sequence alignments and similarity to the human NPY receptors, we chose the more distal cysteine with the CxE sequence to form the conserved disulfide bridge with TM3. The other cysteine residue may be involved in an additional disulfide bridge with the N terminus of the receptor. Accordingly, mutating any of the cysteines in the N terminus or ECL2 of NPR-1 and NPR-11 entailed severe misfolding of the receptor variants (Figures 3a, 4a, 5a, SI: Figure S1, S2), preventing more detailed experimental insights. The structural models of NPR-1 and NPR-11 show common structural features of peptide GPCR, with a wide-open binding pocket, lined by the ECL2 adopting two short antiparallel β-strands.
The peptides were docked into the homology models in an iterative approach that mimics conformational selection and induced fit (see methods for details). Experimentally derived contacts were included in these steps, leading to an energetic penalty if the given restraints are lost. The structures of FLP-21 docked into NPR-1 converged into one well-defined ‘solution’, i.e., structural cluster (Figure 6a). For the docking of FLP-21 and FLP-34–1 into NPR-11, three structural clusters appeared energetically favorable in each case. However, based on the activity data of peptide and receptor variants that were not included as structural restraints during the docking process, only one structural cluster satisfied the experimental data (Figure 6b, 6c). The top panel of Figure 6 shows a backbone superposition of the top 20 scoring models for each docking. The backbone positions of peptides in the best scoring models are very similar, indicating a high convergence of the generated clusters.
Figure 6. Structural models of peptide–receptor binding pockets.
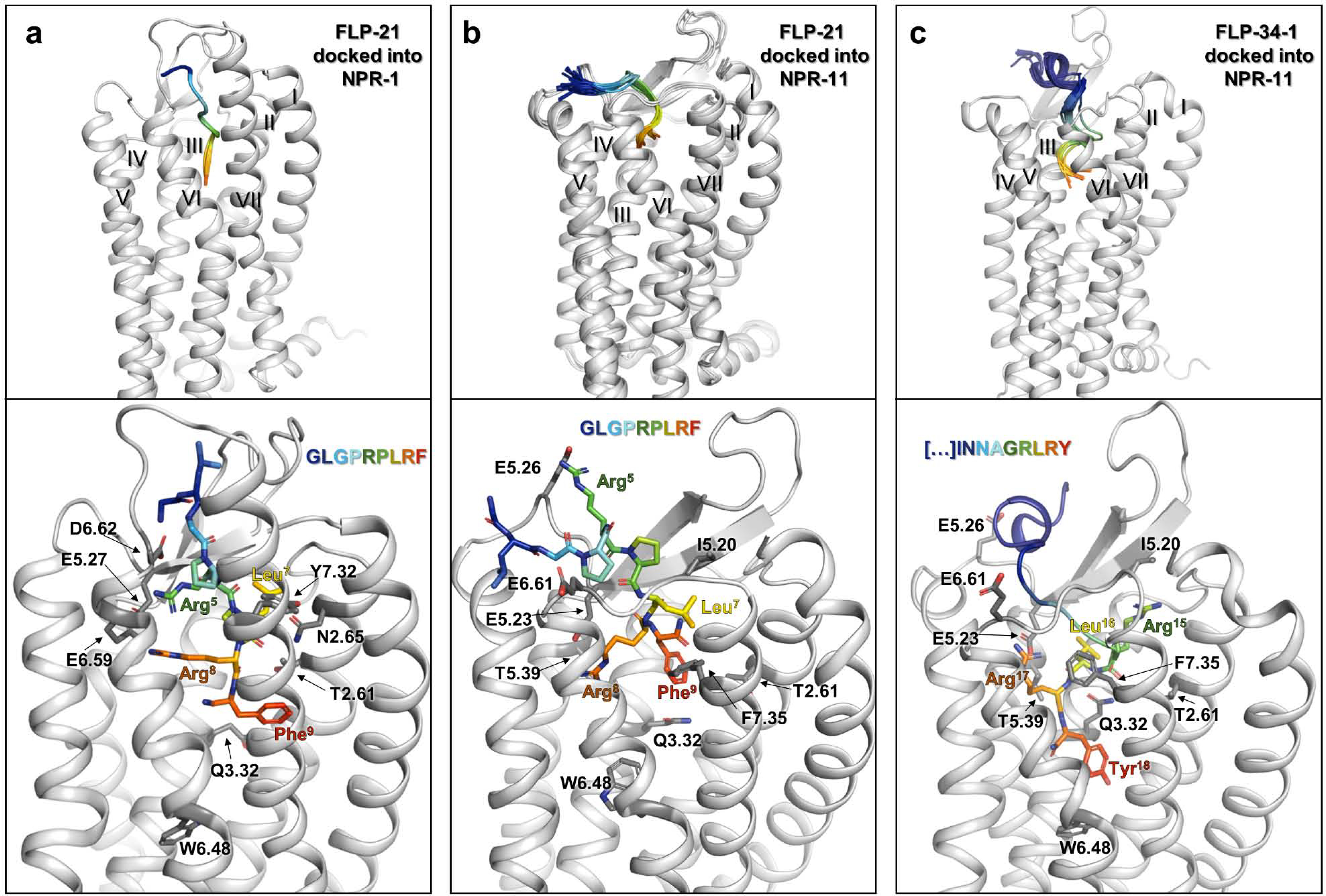
The available functional data was used to guide the docking process of FLP-21 and FLP-34–1 into homology models of NPR-1 (a) and NPR-11 (b, c). See methods for details. a) FLP-21 docked into NPR-1. b) FLP-21 docked into NPR-11. c) FLP-34–1 docked into NPR-11. Receptor backbone is shown in light grey and peptide in rainbow colors (blue to red, N terminus to amidated C terminus, respectively). The top panel shows a superposition of the top 20 scoring models, viewed from ECL3. Lower panel: Close-up view of the top scoring model in the binding pocket from the same angle. Functionally important receptor positions are shown in sticks (dark grey). Atoms are colored by type (oxygen: red; nitrogen: blue). The top20 ensembles for each of the complexes shown in panels a-c are deposited as 3D models in the supplement of this manuscript.
FLP-21 binds NPR-1 in an extended conformation, with the N terminus facing the solvent in proximity to the ECL2 and the C terminus bound in the transmembrane bundle between the ECL2 and the ECL3 (Figure 6a). The in silico energetic analysis reflects the increasing binding contributions towards the conserved C-terminal motif of FLP-21 that were identified by the functional data (SI: Figure S3). The functionally important acidic residues E5.27 as well as E6.59 and D6.62 stick into the binding pocket, resulting in an acidic cluster lining the ECL2 and top of TM6. This cluster is extended by D6.61, which points downward from the top of ECL3 into the binding pocket. Both arginines, R8 and R5 of FLP-21, directly face the acidic cluster, supposedly anchoring the peptide, in line with the drastic loss of binding affinity when mutating the arginines to alanine. R5 of FLP-21 interacts with E6.59 and D6.62. A direct interaction of R5 with D6.62 is not seen in the functional data, but it is well possible that the nearby E6.59, which has the better interaction geometry, compensates the exchange of D6.62 in the double-cycle mutagenesis experiments. R8 of FLP-21 in our model interacts with E5.27, but also E6.59, which reflects the findings from double-cycle mutagenesis (SI: Table S4). L7 reaches towards the opposite side of the binding pocket and interacts with many receptor residues including T2.61, T2.64, N2.65, N2.69 and W2.70. Consistent with this notion, mutagenesis of T2.61 strongly impaired receptor activation. F9 of FLP-21 reaches deeply into the transmembrane bundle of NPR-1 and shows interactions to an overlapping hydrophobic binding pocket including L2.58, T2.61, Q3.32, H7.39, and M7.43. F9 as well as the C-terminal amidation of FLP-21 is in close proximity to Q3.32, but do not reach the putative toggle switch residue W6.48.
For the NPR-11 – FLP-21 combination, we chose the largest cluster, which matched all experimental data and displayed the best interface score (Figure 6b). Similar to binding at NPR-1, FLP-21 binds NPR-11 in a rather extended conformation, with its C terminus reaching into the transmembrane bundle. However, the N terminus of the peptide bends outward between TM5 and TM6, led by the proline-induced kinks (P4 and P6 of FLP-21). In silico energetic analysis highlights increasing contributions to binding affinity towards the C terminus of FLP-21 (SI: Figure S4) similar to FLP-21 binding NPR-1. In agreement with the functional data, R5 of FLP-21 contributes less to binding at NPR-11 compared to NPR-1. We initially hypothesized that R5 of FLP-21 could interact with E6.61 of NPR-11 instead of the missing D/E6.59. However, the E6.61A mutant behaved like wild-type, which is consistent with the observation that E6.61 is directed away from the binding pocket. The missing acidic residue on top of TM6 is apparently compensated by E5.26, a residue in the ECL2 spaced by two residues from E5.23. E5.26 is close to R5 of FLP-21, in line with the observation that deleting the charge at position 5.26 is deleterious for receptor activity. Thus, the acidic cluster in NPR-11 is located in the ECL2 only, and consists of E5.23 and E5.26. Similar to NPR-1, R8 of FLP-21 forms a salt bridge with E5.23 of NPR-11, possibly supported by weaker hydrophilic interactions with H6.55, N6.58 and T6.59. Similar to FLP-21 binding NPR-1, L7 and F9 of FLP-21 bind two slightly overlapping hydrophobic clusters in NPR-11. While L7 of FLP-21 interacts with residues I5.20, V7.32 and F7.35; F9 of FLP-21 interacts with T2.61, W2.70, Q3.32 and F7.35. F9 of FLP-21 reaches down to Q3.32, but not the putative toggle switch residue W6.48, similar to its position in NPR-1. For NPR-11 – FLP-34–1, the size and interface energy of the three structural clusters after docking and refinement was similar. Two clusters displayed a hook-like shape of the C-terminal amino acids of the peptide, which would allow a cyclization reaction of the C terminus, while in the third cluster, the C terminus of the peptide is in an extended conformation and reaches deeply into the transmembrane binding pocket. To experimentally distinguish between these solutions, we prepared a cyclic variant of FLP-34–1, connecting position S8K to the C terminus by a lactam bridge. The resulting cyclized FLP-34–1 variant displayed almost 2000-fold decreased potency in NPR-11 activation relative to the linear variant (SI: Table S6), strongly arguing against this rather exposed, hook-like shape of the C terminus, and favoring the structural ensemble with the extended C terminus reaching into the transmembrane binding pocket (Figure 6c).
Accordingly, in these structures the N-terminal part of FLP-34–1 adopts an α-helical conformation that packs against the ECL2. This is consistent with circular dichroism spectra that demonstrate that FLP-34–1 can adopt an α-helical conformation (SI: Figure S5), similar to NPY [11,22,23]. The orientation of the N-terminal part of FLP-34–1 towards the ECL2, with S8 being close to E5.26 is further supported by the linear [S8K]-FLP-34–1 variant, which displayed a 5-fold gain of function to activate the receptor compared to wild type FLP-34–1 (SI: Table S6).
The importance of the C-terminal -RLRYamide motif of FLP-34–1 is very well reflected in the in silico energetic analysis of the final ensemble (SI: Figure S6). Our model suggest that the interactions of R15 (−4) of FLP-34–1 are split between a few residues, involving side chain and backbone interactions to T2.64, I5.20 and H5.22. I5.20 seems to function as an upper lid of the pocket, in line with the 15-fold loss of potency of the I5.20A mutant (Figure 5a, SI: Table S7). R17 of FLP-34–1 (−2 position, analogous to R8 of FLP-21) forms a salt bridge to E5.23, similar to FLP-21 at NPR-1 and NPR-11. Similar to the other peptide–receptor complexes, L16 (−3) and the C-terminal Y18 bind to slightly overlapping hydrophobic residues in NPR-11 receptor. L16 of FLP-34–1 primarily interacts with Q3.32, L4.51 and H5.22, supported by the functional importance of Q3.32 and L4.51 (Figure 5a, SI: Table S7). Y18 of FLP-34–1 is bound more towards TM7 by T2.61, Q3.32, L6.51, and H7.39. F7.35 functions as the upper border of this subpocket, which is underlined by the drastic loss of function if F7.35 is replaced by smaller residues (Figure 5d, SI: Table S7). Consistent with its delicate role in the binding pockets of both, L16 and Y18, mutation of Q3.32 to histidine makes the binding pocket slightly too tight, thus reducing the binding affinity by 4-fold. Y18 of FLP-34–1 extents down to residue Q3.32, almost reaching W6.48.
Mutation of position Q3.32 differentially affects peptide-binding at NPR-1 and NPR-11 and switches the peptide-binding mode of FLP-21 at NPR-11
Due to its location in the transmembrane binding pocket, Q3.32 is in the direct vicinity of the conserved RF/Yamide C terminus of the peptides, and seems to directly contact F9 of FLP-21, and L16 and Y18 of FLP-34–1, respectively. Mutation of residue Q3.32 showed differential effects in NPR-1 and NPR-11, and led to impaired function or significant gain of function (cf. Figures 3a,4a,5a), respectively, which also appeared to be ligand dependent at NPR-11 receptor. This suggests that this position may be critical for peptide recognition and receptor activation, and motivated an in-depth analysis by combining signaling and binding studies.
Reflecting the tight interaction of F9 of FLP-21 with Q3.32 of NPR-1, the Q3.32H receptor variant significantly loses activity and affinity (Figure 7a,b, left panel; SI: Table S1, S10). We devised NanoBRET ligand binding assays in a double-cycle mutagenesis strategy to confirm the direct contact between Q3.32 and the F9 side chain (Figure 7c, left panel). Indeed, the binding pocket in the wild type NPR-1 is too narrow for [F9Y]-FLP-21 or even a very bulky cyclohexylalanine (F9Cha) substitute, resulting in a strongly decreased affinity of the latter peptide variant (light and dark green curves in Figure 7c, left panel; SI: Table S11). At the Q3.32H receptor mutant, [F9Y]-FLP-21 and [F9Cha] did not further lose affinity and thus had a reduced Ki-shift relative to wild type FLP-21 (Figure 7c, left panel; SI: Table S11), as predicted for a direct contact in a double-cycle mutagenesis experiment. As a control, we used [L7A]-FLP-21, which displayed an additional loss of affinity at the Q3.32H mutant of NPR-1, arguing against a direct interaction (purple curve in Figure 7c, left panel; SI: Table S11). We aimed to explore the effects of further side chains at this position. Unfortunately, alanine and leucine substitutions at position 3.32 of NPR-1 were folding deficient (SI: Figure S1), which precluded further analysis.
Figure 7. Mutation of position Q3.32 differentially affects peptide binding at NPR-1 and NPR-11 and switches the binding mode of FLP-21 at NPR-11.
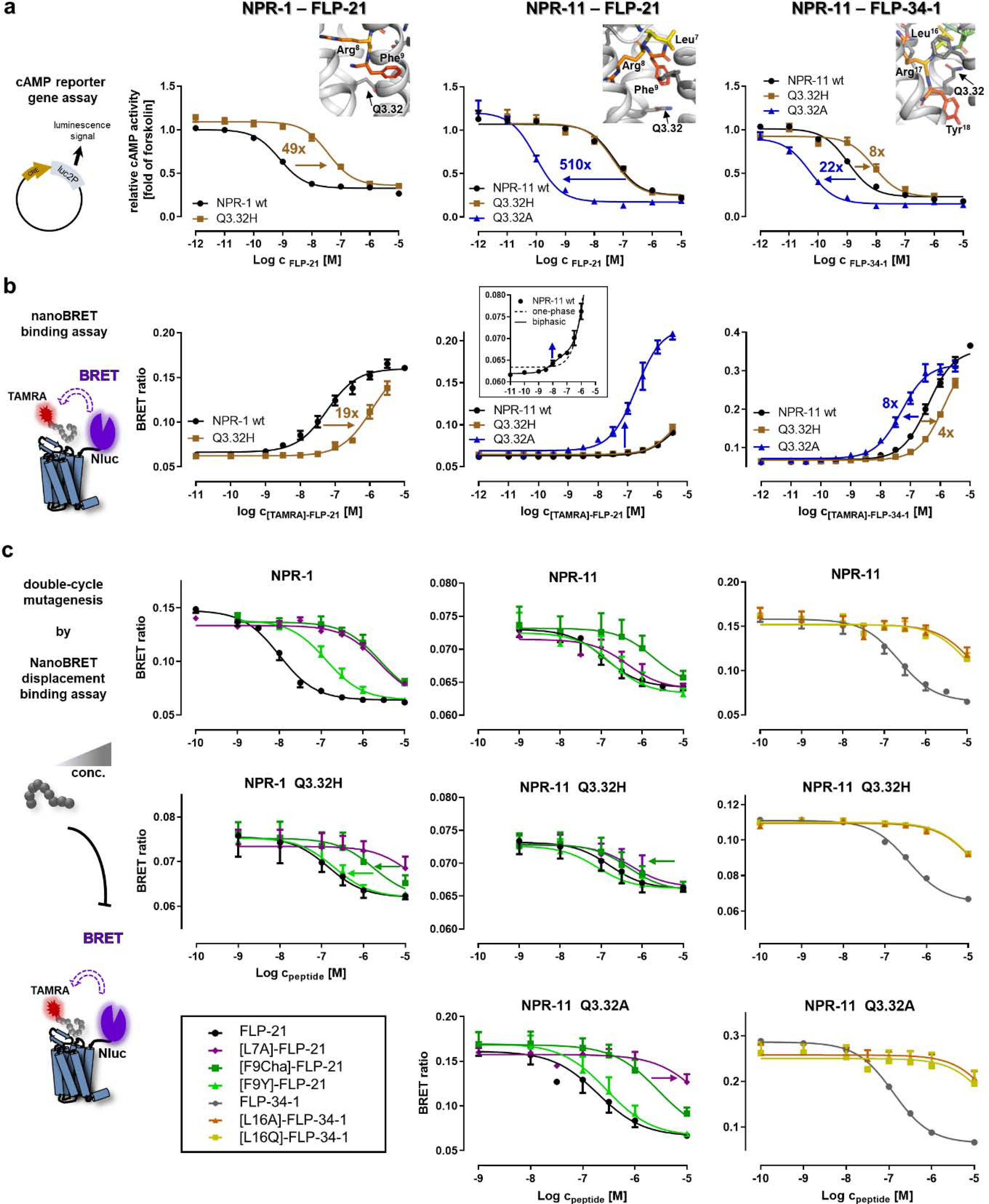
a) Receptor position Q3.32 was tested in a cAMP reporter gene assay (Gi/o) for all three peptide–receptor combinations (from left to right, NPR-1 – FLP-21, NPR-11 – FLP-21 and NPR-11 – FLP-34–1, respectively). The Q3.32A variant of NPR-1 is not correctly folded and was not tested (cf. SI: Figure S1). Insets show the location of Q3.32 in the binding pocket. The Q3.32H variant of NPR-1 displays severely impaired activation. In NPR-11, the Q3.32A exchange induces a dramatic gain of function for FLP-21 and FLP-34–1; while mutation to histidine does not affect activation by FLP-21, but impairs receptor activation by FLP-34–1. b) NanoBRET binding assays to determine peptide affinities at the different receptor variants. Left: NPR-1 mutant Q3.32H displays a weaker affinity to [TAMRA]-FLP-21, in line with the functional data (cf. a). Middle: wild type NPR-11 displays a biphasic behavior for binding of [TAMRA]-FLP-21 with a high-affinity, but low-BRET efficiency component (Kd 15 nM) and a very low-affinity component with a Kd >5 μM. The Q3.32A variant displays a dramatically increased BRET window with a Kd of 190 nM, suggesting a change of binding orientation. Right: The Q3.32H variant of NPR-11 reduces affinity of [TAMRA]-FLP-34–1, while the Q3.32A variant has a moderately increased Kd, without changing the BRET window. c) Displacement binding assays to pinpoint interactions of Q3.32 to the peptides by complementary mutagenesis (cf. Figure 3f). Left: At NPR-1, F9 of FLP-21 interacts with Q3.32, as [F9Y]-FLP-21 (light green triangles) and [F9Cha]-FLP-21 (dark green squares) have a reduced shift of Ki relative to FLP-21 at the Q3.32H variant. In contrast, [L7A]-FLP-21 (purple diamond) has the same Ki shift relative to FLP-21 at both receptor variants. Middle: Binding of FLP-21 variants to NPR-11. F9 of FLP-21 interacts with Q3.32 of NPR-11, as [F9Cha]-FLP-21 (dark green squares) and to a lesser extent [F9Y]-FLP-21 display improved Ki values relative to wild type FLP-21 at the Q3.32H variant of NPR-11. The interactions of L7 of FLP-21 change in the Q3.32A variant of NPR-11 compared to wild type NPR-11 (and Q3.32H variant), as there is a marked rightward shift of the Ki of [L7A]-FLP-21 relative to FLP-21 that is not present at NPR-11 wild type or Q3.32H. Right: L16 of FLP-34 is critical for binding to NPR-11. [L16A/Q]-FLP-34–1 variants (orange triangles and yellow squares, respectively) lose affinity at NPR-11, which is even more pronounced in the Q3.32A mutant. All data represent mean ± SEM of n≥3 independent experiments conducted in technical triplicate.
At NPR-11, the Q3.32H mutant did not measurably affect signaling or binding for wild type FLP-21 (Figure 7, middle panel). Nonetheless, displacement binding assays suggested vicinity to position F9 of FLP-21, similar to the FLP-21 – NPR-1 complex (Figure 7c, middle panel; SI: Table S11). Compared to the wild type NPR-11, the Q3.32H substitution slightly re-arranged the binding pocket and allowed [F9Cha]-FLP-21 and to lesser extent [F9Y]-FLP-21 to bind with better affinities (Ki). The observation that these rather bulky substitutions at the receptor and peptide side were tolerated, and even improved binding further supports the rather large space in the binding pocket seen in the docked models (Figure 6b).
In contrast to NPR-1, a Q3.32A variant of NPR-11 is expressed and correctly folded. Interestingly, this receptor variant displayed a more than 500-fold improved potency of receptor activation by FLP-21 (EC50 0.1 nM compared to 51 nM of the wild type NPR-11, 0.002-fold), and roughly 20-fold increased potency of receptor activation by FLP-34–1, suggesting peptide-dependency. This notion was further supported as mutation of Q3.32 to histidine did not affect the potency of FLP-21, but decreased the potency of FLP-34–1 to activate NPR-11 by 8-fold (Figure 7a, middle and right panels; SI: Table S10).
Direct analysis of ligand binding by NanoBRET revealed a clearly separated biphasic binding of [TAMRA]-FLP-21 at NPR-11 (Figure 7b, middle), containing a high affinity, but low-BRET efficiency component (Kd = 15 nM) and a very low-affinity component with high BRET-efficiency (not saturable up to 10 μM peptide, Kd > 5 μM). Since the efficiency of the resonance energy transfer depends on distance and orientation of the luminescence donor (Nluc fused to the N terminus of the receptor) and acceptor fluorophore (TAMRA attached to the N terminus of the peptide), this directly suggests two distinct binding orientations. In the Q3.32A mutant of NPR-11, [TAMRA]-FLP-21 binds with a Kd of 190 nM, but a drastically increased BRET-window, indicating a novel high-affinity state with changed binding orientation.
In agreement with the saturation binding data, the Ki of unlabeled FLP-21 at the Q3.32A variant in displacement binding assays was 120 nM, comparable to the Ki at wild type NPR-11 (Ki 107 nM). This suggests that the drastically increased potency of receptor activation does not originate from ‘purely’ increased binding affinity, but likely from changed receptor interactions. Double-cycle mutagenesis binding experiments at the Q3.32A variant provided further evidence for a changed binding orientation. Position L7 of FLP-21 is not critical for high-affinity binding to wild NPR-11 (and the Q3.32H variant), as exchange of L7 to alanine resulted in less than 10-fold reduced Ki (purple curve in Figure 7c, middle), in line with the 10-fold reduced potency in signaling assays (Figure 4b, SI: Table S2). At the Q3.32A receptor mutant, however, [L7A]-FLP-21 shows reduced affinity (100-fold reduced Ki, SI: Table S11) and signaling potency (300-fold shifted EC50; SI: Table S5) compared to FLP-21, suggesting that L7 interacts by hydrophobic interactions with Q3.32A in this changed binding conformation, which is disturbed by the L7A exchange. Collectively, these data indicate that the increased receptor signaling of the Q3.32A variant of NPR-11 is based on a different binding orientation of FLP-21 with a higher BRET-efficiency involving a novel interaction of L7 of FLP-21 to position Q3.32A of NPR-11. Finally, signaling of NPR-11 in the Q3.32A mutant is also improved upon stimulation with FLP-34–1, although to a much lesser extent compared to FLP-21 (Figure 7a, right panel; SI: Table S7). In contrast to the situation with FLP-21, this increased potency of receptor activation is paralleled by a similar increase in the binding affinity (Kd of [TAMRA]-FLP-34–1: 388 nM at wild type NPR-11, 49 nM at Q3.32A variant, Figure 7b, right panel), and is not accompanied by a significant change of the BRET efficiency. Similarly, the moderate loss of potency for the Q3.32H mutant (Figure 7a, right panel) matches with a 4-fold reduced binding affinity (Figure 7b, right panel). The structural model suggests that L16 of FLP-34–1 is making hydrophobic contacts to the methylene groups of Q3.32 (−3.4 REU, SI: Figure S6). In line with this, L16A/Q variants of FLP-34–1 show a decreased affinity (Figure 7c, right panel) and potency of receptor activation (SI: Table S6) at all NPR-11 variants. This effect is enlarged at the Q3.32A variant, indicating a high importance of the hydrophobic L16 peptide residue for binding and receptor activation.
DISCUSSION
In the present study, we identified the binding mode of three distinct peptide–receptor interactions of the C. elegans NPY-like FLP/NPR system. Despite the evolutionary far distance, there are remarkable pharmacological and functional similarities between the NPY family of chordates and the FLP/NPR systems in C. elegans that were demonstrated in in vitro and in vivo [13,19,24,25]. For instance, FLP-34–1 is involved in aversive olfactory learning through NPR-11 signaling in vivo [25]. Together with the known involvement of NPR-1 in chemo-avoidance behavior [19], these NPR functions are reminiscent of learning associations of NPY signaling in mammals [7,8], and association to the regulation of food intake [26,27]. Within the expanded repertoire of FLP ligands, there are some variations in the biologically active C-terminal (Rx)RFamide motif, raising questions on a common mechanism for receptor activation, and how receptor recognition and selectivity are regulated. By combining extensive biochemical investigations with molecular docking, we compare the binding modes of the prototypic peptide FLP-21 at NPR-1 and NPR-11, and FLP-34 at NPR-11. We conclude on hallmark features of the peptide–receptor interaction, as well as residues that individually modify the binding mode. We propose these hallmark features are conserved, and set a framework for understanding of the human binding pockets and targeted design of pharmaceuticals.
Hallmark features of NPY-like peptide–receptor interactions
Acidic patches in NPR-1 and NPR-11 are highly important for peptide recognition and receptor activation. The acidic patch in NPR-1 consists of E5.27 in the ECL2, and D6.59, D6.61 and D6.62 on top of TM6, while NPR-11 only has relevant acidic residues in the ECL2, E5.23 and E5.26. Interestingly, D6.61 and D6.62 are required for activation of NPR-1 in vitro and in vivo, but we were unable to pinpoint a direct interaction to R5 or R8 of FLP-21. We propose these residues are important for the initial coarse charge and shape complementarity, consistent with their exposed location in the extracellular loops and the negative cooperativity observed in the double/triple mutants. Accordingly, this patch contributes to ‘drawing’ the peptide into the binding pocket, eventually leading to the salt bridges of R5 to E6.59 and R8 to E5.23. In NPR-11, E5.26 takes over a similar function, as its polarity is absolutely required for receptor activation, but without a direct interaction to any of the arginine residues of the peptides as measured by double-cycle mutagenesis. Interestingly, acidic residues are also enriched in human NPY receptors around D6.59 and in the ECL2 [17]. Nonetheless, the initial recognition of peptides is certainly a highly complex, multistep process [28–31]. In addition to potential direction by surface charge, shape complementarity will likely be of increased importance for larger peptides with a defined secondary structure, such as NPY, and significantly contribute to the observed overall binding geometry. For instance, in the Y2 receptor, extensive hydrophobic interaction of the amphipathic helix of NPY to the ECL2 of the receptor were suggested to take up the peptide from the membrane-bound state [11].
Despite different distributions of acidic residues in the ECLs and variation of the C-terminal arginine motif (RxRFamide versus RxxRFamide), in all three peptide-receptor complexes the structurally conserved acidic residue in the ECL2 (E5.27 in NPR-1, E5.23 in NPR-11, E5.24 in human NPY system, one residue spaced to the cysteine involved in the disulfide bridge, CxE) forms a salt bridge with the absolutely conserved arginine (−2 position) of the RFamide motif, and was by far most sensitive to mutagenesis. The interaction network of E5.23/27 seems to be complex and sensitive, as already the truncation by one methylene group in the corresponding E5.23/27D mutant impaired peptide-induced activation by > 70-fold, which could not be fully restored to the wild type situation when extending the length of the ligand’s side chain. This suggests that E5.23/27 coordinates other interactions (within the receptor or to the ligand), or the positioning of the polar interactions between E5.23/27 and R (−2) need to be at a specific position within the binding pocket.
The other conserved acidic residue, D/E6.59 on top of TM6 was found to interact with the second arginine (R5 of FLP-21) in NPR-1, and also seems to contribute binding energy to the R8 of FLP-21, such that R8 of FLP-21 is ‘sandwiched’ between the ECL2 and TM6. In the extreme case of NPR-11, the absence of the negative charge at position 6.59 still enabled high-affinity peptide binding and receptor activation in a very similar manner. We thus suggest that the structurally conserved E5.23/27 in the ECL2 is a hallmark feature of NPY-like systems, and is at least as important as the well-known D/E6.59. Indeed, a salt bridge between R35 (−2) of NPY and E5.24 is proposed for the evolutionary most ancient human Y2 and Y5 receptors [11,17]. However, in the Y1 and rapidly evolving Y4 receptor [32] subtypes, mutagenesis data suggest that R35 of NPY forms a salt bridge with D6.59, and E5.24 appears to be insensitive to mutation [17], suggesting that the conserved arginine (−2) may switch between either of the two anchor points, depending on the overall peptide conformation.
In agreement with the critical function of E5.23/27 for NPR-1 and NPR-11, the directly contacting R (−2) of the RFamide motif in the peptide has the greatest contribution to affinity and activity in both, FLP-21 and FLP-34–1. Consistent with this notion, FLPs that are short and only contain the minimal RFamide motif without additional Lys/Arg residues in the C terminus (FLP-14, −15–2, −18–5) still have cross-species activity at the human Y2, Y4 and Y5 receptors [13].
The second arginine also contributes to peptide affinity and activity. FLP-21 activated NPR-1 with a potency of < 1 nM and Ki of 5 nM, which is remarkable for a nine amino acid short peptide. FLP-21 also had a significantly higher potency at NPR-1 than, for instance, FLP-15–2 (sequence RGPSGPLRFamide; EC50 22 nM [13]), which does not contain an additional arginine residue in the C terminus, but is otherwise highly similar to FLP-21. This difference in EC50 is consistent with the loss of affinity and activity of [R5A]-FLP-21. Interestingly, the second arginine residue at the −5 position (RxxRFamide) is compatible not only with NPR-1, but FLP-21 also activates NPR-4, −5, −6, and −11 (and human Y2, Y4, and Y5 receptors). On the contrary, FLP-34–1 with a second arginine at the −4 position is more selective, and primarily activates NPR-11 (and human Y1, Y2, Y4 receptors) [13]. However, it remains to be elucidated whether this due to the position of the arginine, or the larger size and secondary structure of the peptide in general.
Functional characterization of FLP-21 and FLP-34–1 highlighted the importance of the conserved C-terminal F/Y residue of the peptides. Interestingly, the C-terminal residue in all FLPs is a phenylalanine, except for FLP-34–1, which carries a tyrosine. The [Y18F]-FLP-34–1 variant showed wild type-like activity at NPR-11, indicating that the hydroxyl group of the tyrosine, which is conserved in the human peptide members of the NPY system, is not (yet) beneficial. Furthermore, all FLP peptides have a hydrophobic residue at the −3 position, which is a leucine in case of FLP-21 (L7) and FLP-34–1 (L16). This contrasts with chordate NPY and PYY, which carry a glutamine at the −3 position (Q34), but is similar to human PP, which has a proline at position −3 (P34). Our docking results suggest that the C-terminal phenylalanine and the leucine at position −3 interact with two partially overlapping hydrophobic pockets, which also slightly differ between NPR-1 and NPR-11. In NPR-1, interactions of the C-terminal F9 and L7 (−3) of FLP-21 overlap at the T2.61 residue, while in NPR-11 the interactions of the same peptide residues of FLP-21 overlap at F7.35 of the receptor. Interestingly, these two residues (T2.61 of NPR-1 and F7.35 of NPR-11) are the very ones that are most important for receptor activation of NPR-1 and NPR-11 (EC50 shift > 150-fold) aside from E5.23/27. This slightly ‘switched’ orientation of the C terminus and leucine (−3) in NPR-1 and NPR-11 with respect to T2.61 and F7.35 indicates some flexibility in the binding pocket. In a situation with a hydrophilic glutamine at position −3, the orientation of this peptide residue would become more restricted, and will likely be oriented towards T2.61 and the nearby Q3.32, which is the most hydrophilic ‘island’ deep in the transmembrane binding pocket. We therefore speculate that an exchange to a hydrophilic residue at peptide position −3, as present in human NPY, would contribute to more specific binding pocket. However, that apparently requires further adjustments in the binding pocket, as [L16Q]-FLP-34–1 displayed impaired activation of NPR-11, which is consistent with the very limited activity of human NPY or human NPFF (FLFQPQRFamide), a short RFamide peptide with a hydrophilic glutamine at position −3, at C. elegans NPRs [13]. According to the currently available sequence information, this switch to glutamine at position −3 may have occurred during early deuterostome evolution, as all protostomes carry a hydrophobic residue, but deuterostomes (including hemichordates) comprise a glutamine [33]. Interestingly, in Echinodermates, which branched off from hemichordates in early deuterostome history, known NPY-like neuropeptides carry a leucine (Ophiopsila aranea, Amphiura filiformis), proline (Asterias rubens) or alanine (Patiria miniata) residue at −3 position [33], which could provide the missing link in this regard.
Plasticity of NPR-11 peptide-binding pocket controls ligand efficacy
In NPR-11, we observed a remarkable plasticity of the binding pocket. A single exchange of Q3.32 to alanine induced a different binding pose of FLP-21 at this receptor, reflected by an increased BRET efficiency of the bound peptide. This extreme flexibility is likely promoted by the absence of D6.59, which allows the peptide to ‘turn’ around the anchor point in the ECL2. Nonetheless, it exemplifies how minimal changes to the binding pocket might perturb the overall binding mode, such that peptides ‘evade’ the restraints imposed by mutation of the peptide-binding pocket. In other words, multiple binding orientations might generally be possible, and the population of these states is shifted by mutagenesis, which might cloud the importance of certain amino acids for a particular pocket when tested in a classic mutagenesis approach. Investigating the peptide orientation by NanoBRET provide an elegant means of studying such effects.
In the case of NPR-11, mutagenesis of Q3.32 in the central binding pocket not only changed the binding orientation of the peptide, but severely affected peptide-dependent receptor activation. Although Q3.32 is involved in a polar network in the transmembrane core, none of the mutations we introduced appeared to affect the basal activity of NPR-11 or NPR-1. This is consistent with previous findings on mutation of Q3.32 in the human Y1 [12] and Y2 [11,34] receptors, which reduce the potency of NPY and the affinity of specific receptor antagonists, but do not increase basal activity. Position 3.32 frequently contacts GPCR ligands [35], but there is no conservation of this residue across different ligand classes or even among peptide-binding GPCRs. Among NPY receptors in chordates and NPRs of C. elegans, however, Q3.32 is extremely conserved, suggesting a functional role. Interestingly, the family-conserved D3.32 in the opioid receptor system was found to contact the free N terminus, thereby positioning the Y1 side chain (‘signaling entity’ of opioid peptides) and mediating receptor activation [36]. This appears like an N-terminal analogy of the NPY system, where the amidated C-terminus and the C-terminal F/Y contact the conserved Q3.32 of the receptor: In all three peptide-receptor combinations of this study, the C-terminal F/Y peptide residue is located in close proximity to Q3.32 of NPR-1 and NPR-11, which suggests a structural basis for the functional importance of this residue. While F9 of FLP-21 lays on top of Q3.32 in NPR-1 and NPR-11 wild type receptors, supported by double-cycle mutagenesis binding data, Y18 of FLP-34–1 even reaches below and is in close proximity to W6.48, coordinated by L16 that is bound between Q3.32 and T2.61. We suggest that the position of the C-terminal tyrosine of FLP-34 in the deep sub-pocket below Q3.32, almost reaching the ‘toggle switch’ W6.48, efficiently contributes to the activation of NPR-11. For FLP-21 in the changed binding mode at the [Q3.32A]–NPR-11 receptor variant, we see an increased influence of L7 of FLP-21, which essentially mirrors the functional data of FLP-34–1 at this receptor. This changed positioning of L7 when bound to the Q3.32A mutant of NPR-11 entails re-positioning of F9 of FLP-21 into the same deep sub-pocket that is occupied by Y18 of FLP-34–1. Accordingly, FLP-21 likely adopts a binding mode very similar to FLP-34 when bound to the Q3.32A variant of NPR-11, leading to more efficient receptor activation despite unchanged ligand affinity to the receptor. For FLP-34–1, the Q3.32A receptor mutation only re-enforces the existing contact of L16 to Q3.32 and/or enables even slightly deeper penetration into the binding pocket, and therefore only modestly increases affinity and activity.
This deep position of the C terminus is seen very similarly for NPY docked into the Y2 receptor [11]. Q3.32 coordinates the orientation of C terminus in the binding pocket by contacts with Q34 and the C-terminal amidation of NPY. Thus, the side chain of Y36 is in proximity to W6.48 [11], which contributes to receptor activation [16]. Very much in agreement with the Y2R, mutation of W6.48 in NPR-1 and NPR-11 reduced receptor activation, but did not abolish signaling, in contrast to several other peptide-activated GPCRs [37]. This suggests that multiple factors converge to activate the receptors, and underlines that several peptide binding modes may principally activate the receptor, albeit with different efficacy.
In conclusion, our study illuminated peptide binding in an ancient NPY-like system. The peptide-binding pockets appeared variable aside from a functionally highly relevant contact of R(−2) of the RFamide motif to a structurally conserved glutamate in the ECL2 (E45.52), which in some case interplays with the conserved D/E6.59 to generate a ‘sandwich’ contact with the arginine. We propose this to be a key interaction for NPY-like systems throughout all bilaterians. Two structurally distinct peptides, FLP-21 and FLP-34–1 occupy rather different binding poses at NPR-11, but have similar binding affinities. However, the deeper penetration of the C terminus of FLP-34–1 into the TM bundle enabled by the contact of L16 (−3) to the conserved Q3.32 leads to more efficacious receptor activation. A single Q3.32A mutation re-orients FLP-21 to resemble the binding mode of FLP-34–1, which in turn drastically enhances receptor activation. We expect that this variability of the binding pockets is not limited to the FLP/NPR system of C. elegans, but rather is a general feature of many peptide-binding pockets.
MATERIALS AND METHODS
Materials
Standard chemicals were obtained from Sigma Aldrich (St. Louis, USA) unless otherwise stated. Enzymes were purchased from ThermoFischer Scientific (Waltham, USA) and cell culture materials from Lonza (Basel, Switzerland).
Cell culture
All in vitro assays were conducted with the commercially available cell line HEK293 (Homo sapiens, female, embryonic kidney; DSMZ ACC 305). Cell cultivation was performed in humidified atmosphere as a monolayer including 37°C and 5% CO2 in T75 cell culture flasks using Dulbecco’s modified Eagle’s medium (DMEM) with Ham’s F12 (1:1), (v/v) supplemented with 15% (v/v) heat inactivated fetal calf serum (FCS).
C. elegans strains
The C. elegans strains used in this study were maintained at 22°C according to Brenner [38] and are listed in SI: Table S12. N2 Bristol and CX4148 (npr-1 (ky13)) [39] were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Strain Ex[npr-1] (APR576, genotype npr-1 (ky13) X; aprEx229 [pnpr-1::npr-1::gfp, pmyo-3::mcherry, pBSK]) was previously generated [13]. All other strains were established during this study.
Peptide synthesis
C-terminally amidated FLP variants with single amino acid exchanges were synthesized as described previously [13]. Briefly, peptides were synthesized on TentaGel R RAM resin (15 μmol) following the Fmoc/tert-butyl strategy (reviewed in [40]) using a Syro II peptide synthesizer (MultiSynTech). Peptides were cleaved off the resin using trifluoroacetic acid (TFA)/H2O/triisopropylsilane (90/5/5, v/v/v). All peptides were purified to > 95% purity by RP-HPLC (Shimadzu) using a Phenomenex Aeris, 100 Å (C18) column. To confirm purity and the correct identity of the synthesized conjugates, analytical RP-HPLC and matrix-assisted laser desorption/time of flight (MALDI-ToF; Ultraflex III MALDI-ToF/ToF, Bruker Daltonics) mass spectrometry analyses were performed, respectively. Further individual FLP derivates are described below.
FLP variants containing unnatural amino acids were synthesized under standard conditions with the exception of the manual introduction of the N‐α‐Fmoc–protected amino acid Homoarginine (hArg) or Cyclohexylalanine (Cha) using 5 eq of 1-hydrobenzotriazole (HOBt), diisopropylcarbodiimide (DIC) and the respective amino acid in DMF at room temperature for at least 4 hours. The peptides were then further elongated to the N terminus by automated synthesis as described above.
N-terminal modification of peptides were made with the standard automated synthesis. Prior to cleavage of peptides from the resin, N-terminal acetylation was carried out on resin using 10 eq of acetanhydrid (Ac2O) and diisopropylethylamin (DIPEA) in dichloromethane (DCM) at room temperature for 15 minutes. Alternatively, the N-terminal modification with the 5(6)-carboxytetramethylrhodamine (TAMRA) fluorophore was carried out using 2 eq of diisopropylethylamine (DIEA) and the respective fluorophore with 1.9 eq (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) in dimethylformamide (DMF) at room temperature overnight in the dark.
The free acid derivative FLP-21-COOH was synthesized using the standard automated synthesis described above, but on Wang resin (15 μmol) instead of TentaGel R RAM. [F9tyramide]-FLP-21, lacking the C-terminal carbonyl group, was prepared analogously to the protocol of Hoffmann et al [41]. Briefly, the peptide sequence was build up from R8 to the G1 (N-terminally Boc-protected) on an acid-sensitive Trityl chloride resin (15 μM). The fully protected FLP-211–8 was cleaved from the resin using glacial acetic acid/TFE/DCM (1:1:8, v/v) twice for 3 hours at room temperature, and was coupled at the free C terminus with 2-phenylethylamine, yielding [F9tyramide]-FLP-21. The residual side chain protecting groups were cleaved off using TFA/H2O/triisopropylsilane (90/5/5, v/v/v) and removed as described [13], and the peptide was further purified under standard conditions.
For the cyclized peptide [cycK8-Cterm]-FLP-34–1, FLP-34–1 was synthesized with an orthogonally protected lysine (Lys(Dde)) introduced at position 8 on Trityl chloride resin (15 μmol). Cleavage of the fully protected peptide from the Trityl chloride resin was performed using glacial acetic acid/TFE/DCM (1:1:8, v/v) twice for 3 hours at room temperature, and the peptide washed very thoroughly with DCM. After the cleavage, Dde was cleaved by addition of 2% hydrazine (v/v) in DMF for 10 min and washing with DMF, repeated 15 times. Cyclization was performed in solution by addition of 5 eq HOBt and DIC in DCM overnight at room temperature. The remaining protecting groups were cleaved using TFA/H2O/triisopropylsilane (90/5/5, v/v/v), and the peptide was further purified as described above.
Generation of plasmids and transgenes
Neuropeptide GPCR constructs for in vitro analysis
The cDNA of NPR-1 and NPR-11 was a genetic fusion of the receptor DNA with a C-terminal enhanced yellow fluorescent protein (eYFP) in pVitro2-hygro-mcs vector (InvivoGen), and was generated as described before [13]. Single amino acid exchanges of the receptor were introduced by using Stratagene QuickChange mutagenesis (Agilent Technologies, Santa Clara, CA, primer pairs listed in SI: Table S13).
For ligand-binding assays based on NanoBRET, a secretable variant of the nanoluciferase (secNluc, Promega, Madison, USA) was genetically fused to the N terminus of the receptor, spaced by a Ser-Gly4-Ser linker (TCAGGCGGTGGAGGTAGT). To promote expression of the fusion protein with enlarged N-terminal domain in the plasma membrane, the sequence contains the secretion sequence of human interleukin-6 at the very N terminus. The genetic fusion was performed by a PCR overlap extension strategy [42]. The 5’ fragment was amplified from the pNL1.3_secNluc plasmid (Promega, Madison, USA), attaching a MluI restriction site and the Ser-Gly4-Ser linker using the primers MluI_secNluc_for (AAAACGCGTGCCACCATGAACTCCTTCTCCACAAGC, with MluI restriction site underscored) and Nluc_Li2_rev (ACTACCTCCACCGCCTGACGCCAGAATGCGTTCGCAC, with Ser-Gly4-Ser- linker underscored). The 3’ fragment was amplified from the NPR-1/NPR-11-eYFP_pVitro2 plasmid using the primers SG4S_NPR-1_for/SG4S_NPR-11_for (TCAGGCGGTGGAGGTAGTATGGAAGTTGAAAATTTTACCGAC/TCAGGCGGTGGAGGTAGTATGGGATCGGTGAATGAATC, Ser-Gly4-Ser linker underscored) and YFP-XbaI-NheI-r (TTTGCTAGCGTGTTACCCCTCTAGACCTG). The fragments were fused in a second PCR reaction via the overlapping Ser-Gly4-Ser linker sequence and amplified using the primers MluI_secNluc_for and YFP-XbaI-NheI-r. The Nluc-NPR-1/11-eYFP sequences was digested with MluI+NheI (NPR-1; NheI has compatible ends with XbaI) or MluI+XbaI (NPR-11) and ligated into the pVitro2 vector backbone digested with MluI+XbaI (all restriction enzymes and T4 DNA ligase from Fermentas/Thermo Fisher Scientific, Waltham, USA).
The sequence identity of all receptor constructs was confirmed by Sanger dideoxy sequencing.
Neuropeptide GPCR constructs for transgenesis
The plasmid containing the genomic npr-1 fused to a GFP with additional 3 kb 5’UTR sequence was a kind gift of Long Ma [19]. To obtain the npr-1 receptor mutants in a C. elegans expression vector, we used this plasmid as template for a site-directed mutagenesis PCR with primer pairs listed in SI: Table S13. After DpnI digestion, the mix was directly transformed into chemically competent E. coli DH5α and correct constructs were confirmed by sequencing (Microsynth AG, Switzerland).
cAMP reporter gene assay
Receptor activation by Gi/o was measured by cAMP reporter gene assays in HEK293 cells as described previously [13]. Briefly, HEK293 cells were transiently co-transfected with vectors encoding the receptor (wild type or mutated) and the cAMP reporter gene plasmid pGL4.29 [luc2P/CRE/Hygro] (Promega; 4 μg total, 1:1 ratio) and re-seeded into white 384-well plates. On the next day, the medium was removed and the cells were stimulated with 20 μl of peptide solution (wildtype or mutated peptides) in serum-free DMEM additionally containing 5 μM forskolin (to elevate cellular cAMP levels). After 4 hours of incubation, the luciferase substrate OneGlo in lysis buffer (Promega) was added and the luminescence was measured in the microplate reader Tecan Spark (Tecan, Männedorf, Switzerland). Data were analyzed with GraphPad Prism 8 and are shown as -fold of forskolin. All data are displayed as mean ± SEM of at least three independent experiments performed in triplicate.
Fluorescence microscopy
Correct folding and expression in the plasma membrane of the C. elegans receptor constructs (wild type or mutated) fused to eYFP was assessed by fluorescence microscopy in human cells. HEK293 cells grown in 8-well μ-slides (Ibiditreat) to 70–80% confluency were transiently transfected with 1 μg vector DNA per well using Lipofectamine2000 (ThermoFisher Scientific) following the manufacturer’s instructions. On the next day, medium was changed to OptiMEM (Invitrogen Life Technologies) and nuclei were stained with 2.5 ng/μl Hoechst33342 (Sigma-Aldrich) for 30 minutes. Cells were examined using an Axiovert Observer Z1 microscope (with Apotome, Plan-Apochromat 63x/1.40 Oil DIC objective, filter sets 02 (365/420), 46 (500/535); Zeiss).
NanoBRET binding assays
The binding affinities of the peptide variants to NPR-1 and NPR-11 was determined by NanoBRET binding assays. As BRET donor, a secretable version of the nanoluciferase (secNluc; Promega, Madison, USA) [43] was genetically fused to the N terminus of the receptor, spaced by a Gly-Ser4-Gly linker (for details on plasmid generation, see above). Wild type peptides labeled with a TAMRA fluorophore were used as a fluorescent tracer and BRET acceptor (for details on peptide synthesis, see above). Membranes of HEK293 cells transiently transfected with the Nluc-NPR-1/11-eYFP constructs were prepared as described [16]. Aliquots containing ~0.5 μg/μl total protein in HEPES buffer, pH 7.4, containing 25 mM CaCl2, 1 mM MgCl2 and Pefabloc SC protease inhibitor (Sigma Aldrich) were stored at −80°C.
The binding assays were conducted using 0.5 μg total protein in a total volume of 100 μl HBSS buffer (pH 7.4) containing 25 mM HEPES, 0.1% bovine serum albumin and Pefabloc SC, denoted as BRET buffer. Upon addition of Coelenterazine H (Nanolight/Prolume, USA) to a final concentration of 4 μM, the total luminescence in the absence of ligand was ~300 000 RLU (430–470 nm filter).
For determination of the Kd of the TAMRA-labeled peptide, 10 μl of a 10x concentrated peptide solution (in H2O + 0.1% BSA) in a concentration range of 10−11 M to 10−4 M (final concentration 10−12 M to 10 −5 M) was incubated with 90 μl BRET buffer containing 0.5 μg total protein in solid black 96 well plates for 10 min under gentle agitation at room temperature.
For competition binding experiments, 10 μl of a 1 μM solution of TAMRA-labeled peptide (in H2O + 0.1% BSA; final concentration 100 nM), 10 μl of a 10x concentrated solution of unlabeled peptide (in H2O + 0.1% BSA; final concentration ranging from 10−10 M to 10−5 M), and 80 μl BRET buffer containing 0.5 μg total protein were incubated in solid black 96 well plates for 90 min under gentle agitation at room temperature.
Directly before the measurements, 10 μl coelenterazine H in BRET buffer (10x stock solution) was added to a final concentration of 4 μM, and BRET was measured in a Tecan Spark plate reader (Tecan, Männedorf, Switzerland) with the following filter settings: luminescence (L) 430–470 nm, fluorescence (F) 550–700 nm. The BRET ratio was calculated by the ratio of F/L. The Kd values were obtained from a three-parameter logistic fit assuming a single binding site or biphasic binding. The IC50 values and Ki values of the competition binding assays were determined with a three-parameter logistic fit in the GraphPad Prism 5 software (GraphPad Software, San Diego, CA, USA), applying the built-in Cheng-Prusoff correction for the Ki values [44].
Generation of transgenic nematodes
All transgenic strains contain stably transmitting extrachromosomal arrays. C. elegans npr-1 knockout mutants expressing the native and mutated npr-1 were generated by microinjection of plasmid DNA into gonads of young adult hermaphrodites. The plasmid mixture contained 10 ng/μl of the plasmid of interest, 20 ng/μl of a mCherry construct driven by a myo-3 promoter (pCFJ104, kind gift from E. Jørgensen) and was filled up to 120 ng/μl with pBluescript II SK+, which served as stuffer DNA. The Eppendorf micromanipulator (InjectMan 4 and FemtoJet 4i) was used for injection. The protocol was adapted from Mello et al [45,46]. Worms were left to regenerate for 4 days at 15 °C and progeny positive for the marker was isolated. F2 individuals stably expressing the marker were established as lines and scored for MeSa avoidance. Multiple independent transgenic lines were established for each transgene tested.
Methyl salicylate (MeSa) avoidance assay
MeSa avoidance was scored according to Ma et al [19]. Worms were synchronized via egg laying till one day after reaching the L4 stage. Around 120 transgenic animals of each strains were collected and washed 3 times in M9. The worms were transferred in 15 μl M9 to the middle of a 10 cm NGM Petri dish divided into four squares. Immediately afterwards, 2 μl MeSa were placed on opposite sites 3.5 cm away from the center and 2 μl ethanol as control on the other two parts. To anesthetize the nematodes on the spot, 2 μl of 0.5 M of sodium azide were added to the substances. Plates were subsequently sealed and left at 22°C until all worms were paralyzed. Animals were scored and the avoidance index calculated ((# on Ethanol - # on MeSa) / by total #).
Comparative modeling
The overall workflow of the comparative modeling involves finding template structures with high identity, threading the target receptor sequence (NPR-1 and NPR-11) onto the template structures and allowing the Rosetta comparative modelling (CM) protocol [21,47,48] to generate models from these threaded structures, which includes an energy minimization step (relax).
Comparative models were created based on three experimentally determined class a GPCR structures (Y1 (PDB code 5ZBQ); ETB (PDB code 5GLI); K-OR-1 (PDB code 4DJH)) with identities of about 29% and 35% for NPR-1 and NPR-11 receptor, respectively. Only a few template structures were used since it was shown that increasing the number of templates results in an average model of the ECL2 conformation and will weaken model accuracy, while using fewer templates can increase the accuracy of the ECL2 by allowing Rosetta to adopt a more sequence-defined structure [48,49]. Initial structure sequence alignments of the templates were obtained from GPCRdb [18], and NPR-1 and NPR-11 sequences were aligned to the template alignment using ClustalW [50]. This sequence alignment was further manually adjusted as previously described (SI: Table S9) [48]. Briefly, the transmembrane helix sequences were aligned starting from the most conserved residue in each α-helix and extended outwards to ensure that highly conserved residue motifs and helix endings remain aligned (e.g.: NPxxY, CWxP). Loop alignments were generated based on common sequence motifs (e.g.: xWxxG in ECL1) and adjusted to maintain secondary structure elements in loop regions (disulfides or β-hairpin in ECL2 of peptide binding GPCR’s [51]). The remaining loop residues were cut in half and each half was joined to the ends of the respective helices. Due to the lack of coordinates in template structures, NPR-1 and NPR 11 sequences were truncated at the N- and C-termini as well as ICL3 (NPR-1: residues 1–7, 228–265, 362–458; NPR-11: residues 1–16, 248–259, 345–402). Programs PSIPRED [52] and OCTOPUS [53] were used to predict transmembrane regions. The sequence of NPR-1 and NPR-11 was threaded onto the aligned coordinates of the template structures. Using the Rosetta comparative modeling (CM) protocol [21] 5,000 models were generated combining segments of the threaded structures. For missing residues without an aligned residue in the template structure, RosettaCM protocol uses sequence-based fragments (3mer and 9mer) to connect the open loops and generate de novo structures. These generated structures were subjected to energy minimization with the Rosetta relax protocol using all-atom restraints. Generated models were clustered based on the backbone root-mean-square deviation (RMSD) of the atomic positions and the top five models of ten clusters were selected for further analysis. While there was almost no difference observed in the transmembrane helices of these receptors, most diversity was detected in the ECL2 and ECL3 region of both modeled receptors, which was kept for the docking experiments to mimic conformational selection. The truncated N terminus was observed to be highly flexible during the modeling step and partially dipped into the binding pocket. These interactions avoided collapse of the ligand-binding pocket, a common problem during homology modeling, and keeps the peptide-binding site intact.
Peptide docking
The overall workflow of the docking process involves creating the peptide with the C-terminal amidation with the RosettaCM protocol [21], docking of the peptide into the comparative models of the receptors using the FlexPepDoc protocol of Rosetta [54] and allowing of the ligand-binding pocket to adjust using the RosettaCM protocol [21]. In the last step, top docked models were refined using the FlexPepDoc protocol [54].
Since the N terminus was observed to dip into the binding pocket and would block peptide binding in the docking step, comparative models were further truncated N-terminally prior to docking to completely open the binding pocket to the peptide (further 9 amino acids for a total of 16 truncated residues in NPR-1 and further 13 for a total of 29 truncated residues in NPR-11). For each receptor (NPR-1 and NPR-11), a set of 50 low-energy comparative models with conformational diversity was used as input structures for the final docking. Conformational diversity was ensured by choosing top 5 models out of 10 different clusters by total score for each receptor.
We first built the peptides (complete sequence of FLP-21; only 9 C-terminal amino acids of FLP-34–1 in the first step) with the C-terminal amidation into the comparative receptor models using the Rosetta CM protocol [21]. In the next step, C-terminally amidated peptides were docked into the comparative models of NPR-1 and NPR-11 with the FlexPepDoc protocol in Rosetta [54]. In this step, peptide backbone conformational sampling takes place using Monte Carlo Metropolis-based rigid body moves, followed by an optimization of the side chains. 50,000 models were generated in this step for each peptide–receptor combination. The docking process was guided by the experimental data, which were implemented as restraints: Direct peptide–receptor contacts identified by double-cycle mutagenesis were translated into AtomPair restraints allowing of 4 Å distance between the distal C atoms, and violation of this restraint resulted in an energetic penalty (BOUNDED function). For peptide or receptor residues with strong relevance for binding/activation but without prior knowledge of the exact contacting residues in the peptide/receptor, ambiguous one-sided restraints were set (SiteConstraint), requiring the Cα atom to be within 6.5 Å of the respective other chain, i.e., peptide or receptor. Violation of the SiteConstraint was penalized using the FLAT_HARMONIC function. For FLP-21 binding to NPR-1, we included the following restraints: AtomPair OE1 164 NH1 315 BOUNDED 0 4 1.0 TAG (R8 to E5.27); AtomPair OE2 164 NH2 315 BOUNDED 0 4 1.0 TAG (R8 to E5.27); AtomPair OE1 247 NH1 312 BOUNDED 0 4 1.0 TAG (R5 to E6.59); AtomPair OE2 247 NH2 312 BOUNDED 0 4 1.0 TAG (R5 to E6.59); AtomPair CD 90 CZ 316 FLAT_HARMONIC 4 1 2.5 (F9 to Q3.32); SiteConstraint CG2 67A B FLAT_HARMONIC 4 1 2.5 (FLP-21 to T2.61); SiteConstraint CG 250A B FLAT_HARMONIC 4 1 2.5 (FLP-21 to D6.62); SiteConstraint CG 314B A FLAT_HARMONIC 4 1 2.5 (L7 to NPR-1).
For FLP-21 binding NPR-11, we included the following restraints: AtomPair OE1 167 NH1 311 BOUNDED 0 4 1.0 TAG (R8 to E5.23); AtomPair OE2 167 NH2 311 BOUNDED 0 4 1.0 TAG (R8 to E5.23); SiteConstraint CG 266A B FLAT_HARMONIC 4 1 2.5 (FLP-21 to F7.35); SiteConstraint CG 312B A FLAT_HARMONIC 4 1 2.5 (F9 to NPR-11).
For FLP-34–1 binding NPR-11, we included the following restraints: AtomPair OE1 167 NH1 311 BOUNDED 0 4 1.0 TAG (R17 to E5.23); AtomPair OE2 167 NH2 311 BOUNDED 0 4 1.0 TAG (R17 to E5.23); AtomPair CD 90 CD1 310 BOUNDED 0 5 1.0 TAG (L16 to Q3.32); SiteConstraint CG 266A B FLAT_HARMONIC 4 1 2.5 (FLP-34–1 to F7.35); SiteConstraint CG 312B A FLAT_HARMONIC 4 1 2.5 (Y18 to NPR-11); SiteConstraint CG 309B A FLAT_HARMONIC 6 1 2.5 (R15 to NPR-11).
The top 2,500 models (top 5%) based on interface score were grouped into five clusters based on the RMSD of the C-terminal five residues of the peptide. Clusters that displayed major distortions (peptide dips under loops) or an extremely unlikely peptide topology with the N terminus of the peptide penetrating into the transmembrane binding pocket were excluded. From the remaining clusters, the top 50 models were subjected to the RosettaCM protocol to allow the binding pocket of the receptor to adjust to the peptide, mimicking an induced fit. 5,000 models were generated in this step and analyzed for a combination of Rosetta total and interface score as well as RMSD of the peptide. The top 20 models by total and interface score were visually inspected for complying with the given experimental restraints. The remaining 13 (FLP-21 – NPR-1), 16 (FLP-21 – NPR-11), or 17 (FLP-34–1 (9 aa) – NPR-11) models were subjected to another round of the FlexPepDoc protocol in Rosetta to generate 50,000 models for refinement of the peptide C terminus in the adjusted binding pocket of the receptor. In case of FLP-34–1 binding to NPR-11, the refinement step was also used for simultaneously prolonging the N-terminal peptide chain of FLP-34–1 for the final 18 amino acids.
The top 500 models (1%) based on interface score of the refinement step were clustered based on RMSD of the peptide, yielding the final structural ensembles. Structural ensembles were inspected and one ensemble was chosen as the proposed binding mode for each combination based on functional data. Top 20 models of the chosen ensemble for each combination were further analyzed to identify putative peptide–receptor interactions by per-residue breakdown of the Rosetta interface energy. Combined interactions (> 0.1 REU) for a score average lower than 0.5 REU are shown in SI: Figure S3, S4 and S6.
Supplementary Material
Many peptide - GPCR signaling systems are evolutionary conserved down to basic animals
We illuminate binding of three distinct neuropeptide Y-like ligands in the FLP/NPR system of C. elegans
We identify E5.23(ECL2) and Q3.32 of the receptors are family-conserved residues critical for affinity and efficacy
NanoBRET based ligand binding assays demonstrate flexibility of peptide ligands in the binding pocket
Comparing binding modes in evolutionary ancient homologs can identify key binding and activation mechanisms
ACKNOWLEDGEMENTS
The authors thank Torsten Schöneberg and Annette G. Beck-Sickinger for helpful discussions, and Tobias Fischer for sharing a script for automatic analysis of the docked models (heatmap). The excellent technical assistance of Christina Dammann, Kristin Löbner, Ronny Müller, Regina Reppich-Sacher, and Janet Schwesinger is gratefully acknowledged. Further, the authors thank E. Jørgenson and L. Ma for kindly sharing reagents.
This work was supported by the Free State of Saxony, Ministry for Higher Education, Research and the Arts (SMWK, grant 100316655 to A.K. and S.P.). The authors acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through CRC 1423, project number 421152132, subprojects B03, C04, Z02 and Z04. Work in the Meiler lab is supported by National Institute of Health, NIH (NIGMS R01 GM080403; NIDA R01 DA046138; NIGMS R01 GM129261).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL DATA
Supplementary information is available for this manuscript and contains 13 tables and 6 figures. The top20 models for each of the peptide–receptor complex are deposited as 3D models.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Jékely G, Global view of the evolution and diversity of metazoan neuropeptide signaling, Proc. Natl. Acad. Sci. 110 (2013) 8702–8707. 10.1073/pnas.1221833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mirabeau O, Joly J-S, Molecular evolution of peptidergic signaling systems in bilaterians, Proc. Natl. Acad. Sci. 110 (2013) E2028–E2037. 10.1073/pnas.1219956110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lee NJ, Enriquez RF, Boey D, Lin S, Slack K, Baldock PA, Herzog H, Sainsbury A, Synergistic attenuation of obesity by Y2- and Y4-receptor double knockout in ob/ob mice, Nutrition. 24 (2008) 892–899. 10.1016/j.nut.2008.06.019. [DOI] [PubMed] [Google Scholar]
- [4].Sato N, Ogino Y, Mashiko S, Ando M, Modulation of neuropeptide Y receptors for the treatment of obesity, Expert Opin. Ther. Pat. 19 (2009) 1401–1415. 10.1517/13543770903251722. [DOI] [PubMed] [Google Scholar]
- [5].Yulyaningsih E, Zhang L, Herzog H, Sainsbury A, NPY receptors as potential targets for anti-obesity drug development, Br. J. Pharmacol. 163 (2011) 1170–1202. 10.1111/j.1476-5381.2011.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carvajal C, Dumont Y, Quirion R, Neuropeptide Y: Role in emotion and alcohol dependence, CNS Neurol Disord Drug Targets. 5 (2006) 181–195, doi: 10.2174/187152706776359592. [DOI] [PubMed] [Google Scholar]
- [7].Tasan RO, Verma D, Wood J, Lach G, Hörmer B, de Lima TCM, Herzog H, Sperk G, The role of Neuropeptide Y in fear conditioning and extinction, Neuropeptides. 55 (2016) 111–126. 10.1016/j.npep.2015.09.007. [DOI] [PubMed] [Google Scholar]
- [8].Gøtzsche CR, Woldbye DPD, The role of NPY in learning and memory, Neuropeptides. 55 (2016) 79–89. 10.1016/j.npep.2015.09.010. [DOI] [PubMed] [Google Scholar]
- [9].Thorsell A, Mathé AA, Neuropeptide Y in Alcohol Addiction and Affective Disorders, Front. Endocrinol 8 (2017) 178. 10.3389/fendo.2017.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pedragosa-Badia X, Stichel J, Beck-Sickinger AG, Neuropeptide Y receptors: how to get subtype selectivity, Front. Endocrinol 4 (2013) 5. 10.3389/fendo.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kaiser A, Müller P, Zellmann T, Scheidt HA, Thomas L, Bosse M, Meier R, Meiler J, Huster D, Beck-Sickinger AG, Schmidt P, Unwinding of the C-Terminal Residues of Neuropeptide Y is critical for Y 2 Receptor Binding and Activation, Angew. Chem. Int. Ed. Engl. 54 (2015) 7446–7449. 10.1002/anie.201411688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang Z, Han S, Keller M, Kaiser A, Bender BJ, Bosse M, Burkert K, Kögler LM, Wifling D, Bernhardt G, Plank N, Littmann T, Schmidt P, Yi C, Li B, Ye S, Zhang R, Xu B, Larhammar D, Stevens RC, Huster D, Meiler J, Zhao Q, Beck-Sickinger AG, Buschauer A, Wu B, Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor, Nature. 556 (2018) 520–524. 10.1038/s41586-018-0046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gershkovich MM, Groß VE, Kaiser A, Prömel S, Pharmacological and functional similarities of the human neuropeptide Y system in C. elegans challenges phylogenetic views on the FLP/NPR system, Cell Commun. Signal. 17 (2019) 123. 10.1186/s12964-019-0436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Peymen K, Watteyne J, Frooninckx L, Schoofs L, Beets I, The FMRFamide-Like Peptide Family in Nematodes, Front. Endocrinol. 5 (2014) 90. 10.3389/fendo.2014.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frooninckx L, Van Rompay L, Temmerman L, Van Sinay E, Beets I, Janssen T, Husson SJ, Schoofs L, Neuropeptide GPCRs in C. elegans, Front. Endocrinol. 3 (2012) 167. 10.3389/fendo.2012.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kaiser A, Hempel C, Wanka L, Schubert M, Hamm HE, Beck-Sickinger AG, G Protein Preassembly Rescues Efficacy of W 6.48 Toggle Mutations in Neuropeptide Y 2 Receptor, Mol. Pharmacol. 93 (2018) 387–401. 10.1124/mol.117.110544. [DOI] [PubMed] [Google Scholar]
- [17].Merten N, Lindner D, Rabe N, Römpler H, Mörl K, Schöneberg T, Beck-Sickinger AG, Receptor Subtype-specific Docking of Asp6.59 with C-terminal Arginine Residues in Y Receptor Ligands, J. Biol. Chem. 282 (2007) 7543–7551. 10.1074/jbc.M608902200. [DOI] [PubMed] [Google Scholar]
- [18].Munk C, Isberg V, Mordalski S, Harpsøe K, Rataj K, Hauser AS, Kolb P, Bojarski AJ, Vriend G, Gloriam DE, GPCRdb: the G protein-coupled receptor database - an introduction, Br. J. Pharmacol. 173 (2016) 2195–2207. 10.1111/bph.13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luo J, Xu Z, Tan Z, Zhang Z, Ma L, Neuropeptide receptors NPR-1 and NPR-2 regulate Caenorhabditis elegans avoidance response to the plant stress hormone methyl salicylate, Genetics. 199 (2015) 523–531. 10.1534/genetics.114.172239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Horovitz A, Double-mutant cycles: a powerful tool for analyzing protein structure and function, Fold. Des. 1 (1996) R121–126. 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- [21].Song Y, DiMaio F, Wang RY-R, Kim D, Miles C, Brunette T, Thompson J, Baker D, High-Resolution Comparative Modeling with RosettaCM, Structure. 21 (2013) 1735–1742. 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Monks SA, Karagianis G, Howlett GJ, Norton RS, Solution structure of human neuropeptide Y, J. Biomol. NMR. 8 (1996) 379–390. 10.1007/BF00228141. [DOI] [PubMed] [Google Scholar]
- [23].Bader R, Bettio A, Beck-Sickinger AG, Zerbe O, Structure and Dynamics of Micelle-bound Neuropeptide Y: Comparison with Unligated NPY and Implications for Receptor Selection, J. Mol. Biol. 305 (2001) 307–329. 10.1006/jmbi.2000.4264. [DOI] [PubMed] [Google Scholar]
- [24].Reddy KC, Hunter RC, Bhatla N, Newman DK, Kim DH, Caenorhabditis elegans NPR-1–mediated behaviors are suppressed in the presence of mucoid bacteria, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 12887–12892. 10.1073/pnas.1108265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fadda M, De Fruyt N, Borghgraef C, Watteyne J, Peymen K, Vandewyer E, Naranjo Galindo FJ, Kieswetter A, Mirabeau O, Chew YL, Beets I, Schoofs L, NPY/NPF-related neuropeptide FLP-34 signals from serotonergic neurons to modulate aversive olfactory learning in Caenorhabditis elegans, J. Neurosci. 40 (2020) 6018–6034. 10.1523/JNEUROSCI.2674-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou J, Martin RJ, Tulley RT, Raggio AM, McCutcheon KL, Shen L, Danna SC, Tripathy S, Hegsted M, Keenan MJ, Dietary resistant starch upregulates total GLP-1 and PYY in a sustained day-long manner through fermentation in rodents, Am. J. Physiol.-Endocrinol. Metab. 295 (2008) E1160–E1166. 10.1152/ajpendo.90637.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nguyen AD, Mitchell NF, Lin S, Macia L, Yulyaningsih E, Baldock PA, Enriquez RF, Zhang L, Shi Y-C, Zolotukhin S, Herzog H, Sainsbury A, Y1 and Y5 Receptors Are Both Required for the Regulation of Food Intake and Energy Homeostasis in Mice, PLOS ONE. 7 (2012) e40191. 10.1371/journal.pone.0040191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE, Pathway and mechanism of drug binding to G-protein-coupled receptors, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 13118–13123. 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Krumm BE, Grisshammer R, Peptide ligand recognition by G protein-coupled receptors, Front. Pharmacol. 6 (2015) 48. 10.3389/fphar.2015.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Saleh N, Hucke O, Kramer G, Schmidt E, Montel F, Lipinski R, Ferger B, Clark T, Hildebrand PW, Tautermann CS, Multiple Binding Sites Contribute to the Mechanism of Mixed Agonistic and Positive Allosteric Modulators of the Cannabinoid CB1 Receptor, Angew. Chem. Int. Ed. Engl. 57 (2018) 2580–2585. 10.1002/anie.201708764. [DOI] [PubMed] [Google Scholar]
- [31].Kaiser A, Coin I, Capturing Peptide-GPCR Interactions and Their Dynamics, Molecules. 25 (2020) 4724. 10.3390/molecules25204724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Larhammar D, Salaneck E, Molecular evolution of NPY receptor subtypes., Neuropeptides. 38 (2004) 141–151. 10.1016/j.npep.2004.06.002. [DOI] [PubMed] [Google Scholar]
- [33].Zandawala M, Moghul I, Yañez Guerra LA, Delroisse J, Abylkassimova N, Hugall AF, O’Hara TD, Elphick MR, Discovery of novel representatives of bilaterian neuropeptide families and reconstruction of neuropeptide precursor evolution in ophiuroid echinoderms, Open Biol. 7 (2017) 170129. 10.1098/rsob.170129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tang T, Hartig C, Chen Q, Zhao W, Kaiser A, Zhang X, Zhang H, Qu H, Yi C, Ma L, Han S, Zhao Q, Beck-Sickinger AG, Wu B, Structural basis for ligand recognition of the neuropeptide Y Y2 receptor, Nat. Commun. 12 (2021) 737. 10.1038/s41467-021-21030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM, Molecular signatures of G-protein-coupled receptors, Nature. 494 (2013) 185–194. 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- [36].Claff T, Yu J, Blais V, Patel N, Martin C, Wu L, Han GW, Holleran BJ, Van der Poorten O, White KL, Hanson MA, Sarret P, Gendron L, Cherezov V, Katritch V, Ballet S, Liu Z-J, Müller CE, Stevens RC, Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists, Sci. Adv. 5 (2019) eaax9115. 10.1126/sciadv.aax9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, Frimurer TM, Schwartz TW, A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors, J. Biol. Chem. 285 (2010) 3973–3985. 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brenner S, The Genetics of Caenorhabditis Elegans, Genetics. 77 (1974) 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].de Bono M, Bargmann CI, Natural Variation in a Neuropeptide Y Receptor Homolog Modifies Social Behavior and Food Response in C. elegans, Cell. 94 (1998) 679–689. 10.1016/S0092-8674(00)81609-8. [DOI] [PubMed] [Google Scholar]
- [40].Mäde V, Els-Heindl S, Beck-Sickinger AG, Automated solid-phase peptide synthesis to obtain therapeutic peptides, Beilstein J. Org. Chem. 10 (2014) 1197–1212. 10.3762/bjoc.10.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hoffmann S, Rist B, Videnov G, Jung G, Beck-Sickinger AG, Structure-affinity studies of C-terminally modified analogs of neuropeptide Y led to a novel class of peptidic Y1 receptor antagonist, Regul. Pept. 65 (1996) 61–70. 10.1016/0167-0115(96)00073-0. [DOI] [PubMed] [Google Scholar]
- [42].Heckman KL, Pease LR, Gene splicing and mutagenesis by PCR-driven overlap extension, Nat. Protoc. 2 (2007) 924–932. 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- [43].Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV, Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate, ACS Chem. Biol. 7 (2012) 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cheng Y, Prusoff WH, Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction, Biochem. Pharmacol. 22 (1973) 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- [45].Mello CC, Kramer JM, Stinchcomb D, Ambros V, Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences, EMBO J. 10 (1991) 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mello C, Fire A, DNA transformation, Methods Cell Biol. 48 (1995) 451–482. [PubMed] [Google Scholar]
- [47].Bender BJ, Vortmeier G, Ernicke S, Bosse M, Kaiser A, Els-Heindl S, Krug U, Beck-Sickinger A, Meiler J, Huster D, Structural Model of Ghrelin Bound to its G Protein-Coupled Receptor, Structure. 27 (2019) 537–544.e4. 10.1016/j.str.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bender BJ, Marlow B, Meiler J, Improving homology modeling from low-sequence identity templates in Rosetta: A case study in GPCRs, PLOS Comput. Biol. 16 (2020) e1007597. 10.1371/journal.pcbi.1007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Larsson P, Wallner B, Lindahl E, Elofsson A, Using multiple templates to improve quality of homology models in automated homology modeling, Protein Sci. 17 (2008) 990–1002. 10.1110/ps.073344908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG, Clustal W and Clustal X version 2.0, Bioinformatics. 23 (2007) 2947–2948. 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- [51].Wheatley M, Wootten D, Conner M, Simms J, Kendrick R, Logan R, Poyner D, Barwell J, Lifting the lid on GPCRs: the role of extracellular loops, Br. J. Pharmacol. 165 (2012) 1688–1703. 10.1111/j.1476-5381.2011.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jones DT, Protein secondary structure prediction based on position-specific scoring matrices, J. Mol. Biol. 292 (1999) 195–202. 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- [53].Viklund H, Elofsson A, OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar, Bioinformatics. 24 (2008) 1662–1668. 10.1093/bioinformatics/btn221. [DOI] [PubMed] [Google Scholar]
- [54].Raveh B, London N, Zimmerman L, Schueler-Furman O, Rosetta FlexPepDock ab-initio: Simultaneous Folding, Docking and Refinement of Peptides onto Their Receptors, PLOS ONE. 6 (2011) e18934. 10.1371/journal.pone.0018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ballesteros JA, Weinstein H, Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors, in: Sealfon SC (Ed.), Methods Neurosci, Academic Press, 1995: pp. 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.