

Abstract
Objective
Serious non-gastrointestinal-tract infections and food poisoning are caused by Bacillus cereus. Vaccination against B. cereus is very important. The aim of this study was to identify and analyze B and T cell epitopes for chromate transporter protein of the bacteria.
Methods
Multiple sequence alignment with the Clustal Omega method was used to identify conserved regions and Geneious Prime was used to produce a consensus sequence. T and B cell epitopes were predicted by various computational tools from the NetCTL and Immune Epitope Database (IEDB), respectively.
Results
Altogether, 6 HTL cells and 11 CTL epitopes were predicted. This vaccine's molecular docking is done with Patch Dock and LigPlot to verify interactions. The immune server (C-IMMSIM) was used to develop In silico immune response in order to assess the multi-epitope vaccine's immunogenic profile.
Conclusion
We designed universal vaccine against B. cereus responsible for food poisoning. The disease may be avoided with the aid of the proposed epitope-based vaccine.
Keywords: Epitope-based vaccine, Bacillus cereus, Food poisoning, Immunity, Bioinformatics
1. Introduction
Acute gastroenteritis with cramps, pain in stomach, vomiting and watery diarrhea is a common symptom of foodborne illness. The consumption of viral or bacterial pathogens (toxic infection) in addition to water or food born bacterial toxins, may cause disease (a state of intoxication). Between 1991 and 2000, the outbreaks of foodborne illness in United States have doubled as reported by Disease Control and Prevention Centers (CDC), which could be credited to improved monitoring processes as well as an increase in the food poisoning rate. Within the genus Bacillus, Bacillus cereus (sensu lato) is a subgroup which is more closely related genetically. B. pseudomycoides, B. thuringiensis, B. mycoides, B. cereus (sensu stricto), B. weihenstephanensis and B. anthracis are the six species that make up this group (Priest and Alexander, 1988, Nakamura et al., 1998). Regardless of their phenotypic differences and pathological consequences, the description of the species has been a point of contention in recent decades. The B. cereus group strains are thought to be one species on the basis of similarity in 16S rRNA, gene material on chromosomes as well as gene synteny (Helgason et al., 2000, Fricker et al., 2011). B. anthracis, B. cereus and B. thuringiensis, in particular, share over 99 percent of their sequences of 16S rRNA in general (Ash et al., 1991). B. cereus could be found in various habitats, such as dirt, sand, sediment as well as on plants (Kotiranta et al., 2000). During a saprophytic life cycle, the germination of spores takes place and can grow vegetatively in soil. Usually spores or bacteria travel rapidly from the natural environment to the field where food is prepared and they cross-contaminate with other ingredients and foods including meat. Therefore, high temperature as well low pH was used to preserve the spores of B. cereus as they are resistant in these conditions (pasteurization, heating). The processes of chemical preservation (Setlow, 2006), facilities used for commercial food processing and catering on a wide scale, in particular, are at high contamination risk if not sanitized in a proper way (Abee et al., 2011).
Aside from the ecological niches of B. cereus as a benevolent organism, it is frequently related to short-term gastroenterological pathologies which are caused by generation of toxin (Stenfors Arnesen et al., 2008). B. cereus, on the other hand, has been linked to a variety of uncommon but potentially lethal, local as well as systemic diseases that are not gastrointestinal in nature (Bottone, 2010). Meningitis (Turnbull et al., 1979, Barrie et al., 1992), fulminant bacteraemia (Hilliard, 2003), serious optical infections (endolphthalmitis and keratitis) (Drobniewski, 1993, Callegan et al., 2002), pneumonia (Avashia et al., 2007), skin infections and endocarditis (Craig and Abeloff, 1974) have been recognized. Infections with B. cereus that are serious and fatal are often related to hospital-acquired infection and colonized catheters that stay in body (Hernaiz et al., 2003). Significant tissue degradation/destruction characteristics all of these pathologies as a consequence of undefined cytolytic and regulation of tissue-reactive enzymes (Bottone, 2010). The aim of this study was to use whole genome analysis of B. cereus to predict the most virulent protein. In addition, the CTL, HTL cell as well as T cell epitopes of a virulent protein were modelled for multi epitope vaccine (MEV) construction. Finally, the current research included the prediction of the structure of a multi epitope dependent vaccine as well as the evaluation of its different properties using immunoinformatic tools.
2. Material and methods
2.1. Retrieval of whole genome FASTA sequences
B. cereus reference protein sequences were retrieved from Protein Database of National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/protein) and the sequences were then saved in FASTA format. Proteins with less than 100 amino acids are too small to predict epitopes, were eliminated. The remaining proteins were used for further study.
2.2. Selection of most antigenic protein through whole genome analysis
VaxiJen is the first server to predict protective antigens without using alignment, overcoming the limitations of alignment-dependent method. VaxiJenv2.0, an online prediction server was used to check antigenicity of SARS-CoV-2 proteins' (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) using default parameters. Threshold for this model was 0.4. Moreover, Secretome P 2.0 webserver http://www.cbs.dtu.dk/services/SecretomeP/ was used to calculate each protein's secretory score. This identification was carried out using the server's default parameter. The secretary proteins were sorted using a score of 0.6 (threshold value for bacterial proteins).
2.3. Multiple sequence alignment of Stage II sporulation protein Q
Following the acquisition of Stage II sporulation protein Q in B. cereus on the basis of antigenicity and secretory score, the sequences of the protein in different strains of the bacteria were retrieved from NCBI. Alignment of multiple sequences (MSAs) were performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Multiple sequence alignments have the advantage of illustrating conserved regions, allowing for the detection of proteins that are distantly related. The conserved region was chosen because it contained the largest number of related and identical amino acids without gaps.
2.4. Generation of consensus sequence
Geneious Prime software aligns sequences first, then produces consensus sequences from the aligned sequences. Geneious is a versatile and detailed collection of molecular biology and next-generation sequencing (NGS) sequence analysis methods. The consensus sequence was generated from the created MSA file by Geneious Prime software.
2.5. Estimation and selection of T-cell epitopes
The epitopes on T-cells may be found on the outer side of the cells presenting antigen and allow MHC molecules to attach to that surface. MHC molecules are classified into two classes: Major Histocompatibility Class I and II (MHC I and MHC II). The peptides, which consist of 8 to 11 amino acids are found in cell epitopes with MHC class I molecules, while peptides ranging from 13 to 17 amino acids in length are found in cell epitopes of molecules include in Major Histocompatibility Class II. To predict the epitopes of T-cell, NetCTL web-based tool was used. For rational vaccine design, the predictions of epitope for Cytotoxic T lymphocyte (CTL) usually required. Using the new NetMHCIIpan 4.0 server, the 15-mer MHC class-II epitopes of T-cell (HTL) were attained. Depending on the percentage value, the epitopes were graded as non-binding, weak binding and strong binding. The percentile requirements are above 10% for non-binding, 2% for strong binding and 10% for weak binding.
2.6. Designing of vaccine construct
In order to construct the vaccine based on the predicted epitopes, GPGPG spacer was chosen over the other potential spacers primarily for two reasons. First, GPGPG extensions near the core binding region reduce binding affinity, leading to the conclusion that GPGPG-containing epitopes will not bind competently unless the spacer was extracted by antigen processing. As a result, this spacer will increase the chances of the related epitopes being regenerated. Second, since areas rich in G and P are correlated with turns, the GPGPG spacer was chosen to build secondary and tertiary structures. Moreover, EAAAK linker was used to bind an adjuvant to first cytotoxic T cell epitope (CTL) to improve the immune response, whereas some epitopes were linked with the GPGPG as well as AAY linkers in order to retain their immunogenic responses that are independent by following the verification of their compatibility of interaction. Since, peptide molecule contains 45 amino acids serves as an antimicrobial and a modulator of the immune system, the defensin was used as an adjuvant in this research.
2.7. Prediction of tertiary Structure, modification and Validation:
After joining epitopes, trRosetta predicts the build vaccine’s structure. trRosetta (https://yanglab.nankai.edu.cn/trRosetta/) is a prediction of protein structure algorithm that is fast and accurate. It constructs the protein structure using a restrained Rosetta and direct energy minimizations. A residual neural network with a deep layer forecasted distributions of orientation and the distance between residues, are among the constraints. trRosetta uses both residue–residue distances and orientations, which offers more structure information than just distances.
2.8. B cell epitope’s prediction
B-cells secrete antibodies that bind to antigens are referred to as epitopes of B-cell. To induce protective immunity, B-cell epitopes of B cell from the pathogen's proteins used linear B-cell epitopes. By using the default settings maximum distance (6) and minimum score (5), Ellipro-tool (IEDB-AR v.2.22) was used for final MEV construct to predict the conformational epitopes of B cells. It calculates the index of residual protrusion (PI), protein form, and neighbor clustering of residues to predict epitopes.
2.9. Enrichment analysis of the predicted vaccine
Antigenicity is the capacity to identify a particular antigen followed by an immune response, while the capability to cause cellular as well as humoral immune responses is immunogenicity. Hence, the vaccine must be immunogenic as well as antigenic. VaxiJen (http://www.jenner.ac.uk/VaxiJen) was used to predict protective antigens without using alignment as a first server. It classifies antigen on the basis of physicochemical characteristics of protein, rather than alignment of specific sequence. The candidate vaccine's allergenicity was tested to ensure that it did not cause any allergic reactions until it was administered to the body. AllerTOP and AllergenFP web servers were used for this purpose. Moreover, ProtParam (http://web.expasy.org/protparam/) was used to detect instability index, theoretical pI, GRAVY, half-life, aliphatic index and hydropathy profiling of stability for the predicted vaccine. Furthermore, codon optimization and cloning (In silico) were performed using JCAT (Java Codon Adaptation Tool) server. This server was used to render MEV optimization of codon compatible with commonly used expression system of prokaryotes (E. coli K12). Extra options were selected to avoid binding sites of prokaryotic ribosome, termination of transcription (rho-independent) and cleavage sites for restriction enzyme. The guanine and cytosine’s quality as well as the adaptation index of codon (CAI) were assessed.
2.10. Immune simulation
For verification of immune response of the predicted MEV, C-ImmSim server (http://150.146.2.1/C-IMMSIM/index.php) was used to perform immune simulation (In silico). The function of this server is to simulate the three components of mammalian system (thymus, lymph node and bone marrow). The ability of the MEV to stimulate cells of immune system like NK cells, HTL, B-cells, CTL, immunoglobulins, cytokines and dendritic cells were tested. In clinical practice, a four-week period between two vaccine doses is recommended. Immune simulation was carried out using a protocol close to this system. In a nutshell, three injections were administered at every four weeks (time-steps parameters which were fixed are 1, 84, as well as 168 and one time-step = 8 h of real life). Altogether, the total simulation steps were 1050.
2.11. Molecular docking
The vaccine’s interacting molecules with receptors of immune system was studied using docking of molecules between immune receptors such as toll like receptors 4 (TLR4), TLR2, and TLR3. If there is an antigen or vaccine interaction appropriately with target immune cells, the host develops an effective immune response. As a result, a docking study at molecular level was accomplished for analysis of MEV's binding to immune receptors of human. The MEV docking with TLR4, TLR2, and TLR3 was implemented using Patchdock (https://bioinfo3d.cs.tau.ac.il/PatchDock/). Protein to protein and the complexes of small molecules with protein are predicted using the PatchDock process. The codes of protein PDB or the structures of protein uploaded are used as inputs to the servers. The LIGPLOT software was used to visualize the docked complex and draw figures. For a given PDB format, the LIGPLOT program produced schematic diagrams of protein–ligand interactions.
3. Results
3.1. Retrieval and alignment of FASTA sequences for B. Cereus
NCBI has FASTA sequences of various strains of B. cereus since it is a completely open database that contains information about proteins from various databases. The alignment of the retrieved sequences was performed to verify the functional and evolutionary relationships between protein sequences. It is a method of arranging nucleotides of DNA or amino acids in proteins in order to find areas of similarity. Custal omega alignment tool was used to align the protein sequences of B. cereus Stage II sporulation protein Q using default settings.
3.2. Consensus sequence generation
The most common residues, present in each location in an alignment of the sequences, either amino acid or nucleotide are referred to as consensus sequence. It denotes the outcomes of alignments of multiple sequences, in which sequences that are connected are compared and identical sequence motifs are determined. Consensus sequences in proteins may reflect entire proteins or short fragments of them that correspond to conserved structural and functional regions. The Geneious Prime Tool was used to establish a consensus sequence of Stage II sporulation protein Q. The generated consensus sequence is as follows:
MRGRNNKKSQKVVHLFQKRWVFPALYIACAAVILMVALWFQGANPKKTPNQDQATPYTQSEDPAVPVTKSSEVVKMPAAANAEVVVQKKFYEDAATEAEQEKALVFYNNTYSPNKGIDIAAKNGKEFNVAAALSGTVTKAEKDSLLGYVVTVDSGNGVAVSYQSLGSVKVEKGARVAQGEVLGKSGLSAMNKEAGSHVHFEVRKDNVAVNPERYLNKSVAEIKADAGAAKATNASGKKADDKSQKEEKSTSTKPESKTEKEEKSTSGSTSDKETNGKQDDKSQKEEKSTSGSTSDKKTNGKQDEEPKKEEKSTNGSTESSNDSSSQE
3.3. Prediction of CTL and HTL epitopes of Stage II sporulation protein Q
NETCTL server was used to forecast the CTL and HTL epitopes. The epitopes were predicted by NetMHCIIpan 4.0 Server. The predicted epitopes are mentioned in Table 1 and Table 2. Altogether, 17 epitopes were predicted. The predicted epitopes consist of 11 CTL epitopes while 6 HTL epitopes.
Table 1.
The predicted CTL epitopes from the consensus sequence of the protein. NETCTL server was used to forecast the CTL epitopes. The epitopes were predicted by NetMHCIIpan 4.0 Server. The predicted epitopes consist of 11 CTL epitopes.
| Residue No. | Peptide sequence | Affinity for MHC binding | Affinity of C terminal cleavage | Efficiency of TAP transport | Prediction score | Antigenicity score |
|---|---|---|---|---|---|---|
| 269(A1) | TSDKETNGK | 0.196 | 0.832 | 0.362 | 0.9786 | 3.1241 |
| 293(A1) | TSDKKTNGK | 0.1832 | 0.7776 | 0.362 | 0.9292 | 2.9245 |
| 33(A2) | ILMVALWFQ | 0.5736 | 0.8551 | 0.049 | 0.8672 | 1.7067 |
| 288(A3) | STSGSTSDK | 0.5685 | 1.0699 | 0.593 | 1.2433 | 2.8255 |
| 25(A24) | LYIACAAVI | 0.6897 | 1.4687 | 1.026 | 1.5281 | 1.0565 |
| 31(A26) | AVILMVALW | 0.3132 | 0.8404 | 1.39 | 1.0106 | 1.2229 |
| 119(B7) | IAAKNGKEF | 0.3111 | 0.6002 | 2.488 | 0.7699 | 0.7111 |
| 201(B8) | EVRKDNVAV | 0.2571 | 0.878 | 0.193 | 0.9402 | 1.3946 |
| 3(B27) | GRNNKKSQK | 0.5253 | 1.3965 | 0.53 | 1.5186 | 2.7186 |
| 96(B44) | TEAEQEKAL | 0.4214 | 1.0444 | 0.969 | 1.1376 | 0.976 |
| 154(B58) | SGNGVAVSY | 0.2252 | 0.5076 | 2.478 | 0.7759 | 1.8013 |
Table 2.
The predicted HTL epitopes from the consensus sequence of the protein.NETCTL server was used to forecast the HTL epitopes. The epitopes were predicted by NetMHCIIpan 4.0 Server. The predicted epitopes consist of 6 HTL epitopes.
| Epitopes | Position | Allele | Score | Strong/weak | Antigenicity score |
|---|---|---|---|---|---|
| GYVVTVDSGNGVAVS | 147 | DRB3_0101 | 0.24 | SB | 1.1891 |
| NTYSPNKGIDIAAKN | 109 | DRB3_0202 | 0.28 | SB | 1.0259 |
| NKSVAEIKADAGAAK | 216 | DRB4_0101 | 0.09 | SB | 1.1495 |
| KSVAEIKADAGAAKA | 217 | DRB4_0101 | 0.28 | SB | 1.0720 |
| MVALWFQGANPKKTP | 35 | DRB5_0101 | 0.6 | SB | 1.1171 |
| VALWFQGANPKKTPN | 36 | DRB5_0101 | 0.95 | SB | 1.1946 |
3.4. Construction of MEV
All 17 predicted epitopes (CTL 11 and HTL 6) were used to build MEV construct. To connect the epitopes, EAAAK linker was used to bind with defensin adjuvant. The adjuvant consists of 45 amino acids and placed at the start (N-terminal) of the construct (highlighted green). EAAAK linker improves stability by severing relations with other protein domains and allowing for effective attachment. With an adjuvant, the vaccine's immunogenicity can improve. Epitopes were fused together in a sequence with AAY linkers for CTL epitopes (highlighted blue) while GPGPG linkers for HTL epitopes (highlighted red) on the basis of compatibility of interaction. GPGPG as well as AAY linkers inhibit the formation of junctional epitopes on junctions which is most important role in development of vaccines with many epitopes. Moreover, they facilitate immunization and presentation of epitope. Finally, the final vaccine construct was consist of 239 amino acids as mentioned below:
GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKKEAAAKILMVALWFQAAYTSDKETNGKAAYTSDKKTNGKAAYSTSGSTSDKAAYLYIACAAVIAAYAVILMVALWAAYIAAKNGKEFAAYEVRKDNVAVAAYGRNNKKSQKAAYTEAEQEKALAAYSGNGVAVSYGPGPGGYVVTVDSGNGVAVSGPGPGNTYSPNKGIDIAAKNGPGPGNKSVAEIKADAGAAKGPGPGKSVAEIKADAGAAKAGPGPGMVALWFQGANPKKTPGPGPGVALWFQGANPKKTPN
3.5. Prediction and assessment of structure of MEV construct
trRosetta was used to predict the structure of vaccine construct. It constructs the protein structure using a restrained Rosetta and direct energy minimizations. Distributions of inter-residue distance and orientation were anticipated by neural network with deep residual, are among the constraints. To increase the precision of simple targets, homologous models are used in the network prediction. Using the pdbsum Ramchandran plot, the overall quality of the structure was determined to be 88.8%. The structure is shown in Fig. 1.
Fig. 1.
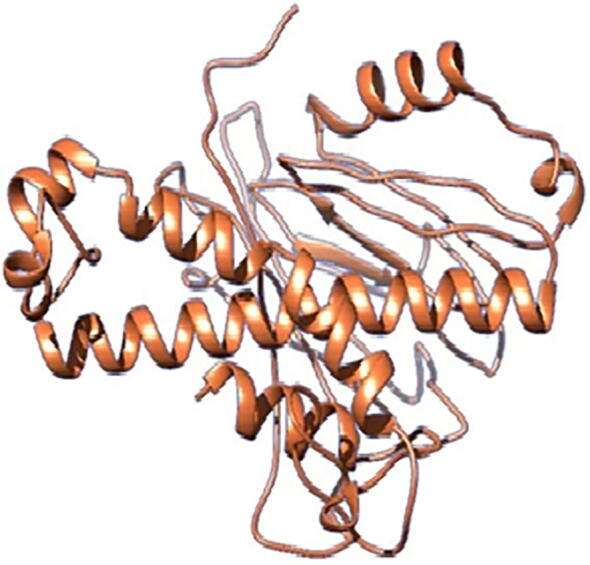
The structure of the predicted protein. The structure is based on the constructed MEV. trRosetta was used to predict the structure of the vaccine construct. The quality of the structure was predicted as 88.8%.
3.6. Prediction of B cell epitopes from constructed MEV
The epitopes related to B-cells can elicit humoral immunity. The presence of these epitopes in the vaccine design is essential for eliciting a successful immune response. To predict B cell epitopes from the constructed MEV, ElliPro webserver was used to predict conformational and linear B cell epitopes using default parameters. The predicted epitopes are mentioned in Table 3 and Table 4.
Table 3.
Conformational or discontinuous B cell epitopes in a vaccine with many epitopes as predicted by ElliPro. The epitopes were predicted using default parameters.
| Discontinuous epitopes | Score |
|---|---|
| A:D225, A:A226, A:G227, A:A228, A:A229, A:K230, A:A231, A:T232, A:N233, A:A234, A:S235, A:G236, A:K237, A:K238, A:A239, A:D240, A:D241, A:K242, A:S243, A:K245, A:E246, A:E247, A:S249, A:T250, A:S251, A:T252, A:K253, A:P254, A:E255, A:S256, A:K257, A:T258, A:E259, A:K260, A:E261, A:E262, A:K263, A:S264, A:T265, A:S266, A:G267, A:S268, A:T269, A:S270, A:D271, A:K272, A:E273, A:T274, A:N275, A:G276, A:K277, A:Q278, A:D279, A:D280, A:K281, A:S282, A:Q283, A:K284, A:E285, A:E286, A:K287, A:S288, A:T289, A:S290, A:G291, A:S292, A:T293, A:S294, A:D295, A:K296, A:K297, A:T298, A:N299, A:G300, A:K301, A:Q302, A:D303, A:E304, A:E305, A:P306, A:K307, A:K308, A:E309, A:E310, A:K311, A:S312, A:T313, A:N314, A:G315, A:S316, A:T317, A:E318, A:S319, A:S320, A:N321, A:D322, A:S323, A:S324, A:S325, A:Q326, A:E327 | 0.777 |
| A:Y91, A:D93, A:A94, A:A95, A:T96, A:E97, A:A98, A:E99, A:Q100, A:E101, A:K102 | 0.634 |
| A:M1, A:R2, A:G3, A:R4, A:N5, A:N6, A:K7, A:K8, A:S9, A:Q10, A:K11, A:V12, A:V13, A:H14, A:L15, A:F16, A:Q17, A:K18, A:R19, A:W20, A:V21, A:F22, A:P23, A:A24, A:L25, A:Y26, A:I27, A:A31, A:E83, A:L104, A:V105, A:F106, A:Y107, A:N108, A:N109, A:T110, A:Y111, A:A121, A:K122, A:N123, A:G124, A:K125, A:K142, A:D143, A:S144, A:L145, A:G147, A:Y148, A:G186, A:L187, A:S188, A:A189, A:N191, A:K192, A:E193, A:A194, A:G195, A:S196 | 0.58 |
| A:A43, A:N44, A:P45, A:K46, A:K47, A:T48, A:P49, A:N50, A:Q51, A:D52, A:Q53, A:A54, A:T55, A:P56, A:Y57, A:T58, A:Q59, A:S60, A:D62, A:P63, A:A64, A:V65, A:P66, A:V67, A:T68, A:N156 | 0.533 |
Table 4.
Linear/continuous B cell epitopes in the Vaccine construct, predicted by ElliPro server using default parameters. The presence of these epitopes in the vaccine design is essential for eliciting a successful immune response.
| Linear epitopes | Position | Score |
|---|---|---|
| MRGRNNKKSQKVVHLFQKRWV | 21 | 0.68 |
| DAATEAEQEKA | 11 | 0.629 |
| VFYNNTYS | 8 | 0.606 |
3.7. Enrichment analysis of MEV construct
The candidate vaccine's safety and efficacy must be determined using a variety of physicochemical properties. With a score of 0.9837, VaxiJen v2.0 approved the antigenicity of vaccine's (a score of greater than 0.4 is considered antigenic). The candidate vaccine's allergenicity was tested to ensure that it did not cause any allergic reactions until it was administered to the body. As expected by the AllerTOP web server, the vaccine was non-allergenic. Moreover, ExPASy ProtParam server determines other physical and chemical characteristics of vaccine, such as molecular weight, aliphatic index, isoelectric point, half-life, instability index and GRAVY ranking. The vaccine construct was consisting of 299 amino acids with molecular weight of 30696.02 Dalton. The estimated half-life of the MEV was 30 h (mammalian reticulocytes, In vitro), more than 20 h (yeast, In vivo) while more than10 hours (Escherichia coli, In vivo). The Instability index of the MEV was 18.12 which classifies the protein as stable. Moreover, aliphatic index of the construct was 67.46 while the GRAVY value was −0.305. The vaccine's theoretical pI was discovered to be 9.97. Furthermore, the reverse translated vaccine had a codon adaptation index (CAI) of 1.0 and a GC content of 50.61 percent.
3.8. Molecular docking analysis for the constructed MEV
The interaction of two or more molecular structures such as drug and protein or an enzyme is known as molecular docking. Docking, in its simplest form, is a molecular simulation technique for predicting the interaction of a protein with small molecules (ligands). The docking analysis highlighted interaction of the predicted protein (Fig. 1) against TLR4 as mentioned in Fig. 2.
Fig. 2.

The docking interaction of MEV protein structure against TLR4. The interaction highlighted the interacting residues among the protein and TLR4.
3.9. Immune simulation analyis
C-IMMSIM immune server was used to assess the immunological profile of the predicted MEV. The responses (secondary and tertiary) produced by the simulation were significantly higher in comparison with primary response as shown in Fig. 3. Furthermore, these secondary and tertiary responses represented decreased concentration of antigen with high level of activity of immunoglobulin. Various B cell isotopes discovered which were long lasting indicating the possibility of creation of memory and switching of isotopes. With the reactivation of immune T cells during vaccination, the helper as well as cytotoxic T cells showed a higher response. The activity of dendritic cell and natural killer cells were observed at constant during the exposure, as was higher macrophage activity. The high level of IL-2 and IFN- induced in simulation helped to generate a strong immune response. In order to check the vaccine's efficiency at day 366 an injection of virus that replicates itself in real time was simulated after vaccination. The antigenic surge is practically absent following vaccination when replicating virus has been injected, suggesting a successful immunological reaction primarily because of defensive effect of higher particular antibodies concentrations. Results could be compared with control simulation comprise of injection of live virus after a year and no vaccination before. The findings show that host cannot retain antigen without prior vaccination, despite an inadequate immune response. Similarly, the vaccine construct was created using sequences that are randomly generated in another control experiment to see how it affected immune response. The sequences which were generated randomly gave results of immunological simulation, as predicted, represents the lack of immune response indicating the failure of vaccine. The lack of antigenic peptides in the random sequence is the simple explanation for this, which in the simulation translates to no presentation of antigens by cells which are trained for antigen presenting.
Fig. 3.

Immune simulation results for the predicted MEV. C-IMMSIM immune server was used to assess the immunological profile of the predicted MEV. The responses (secondary and tertiary) produced by the simulation were significantly higher in comparison with primary response.
4. Discussion
The purpose of this research is to develop a prophylactic vaccination with many epitopes that targets the B. cereus Stage II sporulation Q protein, which is among the most important determining factors of viral antigenicity and host cell entry. A vaccine with many epitopes capable of eliciting cell-mediated as well as humoral immunity was created using a combination of computational methods. Instead of massive proteins or entire genomes, which are commonly used in recombinant vaccine technology, immune responses are produced by the vaccine with many epitopes by using immunogenic sequences of short length. As a result, this method prevents both excessive antigenic load and allergenic responses in the host. To investigate the possibility of interaction with host proteins, immunoinformatic and molecular simulation can be used to examine the entire spectrum of possible antigens.
The Clustal Omega method was used in the alignment of several sequences, which displays conserved regions of all proteins from B. cereus strains. Clustal Omega is a user-friendly software that aligns sequences using a seeded directed tree and the HMM profile algorithm. The consensus sequence was then generated using Geneious prime, a comprehensive method. The useful prime tool provides details on the most common amino acid residues in proteins. The structure of the chromate transporter protein consensus sequence was then predicted using the threading method and the I-TASSER instrument.
The conserved regions of proteins were analyzed for prediction of B-cell epitope whereas the T cell epitope prediction was also done by using conserved regions of proteins. IEDB was used for prediction of epitopes on B cells. The numerous databases are accessible via IEDB. The consensus sequence was used for B cell prediction whereas, the NetCTL server used for forecasting epitopes on T cells. The MHC molecule that belongs to the first class of MHC was used for T cell epitopes since it is expressed by all nucleated cells and introduces the cleaved fragments of antigen to T cells. NetCTL 1.0 is used to predict T cell epitopes since it does not require alleles to produce T cell epitopes (Desai and Kulkarni-Kale, 2014). We were able to acquire two T cell epitopes. On all performance tests, NetCTL-1.2 outperforms MHC-pathway, EpiJen, WAPP and MAPPP in terms of predictive efficiency. However, in none of the studies, the statistical significance of NetCTL-1.2′s superior output over MHC-pathway and EpiJen is important (Sohail Raza et al., 2021). On a broad scale measurement consists of 216 identified HIV epitopes that cover all 12 HLA subtypes, for the peptides that scored in the top 5% the NetCTL-1.2 approach was found sensitive (above 0.72). On this dataset, the most sensitive of the other methods was 0.64.
After that, epitope selection was carried out, and obsolete epitopes were eliminated. There were six T-cell epitopes as well as three B-cell epitopes predicted. Epitopes were attached together by using GPGPG Spacer. There are two explanations why the spacer was chosen. The first is that adding GPGPG lowers the affinity of binding across the key regions that bind. The spacer was chosen for the second reason that has high G and P material, which have been connected to beta turns as well as may generate structures (secondary and tertiary) (Raza et al., 2019). After entering B and T cell epitopes, the structure of B cell and T cell epitopes was predicted by using trRosetta.
In plasma membrane, TLR2 (Toll-Like Receptor 2) and TLR4 (Toll-Like Receptor 4) are the type of receptors that expressed. Usually, vaccine is given in 12 doses over the course of a year. The increase in antigen concentration and relative antibody responses is depicted in (Ai). Two months have passed since the last vaccination, a live-replicating virus has infected the patient. Due to the existence of protective IgGs, the virus is cleared easily, demonstrating the vaccination's efficacy. The corresponding numbers of plasma cells that produce antibodies are represented by Aii, while the behaviour of CTL (cytotoxic T cells in Aiii), HTL (helper T cells in Aiv) and macrophages (Av) is shown in Aiii–Av. (Avi) portrays the cytokine concentration over the course of the simulation, TNF-b, IL-10 and IFN-g levels are all strong, showing pro-inflammatory activity, suggesting a response to vaccine (B) and shows an infection prevention case of a single-injection infection simulation of a virus that replicates itself with no prior vaccination is seen. To make it easier to compare the different plots, the virus is injected simultaneously as seen in panels (Ai–Avi) previously in simulation. In this case, the viral load (Bi) continued to expand unabated, showing that the virus cannot be expelled by a naive host response (Bii–Bvi), proving the effectiveness of vaccine in stopping the viral outbreak. Among other immune cells, macrophages, immature dendritic cells, monocytes and granulocytes all contain the TLR4 (human Toll-Like Receptor 4). 95. TLR4 is activated by CTB because CTB and TLR4 directly interact with each other. 96. The ability of CTAB to cause an inflammatory reaction is lost in macrophages with a TLR4 deficiency, which supports this inference. 96. By direct binding with NF-B in TLR4 receptor cells, CTB may activate it, according to ELISA-based assays. TLR2 is also related to the identification of the viral envelop glycoprotein 93. MyD88 activates the TLR2 signaling pathway, so protein kinase activated by mitogen and NF-B is stimulated as a result, which contributes in central cytokine panel secretion.93. The interaction pattern of vaccine with TLR2 and TLR4 was investigated using docking studies at the molecular level. TLR4 and the vaccine build formed hydrogen bonds (seven) as well as salt bridges (three) when they were interacting, according to docking research. The bridges made of salt were built between Glu68, TLR4′s, Arg41, Asp69 as well as in vaccine's Lys82, Asp113 and Lys85 in the complex docked (Kar et al., 2020). According to docking research, TLR2 and the vaccine build formed hydrogen bonds upto nine and three bridges made of salt when they interact. The bridges made of salt developed were between the Asp520, Asp516 and Arg547 of TLR4 as well as our vaccine's Glu105, Lys85 and Lys82. According to several findings, TLR2 as well as TLR4 are essential in order to generate an efficient immune response (Mutta et al., 2001). In one of the studies, according to Totura et al. mice lacking TLR4 are more vulnerable to infection in comparison to mice with the normal TLR4 gene. In a similar study (Hu et al., 2008), the researchers observed the Toll-Like Receptor’s regulation and expression following infection in monocytic cells from humans. 48. TLR4/TLR2 expression is upregulated 24 h after infection, meaning that it is essential for the development of immune responses, according to their findings. It was also reported by Dosch et al. (2009) that on human macrophages, TLR2 binds to the S protein and causing the body to produce IL-8. The sensitization of TLR2 causes a protein release of inflammatory cytokine-8 (IL-8), which is an essential chemokine needed for the activation of the immune system's natural defenses. The RMSD graph in the simulation of the vaccine construct's molecular dynamics and the results for 10 ns showed only small variations, suggesting the vaccine's long-term viability. The graph (RMSF) revealed high-peaking regions, suggesting vaccine construct's high versatility. The most critical steps in the whole procedure for testing the stability of vaccine by in vivo simulation are simulation of molecular dynamics (MDS). Our MDS data (RMSF and RMSD) are close to those obtained from other research groups that tested the vaccine candidate's stability and versatility under in vivo conditions. 28,87,88. The optimization of codon for engineered vaccine was completed and the vaccine was reverse-translated from its linear construct to cDNA sequence that was unique to it in order to ensure successful expression in the E. coli host. Its GC content was reported as, suggesting that in E. coli cell the vaccine candidate may be expressed efficiently. In addition, the In silico cloning was done by insertion of vaccine in expression vector pET-28a (+), allowing the vaccine to be expressed in a bacterial system. Foroutan et al. (2020) used a similar approach to optimize genetically modified vaccine’s codon prior to the expression in vitro. The studies of immune simulation have shown that our genetically modified vaccine elicited unique immune responses, so following the final injection it was necessary that antigen is cleared after second dose. Actually, our immune simulation research outperformed an experiment on candidate vaccine with many epitopes that was recently released in which to assess the vaccine's efficacy on an exposure to a second antigen, there was no virus that was capable of replicating itself was injected after it was given. Using the AllerTOP v.2.0 server, we predicted that our engineered vaccine will be non-allergenic, which was confirmed by AllergenFP v.1. vexiJen 2.0 predicted the antigenicity score of MEV. ExPASy server's ProtParam platform was used to test the vaccine's other physicochemical properties. The build had a molecular weight of 30696.02 and 18.12 on the instability scale, indicating that vaccine was effective. In general, a score of less than 40 in an instability index suggests that a protein is likely to be stable, whereas one with a higher value (above 40) is predicted to be unstable. The vaccine's theoretical pI was determined to be 9.67. The vaccine's GRAVY index was 0.305 (GRAVY score is inversely proportional to solubility) as a result of the polar existence of vaccine and close proximity to water, implying that it’s solubility is high. The protein's aliphatic index of 67.46 showed it was thermostable. The vaccine's half-life was determined to be thirty hours (In vitro) followed by twenty hours (In vivo) and ten hours (In vivo) in reticulocytes from mammals, yeast and E. coli respectively, indicating the specific time it takes for protein to achieve 50% of its maximum concentration in the cell after it has been synthesized. The structure of NS3-Serine Protease was predicted using homology modelling. Based on this, we must build the crystal structure of our appropriate protein since it does not exist. Computational methods were used to find the inhibitor of NS3 Serine Protease using a molecular modelling technique.
5. Conclusion
B. cereus infections cause substantial morbidity and mortality around the world. Patients are yet to obtain adequate medical preventive measures such as vaccination in order to heal. B. cereus vaccines are available in both live attenuated and inactivated varieties, although they have lower efficacy. The specific approaches (In silico) could be used to create a vaccine that performs well in a shorter amount of time and at a lower cost. Immunoinformatic techniques are used in current research to create a vaccine with many epitopes against B. cereus that contains HTL, CTL, and epitopes of B cells which may elicit powerful immunological reactions. The vaccine with many epitopes was developed and both immunogenic as well as antigenic properties were discovered. Molecular dynamics simulations verified the vaccine's stability, and Molecular Docking experiments confirmed the vaccine's stable interaction with immune receptors. In addition, the expression experiments (In silico) verified that in host cells of bacteria vaccine is expressed and immune simulation studies validated the capability of vaccine to induce an immunological reaction.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
The authors appreciate Taif University Researchers Supporting Project number (TURSP2020/197), Taif University, Taif, Saudi Arabia. This research is also funded by Allcosmos Industries Sdn. Bhd. Arif Efektif Sdn. Bhd., Malaysia with grant Ns. RJ130000.7609.4C187 and RJ130000.7344.4B200.
Footnotes
Peer review under responsibility of King Saud University.
References
- Abee T., Groot M.N., Tempelaars M., Zwietering M., Moezelaar R., van der Voort M. Germination and outgrowth of spores of Bacillus cereus group members: diversity and role of germinant receptors. Food Microbiol. 2011;28(2):199–208. doi: 10.1016/j.fm.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Ash C., Farrow J.A., Dorsch M., Stackebrandt E., Collins M.D. Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S rRNA. Int. J. Syst. Bacteriol. 1991;41(3):343–346. doi: 10.1099/00207713-41-3-343. [DOI] [PubMed] [Google Scholar]
- Avashia S.B., Riggins W.S., Lindley C., Hoffmaster A., Drumgoole R., Nekomoto T. Fatal pneumonia among metalworkers due to inhalation exposure to Bacillus cereus Containing Bacillus anthracis toxin genes. Clinical Infect. Dis. : Off. Publ. Infect. Dis. Soc. Am. 2007;44(3):414–416. doi: 10.1086/510429. [DOI] [PubMed] [Google Scholar]
- Barrie L.A., Gregor D., Hargrave B., Lake R., Muir D., Shearer R. Arctic contaminants: sources, occurrence and pathways. Sci. Total Enviro. 1992;122(1–2):1–74. doi: 10.1016/0048-9697(92)90245-n. [DOI] [PubMed] [Google Scholar]
- Bottone E.J. Bacillus cereus, a volatile human pathogen. Clin. Microbiol. Rev. 2010;23(2):382–398. doi: 10.1128/CMR.00073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callegan M.C., Engelbert M., Parke D.W., 2nd, Jett B.D., Gilmore M.S. Bacterial endophthalmitis: epidemiology, therapeutics, and bacterium-host interactions. Clin. Microbiol. Rev. 2002;15(1):111–124. doi: 10.1128/CMR.15.1.111-124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig T.J., Abeloff M.D. Psychiatric symptomatology among hospitalized cancer patients. Am. J. Psychiatry. 1974;131(12):1323–1327. [PubMed] [Google Scholar]
- Drobniewski F.A. Bacillus cereus and related species. Clin. Microbiol. Rev. 1993;6(4):324–338. doi: 10.1128/cmr.6.4.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai D.V., Kulkarni-Kale U. T-cell epitope prediction methods: an overview. Methods Mol. Biol. 2014;1184:333–364. doi: 10.1007/978-1-4939-1115-8_19. [DOI] [PubMed] [Google Scholar]
- Dosch S.F., Mahajan S.D., Collins A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009;142(1–2):19–27. doi: 10.1016/j.virusres.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M., Skanseng B., Rudi K., Stessl B., Ehling-Schulz M. Shift from farm to dairy tank milk microbiota revealed by a polyphasic approach is independent from geographical origin. Int. J. Food Microbiol. 2011;145(Suppl 1):S24–S30. doi: 10.1016/j.ijfoodmicro.2010.08.025. [DOI] [PubMed] [Google Scholar]
- Foroutan M., Ghaffarifar F., Sharifi Z., Dalimi A. Vaccination with a novel multi-epitope ROP8 DNA vaccine against acute Toxoplasma gondii infection induces strong B and T cell responses in mice. Comp Immunol. Microbiol. Infect. Dis. 2020;69:101413. doi: 10.1016/j.cimid.2020.101413. [DOI] [PubMed] [Google Scholar]
- Hilliard R.E. The effects of music therapy on the quality and length of life of people diagnosed with terminal cancer. J. Music Ther. 2003;40(2):113–137. doi: 10.1093/jmt/40.2.113. [DOI] [PubMed] [Google Scholar]
- Hernaiz C., Picardo A., Alos J.I., Gomez-Garces J.L. Nosocomial bacteremia and catheter infection by Bacillus cereus in an immunocompetent patient. Clin. Microbiol. Infect. : Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2003;9(9):973–975. doi: 10.1046/j.1469-0691.2003.00682.x. [DOI] [PubMed] [Google Scholar]
- Helgason E., Okstad O.A., Caugant D.A., Johansen H.A., Fouet A., Mock M. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis–one species on the basis of genetic evidence. Appl. Environ. Microbiol. 2000;66(6):2627–2630. doi: 10.1128/aem.66.6.2627-2630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Chakravarty S.D., Ivashkiv L.B. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol. Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotiranta A., Lounatmaa K., Haapasalo M. Epidemiology and pathogenesis of Bacillus cereus infections. Microbes Infect. 2000;2(2):189–198. doi: 10.1016/s1286-4579(00)00269-0. [DOI] [PubMed] [Google Scholar]
- Kar T., Narsaria U., Basak S., Deb D., Castiglione F., Mueller D.M. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020;10(1):10895. doi: 10.1038/s41598-020-67749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M., Masuda H., Horii J., Kuma K., Yokoyama N., Ohba T. When overexpressed, a novel centrosomal protein, RanBPM, causes ectopic microtubule nucleation similar to gamma-tubulin. J. Cell Biol. 1998;143(4):1041–1052. doi: 10.1083/jcb.143.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest F.G., Alexander B. A frequency matrix for probabilistic identification of some bacilli. J. Gen. Microbiol. 1988;134(11):3011–3018. doi: 10.1099/00221287-134-11-3011. [DOI] [PubMed] [Google Scholar]
- Raza S., Siddique K., Rabbani M., Yaqub T., Anjum A.A., Ibrahim M. In silico analysis of four structural proteins of aphthovirus serotypes revealed significant B and T cell epitopes. Microb. Pathog. 2019;128:254–262. doi: 10.1016/j.micpath.2019.01.007. [DOI] [PubMed] [Google Scholar]
- Setlow P. Spores of Bacillus subtilis: their resistance to and killing by radiation, heat and chemicals. J. Appl. Microbiol. 2006;101(3):514–525. doi: 10.1111/j.1365-2672.2005.02736.x. [DOI] [PubMed] [Google Scholar]
- Stenfors Arnesen L.P., Fagerlund A., Granum P.E. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 2008;32(4):579–606. doi: 10.1111/j.1574-6976.2008.00112.x. [DOI] [PubMed] [Google Scholar]
- Sohail Raza MTN, Wajeeha Zahir, Muhammad Nabeel Khan, Muhammad Awais, Tahir Yaqub, Masood Rabbani, Muhammad Rashid, Salina Saddick, Muhammad Asif Rasheed. 2021. Analysis of the Spike Proteins Suggest Pangolin as an Intermediate Host of COVID-19 (SARS-CoV-2). Int. J. Agric. Biol. 25(3),639–44.
- Turnbull P.C., Jorgensen K., Kramer J.M., Gilbert R.J., Parry J.M. Severe clinical conditions associated with Bacillus cereus and the apparent involvement of exotoxins. J. Clin. Pathol. 1979;32(3):289–293. doi: 10.1136/jcp.32.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further Reading
- Muta T., Takeshige K. Essential roles of CD14 and lipopolysaccharide-binding protein for activation of toll-like receptor (TLR)2 as well as TLR4 Reconstitution of TLR2- and TLR4-activation by distinguishable ligands in LPS preparations. Eur. J. Biochem. 2001;268(16):4580–4589. doi: 10.1046/j.1432-1327.2001.02385.x. [DOI] [PubMed] [Google Scholar]