

Abstract
目的
对比分析宏基因组测序(mNGS)与常规细菌培养在检测颌面部间隙感染致病微生物方面的差异与一致性,为临床早期明确颌面部间隙感染致病菌提供新的检测方法。
方法
收集2020年3—6月就诊于郑州大学第一附属医院的16例口腔颌面部间隙感染患者的临床资料,分别使用mNGS及常规细菌培养方法对脓液进行检测,对两种方法的检验结果进行分析比较,包括检验周期、阳性检出率、厌氧菌、兼性厌氧菌及需氧菌检出率、病原菌分布、相对物种丰度、耐药基因。
结果
mNGS平均检验周期为(18.81±3.73)h,细菌培养的平均检验周期为(83.25±11.64)h,前者短于后者(P<0.05)。mNGS的阳性检出率100%(16/16),常规细菌培养的阳性检出率31.25%(5/16),前者高于后者(P<0.05)。mNGS厌氧菌检出率为93.75%(15/16),细菌培养厌氧菌检出率为0(0/16),前者高于后者(P<0.05);mNGS的兼性厌氧菌检出率为75.00%(12/16),细菌培养的兼性厌氧菌检出率为25.00%(4/16),前者高于后者(P<0.05);mNGS的需氧菌检出率为25.00%(4/16),细菌培养的需氧菌检出率为12.50%(1/16),前者高于后者(P>0.05)。在mNGS检测出的15种致病菌中,3种为革兰阳性菌,12种为革兰阴性菌;49种非致病菌中,16种为革兰阳性菌,32种为革兰阴性菌,1种为真菌。
结论
与常规细菌培养相比,mNGS具有检验时间短、灵敏度高、准确率高的特点,为临床上早期明确颌面间隙感染致病菌提供新的检测方法,并有利于该疾病的早期临床诊断与治疗。
Keywords: 宏基因组测序, 颌面间隙感染, 病原微生物, 下一代测序技术
Abstract
Objective
This study aimed to compare and analyze the consistency and difference between metagenomic next-generation sequencing (mNGS) and conventional bacterial culture in the detection of pathogenic microorganisms in maxillofacial space infection, as well as to provide a new detection method for the early clinical identification of pathogenic bacteria in maxillofacial space infection.
Methods
The clinical data of 16 patients with oral and maxillofacial space infections in the First Affiliated Hospital of Zhengzhou University from March 2020 to June 2020 were collected. mNGS and conventional bacterial culture methods were used to detect pus. We then analyzed and compared the test results of the two methods, including the test cycle, positive detection rate, anaerobic bacteria, facultative anaerobes and aerobic bacteria detection rates, distribution of pathogenic bacteria, relative species abundance, and resistance genes.
Results
The average inspection period of mNGS was (18.81±3.73) h, and the average inspection period of bacterial culture was (83.25±11.64) h, the former was shorter than the latter (P<0.05). The positive detection rate of mNGS was 100% (16/16), and the positive detection rate of conventional bacterial culture was 31.25% (5/16), the former was higher than the latter (P<0.05). The detection rate of mNGS anaerobic bacteria was 93.75% (15/16), the detection rate of bacterial culture anaerobes was 0 (0/16), the former was higher than the latter (P<0.05). Using mNGS, the detection rate of facultative anaerobes in bacterial culture was 75.00% (12/16), and the detection rate of facultative anaerobes in bacterial culture was 25.00% (4/16), the former was higher than the latter (P<0.05). The detection rate of aerobic bacteria in bacterial culture was 12.50% (1/16), the former was higher than the latter (P>0.05). mNGS detected 15 kinds of pathogenic bacteria, among which 3 were Gram positive, 12 were Gram negative, 49 were non-pathogenic, 16 were Gram positive, and 32 were Gram negative, 1 was fungus.
Conclusion
Compared with conventional bacterial culture, mNGS has the characteristics of short test time, high sensitivity, and high accuracy. Thus, it is a new detection method for the early identification of pathogenic bacteria in maxillofacial space infection and is beneficial to the early clinical diagnosis and treatment of the disease.
Keywords: metagenomic next-generation sequencing, maxillofacial space infection, pathogenic microorganism, next-generation sequencing technology
口腔颌面部感染是口腔颌面外科最常见急症之一,多继发于牙源性及腺源性感染,由于口腔颌面部解剖结构存在上行及下行的潜在通道,感染易向颅内、纵隔扩散,导致脑脓肿、纵隔脓肿、脓毒症等严重并发症的发生,病情凶险威胁患者生命[1],以往资料[2]表明,其死亡率高达61.5%。颌面间隙感染致病菌的早期确定及针对性用药尤为重要,然而传统病原微生物检测手段虽具有快速、简单、成本及技术要求低等特点,但无法对标本所含病原微生物做出整体分析。尤其是病毒、未知或难培养的病原微生物,目前的检测方法为涂片、培养、聚合酶链式反应(polymerase chain reaction,PCR)等,均具有较大局限性[3]–[4]。近年来,宏基因组测序(metagenomic next-generation sequencing,mNGS)技术越来越受到重视,可以在不使用任何引物或探针的情况下,对待测样品所有DNA片段进行扩增和测序[5]。理论上,mNGS可以在一次实验中识别所有潜在的病原体,对标本中可能存在的病原体进行全面检测,从而有助于颌面部间隙感染的早期诊断。
1. 材料和方法
1.1. 一般资料
收集2020年3—6月郑州大学第一附属医院口腔颌面外科收治的符合入选排除标准的16例颌面部间隙感染患者的临床资料,其中男性12例,平均年龄为(54.08±17.21)岁;女性4例,平均年龄为(71.15±12.50)岁。感染部位分别为单间隙9例,多间隙7例。感染来源分别为牙源性12例,非牙源性4例。纳入标准:1)年龄>18岁;2)确诊为颌面部间隙感染患者;3)应用抗生素前已留取病变局部抽吸脓液;4)患者或其家属已签署知情同意书。排除标准:1)妊娠期妇女;2)合并颌面部外其他部位感染;3)送检样本的微生物核酸含量<100 copies·mL−1。
1.2. 样本采集
应用抗生素前收集患者颌面部局部肿胀抽吸液2份,同时送医院微生物培养以及临床精准用药中心进行病原体mNGS。样本量:医院微生物培养需要抽吸液样本至少1 mL,及时送检;mNGS样本需抽吸液至少2 mL,4~20 °C暂存,2 h内送检,如不能及时送检,−20 °C保存,24 h内送检。
1.3. 检测方法
1.3.1. 常规细菌培养
采用梅里埃VITEK 2 Compact 全自动细菌鉴定及药敏分析系统对脓液进行细菌鉴定及药敏试验分析,VITEK试验卡中的GN卡用于革兰阴性杆菌鉴定,GP卡用于革兰阳性球菌鉴定,采用K-B法药敏试验进行补充分析。
1.3.2. mNGS
1)核酸提取和文库构建:提取2~3 mL脓液标本,将标本与玻璃珠混合震荡,按照MinION试剂盒的标准建库流程提取DNA和构建测序文库;2)测序与数据预处理:测序过程基于牛津纳米孔技术(Oxford nanopore technologies,ONT)平台和ONT MinKNOW软件v3.3.2完成,通过ONT Guppy软件v3.0.3将原始序列数据(fast5文件)进行碱基读取得到存储测序数据(fastq测序文件),然后利用比对软件blastn-short v2.7.1+进行样本拆分;3)数据清洗:去除读长≤500碱基的序列、质量控制值<8的低质量序列;4)去宿主和菌种鉴定:使用比对软件Minimap2 v2.14-r883将序列与人类参考基因组(GRCh38)进行比对来去除宿主序列。使用比对软件Centrifuge v1.0.4将其余序列比对到微生物序列,并通过比对软件Megablast v2.7.1进行验证。剔除比对到多种细菌的序列,然后计算对应微生物的物种丰度(比对到该物种的序列/比对到所有微生物的序列数);5)耐药分析:采用耐药基因数据库(comprehensive antibiotic research database,CARD)v2.0.2中耐药基因序列作为参考序列,使用ONT测序质控后的长序列进行比对,将比对上的序列进行局部组装后,获取对应的基因型,从而进行耐药基因的精确鉴定,进而得到相关耐药药物种类、具体耐药药物、基因家族和耐药机制等注释信息。
1.4. mNGS微生物阳性判定方法
细菌的检出序列数(Reads)>200,同时细菌检出丰度(检出序列数占所有检出微生物序列数的比例)>0.5[6]。
1.5. 观察指标
观察2种方法的检验周期、阳性检出率、厌氧菌、兼性厌氧菌以及需氧菌检出率、相对物种丰度、耐药基因型等。
1.6. 统计分析
采用SPSS 24.0软件对所得数据进行统计分析,计量资料以均数±标准差表示,组间比较采用t检验;计数资料以例数或百分比表示,比较采用χ2检验,以P<0.05为差异有统计学意义。
2. 结果
2.1. 部分因素与革兰阳性菌和阴性菌比例的关系
12例男性中,革兰阳性菌与阴性菌检出比例为39.1%(9/23),4例女性中,革兰阳性菌与阴性菌检出比例为50%(5/10),其差异无统计学意义(P>0.05);9例单间隙中,革兰阳性菌与阴性菌检出比例为40.91%(9/22),7例多间隙中,革兰阳性菌与阴性菌检出比例为45.45%(5/11),差异无统计学意义(P>0.05);12例牙源性中,革兰阳性菌与阴性菌检出比例为38.46%(10/26),4例非牙源性中,革兰阳性菌与阴性菌检出比例为57.14%(4/7),差异无统计学意义(P>0.05)。
2.2. 检验周期、阳性检出率的比较
常规细菌培养的检验平均时间为(83.25±11.64)h,mNGS的平均检验时间为(18.81±3.73)h,mNGS较细菌培养的检验周期明显缩短(P<0.05);常规细菌培养的阳性检出率为31.25%(5/16),mNGS的阳性检出率为100%(16/16),差异有统计学意义(P<0.05)。
2.3. 厌氧菌、兼性厌氧菌及需氧菌检出率的比较
常规细菌培养的厌氧菌检出率为0(0/16),mNGS厌氧菌的检出率为93.75%(15/16),差异有统计学意义(P<0.05);常规细菌培养的兼性厌氧菌检出率为25.00%(4/16),mNGS的兼性厌氧菌检出率为75.00%(12/16),差异有统计学意义(P<0.05);常规细菌培养的需氧菌检出率为12.50%(1/16),mNGS需氧菌的检出率为25.00%(4/16),差异无统计学意义(P>0.05)。
2.4. mNGS致病菌分布状况
16例样本检测出15种致病菌,其中3种为革兰阳性菌,从高到低依次为咽峡炎链球菌占68.75%(11/16)、星座链球菌占50.00%(8/16)、化脓链球菌占12.50%(2/16);12种为革兰阴性菌,从高到低依次为中间普雷沃菌占81.25%(13/16)、牙龈卟啉单胞菌占68.75%(11/16)、产黑色素类杆菌占43.75%(7/16)、核粒梭菌占43.75%(7/16)、齿垢密螺旋体占25.00%(4/16)、坏死梭杆菌占25.00%(4/16)、中间链球菌占18.75%(3/16)、福赛斯坦纳菌占18.75%(3/16)、肺炎克雷伯杆菌12.50%(2/16)、具核梭杆菌占12.50%(2/16)、大肠杆菌占6.25%(1/16)、流感嗜血杆菌占6.25%(1/16)(图1)。在本次研究中表达较高的关键致病菌(占比在50%以上)依次为:中间普雷沃菌、咽峡炎链球菌、牙龈卟啉单胞菌、星座链球菌;其中,中间普雷沃菌的物种丰度在性别方面的差异有统计学意义(P<0.05),咽峡炎链球菌、牙龈卟啉单胞菌、星座链球菌的物种丰度在性别方面的差异无统计学意义(P>0.05);中间普雷沃菌、咽峡炎链球菌、牙龈卟啉单胞菌、星座链球菌的物种丰度在感染来源(牙源性和非牙源性)方面的差异无统计学意义(P>0.05);中间普雷沃菌、咽峡炎链球菌、牙龈卟啉单胞菌、星座链球菌的物种丰度在多间隙与单间隙中的差异无统计学意义(P>0.05)。
图 1. mNGS致病菌分布状况.

Fig 1 Distribution of pathogenic bacteria in mNGS
2.5. mNGS非致病菌分布状况
在16例样本中,13例检测出49种非致病菌(图2),其中16种革兰阳性菌,32种革兰阴性菌,1种真菌。
图 2. mNGS非致病菌分布状况.
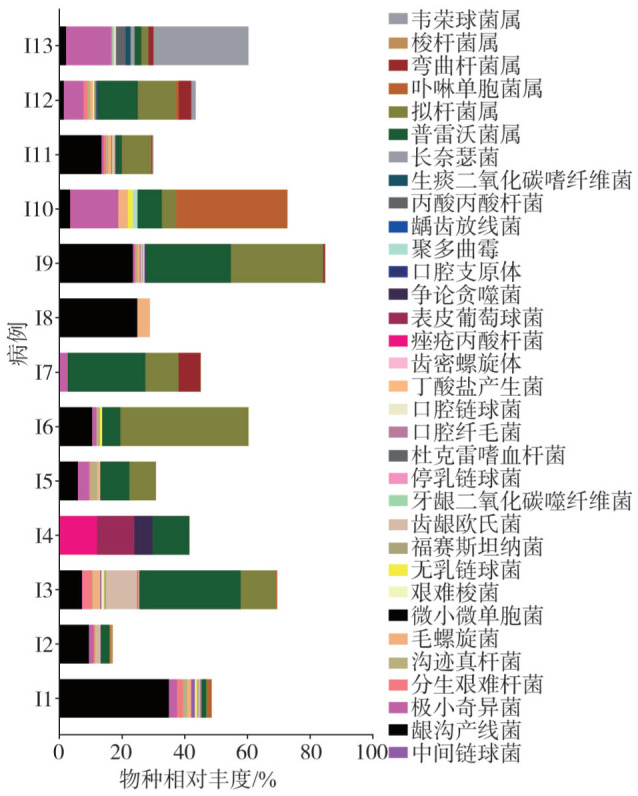
Fig 2 Distribution of non-pathogenic bacteria in mNGS
16种革兰阳性菌从高到低依次为:微小微单胞菌、极小奇异菌、龈沟产线菌、毛螺旋菌、沟迹真杆菌、分生艰难杆菌、中间链球菌、艰难梭菌、无乳链球菌、齿龈欧氏菌、停乳链球菌、口腔链球菌、丁酸盐产生菌、痤疮丙酸杆菌、表皮葡萄球菌、丙酸丙酸杆菌。
32种革兰阴性菌从高到低依次为:普雷沃菌属(栖居普雷沃菌、栖牙普雷沃菌、中间普雷沃菌、深黑色普雷沃菌、靶心普雷沃菌、产黑色素普雷沃菌)、拟杆菌属(解肝素拟杆菌、动胶拟杆菌、脆弱拟杆菌、多氏拟杆菌、普通拟杆菌、副拟杆菌、卵形拟杆菌)、卟啉单胞菌属(牙龈卟啉单胞菌、不解糖卟啉单胞菌)、弯曲杆菌属(简明弯曲菌、纤细弯曲菌)、福赛斯坦纳菌、牙龈二氧化碳噬纤维菌、杜克雷嗜血杆菌、梭杆菌属(牙周梭杆菌、具核梭杆菌、坏死梭杆菌)、韦荣球菌属(小韦荣球菌、啮齿韦荣氏球菌)、齿密螺旋体、争论贪噬菌、口腔纤毛菌、口腔支原体、龋齿放线菌、生痰二氧化碳噬纤维菌、长奈瑟菌。
1种真菌为聚多曲霉。
2.6. 耐药基因型的分布状况
本研究发现了8种抗菌药物耐药类型,在耐药基因型中,大环内酯类耐药5种,林可酰胺类耐药4种,四环素类耐药9种,头霉素类耐药5种,链霉素类耐药1种,氟喹诺酮类耐药7种,头孢菌素类耐药4种,碳青霉烯类耐药2种(表1)。
表 1. 耐药基因型的分布状况.
Tab 1 Distribution of resistant genotypes
| 耐药药物种类 | 耐药基因 |
| 大环内酯类 | ErmF、ErmB、lsaC、Erm(35)、H-NS |
| 林可酰胺类 | ErmF、ErmB、lsaC、Erm(35) |
| 四环素类 | tetM、tetS、lsaC、tetQ、emrY、AcrS、acrB、LAP-2、H-NS |
| 头霉素类 | CfxA、CfxA2、CfxA3、CfxA5、H-NS |
| 链霉素类 | marA |
| 氟喹诺酮类 | H-NS、mdtH、emrA、emrR、acrB、LAP-2、QnrS1 |
| 头孢菌素类 | H-NS、marA、AcrS、acrB |
| 碳青霉烯类 | lsaC、marA |
3. 讨论
在颌面部间隙感染中细菌培养是临床中最常用的检测致病菌的方法,但细菌培养时间普遍较长,且许多细菌不易培养,多重感染易被忽视,PCR检测具有极高的灵敏度和特异性,但无法完成宏基因组筛查,检出率较低,对于重症的颌面多间隙感染患者来说,因传统检测方法无法及时确定病原体信息,不能提早针对性用药,从而易使病情恶化[7]。因此,快速、特异的病原体检测方法,对有颌面间隙感染的早期诊断和有针对性的用药控制感染的扩散具有重要的意义。
mNGS可以分析病原体群体基因组成及功能,解读病原体群体的多样性与丰度,探求病原体与环境、宿主之间的关系,挖掘具有应用价值的基因资源,有利于开发新的病原体活性物质[7]。而且该技术不依赖于传统的微生物培养,直接对临床样本中的核酸进行mNGS,然后与数据库进行比对分析,根据比对到的序列信息来判断样本包含的病原微生物种类,能够快速、客观地检测临床样本中较多的病原微生物(包括病毒、细菌、真菌、寄生虫),且无需特异性扩增[8]–[12],该方法不仅可以避开传统方法(培养法、血清法和PCR法)的局限性,快速地检测出已知病原体,还可以发现许多未知病原体,直接分析出样本中的病原体谱、丰度和分布情况,了解潜在病原体的变化情况,有助于诊断一些不明病原体引发的感染性疾病[13]–[16]。
本研究统计了细菌培养及mNGS两种方法的检验周期,结果发现,mNGS的检测时间较细菌培养显著缩短。Fan等[17]使用mNGS在48 h内快速诊断中枢神经系统布氏杆菌病,而培养法则需耗时7 d。Grumaz等[18]将mNGS用于重症血流感染患者的病原体检测,平均报告时间从常规血培养的5 d缩短至30 h以内,上述报道均与本研究结果相符。而在重症的颌面多间隙感染患者中,快速获取病原体相关信息的意义更为突出,缩短检测时间可为患者争取宝贵的治疗时间,有效提高抢救的成功率。mNGS对于病原体检出的阳性率远高于细菌培养,Parize等[19]在免疫抑制的患者中,通过mNGS发现了较传统方法更高比例的细菌及病毒感染;Street等[20]对关节假体感染进行mNGS检测,观察到该技术对病原体种水平的检出敏感性为93%,上述研究均提示,mNGS较细菌培养的敏感性更高,与本研究结果一致。由于前期经验性广谱抗菌药物的使用、病原体生长缓慢或病原体对生存条件要求苛刻等因素,往往会导致培养阳性率低,最终带来治疗延误、住院时间延长、病死率增加等问题[21]。mNGS的高敏感性使它能够检测到临床罕见、培养困难、甚至既往未知的病原体,对明确感染性疾病的诊断意义重大。
本次研究利用mNGS,在检测出的致病菌中,革兰阴性菌依次为中间普雷沃菌、牙龈卟啉单胞菌、产黑色素类杆菌、肺炎克雷伯菌,革兰阳性菌依次为咽峡炎链球菌、星座链球菌、化脓链球菌;在非致病菌中,革兰阴性菌依次为普雷沃菌属、拟杆菌属、卟啉单胞菌属,革兰阳性菌依次为微小微单胞菌、极小奇异菌、龈沟产线菌。常规细菌培养检测出的病原菌,革兰阳性菌依次为星座链球菌、咽峡炎链球菌,革兰阴性菌为肺炎克雷伯菌。在两种方法检出的致病菌中,中间普雷沃菌、牙龈卟啉单胞菌、产黑色素类杆菌为革兰阴性厌氧菌,咽峡炎链球菌、星座链球菌、化脓链球菌为革兰阳性兼性厌氧菌,肺炎克雷伯菌为革兰阴性需氧菌,本研究显示,mNGS对于厌氧菌及兼性厌氧菌检出阳性率远高于细菌培养。李冰等[22]利用mNGS,对厌氧菌检测的阳性率为82.76%,证明了mNGS在厌氧菌感染的精准化诊断方面相比细菌培养具有优势。周围等[23]对2例重症医学科感染性休克患者,采用mNGS进行感染性微生物检测,且最终根据mNGS检测结果确诊为厌氧菌感染。上述研究均显示,mNGS相比细菌培养,在厌氧菌的诊断方面上更具有优势,与本研究结果一致。本次纳入研究的16位颌面间隙感染患者,在入院后均抽取其感染部位脓液,同时进行mNGS及细菌培养,对符合切开引流指征的患者行局部切开引流,因细菌培养阳性检出率低,无法准确指导临床用药,故本研究根据mNGS检出致病菌及耐药基因情况并参照相关用药指南[24]–[26]对每个患者制定了个性化用药方案,本研究的样本检测分析及指南的临床用药建议可选择及推荐的抗生素药物主要有:氨苄西林舒巴坦、青霉素G结合甲硝唑、头孢菌素头孢曲松结合甲硝唑等。经过治疗后,16位患者的临床症状及炎症指标均明显改善,因此mNGS有助于临床的精准用药,并且在耐药基因检出方面,mNGS可解决很多现有的技术无法处理的问题[27]。
本研究的不足之处在于样本量有限,对分析部分因素与革兰阳性菌、阴性菌比例的关系,以及关键致病菌的丰度对性别、感染来源及扩散部位的相关性分析缺乏可靠性,今后需扩大样本量进一步统计分析,增加结论的可靠性。
尽管mNGS敏感度较高,但仍有很多不足之处,mNGS检测结果是一个病原体列表,可能同时呈现多项阳性结果,临床医师难以仅仅依靠测序结果确定真正的病原菌,因此,检测结果需要跨学科的全方位解读[28]–[29],而且不同的检测机构,在病原菌鉴定以及相对丰度计算方面会出现差别[30],目前,mNGS生物信息分析流程缺乏统一标准,研究人员基于个人经验、可及性以及简便性选用分析软件,会对病原检测结果的可重复性及准确性造成影响,成为mNGS临床应用标准化的障碍。
综上所述,与常规细菌培养相比mNGS具有检验时间短、灵敏度高、准确率高的特点,为颌面部间隙感染病原微生物的确定提供新方法,并有利于该疾病的早期临床诊断与治疗。
Funding Statement
[基金项目] 河南省自然科学基金面上项目(212300410391);河南省医学科技攻关计划联合共建项目(LHGJ20200270);河南省医学科技攻关项目(SBGJ202002071);河南省中青年卫生健康科技创新人才项目(YXKC2020030)
Supported by: General Project of Henan Provincial Natural Science Foundation (212300410391); Joint Co-construction Project of Henan Medical Science and Technology Research Plan (LHGJ20200270); Henan Province Medical Science and Technology Research Project (SBGJ202002071); Henan Province Young and Middle-aged Health Science and Technology Innovation Talent Project (YXKC2020030).
Footnotes
利益冲突声明:作者声明本文无利益冲突。
References
- 1.张 春旭, 梁 新华. 颌面部间隙感染的研究进展[J] 国际口腔医学杂志. 2009;36(1):55–57. [Google Scholar]; Zhang CX, Liang XH. Research progress of maxillofacial space infection[J] Int J Stomatol. 2009;36(1):55–57. [Google Scholar]
- 2.Chen KC, Chen JS, Kuo SW, et al. Descending necrotizing mediastinitis: a 10-year surgical experience in a single institution[J] J Thorac Cardiovasc Surg. 2008;136(1):191–198. doi: 10.1016/j.jtcvs.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 3.Crama N. Real-time polymerase chain reaction in the diagnosis of acute postoperative endophthalmitis[J] Am J Ophthalmol. 2012;154(6):1002–1003. doi: 10.1016/j.ajo.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Goldschmidt P, Degorge S, Benallaoua D, et al. New test for the diagnosis of bacterial endophthalmitis[J] Br J Ophthalmol. 2009;93(8):1089–1095. doi: 10.1136/bjo.2008.152181. [DOI] [PubMed] [Google Scholar]
- 5.Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection[J] Annu Rev Pathol. 2019;14:319–338. doi: 10.1146/annurev-pathmechdis-012418-012751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horiba K, Kawada JI, Okuno Y, et al. Comprehensive detection of pathogens in immunocompromised children with bloodstream infections by next-generation sequencing[J] Sci Rep. 2018;8(1):3784. doi: 10.1038/s41598-018-22133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.李 林海, 陈 丽丹, 肖 斌, et al. 宏基因组测序在感染性疾病病原体检测中的应用[J] 传染病信息. 2018;31(1):15–18. [Google Scholar]; Li LH, Chen LD, Xiao B, et al. Application of metagenomic sequencing in detecting the pathogens of infectious diseases[J] Infect Dis Info. 2018;31(1):15–18. [Google Scholar]
- 8.张 晖, 弓 孟春, 徐 军, et al. 中国精准急诊医学的应用体系规划探索[J] 中华急诊医学杂志. 2016;25(10):1219–1223. [Google Scholar]; Zhang H, Gong MC, Xu J, et al. Application system of precision emergency medicine in China[J] Chin J Emerg Med. 2016;25(10):1219–1223. [Google Scholar]
- 9.Goldberg B, Sichtig H, Geyer C, et al. Making the leap from research laboratory to clinic: challenges and opportunities for next-generation sequencing in infectious disease diagnostics[J] mBio. 2015;6(6):e01888–15. doi: 10.1128/mBio.01888-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westblade LF, van Belkum A, Grundhoff A, et al. Role of clinicogenomics in infectious disease diagnostics and public health microbiology[J] J Clin Microbiol. 2016;54(7):1686–1693. doi: 10.1128/JCM.02664-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lusk RW. Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data[J] PLoS One. 2014;9(10):e110808. doi: 10.1371/journal.pone.0110808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.周 永召, 李 亚伦, 范 红, et al. 临床宏基因组学在呼吸感染性疾病精准诊疗中的疑问解析[J] 中国呼吸与危重监护杂志. 2018;17(6):539–543. [Google Scholar]; Zhou YZ, Li YL, Fan H, et al. Interrogative analysis of clinical metagenomics in the application of precision diagnosis and treatment of respiratory infectious disease[J] Chin J Respirat Crit Care Med. 2018;17(6):539–543. [Google Scholar]
- 13.Lindahl BD, Nilsson RH, Tedersoo L, et al. Fungal community analysis by high-throughput sequencing of amplified markers-a user's guide[J] New Phytol. 2013;199(1):288–299. doi: 10.1111/nph.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Lei L, Yang B, et al. Direct electron transfer from electrode to electrochemically active bacteria in a bioelectrochemical dechlorination system[J] Bioresour Technol. 2013;148:9–14. doi: 10.1016/j.biortech.2013.08.108. [DOI] [PubMed] [Google Scholar]
- 15.Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd[J] Science. 1995;269(5223):496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 16.Simner PJ, Miller S, Carroll KC. Understanding the promises and hurdles of metagenomic next-generation sequencing as a diagnostic tool for infectious diseases[J] Clin Infect Dis. 2018;66(5):778–788. doi: 10.1093/cid/cix881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan S, Ren H, Wei Y, et al. Next-generation sequencing of the cerebrospinal fluid in the diagnosis of neurobrucellosis[J] Int J Infect Dis. 2018;67:20–24. doi: 10.1016/j.ijid.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 18.Grumaz S, Stevens P, Grumaz C, et al. Next-generation sequencing diagnostics of bacteremia in septic patients[J] Genome Med. 2016;8(1):73. doi: 10.1186/s13073-016-0326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parize P, Muth E, Richaud C, et al. Untargeted next-generation sequencing-based first-line diagnosis of infection in immunocompromised adults: a multicentre, blinded, prospective study[J] Clin Microbiol Infect. 2017;23(8):574.e1–574.e6. doi: 10.1016/j.cmi.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Street TL, Sanderson ND, Atkins BL, et al. Molecular diagnosis of orthopedic-device-related infection directly from sonication fluid by metagenomic sequencing[J] J Clin Microbiol. 2017;55(8):2334–2347. doi: 10.1128/JCM.00462-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bleeker-Rovers CP, Vos FJ, de Kleijn EMHA, et al. A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol[J] Medicine (Baltimore) 2007;86(1):26–38. doi: 10.1097/MD.0b013e31802fe858. [DOI] [PubMed] [Google Scholar]
- 22.李 冰, 缪 青, 金 文婷, et al. 宏基因二代测序技术对厌氧菌感染精准化诊断的临床价值[J] 中华医院感染学杂志. 2019;29(13):1927–1930, 1953. [Google Scholar]; Li B, Miao Q, Jin WT, et al. Clinical value of metagenomic next-generation sequencing in precise diagnosis of anaerobic bacteria infection[J] Chin J Nosocomiol. 2019;29(13):1927–1930, 1953. [Google Scholar]
- 23.周 围, 史 伟峰, 吴 玉敏, et al. 高通量测序技术在厌氧菌感染诊断中的作用[J] 检验医学. 2020;35(4):390–392. [Google Scholar]; Zhou W, Shi WF, Wu YM, et al. The role of high-throughput sequencing in the diagnosis of anaerobic infection[J] Laborat Med. 2020;35(4):390–392. [Google Scholar]
- 24.桑 福德. In: 热病: 桑德福抗微生物治疗指南[M] 范 洪伟, translator. 北京: 中国协和医科大学出版社; 2018. [Google Scholar]; Sanford JP. In: Guidelines for antimicrobial treatment of fever Sandford[M] Fan HW, translator. Beijing: China Peking Union Medical University Publishing House; 2018. [Google Scholar]
- 25.Kas per DL, Fauci AS. In: 哈里森感染病学[M] 胡 毕节, 潘 珏, 高 晓东, translators. 上海: 上海科学技术出版社; 2019. [Google Scholar]; Kasper DL, Fauci AS. In: Harrison infectious diseases[M] Hu BJ, Pan J, Gao XD, translators. Shanghai: Shanghai Science and Technology Publishing house; 2019. [Google Scholar]
- 26.王 明贵. 广泛耐药革兰阴性菌感染的实验诊断、抗菌治疗及医院感染控制: 中国专家共识[J] 中国感染与化疗杂志. 2017;17(1):82–92. [Google Scholar]; Wang MG. Laboratory diagnosis, clinical management and infection control of the infections caused by extensively drug-resistant Gram-negative bacilli: a Chinese consensus statement[J] Chin J Infect Chemother. 2017;17(1):82–92. doi: 10.1016/j.cmi.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 27.McDermott PF, Tyson GH, Kabera C, et al. Whole-genome sequencing for detecting antimicrobial resistance in Nontyphoidal Salmonella[J] Antimicrob Agents Chemother. 2016;60(9):5515–5520. doi: 10.1128/AAC.01030-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Couto N, Schuele L, Raangs EC, et al. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens[J] Sci Rep. 2018;8(1):13767. doi: 10.1038/s41598-018-31873-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasman H, Saputra D, Sicheritz-Ponten T, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples[J] J Clin Microbiol. 2014;52(1):139–146. doi: 10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ai JW, Weng SS, Cheng Q, et al. Human endophthalmitis caused by Pseudorabies virus infection, China, 2017[J] Emerg Infect Dis. 2018;24(6):1087–1090. doi: 10.3201/eid2406.171612. [DOI] [PMC free article] [PubMed] [Google Scholar]