

Abstract
Recent work indicates that PPARα is required for perfluorooctanoic acid (PFOA)-induced postnatal lethality resulting from prenatal exposure. The present study tested the hypothesis that relatively modest activation of PPARα during prenatal development will cause postnatal lethality, similar to that observed with PFOA, a relatively low affinity PPARα agonist. Female wild-type and Pparα-null mice were mated overnight with males of the same genotype. The presence of a copulatory plug on the morning after mating was indicative of pregnancy and considered gestation day (GD) 0. Plugged female mice were fed either a control diet or one containing clofibrate (0.5%) or Wy-14,643 (0.005%) until GD18 or until parturition. Mice were examined on GD18 or on postnatal day (PND) 20 following the prenatal exposure period. Dietary administration of clofibrate or Wy-14,643 did not affect maternal weight or weight gain, the average number of implantations, the percentage of litter loss, the average number of live/dead fetuses, average crown-rump length, or the average fetal weight on GD18 in either genotype. An increase in relative maternal liver weight and elevated expression of PPARα target genes in maternal and fetal livers on GD18 were observed, indicative of PPARa-dependent changes in both the maternal and fetal compartments. However, no defects in postnatal development were observed by either clofibrate or Wy-14,643 in either genotype by PND20. These results demonstrate that relatively low level activation of PPARα by clofibrate or Wy-14,643 during prenatal development does not cause postnatal lethality.
Keywords: Peroxisome proliferator-activated, receptor-α, Postnatal development, Nuclear receptor, Prenatal exposure
1. Introduction
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated, soluble nuclear receptors that include three isoforms: PPARα, PPARβ (also referred to as PPARδ or PPARβ/γ) and PPARγ. PPARα is expressed in most tissues but is noticeably higher in liver, kidney and heart (Auboeuf et al., 1997; Braissant et al., 1996; Braissant and Wahli, 1998) where it is known to regulate expression of proteins required for fatty acid transport, catabolism, and energy homeostasis (Peters et al., 2005). The fibrate class of hypolipidemic drugs, phthalate monoesters and perfluorinated compounds are all known to activate PPARa (Bility et al., 2004; Forman et al., 1997; Maloney and Waxman, 1999; Wolf et al., 2008a). In addition to its known essential role in the regulation of lipid homeostasis, activation of PPARα also causes an increase in hepatocyte proliferation leading to hepatocellular carcinoma in rodents (Hays et al., 2005; Peters et al., 1998, 1997; Reddy et al., 1980); humans appear to be refractory to these effects (Gonzalez and Shah, 2008; Klaunig et al., 2003; Peters, 2008; Peters et al., 2005). More recently, evidence has also surfaced suggesting that PPARα is essential for modulating postnatal lethality observed in rodents exposed to perfluorooctanoic acid (PFOA) during prenatal development (Abbott et al., 2007).
PFOA is one of a number of perfluorinated compounds that are capable of causing activation of PPARa (Wolf et al., 2008a). Perfluorinated compounds are not extensively metabolized in vivo because of the strong covalent bond between carbon and fluorine atoms (Ullrich and Diehl, 1971) and are hence environmentally persistent (Liou et al., 2010). Recent studies show that exposure to PFOA during prenatal development results in dose-dependent full-litter resorptions, as well as delayed development and postnatal lethality in CD-1 mice (Lau et al., 2006) and 129/Sv mice (Abbott et al., 2007). These effects are mediated by PPARα, as they are found in wild-type mice but not in Pparα-null mice (Abbott et al., 2007). Evidence also exists suggesting that these effects are due to gestational exposure to PFOA that may cause alterations in mammary gland function but are not due to lactational exposure of PFOA (Lau et al., 2006; White et al., 2007; Wolf et al., 2007). The present study was designed to test the hypothesis that relatively modest activation of PPARα during prenatal development will cause postnatal lethality, similar to that observed with PFOA, a relatively low affinity PPARα agonist.
2. Materials and methods
2.1. Animal studies
Animal experiments were approved by the Institutional Animal Care and Use Committee at The Pennsylvania State University, which conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Male and female wild-type and Pparα-null mice (Lee et al., 1995) on a 129/Sv genetic background (Akiyama et al., 2001) were used for this study.
2.1.1. Study design
Female wild-type or Pparα-null mice were mated overnight with male mice of the same genotype, and examined for the presence of a copulatory plug after mating. The presence of a copulatory plug was considered indicative of successful mating and designated gestation day (GD) 0. Pregnant female mice were weighed and randomly assigned to one of three groups and fed either a control diet, a diet containing 0.5% clofibrate (Dyets, Inc., Bethlehem, PA) or a diet containing 0.005% Wy-14,643 (Dyets, Inc., Bethlehem, PA). Mice were fed these diets until GD18 or until parturition. After parturition, all groups of mice were fed the control diet. Mice were examined on either GD18 or on postnatal day (PND) 20. The dietary concentrations of PPARα agonists were chosen in an attempt to model the relative ability of PFOA to activate PPARα in the liver and cause approximately a doubling of relative liver weight as shown by previous studies in rodent models (Lee et al., 1995; Marsman et al., 1992; Wolf et al., 2008b), an effect which is known to be associated with increased developmental delays and neonatal lethality (Abbott et al., 2007). The rationale that this approach would achieve low level activation of PPARα is based in part on several relationships. Dietary clofibrate at a dose of 0.5% causes an increase in rat liver weight of ~1.5-fold after 3 weeks of treatment, while 0.005% Wy-14,643 causes an increase in rat liver weight of ~2-fold after 3 weeks of treatment (Marsman et al., 1992). This is consistent with the fact that clofibrate is less effective for increasing PPARα-dependent reporter activity as compared to Wy-14,643 (Shearer and Hoekstra, 2003). It is also known that PFOA is less effective at activating PPARα as compared to Wy-14,643 (Maloney and Waxman, 1999) and that doses of PFOA, capable of causing a modest (~1.5–2-fold) increase in maternal liver weight, cause marked developmental delay and neonatal lethality (Abbott et al., 2007). Clofibrate was chosen as one model PPARα agonist because it is a relatively less effective agonist (e.g. one that would cause low level activation) as compared to Wy-14,643 based on cell based reporter assays, and is more comparable with the PFOA in terms of activating PPARα based on similar cell based reporter assays (Maloney and Waxman, 1999; Shearer and Hoekstra, 2003). The very low dietary level of the PPARα agonist Wy-14,643 was selected in part because it is more effective at activating PPARα, and should thus more closely model PPARα activation observed in response to PFOA. These relationships were collectively used to establish a dosing paradigm that was predicted to cause low level activation of PPARα.
For GD18 analyses, pregnant mice were euthanized by overexposure to carbon dioxide, and livers were carefully dissected and snap frozen until later use. Gravid uterine weights were recorded. For each litter, the number of live fetuses, dead fetuses and resorption sites were counted. The sex of each fetus was determined, crown to rump length was measured, and fetal and fetal liver weights were recorded. Fetal livers were snap frozen after weighing for RNA analysis.
For PND20 analysis, pregnant mice were allowed to deliver their litters and day of parturition was recorded. Pups were weighed on the day of delivery and on PND7, PND14 and PND20. The pups were observed daily to determine postnatal lethality, and the onset of eye opening was examined as a measure of postnatal development. Dams and pups were euthanized by overexposure to carbon dioxide on PND20 and livers were obtained by dissection and snap frozen after weighing for RNA analysis.
2.2. Quantitative real-time PCR (qPCR) analysis
Total RNA was isolated from liver samples using Ribozol (Amresco, Solon, OH). For maternal liver, four independent samples from four mice from each group were used. For fetal liver, samples from one fetus randomly chosen from each of four individual litters were used. For neonatal liver, samples from one pup representing each of four litters were used. The cDNA was generated using 2.5 μg total RNA with Multiscribe Reverse Transcriptase kit (Applied Biosystems, Foster City, CA). The mRNAs encoding the known PPARα target genes, cytochrome P450 4a 10 (Cyp4a10) and acyl-CoA oxidase 1 (Aco), were measured using qPCR analysis. The sequence for the forward and reverse primers used to quantify mRNAs for Cyp4a10, Aco and internal control, glyceraldehyde 3-phosphate dehydrogenase (Gapdh) are described previously (Foreman et al., 2009). PCR reactions were carried out using SYBR® Green Supermix for IQ.(Quanta Biosciences, Gaithersburg, MD) in the iCycler and detected using the MyiQ. Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). The conditions used for PCR were 95 °C for 15 s, 94°C for 10 s, 60°C for 30s, and 72 °C for 30 s, repeated for 45 cycles. The PCR included a no template reaction control for detecting contamination and genomic amplification. All reactions had >85% efficiency. Relative expression levels of mRNA were analyzed for statistical significance using ANOVA and post hoc tests.
2.3. Statistical analysis
Data were analyzed for statistical significance using analysis of variance and the Tukey’s post hoc test (Prism 5.0a, GraphPad Software Inc., San Diego, CA). The criterion used to determine statistical significance was P≤0.05. For fetal and neonatal endpoints, statistical analysis revealed essentially identical results when the individual or litter was used as the statistical unit (data not shown). Figure legends indicate whether the individual or litter was used as the statistical unit.
3. Results
3.1. Effect of prenatal PPARα agonism on maternal and fetal endpoints on GD18
Prenatal exposure to PFOA in pregnant female mice causes an increase in resorptions and postnatal lethality in surviving offspring (Abbott et al., 2007; Lau et al., 2006). The increase in postnatal lethality in mice was associated with doses of PFOA where relative liver weight is twice that of control as observed in non-pregnant mice (Wolf et al., 2008b). Thus, the effect of prenatal exposure to the PPARα agonists clofibrate and Wy-14,643 at doses that are also associated with causing approximately a doubling of liver weight in non-pregnant mice and rats (Lee et al., 1995; Marsman et al., 1992), was determined in wild-type and Pparα-null mice. Average maternal weight and average maternal weight gain during pregnancy were not influenced by exposure to 0.5% clofibrate or 0.005% Wy-14,643 as compared to controls in both genotypes (Table 1). No differences in the average number of implants per dam, the average number of live or dead fetuses per litter, the average number of resorptions per litter, the percentage of litter loss, the average fetal weight or the average crown to rump length were observed in litters examined from mice of both genotypes treated with either clofibrate or Wy-14,643 as compared to control (Table 2). Additionally, no difference in the distribution of male and female fetuses was observed by either treatment in either genotype compared to control (Table 2).
Table 1.
Effect of prenatal PPARα agonism on pregnancy outcome on GD18.
| Genotype Diet | Wild-type |
Pparα-null |
||||
|---|---|---|---|---|---|---|
| Control | Clofibrate | Wy-14,643 | Control | Clofibrate | Wy-14,643 | |
|
| ||||||
| Number of dams | 9 | 8 | 7 | 8 | 10 | 13 |
| Maternal weight (g) on GD18 | 34.3 ± 2.6a | 32.5 ± 4.0a | 35.5 ± 6.7a | 30.8 ± 3.5a | 30.4 ± 4.9a | 30.7 ± 2.8a |
| Maternal weight gain (g) on GD18 | 12.4 ± 2.3a | 10.6 ± 1.8a | 12.9 ± 7.3a | 10.3 ± 2.2a | 9.1 ± 3.7a | 11.5 ± 2.5a |
| Gravid uterus weight (g) on GD18 | 9.7 ± 1.5a | 7.4 ± 1.1a | 8.7 ± 3.9a | 7.0 ± 1.9a | 6.3 ± 3.4a | 8.1 ± 2.7a |
| Implants per uterus (I) | 8.2 ± 2.1a | 5.4 ± 2.1a | 7.0 ± 2.2a | 6.6 ± 1.5a | 7.1 ± 2.6a | 6.4 ± 2.4a |
| Number of live fetuses per litter | 7.0 ± 1.2a | 4.6 ± 2.0a | 5.7 ± 2.8a | 5.0 ± 1.7a | 4.7 ± 2.8a | 5.8 ± 2.2a |
| Number of dead fetuses per litter (D) | 0.0 ± 0.0a | 0.0 ± 0.0a | 0.1 ± 0.4a | 0.1 ± 0.4a | 0.3 ± 0.7a | 0.1 ± 0.3a |
| Number of resorptions per litter (R) | 1.2 ± 1.6a | 0.8 ± 1.0a | 1.1 ± 1.2a | 1.5 ± 1.5a | 2.1 ± 2.0a | 0.7 ± 1.0a |
| % Litter loss = [(D + R)/I*100] | 12.4 ± 15.2a | 12.3 ± 17.4a | 25.1 ± 34.6a | 23.6 ± 20.8a | 36.1 ± 33.8a | 9.4 ± 13.0a |
Values represent the mean ± S.E.M. Values within a row with different letters are significantly different, P ≤ 0.05.
Table 2.
Effect of prenatal PPARα agonism on fetal endpoints on GD18.
| Genotype | Diet | Number of fetuses/littera | Crown to rump length (mm)b | Body weight (g)b | Ratio of female fetuses to total number of fetusesa | Ratio of male fetuses to total number of fetusesa |
|---|---|---|---|---|---|---|
|
| ||||||
| Wild-type | Control | 7.0±1.2a | 20.1±1.4a | 1.0±0.2a | 47±5a | 53±5a |
| Clofibrate | 4.6±2.0a | 19.6±1.2a | 1.0±0.2a | 38±7a | 62±7a | |
| Wy-14,643 | 5.7±2.8a | 20.1±1.1a | 1.0±0.1a | 40±7a | 40±7a | |
| PPAR α-null | Control | 5.0±1.7a | 20.2±0.9a | 1.0±0.2a | 61±8a | 39±8a |
| Clofibrate | 4.7±2.8a | 18.9±1.5a | 0.9±0.1a | 49±9a | 51±9a | |
| Wy-14,643 | 5.8±2.2a | 20.2±1.0a | 1.0±0.1a | 51±5a | 49±5a | |
Values represent the mean ± S.E.M. Values within a column with different letters are significantly different, P ≤ 0.05.
The statistical unit was the litter.
The statistical unit was the individual.
PPARα agonists are known to increase replicative DNA synthesis and hyperplasia in the liver through a PPARα-dependent mechanism (Peters et al., 1998). Compared to controls, relative maternal liver weight on GD18 was increased by clofibrate and Wy-14,643 in wild-type mice but not in similarly treated Pparαnull mice (Fig. 1A). In contrast, relative fetal liver weight on GD18 was increased only modestly in wild-type mice by clofibrate but not by Wy-14,643 as compared to control, while relative fetal liver weight was unchanged by clofibrate and Wy-14,643 in Pparα-null mice (Fig. 1B). To determine the relative efficacy of clofibrate and Wy-14,643 to activate PPARα in maternal and fetal liver, expression of the well characterized PPARα target genes Aco and Cyp4a10 was quantified. Expression of Aco and Cyp4a10 mRNA was increased by clofibrate and Wy-14,643 in both maternal liver and fetal liver as compared to control, and these effects were not found in similarly treated Pparα-null mice (Fig. 1C–F). Interestingly, the relative increase in expression of Aco and Cyp4a10 mRNA was higher in Wy-14,643-treated fetuses as compared to the increase observed in maternal liver (Fig. 1C–F). These data clearly demonstrate that the doses of clofibrate and Wy-14,643 effectively activated PPARα causing modest maternal liver hepatomegaly and increased expression of target genes known to modulate lipid catabolism.
Fig. 1.
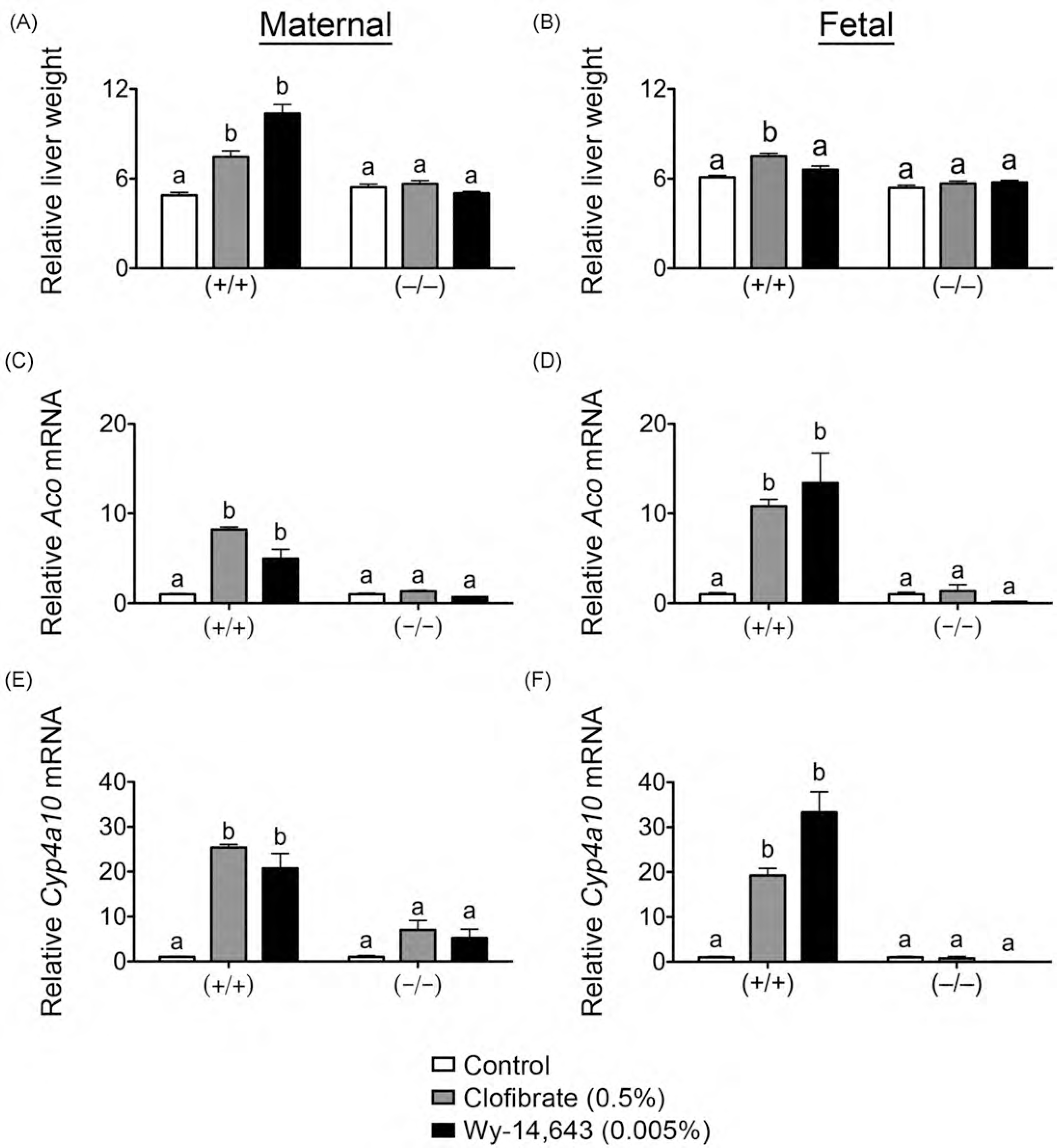
Effect of prenatal PPARα agonism on maternal and fetal endpoints on GD18. Pregnant female wild-type (+/+) or Pparα-null (−/−) mice were fed either a control diet or one containing clofibrate (0.5%) or Wy-14,643 (0.005%) until GD18. Relative maternal (A) and fetal (B) liver weight (liver weight (g)/body weight (g) × 100) on GD18. Relative expression of the PPARα target genes Aco (C and D) and Cyp4ct10 (E and F) in maternal (C and E) and fetal (D and F) liver on GD18 was measured by qPCR as described in Section 2. Values are the average normalized fold change as compared to vehicle control and represent the mean± S.E.M. The statistical unit was the individual. Values with different letters are significantly different, P≤ 0.05, as determined by ANOVA and Tukey’s test.
3.2. Effect of prenatal PPARα agonism on postnatal development
Since prenatal exposure to PFOA led to reduced survival of pups and delayed development (as assessed by the onset of eye opening) in wild-type mice but not in Pparα-null mice (Abbott et al., 2007), postnatal development was assessed in the present study following prenatal exposure to either clofibrate or Wy-14,643. The day of parturition was not affected by prenatal exposure to either clofibrate or Wy-14,643 in either genotype (Table 3). Postnatal lethality of pups up until PND20 was not different between clofibrate or Wy-14,643-treated wild-type or Pparα-null mice as compared to controls (Fig. 2A). Similarly, the onset of eye opening and postnatal weight gain was not influenced by prenatal exposure to either clofibrate or Wy-14,643 in either genotype as compared to controls (Fig. 2B and C). Additionally, no differences in the distribution of male and female pups were observed by either treatment (data not shown) and no changes in postnatal weight gain between male and female pups in the different treatment groups were observed (Table 3). Relative maternal liver weight (data not shown) and relative pup liver weight (Fig. 3) were not changed on PND20 following prenatal exposure to either clofibrate or Wy-14,643 in either genotype as compared to control. Similarly, relative expression of the PPARa target genes Aco and Cyp4a10 in maternal liver was not different on PND20 following prenatal exposure to either clofibrate or Wy-14,643 in either genotype as compared to control (data not shown). Compared to control, relative expression of Aco and Cyp4a10 mRNA in pup liver was not different on PND20 following prenatal exposure to either clofibrate or Wy-14,643 in wild-type or Pparoα-null mice (Fig. 3B and C).
Table 3.
Effect of prenatal PPARα agonism on pregnancy outcome and postnatal weight gain.
| Genotype | Treatment group | Day of parturitiona | Number of litters | Average number of pups/litterb | Pup weights (g) |
||||
|---|---|---|---|---|---|---|---|---|---|
| PND0b | Female |
Male |
|||||||
| PND14a | PND21a | PND14a | PND21a | ||||||
|
| |||||||||
| Wild-type | Control | 19.4±0.2a | 12 | 4.3±0.6 | 1.4±0.3a | 6.6±0.2a | 7.1±0.4a | 6.2±0.2a | 6.7±0.4a |
| Clofibrate | 19.4±0.3a | 7 | 3.3±0.3 | 1.5±0.2a | 8.6±0.3a | 10.0±0.3a | 8.3±0.2a | 9.5±0.3a | |
| Wy-14,643 | 19.4±0.2a | 7 | 4.0±0.9 | 1.5±0.1a | 7.2±0.2a | 8.4±0.3a | 7.2±0.3a | 8.3±0.4a | |
| Pparα-null | Control | 19.6±0.2a | 8 | 5.3±0.7 | 1.5±0.3a | 6.1±0.2a | 6.7±0.2a | 6.5±0.2a | 7.3±0.3a |
| Clofibrate | 19.3±0.2a | 8 | 4.6±0.4 | 1.4±0.2a | 7.4±0.3a | 8.5±0.2a | 7.0±0.2a | 8.2±0.2a | |
| Wy-14,643 | 19.6±0.2a | 11 | 5.6±0.4 | 1.5±0.2a | 5.9±0.1a | 7.2±0.2a | 6.0±0.1a | 7.4±0.3a | |
Mice were fed either a control diet or one containing 0.5% clofibrate or 0.005% Wy-14,643 during gestation. Mice were allowed to deliver and the day of parturition was recorded. After parturition, mice were provided only the control diet. The number of pups born per litter and pup weight was recorded on PND0 (day of delivery). Body weight was measured for male and female pups until PND21. Values represent the mean ± S.E.M. Values within a column with different letters are significantly different, P ≤ 0.05.
The statistical unit was the individual.
The statistical unit was the litter.
Fig. 2.
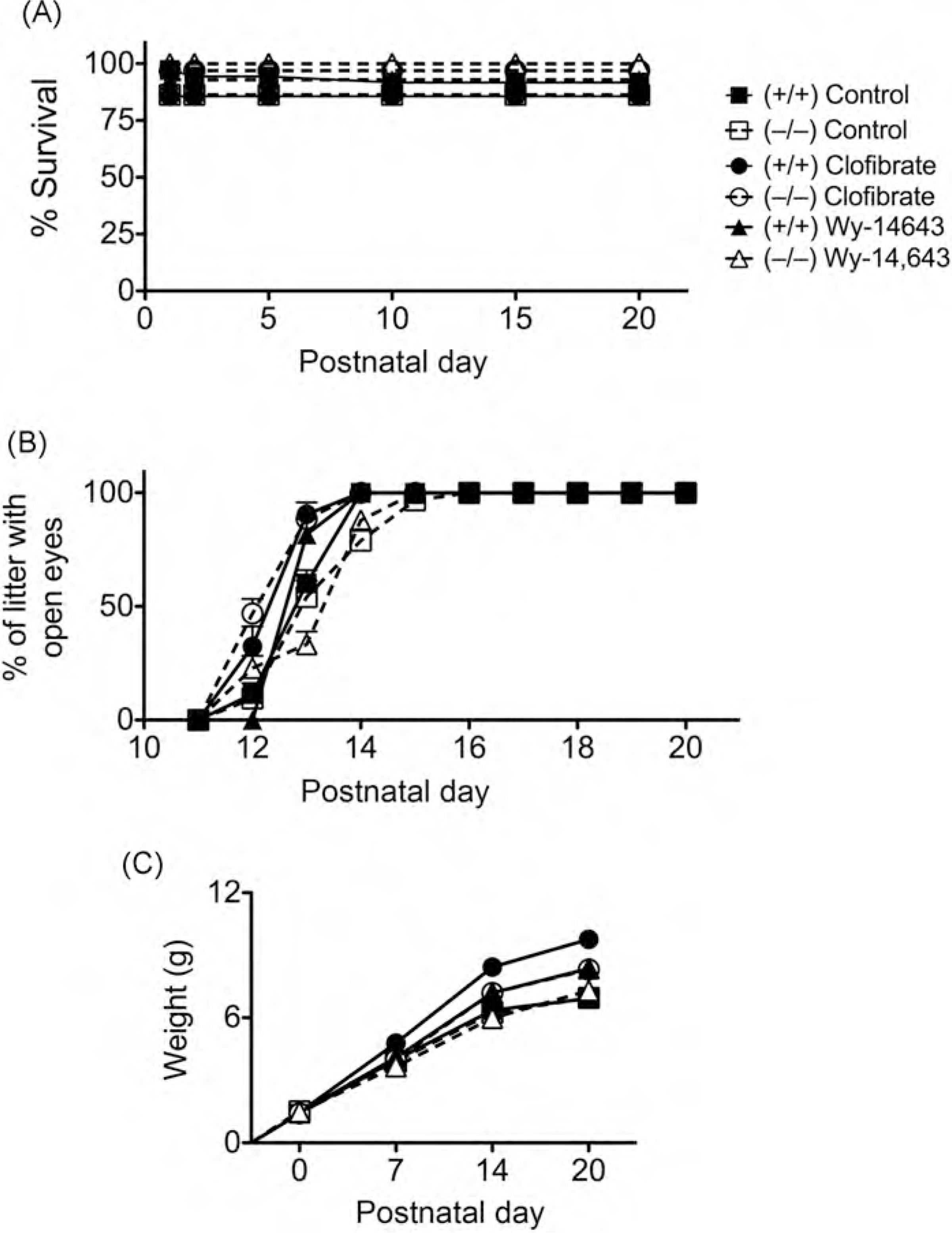
Effect of prenatal PPARα agonismon postnatal development. Pregnant female wild-type (+/+) or Pparα-null (−/−) mice were fed either a control diet or one containing clofibrate (0.5%) or Wy-14,643 (0.005%) until parturition, after which mice were fed control diet until PND20. Mice were observed daily for (A) postnatal lethality and (B) the onset of eye opening. The pups were weighed on PNDO, 7,14 and 20 (C). Values are the average normalized fold change as compared to vehicle control and represent the mean ± S.E.M. The statistical unit for (A) and (B) was the litter, the statistical unit for (C) was the individual. Values with different letters are significantly different, P≤ 0.05, as determined by ANOVA and Tukey’s test.
Fig. 3.
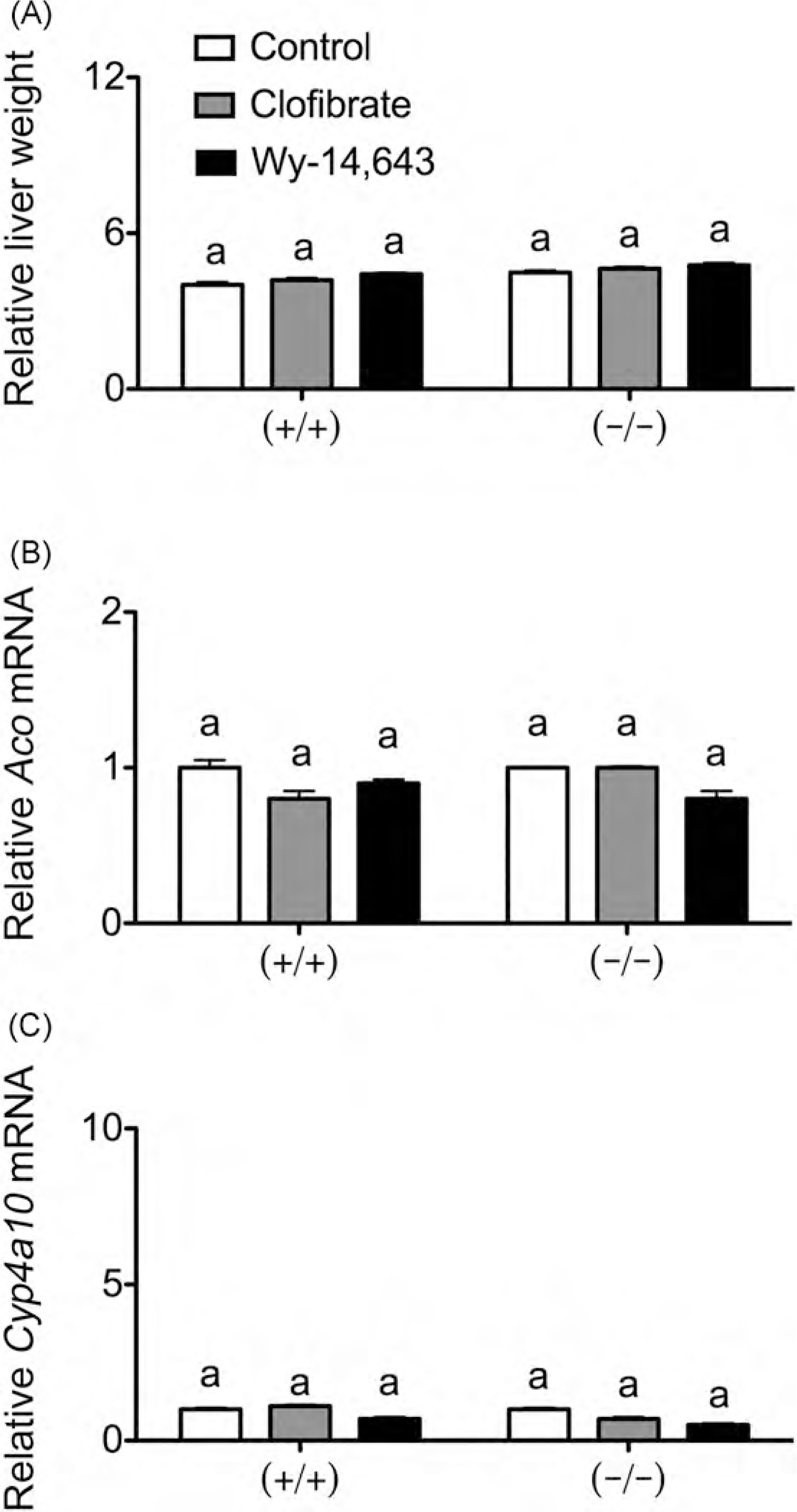
Effect of prenatal PPARα agonism on pup liver endpoints on PND20. Pregnant female wild-type (+/+) or Pparα-null (−/−) mice were fed either a control diet or one containingclofibrate (0.5%) or Wy-14,643 (0.005%) until parturition. (A) Relative pup liver weight (liver weight (g)/body weight (g) × 100) on PND20. Relative expression of the PPARα target genes Aco (B) and Cyp4ct10 (C) in pup liver was measured on PND20 by qPCR as described in Section 2. Values are the average normalized fold change as compared to vehicle control and represent the mean ± S.E.M. The statistical unit was the individual. Values with different letters are significantly different, P≤ 0.05, as determined by ANOVA and Tukey’s test.
4. Discussion
Previous studies demonstrated that prenatal exposure to PFOA results in dose-dependent full-litter resorptions, delayed development and postnatal lethality in mice (Abbott et al., 2007; Lau et al., 2006). As these effects are found in wild-type mice but not in Pparoα-null mice, this demonstrates that these effects are mediated by PPARa (Abbott et al., 2007). Cross-fostering studies established that gestational exposure to PFOA, rather than lactational exposure to PFOA, is required to elicit defects in postnatal development including delays in the onset of eye opening and early lethality (Wolf et al., 2007). Since PFOA is known to cause activation of PPARα, the present study tested the hypothesis that relatively low level activation of PPARα during prenatal development will cause postnatal lethality, similar to that observed with PFOA, a relatively low affinity PPARα agonist. Dietary administration of clofibrate and Wy-14,643 during prenatal development caused a PPARα-dependent increase in maternal liver, consistent with the known mitogenic activity associated with PPARα activation in liver (Peters et al., 1998). Similarly, a PPARα-dependent increase in expression of the PPARα target genes, Aco and Cyp4a10, was also observed in both maternal and fetal liver on GD18 providing direct evidence that PPARα activity was increased in both maternal and fetal compartments. Surprisingly, prenatal exposure to the PPARα agonists clofibrate or Wy-14,643 did not cause any developmental anomalies assessed on GD18, nor did it cause any developmental delays in eye opening or postnatal lethality of pups. These results are similar to those previously reported with perfluorobutyrate (PFBA) where no adverse developmental toxicity was observed following prenatal exposure (Das et al., 2008). This is of interest because PFBA is a short-chain perfluorinated chemical that has shorter half-life than PFOA and a weaker potency for PPARα activation as compared to PFOA (Chang et al., 2008; Wolf et al., 2008a).
These studies do not dispute the fact that prenatal PFOA exposure in mice causes neonatal lethality through a PPARα-dependent mechanism (Abbott et al., 2007). Moreover, the reason why prenatal exposure to PFOA causes PPARα-dependent postnatal lethality, while prenatal exposure to either clofibrate or Wy-14,643 does not, cannot be determined from this study. This disparity could be due in part to differences in gene expression resulting from prenatal exposure to the different compounds. It is also possible that this disparity is due in part to differences in bioaccumulation. PFOA is known to persist in environment and is not metabolized extensively in vivo because of the strong covalent bond between carbon and fluorine (Ullrich and Diehl, 1971). In mice, the half-life of PFOA has been estimated to be 15.6 days (Lou et al., 2009) whereas clofibrate and Wy-14,643 have comparatively shorter half-lives. For example, the half-life of clofibrate in humans is 15 h because it is readily absorbed from gastrointestinal tract, metabolized by CYP3A4, and excreted (Miller and Spence, 1998). Thus, prenatal exposure to PFOA could cause accumulation of PFOA in fetal liver that subsequently influences postnatal development due to more sustained PPARα activity, while clofibrate and Wy-14,643 are less likely to result in this effect. This idea is supported by the observed PPARα-dependent increase in relative liver weight in PND22 pups from PFOA-exposed dams at doses ≤1.0 mg/kg (Abbott et al., 2007). In contrast, results from the present studies show that relative liver weight in PND20 pups from clofibrate or Wy-14,643-exposed dams is not different than controls and no changes in expression of the PPARα target genes Aco and Cyp4a10 levels are found. Combined, these findings suggest that prenatal exposure to PFOA could cause accumulation in fetal liver that influences postnatal development through PPARα-dependent mechanisms, while clofibrate and Wy-14,643 do not.
Several studies have examined the effects of either prenatal or neonatal exposures to lactating rodents treated with various PPARα ligands, including Wy-14,643, nafenopin, clofibrate, ciprofibrate, and diethylhexyl phthalate (DEHP) (Cibelli et al., 1988; Cimini et al., 1994; Fahl et al., 1983; Singh and Lazo, 1992; Stefanini et al., 1989, 1999, 1995; Wilson et al., 1991). Collectively, these studies show that exposure to PPARα agonists induces both peroxisome proliferation and increased expression of PPARα target genes (e.g. Aco, Cyp4a10) in fetal and neonatal rodents. Interestingly, 14-day-old rat pups exhibit enhanced sensitivity to PPARα activity as compared to older rat pups (Dostal et al., 1987). This is the first evidence suggesting that neonatal rodents are more sensitive than adults to PPARα activation. Results from the present studies are consistent with this idea because the relative increase in expression of Aco mRNA resulting from prenatal exposure to both clofibrate and Wy-14,643 was higher in fetal liver on GD18 as compared to maternal liver. While this effect was not found with the increase in expression of Cyp4a10 mRNA following prenatal exposure to clofibrate, relatively higher Cyp4a10 mRNA was found in fetal liver on GD18 as compared to maternal liver as a result of prenatal exposure to Wy-14,643. The significance of this apparent difference in sensitivity to PPARα agonism remains to be determined.
Acknowledgements
The studies were supported by unrestricted gifts from the 3M Company and Dupont Haskell Global Centers of Health and Environmental Sciences.
Footnotes
Conflict of interest statement
JMP has been retained as an expert consultant by the 3M Company. PSP, CRA, CHF and FJG have no competing interests.
References
- Abbott BD, Wolf CJ, Schmid JE, Das ICP., Zehr RD., Helfant L., Nakayama S., Lindstrom AB., Strynar MJ., Lau C., 2007. Perfluorooctanoic acid inducec developmental toxicity in the mouse is dependent on expression of peroxisome proliferator activated receptor-α. Toxicol. Sci. 98, 571–581. [DOI] [PubMed] [Google Scholar]
- Akiyama TE, Nicol CJ, Fievet C, Staels B, Ward JM, Auwerx J, Lee SS, Gonzalez FJ., Peters JM., 2001. Peroxisome proliferator-activated receptor-α regulate: lipid homeostasis, but is not associated with obesity: studies with congenic mouse lines. J. Biol. Chem. 276, 39088–39093. [DOI] [PubMed] [Google Scholar]
- Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, Staels B, Auwerx J, Laville M, Vidal H, 1997. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and live] X receptor-α in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes 48,1319–1327. [DOI] [PubMed] [Google Scholar]
- Bility M, Thompson JT, McKee RH, David RM, Butala JH, Vanden Heuvel JP Peters JM., 2004. Activation of mouse and human peroxisome proliferator-activated receptors (PPARs) by phthalate monoesters. Toxicol. Sci. 82,170–182 [DOI] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W, 1996. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distributor of PPAR-α-β, and -γ in the adult rat. Endocrinology 137,354–366. [DOI] [PubMed] [Google Scholar]
- Braissant O, Wahli W, 1998. Differential expression of peroxisome proliferator-activated receptor-α-β, and -γ during rat embryonic development. Endocrinol ogy 139, 2748–2754. [DOI] [PubMed] [Google Scholar]
- Chang SC, Das IC, Ehresman DJ, Ellefson ME, Gorman GS, Hart JA, Noker PE Tan YM., Lieder PH., Lau C., Olsen GW., Butenhoff JL, 2008. Comparative pharmacokinetics of perfluorobutyrate in rats, mice, monkeys, and humans anc relevance to human exposure via drinking water. Toxicol. Sci. 104,40–53. [DOI] [PubMed] [Google Scholar]
- Cibelli A, Stefanini S, Ceru MP, 1988. Peroxisomal beta-oxidation and catalase activities in fetal rat liver: effect of maternal treatment with clofibrate. Cell Mol Biol. 34,191–205. [PubMed] [Google Scholar]
- Cimini AM, Sulli A, Stefanini S, Serafini B, Moreno S, Rossi L, Giorgi M, Ceru MP., 1994. Effects of di-(2-ethylhexyl)phthalate on peroxisomes of liver, kidney and brain of lactating rats and their pups. Cell Mol. Biol. (Noisy-le-grand) 401063–1076. [PubMed] [Google Scholar]
- Das KP, Grey BE, Zehr RD, Wood CR, Butenhoff JL, Chang SC, Ehresman DJ Tan YM., Lau C., 2008. Effects of perfluorobutyrate exposure during pregnanq in the mouse. Toxicol. Sci. 105,173–181. [DOI] [PubMed] [Google Scholar]
- Dostal LA., Jenkins WL., Schwetz BA., 1987. Hepatic peroxisome proliferation anc hypolipidemic effects of di(2-ethylhexyl)phthalate in neonatal and adult rats Toxicol. Appl. Pharmacol. 87, 81–90. [DOI] [PubMed] [Google Scholar]
- Fahl WE, Lalwani ND, Reddy MK, Reddy JK, 1983. Induction of peroxisoma enzymes in livers of neonatal rats exposed to lactating mothers treated with hypolipidaemic drugs. Role of drug metabolite transfer in milk. Biochem. J. 210875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman JE, Chang SC, Ehresman DJ, Butenhoff JL, Anderson CR, Palkar PS Kang BH., Gonzalez FJ., Peters JM., 2009. Differential hepatic effects of perfluorobutyrate mediated by mouse and human PPAR-alpha. Toxicol. Sci. 110204–211. [DOI] [PubMed] [Google Scholar]
- Forman BM, Chen J, Evans RM, 1997. Hypolipidemic drugs, polyunsaturatec fatty acids, and eicosanoids are ligands for peroxisome proliferator-activatec receptors α and β. Proc. Natl. Acad. Sci. U.S.A. 94,4312–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez FJ, Shah YM, 2008. PPARα: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology 246,2–8. [DOI] [PubMed] [Google Scholar]
- Hays T, Rusyn L, Burns AM, Kennett MJ, Ward JM, Gonzalez FJ, Peters JM., 2005. Role of peroxisome proliferator-activated receptor-α (PPARα) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis 26219–227. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG., Lai DY., McKee RH., Peters JM., Roberts RA., Fenner-Crisp PA., 2003PPARα agonist-induced rodent tumors: modes of action and human relevance Crit. Rev. Toxicol. 33,655–780. [DOI] [PubMed] [Google Scholar]
- Lau C, Thibodeaux JR, Hanson RG, Narotsky MG, Rogers JM, Lindstrom AR, Strynar MJ, 2006. Effects of perfluorooctanoic acid exposure during pregnancy in the mouse. Toxicol. Sci. 90, 510–518. [DOI] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ, 1995. Targeted disruption of the α isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell Biol. 15, 3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou JS, Szostek B, Derito CM, Madsen EL, 2010. Investigating the biodegradability of perfluorooctanoic acid. Chemosphere 80,176–183. [DOI] [PubMed] [Google Scholar]
- Lou I, Wambaugh JF, Lau C, Hanson RG, Lindstrom AB, Strynar MJ, Zehr RD, Setzer RW, Barton HA, 2009. Modeling single and repeated dose pharmacokinetics of PFOA in mice. Toxicol. Sci. 107,331–341. [DOI] [PubMed] [Google Scholar]
- Maloney EK, Waxman DJ, 1999. trans-Activation of PPARα and PPARγ by structurally diverse environmental chemicals. Toxicol. Appl. Pharmacol. 161, 209–218. [DOI] [PubMed] [Google Scholar]
- Marsman DS, Goldsworthy TL, Popp JA., 1992. Contrasting hepatocytic peroxisome proliferation, lipofuscin accumulation and cell turnover for the hepatocarcinogens Wy-14,643 and clofibricacid. Carcinogenesis 13,1011–1017. [DOI] [PubMed] [Google Scholar]
- Miller DB, Spence JD, 1998. Clinical pharmacokinetics of fibric acid derivatives (fibrates). Clin. Pharmacokinet. 34,155–162. [DOI] [PubMed] [Google Scholar]
- Peters JM, 2008. Mechanistic evaluation of PPAR-mediated hepatocarcinogenesis: are we there yet? Toxicol. Sci. 101,1–3. [Google Scholar]
- Peters JM, Aoyama T, Cattley RC, Nobumitsu U, Hashimoto T, Gonzalez FJ, 1998. Role of peroxisome proliferator-activated receptor α in altered cell cycle regulation in mouse liver. Carcinogenesis 19,1989–1994. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cattley RC, Gonzalez FJ, 1997. Role of PPAR α in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 18, 2029–2033. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cheung C, Gonzalez FJ, 2005. Peroxisome proliferator-activated receptor-α and liver cancer: where do we stand? J. Mol. Med. 83, 774–785. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Azarnoff DL, Hignite CE, 1980. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 283,397–398. [DOI] [PubMed] [Google Scholar]
- Shearer BG, Hoekstra WJ, 2003. Recent advances in peroxisome proliferator-activated receptor science. Curr. Med. Chem. 10,267–280. [DOI] [PubMed] [Google Scholar]
- Singh I, Lazo O, 1992. Peroxisomal enzyme activities in brain and liver of pups of lactating mothers treated with ciprofibrate. Neurosci. Lett. 138, 283–286. [DOI] [PubMed] [Google Scholar]
- Stefanini S, Mauriello A, Farrace MG, Cibelli A, Ceru MP, 1989. Proliferative response of foetal liver peroxisomes to clofibrate treatment of pregnant rats. A quantitative evaluation. Biol. Cell 67,299–305. [PubMed] [Google Scholar]
- Stefanini S, Nardacci R, Farioli-Vecchioli S, Pajalunga D, Sartori C, 1999. Liver and kidney peroxisomes in lactating rats and their pups after treatment with ciprofibrate. Biochemical and morphometric analysis. Cell Mol. Biol. (Noisy-legrand) 45,815–829. [PubMed] [Google Scholar]
- Stefanini S, Serafini B, Nardacci R, Vecchioli SF, Moreno S, Sartori C, 1995. Morphometric analysis of liver and kidney peroxisomes in lactating rats and their pups after treatment with the peroxisomal proliferator di-(2-ethylhexyl)phthalate. Biol. Cell 85,167–176. [DOI] [PubMed] [Google Scholar]
- Ullrich V, Diehl H, 1971. Uncoupling of monooxygenation and electron transport by fluorocarbons in liver microsomes. Eur. J. Biochem. 20, 509–512. [DOI] [PubMed] [Google Scholar]
- White SS, Calafat AM, Kuklenyik Z, Villanueva L, Zehr RD, Helfant L, Strynar MJ, Lindstrom AB, Thibodeaux JR, Wood C, Fenton SE, 2007. Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol. Sci. 96,133–144. [DOI] [PubMed] [Google Scholar]
- Wilson GN, King T, Argyle JC, Garcia RF, 1991. Maternal clofibrate administration amplifies fetal peroxisomes. Pediatr. Res. 29,256–262. [DOI] [PubMed] [Google Scholar]
- Wolf CJ, Fenton SE, Schmid JE, Calafat AM, Kuklenyik Z, Bryant XA, Thibodeaux J, Das KP, White SS, Lau CS, Abbott BD, 2007. Developmental toxicity of perfluorooctanoic acid in the CD-1 mouse after cross-foster and restricted gestational exposures. Toxicol. Sci. 95,462–473. [DOI] [PubMed] [Google Scholar]
- Wolf CJ, Takacs ML, Schmid JE, Lau C, Abbott BD, 2008a. Activation of mouse and human peroxisome proliferator-activated receptor α by perfluoroalkyl acids of different functional groups and chain lengths. Toxicol. Sci. 106, 162–171. [DOI] [PubMed] [Google Scholar]
- Wolf DC, Moore T, Abbott BD, Rosen MB, Das KP, Zehr RD, Lindstrom AB, Strynar MJ, Lau C, 2008b. Comparative hepatic effects of perfluorooctanoic acid and WY 14,643 in PPAR-alpha knockout and wild-type mice. Toxicol. Pathol. 36, 632–639. [DOI] [PubMed] [Google Scholar]