

Abstract
Recycling of all-trans-retinal to 11-cis-retinal through the visual cycle is a fundamental metabolic pathway of vision. A potent retinoid isomerase (RPE65) inhibitor, (R)-emixustat, has been developed and tested in several clinical trials; however, it has not received regulatory approval for use in any specific retinopathy. Rapid clearance of this drug presents challenges to maintaining therapeutic concentrations in eyes within a therapeutic window. To address this pharmacokinetic inadequacy, we rationally designed and synthesized a series of emixustat derivatives with strategically placed fluorine and deuterium atoms to slow the key metabolic transformations known for emixustat. Crystal structures and quantum chemical analysis of RPE65 in complex with the most potent emixustat derivatives revealed the structural and electronic basis for how fluoro substituents can be favorably accommodated within the active site pocket of RPE65. We found a close (~3.0 Å) F∙∙π interaction that is predicted to contribute ~2.4 kcal/mol to the overall binding energy.
Graphical Abstract
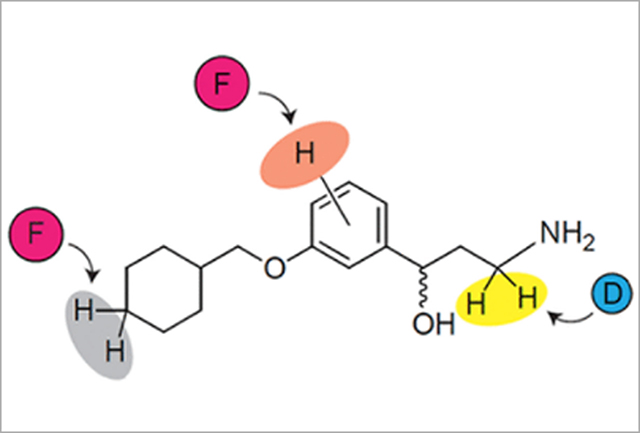
INTRODUCTION
Retinal photoreceptor cells can respond to light throughout life because they continuously regenerate a light-sensitive chromophore and photoreceptor structures. Defects in various proteins involved in these processes cause photoreceptor degeneration.1 Light-detection is mediated by a group of G protein-coupled receptor proteins called opsins located in rod and cone photoreceptor cells of the retina. The light-absorbing chromophore of most vertebrate opsins is 11-cis-retinal. Absorption of a photon by an opsin pigment causes isomerization of the chromophore to all-trans-retinal. Regeneration of the visual chromophore following light exposure is dependent upon an enzymatic pathway referred to as the visual cycle (Figure 1A).2 To understand why human vision declines with age, considerable research has focused on the retina, especially the layer of rod and cone photoreceptor cells that convert light into electrical signals. However, age-related decreases in retinal photoreceptor cell function cannot be explained alone by rod/cone cell loss, abnormal retinal plasticity, or any acute signs of retinal disease. Rather, there are pathological events that take place over time, including the aberrant metabolism of all-trans-retinal that can interfere with normal photoreceptor function (Figure 1A). All-trans-retinal, when released from rhodopsin, primarily re-enters the visual cycle. However, in some individuals, it also can persist as an unbound potentially toxic aldehyde or react with other molecules to create toxic compounds such as N-retinylidene-N-retinylethanolamine (A2E) and retinal dimers.3 These events are thought to contribute to the etiology of blinding diseases such as age-related macular degeneration (AMD) and Stargardt disease.4
Figure 1. Pharmacodynamics and pharmacokinetics of visual cycle modulators.
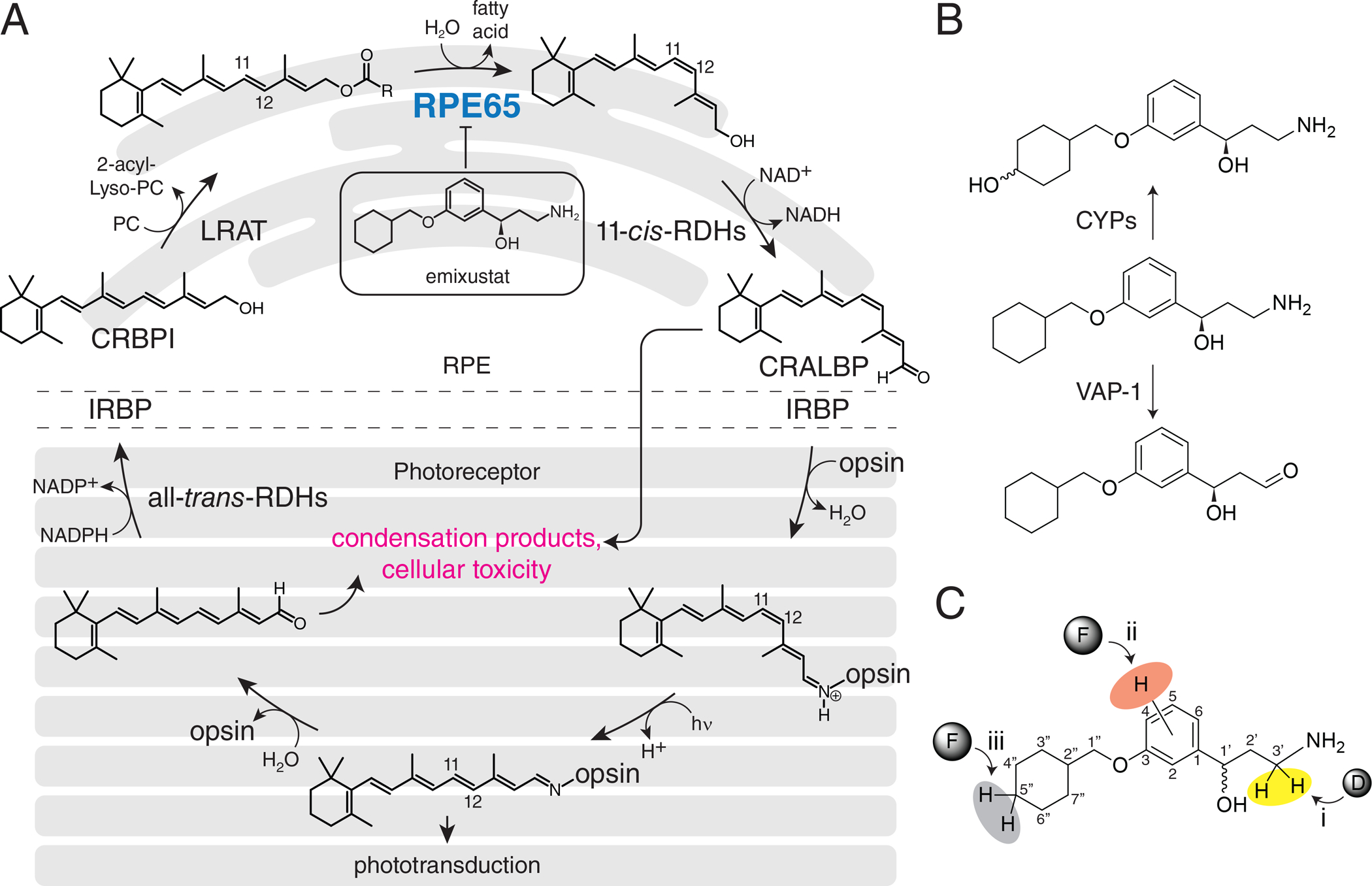
A) Visual cycle modulators such as emixustat inhibit RPE65, thus blocking the key trans/cis isomerization step of the visual cycle and reducing the formation of toxic retinaldehyde condensation products. B) Sites of emixustat modification in vivo and strategies to modify its metabolism. C) Three strategies for modifying emixustat metabolism were investigated in this work.
Various therapeutic strategies have been developed to combat the aberrant metabolism of retinoids including all-trans-retinal. One of these involves the inhibition of the retinoid isomerase (RPE65), a key enzyme of the visual cycle (Figure 1A), to slow the generating of all-trans-retinal without significantly impairing vision. (R)-emixustat, derived from retinylamine (Figure 1B),5 is currently undergoing clinical development as a potent RPE65 inhibitor.6–14 However, complete suppression of RPE65 activity has undesirable blinding consequences, thus only a narrow concentration range of the drug is tolerable. Effective use of emixustat is further complicated because it is rapidly metabolized,15–18 limiting the duration of its pharmacological effects. A second strategy is to lower the toxic effects of unbound all-trans-retinal and temporarily sequester it by adduction to a primary amine.19, 20 Since most RPE65 inhibitors contain an amino group, they can play a dual role to both slow the metabolism of all-trans-retinal as well as sequester it.19
Here, we describe the synthesis of several derivatives of emixustat, involving strategic incorporation of deuterium or fluorine to investigate three areas of the emixustat structure that could modulate its potency and metabolism (Figure 1C). Regioselective incorporation of fluorine is known to have a broad range of potential impacts on the properties of small molecules. These include pKa modulation; alteration of target selectivity through conformational variations or changes in specific hydrophobic interactions; and alteration of tissue-specific penetration (e.g., CNS), through modification of lipophilicity.21 These effects of fluorination are in addition to the well-established strategy of replacing metabolically labile hydrogens with C-F bonds.22 Regioselective incorporation of deuterium was utilized to investigate the role of amine oxidation in the rapid metabolic elimination of emixustat. We hypothesized that we could attenuate this metabolic process via engineering a localized primary isotope effect.23 Collectively, we envisioned a collection of rationally designed compounds that could improve the potency, absorption, selectivity, and metabolism of drug candidates to mitigate the toxicity of all-trans-retinal in age-related blindness.
RESULTS
Synthesis
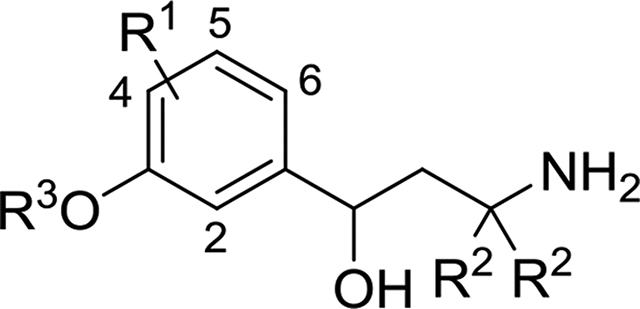
Here, we report three families of molecules where we employed a common strategy, constructing a variety of alkoxy substituents of a central aryl core initially, and then adding a γ- hydroxyalkylamine moiety, using both established and novel methods. We considered two general approaches that are strategically differentiated by the directionality of molecule assembling with respect to the aryl core (Schemes 1 and 2). Additionally, a possibility of the construction the chiral γ-hydroxyalkylamine framework using assistance of removable chiral auxiliary (Scheme 1) as well as an option for intentional α-deuteration toward amino-group (Scheme 2) were considered. In the very beginning of both general synthetic routes the alkyl substituents were installed onto either properly fluorinated hydroxyacetophenone (Scheme 1) or hydroxybenzaldehyde backbone (Scheme 2) through conventional alkylation of the phenolic hydroxyls with the corresponding alkyl bromides or mesylates.
Scheme 1.
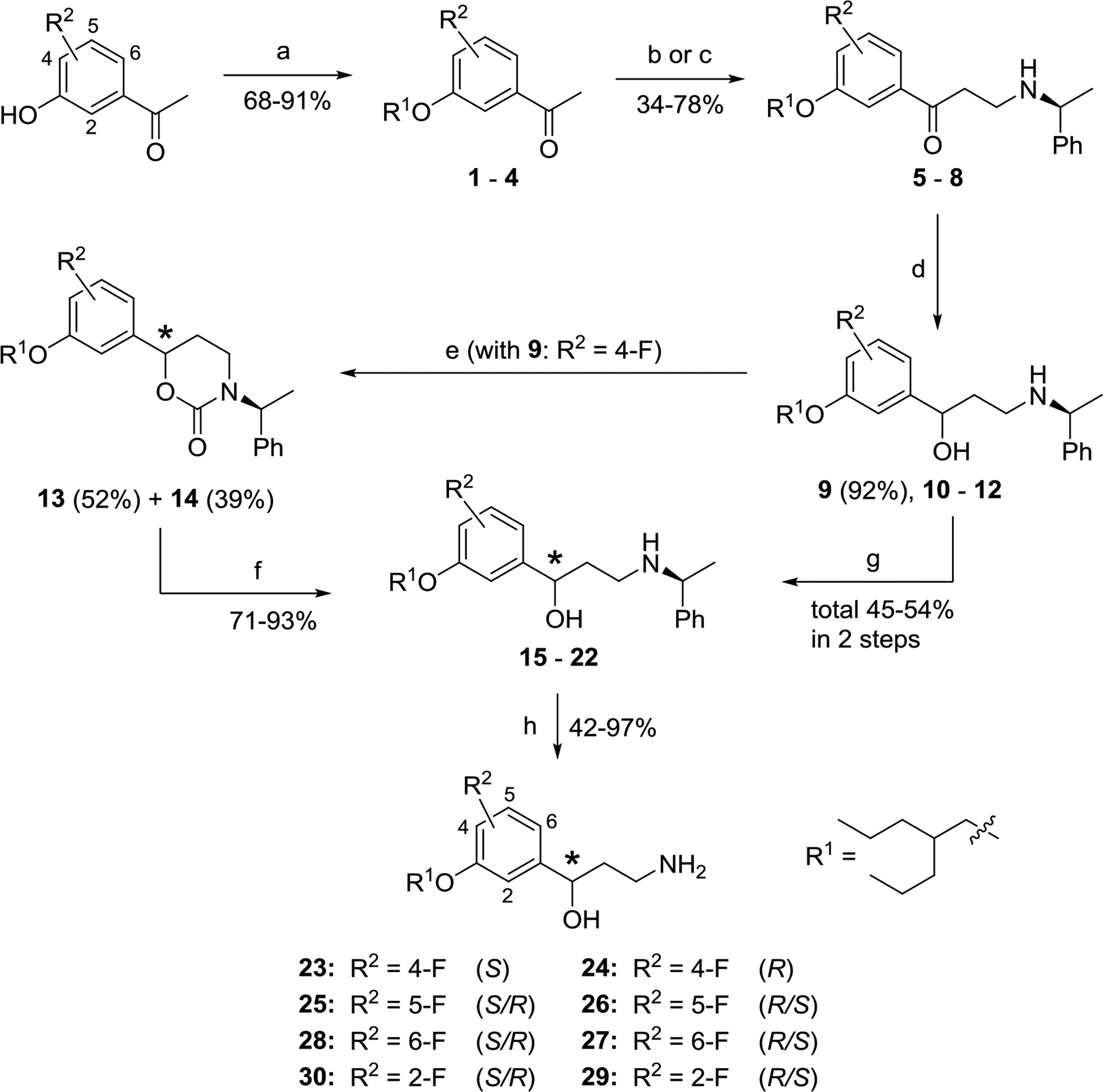
Synthesis and asymmetric resolution of (2-propylpentyl)oxyfluorophenyl analogs of emixustat 23–30. a
aReagents and conditions: (a) 2-Propylpentylmesylate, K2CO3, DMF, 85 °C, 2 – 4 h. (b) Paraform, (S)-(‒)-methylbenzylamine, conc. HCl (cat.), 1,4-dioxane, MW, 100 – 130 °C, 5 min. (c) 1,3,5-Trioxane, (S)-(‒)-methylbenzylamine, conc. HCl (cat.), 1,4-dioxane, sealed tube, 110 °C, overnight. (d) NaBH4, MeOH, r.t., 1 h. (e) (i) CDI, THF, reflux, overnight; (ii) separation of diastereomers by FC on SiO2. (f) KOH, EtOH, reflux, overnight. (g) Separation of diastereomeric mixtures 10 – 12 by chiral HPLC. (h) Ammonium formate, Pd/C (cat.), MeOH, reflux, 30 min. – overnight.
Scheme 2.
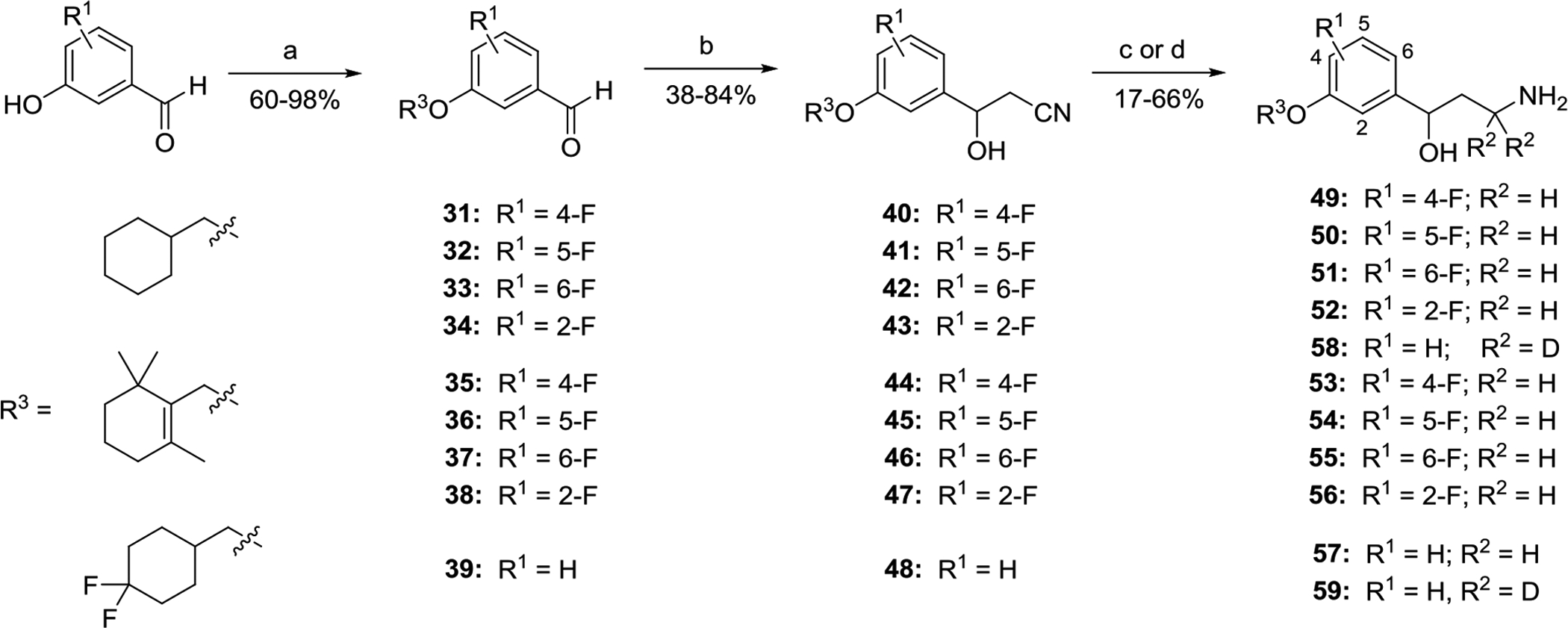
Synthesis of racemic fluorinated emixustat analogs 49–59 via addition of acetonitrile to functionalyzed benzaldehydes.
a Reagents and conditions: (a) R3-Br or R3-OMs, K2CO3, DMF, 80–90 °C, overnight. (b) MeCN, t-BuOK, THF, −50 °C, or LDA, THF, −78 °C; 1.5 – 3.0 h. (c) LAH, THF, 0 °C, 0.5 – 1.0 h. (d) LAD, THF, 0 °C, 1 h.
Synthesis of non-racemic (2-propylpentyl)oxyfluorophenyl-series of γ-aminoalcohols 23–30 by means of capitalization of the chiral removable appendage for resolution of the diastereomeric intermediates is shown in Scheme 1. Indeed, recently developed for synthesis of enantiopure γ-oxo-α-amino acids24, 25 and β-aminoketones 3-component Mannich reaction with chiral benzylamines,26 being applied to fluorinated in the benzene ring alkoxyacetophenones 1–4, (S)-(−)-α-methylbenzylamine and formaldehyde precursors afforded a suitable access to the corresponding β-aminoketones 5–8. The HCl-catalyzed Mannich reactions of fluorinated acetophenones 1–4 with (S)-(−)-α-methylbenzylamine, leading to the chiral β-aminoketones 5–8, were initially performed using paraformaldehyde and MW-irradiation (Method 1): Efficient large scale microwave-assisted Mannich reactions using substituted acetophenones.27 While this method provided reasonable yield of β-aminoketones 5 and 7 (47% in both cases) from the ketones 1 and 3, respectively, the reactions of the acetophenones 2 and 4 with the ortho-positioned fluoro and alkoxy-substituents under the MW-conditions repeatedly resulted in insufficient production of aminoketones 6 and 8 (<20%). To our delight, the more traditional method for the Mannich condensation (heating of 2 and 4 with 1,3,5-trioxane in a sealed pressure tube), Method 2,28 was found to be more efficient for preparation of the chiral β-aminoketones 6 and 8 (34% and 78%, respectively).
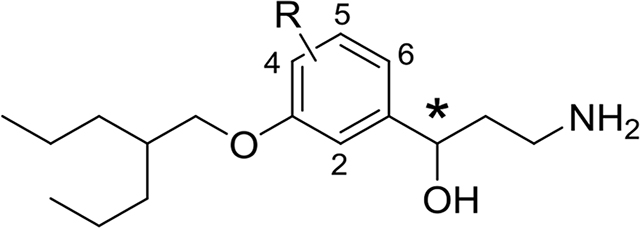
The chiral β-aminoketones 5–8, prepared through the Mannich reaction, were reduced by sodium borohydride in MeOH to give the corresponding N-protected γ-aminoalcohols 9–12 as a mixture of diastereomers in each case. All of the mixtures of diastereomeric γ-aminoalcohols 9–12 could be separated to single diastereomers 15–22 using preparative chiral HPLC as detailed in the Supporting Information. The final N-deprotection of individual diastereomers 15–22 through Pd/C-catalyzed transfer hydrogenation with ammonium formate as a hydrogen source afforded the target γ-aminoalcohols 23–30 as single enantiomers, which were studied biologically without further characterization of absolute stereochemistry. For more detailed biological evaluation, the most active chiral 3-alkyloxy-4-fluoro-derivative 24 as well as its enantiomer 23 were synthesized by a more practical method avoiding laborious and time-consuming preparative HPLC separation. Thus, a mixture of diastereomeric N-benzylated γ-aminoalcohols 9 was treated with 1,1’-carbonyldiimidazole (CDI)29 giving almost quantitatively a mixture of chiral diastereomeric cyclic carbamates, which was readily separated to single components 13 (52%) and 14 (39%) by routine flash chromatography (FC) on silica gel. The cyclic carbamates 13 and 14 were separately hydrolyzed with KOH30, thus resolving acyclic N-benzylated γ-aminoalcohols 15 (93%) and 16 (71%). The γ-aminoalcohols 15 and 16 were N-deprotected as above (ammonium formate in MeOH, Pd/C-catalyst) giving the corresponding enantiomeric γ-aminoalcohols 23 (91%) and 24 (93%). A comparison of the duplicated specific rotation values [α]D25 −21.32° (c = 0.0003, CH2Cl2) in 23 and +21.56° (c = 0.0002, CH2Cl2) in 24 with the reported data [α]D26.7 +19.66 (c = 0.01125, EtOH) for (R)-emixustat31 tentatively favor assignment of the absolute R-configuration for 24 and the opposite S-configuration for 23. However, for better understanding of the SAR additional structural study of the chiral γ-aminoalcohols 23–30 would be desirable.
The synthesis of the racemic fluorinated emixustat analogs 49–59 was performed through an alternative 3-step synthetic route (Scheme 2) similarly to the method elaborated for the parent emixustat.5, 19, 31–33 After alkylation of fluorinated hydroxybenzaldehydes to give the alkoxy-derivatives 31–39, treatment of the latter with deprotonated acetonitrile led to a 3 atom elongation with formation of the corresponding β-hydroxy-β-arylpropionitriles 40–48 (38 – 84%). The nitrile groups in these advanced intermediates 40–48 were either reduced with LAH to give racemic γ-aminoalcohols 49–57 or reduced and α-bis-deuterated with LAD to give the bis-deuterio-analogs 58 and 59 in variable yields. Although β-hydroxypropionitriles 40–48 might be potentially converted to enatioenriched or enantiopure state using an additional oxidation/asymmetric reduction sequence as described for emixustat,31, 34 in this study the fluorinated emixustat-analogs 49–59 were preliminary evaluated in the racemic form, as mixtures of their (S)- and (R)-enantiomers.
Inhibitory properties and pharmacokinetics of the tested compounds
A total of nineteen novel emixustat derivatives were synthesized, and their inhibitory effects on RPE65 in vitro were characterized by the decrease of 11-cis-retinol production by bovine RPE microsomes (Figure 2, A and B; Table 1, Supporting Table 1).5 All of the synthesized primary amines showed intense inhibition of RPE65 activity at submicromolar concentrations. Compared to emixustat and MB-001, a single fluorination at C4 of the phenyl ring significantly increases the inhibitory potency. Pronounced enhancement is seen with compound 24 (IC50 = 50 ± 9 nM), and is the most potent RPE65 inhibitor that we have identified to date, with an inhibitory potency 3 times greater than the racemic emixustat (IC50 = 172± 29 nM), and twice that of (R)-MB-004 (IC50 =106 ± 16 nM)33 which is the parent molecule of compound 24 lacking the 4-fluorine substitution. The gem-difluorination of the cyclohexyl ring in 57 also enhanced the inhibition of 11-cis-retinol production. The inhibition curve of 57 was found to exhibit a double sigmoidal feature indicative of two distinct modes of inhibitor binding to RPE65.
Figure 2.
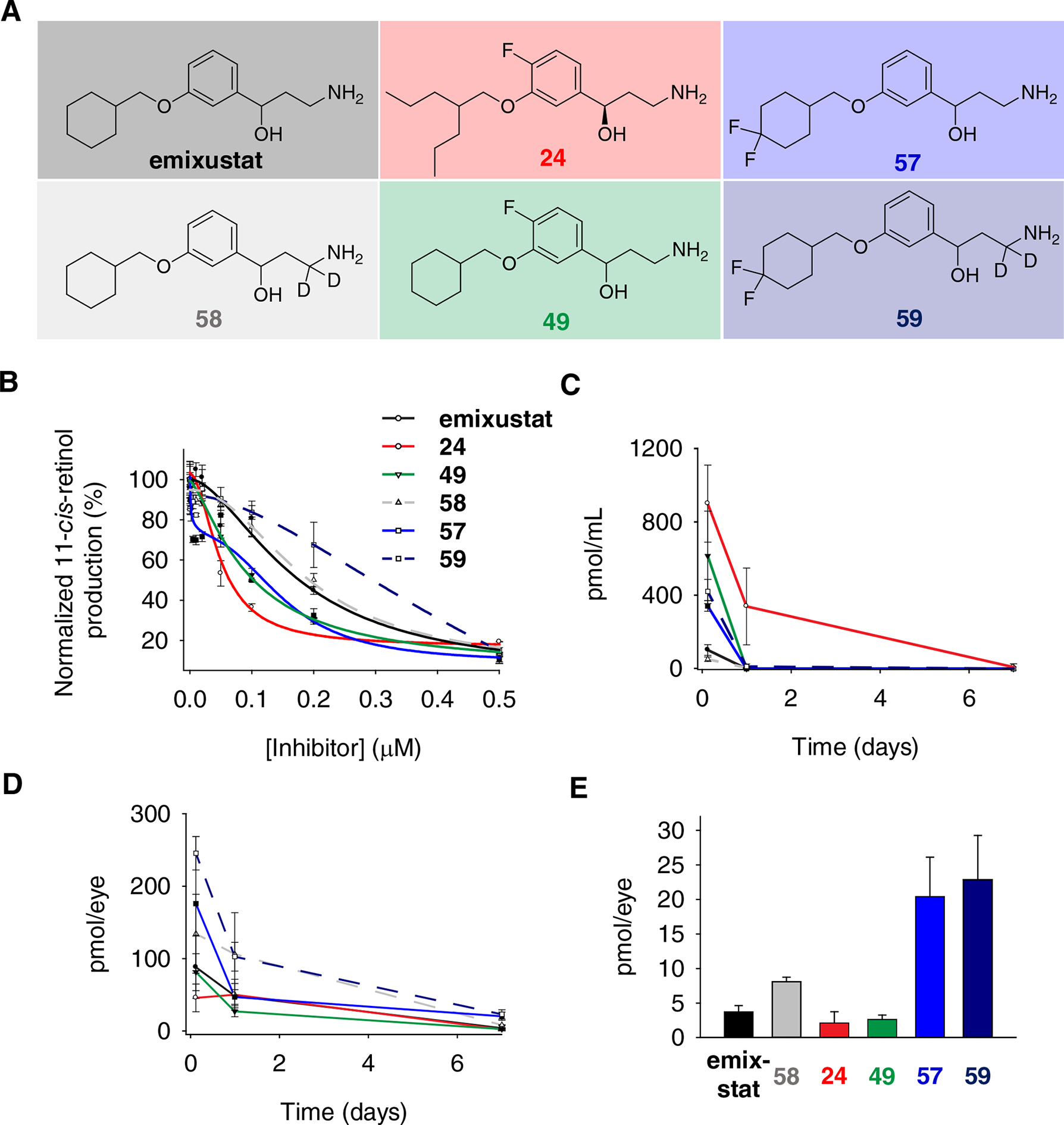
A) Structures of visual cycle modulators used for pharmacokinetics studies. B) Evaluation of the inhibitory effects of selected visual cycle modulators on 11-cis-retinol production by bovine RPE microsomes. Data points are shown as mean ± s.d.; n = 3. C, D) The progress of visual cycle modulator elimination in mouse serum (C) and eyes (D) after a single dose of intraperitoneal injection. Data points are shown as mean ± s.d.; n = 4–5. E) The primary amine levels in mouse eyes on day 7 after the treatments.
Table 1.
Mannich derived γ-hydroxyamines.
| Compound | * | R | IC50 (nM) | Compound | * | R | IC50 (nM) |
|---|---|---|---|---|---|---|---|
| 23 | S | 4-F | 50 ± 9 | 24 | S | 4-F | 177 ± 11 |
| 25 | R/S | 5-F | 295 ± 132 | 26 | R/S | 5-F | 184 ± 24 |
| 28 | R/S | 6-F | 132 ± 19 | 27 | R/S | 6-F | 157 ± 24 |
| 30 | R/S | 2-F | 274 ± 47 | 29 | R/S | 2-F | 287 ± 28 |
Oxidative deamination, and hydroxylation on the cyclohexyl ring are two pathways for emixustat elimination in vivo;17 the former being more dominant in the vasculature as compared to the liver.17 By analogy to studies of oxidative deamination of other compounds,35 deuteration at the position α to the amino group of emixustat would be predicted to slow down its metabolic elimination due to a primary kinetic isotope effect. We explored this possibility by comparing the rate of oxidative deamination of emixustat and its α-deuterated derivative of emixustat (compound 58) in an HPLC-based activity assay using mouse aorta homogenates as the source of VAP-1 enzyme (Figure 3A). We monitored formation of the dehydrated aldehyde product of VAP-1 catalysis (ACU-5201) by reversed phase HPLC (Figure 3B) whose identity and absolute amount were determined by comparison to an authentic synthetic standard (Figure 3, B and C). As compared to emixustat, the formation of ACU-5201 from compound 58 was reduced by approximately 66% at both time points examined (Figure 3D). This data is consistent with the idea that proton abstraction from the amine α-carbon is a rate-limiting step in the catalytic mechanism of emixustat deamination by VAP-1.
Figure 3. Impact of emixustat deuteration on VAP-1 metabolic susceptibility.
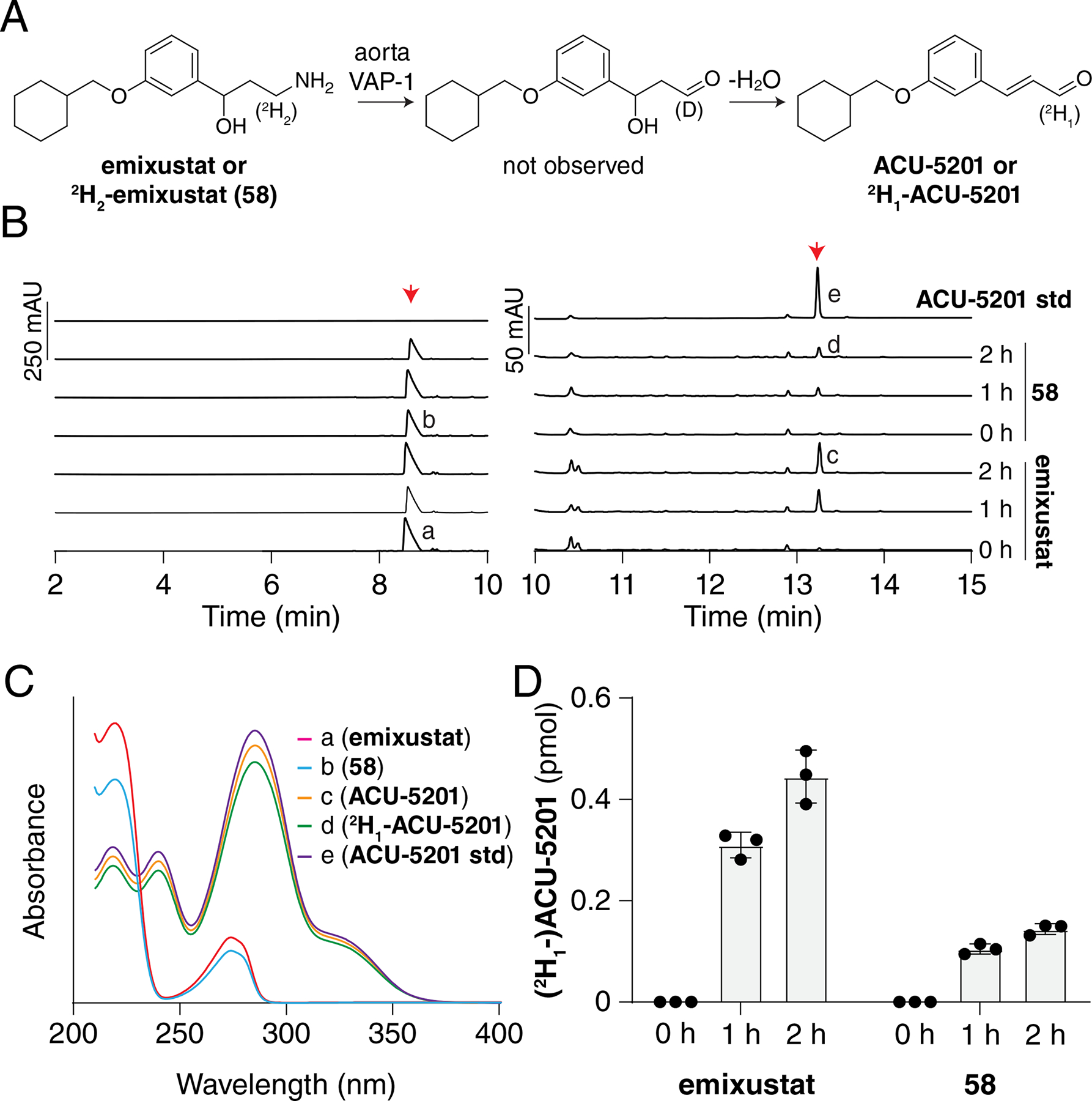
A) Scheme showing the oxidation of emixustat or 58 (2H-emixustat) by VAP-1 present in the aorta homogenates used for the assay. B) HPLC chromatographs showing the formation of ACU-5201 (right red arrow) over time, after incubation of emixustat or 58 (left red arrow) with mouse aorta homogenates containing VAP-1. The HPLC traces are split at 10 min with two different scales used to aid in visualization. C) The identities of the product peaks were verified by comparison of their retention times and UV/Vis absorbance spectra to that of authentic ACU-5201 standard. D) Quantification of product formation showed that the deuteration at the 3-position reduced product formation nearly three-fold at both 1 h and 2 h time points. Mean values ± SDs and individual data points are shown in the graph.
To investigate the impact of fluorination and deuteration on the elimination of emixustat in mouse eyes, a single dose of 380 nmol of emixustat, 24, 49, 57, 58 or 59 was individually administrated to wild type mice by intraperitoneal injection. The concentrations of the administered compounds found in mouse eyes were quantified by a mass spectrometry method (Figure 2A, D and E).19 Most compounds were monitored for the loss of a water (−18 Da) in the LC-MS/MS-ESI fragmentation spectra. An exception was 58, in which loss of a water plus a methylene imine (−47 Da) was monitored due to a high background effect of the fragmentation at minus 18.
We first measured the total plasma concentrations of these compounds 3 h, 1 d, or 7 d after compound administration (Figure 2C). Interestingly, we observed that compounds 24 and 49, which both contain a 4-fluoro substituents, exhibited the highest initial plasma levels suggesting that this modification can deactivate elimination pathway(s) of relevance in mice. Unexpectedly, we observed that 2H2-emixustat had an initial serum concentration comparable to that of emixustat, which suggests that VAP-1 may not be a major pathway of emixustat clearance in the mouse. At 1 d after injection all compounds besides 24 were below the limits of detection. The longer plasma half-life of compound 24 is likely due to its more flexible alkyl substituents, which may be less susceptible to oxidation.
Next, we measured the distribution and retention of these compounds into the mouse eye. Emixustat efficiently distributed to ocular tissue reaching 88 ± 43 pmol/eye at 3 h after i.p. drug administration, nearly equal to 15% of the rhodopsin content in mouse eyes. 5”-gem-difluorination of the emixustat cyclohexyl (i.e. compound 57) further enhanced ocular distribution. Compound 57 reached a concentration of 176 ± 93 pmol/eye, almost twice that of emixustat at the same time after administration. Interestingly, we observed that compounds 24 and 49, which displayed the highest initial plasma concentrations, exhibited the poorest distribution within ocular tissue. These data point to a possible ocular uptake mechanism for emixustat that is disrupted by 4-fluorination. Scrutiny of the mass spectra for 24 and 59 standards revealed that their peak intensities in methanol were much weaker than (about 5–10%) the peak intensities of emixustat and 57 standards in methanol at the same concentration. In the tandem mass spectra, 24 and 49 were also resistant to losing the methylene imine fragment. The difficulty of ionization and fragmentation of 24 and 49 in mass spectrometry imply that 4-fluorination on the phenyl ring might intensify the intra-molecular N∙∙H∙∙O hydrogen bonding interaction and reduce the molecule flexibility to hamper drug delivery to the eye. In our previous studies, most drug candidates against light-induced retinal degeneration decayed to negligible levels just 24 h after a single-dose intraperitoneal injection.36 By contrast, emixustat and most of its fluorinated derivatives only moderately declined to around 50 pmol/eye during the same period, approximating 10% of the rhodopsin content. On day 7, the level of emixustat further dropped to about 4 pmol/eye, which is too low for all-trans-retinal sequestration, but still sufficient to inhibit RPE65 activity and visual chromophore recovery.19
The level of deuterated emixustat, 58, in the eye was approximately two-fold higher than that of emixustat. In light of the plasma level data presented above we cannot attribute this elevation to high circulating concentrations favoring distribution into the ocular tissue. Instead, it is possible that deuteration could positively impact ocular drug retention or affect metabolism within the eye by as-yet undefined processes. On the other hand, compound 57, in which hydroxylation was partially blocked due to the gem-difluoro substitution on C-5” position, was found at a concentration of 21 ± 5.7 pmol in mouse eyes even after 7 days of a single intraperitoneal injection (Figure 2, D and E). This amount is 5 times higher than that for emixustat, and 10 times higher than for compounds 24 and 49; and it is an effective amount for sequestration of excessive all-trans-retinal36. These results suggest that compound 57 is a promising next-generation visual cycle modulator against retinal degeneration due to its higher inhibition potency, more specific delivery, and slower decay in the eyes.
Crystal structures of RPE65 in complex with fluorinated visual cycle modulators
To gain an understanding of the factors underlying the enhanced potency of compounds 24, 49, and 57 compared to their parent molecules, we determined the crystal structures of RPE65 in complex with each of these compounds. The crystals were isomorphous to prior RPE65 structures in space group P65 and diffracted X-rays to resolutions of 1.95, 2.15, and 1.90 Å, respectively (Supplementary Table 2). The structures were refined to overall Rfree values of 21.1%, 21.7%, and 20.1% with excellent geometrical and clash score statistics (Supplementary Table 2). A 2.1 Å resolution structure of apo-RPE65 was also determined for comparison (Supplementary Table 2). Unbiased residual maps obtained after the first rounds of refinement in the absence of modeled ligands revealed a clear |Fo|-|Fc| density for the bound visual cycle modulators in the proximal active site region, including well-defined features corresponding to the fluoro substituents (Figure 4). Residual electron density was also present in the distal cavity corresponding to a palmitate molecule forming a coordinate bond with the iron prosthetic group via its carboxylate moiety, consistent with prior findings.32, 33 Electron density maps following the inclusion of the inhibitor ligand in the model showed a well-defined density for the aryl γ-hydroxypropylamine moiety in all cases. The β,β-dipropylethoxy group of 24 was similarly well defined whereas the densities for the cyclohexylmethoxy groups of 49 and 57 were comparatively much weaker (Figure 4). The relative quality of the electron density support for these different moieties is consistent with prior studies on emixustat and MB-004.32, 33 The crystal structure of apo-RPE65 featured an active site |Fo|-|Fc| density consistent with hexaethylene glycol monooctyl ether (C8E6).
Figure 4.
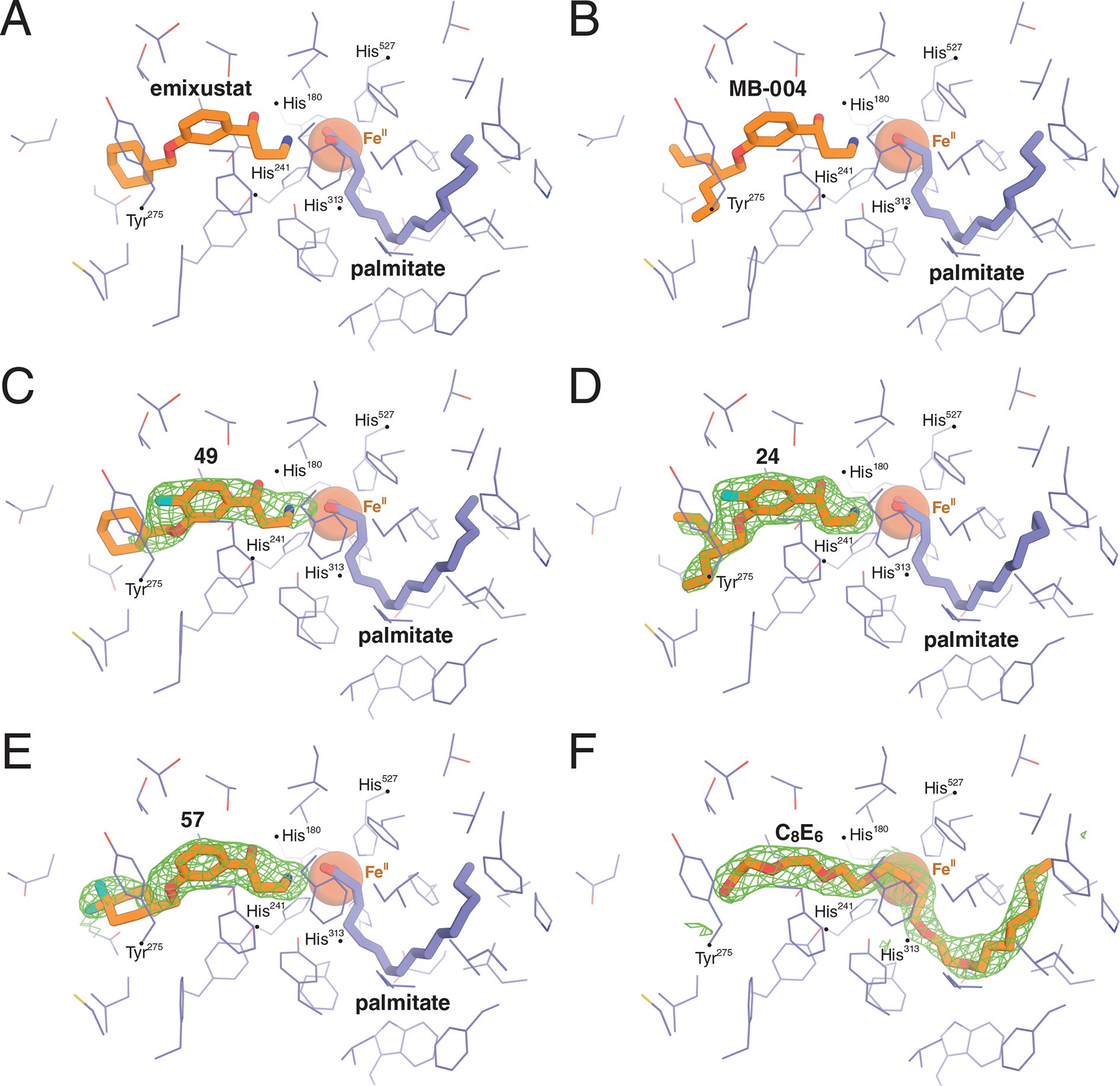
Crystal structures of bovine RPE65 in complex with A) emixustat (PDB accession code 4RSC)32 B) MB-004 (PDB accession code 5UL5,33), C) C-4-fluoro-emixustat (compound 49), D) C-4-fluoro-MB-004 (compound 24), E) C-4-difluoro-emixustat (compound 57), and F) the detergent hexaoxyethylene monooctyl ethyl (C8E6). The green mesh represents unbiased sigma-A weighted |Fo|-|Fc| electron density contoured at 3 RMSD calculated prior to modeling the ligand.
Linear detergents are known to inhibit the activity of RPE65,37, 38 and related carotenoid oxygenase enzymes.39, 40 The structure of apo-RPE65 solved with C8E6 bound to its active site provides evidence that such inhibition can be partially attributed to direct active site binding.37 Despite the high concentration of detergent in the crystallization mother liquor (>16 mM), compounds 24, 49, and 57 were able to outcompete the detergent for binding to the RPE65 active site consistent with their strong active site-binding affinities.
The binding modes for compounds 24, 49, and 57 largely overlap with those of MB-004 and emixustat with a few notable exceptions (Figure 4). The binding mode of the aryl γ-hydroxypropylamine moiety of compound 57 was similar to that of emixustat (Figure 4, A and E), whereas its terminal ring was rotated by ~90° as evidenced by clear electron density for its gem-difluoro moiety. This change in cyclohexyl ring position was likely driven by fluorine-associated steric effects. Notably, a similar ring positioning was observed previously for emixustat in a P6522 RPE65 crystal form (PDB accession code 4RYX). In this position, the gem-difluoro moiety engages in only a single van der Waals interaction with the Asn194 side-chain oxygen atom. Although the bi-phasic IC50 curve for 57 suggested two modes of RPE65 binding (Supporting Table 1), we could not discern such behavior from the structural data, although it is notable that RPE65 is more poorly ordered near the active site opening in the structure of the RPE65-compound 57 complex as compared to the structure with emixustat bound. It is thus possible that multiple conformations, one of which has a higher affinity for 57, could be responsible for the bi-phasic inhibition results. In the case of 49, the C1”-O-C3-C2 dihedral angle is rotated by ~49° compared to the corresponding angle in emixustat, which is nearly planar. The analogous dihedral angle in 24 similarly deviates from the planarity seen in MB-004 by ~31°. These differences can be attributed to the presence of the nearby 4-fluoro group giving rise to two effects. First, the presence of the fluorine causes a ~0.4 Å downward shift in the binding position of the aryl ring, which may necessitate a corresponding rotation in the C3”-O bond to avoid steric clashes with the oxy-linked alkane moieties. Second, the rotation could also be driven by electrostatic effects between the electron-dense fluoro moiety and the lone pair electrons on the O-atom of the aryl ether. In addition, the structure of the enzyme-compound 24 complex revealed a conformational difference in one of the propyl groups of 24 as compared to the complex with MB-004.
The enzyme environment around the 4-fluoro substituent of the bound modulator is likely an important factor that could help explain the greater potency of the 4-fluoro compounds 24 and 49 compared to their parent molecules. The dominant interaction occurs with Tyr275 where the fluoro group makes a close (~3–3.1 Å) en face contact with the aromatic ring. Compared to structures with the parent molecules bound, the Tyr275 side-chain is rotated by ~8° around the Cβ-Cγ bond, likely to alleviate steric clashes with the fluoro moiety or to facilitate the en face interaction. Redmond’s lab has mutagenized position Tyr275 previously.41 Only aromatic reside Phe was party active (44%), whereas other substitutions were inactive.
Quantum chemical analysis of the aromatic-fluoro interaction observed in crystals
To further elucidate the energetics of the aromatic-fluoro interaction observed in the crystal structures, we employed quantum chemistry calculations. One would expect the ~3 Å interaction to be repulsive in the first approximation as a highly electronegative F atom is brought in the close vicinity of an electron-rich π cloud. However, analogous stabilizing Cl∙∙π dispersion-driven interactions are known in protein chemistry.42 Their strength is on the order of 2 kcal/mol and are thought to be dispersion-driven forces. In contrast, F∙∙π contacts have not been investigated thoroughly. Experiments based on synthetic models provide estimates of the stabilizing interaction of roughly 1.6 kcal/mol.43 In recent work, Li and co-workers44 found that the stability of the F∙∙π interaction increases with positive charge accumulation in the π-system. Thus, the interaction seems to be mostly electrostatically-driven.
Using dimer model systems obtained from the crystal structures (Figure 5), we estimated the strength of the interaction (ΔEint) between Tyr275 and each of the two fluorinated compounds 49 and 24 to be −2.34 and −2.69 kcal/mol, respectively, using the state-of-the-art DLPNO-CCSD(T) method at the basis set limit.45, 46 The interaction energy was broken down into various contributions using the local energy decomposition (LED) scheme.47 According to Table 3, the overall electrostatic interaction has a repulsive characteristic (ΔEHF-CCSD(electro) > 0). Here, key stabilizing factors within this contribution are the electrostatic attraction between ‘prepared’ wave-functions of the fragments ΔEHF(elstat) and charge-transfer contributions ΔECCSD(CT). Both are significantly more negative for compound 24. Within the latter, 24 → Tyr double excitations have a significant contribution (−3.70 kcal/mol). Overall repulsive electrostatic effects are compensated by the dispersion interactions ΔECCSD(disp) of −4.28 and −8.23 kcal/mol for compounds 49 and 24, respectively. The magnitudes of ΔEHF(elstat) and ΔECCSD(disp) are similar, thus both electrostatic and van der Waals forces are driving the examined interactions.
Figure 5.
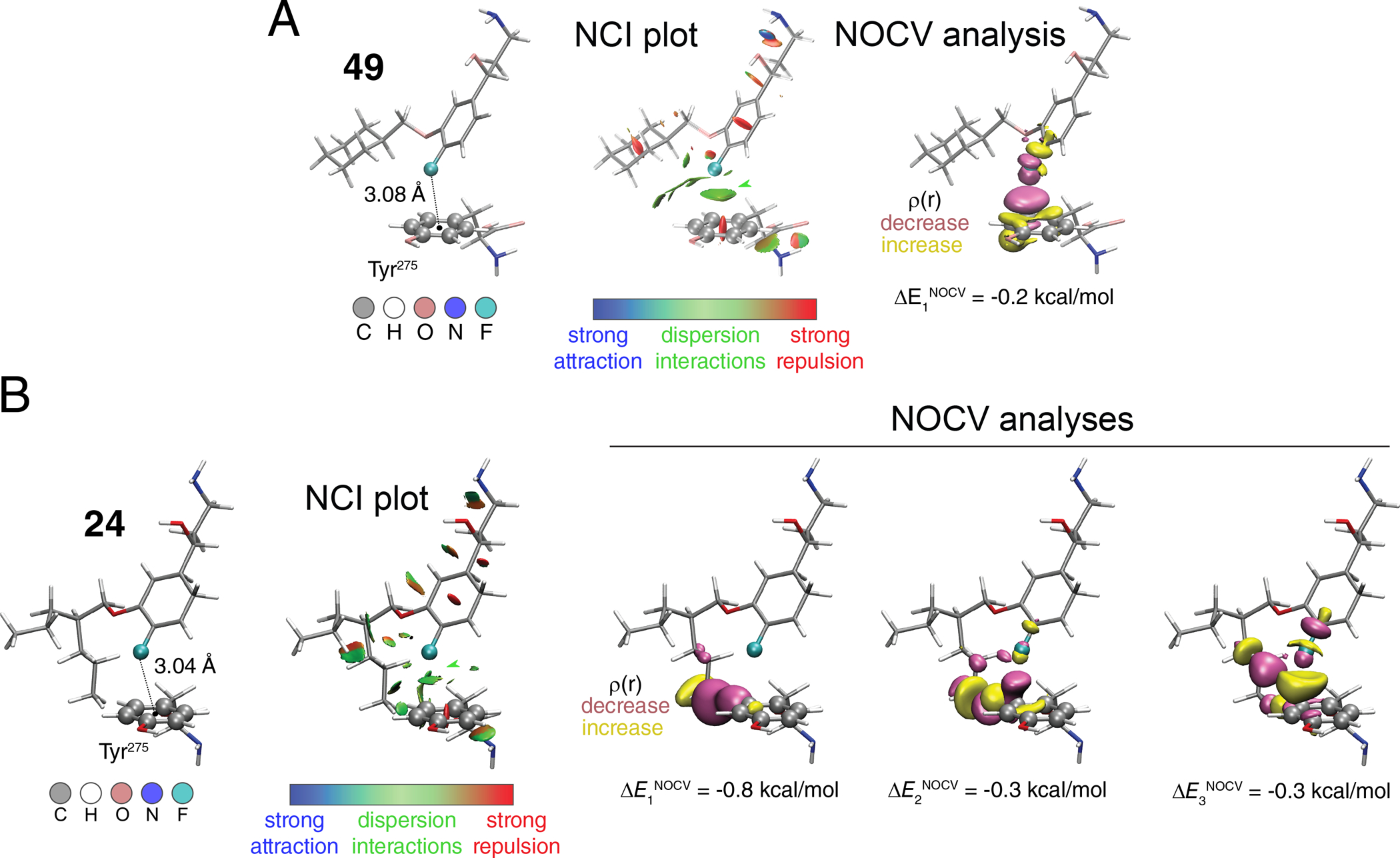
Theoretical analyses of the interactions of compound 49 (A) and compound 24 (B) with Tyr275, using a model dimer system.
Table 3.
Local energy decomposition analysis of the DLPNO-CCSD(T) total interaction energy (ΔEint) in the crystal structure-derived dimers of compounds 49 or 24 with Tyr275. All values are in kcal/mol.
| ΔE [kcal/mol] | 49 | 24 |
|---|---|---|
| ΔEHF-CCSD(electro) | 1.87 | 5.80 |
| including: | ||
| ΔEHF(el-prep) | 8.46 | 27.11 |
| ΔECCSD(el-prep) | 1.51 | 5.33 |
| ΔEHF(elstat) | −4.77 | −15.21 |
| ΔEHF(exch) | −1.58 | −5.71 |
| ΔECCSD(CT EmixF → Tyr) | −1.58 | −3.70 |
| ΔECCSD(CT Tyr → EmixF) | −0.17 | −2.02 |
| ΔECCSD(disp) | −4.28 | −8.23 |
| ΔE(T) | −0.47 | −1.10 |
| ΔE(CBS) | 0.54 | 1.01 |
| ΔEint | −2.34 | −2.69 |
To further investigate the nature of the interaction between Tyr275 and compounds 49 and 24 in 3D space we performed a non-covalent interaction (NCI) analysis (Figure 5).48 Briefly, the NCI plot shows low-density and low-gradient regions that are associated with non-covalent interactions colored according to one of the components of the density Laplacian (λ2): strong attractive interactions appear at λ2<0 (e.g. H-bonds; blue in Figure 5), while steric repulsion is associated with positive values of λ2 (red in Figure 5). Dispersion forces appear with small negative values around λ2≈0 (green in Figure 5). The examined systems’ NCI plots revealed a significant dispersion interaction region in the middle between the F atom of both 24 and 49 and the π-plane of Tyr275 (Figure 5: isovalue of s = 0.6 a.u., colored according to −0.035 < sign(λ2)ρ < 0.02).). Another way to look at intermolecular interactions is to study natural orbitals for chemical valence (NOCV)49 within the density functional theory (DFT, here we used ωB97X-D3BJ functional).50, 51 Briefly, we start by considering isolated fragments 1 and 2 at the geometry of a dimer. The two are characterized with the electron densities ρ1 and ρ2, respectively. Simple union of these densities yields promolecular density ρpro = ρ1 + ρ2 along with an associated promolecular wavefunction Ψpro. Self-consistent optimization of the latter provides Ψopt with the optimal density ρopt. We then define deformation density Δρ as the difference between promolecular density and self-consistently converged density Δρ = ρpro – ρopt. Eigenorbitals of the corresponding deformation density operator are called NOCVs and typical bonding and antibonding pairs are described with complementary NOCVs (φ±n).
With each such pair, we associate the orbital deformation density Δρorbn:
where v is a corresponding NOCV eigenvalue. By using extended transition state theory (ETS),52 one assigns a particular energy portion to such orbital interaction. By summing up all NOCVs interaction energies, one obtains the so-called orbital-interaction energy. In the case of our dimers this yields −0.7 and −2.4 kcal/mol for 49 and 24, respectively. The orbital-interaction energies should be attributed mainly to electrostatic stabilization of the electron density in the dimers and less to dispersion. The latter is added a posteriori in our calculations and it does not influence the density distribution directly – total interaction energy (including dispersion correction) is −2.3 kcal/mol for 49 and −3.4 kcal/mol for 24. In this context, up to three significant NOCVs complementary pairs yield deformation densities shown in Figure 5 (±0.005 a.u. isosurface) that account for up to 60% of the orbital interaction energies in both cases. In the case of 24, the key ingredients are the interactions between the n-propyl chain and the Tyr275 phenyl ring (Δρorb1, Δρorb2). The inspection of Δρorb3 shows that the region between the F atom and phenyl ring gains some electron density. At the same time, electron density reorganization takes place at both fragments within σ(C-F) /σ*(C-F)/n(F) and π(phenyl)/π*(phenyl) orbitals of 24 and Tyr275, respectively. The electron density reorganization for 49 compared to 24 is less pronounced and is reflected in the overall diminished interaction energy.
DISCUSSION AND CONCLUSIONS
Emixustat, a first-in-class visual cycle modulation drug candidate has displayed promising in vitro and in vivo properties for the treatment of a variety of retinal many diseases. However, emixustat suffers from sub-optimal efficacy, problematic side effects, and rapid metabolism which cloud its clinical future.17
This work investigates whether fluorination and/or deuteration of emixustat can eliminate the pharmacokinetic shortcomings of this clinical candidate. We developed novel synthetic approaches to produce specific chiral products as emixustat inhibitory activity towards RPE65 was previously shown to depend on the C1 stereochemistry.19 We present an advanced three-component Mannich reaction utilizing (S)-(−)-α-methylbenzylamine to produce the desired stereoisomer. In this reaction scheme, the resultant from subsequent reduction step γ-hydroxypropylaminobenzyl product(s) consists of a mixture of diastereomers that can be carbamylated and separated using conventional flash column chromatography on silica gel as opposed to chiral chromatography required for separation of the parent enantiomers. Facile hydrolysis of the carbamate succeeding by debenzylation regenerates the key γ-hydroxypropylamine framework with the defined stereochemistry. This method provides clear advantages in terms of cost and scalability over chiral separation methods and perhaps can extend to other similar compounds.
We investigated the impact of these emixustat and emixustat derived compounds 57, 58, and 59 on RPE65 activity in vitro and in live mice. Our first observation was that RPE65 inhibition does not correlate with accumulation in the eye. 57, 58, and 59 accumulate more in the serum of the eye than emixustat, as shown in Figure 2, possibly due to an increase in metabolic stability or selective uptake or retention by an as-yet undefined ocular component. The half-life of the gem-difluoro molecules, 57 and 59, are higher than 58 which is at least in part due to greater metabolic stability from the abrogation of known oxidation pathways of the cyclohexyl moiety, it is possible that other factors are at play. Another possible explanation of this prolonged half-life effect is that the gem-difluoro molecules could be transported to or retained within ocular tissues (for example by promoting melanin binding) more efficiently than their non-fluorinated counterparts. Practically, 57 or gem-difluoro-emixustat have better pharmacokinetic properties than emixustat in terms of elimination half-life and consequent swings in eyes levels solving some of the problems associated with the use of emixustat in humans.
VAP-1 is a lesser known phase 1 metabolic pathway53 important for primary amine oxidation of clinically used drugs including primaquine54 and tresperimus55 in addition to emixustat.15 Our results here suggest that deuteration of primary amines susceptible to VAP-1 oxidation could be a generally effective approach to prolonging their in vivo activity. The impact of alpha deuterium substitution of amines on VAP-1 metabolic susceptibility was previously studied in vitro using benzylamine and various phenylethylamines as test substrates.56 A variety of para-substituted phenylethylamines, whose amine protons are in an environment similar to those of emixustat, all displayed kinetic isotope effects on kcat of ~5–8, which is within the theoretically expected primary kinetic isotope effect (i.e. KIE = 3–7),57–60 for a reaction with a rate-limiting proton abstraction step. Our data showing impaired oxidation of deuterated emixustat as compared to emixustat is consistent with hydrogen abstraction being at least partially rate-limiting in the mechanism of emixustat oxidation by VAP-1.
The structure-activity relationships presented here are largely consistent with previous observations including higher inhibitory activity for the R- vs S-isomer of the γ-aminoalcohol (Table S1, compare 24 to 23),19 as well as compounds with a β,β-dipropylethoxy substituent as opposed to a cyclohexyl or β -ionone moiety.33 Our structural biology results support our previous findings that the β,β-dipropylethoxy group19, 33 can engage binding pockets within the RPE65 active site cavity that are not accessible to a cyclohexyl group, which likely improves the binding affinity of these compounds. We expand the known SAR of RPE65 inhibitors to include the effects of fluorine substitution throughout the base structures of emixustat, MB-001 and MB-004. We found that fluorine substitution on the aryl moiety could have either a positive or negative impact on inhibitory activity. A notable favorable impact was the 4-fluoro derivative of MB-004 (compound 24) and 4-fluoro-emixustat (49) which exhibited an IC50 value (50 nM) approximately two-fold lower than MB-004 and emixustat, respectively. Structurally, the enhanced affinity appears to result from an energetically favorable interaction between the fluoro substituent and an active site tyrosyl side chain (Tyr275). According to our calculations, the key stabilization is provided by van der Waals42 contributions as opposed to electrostatic interactions consistent with the electron rich nature of the phenolic side chain. Introduction of a gem-difluoro group at the 5” position of the cyclohexyl ring of emixustat (compound 57) also strengthened inhibitory activity although the structural basis for this effect was less clear given that the gem-difluoro group interacts minimally with the RPE65 active site pocket. The gem-difluoro substitution are both pharmacokinetically and pharmacodynamically favorable and should be considered in the future development of other emixustat derivatives to improve in vivo activity.
EXPERIMENTAL SECTION
Chemistry.
All reactions were performed in oven-dried glassware, under dry argon or nitrogen atmosphere. Synthesis under microwave irradiation was performed with Biotage, Initiator+ Robot Eight – microwave system (Charlotte, NC). Lithium aluminum deuteride (98 atom% 2H(D)) was purchased from Millipore Sigma Isotopes (Miamisburg, OH). All other reagents were used as supplied without purification. Analytical thin-layer chromatography (TLC) was performed on 0.25 mm glass-backed EMD Millipore 60 F254 plates. Visualization of the developed chromatogram was accomplished with UV light (254 nm) and stained with either ethanolic phosphomolybdic acid (PMA), ceric ammonium molybdate, or permanganate (KMnO4). Liquid chromatography was performed using a forced air-flow (flash chromatography) on silica gel (Merck, 230–400 mesh), using eluting solvents (reported as V:V ratio mixture). The 1H, 13C61/DEPT,19F NMR, and 2D-NMR spectra were recorded at 25 °C on Bruker Avance NMR spectrometers operating at 300, 400, 500, 600, and 700 MHz for the 1H channel and were in accordance with the assigned structures. 19F NMR spectra were recorded without decoupling from protons. Chemical shifts reported in δ units, part per million (ppm) with reference to the residual solvent peak CDCl3 (δ 7.26), CD3OD (δ 3.31), C6D6 (δ 7.16) for 1H and (δ 77.36), CD3OD (δ 49.00), C6D6 (δ 128.06) for 13C spectra, respectively or TMS (δ 0.00). NMR data are presented in the following order: chemical shift, peak multiplicity (b = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, ddd = doublet of doublet of doublet, ddt = doublet of doublet of triplet, dq = doublet of quartet, dm = doublet of multiplet, br = broad), coupling constant (in Hz). The specific light rotations were measured with a JASCO digital polarimeter (Model P-1010, λ = 589 nm, ±0.05° accuracy) using a cylindrical quartz cell (5 mL, l = 5 cm) at 25 °C. Mass spectra were recorded in positive ionization mode on an Agilent 6545 QTOF mass spectrometer (Agilent technologies, USA), equipped with an electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI) interfaces, coupled to an Agilent 1260 Ultra High-Pressure Liquid Chromatography UHPLC (Agilent Technologies, Santa Clara, CA, USA). The Agilent 1260 series system consists of a G4204A quaternary pump, G4226A ALS auto-sampler, and G1316C column compartment. UHPLC was carried out on ZORBAX RRHD Eclipse Plus C18, 95 Å, 2.1 × 50 mm, 1.8 μm column (Agilent Technologies, USA) column with H2O (0.1% formic acid)-acetonitrile gradient elution from 5% to 95% acetonitrile in the course of 10 min at a flow rate of 0.5 mL/min. Preparative and analytical HPLC (Young Lin Instruments, Anyang, Korea) were performed on LUNA C18(2) (10 μm, 250 mm × 21.2 mm) column and chiral column, LUX Amylose-1 (5 μm, 250 mm × 21.2 mm), for preparative purification, and (5 μm, 250×4.6 mm) column for analysis, all from Phenomenex, Inc. (Torrance, CA). Acetonitrile and double distilled water were used as an eluent in different ratios. The final biological tested compounds displayed ≥95% purity (confirmed using analytical HPLC). Compound 57 was obtained of 90 % purity. Synthesis of the final emixustat analogs is detailed herein while synthesis of all the precursors is described in the Supporting Information.
(S)-3-Amino-1-(4-fluoro-3-((2-propylpentyl)oxy)phenyl)propan-1-ol (23):
General Procedure:
The N-protected aminoalcohol 15 (280 mg, 0.70 mmol) was suspended with 10% Pd/C (100 mg) in dry MeOH (4 mL) under a nitrogen atmosphere. After 30 min, solid ammonium formate (1.29 mmol) was added. The resulting mixture was refluxed for 30 min - overnight (TLC monitoring), cooled to 0 °C, filtered through a celite pad and washed with chloroform. The filtrate was concentrated under reduced pressure to afford the corresponding title γ-aminoalcohol 23 (188 mg, 91%) as a colorless oil. [α]D25 = −21.32° (c = 0.0003, CH2Cl2). 1H NMR (300 MHz, CDCl3): δ 7.23 – 6.85 (m, 2H), 6.80 (ddd, J = 2.0, 4.0, 8.0 Hz, 1H), 4.82 (dd, J = 3.0, 8.5 Hz, 1H), 3.89 (d, J = 5.5 Hz, 2H), 3.64 (bs, 2H), 3.37 – 2.96 (m, 1H), 2.96 – 2.59 (m, 1H), 2.07 – 1.57 (m, 3H), 1.56 – 1.19 (m, 8H), 0.91 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, CDCl3): δ 151.7 (d, J = 244.5 Hz), 147.2 (d, J = 10.5 Hz), 141.4 (d, J = 3.5 Hz), 117.5 (d, J = 7.0 Hz), 115.5 (d, J = 18.5 Hz), 112.2, 74.3, 72.3, 40.1, 39.8, 37.6, 33.6, 19.9, 14.4. 19F NMR (376 MHz, CDCl3): δ ‒137.5 (m). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2172.
(R)-3-Amino-1-(4-fluoro-3-((2-propylpentyl)oxy)phenyl)propan-1-ol (24):
According to general procedure, N-benzylated compound 16 (458 mg, 1.14 mmol) was deprotected to give the title γ-aminoalcohol 24 (314.2 mg, 93%) as a colorless oil. [α]D25 = +21.56° (c = 0.0002, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 7.06 – 6.94 (m, 2H), 6.90 −6.70 (m, 1H), 4.82 (dd, J = 3.0, 8.5 Hz, 1H), 3.89 (d, J = 5.5 Hz, 2H), 3.5 (bs, 2H), 3.20 – 3.02 (m, 1H), 3.00 – 2.90 (m, 1H), 1.89 – 1.80 (m, 2H), 1.78 – 1.68 (m, 1H), 1.49 – 1.20 (m, 8H), 0.91 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 151.8 (d, J = 244.5 Hz), 147.3 (d, J = 10.5 Hz), 141.2 (d, J = 3.5 Hz), 117.6 (d, J = 7.0 Hz), 115.6 (d, J = 18.5 Hz), 112.3, 74.7, 72.4, 40.2, 39.4, 37.6, 33.6, 19.9, 14.4. 19F NMR (376 MHz, CDCl3): δ ‒137.5 (m). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2173.
(R/S)-3-Amino-1-(3-fluoro-5-((2-propylpentyl)oxy)phenyl)propan-1-ol (25):
According to general procedure, N-benzylated compound 17 (236 mg, 0.59 mmol) was deprotected giving the chiral γ-aminoalcohol 25 (170 mg, 97%) as a colorless oil. 1H NMR (600 MHz, C6D6): δ 7.10 – 7.02 (m, 1H), 6.99 – 6.85 (m, 1H), 6.68 – 6.54 (m, 1H), 5.14 – 4.61 (m, 1H), 3.56 (d, J = 5.5 Hz, 2H), 2.50 – 2.35 (m, 1H), 2.34 – 2.14 (m, 1H), 1.70 – 1.62 (m, 1H), 1.49 – 1.19 (m, 10H), 0.87 (t, J = 7.0 Hz, 6H). 13C NMR (150 MHz, C6D6): δ 164.3 (d, J = 244.0 Hz), 161.2 (d, J = 11.0 Hz), 150.0 (d, J = 8.5 Hz), 108.3 (d, J = 2.5 Hz), 105.0 (d, J = 22.0 Hz), 100.3 (d, J = 25.0 Hz), 75.1, 70.9, 40.7, 39.7, 37.8, 34.0, 20.3, 14.6. 19F NMR (376 MHz, C6D6): δ ‒112.89 (t, J = 10.0 Hz). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2179.
(R/S)-3-Amino-1-(3-fluoro-5-((2-propylpentyl)oxy)phenyl)propan-1-ol (26):
Deprotection of N-benzylated compound 18 (46 mg, 0.11 mmol) according to general procedure afforded the chiral γ-aminoalcohol 26 (14.2 mg, 42%) as a colorless oil. 1H NMR (400 MHz, CDCl3 + CD3OD): δ 6.72 – 6.67 (m, 1H), 6.67 – 6.59 (m, 1H), 6.54 – 6.43 (m, 1H), 4.76 (dd, J = 4.0, 8.0 Hz, 1H), 3.80 (d, J = 5.5 Hz, 2H) 3.77 (bs, 2H), 3.07 – 2.70 (m, 2H), 1.86 – 1.73 (m, 3H), 1.50 – 1.24 (m, 8H), 0.91 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, CDCl3 + CD3OD): δ 162.7 (d, J = 244.0 Hz), 160.7 (d, J = 11.0 Hz), 148.25 (d, J = 8.5 Hz), 107.7 (d, J = 2.5 Hz), 104.5 (d, J = 22.0 Hz), 100.6 (d, J = 25.0 Hz), 73.1, 71.3, 40.0, 39.2, 37.5, 33.7, 20.0, 14.4. 19F (376 MHz, CDCl3 + CD3OD): δ ‒112.74 (t, J = 10.0 Hz). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2186.
(R/S)-3-Amino-1-(2-fluoro-5-((2-propylpentyl)oxy)phenyl)propan-1-ol (27):
According to general procedure, N-benzylated compound 20 (30 mg, 0.07 mmol) was deprotected giving the chiral γ-aminoalcohol 27 (13.4 mg, 61%) as a colorless oil. 1H NMR (400 MHz, C6D6): δ 7.69 (ddd, J =1.0, 3.0, 6.0 Hz, 1H), 6.83 (t, J = 9.5 Hz, 1H), 6.70 – 6.58 (m, 1H), 5.42 (dd, J = 1.0, 8.5 Hz, 1H), 3.70 (d, J = 5.5 Hz, 2H), 2.51 – 2.40 (m, 1H), 2.37 – 2.29 (m, 1H), 1.84 – 1.76 (m, 1H), 1.75 – 1.65 (m, 1H), 1.54 −1.45 (m, 1H), 1.44 – 1.21 (m, 8H), 0.87 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, C6D6): δ 156.4, 154.2 (d, J = 236.0 Hz), 134.3 (d, J = 14.5 Hz), 115.6 (d, J = 23.5 Hz), 114.0 (d, J = 8.0 Hz), 113.4 (d, J = 4.5 Hz), 71.3, 70.1, 40.0, 38.6, 38.0, 34.1, 20.3, 14.6. 19F NMR (376 MHz, C6D6): δ ‒131.25 (m). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2178.
(R/S)-3-Amino-1-(2-fluoro-5-((2-propylpentyl)oxy)phenyl)propan-1-ol (28):
Following the general procedure, N-benzylated compound 19 (33 mg, 0.08 mmol) was deprotected giving the chiral γ-aminoalcohol 28 (15.2 mg, 61%) as a colorless oil. 1H NMR (700 MHz, C6D6): δ 7.71 (dd, J = 3.0, 6.0 Hz, 1H), 6.83 (t, J = 9.5 Hz, 1H), 6.71 – 6.56 (m, 1H), 5.43 (dd, J = 1.0, 8.5 Hz, 1H), 3.70 (d, J = 5.5 Hz, 2H), 2.52 – 2.37 (m, 1H), 2.37 – 2.29 (m, 1H), 1.85 – 1.75 (m, 1H), 1.75 – 1.63 (m, 1H), 1.51 – 1.43 (m, 1H), 1.43 – 1.20 (m, 8H), 0.86 (t, J = 7.0 Hz, 6H). 13C NMR (176 MHz, C6D6): δ 156.3, 154.2 (d, J = 236.0 Hz), 134.3 (d, J = 14.5 Hz), 115.6 (d, J = 23.5 Hz), 114.0 (d, J = 8.0 Hz), 113.3 (d, J = 4.5 Hz), 71.3, 70.2, 41.0, 38.56, 38.01, 34.08, 20.35, 14.66. 19F NMR (376 MHz, C6D6): δ ‒131.25 (m). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2177.
(R/S)-3-Amino-1-(2-fluoro-3-((2-propylpentyl)oxy)phenyl)propan-1-ol (29):
According to general procedure, N-benzylated compound 22 (36 mg, 0.09 mmol) was deprotected to give the chiral γ-aminoalcohol 29 (14.1 mg, 52%) as a colorless oil. 1H NMR (700 MHz, C6D6): δ 7.60 (t, J = 8.0 Hz, 1H), 7.07 (t, J = 8.0 Hz, 1H), 6.68 (dd, J = 1.0, 8.0 Hz, 1H), 5.44 (dd, J = 2.0, 8.5 Hz, 1H), 3.68 (dd, J = 2.0, 5.5 Hz, 2H), 2.45 – 2.39 (m, 1H), 2.33 – 2.29 (m, 1H), 1.80 – 1.70 (m, 2H), 1.55 – 1.47 (m, 1H) 1.46 – 1.23 (m, 8H), 0.88 (t, J = 7.0 Hz, 6H). 13C NMR (176 MHz, C6D6): δ 150.0 (d, J = 244.5 Hz), 147.6 (d, J = 11.0 Hz), 134.6 (d, J = 10.5 Hz), 123.9 (d, J = 4.0 Hz), 119.3 (d, J = 3.5 Hz), 113.2, 72.2, 69.8 (d, J = 2.5 Hz), 40.8, 38.5, 38.0, 34.0, 20.3, 14.6. 19F NMR (376 MHz, C6D6): δ ‒142.05 (t, J = 7.0 Hz). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2187.
(R/S)-3-Amino-1-(2-fluoro-3-((2-propylpentyl)oxy)phenyl)propan-1-ol (30):
Following the general procedure, N-benzylated compound 21 (35 mg, 0.09 mmol) was deprotected giving the chiral γ-aminoalcohol 30 (13.6 mg, 52%) as a colorless oil. 1H NMR (400 MHz, C6D6): δ 7.60 (t, J = 1.0, 8.0 Hz, 1H), 7.01 (t, J = 8.0 Hz, 1H), 6.69 (t, J = 8.0 Hz, 1H), 5.44 (dd, J = 2.0, 8.5 Hz, 1H), 3.68 (d, J = 5.5 Hz, 2H), 2.40 – 2.38 (m, 1H), 2.34 – 2.27 (m, 1H), 1.82 – 1.69 (m, 2H), 1.54 – 1.48 (m, 1H) 1.46 – 1.21 (m, 8H), 0.88 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, C6D6): δ 150.12 (d, J = 244.5 Hz), 147.6 (d, J = 11.0 Hz), 134.6 (d, J = 10.5 Hz), 123.9 (d, J = 4.0 Hz), 119.3 (d, J = 3.5 Hz), 113.2, 72.2, 69.8 (d, J = 2.5 Hz), 40.8, 38.6, 38.0, 34.0, 20.3, 14.6. 19F (376 MHz, C6D6): δ ‒142.11 (t, J = 7.0 Hz). HRMS (ESI): calcd for C17H28FNO2 [M + H]+, 298.2176; found, 298.2178.
3-Amino-1-(3-(cyclohexylmethoxy)-4-fluorophenyl)propan-1-ol (49):
General Procedure:
To a solution of compound 40 (8 g, 28.8 mmol, 1 eq) in dry THF (55 mL) was added lithium aluminum hydride (2.46 g, 64.9 mmol, 2.25 eq) in portions under nitrogen at 0 °C, and then the reaction mixture was stirred at 0 °C for another 0.5 h. The reaction was quenched with adding water (2.46 mL), sodium hydroxide solution (10%, 2.46 mL) and water (7.38 mL) sequentially. The resulting mixture was filtered and concentrated under vacuum. The residue was purified by silica gel chromatography with dichloromethane/ methanol (10/1) as eluent to give 49 (1.5 g, 18%) as a yellow gum. 1H NMR (600 MHz, CDCl3): δ 7.08 – 6.88 (m, 2H), 6.85 – 6.72 (m, 1H), 4.82 (d, J = 6.9 Hz, 1H), 4.50 (bs, 3H), 3.77 (d, J = 6.4 Hz, 2H), 3.18 – 3.00 (m, 1H), 3.00 – 2.83 (m, 1H), 1.94 – 1.82 (m, 3H), 1.82 – 1.76 (m, 2H), 1.76 – 1.70 (m, 2H), 1.70 – 1.65 (m, 1H), 1.33 – 1.22 (m, 2H), 1.22 – 1.13 (m, 1H), 1.07 – 0.94 (ddd, J = 2.4, 12.2, 24.1 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 151.8 (d, J = 244.8 Hz), 147.3 (d, J = 10.6 Hz), 141.1 (d, J = 3.1 Hz), 117.6 (d, J = 6.8 Hz), 115.7 (d, J = 18.4 Hz), 112.3 (d, J = 1.0 Hz), 74.9, 74.0, 39.6, 38.6, 37.8, 29.9, 26.5, 25.8. 19F (565 MHz, CDCl3): δ ‒136.8 – ‒136.95 (m). HRMS (ESI): calcd for C16H24FNO2 [M + H]+, 282.1869; found, 282.1862.
3-Amino-1-(3-(cyclohexylmethoxy)-5-fluorophenyl)propan-1-ol (50):
General procedure followed with nitrile 41 (5 g, 35.6 mmol, 1.1 eq) yielding a yellow gum (1.60 g, 51%). 1H NMR (600 MHz, CDCl3): δ 6.73 – 6.68 (m, 1H), 6.65 (d, J = 9.3 Hz, 1H), 6.46 (dt, J = 2.2, 10.7 Hz, 1H), 4.87 (dd, J = 2.6, 8.4 Hz, 1H), 3.71 (d, J = 6.4 Hz, 2H), 3.29 (bs, 3H), 3.10 – 3.03 (m, 1H), 3.00 −2.89 (m, 1H), 1.88 – 1.80 (m, 3H), 1.79 – 1.73 (m, 3H), 1.73 – 1.64 (m, 2H), 1.32 – 1.24 (m, 2H), 1.23 – 1.15 (m, 1H), 1.03 (ddd, J = 3.2, 12.3, 24.3 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 163.6 (d, J = 244.3 Hz), 160.6 (d, J = 11.2 Hz), 148.47 (d, J = 8.6 Hz), 117.6 (d, J = 1.9 Hz), 104.6 (d, J = 22.2 Hz), 100.6 (d, J = 25.0 Hz), 74.8, 73.8, 40.3, 39.2, 37.7, 29.9, 26.6, 25.8. 19F (565 MHz, CDCl3): δ ‒112.20 (t, J = 10.1 Hz). HRMS (ESI): calcd for C16H24FNO2 [M + H]+, 282.1869; found, 282.1874.
3-Amino-1-(5-(cyclohexylmethoxy)-2-fluorophenyl)propan-1-ol (51):
General procedure followed with compound 42 (3.5 g, 12.6 mmol, 1 eq) giving a yellow gum (800 mg, 22%) 1H NMR (600 MHz, CDCl3): δ 7.11 (dd, J = 3.1, 6.0 Hz, 1H), 6.87 (dd, J = 9.0, 9.7 Hz, 1H), 6.69 (dt, J = 3.6, 9.0 Hz, 1H), 5.20 (dd, J = 3.0, 8.4 Hz, 1H), 3.75 – 3.69 (m, 2H), 3.47 – 2.98 (m, 4H), 2.98 – 2.94 (m, 1H), 1.94 – 1.88 (m, 1H), 1.87 – 1.82 (m, 2H), 1.80 – 1.72 (m, 4H), 1.70 – 1.66 (m, 1H), 1.32 – 1.24 (m, 2H), 1.23 – 1.15 (m, 1H), 1.03 (ddd, J = 3.3, 12.3, 24.2 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 155.7 (d, J = 1.7 Hz), 153.6 (d, J = 237.4 Hz), 132.8 (d, J = 14.9 Hz), 115.5 (d, J = 23.5 Hz), 113.9 (d, J = 8.0 Hz), 112.7 (d, J = 4.5 Hz), 74.2, 69.5 (d, J = 1.6 Hz), 40.5, 38.1, 37.9, 30.0, 30.0, 26.6, 25.9. 19F (565 MHz, CDCl3): δ ‒130.78 – ‒130.88 (m). HRMS (ESI): calcd for C16H24FNO2 [M + H]+, 282.1869; found, 282.1864.
3-Amino-1-(3-(cyclohexylmethoxy)-2-fluorophenyl)propan-1-ol (52):
The general procedure was followed using nitrile 43 (3 g, 10.8 mmol, 1 eq) resulting in isolation of a yellow gum (2.1 g, 66%). 1H NMR (600 MHz, CDCl3): δ 7.1 (t, J = 6.7 Hz, 1H), 7 (t, J = 8.0 Hz, 1H), 6.82 (t, J = 8.0 Hz, 1H), 5.25 (d, J = 8.3 Hz, 1H), 4.25 (bs, 3H), 3.77 (d, J = 6.3 Hz, 2H), 3.15 – 3.06 (m, 1H), 3.06 – 2.96 (m, 1H), 2.00 – 1.95 (m, 1H), 1.89 – 1.84 (m, 3H), 1.83 – 1.79 (m, 1H), 1.76 – 1.72 (m, 2H), 1.7 – 1.66 (m, 1H), 1.32 – 1.24 (m, 2H), 1.22 – 1.14 (m, 1H), 1.07 – 1.00 (ddd, J = 3.3, 12.4, 24.2 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 149.4 (d, J = 244.7 Hz), 147.1 (d, J = 10.7 Hz), 132.6 (d, J = 10.7 Hz), 123.8 (d, J = 4.3 Hz), 118.4 (d, J = 3.2 Hz), 113.4, 74.9, 68.7, 39.8, 37.7, 37.1, 29.1, 26.6, 25.8. 19F (565 MHz, CDCl3): δ ‒141.76 – ‒141.84 (m). HRMS (ESI): calcd for C16H24FNO2 [M + H]+, 282.1869; found, 282.1869.
3-Amino-1-(4-fluoro-3-((2,6,6-trimethylcyclohex-1-en-1-yl)methoxy)phenyl)propan-1-ol (53):
Steps were carried out according to the general procedure using compound 44 (6 g, 18.9 mmol, 1 eq) to give title compound 53 (1.40 g, 22%) as a yellow gum. 1H NMR (600 MHz, CDCl3): δ 7.12 (dd, J = 1.54, 8.0 Hz, 1H), 6.99 (dd, J = 8.3, 11.0 Hz, 1H), 6.87 – 6.81 (m, 1H), 4.90 (dd, J = 2.2, 8.8 Hz, 1H), 4.50 (s, 2H), 3.62 (bs, 3H), 3.15 – 3.06 (m, 1H), 3.01 – 2.91 (m, 1H), 2.03 (t, J = 6.2 Hz, 2H), 1.89 – 1.82 (m, 1H), 1.78 – 1.74 (m, 1H), 1.72 (s, 3H), 1.66 – 1.61 (m, 2H), 1.53 – 1.45 (m, 2H), 1.05 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 152.1 (d, J = 244.6 Hz), 147.3 (d, J = 11.1 Hz), 141.4 (d, J = 3.3 Hz), 136.2, 132.9, 118.07 (d, J = 6.7 Hz), 115.7 (d, J = 18.6 Hz), 113.2 (d, J = 1.9 Hz), 74.9, 66.4, 40.4, 39.4, 39.3, 34.1, 33.0, 28.5, 19.9, 19.3. 19F (565 MHz, CDCl3): δ ‒135.72 – ‒135.82 (m). HRMS (ESI): calcd for C19H28FNO2 [M + H]+, 322.2182; found, 322.2176.
3-Amino-1-(3-fluoro-5-((2,6,6-trimethylcyclohex-1-en-1-yl)methoxy)phenyl)propan-1-ol (54):
Utilizing the general procedure with compound 45 (6 g, 18.9 mmol, 1 eq) led to successful isolation of a yellow gum (1.50 g, 18%). 1H NMR (600 MHz, CDCl3): δ 6.82 – 6.72 (m, 1H), 6.70 – 6.62 (m, 1H), 6.57 – 6.50 (m, 1H), 4.88 (dd, J = 2.5, 8.4 Hz, 1H), 4.59 (bs, 3H), 4.39 (s, 2H), 3.18 – 3.06 (m, 1H), 3.06 – 2.84 (m, 1H), 2.07 – 2.00 (m, 2H), 1.97 – 1.86 (m, 1H), 1.83 – 1.75 (m, 1H), 1.67 (s, 3H), 1.66 – 1.56 (m, 2H), 1.53 – 1.42 (m, 2H), 1.02 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 163.6 (d, J = 242.9 Hz), 160.7 (d, J = 10.6 Hz), 148.0 (d, J = 8.4 Hz), 136.1, 132.8, 107.8 (d, J = 1.4 Hz), 104.7 (d, J = 22.7 Hz), 100.9 (d, J = 24.8 Hz), 74.1, 64.9, 39.6, 39.3, 38.2, 34.1, 32.9, 28.5, 19.9, 19.3. 19F (565 MHz, CDCl3): δ −111.89 (t, J = 9.9 Hz). HRMS (ESI): calcd for C19H28FNO2 [M + H]+, 322.2182; found, 322.2173.
3-Amino-1-(2-fluoro-5-((2,6,6-trimethylcyclohex-1-en-1-yl)methoxy)phenyl)propan-1-ol (55):
The general procedure was carried out with 46 (3.0 g, 9.45 mmol, 1 eq) giving amine 55 (1.1 g, 34%) as a yellow gum. 1H NMR (600 MHz, CDCl3): δ 7.18 (dd, J = 3.0, 5.8 Hz, 1H), 6.88 (t, J = 9.4 Hz, 1H), 6.79 – 6.75 (m, 1H), 5.22 (dd, J = 2.0, 8.3 Hz, 1H), 4.42 (d, J = 9.7 Hz, 1H), 4.38 (d, J = 9.7 Hz, 1H), 4.14 (bs, 3H), 3.16 – 3.06 (m, 1H), 3.06 – 2.95 (m, 1H), 2.03 (t, J = 5.9 Hz, 2H), 1.99 – 1.93 (m, 1H), 1.82 −1.76 (m, 1H), 1.69 (s, 3H), 1.66 – 1.61 (m, 2H), 1.53 – 1.45 (m, 2H), 1.03 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 155.8 (d, J = 1.4 Hz), 153.7 (d, J = 237.4 Hz), 135.7, 133.2, 132.6 (d, J = 14.8 Hz), 115.5 (d, J = 23.7 Hz), 114.4 (d, J = 7.9 Hz), 112.8 (d, J = 4.4 Hz), 69.1, 65.2, 40.1, 39.3, 37.4, 34.2, 32.9, 28.5, 28.5, 19.9, 19.4. 19F (565 MHz, CDCl3): δ −130.41 – −130.48 (m). HRMS (ESI): calcd for C19H28FNO2 [M + H]+, 322.2182; found, 322.2179.
3-Amino-1-(2-fluoro-3-((2,6,6-trimethylcyclohex-1-en-1-yl)methoxy)phenyl)propan-1-ol (56):
The actions of the general procedure were carried out with nitrile 47 (6.5 g, 20.5 mmol, 1 eq) to give a brown gum (1.06 g, 16%). 1H NMR (600 MHz, CDCl3): δ 7.14 (t, J = 6.8 Hz, 1H), 7.04 (t, J = 8.0 Hz, 1H), 6.94 (t, J = 8.0 Hz, 1H), 5.24 (d, J = 7.2 Hz, 1H), 4.48 (d, J = 9.8 Hz, 1H), 4.45 (d, J = 9.8 Hz, 1H), 4.24 (bs, 3H), 3.12 – 3.03 (m, 1H), 3.03 – 2.92 (m, 1H), 2.03 (t, J = 6.0 Hz, 2H), 1.99 – 1.92 (m, 1H), 1.86 – 1.78 (m, 1H), 1.72 (s, 3H), 1.66 – 1.60 (m, 2H), 1.52 – 1.46 (m, 2H), 1.05 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 149.8 (d, J = 244.3 Hz), 147.1 (d, J = 11.2 Hz), 136.1, 133.0, 132.9 (d, J = 11.1 Hz), 123.8 (d, J = 4.2 Hz), 118.9 (d, J = 3.2 Hz), 114.5, 68.7, 66.5, 39.9, 39.3, 37.4, 34.1, 33.0, 28.5, 19.9, 19.3. 19F (565 MHz, CDCl3): δ ‒140.35 – ‒140.46 (m). HRMS (ESI): calcd for C19H28FNO2 [M + H]+, 322.2182; found, 322.2173.
3-Amino-1-(3-((4,4-difluorocyclohexyl)methoxy)phenyl) propan-1-ol (57):
Using compound 48 (3.0 g, 10.1 mmol, 1 eq) and following the general procedure gave amine 57 (580 mg, 17% yield) as a yellow gum. 1H NMR (600 MHz, CDCl3): δ 7.19 (t, J = 7.9 Hz, 1H), 6.92 – 6.89 (m, 1H), 6.89 – 6.86 (m, 1H), 6.74 (dd, J = 2.0, 8.2 Hz, 1H), 5.80 (bs, 4H), 4.84 (dd, J = 2.6, 9.0 Hz, 1H), 3.76 (d, J = 6.3 Hz, 2H), 3.12 – 3.04 (m, 1H), 3.01 – 2.94 (m, 1H), 2.15 – 2.07 (m, 2H), 1.97 – 1.86 (m, 4H), 1.84 – 1.81 (m, 1H), 1.81 – 1.64 (m, 2H), 1.43 – 1.35 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 159.2, 146.3, 129.5, 123.6 (t, J = 240.0 Hz), 118.0, 113.2, 111.8, 73.2, 71.8, 38.8, 37.4, 36.0, 33.2 (dd, J = 23.2, 24.7 Hz), 25.9, 25.8. 19F (565 MHz, CDCl3): δ ‒91.39 (d, J = 235.8 Hz), ‒101.99 (d, J = 235.8 Hz). HRMS (ESI): calcd for C16H23F2NO2 [M + H]+, 300.1775; found, 300.1761.
3-Amino-1-(3-(cyclohexylmethoxy)phenyl)propan-3,3-d2-1-ol (58):
3-(3-(cyclohexylmethoxy) phenyl)-3-hydroxypropanenitrile19 (200 mg, 0.77 mmol) was reduced with lithium aluminium deuteride (LAD) (100 mg, 2.38 mmol). Following the common workup procedure, the crude product was purified by flash chromatography (silica, 70:30:0 – 90:10:4 – 70:30:4 DCM/MeOH/NH4OH) giving a yellow oil 58 (69 mg, 34%). 1H NMR (500 MHz, CDCl3) δ 7.23 (t, J = 7.9 Hz, 1H), 6.96 (s, 1H), 6.91 (d, J = 7.6 Hz, 1H), 6.77 (dd, J = 8.4, 2.6 Hz, 1H), 4.94 (dd, J = 8.6, 3.2 Hz, 1H), 3.76 (d, J = 6.4 Hz, 2H), 2.45 (s, 3H), 1.91 – 1.82 (m, 3H), 1.79 – 1.65 (m, 5H), 1.35 – 1.14 (m, 3H), 1.05 (qd, J = 12.2, 3.4 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 159.6, 146.9, 129.3, 117.8, 113.2, 111.8, 75.6, 73.5, 39.5, 37.9, 30.1, 26.6, 25.9. HRMS (ESI): m/z calcd for C16H24D2NO2 [M+H]+ 266.2084; found 266.2083.
3-Amino-1-(3-((4,4-difluorocyclohexyl)methoxy)phenyl)propan-3,3-d2-1-ol (59):
The general procedure for reduction of nitrile was followed using nitrile 48 (200 mg, 0.77 mmol) and lithium aluminium deuteride (LAD) (100 mg, 2.38 mmol). Following purification by flash chromatography (silica, 90:10:0 – 70:30:0 – 90:10:4 – 70:30:4 DCM/MeOH/NH4OH) gave a colorless syrup 59 (140 mg, 45%). 1H NMR (500 MHz, CDCl3) δ 7.23 (d, J = 7.8 Hz, 1H), 6.97 (s, 1H), 6.93 (d, J = 7.6 Hz, 1H), 6.77 (dd, J = 8.1, 2.5 Hz, 1H), 4.95 (dd, J = 8.7, 3.0 Hz, 1H), 3.83 (dd, J = 6.5, 2.1 Hz, 2H), 2.18 – 2.10 (m, 3H), 2.00 – 1.93 (m, 4H), 1.92 – 1.84 (m, 4H), 1.81 – 1.66 (m, 5H), 1.47 – 1.38 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 159.2, 147.1, 129.4, 118.2, 113.2, 111.7, 75.6, 71.8 (d, J = 2.8 Hz), 39.5, 36.1, 33.2 (dd, J = 25.5, 22.8 Hz), 25.9 (d, J = 9.7 Hz). 19F NMR (471 MHz, CDCl3) δ −91.40 (d, J = 236.4 Hz), −102.00 (d, J = 236.2 Hz). HRMS (ESI): m/z calcd for C16H21D2F2NO2 [M]+ 301.1820; found 301.1822.
RPE65 crystallization and structure determination.
Crystals of RPE65 in complex with 4-fluoro-emixustat (49), C-2’-fluoro-MB-004 (24), or C-4 gem-difluoro-emixustat (57) were obtained using previously described procedures.32, 33 Briefly, isolated bovine RPE membranes were incubated with 1 mM of each compound (delivered in DMF) for 15 min prior to solubilization with 24 mM hexaethylene glycol monooctyl ether (C8E6). After anion exchange chromatography, purified RPE65 was concentrated to 10–15 mg/mL and the test compounds were again to a concentration of 1 mM prior to crystallization. An RPE65 sample was also prepared in the absence of added inhibitors. Crystals were grown by the hanging-drop vapor-diffusion method by mixing 2 μL of a 10 mg/mL RPE65 sample with 2 μL of one of the following crystallization solutions: 100 mM 2-(cyclohexylamino)ethanesulfonic acid–NaOH, pH 9.5, containing 40% (v/v) polyethylene glycol 300 and 200 mM NaCl, which was used for the samples containing 49 or no added inhibitor, or 100 mM Tris-HCl, pH 8.5, containing 30% (v/v) polyethylene glycol 200 and 200 mM ammonium phosphate dibasic, which was used for the samples containing 24 or 57. In both cases, the drops were incubated over a well solution consisting of 100 mM 2-(cyclohexylamino)ethanesulfonic acid–NaOH, pH 9.5, containing 40% (v/v) polyethylene glycol 300 and 200 mM NaCl at 8 °C. Crystals of approximately 100 × 100 × 300 μm in size were obtained after 1–2 weeks of incubation. Mature crystals were harvested directly into liquid nitrogen for X-ray data collection.
X-ray diffraction data were collected at the SSRL 12–2, the APS NE-CAT 24-ID-E, or the NSLS-II FMX beamLines. Data were processed using XDS62and the initial model was obtained by direct refinement using published RPE65 coordinates in which ligands had been removed32 (PDB accession codes: 4RSE and 4RSC). The structures were refined by alternating reciprocal space refinement in REFMAC63and manual building and adjustments in Coot.64 Ligand coordinates and geometry dictionary files were generated using the Grade server (http://grade.globalphasing.org/cgi-bin/grade/server.cgi). The models were validated using Molprobity65 and the wwPDB validation server.66
RPE65 retinoid isomerase activity assay.
Primary amine listed in Table 1, Table 2, and Supplementary Table 1 in DMF (1 μL) was added into a suspension containing 300 μg of RPE microsomal proteins, 1% bovine serum albumin (BSA), 2 mM disodium pyrophosphate, and 25 μM human apo-cellular retinaldehyde-binding protein (CRALBP) in 10 mM BTP buffer (200 μL) to a final concentration from 0 to 2 μM. After incubation at room temperature for 5 min, the resulting mixture was mixed with half microliter of all-trans-retinol (5 mM) in DMF, and then incubated at 37 °C for 1 h. The reaction was quenched by adding 400 μL of methanol (Fisher Chemical, Fair Lawn, NJ), and the products were extracted with 400 μL of hexanes. Production of 11-cis-retinol was quantified by normal phase HPLC using a Zorbax Rx-SIL column (5 μm, 4.6 × 250 mm, Agilent, Santa Clara, CA) with 10% (v/v) ethyl acetate in hexanes as the eluent at a flow rate of 1.4 mL·min−1. Retinoids were detected by monitoring their absorbance at 325 nm and quantified based on a standard curve representing the relationship between the amount of 11-cis-retinol and the area under the corresponding chromatographic peak.
Table 2.
α-Cyano alcohol derived γ-hydroxyamines.
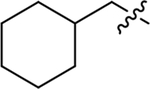
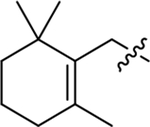
| Compound | R3 | R1 | R2 | IC50 (nM) | Compound | R3 | R1 | R2 | IC50 (nM) |
|---|---|---|---|---|---|---|---|---|---|
| Racemic emixustat |
|
H | H | 172 ± 29 | (R)-MB-001 |
|
H | H | 323 ± 145 |
| 49 | 4-F | H | 95 ± 5 | 53 | 4-F | H | 284 ± 20 | ||
| 50 | 5-F | H | 201 ± 23 | 54 | 5-F | H | 292 ± 89 | ||
| 51 | 6-F | H | 145 ± 9 | 55 | 6-F | H | 124 ± 21 | ||
| 52 | 2-F | H | 358 ± 42 | 56 | 2-F | H | 169 ± 13 | ||
| 58 | H | D | 190 ± 15 | 57 |
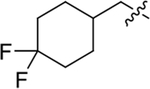
|
H | H | 103 ± 29 | |
| 59 | H | D | 269 ± 40 |
Quantification of representative primary amine compound levels in serum and eyes of mice after treatments.
Eight weeks old BL/6J mice were treated with 380 nmol visual cycle modulator 24, 49, 57, 58, 59 or emixustat in DMSO (50 μL) by intraperitoneal injection, and sacrificed at 3 h, 1 day, or 7 days later. Blood and eyeball samples were collected immediately. After clotting at room temperature for 30 min, the blood samples were centrifuged for 10 minutes at 17,000g in a temperature-controlled benchtop centrifuge (Eppendorf AG). Each serum sample (100 μL) was carefully removed to avoid disturbing loose clots, precipitated with 400 μL of pre-cooled methanol, and centrifuged at 17,000g for 15 min at 4 °C. The supernatant was carefully transferred to a SpinX centrifuge tube filter with a 0.45 μm cellulose acetate membrane (Costar, Salt Lake City, UT), and centrifuged at 7,000 g for 2 min. Filtered samples were dried under vacuum, reconstituted in 100 μL 50% methanol/water, and centrifuged at 17,000g for 15 min at 4 °C. The resulting supernatants were ready for Liquid Chromatography/Mass Spectrometry analyses. The two eyeballs from each mouse were homogenized in acetonitrile (2 × 800 μL). The resulting mixture was centrifuged at 17,000g for 15 min at 4 °C. The supernatant was dried under vacuum, reconstituted in 100 μl 50% methanol/water, and centrifuged at 17,000g for 15 min at 4 °C. Twenty microliters of the supernatant extracted from serum or eye samples was injected into an Ultimate 3000 HPLC system coupled with LXQ mass spectrometer (ThermoFisher Scientific, Waltham, MA) with an electrospray ionization unit. The separation was performed on a Proshell EC-18 column (2.7 μm, 3.0 × 150 mm, Agilent, Santa Clara, CA) using a mobile phase consisting of 0.1% aqueous formic acid (A) and acetonitrile (B) at a flow rate of 600 μL·min−1 and the mobile phase gradients and time course were as follows: 0–2 min, 95% A / 5% B; 2–10 min, 95%−15% A / 5%−85% B. The signals were detected in the selected reaction monitoring (SRM) mode at conditions described in Supplementary Table 3 and quantified based on the standard curves representing the relationship between the amounts of primary amine standards and the areas under the corresponding chromatographic peaks.
VAP-1 oxidation assay.
Mouse aorta homogenates were used as the source of vascular adhesion protein-1 (VAP-1) for this study. Aortas were removed from mice (4–6 week-old) that had been euthanized by CO2 asphyxiation followed by cervical dislocation. The aorta was dissected and the blood was removed by rinsing the tissue with phosphate-buffered saline. Aorta samples were used immediately or stored at −80 °C until needed. Two aortas were minced using a stainless steel single edge blade and homogenized in a KONTES Potter-Elvehjem tissue grinder/homogenizer glass pestle in 1 mL of 10 mM HEPES-NaOH, pH 7.6. The homogenate was collected into a 1.5 mL Eppendorf tube. Five μl of a 20 mM ethanolic stock solution of emixustat or d2-emixustat (58) were added to the aorta homogenate to give a final substrate concentration of 100 μM. The sample was mixed and then incubated at 28 °C with 300 RPM shaking in an Eppendorf Thermomixer. 200 μL samples were taken at 0, 1, and 2 h after initiation of the reaction. At each time point, the reactions were immediately quenched with 100 μL of 100% MeOH, vortexed for 3 sec, and stored at −20 °C. After samples from all time points were collected and frozen, the samples were thawed and centrifuged at 15,000 RPM for 10 min. 250 μL of each supernatant was collected, placed into a borosilicate tube, and dried in a Speedvac rotoevaporator. Each dried sample was redissolved in 300 μL of a 1:1 MeOH/H2O solution, centrifuged to remove particulates, and then transferred to an HPLC vial. 50 μL of the sample was used for analysis on an Agilent 1260 Infinity series HPLC equipped with a Proshell EC-18 column and a diode array detector. The sample was separated using a mobile phase consisting of 0.1% (v/v) formic acid in H2O and acetonitrile at the following ratios and time intervals: 95:5 for 2 min, a gradient from 95:5 to 15:85 over 8 min, a gradient from 15:85 to 2:98 over 0.5 min, continue 2:98 for 4 min, and then a gradient from 2:98 to 95:5 over 0.5 min. The reaction substrate and product were assessed by monitoring absorbance at 275 nm. Emixustat and d2-emixustat eluted at ~8.5 min, while the assay product (ACU-5201) eluted at ~13.25 min. A dilution series of known concentrations of authentic ACU-5201 in 1:1 MeOH/H2O was run to generate a standard curve and facilitate the conversion of product AUCs to absolute mass.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a Bar-Ilan University new faculty grant (to A.G). K. Palczewski is the Irving H. Leopold Chair of Ophthalmology at the Gavin Herbert Eye Institute, Department of Ophthalmology, University of California, Irvine. This research was supported in part by grants to K.P. from the National Institutes of Health (NIH) (EY009339, EY027283, EY030873); to P.D.K. from the U.S. Department of Veterans Affairs (I01BX004939); and to G.P.T. from the National Science Foundation (NSF-CHE Award No. 1904530) and the Department of Defense (DOD-CDMRP Award No. W81XWH-16-1-0699). The authors also acknowledge support from an RPB unrestricted grant to the Department of Ophthalmology, University of California, Irvine. We also thank the Interdisciplinary Centre for Mathematical and Computational Modelling in Warsaw, Poland, under grant GB79–5, for access to high-performance computing resources.
ABBREVIATIONS
- A2E
N-retinylidene-N-retinylethanolamine
- AMD
age-related macular degeneration
- C8E6
hexaoxyethylene monooctyl ether
- CRALBP
cellular retinaldehyde-binding protein
- DLPNO-CCSD(T)
domain-based local pair natural orbital coupled-cluster with single, double, and perturbative triples excitation
- ETS
transition state theory
- KIE
kinetic isotope effect
- LED
local energy decomposition
- NCI
non-covalent interaction
- NOCV
natural orbitals for chemical valence
- RPE65
retinoid isomerase
- VAP-1
vascular adhesion protein-1
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.
Synthesis, characterization, copies of the NMR spectra and purity HPLC traces for the final synthesized compounds and intermediates.
Accession Codes:
The X-ray crystallography datasets reported in this article have been deposited in the Protein Data Bank (ID codes: 7K88 (C8E6 complex), 7K89 (compound 49 complex), 7K8G (compound 24 complex), and 7L0E (compound 57 complex)).
The authors declare no competing financial interest.
REFERENCES
- 1.Travis GH; Golczak M; Moise AR; Palczewski K Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol 2007, 47, 469–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palczewski K; Kiser PD Shedding new light on the generation of the visual chromophore. Proc Natl Acad Sci U S A 2020, 117, 19629–19638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sparrow JR; Gregory-Roberts E; Yamamoto K; Blonska A; Ghosh SK; Ueda K; Zhou J The bisretinoids of retinal pigment epithelium. Prog Retin Eye Res 2012, 31, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sparrow JR Bisretinoids of RPE lipofuscin: trigger for complement activation in age-related macular degeneration. Adv Exp Med Biol 2010, 703, 63–74. [DOI] [PubMed] [Google Scholar]
- 5.Golczak M; Kuksa V; Maeda T; Moise AR; Palczewski K Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A 2005, 102, 8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubota R; Birch DG; Gregory JK; Koester JM Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease. Br J Ophthalmol 2020, 0, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubota R; Jhaveri C; Koester JM; Gregory JK Effects of emixustat hydrochloride in patients with proliferative diabetic retinopathy: a randomized, placebo-controlled phase 2 study. Graefes Arch Clin Exp Ophthalmol 2021, 259, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubota R; Gregory J; Henry S; Mata NL Pharmacotherapy for metabolic and cellular stress in degenerative retinal diseases. Drug Discov Today 2020, 25, 292–304. [DOI] [PubMed] [Google Scholar]
- 9.Kubota R; Calkins DJ; Henry SH; Linsenmeier RA Emixustat reduces metabolic demand of dark activity in the retina. Invest Ophthalmol Vis Sci 2019, 60, 4924–4930. [DOI] [PubMed] [Google Scholar]
- 10.Rosenfeld PJ; Dugel PU; Holz FG; Heier JS; Pearlman JA; Novack RL; Csaky KG; Koester JM; Gregory JK; Kubota R Emixustat hydrochloride for geographic atrophy secondary to age-related macular degeneration: a randomized clinical trial. Ophthalmology 2018, 125, 1556–1567. [DOI] [PubMed] [Google Scholar]
- 11.Bavik C; Henry SH; Zhang Y; Mitts K; McGinn T; Budzynski E; Pashko A; Lieu KL; Zhong S; Blumberg B; Kuksa V; Orme M; Scott I; Fawzi A; Kubota R Visual cycle modulation as an approach toward preservation of retinal integrity. PLoS One 2015, 10, e0124940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dugel PU; Novack RL; Csaky KG; Richmond PP; Birch DG; Kubota R Phase ii, randomized, placebo-controlled, 90-day study of emixustat hydrochloride in geographic atrophy associated with dry age-related macular degeneration. Retina 2015, 35, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubota R; Al-Fayoumi S; Mallikaarjun S; Patil S; Bavik C; Chandler JW Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina 2014, 34, 603–609. [DOI] [PubMed] [Google Scholar]
- 14.Kubota R; Boman NL; David R; Mallikaarjun S; Patil S; Birch D Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina 2012, 32, 183–188. [DOI] [PubMed] [Google Scholar]
- 15.Reid MJ; Eyre R; Podoll T Oxidative deamination of emixustat by human vascularadhesion protein-1/semicarbazide-sensitive amine oxidase. Drug Metab Dispos 2019, 47, 504–515. [DOI] [PubMed] [Google Scholar]
- 16.Podoll T; Geisler L; Parys MV; Hanson G; Reid MJ Validation and reproducibility of an LC-MS/MS method for emixustat and its three deaminated metabolites in human plasma. Bioanalysis 2018. [DOI] [PubMed] [Google Scholar]
- 17.Fitzsimmons ME; Sun G; Kuksa V; Reid MJ Disposition, profiling and identification of emixustat and its metabolites in humans. Xenobiotica 2018, 48, 592–604. [DOI] [PubMed] [Google Scholar]
- 18.Miao Z; Farnham JG; Hanson G; Podoll T; Reid MJ Bioanalysis of emixustat (ACU-4429) in whole blood collected with volumetric absorptive microsampling by LC-MS/MS. Bioanalysis 2015, 7, 2071–2083. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J; Kiser PD; Badiee M; Palczewska G; Dong Z; Golczak M; Tochtrop GP; Palczewski K Molecular pharmacodynamics of emixustat in protection against retinal degeneration. J. Clin. Invest 2015, 125, 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeda A; Golczak M; Chen Y; Okano K; Kohno H; Shiose S; Ishikawa K; Harte W; Palczewska G; Maeda T; Palczewski K Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat Chem Biol 2011, 8, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller K; Faeh C; Diederich F Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 22.Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of fluorine in medicinal chemistry. J Med Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 23.Gant TG Using deuterium in drug discovery: leaving the label in the drug. J Med Chem 2014, 57, 3595–3611. [DOI] [PubMed] [Google Scholar]
- 24.Jakubec P; Petras P; Duris A; Berkes D The first example of a crystallization-induced asymmetric transformation (CIAT) in the Mannich reaction. Tetrahedron-Asymmetr 2010, 21, 69–74. [Google Scholar]
- 25.Valachova D; Ferko B; Puchl’ova E; Caletkova O; Berkes D; Kolarovic A; Jakubec P Stereoselective Synthesis of syn-gamma-Hydroxynorvaline and Related alpha-Amino Acids. Synthesis-Stuttgart 2019, 51, 4568–4575. [Google Scholar]
- 26.Cierna M; Markus J; Dohanosova J; Moncol J; Jakubec P; Berkes D; Caletkova O Stereoselective Mannich Reaction Driven by Crystallization. Eur J Org Chem 2020, 2020, 5685–5689. [Google Scholar]
- 27.Lehmann F; Pilotti A; Luthman K Efficient large scale microwave assisted Mannich reactions using substituted acetophenones. Mol. Divers 2003, 7, 145–152. [DOI] [PubMed] [Google Scholar]
- 28.Wang J; Wang Y; Liu D; Zhang W Asymmetric hydrogenation of bsecondary amino ketones catalyzed by a ruthenocenyl phosphino-oxazoline-ruthenium complex (RuPHOX-Ru): the synthesis of g-secondary amino alcohols. Adv. Synth. Catal 2015, 357, 3262–3272. [Google Scholar]
- 29.Ella-Menye JR; Sharma V; Wang G New synthesis of chiral 1,3-oxazinan-2-ones from carbohydrate derivatives. J. Org. Chem 2005, 70, 463–469. [DOI] [PubMed] [Google Scholar]
- 30.Davies SG; Haggitt JR; Ichihara O; Kelly RJ; Leech MA; Price Mortimer AJ; Roberts PM; Smith AD Asymmetric total synthesis of sperabillins B and D via lithium amide conjugate addition. Org. Biomol. Chem 2004, 2, 2630–2649. [DOI] [PubMed] [Google Scholar]
- 31.Scott IL; Kuksa V; Orme MW; Little T; Gall A; Hong F Alkoxy compounds for disease treatment. Patent2014, US8829244B2, 1–229.
- 32.Kiser PD; Zhang J; Badiee M; Li Q; Shi W; Sui X; Golczak M; Tochtrop GP; Palczewski K Catalytic mechanism of a retinoid isomerase essential for vertebrate vision. Nat Chem Biol 2015, 11, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiser PD; Zhang J; Badiee M; Kinoshita J; Peachey NS; Tochtrop GP; Palczewski K Rational tuning of visual cycle modulator pharmacodynamics. J Pharmacol Exp Ther 2017, 362, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott IL; Kuksa V; Orme MW; Little T; Gall A; Hong F Preparation of alkoxybenzene compounds for disease treatment. PCT INt Appl. 2009, 0454479. [Google Scholar]
- 35.Yu PH Three types of stereospecificity and the kinetic deuterium isotope effect in the oxidative deamination of dopamine as catalyzed by different amine oxidases. Biochem Cell Biol 1988, 66, 853–861. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Q; Sun W; Okano K; Chen Y; Zhang N; Maeda T; Palczewski K Sponge transgenic mouse model reveals important roles for the microRNA-183 (miR-183)/96/182 cluster in postmitotic photoreceptors of the retina. J. Biol. Chem 2011, 286, 31749–31760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiser PD; Farquhar ER; Shi W; Sui X; Chance MR; Palczewski K Structure of RPE65 isomerase in a lipidic matrix reveals roles for phospholipids and iron in catalysis. Proc Natl Acad Sci U S A 2012, 109, E2747–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golczak M; Kiser PD; Lodowski DT; Maeda A; Palczewski K Importance of membrane structural integrity for RPE65 retinoid isomerization activity. J. Biol. Chem 2010, 285, 9667–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sui X; Kiser PD; Che T; Carey PR; Golczak M; Shi W; von Lintig J; Palczewski K Analysis of carotenoid isomerase activity in a prototypical carotenoid cleavage enzyme, apocarotenoid oxygenase (ACO). J. Biol. Chem 2014, 289, 12286–12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kloer DP; Ruch S; Al-Babili S; Beyer P; Schulz GE The structure of a retinal-forming carotenoid oxygenase. Science 2005, 308, 267–269. [DOI] [PubMed] [Google Scholar]
- 41.Redmond TM; Poliakov E; Kuo S; Chander P; Gentleman S RPE65, visual cycle retinol isomerase, is not inherently 11-cis-specific: support for a carbocation mechanism of retinol isomerization. J. Biol. Chem 2010, 285, 1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imai YN; Inoue Y; Nakanishi I; Kitaura K Cl-pi interactions in protein-ligand complexes. Protein Sci. 2008, 17, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jennings WB; O’Connell N; Malone JF; Boyd DR An evaluation of substituent effects on aromatic edge-to-face interactions and CF-pi versus CH-pi interactions using an imino torsion balance model. Org Biomol Chem 2013, 11, 5278–5291. [DOI] [PubMed] [Google Scholar]
- 44.Li P; Maier JM; Vik EC; Yehl CJ; Dial BE; Rickher AE; Smith MD; Pellechia PJ; Shimizu KD Stabilizing fluorine-pi interactions. Angew. Chemie, Int. Ed 2017, 56, 7209–7212. [DOI] [PubMed] [Google Scholar]
- 45.Riplinger C; Neese F An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys 2013, 138, 034106. [DOI] [PubMed] [Google Scholar]
- 46.Riplinger C; Sandhoefer B; Hansen A; Neese F Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys 2013, 139, 134101. [DOI] [PubMed] [Google Scholar]
- 47.Schneider WB; Bistoni G; Sparta M; Saitow M; Riplinger C; Auer AA; Neese F Decomposition of intermolecular interaction energies within the local pair natural orbital coupled cluster framework. J. Chem. Theory Comput 2016, 12, 4778–4792. [DOI] [PubMed] [Google Scholar]
- 48.Johnson ER; Keinan S; Mori-Sanchez P; Contreras-Garcia J; Cohen AJ; Yang W Revealing noncovalent interactions. J. Am. Chem. Soc 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitoraj M; Michalak A Natural orbitals for chemical valence as descriptors of chemical bonding in transition metal complexes. J Mol Model 2007, 13, 347–355. [DOI] [PubMed] [Google Scholar]
- 50.Lin YS; Li GD; Mao SP; Chai JD Long-range corrected hybrid density functionals with improved dispersion corrections. J. Chem. Theory Comput 2013, 9, 263–272. [DOI] [PubMed] [Google Scholar]
- 51.Grimme S; Ehrlich S; Goerigk L Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 52.Ziegler T; Rauk A Calculation of bonding energies by Hartree-Fock Slater method .1. Transition-sate method. Theor Chim Acta 1977, 46, 1–10. [Google Scholar]
- 53.Gong B; Boor PJ The role of amine oxidases in xenobiotic metabolism. Expert Opin Drug Metab Toxicol 2006, 2, 559–571. [DOI] [PubMed] [Google Scholar]
- 54.Benedetti MS Biotransformation of xenobiotics by amine oxidases. Fundam Clin Pharmacol 2001, 15, 75–84. [DOI] [PubMed] [Google Scholar]
- 55.Claud P; Padovani P; Guichard JP; Artur Y; Laine R Involvement of semicarbazide-sensitive amine oxidase in tresperimus metabolism in human and in rat. Drug Metab Dispos 2001, 29, 735–741. [PubMed] [Google Scholar]
- 56.Heuts DP; Gummadova JO; Pang J; Rigby SE; Scrutton NS Reaction of vascular adhesion protein-1 (VAP-1) with primary amines: mechanistic insights from isotope effects and quantitative structure-activity relationships. J. Biol. Chem 2011, 286, 29584–29593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Leary MH Multiple isotope effects on enzyme-catalyzed reactions. Annu Rev Biochem 1989, 58, 377–401. [DOI] [PubMed] [Google Scholar]
- 58.Northrop DB The expression of isotope effects on enzyme-catalyzed reactions. Annu Rev Biochem 1981, 50, 103–131. [DOI] [PubMed] [Google Scholar]
- 59.O’Leary MH Determination of heavy-atom isotope effects on enzyme-catalyzed reactions. Methods Enzymol 1980, 64, 83–104. [DOI] [PubMed] [Google Scholar]
- 60.Schowen KB; Schowen RL The use of isotope effects to elucidate enzyme mechanisms. Bioscience 1981, 31, 826–831. [Google Scholar]
- 61.Trifonov L; Afri M; Palczewski K; Korshin EE; Gruzman A An Expedient Synthesis of CMF-019: (S)-5-Methyl-3-{1-(pentan-3-yl)-2- (thiophen-2-ylmethyl)-1H-benzo[d]imidazole-5-carboxamido}hexanoic Acid, a Potent Apelin Receptor (APJ) Agonist. Med Chem 2018, 14, 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabsch W Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 2010, 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams CJ; Headd JJ; Moriarty NW; Prisant MG; Videau LL; Deis LN; Verma V; Keedy DA; Hintze BJ; Chen VB; Jain S; Lewis SM; Arendall WB 3rd; Snoeyink J; Adams PD; Lovell SC; Richardson JS; Richardson DC MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Read RJ; Adams PD; Arendall WB 3rd; Brunger AT; Emsley P; Joosten RP; Kleywegt GJ; Krissinel EB; Lutteke T; Otwinowski Z; Perrakis A; Richardson JS; Sheffler WH; Smith JL; Tickle IJ; Vriend G; Zwart PH A new generation of crystallographic validation tools for the protein data bank. Structure 2011, 19, 1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.