

Abstract
Toll-like receptor (TLR) signaling is a well characterized, innate immue cellular defense mechanism used to detect and respond to pathogen-associated molecular patterns (PAMPs). TLR signaling is highly conserved and has evolved to have both extracellular and endosomal receptors that recognize PAMPs from a wide range of microbial pathogens. Recent literature has emerged to show that activation of TLRs not only leads to the upregulation of cellular defense mechanisms, but also results in upregulation of DNA repair genes and increased functional DNA repair. Endosomal TLR agonists result in increased survival and repair after both ionizing and ultraviolet (UV) radiation, suggesting that the repair pathways for single and double strand breaks are affected. This review brings together these and other experimental findings to examine how DNA repair pathways may be linked to TLR signaling. Also discussed are the varied outcomes and related physiological implications that increased DNA repair after injury might have.
Keywords: toll-like receptor, DNA repair, AP-1, DNA damage
Introduction
Toll-like receptors (TLR) are a main component of the innate immune system, which serves as a first line of defense against a wide range of injurious substances. TLR activation not only initiates an inflammatory response that works to combat pathogens, but also potentially results in tissue damage that must be repaired after the infection is neutralized. Among humans and mice, there are currently thirteen known TLRs, some of which localize to the cell membrane and are meant to defend against extracellular pathogens, while others are located in the endosome and detect intracellular pathogens. These receptors function by recognizing pathogen associated molecular patterns (PAMPs). Each TLR responds to very specific PAMPs and initiates the appropriate cellular response to defend against the pathogen that it recognized. The elegant and complex TLR signaling pathways have been the subject of many well written and comprehensive reviews, as such, we will only give a brief overview (Barton and Kagan, 2009; Kawai and Akira, 2011; Trinchieri and Sher, 2007). Teleologically, it may be of survival value to damaged cells and the surrounding tissue to have a way of repairing damage created by TLR activating agonists, the subject of this review.
All TLRs contain two main signaling domains. The first is an extracellular or endosomal pathogen sensing domain that consists of leucine rich repeats (LRR) in the conformation of a shepherd’s crook, and the second is an intracellular TIR domain that interacts with signaling and adapter molecules to relay the message that a pathogen has been sensed. These trans-membrane proteins function by either homo- or hetero-dimerizing to create a functional sensing and signaling complex. TLRs 1/2, 2/6, 4/4, and 5/5 are known to be extracellular sensors in that reside in the plasma membrane of the cell, while TLRs 3/3, 7/7, 8/8, and 9/9 are considered intracellular sensors and reside in the membrane of the endosome. There are also reports of TLRs 2 and 4 localizing to and functioning from the endosome (Dietrich et al., 2010; Shibata et al., 2011). MyD88 is the necessary adapter molecule that interacts with the TIR domain of all TLRs except for TLR3 which utilizes TRIF as its sole adapter molecule to lead to downstream signaling. TLR4 can use either MyD88 or TRIF to relay its signal, which is thought to depend on where it is localized (McGettrick and O’Neill, 2010). The varied location of and large number of possible signals from TLR4 makes it the most functionally diverse and widely studied TLR. Using distinct combinations of adapter and signaling molecules, each TLR elicits a tailored response to the pathogen that it senses. For an extensive overview of TLR ligands, both synthetic and naturally occurring, please refer to the review by Kawai et al. (Kawai and Akira, 2011). The pathogen-specific response of TLRs involves the release of cytokines tailored for neutralization of the activating pathogen, recruitment of necessary immune components by release of chemokines and up-regulation of costimulatory molecules, regulation of cell cycle, and when appropriate mediation of inflammation (Loegering and Lennartz, 2011; Prince et al., 2011; Waltz et al., 2011).
TLRs are not only able to sense PAMPs, but also respond to damage-associated molecular patterns (DAMPs), that are endogenous signaling components released by cells upon acute damage and/or infection. DAMPs are also sensed by TLRs present on surrounding cells. The notion that TLRs are able to recognize and respond to these DAMPs has become widely recognized and the number of known DAMPs has grown rapidly over the past few years (Piccinini and Midwood, 2010). These DAMPs are most notably created under conditions of oxidative stress, DNA damage, viral infection, and ischemia/reperfusion (Huang et al., 2011; Marichal et al., 2011; Tang et al., 2011; Wheeler et al., 2009). Comprised of many categories of molecules, the list of TLR-activating DAMPs contains proteins/peptides, fatty acids/lipoproteins, proteoglycans/glycosaminoglycans, and nucleic acids/nucleic acid-protein complexes. Endogenous DAMPs have now been identified for all of the TLRs, which are the subject of many previous reviews that compile lists of known DAMPs associated with many different environmental and disease conditions (Goh and Midwood, 2012; Krysko et al., 2011; Kuipers et al., 2011; Miller et al., 2011; Nace et al., 2012).
Among the currently identified DAMPs that are known to signal through TLRs there are a few that are particularly relevant to this review because they are associated with DNA damage and DNA damaging conditions (Table 1). In a mouse model of hepatic ischemia/reperfusion. endogenous histone proteins, which are released during DNA damaging conditions, are shown to activate TLR9 (Huang et al., 2011). Another DNA related DAMP is HMGB-1, a chromatin binding protein, that is a well-recognized DAMP capable of signaling through TLRs 2,4,and 9. This DNA associated molecule is implicated in the pathogenesis of a wide range of diseases including skin cancer, epilepsy, and sepsis (Maroso et al., 2010; Mittal et al., 2010; Yu et al., 2006). Chromatin and other self-DNA complexed with proteins/peptides, when released from the cell, are involved in the pathogenesis of autoimmune disorders, such as systemic lupus erythematosus (SLE) and Sjogren’s syndrome, by serving as antigen for autoantibody production, antibodies complexed with self-nucleic acid can then signal through TLRs 7 and 9 to exacerbate pathogenic inflammation (Celhar et al., 2012; Rubin et al., 1986). The antimicrobial peptide LL37 has also been shown to complex with free self nucleic acids and serve as a TLR 7 and 8 agonist (Ganguly et al., 2009; Lande et al., 2007). This aberrant TLR activation has been implicated in the pathogenesis of psoriasis. Interestingly, a recent publication shows that UVB irradiated keratinocytes release damaged self non-coding RNAs that serve as potent TLR3 agonists (Bernard et al., 2012). UVB causes extreme DNA damage that must be repaired, further experiments should be performed to determine if possibly the described TLR3 activation leads to increased DNA repair. Simple mislocalization and/or aberrant release of these ubiquitously present DAMP molecules can stimulate potentially harmful inflammation through recognition by TLRs and other pattern recognition receptors. The ability of the cell to differentiate a genuine danger signal from a normally functioning molecule speaks to how tightly regulated the TLR signaling process is. This review will focus on how activation of TLR pathways might stimulate increased DNA repair, the aforementioned DAMPs serve as intriguing venues to investigate if DAMPs arising from DNA damaging conditions may be helping to initiate repair by signaling through TLRs.
Table 1.
Selected DAMPs associated with DNA damage and DNA damaging conditions.*
| DAMP | TLR(s) Antagonized | Conditions When Released | Associated Organ/Disease | Reference(s) |
|---|---|---|---|---|
| damaged self non-coding RNAs | TLR3 | UVB irradiation | skin | Bernard et al., 2012 |
| histone proteins | TLR9 | ischemic injury | hepatic ischemia/reperfusion | Huang et al., 2011 |
| HMGB-1 | TLR2, TLR4 | oxidative stress, physical injury, ischemic injury, inflammation | skin cancer, epilepsy, sepsis | Maroso et al., 2010; Mittal et al., 2010; Yu et al., 2006 |
| chromatin-protein complexes | TLR7, TLR9 | unknown | SLE, Sjogren’s syndrome | Celhar et al., 2012; Rubin et al., 1986 |
| antimicrobial peptide-nucleic acid complex | TLR7, TLR8 | cell death | psoriasis | Ganguly et al., 2009; Lande et al., 2007 |
for a more extensive list of DAMPs and their receptor molecules please refer to the following reviews: (Goh and Midwood, 2012; Krysko et al., 2011; Kuipers et al., 2011; Miller et al., 2011; Nace et al., 2012; Piccinini and Midwood, 2010)
With all of the opportunity present for these TLR signaling pathways to go awry, it is not surprising that they are implicated in the pathogenesis of many diseases and are a target for therapeutics seeking to control the immune response stimulated by TLR agonists. TLR antagonist therapies are routinely used and continue to be under development for treatment of a wide range of diseases including viral infection, lymphoma, melanoma, SLE, and rheumatoid arthritis (RA) (Alexandrescu et al., 2010; Bourquin et al., 2011; Sun et al., 2007; Ubol and Halstead, 2010). These drugs target many different parts of the signaling pathway including controlling TLR signaling by competitive inhibition of the ligand binding site, and suppression of homodimerization of the receptors (Lee et al., 2011; Park and Youn, 2010). Understanding the many complicated and varied mechanisms of regulation and outcomes from the TLR signaling pathways will allow for more specific and appropriate immune modulation therapy in diseased tissue.
TLR Signaling Leads to Increased DNA Repair
Recently, a body of evidence has emerged that shows the up-regulation of DNA repair genes and functional DNA repair after treatment with TLR agonists. This is initially surprising because TLR signaling is traditionally thought to defend against exogenous pathogens by mobilizing an inflammatory response and interfacing with adaptive immunity, rather than lead to increased repair of self-DNA. Although, with our growing knowledge of how TLRs respond to DAMPs, perhaps increased repair is a logical downstream effect.
Synthetic versions of CpG DNA are the most widely studied TLR9 ligand because of their clinical relevance and therapeutic use. In 2008, Zheng et al., while investigating the outcomes of TLR9 engagement on γ-irradiated CD4+ T cells as it relates to cancer therapeutics, observed that CpG DNA stimulated CD4+ T cells exhibited increased DNA repair rates after irradiation (Zheng et al., 2008). They also showed an increase in the number of cells in the G2 cell-cycle phase and a decrease in apoptotic cells as measured by Annexin V staining. This accumulation of cells in the G2 phase of the cell-cycle may allow for the increase that is seen in functional double strand break (DSB) repair which ultimately leads to increased survival and a decrease in programmed cell death. More recently, a global gene expression analysis was conducted to investigate which families of genes are up- and down-regulated in vivo upon TLR9 stimulation (Klaschik et al., 2010). This study finds that, along with several other gene families, there is a significant increase in mouse splenic mRNAs that code for proteins involved in the regulation of cell cycle and DNA repair five days after an intraperitoneal (i.p.) injection of CpG DNA. While splenomegaly was observed on day five of these experiments, the ratios of B cells, T cells, and monocyte/macrophages were similar to those of an untreated control spleens, allowing for useful interpretation of gene regulation results. An in silico gene expression study, using data sets collected both after i.p. injection of CpG DNA and intranasal inoculation found similar results, i.e., that in immune cells, genes encoding for DNA repair molecules are up-regulated (Sommariva et al., 2011). The up-regulation of these cell cycle and DNA repair genes helps to support and explain previous functional findings, and adds to evidence that stimulation of TLR9 not only leads to a pro-inflammatory response, but also results in repair of DNA damage.
Although TLRs 7 and 8 are expressed at varied levels in different tissues and have been shown to activate dendritic cells in distinct ways, both reside in the membrane of the endosome and are known to recognize single stranded RNAs (Larange et al., 2009). The TLR7 and TLR8 agonist, Imiquimod, is commonly used as an immune modulating topical therapeutic agent used to treat dermatologic diseases such as HPV infection, non-melanoma skin cancer, and actinic keratosis (Edwards et al., 1998; Hanke et al., 2010; Oster-Schmidt, 2004; Swanson et al., 2010). This intracellular TLR agonist has been found to enhance DNA repair gene expression as well as functional repair of DNA damage mechanisms when tested by topical application on the skin of UV-irradiated mice (Fishelevich et al., 2011). After in vitro treatment of bone marrow-derived cell lines with Imiquimod, nucleotide excision repair (NER) gene expression is found to be increased along with nuclear localization of the repair molecule Xeroderma pigmentosum, complementation group A (XPA). Paired with in vivo experiments that show increased resolution of cyclobutane pyrimidine dimers (CBPD) in UV-irradiated and Imiquimod-treated mice compared to control-irradiated mice, these findings support a more universal role of TLR stimulation in up-regulating functional DNA repair.
Aside from up-regulation of traditional DNA repair, the TLR7 agonist, 8-mercaptoguanosine (8-SGuo), in conjunction with IL-4, has been implicated in an increase of class switch recombination and IgG1 production by B cells (Tsukamoto et al., 2009). While it remains unclear if 8-SGuo signaling contributes to the DSB repair function necessary for successful class switch recombination, the TLR7 agonist helps to initiate B cell differentiation and maturation. This observation adds to the growing evidence that TLR signaling is involved in many more complex processes than those previously understood as immunomodulatory functions. Treatment with intracellular TLR agonists for TLR7 and TLR9 seem to result in increased DNA repair processes by several mechanistic readouts. It is of note that there are no recent reports of the same findings using extracellular TLR agonists. However, it has been shown that pretreatment with Lactobacillus bifidus extract causes an increase in DNA repair after UV irradiation which may be a result of extracellular TLR stimulation by the bacterial extract (Born and Born, 1987). Although bifidus is commonly included in anti-aging creams for its ability to increase DNA repair, this property of the compound has still not been sufficiently confirmed in peer-reviewed literature (Natarajan et al., 1988). More research is needed to determine if this phenomenon of TLR induced DNA repair is limited to intracellular TLRs or if perhaps this result can also be induced by extracellular TLR agonists.
Mechanisms by Which TLR Signaling Might Result in Increased DNA Repair
The promoter regions of genes known to be involved in the NER pathway, which repairs single strand breaks (SSB), have been investigated and have uncovered a possible link between DNA repair and TLR signaling. The 5’ promoter regions of all of the NER genes studied (ERCC1, XPA, XPB, XPD, XPF, and XPG) contain at least one putative AP-1 binding site (Zhong et al., 2000). AP-1 is a transcription factor known to be induced by TLR signaling and the existence of its binding sites in promoter regions of many DNA repair genes suggests evidence of an association between TLR signaling and increased functional DNA repair (Figure 1). Although, the existence of AP-1 binding sites is not necessarily the only mechanism of gene regulation and may not be directly correlated with the observed functional increase in DNA repair. Experimental models deficient in TLR and/or AP-1 need to be utilized to determine the importance of the AP-1 binding site in DNA repair genes, and also to investigate if TLR deficient mice have impaired DNA repair. The TLR signaling pathway, along with its many other known functions, may represent a mechanism that the cell has evolved to control all of the genes involved in a necessary response to acute damage coordinately. This phenomenon has been observed in lower prokaryotic organisms, for example Saccharomyces cerevisiae, in which there is a conserved transcription factor binding sequence in the promoter of at least eleven DNA repair and metabolism genes (Xiao et al., 1993). The ability of TLRs to sense DAMPs makes them ideal candidates for overarching regulation of the DNA repair process. However, somehow the signal to up-regulate repair must be differentiated from, and balanced between, the signal to induce a tissue destructive inflammatory response. Much further research is needed to elucidate which molecules are involved in this repair response after TLR signaling and how it might be regulated.
Figure 1. Possible direct and indirect mechanisms of transcriptional activation of DNA repair machinery induced by TLR signaling pathways.
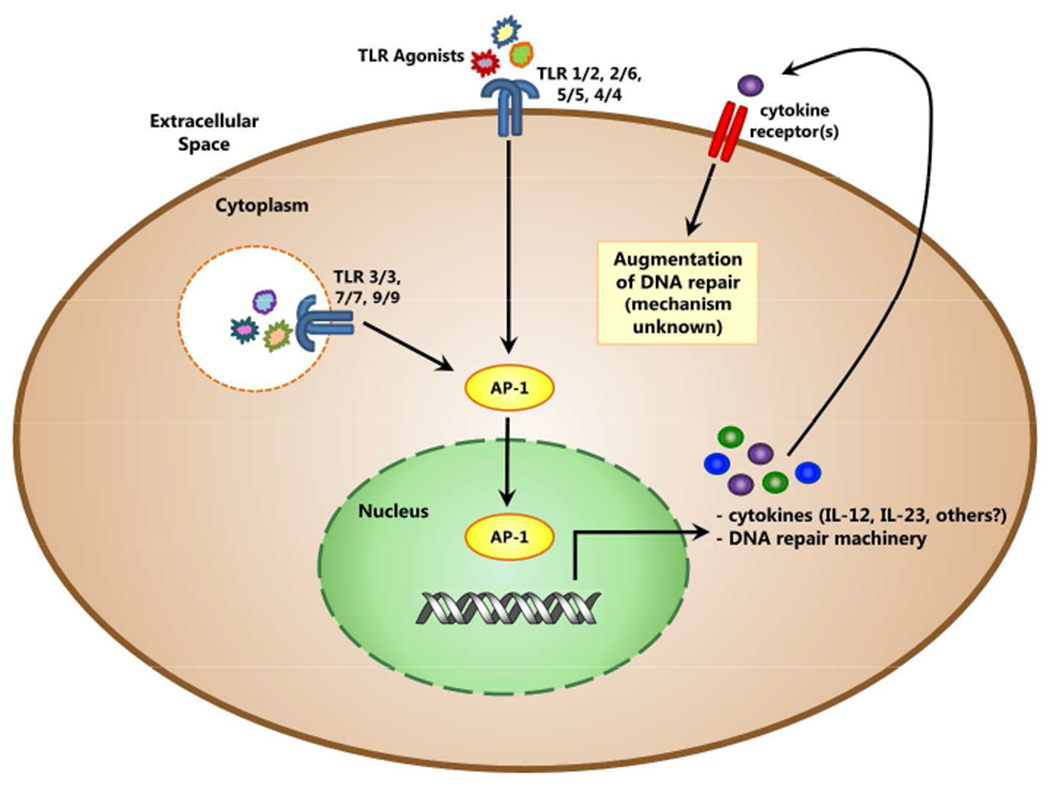
Many genes involved in DNA repair have AP-1 binding sites in their promoter regions. Activation of the AP-1 transcription factor by signaling through the TLR pathway may be a molecular link between TLR agonist treatment and transcriptional control of DNA repair. Autocrine signaling by cytokines, such as IL-12, made in response to TLR agonist sensing, represents an indirect mechanism by which TLR signaling may increase DNA repair. The cytokines produced may signal through their appropriate cytokine receptor in an autocrine or paracrine manner to increase transcription of DNA repair genes.
Another way TLR activation may up-regulate DNA repair is by initiating the production of cytokines that act on the cell in an autocrine or paracrine manner (Figure 1). There are reports that mice with UV-induced DNA damage show increased repair of this damage when exogenous IL-12 or IL-18 is injected into to the skin (Schwarz and Schwarz, 2009). The skin of treated and control mice show the same amount of DNA damage immediately after UV irradiation, measured by amount of CBPD, but twenty four hours after UV exposure, the CBPD level is significantly lower in the skin of cytokine-treated mice versus control. Increased DNA repair is also seen when exogenous IL-23 is added to UV irradiated mice, which might be expected because of the shared p40 subunit between IL-12 and IL-23 (Majewski et al., 2010). The presence of IL-12, IL-18, or IL-23 is able to stimulate DNA repair mechanisms that result in increased functional repair and signaling by many TLRs is known to initiate production of these, among many other, cytokines. Interestingly it has been found that PBMC from Xeroderma pigmentosum (XP) patients have significantly attenuated IFN-γ production following poly I:C, a TLR3 agonist, stimulation (Gaspari et al., 1993). These data can be interpreted as suggesting impaired TLR3 signaling in XP patients. This raises the intriguing possibility that impaired TLR signaling may in some way contribute to the profound DNA repair defects observed in these patients. It is of note that the TLR3 signaling pathway uses TRIF as the sole signaling adaptor molecule, this data may suggest that the TRIF signaling pathway as well as the MyD88 signaling pathway is involved in the up-regulation of DNA repair. Up-regulation of DNA repair machinery after autocrine signaling by TLR-induced cytokines may represent a mechanism by which TLR agonist treatment results in increased DNA repair.
UV radiation-induced cellular damage has been implicated in inducing cell signaling pathways that produce pro-inflammatory cytokines that may signal in the previously described autocrine fashion. However, the most commonly observed UV-induced phenotype is one of immune suppression (Muthusamy and Piva, 2010; Norval and Halliday, 2011). One of the critical hallmarks of the UV effect that results in cutaneous immunosuppression is the migration of antigen presenting cells (APC) impaired by DNA damage to the local lymph node (Vink et al., 1996). Using a mouse contact hypersensitization model, treatment with Imiquimod has been shown to decrease UV-induced immunosuppression (Thatcher et al., 2006). UV-irradiated animals that are treated with the topical TLR7 agonist, Imiquimod, have less DNA damage in the local draining lymph node when compared to control-irradiated mice. Increased repair that leads to successful antigen presentation may be a mechanism whereby this TLR7 agonist is able to counteract UV immunosuppression.
Categories of DNA Damage and Associated Modulation by TLR Agonists
DNA damage can occur through many routes, including UV radiation, ionizing radiation, chemical agents, oxidative stress, and defective receptor editing (Aziz et al., 2012; Azzam et al., 2011; Dizdaroglu, 2012; Rastogi et al., 2010). These injurious circumstances result in several types of damage that lead to varied phenotypic outcomes in the cell (Table 2). As discussed before, it has becoming evident that repair of this range of DNA damage may be increased upon activation of TLR signaling. UV and ionizing radiation (γ-irradiation) most classically result in SSB and DSB, respectively. CBPD are another main damage associated mainly with UVB irradiation (290-320 nm). These dimers require the NER complex to be excised and corrected, and once this process occurs, the integrity of the affected DNA is maintained. Because γ-irradiation results in DSB that cannot be repaired using a complementary template, it is associated with mutagenesis and chromosome abnormalities that often lead to cell death. Repair of both UV- and γ-irradiation have been shown to be increased after treatment with an endosomal TLR agonists (Fishelevich et al., 2011; Zheng et al., 2008), implying that both the NER and DSB repair complexes are stimulated.
Table 2.
Mechanism, Type, and Possible Effectors of DNA Damage
| Classical Phenotype | Hallmark Types of Damage Induced | TLR Effect | |
|---|---|---|---|
| UV Radiation | impaired antigen presentation, immune suppression, loss of contact hypersensitization (CHS), gradual formation of mutations | SSB, CBPD | TLR 7 agonist Imiquimod may enhance repair, and maintain CHS (Fishelevich et al., 2011) |
| Ionizing Radiation | depletion of cells by apoptosis, immune suppression, heritable gene mutations | DSB, chromosome abnormalities, mutagenesis | TLR 9 agonist Cpg DNA increases DNA repair and cell survival (Zheng et al., 2008) |
| Chemical Agents | cytotoxicity | SSB, DSB | anti-tumor synergy with TLR 7 and 8 agonist and cyclophosphamide (Dumitru et al., 2010) |
| Oxidative Stress | tissue and organ damage related to inflammation | SSB, histone instability | not yet investigated |
| Receptor Editing | defects cause: clonal deletion during maturation, decreased Ab repertoire limited adaptive immune response | abasic site, DSB | TLR 7 agonist 8-S-Guo induces class switch and IgG1 production (Tsukamoto et al., 2009) |
Pathogens, although they are not classically known to directly cause host DNA damage, can initiate an inflammatory immune response through TLR signaling that leads to an environment with increased oxidative stress. This oxidative stress and associated reactive oxygen species (ROS) are a prominent cause of DNA damage, most notably SSB and instability of histones due to methylation inhibition (Evans and Cooke, 2004). These conditions are well known to lead to the release of DAMPs that signal through TLRs on self and surrounding cells. ROS induced DAMP signaling classically leads to inflammation and associated tissue damage, however conditions of oxidative stress also lead to the eventual repair of affected tissue (Gill et al., 2010). It is possible that when a PAMP is sensed by a TLR, DNA damaging ROS are released, which are then followed by the release of DAMPs that signal through TLRs to help promote repair; however, much research is needed to confirm this hypothesis.
Outcomes of Increased DNA Repair
After unintentional damage by exposure to a genotoxic agent, increased repair can be a positive outcome for a cell that has an important putative role in host defense, for example an APC. However, increased repair in cells that have been programmed for death, are infected with a pathogen, or that are being deliberately targeted for death using a chemotherapeutic drug, may result in an undesirable outcome. For example, if a severely UV-damaged skin cell repairs itself enough to surpass the DNA damage check point during cell cycle, growth will not be arrested and the cell may divide, allowing for propagation of a mutation that could render the cell cancerous. In this scenario, it might be advantageous to inhibit TLR signaling so that repair will not be increased, damage can be recognized, and the cell deleted. Lewis et al. found that TLR4 signaling contributes to UV immune suppression by promoting T-regulatory cell formation (Lewis et al., 2011). They therefore, proposed that TLR inhibition therapy may prevent dampening of a necessary immune response. They did not investigate how UV irradiation activated TLR4 signaling, or how the induction of DAMPs may be involved. The study also did not resolve, on a molecular level, how TLR4 enhanced immune suppression. Nonetheless, this intriguing finding suggests that TLR and the PAMPs/DAMPs that engage them could present opposing signals in the setting of genotoxic challenges. Further research is needed to clarify these apparently divergent outcomes of TLR signaling.
Cancer chemotherapy is another example of a situation where increased DNA repair is not a favorable outcome. Such therapy may use DNA damaging chemotherapeutic agents targeted to tumor cells to cause cell death. In this scenario, if repair is increased, the undesirable outcome of increased survival of tumor cells would be expected. However, just the opposite has been observed. TLR agonists have been shown to work synergistically with the chemotherapeutic drug cyclophosphamide (a known genotoxic agent) (Dumitru et al., 2010). By mechanisms not yet elucidated, the TLR agonist sensitizes the tumor cells to DNA damage that, given the previously discussed findings, is not what would be expected. The same phenomenon is seen in work by Sommariva et al. in which TLR9 agonist treatment caused the upregulation of DNA repair genes in immune cells, but exactly the opposite in tumor cells which aided in their death (Sommariva et al., 2011). Up- or down-regulation of DNA repair may represent a molecular biological explanation for the synergy seen when chemotherapy is administered with TLR agonists (Larange et al., 2009). These studies suggest that the tumor microenvironment evolves into an area that is advantageous for treatment with agents that stimulate TLR signaling. In this environment, DNA repair in non-diseased cells might be increased, and in tumor cells DNA repair might be decreased. These phenomenon need further investigation to better understand how immune versus stromal cells, or normal versus cancerous cells respond to TLR agonists and associated DNA repair/cell survival.
Discussion
The paradoxical data discussed in this review implies that TLR signaling in some environments can lead to increased DNA repair, while in others the opposite might be true. However, overall the evidence presented suggests that the TLR signaling pathways and cell fate might be connected through the DNA repair pathway. Supporting this hypothesis, a recent study finds that TLR expression is regulated by DNA metabolic stress and the p53 pathway (Menendez et al., 2011). Pathogens are well known to signal through TLRs and cause the subsequent up-regulation of TLR expression, fact that genotoxic stress and DNA damage might have the same effect on the TLR pathways is consistent with the data discussed in this review. If the molecular signaling pathway of a TLR that leads to promotion or inhibition of DNA repair, or visa versa, and the conditions necessary for each outcome can be elucidated, then TLR modulation could represent a breakthrough in how to control cell fate, either death or survival, where appropriate. Because multiple drugs involved in TLR modulation are already approved for use, elucidation of this pathway and how it might be related to a wide range of diseases presents a unique opportunity for rapid treatment and observation of a successful clinical outcome.
More work is needed to determine if all TLRs are involved in augmenting DNA repair. TLR and their numerous agonists (PAMPs and DAMPs) present fine-tuned, tailored signals at the cellular level, that have profound effects on shaping the type of immune response that is elicited. Many more experiments are necessary to investigate if in the case of TLR and DNA repair, the same types of diversity may be present, resulting in enhanced or decreased DNA repair, control of cell survival or death, emergence or deletion of cancer cells, and immune competence or immune suppression. Figuring out how DNA repair may be involved in resolving this cell fate paradox presents an exciting opportunity to better understand a novel attribute of TLR signaling.
Acknowledgements
The authors would like to thank Drs. Stefanie Vogel and Darren Perkins for their help in reviewing and editing this paper.
Footnotes
The authors declare no conflict of interest.
References
- Alexandrescu DT, Ichim TE, Riordan NH, Marincola FM, Di Nardo A, Kabigting FD, et al. (2010) Immunotherapy for melanoma: current status and perspectives. Journal of immunotherapy 33:570–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz K, Nowsheen S, Pantelias G, Iliakis G, Gorgoulis VG, Georgakilas AG (2012) Targeting DNA damage and repair: Embracing the pharmacological era for successful cancer therapy. Pharmacology & therapeutics 133:334–50. [DOI] [PubMed] [Google Scholar]
- Azzam EI, Jay-Gerin JP, Pain D (2011) Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer letters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Kagan JC (2009) A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nature reviews Immunology 9:535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, Muehleisen B, Muto J, Borkowski AW, et al. (2012) Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nature medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born AL, Born W (1987) Replicative and repair DNA synthesis after solar damage. Acta dermato-venereologica Supplementum 134:40–2. [PubMed] [Google Scholar]
- Bourquin C, Hotz C, Noerenberg D, Voelkl A, Heidegger S, Roetzer LC, et al. (2011) Systemic cancer therapy with a small molecule agonist of toll-like receptor 7 can be improved by circumventing TLR tolerance. Cancer research 71:5123–33. [DOI] [PubMed] [Google Scholar]
- Celhar T, Magalhaes R, Fairhurst AM (2012) TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunologic research. [DOI] [PubMed] [Google Scholar]
- Dietrich N, Lienenklaus S, Weiss S, Gekara NO (2010) Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PloS one 5:e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M (2012) Oxidatively induced DNA damage: Mechanisms, repair and disease. Cancer letters. [DOI] [PubMed] [Google Scholar]
- Dumitru CD, Antonysamy MA, Tomai MA, Lipson KE (2010) Potentiation of the anti-tumor effects of imidazoquinoline immune response modifiers by cyclophosphamide. Cancer biology & therapy 10:155–65. [DOI] [PubMed] [Google Scholar]
- Edwards L, Ferenczy A, Eron L, Baker D, Owens ML, Fox TL, et al. (1998) Self-administered topical 5% imiquimod cream for external anogenital warts. HPV Study Group. Human PapillomaVirus. Archives of dermatology 134:25–30. [DOI] [PubMed] [Google Scholar]
- Evans MD, Cooke MS (2004) Factors contributing to the outcome of oxidative damage to nucleic acids. BioEssays : news and reviews in molecular, cellular and developmental biology 26:533–42. [DOI] [PubMed] [Google Scholar]
- Fishelevich R, Zhao Y, Tuchinda P, Liu H, Nakazono A, Tammaro A, et al. (2011) Imiquimod-induced TLR7 signaling enhances repair of DNA damage induced by ultraviolet light in bone marrow-derived cells. Journal of immunology 187:1664–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. (2009) Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. The Journal of experimental medicine 206:1983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspari AA, Fleisher TA, Kraemer KH (1993) Impaired interferon production and natural killer cell activation in patients with the skin cancer-prone disorder, xeroderma pigmentosum. The Journal of clinical investigation 92:1135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Tsung A, Billiar T (2010) Linking oxidative stress to inflammation: Toll-like receptors. Free radical biology & medicine 48:1121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh FG, Midwood KS (2012) Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatology (Oxford) 51:7–23. [DOI] [PubMed] [Google Scholar]
- Hanke CW, Beer KR, Stockfleth E, Wu J, Rosen T, Levy S (2010) Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: results of two placebo-controlled studies of daily application to the face and balding scalp for two 3-week cycles. Journal of the American Academy of Dermatology 62:573–81. [DOI] [PubMed] [Google Scholar]
- Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. (2011) Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology 54:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–50. [DOI] [PubMed] [Google Scholar]
- Klaschik S, Tross D, Shirota H, Klinman DM (2010) Short- and long-term changes in gene expression mediated by the activation of TLR9. Molecular immunology 47:1317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. (2011) Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends in immunology 32:157–64. [DOI] [PubMed] [Google Scholar]
- Kuipers MT, van der Poll T, Schultz MJ, Wieland CW (2011) Bench-to-bedside review: Damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care 15:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–9. [DOI] [PubMed] [Google Scholar]
- Larange A, Antonios D, Pallardy M, Kerdine-Romer S (2009) TLR7 and TLR8 agonists trigger different signaling pathways for human dendritic cell maturation. Journal of leukocyte biology 85:673–83. [DOI] [PubMed] [Google Scholar]
- Lee KH, Liu YJ, Biswas A, Ogawa C, Kobayashi KS (2011) A novel aminosaccharide compound blocks immune responses by Toll-like receptors and nucleotide-binding domain, leucine-rich repeat proteins. The Journal of biological chemistry 286:5727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis W, Simanyi E, Li H, Thompson CA, Nasti TH, Jaleel T, et al. (2011) Regulation of ultraviolet radiation induced cutaneous photoimmunosuppression by toll-like receptor-4. Archives of biochemistry and biophysics 508:171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loegering DJ, Lennartz MR (2011) Protein kinase C and toll-like receptor signaling. Enzyme research 2011:537821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski S, Jantschitsch C, Maeda A, Schwarz T, Schwarz A (2010) IL-23 antagonizes UVR-induced immunosuppression through two mechanisms: reduction of UVR-induced DNA damage and inhibition of UVR-induced regulatory T cells. The Journal of investigative dermatology 130:554–62. [DOI] [PubMed] [Google Scholar]
- Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, et al. (2011) DNA released from dying host cells mediates aluminum adjuvant activity. Nature medicine 17:996–1002. [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. (2010) Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nature medicine 16:413–9. [DOI] [PubMed] [Google Scholar]
- McGettrick AF, O’Neill LA (2010) Localisation and trafficking of Toll-like receptors: an important mode of regulation. Current opinion in immunology 22:20–7. [DOI] [PubMed] [Google Scholar]
- Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, Resnick MA (2011) The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS genetics 7:e1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. (2011) Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circulation research 108:235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal D, Saccheri F, Venereau E, Pusterla T, Bianchi ME, Rescigno M (2010) TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. The EMBO journal 29:2242–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthusamy V, Piva TJ (2010) The UV response of the skin: a review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Archives of dermatological research 302:5–17. [DOI] [PubMed] [Google Scholar]
- Nace G, Evankovich J, Eid R, Tsung A (2012) Dendritic cells and damage-associated molecular patterns: endogenous danger signals linking innate and adaptive immunity. Journal of innate immunity 4:6–15. [DOI] [PubMed] [Google Scholar]
- Natarajan AT, van Zeeland AA, Obe G (1988) Some studies on the DNA-repair-eliciting and genotoxic activity of cell-free extracts of Lactobacillus bifidus. Mutation research 206:47–54. [DOI] [PubMed] [Google Scholar]
- Norval M, Halliday GM (2011) The consequences of UV-induced immunosuppression for human health. Photochemistry and photobiology 87:965–77. [DOI] [PubMed] [Google Scholar]
- Oster-Schmidt C (2004) Two cases of squamous cell carcinoma treated with topical imiquimod 5%. Journal of the European Academy of Dermatology and Venereology : JEADV 18:93–5. [DOI] [PubMed] [Google Scholar]
- Park SJ, Youn HS (2010) Suppression of homodimerization of toll-like receptor 4 by isoliquiritigenin. Phytochemistry 71:1736–40. [DOI] [PubMed] [Google Scholar]
- Piccinini AM, Midwood KS (2010) DAMPening inflammation by modulating TLR signalling. Mediators of inflammation 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince LR, Whyte MK, Sabroe I, Parker LC (2011) The role of TLRs in neutrophil activation. Current opinion in pharmacology 11:397–403. [DOI] [PubMed] [Google Scholar]
- Rastogi RP, Richa, Kumar A, Tyagi MB, Sinha RP (2010) Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. Journal of nucleic acids 2010:592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin RL, Tang FL, Chan EK, Pollard KM, Tsay G, Tan EM (1986) IgG subclasses of autoantibodies in systemic lupus erythematosus, Sjogren’s syndrome, and drug-induced autoimmunity. Journal of immunology 137:2528–34. [PubMed] [Google Scholar]
- Schwarz T, Schwarz A (2009) DNA repair and cytokine responses. The journal of investigative dermatology Symposium proceedings / the Society for Investigative Dermatology, Inc [and] European Society for Dermatological Research 14:63–6. [DOI] [PubMed] [Google Scholar]
- Shibata T, Motoi Y, Tanimura N, Yamakawa N, Akashi-Takamura S, Miyake K (2011) Intracellular TLR4/MD-2 in macrophages senses Gram-negative bacteria and induces a unique set of LPS-dependent genes. International immunology 23:503–10. [DOI] [PubMed] [Google Scholar]
- Sommariva M, De Cecco L, De Cesare M, Sfondrini L, Menard S, Melani C, et al. (2011) TLR9 agonists oppositely modulate DNA repair genes in tumor versus immune cells and enhance chemotherapy effects. Cancer research 71:6382–90. [DOI] [PubMed] [Google Scholar]
- Sun S, Rao NL, Venable J, Thurmond R, Karlsson L (2007) TLR7/9 antagonists as therapeutics for immune-mediated inflammatory disorders. Inflammation & allergy drug targets 6:223–35. [DOI] [PubMed] [Google Scholar]
- Swanson N, Abramovits W, Berman B, Kulp J, Rigel DS, Levy S (2010) Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: results of two placebo-controlled studies of daily application to the face and balding scalp for two 2-week cycles. Journal of the American Academy of Dermatology 62:582–90. [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Zeh HJ 3rd, Lotze MT (2011) High-mobility group box 1, oxidative stress, and disease. Antioxidants & redox signaling 14:1315–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher TH, Luzina I, Fishelevich R, Tomai MA, Miller RL, Gaspari AA (2006) Topical imiquimod treatment prevents UV-light induced loss of contact hypersensitivity and immune tolerance. The Journal of investigative dermatology 126:821–31. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Sher A (2007) Cooperation of Toll-like receptor signals in innate immune defence. Nature reviews Immunology 7:179–90. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Nagai Y, Kariyone A, Shibata T, Kaisho T, Akira S, et al. (2009) Toll-like receptor 7 cooperates with IL-4 in activated B cells through antigen receptor or CD38 and induces class switch recombination and IgG1 production. Molecular immunology 46:1278–88. [DOI] [PubMed] [Google Scholar]
- Ubol S, Halstead SB (2010) How innate immune mechanisms contribute to antibody-enhanced viral infections. Clinical and vaccine immunology : CVI 17:1829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink AA, Strickland FM, Bucana C, Cox PA, Roza L, Yarosh DB, et al. (1996) Localization of DNA damage and its role in altered antigen-presenting cell function in ultraviolet-irradiated mice. The Journal of experimental medicine 183:1491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltz P, Carchman EH, Young AC, Rao J, Rosengart MR, Kaczorowski D, et al. (2011) Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 7:315–20. [DOI] [PubMed] [Google Scholar]
- Wheeler DS, Chase MA, Senft AP, Poynter SE, Wong HR, Page K (2009) Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respiratory research 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Singh KK, Chen B, Samson L (1993) A common element involved in transcriptional regulation of two DNA alkylation repair genes (MAG and MGT1) of Saccharomyces cerevisiae. Molecular and cellular biology 13:7213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. (2006) HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26:174–9. [DOI] [PubMed] [Google Scholar]
- Zheng L, Asprodites N, Keene AH, Rodriguez P, Brown KD, Davila E (2008) TLR9 engagement on CD4 T lymphocytes represses gamma-radiation-induced apoptosis through activation of checkpoint kinase response elements. Blood 111:2704–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Thornton K, Reed E (2000) Computer based analyses of the 5’-flanking regions of selected genes involved in the nucleotide excision repair complex. International journal of oncology 17:375–80. [DOI] [PubMed] [Google Scholar]