

ABSTRACT
BCMA-targeting chimeric antigen receptor (CAR)-T cell therapy has shown remarkable clinical efficacy against multiple myeloma, yet antigen escape and tumor relapse still occur after the use of these therapies. Designing CAR-T therapies that targets multiple antigens simultaneously seems a feasible way to avoid antigen escape, and it has been extensively studied elsewhere. Here, we report novel BCMA-OR-CD38 Tan CAR T cells that can trigger robust cytotoxicity against target cells expressing either BCMA or CD38. We demonstrate that, in in vitro studies, these BCMA-OR-CD38 Tan CAR T cells exhibit similar CAR expression, superior cytotoxicity and antigen-stimulated T cell proliferation as compared to single-targeted CAR T cells or CD38-OR-BCMA Tan CAR T cells. Importantly, these BCMA-OR-CD38 Tan CAR-T cells can achieve complete tumor clearance in myeloma-bearing mice with no relapse observed through the course of these experiments. Finally, this BCMA-OR-CD38 Tan CAR was fully compatible with existing clinical grade T cell manufacturing procedures and can be implemented using current clinical protocols. Taken together, our results present an effective solution to the challenge of antigen escape in BCMA CAR T-cell therapies.
KEYWORDS: Tandem-CAR T, CD38 CAR, BCMA CAR, multiple myeloma, antigen escape
Introduction
Multiple myeloma (MM) is the second most common hematological malignancy worldwide, and resulted in 113,474 deaths globally in 2019.1 As a cancer of plasma cells, MM is characterized by the infiltration of malignant plasma cells in the bone marrow and is associated with high levels of monoclonal (M) protein in the blood and/or serum.2. In recent years, the introduction of drugs targeting MM to its microenvironments, such as the proteasome inhibitor bortezomib and the immunomodulatory drugs (IMiDs) thalidomide and lenalidomide, have been used in initial, consolidation, maintenance, and salvage therapies, and have markedly improved patient outcomes and survival rates.3 However, MM remains largely incurable, due to the development of resistant and refractory disease.4
Chimeric antigen receptor T cell (CAR-T) therapy has emerged as a novel immunotherapy that has tremendous potential for long-term disease control in some hematologic malignancies,5–7 which has encouraged the development of CAR-T cells for treating MM, especially in the context of relapsed/refractory disease.8 Among the several CAR T-cell therapies, major efforts have been devoted to CAR T-cells targeting B-cell maturation antigen (BCMA), and more than 20 early-phase clinical studies are currently performed.9,10 Notably, the bb2121 anti-BCMA CAR-T Cell (Idecabtagene Vicleucel, ide-cel) from Bluebird Bio was approved by the Food and Drug Administration (FDA) as the first BCMA CAR T cell for the treatment of adult patients with relapsed or refractory MM on Mar 26th, 2021.11 However, several clinical trials have reported patients’ relapse involving tumor cell BCMA antigen loss or expression down-regulation below the threshold required for CAR T cell activity.12–15 Furthermore, a recent report demonstrated that biallelic loss of BCMA is one of the resistance mechanisms to Idecabtagene Vicleucel therapy.16 T cells expressing a single-chain bispecific CAR, also known as “Tan CAR”, was previously proposed in a proof-of-concept approach to increase the specificity of effector cells and to offset antigen escape.17 Subsequently, multiple studies have demonstrated that Tan CARs reduced the likelihood of antigen escape and potentially improved the efficacy of CAR-T cell therapies.18–20 The design of Tan CARs is based on an “OR”-gated signal computation, and when either of two different antigens is present on a target cell, the T-cells will be activated, thus, tumor cells have to lose both antigens simultaneously to escape T cell surveillance.17,21 To construct tandem dual CARs for the treatment of MM, it is necessary to choose another antigen that is widely expressed in MM cells. In addition to BCMA, CD38 has recently been identified as a feasible antigen target for MM, since myeloma cells show strong and uniform expression of CD38 at different disease stages.22,23 Moreover, anti-CD38 CAR-T-cell therapy is currently being tested in the clinic trials (NCT03464916, NCT03754764). In a previous study, we identified an anti-CD38 scFv from an scFv phage display library, and the therapeutic effects of CD38 CAR-T cells on myeloma cells has been demonstrated with in vivo and in vitro experiments (a manuscript during revision). We thus reason that simultaneous targeting of BCMA and CD38 would take advantage of the therapeutic efficacy of BCMA targeting while preventing tumor escape due to BCMA loss.
Here, we describe two novel Tan CARs which were composed of an antigen recognition domain (an anti-CD38 and an anti-BCMA scFv in tandem), CD8 hinge and transmembrane domain, CD28 costimulatory domain, and the signaling portion of CD3ζ. Our results found that Tan-CAR T cells could trigger robust T cell-mediated cytotoxicity and cytokine production when either BCMA or CD38 was present on a target cell. Moreover, these Tan-CAR-T cells had significantly higher cytotoxicity and proliferation than single-targeted CAR T cells when encountering BCMA and CD38 antigens simultaneously. Notably, BCMA-OR-CD38 Tan CAR-T cells exhibited more potent antitumor and proliferative activity than CD38-OR-BCMA Tan CAR-T cells. Next, we assessed the efficacy of BCMA-OR-CD38 Tan CAR-T cells against multiple myeloma cells in vivo. The results showed that BCMA-OR-CD38 Tan CAR-T cells had robust therapeutic effects on an NPG mouse model bearing myeloma, and could achieve complete tumor clearance four days after the second dose of Tan-CAR T cells. Thus, our results suggest that this tandem-dual antigen targeting strategy represents an effective anti-neoplastic therapy and may ultimately provide an effective safeguard against antigen escape of BCMA in adoptive T-cell therapies for MM.
Results
Construction and generation of Tan-CAR-T cells
The antigen-binding domain of BCMA-OR-CD38 Tan-CAR or CD38-OR-BCMA Tan-CAR was composed of an anti-BCMA scFv (VL-linker-VH) and an anti-CD38 scFv (VH-linker-VL) in tandem connected via a long, flexible linker peptide (Gly4Ser)4 (Figure 1(b)). The difference between these two Tan CARs is the order of the two scFv domains (Figure 1(c)). The corresponding single-targeted CARs contained only the respective scFvs for antigen binding (Figure 1(a)). The retroviral vector titers of Tan-CAR were high (BCMA-OR-CD38 CAR: 3.59 × 108 CFU/mL; CD38-OR-BCMA CAR: 2.82 × 108 CFU/mL) though this was lower than the CD19 CAR (as retroviral vector standard), BCMA-CAR and CD38-CAR groups (Figure 1(e)). The transduction efficiency of BCMA-OR-CD38 Tan-CAR + T was 85.7%, CD38-OR-BCMA Tan-CAR + T was 74.0%, BCMA-CAR+ T was 78.2% and CD38-CAR+ T was 87.3% (Figure 1(g)). The Tan CARs were expressed with a similar efficiency as the BCMA CAR or CD38 CAR, implying that the targeting of this dual-antigen CAR modification did not affect the transduction efficiency of this CAR. Notably, the retroviral vector titers and T cell transduction efficiency of BCMA-OR-CD38 CAR were higher than that of CD38-OR-BCMA Tan-CAR. Transgenic copy counts, or vector copy numbers (VCNs), are one of the indicators for evaluating the safety of CAR T cell products. The FDA recommends that the integration copy number should be < 5 copies per genome.24 In our results, the copy number of the CAR molecule (BCMA-CAR, CD38-CAR, or Tan-CARs) in the corresponding CAR-T cells were all less than 5 copies/ CAR-T cell (Figure 1(f)).
Figure 1.
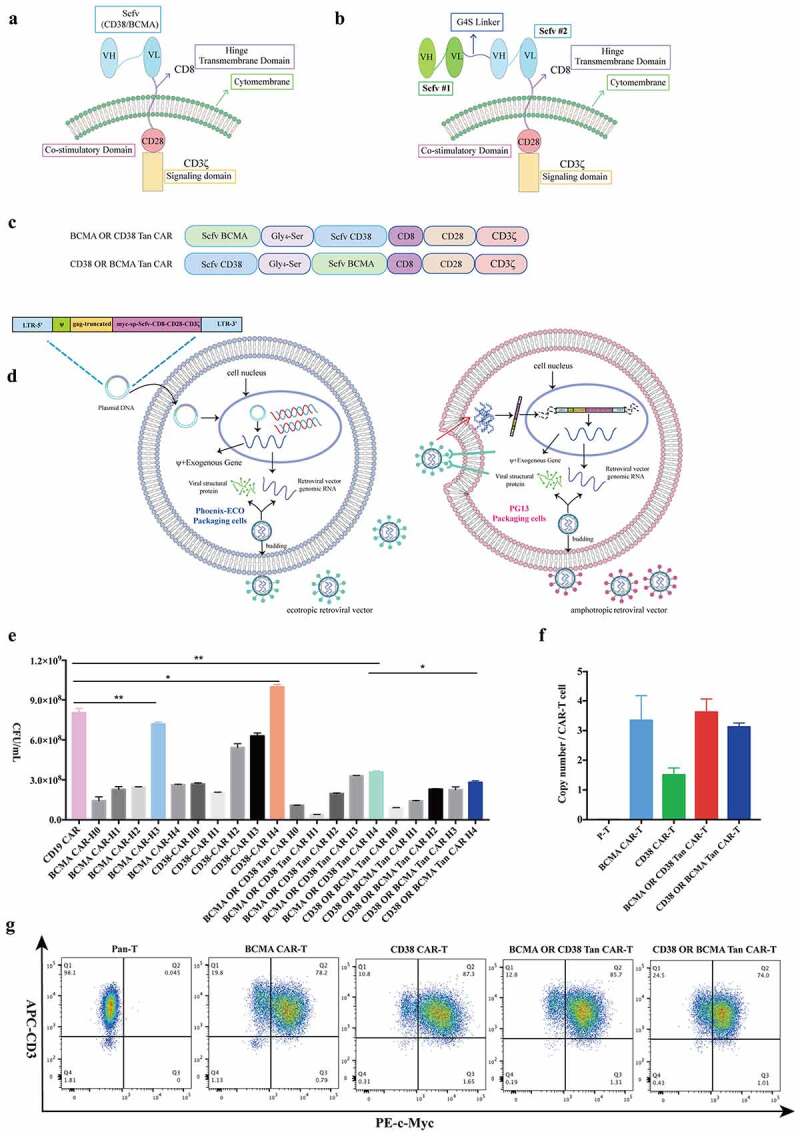
Construction and generation of retroviral vectors. (a) Schematic of a second-generation, single-targeted CAR (BCMA-CAR or CD38-CAR). (b) Schematic of a bi-specific, OR-gate tandem-CAR. (c) Schematic of two Tan CARs containing variations in the order of scFv domains. CD8 is the hinge and transmembrane domain, CD28 is the costimulatory domain. (d) Schematic of the retroviral vector packaging process. First, the Phoenix-ECO cells produced ecotropic retroviral vectors by transient transfection and then integrated them into the packaging cell line PG13 to produce amphotropic retroviral vectors that can transduce human primary T cells. (e) The titer detection of amphotropic retroviral vectors by Q-PCR. The amphotropic retroviral vector supernatants from CD38-CAR, BCMA-CAR, or Tan-CARs were harvested every 24 h for a total of 5 days, named H0, H1, H2, H3, and H4. (f) Detection of the copy number of CAR molecules. Q-PCR was performed using primers specific for this transgene, and genomic DNA extracted from the isolated and activated human T cells 48 h after transducing was used as templates. (g) Analysis of transduction efficiency. The retroviral vectors BCMA-CAR, CD38-CAR, or Tan-CARs transduced the isolated and activated human T cells, and the transduction efficiency was evaluated by detection of c-Myc protein expression using flow cytometry. The data shown is the mean ± standard deviation (SD). The results were analyzed with a student’s t test. Error bars represent the SD, * p < .05, **p < .01
Detection of effective Tan CAR-T targeting to cells expressing various antigens
To evaluate specific cytotoxicity of Tan-CAR T cells on myeloma cells, the BCMA-OR-CD38 Tan CAR-T, the CD38-OR-BCMA Tan CAR-T, BCMA CAR-T, CD38 CAR-T and Pan-T cells were co-incubated respectively with BCMA-K562 or CD38-Raji cells for 8 hours. Annexin-V staining was used to determine the apoptosis of tumor cells. The Tan CAR-T cells showed significant cytotoxicity toward BCMA-K652 and CD38-Raji cells, while single-targeted CAR-T cells only respond to BCMA+ or CD38+ target cells, respectively (Figure 2(a,c)). Notably, the cytotoxicity of BCMA-OR-CD38 Tan CAR-T associated with BCMA-K652 and CD38-Raji cells was significantly higher than CD38-OR-BCMA Tan CAR-T cells (Figure 2(a)). The effective targeting of Tan CAR-T cells toward CD38 and BCMA antigens simultaneously was investigated based on two distinct strategies, the apoptosis or survival rate analysis of target cells after them co-cultured with CAR-T cells for 8 hours. The target cells consisted of BCMA-K562 and CD38-Raji cells mixed in a ratio of 1:1. The results showed that Tan CAR-T cells efficiently lysed both BCMA-K562 and CD38-Raji cells, and the cytotoxicity associated with these cells was significantly higher than single-targeted CAR-T cells (Figure 2(a,c)). We also carried out another experiment to further compare the cytolytic efficiency of single-targeted CAR-T cells with Tan CAR-T cells. RPMI-Luc cells expressing both CD38 and BCMA antigens were utilized for co-culturing with different CAR-T cells, after 8 hours of incubation, the BCMA-OR-CD38 Tan-CAR-T cells showed more significant cytotoxicity than single-targeted CAR-T cells and CD38-OR-BCMA Tan-CAR-T cells (Figure 2(b)). The un-modified K562 cells (CD38−BCMA−) were negative control cells and there was no significant cytotoxicity in this background (Figure 2(a)). The Pan-T cells (un-transduced T cells) were used as control effective T cells, and their responses to target cells were limited (Figure 2(a-c)).
Figure 2.
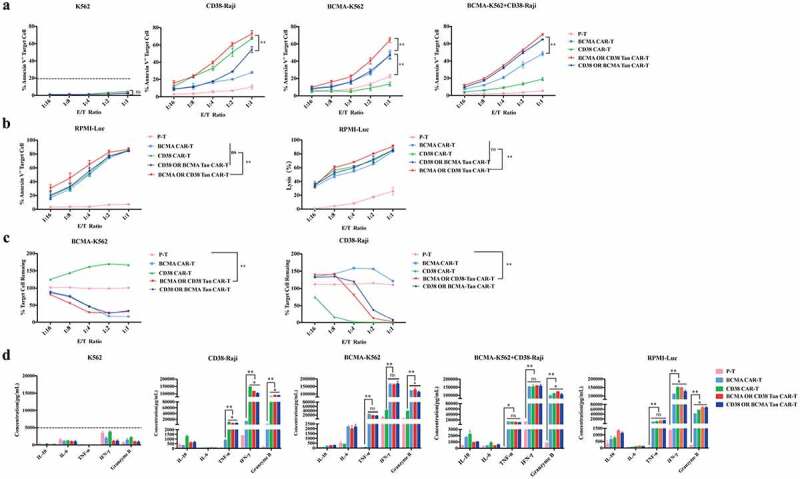
The specific cytotoxicity of Tan CAR-T cells after antigen stimulation. (a) The cytotoxicity of single-targeted CAR-T and Tan CAR-T cells after 8 hours of co-incubation with BCMA-K562, CD38-Raji, or BCMA-K562+ CD38-Raji cells (mixed in 1:1) at different effect-to-target ratios. Apoptosis was determined using an Annexin-V apoptosis Kit. (b) The cytotoxicity of single-targeted CAR-T and Tan CAR–T cells after 8 hours of co-incubation with RPMI-Luc was detected with two different methods, Annexin-V apoptosis (left panel) and luciferase assay (right panel). (c) The specific cytotoxicity of Tan CAR-T cells for BCMA-K562 and CD38-Raji cells. BCMA-K562+ CD38-Raji cells (mixed in 1:1) were co-cultured with single-targeted CAR-T and Tan CAR-T cells for 48 hours, and the survival of BCMA-K562 or CD38-Raji cells was determined. (d) Cytokine release from Tan-CAR T cells in response to target cells as indicated, with the cytokines evaluated including IL-6, IL-10, TNF-α, IFN-γ, and Granzyme B. Data shown is the mean ± standard deviation (SD). The results were analyzed by two-way ANOVA, and error bars represent the SD, * p < .05, **p < .01
The release of proinflammatory cytokines by CAR T cells was then measured using the Human CD8/NK Panel CBA Kit. After 8 hours of incubation of effector and target cells together, the cell culture supernatant was harvested and the levels of cytokines IL-10, IL-6, TNF-α, IFN-γ and Granzyme B were measured by flow cytometry. The levels of TNF-α, IFN-γ and Granzyme B released by Tan CAR T cells significantly increased after target cell stimulation to levels that was comparable to that generated by BCMA-CAR or CD38 CAR T cells in response to their respective cognate antigens (Figure 2(d)). The K562 cells that did not express CD38 or BCMA antigen were used as negative control cells. The results showed that Tan CAR T cells released limited proinflammatory cytokines after co-incubated with K562 cells (Figure 2(d)).
Tan CAR-T-cell proliferation in response to CD38+ and BCMA+ tumor cells
To investigate if tumor cells expressing specific antigens could stimulate CAR-T cell proliferation, we performed a CFSE proliferation assay. CAR-T and Pan-T cells were labeled with CFSE and co-cultured with BCMA K562+ CD38 Raji (mixed in 1:1) or PRMI-Luc cells, and the T-cell proliferation was evaluated via detection of decreasing CFSE fluorescence intensity using flow cytometry. The results showed that Tan-CAR T cells, particularly BCMA-OR-CD38 Tan-CAR T cells, proliferated significantly more than single-targeted CAR-T cells and CD38-OR-BCMA Tan-CAR T cells in response to activation by target cells at a 1:1 effect-to-target ratio (Figure 3(a)). Furthermore, we also plotted the T cell proliferation and viability curves without target cell stimulation. These showed that tan-CAR T cells proliferated to an extent that was similar to single-targeted CAR-T cells (Figure 3(b)).
Figure 3.
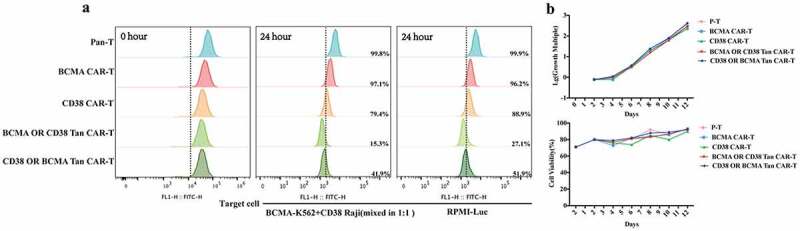
T cell proliferation detection. (a) CAR T cells labeled with CFSE were co-cultured with BCMA K562+ CD38 Raji (mixed in 1:1) or PRMI-Luc cells for 24 hours at a 1:1 effect-to-target ratio, and the CFSE fluorescence intensity was determined by flow cytometry. (b) Pan-T and CAR- T cells were counted every 2 days using an automated cell counter and cell growth and viability curves were plotted here
BCMA-OR-CD38 Tan CAR-T cells are effective in the treatment of tumors in vivo
As illustrated above, BCMA-OR-CD38 Tan CAR-T cells exhibited more potent antitumor and proliferative activity than CD38-OR-BCMA Tan CAR-T cells. Thus, we next assessed the efficacy of BCMA-OR-CD38 Tan CAR-T cells against multiple myeloma cells in vivo in an intravenous RPMI-Luc tumor xenograft model. After tumor injection, tumor-bearing mice with similar tumor sizes were randomly divided into 5 groups (n = 8 per group), and Pan- T, BCMA CAR-T, CD38 CAR-T, or BCMA-OR- CD38 Tan CAR-T cells were provided by intravenous injection on days 12 and 19 (Figure 4(a)). Bioluminescence imaging revealed that either single-targeted CAR-T cells or BCMA-OR-CD38 Tan CAR-T cells yielded complete tumor clearance by day 23 (Figure 4(b)). Correspondingly, we also detected tumor cell residues in the bone marrow (BM), liver, spleen, and blood of mice by flow cytometry. The result showed that there were almost no tumor cells observable in the mice receiving BCMA-OR-CD38 Tan CAR-T or single-targeted CAR-T treatment (Figure 4(c)). Notably, two weeks after the second CAR-T injection, we measured the levels of human IFN-γ in the blood serum using ELESA assay, which were significantly higher for BCMA-OR-CD38 Tan CAR-T cell-treated mice than for single-targeted CAR-T cell-treated mice, indicative of greater IFN-γ production by the Tan CAR-T cells (Figure 4(e)). Next, we evaluated the expansion of CAR-T cells by detecting the percentage of CD3+ T cells in the peripheral blood. The results indicated that BCMA-OR-CD38 Tan CAR-T cells had experienced more extensive expansion in mice than either the Pan-T or CD38 CAR-T cells did (Figure 4(d)). Finally, the safety of this Tan CAR-T cell therapy was evaluated, and the results showed that mice in the BCMA-OR-CD38 Tan CAR-T cells-treated groups had significantly longer median survival times than those from the Untreated or Pan-T groups (Figure 4(f)). However, there was no significant difference between BCMA-OR-CD38 Tan CAR-T and single-targeted CAR-T cells in terms of tumor clearance or median survival times.
Figure 4.
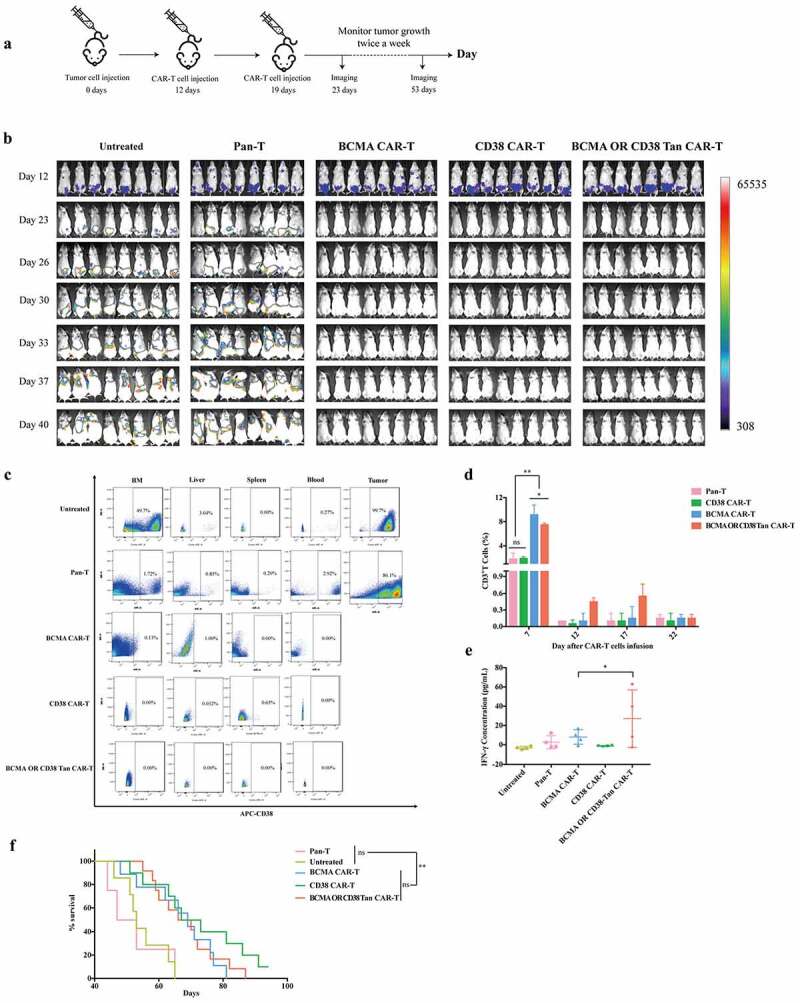
BCMA-OR-CD38 Tan CAR-T cells showed significant anti-tumor activity in vivo. (a) Outline of in vivo xenograft tumor model treatment schedule: NPG mice were i.v. injected with 2 × 106 RPMI-Luc cells on day 0. After that, tumor-bearing mice were randomly divided into 5 groups (n = 8 per group) and received Pan-T, BCMA CAR-T, CD38 CAR-T, or BCMA-OR-CD38 Tan CAR-T cells at an i.v. dose of 1 × 108/kg on days 12 and 19. Tumor growth was followed by MIIS living imaging every 3 days beginning on day 4 after the second CAR-T injection. (b) The mouse tumor burden at specific time points is indicated by bioluminescence radiance (n = 8 mice per group), and the results were pooled from two independent experiments, with the CAR-T cells being prepared with PBMCs obtained from two health donors. (c) Mouse BM, liver, spleen, blood and tumor cells were stained with anti-human CD38 APC to evaluate whether there were still residual RPMI-Luc cells in mice after CAR-T cells infusion. (d) The percentage of CD3+ T cells in peripheral blood was used to evaluate the expansion of CAR T cells. Blood was collected by tail vein and stained with the anti-human CD3 BV786. (e) Two weeks after the second CAR-T injection, the levels of IFN-γ in serum from each group were measured using ELISA assay. (f) Survival analysis of mice treated with Pan-T, BCMA CAR-T, CD38 CAR-T, or BCMA-OR-CD38 Tan CAR-T cells. Kaplan-Meier survival curves were tested using the Mantel–Cox log rank test. The data is shown as the mean ± standard deviation (SD). The results were analyzed by two-way ANOVA, error bars represent the SD, and * p < .05, **p < .01
Materials and methods
Plasmid construction and production of retroviral vectors
The tandem-CARs retroviral vector consisted of the following components in-frame from the 5ʹ end to the 3ʹ end: a signal peptide sequence derived from the murine Ig-H (immunoglobulin heavy chain), a human c-Myc tag, an anti-BCMA-scFv derived from the C11D5.3 monoclonal antibody (mAb),25 an anti-CD38 scFv identified from an scFv phage display library in our previous study (a manuscript during revision), the hinge and transmembrane regions of the CD8 molecule, the costimulatory domain of CD28, and the CD3ζ signaling domain. The codon optimization and sequence synthesis for this was carried out at General Biosystems (China). The anti-CD38 and anti-BCMA scFv fragments were assembled by overlap extension PCR and cloned to an MFG retroviral backbone, which is referred to as the BCMA-OR-CD38 Tan-CAR or CD38-OR-BCMA Tan-CAR retroviral vector. The difference between these two Tan CARs is the order of the two scFv domains. We also prepared CD38-CAR and BCMA-CAR retroviral vectors, which had a single CD38 or BCMA scFv domain. The process for the production of Tan CARs and single-targeted CARs retroviral vectors was consistent with the manufacture of clinical grade anti-CD19-CAR retroviral vectors as previously described.26,27 Retroviral vectors producer cell lines were established using two different packing cell lines. The plasmids encoding these CAR genes were introduced into the human-derived ecotropic packaging cell line Phoenix-eco, and the retroviral vector supernatant transiently produced by these transfected cells was harvested and integrated stably into the genomic DNA of the murine-derived amphotropic packaging cell line PG13 (Figure 1(d)). The retroviral vector supernatant produced by PG13 cells was harvested every 24 hours for a total of five days. The collected samples were called H0, H1, H2, H3, and H4, and were utilized for the transduction of human primary T cells.
Cell-line generation and maintenance
K562 cells were obtained from the Cell Resource Center, Peking Union Medical College (Beijing, China). Raji cells are a CD38+ human B lymphocyte cell line that was obtained from the American Type Culture Collection (ATCC, United States). BCMA+ GFP+-K562 cells were a generous gift from Dr. Wu (China Agricultural University) received in 2018. GFP-Luciferase RPMI8226 cells (RPMI-Luc) were a CD38+ and BCMA+ myeloma cell line that were obtained from the ATCC. The PG13 gibbon ape leukemia virus packaging cell line and the human ecotropic packaging cell line Phoenix-ECO were obtained from ATCC. K562, Raji, and RPMI cells were maintained in RPMI-1640 medium (Gibco, United States) supplemented with 10% fetal bovine serum (FBS) (Gibco, United States), with 1% penicillin-streptomycin (P/S) solution (Gibco, United States). PG13 and Phoenix ECO cells were cultured in DMEM (Gibco, United States) supplemented with 10% FBS and 1% P/S. All cells were cultured in an incubator (Thermo Fisher, United States) at 37°C and 5% CO2. All cell lines were tested negative for the mycoplasma contamination and cell-surface markers for these cell lines were validated by flow cytometry.
The generation of CAR-T cells
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors by gradient centrifugation using LymphoprepTM (STEMCELL Technologies, Canada). The use of human PBMCs was approved by the Ethics Committee of Beijing University of Chinese Medicine, and all donors gave informed consent. After stimulating these PBMCs with 100 ng/mL of the anti-CD3 monoclonal antibody OKT3 (Sino Biological, China) and 100 U/mL IL-2 (Sino Biological, China) for 48 hours, these T cells were transduced using Tan CARs and single-targeted CARs retroviral vectors. The transduction efficiency was determined using flow cytometry. All T cells were expanded in AIM V medium (Gibco, United States) supplemented with 10% FBS, 1% P/S and IL-2 (100 U/mL), which was renewed every 2 days or as necessary.
Quantitative real-time PCR (Q-PCR)
The titers of retroviral vectors produced by PG13 were determined using Q-PCR. The RNA from retroviral vectors (H0, H1, H2, H3, and H4) was extracted using a QIAamp Viral RNA Mini Kit (QIAGEN, Germany) and reverse transcribed using the QuantiNova Reverse Transcription Kit (QIAGEN, Germany), all of which followed the protocol described by the manufacturer. A five-point standard curve that consisted of 104 to 108 copies/μL of CAR plasmid DNA was prepared, and the copy number of these retroviral vectors were calculated by the absolute quantitative method. A 95 bp fragment containing portions of the MFG retroviral backbone sequence was amplified using a forward primer (5´-GACACCAGACTAAGAACCTAGAAC-3´) and a reverse primer (5´- AGCTGCGATGCCGTCTACTTTGAG-3´).
The copy number of the CAR gene was quantified by Q-PCR according to the protocol described previously.28 The genomic DNAs from PBMCs 48 h post transduction were extracted using the QIAamp DNA Mini and Blood Mini Kit (Qiagen, Germany). A five-point standard curve was generated for analyses, consisting of 104 to 108 copies/μL of CAR plasmid DNA spiked into 100 ng of non-transduced T cells genomic DNA to control for background signals. The amplification of GAPDH was used as an internal control for the normalization of DNA quantities. The CAR transgene forward primer used was: 5ʹ-ATCGCTCACAACCAGTCG-3ʹ; while the reverse primer used was: 5ʹ-GGTCAGGGAAGTTTACAAGG-3ʹ. Q-PCR was performed with 100 ng of genomic DNA in each reaction using a QuantStudioTM 6 Flex Real-Time PCR System (Life technologies, United States).
Flow cytometry
For the determination of the transduction efficiency of T cells and retroviral vector titers, one million CAR-T cells or PG13 cells were harvested and stained with human c-Myc PE-conjugated antibody (R&D System, United States) for 30–60 mins at 4°C. The uncombined antibody solution was washed away using phosphate-buffered saline (PBS) (Gibco, United States). Fluorescent signals were detected using CytoFLEX (Beckman, United States) and analyzed in CytoFLEX analysis software.
Cytotoxicity assays
BCMA-K562, CD38-Raji, BCMA-K562+ CD38-Raji (mixed in 1:1) or RPMI-Luc target cells were seeded in a 96-well plate at 8 × 104 cells/well and co-incubated with Pan-T (un-transduced T cells), CD38 CAR-T, BCMA CAR-T, BCMA-OR-CD38 Tan CAR-T, or CD38-OR-BCMA Tan CAR-T cells at different effect-to-target ratios (1:16, 1:8. 1:4, 1:2, and 1:1). The effector cell seeding density was based on CAR+ cell counts. After 8 hours of incubation, these cells were stained with CD3-BV421 (BD, United States) for 60 mins. The cells were then washed with PBS and stained with Annexin V-Alexa Fluor 647 (Bio Friend, China) for 30 mins at room temperature. These cells were immediately analyzed by CytoFLEX flow cytometry.
The survival rates of BCMA-K562 or CD38-Raji cells were analyzed after BCMA-K562+ CD38-Raji cells (mixed in 1:1) were co-cultured with Pan-T and CAR-T cells at different effect-to-target ratios. After 48 hours of incubation, cells were harvested and stained with CD3-BV421 or CD38-APC (Invitrogen, United States) for 60 mins. Fluorescent signals were detected using a CytoFLEX (Beckman, United States) and analyzed using FlowJo software (FlowJo LLC).
For RPMI-Luc cells, a luciferase assay was carried using the ONE-GloTM EX Luciferase Assay System (Promega, United States) after co-culturing with CAR-T cells for 8 hours. The cells were reacted with substrate solution for 3 mins, and the relative luminescence units (RLU) were determined using a SpectraMax i3x Multi-Mode Microplate Reader (Molecular Devices, United States). The lysis rate of tumor cells was calculated according to the following formula:
.
Cytokine release assays
Target cells were seeded in 96-well plates at 8 × 104 cells/well and co-cultured with Pan-T or CAR-T cells at a ratio of 1:1 for 8 hours. The effector cell seeding density was based on the CAR+ cells count. The cell culture supernatant samples were harvested, and the levels of various cytokines were evaluated using the LEGENDplexTM Multi-Analyte Flow Assay Kit (Human CD8/NK Panel) (BioLegend, United States). Capture beads consisting of IL-10, IL-6, TNF-α, IFN-γ, and Granzyme B were mixed and co-incubated with supernatant samples for 2 hours at room temperature on a shaker. After washing, the detection antibody cocktail and streptavidin-phycoerythrin (SA-PE) were added sequentially. The concentration of cytokines was determined using a standard curve generated in the same assay. Fluorescence signals were detected using a BD LSRFortessa™ Cell Analyzer (BD, United States) and flow data was analyzed using LEGENDplex™ online analysis software.
T cell proliferation assays
BCMA-K562+ CD38-Raji (mixed in 1:1) or RPMI-Luc target cells were seeded in 96-well plates at 8 × 104 cells/well. These Pan-T and CAR-T cells were stained with Carbo-xyfluorescein Diacetate Succinimidyl Ester (CFSE) (BD, United States) for 30–60 mins at 37°C. After washing with AIM-V medium, these CAR-T cells were incubated with target cells at a ratio of 1:1. The effector cell seeding density was based on the CAR+ cells count. After 24 h of incubation, the cells were stained with CD3-APC, the CFSE fluorescence intensity was determined using a CytoFLEX and the data was analyzed using FlowJo software. The Pan-T and CAR-T cell counts were performed using a CountessTM II Automated Cell Counter (Thermo Fisher, United States) every 2 days, and the T cell proliferation and viability curves were plotted using GraphPad Prism 7.0a software.
CAR-T cell activity detection in vivo
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of Beijing University of Chinese Medicine. Six- to seven-week-old female NOD.Cg-Prkdcscid Il2rgtmVst/Vst (NPG) mice were obtained from Vitalstar Biotechnology Co. (Beijing, China). To establish xenograft models, these NPG mice were intravenously injected (i.v.) with a RPMI-Luc tumor cell line (2 × 106 cells/mouse) on day 0. Then these mice were randomly divided into five groups (Untreated, Pan-T, CD38 CAR-T, BCMA CAR-T, BCMA-OR-CD38 Tan CAR-T and CD38-OR-BCMA Tan CAR-T) on day 12 after tumor injection. CAR T cells were injected intravenously into tumor-bearing mice at a dose of 1 × 108/kg on days 12 and 19. The tumor progression was monitored using the MIIS living image system (Molecular Devices, United States) every 3 days beginning on day 4 after the second CAR-T injection. For in vivo imaging, the mice were injected intraperitoneally with 150 mg/kg VivoGlo™ Luciferin (Promega, United States) and imaged under isoflurane anesthesia using the MIIS living imaging system. We also utilized flow cytometry to further evaluate whether there were still residual tumor cells in these mice. In this experiment, blood was collected by removing the eyeball, liver, spleen, and bone marrow, and tumor tissues were isolated after these mice were sacrificed. The liver, spleen and tumor cells were obtained by grinding tissues carefully, while bone marrow cells were aspirated from femurs and tibias, and the lymphocytes were isolated by lysing red blood cells. Next, the cells were stained with anti-human CD38-APC (BD, United States) for 60 mins. After washing with PBS, the cells were stained with 7-AAD (5 µl/test) (BD, United States) for 10 mins before flow cytometry (BD LSRFortessa™ Cell Analyzer) analysis. At different time points after the second CAR-T cell infusion, the percentage of CD3+ T cells in the peripheral blood was evaluated by flow cytometry. The blood was collected from the tail vein and stained with an anti-human CD3-BV786 antibody (BD, United States) and 7-AAD, this detection was carried out using a BD LSRFortessa™ Cell Analyzer. Two weeks after the second CAR-T injection, blood was collected via the tail vein, after at least 2 hours standing, the serums were obtained by centrifuging at 3000 × g for 20 minutes at 4°C. The levels of human IFN-γ in serum from each group were measured using Human IFN-gamma ELISA Kit (Proteintech, United States). Finally, the mice were sacrificed when they lost the ability to eat and exercise spontaneously, and the survival time for each mouse was recorded to plot Kaplan-Meier survival curves.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 7.0a software. For the comparisons of two groups, a two-tailed Student t test or nonparametric test was performed. When comparing multiple groups, a one-way ANOVA or a two-way ANOVA test, as appropriate, was applied. The significances of the differences in the Kaplan-Meier survival curves were analyzed using the Mantel–Cox log rank test. A P value of < 0.05 was considered statistically significant.
Discussion
Inspired by the success of CD19 CAR T-cells therapy for B-cell leukemia and lymphoma, several CAR T-cell therapies are also being developed for cancer, among which BCMA CAR-T has received much attention.29,30 BCMA, also known as tumor necrosis factor receptor superfamily member 17 (TNFRSF17), is normally expressed solely on a subset of mature B-cells, whereas it is uniformly expressed by malignant plasma cells in many cases of MM.31 However, recent clinical trials have reported that individual patients with MM who received BCMA-targeted CAR-T cell therapy suffered relapses due to the loss of the BCMA antigen.12–15 To find other target antigens for CAR-T cells and develop new strategies for MM treatment, previously, we identified an anti-CD38 scFv from a scFv phage display library, and found that novel CD38 CAR-T cells had robust therapeutic effects on myeloma cells. Based on this, we further engineered single-chain bispecific (OR-gate) CARs that efficiently targeted not only BCMA, but also CD38, and evaluated the specificity and efficacy of these tan-CAR T cells in vitro and in vivo.
Human CD38 antigen (45 kDa) is a single-chain type II transmembrane glycoprotein, and > 90% of malignant plasma cells from patients with MM show surface expression of CD38.32 In addition, CD38 is also expressed by cells of the immune system, including T-cells, B-cells, NK-cells, macrophages, and dendritic cells,33 but the expression levels are lower compared to MM cells.34 Thus, CD38 has been regarded as a feasible target for the treatment of MM, and several studies have indicated the efficacy and safety of CD38 monoclonal antibodies in clinical applications.35,36 Remarkably, an anti-CD38 antibody (Daratumumab) has been approved for the treatment of RRMM by the FDA.37 Encouraged by these results, many researchers have explored the feasibility of developing CAR-T cell therapy targeting CD38 molecules. Early preclinical studies had shown that CD38 CAR-T cells were capable of proliferating, producing cytokines and effectively eliminating CD38+ myeloma cells.22 Recently, several clinical trials on anti-CD38 CAR-T-cell treatments of MM are in progress (NCT03464916, NCT03754764). Our study found that CD38-specific CAR-T cells showed significant lytic activity against myeloma cells in vivo and in vitro, and these cells showed no defects in ex vivo expansion.
Currently, tandem-dual CARs targeting two distinct antigens has become an effective way that can avoid tumor relapse due to antigen escape, potentially improve the efficacy issues with anti-BCMA CAR-T.38 As illustrated above, CD38 is an attractive target antigen that is combinable with BCMA CAR in dual CAR T-cell targeting strategies to prevent MM relapse caused by loss of BCMA antigen expression. Thus, we designed Tan CARs that target BCMA and CD38, and evaluated the anti-tumor activity of these CAR T-cells against multiple myeloma in vitro and in vivo. The dual target specificity of Tan CAR was achieved via inserting dual-antigen recognition domains into a single CAR molecule. Besides Tan CAR, there are multiple approaches to achieving bi-specific signal recognition, such as co-expressing two different CARs in one T cell (called Dual CAR)39 or mixing two CAR-T cell lines, each targeting a different antigen (called CAR pool).40 Compared to Dual CAR, Tan CAR has a significantly smaller genetic payload (~40% smaller in DNA length), which leads to more efficient viral vector packaging, higher transduction efficiency, and increased antigen-stimulated proliferation.20,41,42 These factors are beneficial for clinical T cell production and genetic modification of CAR-T cells.27,43 A CAR pool strategy avoids the issue of poor transduction efficiency, but requires manufacturing two CAR-T products that significantly increases treatment cost and reduces the probability of successful T-cell production within a short clinical timeframe. More importantly, the two engineered T-cell populations may compete for the limited nutrients and homeostatic cytokines available in circulation.19 Finally, as mentioned above, CD38 is also expressed on the surface of T cells, which may cause the fratricide of CAR-T cells. Therefore, it is possible that co-administering BCMA and CD38 CAR-T cell populations might result in the disproportionate expansion of CD38 CAR-T cells at the expense of BCMA CAR-T cells, thereby compromising this strategy’s ability to safeguard against the loss of BCMA. For this reason, we chose to engineer Tan CARs capable of dual-antigen recognition by attaching two tandem scFv domains to a single CAR molecule. Our data indicates that Tan-CAR T cells were indeed insensitive to the loss of BCMA on target cells, and triggered robust target-cell lysis capabilities in response to either BCMA or CD38 stimulation.
In this study, we designed two Tan CARs that had the antigen recognition domain from BCMA scFv and CD38 scFv in tandem via glycine and serine residues, a long, flexible linker peptide that has been utilized in the clinic since it has lower immunogenicity than other linkers.44,45 The difference between these two Tan CARs is the order of the two scFv domains. These Tan CAR T cells were transduced with retroviral vectors harvested from the PG13 retroviral vector producer cell line and the transduction efficiency exceeded 70%. We evaluated the effective targeting of Tan CAR-T on BCMA and CD38 both in vivo and in vitro. Our results showed that Tan-CAR-T cells could trigger robust T cell-mediated cytotoxicity and cytokine production after being stimulated with either BCMA or CD38. Moreover, these Tan-CAR-T cells had significantly higher cytotoxicity and proliferation than single-targeted CAR-T cells when encountering BCMA and CD38 antigens simultaneously. Notably, BCMA-OR- CD38 Tan CAR-T cells exhibited more potent antitumor and proliferative activity than CD38-OR-BCMA Tan CAR-T cells when they interacted with BCMA-K562, CD38-Raji, BCMA-K562+ CD38-Raji (mixed in 1:1), or RPMI-Luc cells. Thus, the therapeutic effect of BCMA-OR-CD38 Tan CAR T-cells was investigated using an immunodeficient mouse model bearing RPMI-Luc tumor cells. Our data showed that BCMA-OR-CD38 Tan CAR-T cells could achieve complete clearance of myeloma cells with no tumor relapse observed. Moreover, the levels of IFN-γ in serum were significantly higher for BCMA-OR-CD38 Tan CAR-T cell-treated mice than for single-targeted CAR-T cell-treated mice. Unfortunately, due to the lack of corresponding target cells, we were unable to further verify the specificity of BCMA-OR-CD38 Tan-CAR-T cells in vivo. Finally, the overall survival rate analysis showed that BCMA-OR-CD38 Tan CAR-T cells had similar safety as single-targeted CAR-T cells, no additional toxicities were observed. However, for a more reliable outcome further research has to include more detailed validation experiments. Since CD38 is also expressed by some key immune cell population, the on-target off-tumor effects of CD38-targeted CAR-T cells including Tan CAR-T in the treatment of MM has become a problem that cannot be neglected. Researchers have made many attempts to address this issue, such as optimizing the design of the antigen recognition domain of CD38 CARs,46 designing doxycycline (DOX) inducible Tet-on CD38-CARs,47 knocking out48 or block CD38 molecules.49 In previous research, we found that shRNA-mediated knockdown of CD38 for CD38 CAR-T cells exhibited similar proliferative capacity with CD38 CAR-T cells, possibly due to the lack of expression of CD38 in CD38 CAR-T cells (unpublished observation).
Recently, studies about Tan-CAR-T cells targeting CD38 and BCMA for the treatment of MM have been increasing, and some of them have being test in clinical trials (ChiCTR1800018143, ChiCTR1900026286, NCT03767751). Notably, a clinical trial demonstrated that the bispecific anti-BCMA/anti-CD38 CAR-T cells had an improved efficacy and manageable safety profile for the therapy of relapsed or refractory multiple myeloma (RRMM).50 In this study, we reported a novel BCMA-OR-CD38 Tan CAR T cell line that can robustly eliminate MM cells in vitro and in vivo. This BCMA-OR-CD38 Tan CAR-T may provide an effective and clinically applicable solution to the challenge of antigen escape, which has been observed in several clinical trials of BCMA CAR-T cell therapies. The process of the production of BCMA-OR-CD38 Tan CAR-T cells was consistent with the current clinical grade anti-CD19-CAR T cell manufacturing process, without the extra burden caused by large viral packaging and transduction payloads, which enabled the production of a single T-cell product targeting two clinically validated antigens associated with MM. In the future, after the efficacy and safety of BCMA-OR-CD38 Tan CAR-T cells in vitro and in vivo were sufficiently assessed, we also wish that the therapeutic effect of BCMA-OR-CD38 Tan CAR-T cells on MM can be tested in clinical trials.
Acknowledgments
We thank Dr. Sen Wu from the College of Biological Sciences, China Agricultural University for providing the BCMA+GFP+-K562 cell line. We thank Mr. Tieshan Wang from the Institute of Chinese Medicine, Beijing University of Chinese Medicine for the assistance with flow cytometry.
Funding Statement
This study was supported by “Double First-Class” guidance special faculty construction project of The Chinese Education Commission (100004150051)
Disclosure statement
The authors declared they had no conflict of interests.
Data availability statement
All of the data and materials are available from the corresponding author upon reasonable ask.
Authors’ contributions
Jianxun Wangand Yaru Feng conceived and designed this study; Yaru Feng wrote the manuscript; Yaru Feng and Xiuying Liu performed the most experiments; Xiaorui Li, Yating Zhou, Zhiru Song, Jing Zhang and Bingjie Shi assisted in the experiments. All the authors read and approved the final version of the manuscript.
References
- 1.Zhou L, Deng Y, Li N, Zheng Y, Tian T, Zhai Z, Yang S, Hao Q, Wu Y, Song D, et al. Global, regional, and national burden of Hodgkin lymphoma from 1990. to 2017: estimates from the; 2017. global burden of disease study. J Hematol Oncol. 2019;12(1):107. doi: 10.1186/s13045-019-0799-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC.. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–11. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi G, Richardson PG, Anderson KC.. Promising therapies in multiple myeloma. Blood. 2015;126(3):300–310. doi: 10.1182/blood-2015-03-575365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA, Giralt S, Mateos MV, Leleu X, Anderson KC, et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2018;32(2):252–262. doi: 10.1038/leu.2017.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. Chimeric antigen receptor T cells in refractory B-Cell Lymphomas. N Engl J Med. 2017;377(26):2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, Wood B, Lozanski A, Byrd JC, Heimfeld S, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-Specific chimeric antigen receptor-Modified T cells after failure of Ibrutinib. J Clin Oncol. 2017;35(26):3010–3020. doi: 10.1200/JCO.2017.72.8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikkilineni L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. 2017;130(24):2594–2602. doi: 10.1182/blood-2017-06-793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gagelmann N, Riecken K, Wolschke C, Berger C, Ayuk FA, Fehse B, Kröger N. Development of CAR-T cell therapies for multiple myeloma. Leukemia. 2020;34(9):2317–2332. doi: 10.1038/s41375-020-0930-x. [DOI] [PubMed] [Google Scholar]
- 10.Gagelmann N, Ayuk F, Atanackovic D, Kroger N. B cell maturation antigen-specific chimeric antigen receptor T cells for relapsed or refractory multiple myeloma: a meta-analysis. Eur J Haematol. 2020;104(4):318–327. doi: 10.1111/ejh.13380. [DOI] [PubMed] [Google Scholar]
- 11.Mullard A. FDA approves first BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discov. 2021. doi: 10.1038/d41573-021-00063-1. [DOI] [PubMed] [Google Scholar]
- 12.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan C, Pavletic S, et al. T cells genetically modified to express an Anti-B-Cell maturation antigen chimeric antigen receptor cause remissions of poor-Prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267–2280. doi: 10.1200/JCO.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, Brudno JN, Stetler-Stevenson M, Feldman SA, Hansen BG, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128(13):1688–1700. doi: 10.1182/blood-2016-04-711903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, Raje N, Lin Y, Siegel D, Oriol A, et al. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 15.Green DJ, Pont M, Sather BD, Cowan AJ, Turtle CJ, Till BG, Nagengast AM, Libby EN, Becker PS, Coffey DG, et al. Fully human Bcma targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood. 2018;132(Supplement 1):1011. doi: 10.1182/blood-2018-99-117729. [DOI] [Google Scholar]
- 16.Samur MK, Fulciniti M, Aktas Samur A, Bazarbachi AH, Tai Y-T, Prabhala R, Alonso A, Sperling AS, Campbell T, Petrocca F, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. 2021;12(1):868. doi: 10.1038/s41467-021-21177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schönfeld K, Koch J, Dotti G, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, Han X, Ti D, Dai H, Wang C, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood. 2020;136(14):1632–1644. doi: 10.1182/blood.2020005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KKH, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zah E, Nam E, Bhuvan V, Tran U, Ji BY, Gosliner SB, Wang X, Brown CE, Chen YY. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat Commun. 2020;11(1):2283. doi: 10.1038/s41467-020-16160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 22.Drent E, Groen RW, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PWHI, Kuball J, Sebestyen Z, Yuan H, de Bruijn J, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica. 2016;101(5):616–625. doi: 10.3324/haematol.2015.137620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, de Jong-Korlaar R, Yuan H, Noort WA, Klein SK, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia. 2015;29(10):2039–2049. doi: 10.1038/leu.2015.123. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Stepto H, Schneider CK. Development of the first World Health Organization lentiviral vector standard: toward the production control and standardization of lentivirus-based gene therapy products. Hum Gene Ther Methods. 2017;28(4):205–214. doi: 10.1089/hgtb.2017.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lam N, Trinklein ND, Buelow B, Patterson GH, Ojha N, Kochenderfer JN. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat Commun. 2020;11(1):283. doi: 10.1038/s41467-019-14119-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tumaini B, Lee DW, Lin T, Castiello L, Stroncek DF, Mackall C, Wayne A, Sabatino M. Simplified process for the production of anti-CD19-CAR-engineered T cells. Cytotherapy. 2013;15(11):1406–1415. doi: 10.1016/j.jcyt.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, Wilson WH, Rosenberg SA. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32(7):689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119(17):3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, Liedtke M, Rosenblatt J, Maus MV, Turka A, et al. Anti-BCMA CAR T-Cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao WH, Liu J, Wang BY, Chen Y-X, Cao X-M, Yang Y, Zhang Y-L, Wang F-X, Zhang P-Y, Lei B, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141. doi: 10.1186/s13045-018-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, Gress RE, Hakim FT, Kochenderfer JN. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leo R, Boeker M, Peest D, Hein R, Bartl R, Gessner JE, Seibach J, Wacker G, Deicher H. Multiparameter analyses of normal and malignant human plasma cells: CD38++, CD56+, CD54+, cIg+ is the common phenotype of myeloma cells. Ann Hematol. 1992;64(3):132–139. doi: 10.1007/bf01697400. [DOI] [PubMed] [Google Scholar]
- 33.Funaro A, De Monte LB, Dianzani U, Forni M, Malavasi F. Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. Eur J Immunol. 1993;23(10):2407–2411. doi: 10.1002/eji.1830231005. [DOI] [PubMed] [Google Scholar]
- 34.Bonello F, D’Agostino M, Moscvin M, Cerrato C, Boccadoro M, Gay F. CD38 as an immunotherapeutic target in multiple myeloma. Expert Opin Biol Ther. 2018;18(12):1209–1221. doi: 10.1080/14712598.2018.1544240. [DOI] [PubMed] [Google Scholar]
- 35.Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, Minnema MC, Lassen U, Krejcik J, Palumbo A, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med. 2015;373(13):1207–1219. doi: 10.1056/NEJMoa1506348. [DOI] [PubMed] [Google Scholar]
- 36.Mikhael J, Richter J, Vij R, Cole C, Zonder J, Kaufman JL, Bensinger W, Dimopoulos M, Lendvai N, Hari P, et al. A dose-finding Phase 2 study of single agent isatuximab (anti-CD38 mAb) in relapsed/refractory multiple myeloma. Leukemia. 2020;34(12):3298–3309. doi: 10.1038/s41375-020-0857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhatnagar V, Gormley NJ, Luo L, Shen YL, Sridhara R, Subramaniam S, Shen G, Ma L, Shord S, Goldberg KB, et al. FDA approval summary: daratumumab for treatment of multiple myeloma after one prior therapy. The Oncologist. 2017;22(11):1347. doi: 10.1634/theoncologist.2017-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Schans JJ, van de Donk N, Mutis T. Dual targeting to overcome current challenges in multiple myeloma CAR T-Cell treatment. Front Oncol. 2020;10:1362. doi: 10.3389/fonc.2020.01362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21(11):2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, Fisher WE, Heslop HE, Rooney CM, Brenner MK, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. 2014;22(3):623–633. doi: 10.1038/mt.2013.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bos TJ, De Bruyne E, Van Lint S, Heirman C, Vanderkerken K. Large double copy vectors are functional but show a size-dependent decline in transduction efficiency. J Biotechnol. 2010;150(1):37–40. doi: 10.1016/j.jbiotec.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 42.Kumar M, Keller B, Makalou N, Sutton RE. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther. 2001;12(15):1893–1905. doi: 10.1089/104303401753153947. [DOI] [PubMed] [Google Scholar]
- 43.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weimer T, Wormsbacher W, Kronthaler U, Lang W, Liebing U, Schulte S. Prolonged in-vivo half-life of factor VIIa by fusion to albumin. Thromb Haemost. 2008;99(4):659–667. doi: 10.1160/TH07-08-0525. [DOI] [PubMed] [Google Scholar]
- 45.Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res. 2016;4(6):498–508. doi: 10.1158/2326-6066.CIR-15-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, Martens ACM, Zweegman S, van de Donk NWCJ, Groen RWJ, et al. A rational strategy for reducing On-Target off-tumor effects of CD38-Chimeric antigen receptors by affinity optimization. Mol Ther. 2017;25(8):1946–1958. doi: 10.1016/j.ymthe.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drent E, Poels R, Mulders MJ, van de Donk N, Themeli M, Lokhorst HM, Mutis T. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS One. 2018;13(5):e0197349. doi: 10.1371/journal.pone.0197349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gurney M, Stikvoort A, Nolan E, Kirkham-mccarthy L, Khoruzhenko S, Shivakumar R, Zweegman S, Van de Donk NWCJ, Mutis T, Szegezdi E, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020. Online ahead of print. doi: 10.3324/haematol.2020.271908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao Z, Tong C, Wang Y, Chen D, Wu Z, Han W. Blocking CD38-driven fratricide among T cells enables effective antitumor activity by CD38-specific chimeric antigen receptor T cells. J Genet Genomics. 2019;46(8):367–377. doi: 10.1016/j.jgg.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 50.Li C, Mei H, Hu Y, Guo T, Liu L, Jiang H, Tang L, Wu Y, Ai L, Deng J, et al. A bispecific CAR-T cell therapy targeting Bcma and CD38 for relapsed/refractory multiple myeloma: updated results from a phase 1 dose-climbing trial. Blood. 2019;134(Supplement_1):930. doi: 10.1182/blood-2019-130340. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the data and materials are available from the corresponding author upon reasonable ask.