

ABSTRACT
Background: In recent years, innovation in oncology has created new challenges for pricing and reimbursement systems. Oncology medicines with multiple indications face a number of access challenges: (1) the number of assessments and administrative burden; (2) aligning price to different values of the same product; (3) managing clinical uncertainty at time of launch; and (4) managing budget uncertainty. These challenges impact a range of stakeholders and can result in delayed patient access to life-saving treatments. Consequently, countries have taken steps to facilitate patient access.
Methods: Drawing on the experience across Europe we have reviewed different mechanisms countries have adopted that address these challenges. These include approaches aimed directly at the issue, multi-year-multi-indication (MYMI) agreements (BE, NL), and other approaches to manage access: flexible access agreements for new indications with clinical uncertainty (UK); development of a new agreement for each new indication (IT); and immediate access for new indications and bundled assessments (DE).
Results: MYMI agreements are valuable where existing rules mean that every indication faces the same upfront evaluation process that delays patient access. They are also useful in managing budget impact and uncertainty. Other approaches that adopt an indication-specific approach helps manage clinical uncertainty at the time of launch and realise different values for the same product. They can help align price to value, even though indication-based pricing does not exist. Bundled assessments reduce the administrative burden for stakeholders, and the benefits of immediate reimbursement is that patient access is not delayed.
Conclusion: The challenges for medicines with multiple indications impact a range of stakeholders and can result in delayed patient access to life-saving treatments. MYMI agreements have created a more pragmatic approach to HTA for medicines with multiple indications to ensure both fast and broad patient access. Continued innovation in oncology will require further innovative approaches in pricing and reimbursement. It is important that policymakers, payers and manufacturers engage in early discussions and are willing to find new solutions to help accelerate patient access to innovative therapies.
KEYWORDS: Patient Access, Pricing and Reimbursement, Multiple Indications, Europe, Oncology, Immuno-oncology
Introduction
Background
Over the past six years, an increasing number of oncology medicines have received additional indications after launch. In 2014 approximately 50% of oncology medicines were effective in more than one indication. By 2020 this proportion has reached over 75% [1,2]. This is primarily due to the discovery of immune checkpoint pathways and the associated developments in immuno-oncology (I-O) medicines [3]. Immune checkpoints play important roles in immune regulation and blocking immune checkpoints on the cell membrane has now been recognised as an effective strategy in the treatment of cancer [4]. PD-1 (programmed death 1) is an example of an immune checkpoint receptor protein found on the surface of T-cells. Immune checkpoint inhibitors, such as the PD-1/L1 inhibitors, prevent the interaction between PD-L1 on tumour cells and PD-1 on T-cells, allowing the immune system to launch an anti-tumour response [5]. Many tumour cells express PD-L1, meaning this class of inhibitors has promise across multiple tumour types [6].
The first launches in the anti-PD-1/L1 class were in 2014, and as of June 2020 there are 6 PD-1 or PD-L1 inhibitors approved for marketing authorisation in Europe (pembrolizumab, MSD; nivolumab, BMS; atezolizumab, Roche; durvalumab, AstraZeneca; avelumab, Pfizer and Merck KGaA; cemiplimab, Sanofi and Regeneron) [7]. Due to their broad range of tumour activity, PD-1/L1 inhibitors are currently used to treat patients in over 30 indications with numerous still under research [8]. In the coming years, these innovations will continue to evolve, launching in new indications, being used in combinations and making greater use of biomarkers to improve treatment outcomes. As of November 2019, there were 2,975 active trials examining PD-1/L1 inhibitors against over 300 targets (Figure 1).
Figure 1.
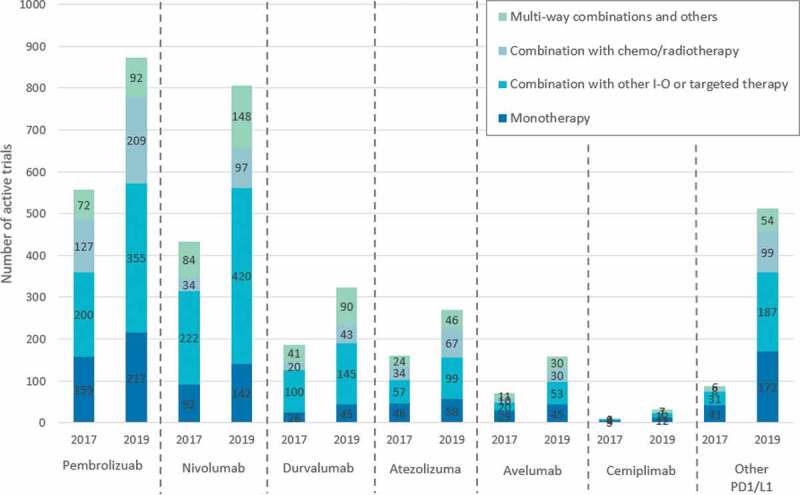
Anti-PD-1/L1 clinical trial landscape as of November 2019 Source: Cancer Research Institute.
There have been many studies examining the survival benefits of I-O therapies and many have quickly established new standards of care for numerous oncology indications [9,10]. Given these medicines are indicated for advanced tumour types, any delays in patient access can have a significant impact on health outcomes. In Sweden for example, between 2011 and 2015, approximately 1,630 patients would have been eligible for ipilimumab; of these, only about 230 (14%) received treatment. It has been estimated that this corresponds to at least 840 years of lost survival for the approximately 280 patients (20%) who could have lived 3 years or longer with treatment [11].
The OECD highlights that there are high inequalities in access to oncology medicines across EU countries, and the time from first marketing approval to coverage in a given country ranges from 1 to 66 months [12]. The European Federation of Pharmaceutical Industries and Associations (EFPIA) maintains a database known as Patients Waiting to Access Innovative Therapies (Patients WAIT), which monitors delays between the date of a drug’s marketing authorisation under the European Medicines Agency’s centralised procedure and the date of actual patient access to the product. The sample for each year includes drugs that were authorised in the preceding 4 years. The data suggests that patient access delays to new oncology medicines is higher than other medicines [13].
Access challenges for medicines with multiple indications
In healthcare systems with formal health technology assessment (HTA) processes, new oncology medicines must be assessed and approved on an individual indication basis before being reimbursed. This holds even when a medicine has received prior reimbursement for other indications [14]. There is a risk of availability for patients and access delays to these types of medicines. This is due to the following challenges:
The number of assessments increases the administrative burden and may delay access: Many HTA agencies require an evaluation of each oncology product by indication [15]. In principle, this process means payers can assess if additional indications represent good value for money. However, medicines with multiple indications will drastically increase the administrative burden of appraisal annually. If each of the six currently approved PD-1/L1 inhibitors launches a new indication every 6 months, and P&R authorities conduct the assessment within the 180-day period required under the Transparency Directive [16], it will mean 72 months of assessments are required for just the anti-PD-1/L1 class per country. This adds a substantial workload for HTA agencies.
Aligning price to different values of the same product: The assessment of a medicine with multiple indications is further complicated by the likelihood that the ‘value’ of this product to patients and the healthcare system is different for different indications [17]. This will require different comparators and end points relevant to the new indication and patient population. It is likely that clinical and cost effectiveness of the new indication will differ from the initial indications. This creates a challenge, as most countries’ reimbursement agencies are not equipped to handle different prices per indication level [18]. Many countries apply a single price per product, and this creates challenges. Although value assessment is done at indication level, there will be a single price and in reality, increased volumes lead to an expectation of price decreases. This may discourage manufacturers from expanding into indications that would, due to their additional value to patients and the healthcare system, warrant a higher price if a standalone indication [19].
Managing clinical uncertainty at time of launch: New indications may enter the market with clinical uncertainty. The EMA has become more flexible in evidence needed to grant marketing authorisation and are willing to accept Phase II single-arm trials as part of expedited regulatory approval schemes [20]. Indeed, products with multiple indications approved in quick succession bring about new uncertainties for HTA agencies [21]. Early or conditional access decisions are challenging for payers due to the limited clinical evidence available to quantify the benefit–risk ratio, efficacy and relative cost-effectiveness of new oncology medicines [22]. Randomised-controlled trials are regarded as the ‘gold standard’ trial for evaluating the effectiveness of interventions and HTA agencies also favour patient-relevant clinical outcomes, such as overall survival (OS). Consequently, while regulators may accept surrogate endpoints that are likely to predict clinical benefit, such as overall response rate (ORR), payers and HTA bodies have been more reluctant to accept them as a basis to inform pricing and reimbursement decisions [23]. This is particularly relevant for indications addressing high unmet need that have been approved earlier, or small volume indications where obtaining statistical significance in a clinical trial is more challenging. The inability of healthcare systems to manage clinical uncertainty could disincentivize manufacturers to launch an indication [24].
Managing budget impact and uncertainty: Medicines with multiple indications also introduce budget uncertainties and difficulties for payers due to the expansion of the potential patient population [25]. Increasing numbers of future indications leads to increased volumes of medicine sold. Though countries engage in pipeline reviews, this typically reviews upcoming products the healthcare system needs to consider within the next annual budget cycle, rather than anything longer-term.
The challenges surrounding medicines with multiple indications impact stakeholders across the healthcare system. Long HTA and P&R processes are resource intensive for HTA agencies, manufacturers and other stakeholders; this would lead to delays in access to medicines that offer significant survival benefits to patients. The budget uncertainty due to the addition of new indications, and the inability to value a product across multiple indications are particular challenges for payers and HTA agencies. As such, policymakers are being asked to come up with novel solutions that can address these various challenges. This review draws on the experience across Europe to understand solutions policymakers are adopting to provide faster access to oncology medicines with multiple indications, while balancing affordability and healthcare system sustainability.
Methods
In Europe, countries have adopted different mechanisms to address some of the challenges facing oncology medicines with multiple indications to accelerate patient access. These include approaches aimed directly at the issue, multi-year-multi-indication agreements, which have been explored in Belgium and the Netherlands. To investigate the pros and cons of multi-year-multi-indication agreements we compared them to markets adopting alternative approaches to manage access. These include: (1) Flexible access agreements for new indications with clinical uncertainty, as supported by the Cancer Drugs Fund in England; (2) Development of a new agreement for each new indication, under the Italian reimbursement system; and (3) Immediate access for new indications and bundled assessments, as experienced in Germany.
To review the pros and cons of these agreements government publications; academic literature; online newspaper articles; blogs; and consultancy reports were searched using key words, including ‘multi-indication reimbursement/assessment challenges’, ‘multi-indication pricing challenge’, ‘multi-indication cancer drug’, ‘multi-indication contracts/ agreements’, ‘multi-indication budget impact’ and ‘the future of multi-indication value assessments’. Research was conducted in both English and local country languages. The academic literature included peer-reviewed articles available in academic and open-source databases (including PubMed, Springer, EconLit, and Google Scholar). Titles and abstracts were scanned to narrow selection to relevant documents. This literature review was first undertaken in late 2018 and updated in June 2020.
Results
Multi-year-multi-indication agreements
Multi-year-multi-indication (MYMI) agreements are a new form of agreement between payers and manufacturers that goes across multiple indication and years. Instead of treating each indication differently they create a comprehensive framework which covers multiple indications in terms of value assessment, pricing and reimbursement. One of the most important features of MYMI agreements has been the push to ensure accelerated patient access for upcoming indications. In addition to this, the process aims to reduce uncertainty and improve predictability for both payers and manufacturers as prices are not renegotiated following the launch of a new indication. Once the new indication is available clinicians can freely prescribe the products within that indication.
MYMI agreements can incorporate a variety of components [26]:
A pricing arrangement that covers upcoming indications: This may allow adjustment of the price as new indications result in higher volume but through a predefined agreement. By anticipating upcoming indications, this encourages the launch of all indication as quickly as possible. This directly addresses the challenge of launch sequencing.
Abbreviated upfront value assessment or no assessment for new indications: Once a new indication has regulatory approval there may be a lighter HTA process, reducing the administrative cost burden while enabling patients to access new indications shortly after regulatory approval. Alternatively, no upfront assessment means an indication is automatically reimbursed and patients are able to access the new indication as soon as it is approved. In both cases there may be periodic re-evaluations of the indications, including real-world evidence (RWE) in countries where registries exist, to allow payers to re-evaluate market access recommendations and price to ensure value for money. Periodic re-evaluations can be important as they help deal with new or unexpected effects within a specific class.
Pre-launch agreement to reimburse new indications over a specific period: By allowing for immediate or accelerated access, the agreement ensures patient access to all possible indications.
Budget allocation: The agreement can incorporate an individual product or product-class budget through the period with reference to horizon scanning. Budgeting allows for additional payer predictability, however, ensure flexibility to make sure there is no constraint in accessing new indications.
Over the last few years several European countries have experimented with MYMI agreements. To review the overall experience and impact of MYMI agreements we have looked specifically at Belgium and the Netherlands.
Belgium
The traditional P&R process in Belgium starts with the submission of an application to the National Institute for Health and Disability Insurance (NIHDI) for each indication of a product. The Commission for Reimbursement of Medicines (CRM) evaluates the application and delivers its opinion to the Minister of Social Affairs [27]. Each new indication is assessed individually, and the manufacturer has the option of proposing a MEA. These formal agreements have been possible since 2010 and are known as ‘conventions’ concluded between the pharmaceutical companies and the NIHDI. Conventions aim to address either clinical evidence or budgetary uncertainties [28]. Conventional agreements are valid for up to three years and can be extended based on subsequent negotiations or following a re-assessment of the value of the product by the CRM [29].
In 2015, a new agreement between the pharmaceutical industry and government known as the ‘Pact of the Future’ was developed [30]. The Pact was a wide-ranging framework that covered several areas from research to budget sustainability, including enabling faster and more widespread patient access to innovative therapies. The authorities showed the willingness to adopt more innovative contracting. For PD-1/L1 inhibitors this led to an accelerated reimbursement procedure, in the framework of a MYMI agreement [31]. The terms of the agreement were that all PD-1/L1 inhibitors approved for reimbursement by the Minister of Social Affairs will have new indications reimbursed within one month after approval by the EMA.
Netherlands
The Netherlands had gone through broader changes to the pricing and reimbursement process prior to development of MYMI agreements. This used to be an open access system for in-hospital products, however from 2015 a new set of rules were introduced for high-budget-impact medicines. This meant any product expected to cost over €50,000 per patient per year with a budget impact of €10 m, or with an overall budget impact of €40 m or more per year – is placed in a ‘lock’ [32]. As such, until further negotiation and additional financial arrangements, the high-budget-impact product is excluded from the basic insurance package. At this point, the National Health care Institute (ZiN) will carry out an HTA assessment and will produce a recommendation to be used as starting point of the negotiation [33]. In order to move out of the lock, the manufacturer and the MoH must negotiate an agreement which enables reimbursement for eligible (or selected) patients at a socially acceptable price and budget impact. At this time, the first I-O products on the market (Keytruda and Opdivo) were placed in the lock until an alternative arrangement was made [34,35]. In addition, for oncology products, the scientific committee of the Dutch association for medical oncology or Commissie Beoordeling Nieuwe Oncologische Middelen (CieBOM) will also input their clinical opinion that can influence the uptake [36].
Since 2016, manufacturers have been able to negotiate and implement multi-year contracts for products with a single indication. Following this there have been negotiations between industry and the Ministry of Health to develop MYMI agreements on a per product basis [37]. The terms of the MYMI agreement in the Netherlands were each manufacturer has a separate MYMI contract, and the terms of these contracts remain confidential. However, it is understood that all agreements take the form of a price-volume agreement [38]. A product-specific budget is also set, this is revised annually and also remains confidential to each manufacturer. Under this agreement, all EMA-approved indications do not need to go into the normal HTA assessment by ZiN, however a positive recommendation by the oncology appraisal committee, CieBOM, is required for the indication to be reimbursed. For all indications that launch under MYMI agreement, a retrospective full HTA re-evaluation is a possibility, and ZiN can conduct additional reassessments, especially if clinical uncertainties remain.
Managed access for new indications with clinical uncertainty
Mechanisms exist to specifically fund indications earlier, that would otherwise be delayed because clinical uncertainties related to the immaturity of data make it difficult for HTA agencies to recommend these indications using standard methods. The Cancer Drugs Fund (CDF) is a source of funding for oncology medicines in England. The current CDF is used where there is plausible potential for a medicine to satisfy the criteria for routine use within the National Health Service, but where there is currently too much uncertainty surrounding the clinical data, and consequently the cost-effectiveness estimates to make such a recommendation [39]. This mechanism is particularly useful in England because of the methodology utilised by the National Institute of Health and Care Excellence (NICE). NICE determines the scope for each appraisal indicating both the patient population and the relevant comparators to be studied. The manufacturer then presents a submission adapted to the NICE scope that reviews all the available effectiveness and cost-effectiveness evidence related to the technology [40]. Uncertainties in the evidence supporting clinical- and/or cost-effectiveness can lead to NICE not recommending funding for new treatments until further evidence is available.
All new oncology indications are appraised by NICE, and NICE can make one of three recommendations: recommended for routine commissioning; not recommended for routine commissioning or recommended for use within the CDF. Several criteria are used to consider if a drug can be recommended for use within the CDF, including whether the drug has plausible potential to be cost-effective at the current price, further data collection could reduce clinical uncertainty and CDF data collection is feasible [41]. For drugs recommended for use within the CDF, a Managed Access Agreement will need to be agreed between the manufacturer and NHS England. This consists of two key components: (1) Data Collection Arrangement – this sets out the outcomes that need to be collected in order to resolve the key areas of clinical uncertainty, generally for a 2-year period; and (2) CDF Commercial Agreement – this determines the cost of the drug during the managed access period.
A key objective of the CDF is to provide patients with access to the most promising cancer drugs at the earliest opportunity [39]. Under routine appraisal, any drug that receives a routine commissioning recommendation from NICE receives funding 90 days after the publication of NICE’s Final Guidance. However, under the CDF, interim funding will be available from the point of marketing authorisation once a recommendation has been made. All approved anti-PD-1/L1s have been recommended for use within the CDF for at least one of their indications [42]. While this mechanism is helpful in addressing clinical- and/or cost-effectiveness uncertainty at the time of launch, and therefore reducing time to access, each new indication still results in a new assessment by NICE and a new Managed Access Agreement for use in the CDF.
Development of a new agreement for each new indication
In this case, the infrastructure for indication-specific agreements is developed to such an extent that a new agreement is created for each indication. Italy is commonly seen as an example of this type of system. In the case of innovative oncology medicines, the Italian system requires a monitoring registry and there is the possibility of using a Managed Entry Agreement (MEA), typically when the outcomes are uncertain [43]. The negotiated agreement (including any agreed MEA) lasts for 2 years and is renewed unless either party wants to renegotiate the terms [44]. Italy primarily uses three types of MEAs: risk-sharing agreements (to share cost between the company and national healthcare), payment-by-results or performance-based (company provides payback depending on results of the treatment per patient) and fee for efficacy (company receives payment when and if the treatment is regarded as effective) – however, the last of these is rarely used.
Companies can enter agreements at national or regional level. Since 2005, Italy has established national treatment registries to track the performance of various products. The infrastructure for data collection and analysis is crucial and enables the implementation of MEAs [45]. Based on this, individual payback schemes are agreed on for the same product for each one of its indications between the AIFA and the company [46]. A manufacturer gets reimbursed based on the net price for each indication for the volume of the product sold and may need to pay back depending on the performance of the product, which is monitored through the registries [47]. As a result, the majority (over 55%) of the MEAs in Italy are performance-based and most of the financial details of the agreements are confidential [33]. Following the approval of CAR-T therapies, AIFA introduced a new registry for these therapies to support a staged payment scheme. Under this scheme, payments (adjusted for a confidential discount on the list price) will be made in instalments, provided the agreed outcome(s) have been achieved and sustained [48].
In April 2017, AIFA announced a new reimbursement scheme for innovative products – to reward innovation and make the novel products accessible while at the same time managing its budget. The Italian Ministry of Health allocated a €1b fund for innovative therapies, with 50% dedicated to oncology medicines, and a new algorithm was created for AIFA’s CTS to assess the innovativeness of a product. The main criteria of this assessment are unmet therapeutic needs, added therapeutic value, and quality of the evidence from the clinical trials [49]. AIFA designates a product as one of three innovative classes based on how it scores on these criteria: innovative, not innovative or conditionally innovative. Those products that earn the innovative designation get access to the innovative drugs’ fund and are immediately included in regional formularies for a period of 36 months, while the conditionally innovative ones only receive the regional formularies inclusion benefit [50].
There are no allowances for medicines with multiple indications. A new indication will result in a new negotiation, and an agreed MEA. This may take the form of updating the existing MEA. For example, consider the case of Bevacizumab, which has a different scheme for each of its five indications. For the first-line treatment of colorectal cancer it used a financial-based agreement, while a performance-based agreement was applied for second-line treatment of the same disease. We understand that for PD-1s, different approaches have been used. For example, in some cases a series of financial MEAs have been implemented. In other cases, a single price-volume agreement along with a payback mechanism was achieved between the manufacturer and the AIFA [51].
Immediate access for new indications and bundled assessments
Through this mechanism, once a new indication is approved it is immediately reimbursed by the healthcare system. The payer subsequently conducts an assessment and takes a weighted average across all indications and derives a new overall value and a new price. The German system is an example. In Germany all new products are automatically reimbursed for up to 1 year following their inclusion in the official list of launched products (Lauer Taxe), and the manufacturer is allowed free pricing during this time. However, in order to continue to be reimbursed after this period, the new product must go through the centralised AMNOG P&R process, with the net price negotiated with the Federal Association of Sick Funds (GKV) [52]. New indications must go through the same process and this will result in renegotiation of the existing reimbursement price [53].
In contrast to other countries, the German system with its post-launch assessment ensures early access for single- and multi-indication medicine so that many of the challenges mentioned above are resolved. In 2017, AMNOG made a series of changes to increase flexibility. In some circumstances it is possible to exclude certain subpopulations from the reimbursement application and price negotiations if the G-BA agrees on it. The only documented example to date of price renegotiation due to indication/patient subpopulation restrictions is the case of PCSK9 inhibitors, where the price was renegotiated after their reimbursement was restricted to heterozygous familial hypercholesterolaemia and patients who cannot tolerate statins [54].
With respect to PD-1/L1s, new indications go through the same process, and each new indication launch leads to a renegotiation of the existing reimbursement price. To account for the different value of the drug across all indications or subgroups, a single price is maintained but this single price represents a volume-weighted average price per indication [55]. Since 2017, successive indications of the same drug that are expected to receive approval within 6 months of each other can undergo a joint/single procedure (‘bundling’ procedure) in accordance with the application of the pharmaceutical company, if agreed by G-BA [53]. In the case of Keytruda, EMA approval was granted for first-line treatment of renal cell carcinoma (RCC) in combination with axitinib on 30 August 2019, followed by first-line treatment for head and neck cancer on 21 November 2019. The G-BA did not start the procedure for both of these indications until 1 December 2019, however a separate evaluation was still published for both indications [56,57].
Discussion
This literature review presents publicly available information on approaches policymakers have used to provide faster access to oncology medicines with multiple indications. Looking at the outcome of MYMI, that there is evidence that agreements have had a positive impact on time to patient access for new I-O indications (Figure 2). On average, time to patient access was reduced in Belgium from 395 days to 30 days, and from 220 days to 120 days in the Netherlands. Considering the access challenges identified for medicines with multiple indications we can assess the extent MYMI agreements address these challenges compared to other approaches to expedite access.
Figure 2.
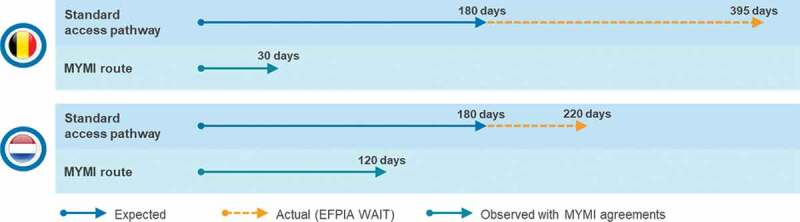
Impact of MYMI agreements on speed to patient access in Belgium and the Netherlands Source: 2019 EFPIA WAIT indicator
In terms of addressing the number of assessments needed for multiple indications, MYMI agreements can expedite value assessment, reducing the upfront workload of HTA bodies. In the Netherlands this takes the form of a clinical value assessment. In Belgium this has created an ‘ex-post assessment’ system – this resembles a German-style system that ensures immediate patient access, and then has later evaluations once the medicines are on the market. In Germany, new indications are available shortly after market authorisation and all indications are launched as a requirement of AMNOG. A bundled assessment reduces the administrative burden for both HTA bodies and manufacturers, and the benefits of immediate reimbursement is that patient access is not restricted by delaying evaluations and grouping these together. In both England and Italy, a new assessment is needed for each indication. MYMI can reduce the number of assessments in market that would otherwise undertake an assessment of every indication.
In principle, a system that assess individual indications aligns the price more closely to value of product. For example, England and Italy do this more effectively due to their indication-specific approach to establishing new agreements. This approach helps realise different values for the same product, and financial-based agreements can help align price to different values of the same product. This, however, does require monitoring by indication. The use of MEAs in the Italian system has created a formal process requiring a monitoring patient registry and the negotiation of an agreement reflecting each new indication. However, in practice indication-based pricing does not exist in many markets, and there are significant limitations in number of agreements per product [58]. In contrast, MYMI agreements create one value-based price across all indications, thus the price can still be aligned to the value of the product. Overall, although MEAs have improved availability of medicines and accelerated patient access, where a product has many indication, a more flexible administrative framework may be needed so that the implementation of complex agreements does not become a barrier to access [59]. Therefore, going forward MYMI agreements may be particularly relevant for medicines, such as immuno-oncology products, with will have an increasing number of indications over time.
Managing clinical uncertainty at time of launch is also supported by Italy’s performance-based agreements as the registry system supports ongoing evidence collection. In England, the CDF has been particularly useful in facilitating patient access while enabling RWE data collection. Between July 2016 and August 2019, the CDF has funded 78 drugs treating 155 different cancer indications. Over 37,300 patients have been registered to receive a CDF funded treatment, of which 8,200 patients have been registered to receive treatment sooner than they previously would have through interim funding arrangements in the CDF [60]. By allowing re-assessments after launch, MYMI agreements also ensure clinical uncertainty does not restrict patient access. This allows manufacturers to collect post-marketing data and RWE, which is playing an increasing role in healthcare decision-making. Indeed, in 2019 the US FDA expanded the use of palbociclib to include use in breast cancer in men based on a filing that drew heavily on post-marketing data and RWE [61]. By contrast, Germany has traditionally based its assessment and quantification of the additional benefit largely, on evidence of the highest level and quality and on measurements of “hard” patient-relevant clinical outcomes [62]. There is a risk that the HTA rigidity to manage clinical uncertainty upfront could limit patient access given current trends. A new regulation in Germany for more safety in drug supply (GSAV) means the Federal Joint Committee (G-BA) may now mandate RWE collection through indication-based registries for the national benefit assessment of new medicines that can only show “limited scientific evidence” [63]. Overall, the approach to re-assessment by MYMI is clearly helpful in addressing clinical uncertainty.
Lastly, MYMI agreements appear to be useful in managing budget impact and uncertainty. In the Netherlands, the agreement has resulted in net cost reductions for the government across multiple products and has helped the government provide treatment to broader patient populations across multiple indications [64]. Individual access agreements can partly address this challenge. In Italy, the MEA infrastructure in combination with the Innovation Fund has helped secure budget and ensure faster access by overcoming regional restrictions [50]. However, it is the multi-year nature of MYMI agreements that really improves predictability of prices, benefiting payers and reducing risk for manufacturers.
Figure 3 compares the extent that each of these solutions address the challenges for medicines with multiple indications.
Figure 3.
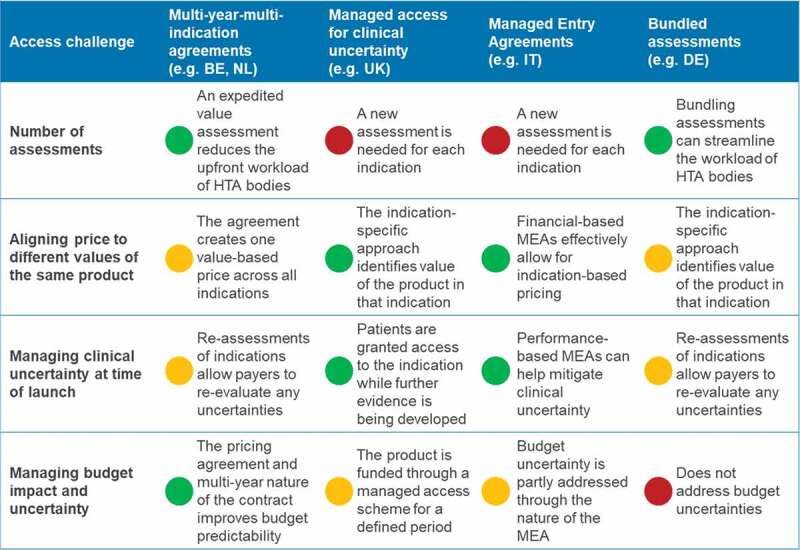
Extent that solutions address the challenges for medicines with multiple indications Notes: Red – Mechanism does not address access challenge; Amber – Mechanism partly addresses access challenge; Green – mechanism addresses access challenge
MYMI agreements are valuable where existing rules mean that every indication faces the same upfront evaluation process that delays patient access to innovative therapies. Establishing the MYMI agreement can be take a significant period of time (in the experiences so far, over two years) and requires significant support from multiple stakeholders, such as patients and clinicians. Once contracts are in place there is also the need for renegotiating the terms of the new contract. This does not mean necessarily mean a significant reduction in resources devoted to value assessment as the product may be subject to re-assessment across all indications at a later stage. While the existing approaches can also be time intensive, the initial period of negotiation is arguably shorter within a known framework (as each new indication will trigger a new assessment). Overall, the great advantage of MYMI agreements is their flexibility and ability to support faster patient access with significant reduced timelines for subsequent indications.
Conclusion
In recent years, innovation in oncology has created new challenges for P&R systems. The challenges for medicines with multiple indications impact a range of stakeholders and can result in delayed patient access to life-saving treatments. Considering these challenges European countries have taken steps to facilitate patient access. We find that MYMI agreements have adopted a more pragmatic approach to HTA for medicines with multiple indications to ensure both fast and broad patient access. Though prices are not necessarily perfectly aligned to value and there is less flexibility compared to individual agreements, there is budget predictability for the payer and a reduction in resources devoted to upfront assessment. Continued innovation in oncology (complex combinations and CAR-Ts) will require further innovative approaches in pricing and reimbursement. It is important that policymakers, payers and manufacturers engage in early discussions and are willing to find new solutions to help accelerate patient access to innovative therapies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
E. Darquennes, D. Hemelsoet, J. Huismans, R.Normand & A. Roediger are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ USA, who may own stock and/or hold stock options in the Company.
References
- [1].Barham LMulti-indication pricing: no longer mission impossible? 2016. Available at: http://deep-dive.pharmaphorum.com/deep-dive-market-access-july-2016#!/leela-barham-article.
- [2].IQVIA. Global Oncology Trends: innovation, Expansion and Disruption. 2019. Available at: https://www.iqvia.com/institute/reports/global-oncology-trends-2018#reportcharts.
- [3].Pan C, Liu H, Robins E, et al. Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol. 2020;13(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gu D, Ao X, Yang Y, et al. Soluble immune checkpoints in cancer: production, function and biological significance. J Immunother Cancer. 2018Dec;6(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Thurston DE.Immuno-oncology agents for cancer therapy. The Pharmaceutical Journal, PJ May2020online, online 10.1211/PJ.2020.2020782 [DOI] [Google Scholar]
- [6].Wu Y, Chen W, Xu ZP, et al. PD-L1 Distribution and Perspective for Cancer Immunotherapy—Blockade, Knockdown, or Inhibition. Frontiers in Immunology. 2019;10:2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cancer Research Institute . PD-1/PD-L1 Landscape. Available at: https://www.cancerresearch.org/scientists/immuno-oncology-landscape/pd-1-pd-l1-landscape#landscape
- [8].PhRMA . Medicines in Development for Immuno-Oncology. 2017. Available at: https://www.phrma.org/Report/Medicines-in-Development-for-Immuno-Oncology-2017-Report
- [9].Gauci M-L, Lanoy E, Champiat S, et al. Long-term survival in patients responding to anti–PD-1/PD-L1 therapy and disease outcome upon treatment discontinuation. Clin Cancer Res. 2019Feb1;25(3):946–956. [DOI] [PubMed] [Google Scholar]
- [10].Anti–PD-1/PD-L1 therapy ‘rapidly becoming’ standard of care for Merkel cell carcinoma. Available at: https://www.healio.com/news/hematology-oncology/20170325/antipd1pdl1-therapy-rapidly-becoming-standard-of-care-for-merkel-cell-carcinoma
- [11].Hansson J, Wilking U. Medical breakthrough that did not reach the cancer patients. Läkartidningen. 2017; 114: EL7SAvailable at: https://lakartidningen.se/Opinion/Debatt/2017/05/Medicinskt-genombrott-som-inte-nadde-cancerpatienterna/
- [12].OECD . Addressing Challenges in Access to Oncology Medicines. 2020. Available at: https://www.oecd.org/health/health-systems/Addressing-Challenges-in-Access-to-Oncology-Medicines-Analytical-Report.pdf
- [13].IQVIA . EFPIA Patient W.A.I.T. Indicator 2018 survey. 2019
- [14].Medicines Australia . PD-1 and PD-L1 checkpoint inhibitor immunotherapies: options for subsidy consideration for multiple cancer types. 2018. Available at: https://medicinesaustralia.com.au/wp-content/uploads/sites/52/2018/08/submission-27-medicines-australia.pdf
- [15].Adkins EM, Nicholson L, Floyd D, et al. Oncology drugs for orphan indications: how are HTA processes evolving for this specific drug category? 2017, June10. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5473500/. [DOI] [PMC free article] [PubMed]
- [16]. Council Directive 89/105/EEC of relating to the transparency of measures regulating the prices of medicinal products for human use and their inclusion in the scope of national health insurance systems. 1988 December 21;
- [17].Persson U, Norlin JM. Multi-indication and Combination Pricing and Reimbursement of Pharmaceuticals: opportunities for Improved Health Care through Faster Uptake of New Innovations. Appl Health Econ Health Policy. 2018;16(2):157‐165. [DOI] [PubMed] [Google Scholar]
- [18].Campillo-Artero C, Puig-Junoy J, Segú-Tolsa JL, et al. Price Models for Multi-indication Drugs: a Systematic Review. Appl Health Econ Health Policy. 2020;18(1):47‐56. [DOI] [PubMed] [Google Scholar]
- [19].Merrill J. Multi-indication pricing: big hurdles and actionable options. Pharma Intelligence Informa. 2016,78(22). Available athttps://pink.pharmaintelligence.informa.com/-/media/supporting-documents/pink-issue-pdfs/p160530.pdf?la=en [Google Scholar]
- [20].Nagai S. Flexible and Expedited Regulatory Review Processes for Innovative Medicines and Regenerative Medical Products in the US, the EU, and Japan. Int J Mol Sci. 2019Jan;20(15):3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].OECD. Addressing Challenges in Access to Oncology Medicines. 2020Available at: https://www.oecd.org/health/health-systems/Addressing-Challenges-in-Access-to-Oncology-Medicines-Analytical-Report.pdf
- [22].Martinalbo J, Bowen D, Camarero J, et al. Early market access of cancer drugs in the EU. Ann Oncol. 2016Jan1;27(1):96–105. [DOI] [PubMed] [Google Scholar]
- [23].Wilsdon T, Barron A, Edwards G, Lawlor R. The benefits of personalised medicine to patients, society and healthcare systems. EFPIA. 2018. Available at: https://www.efpia.eu/media/362040/cra-ebe-efpia-benefits-of-pm-final-report-6-july-2018.pdf [Google Scholar]
- [24].Campillo-Artero C. Is indication-based drug pricing used in practice? PharmacoEcon Outcomes News. 2019Oct;838:14–15. [Google Scholar]
- [25].Neri M, Towse A, Garau M. Multi-Indication Pricing (MIP): practical Solutions and Steps to Move Forward. Office of Health Economics. 2018Dec1. [Google Scholar]
- [26].ISPOR Warsaw 2019. MSD Symposium. Available at: https://www.ispor.org/docs/default-source/events/warsaw-2019/1-327_msd-ed-symp.pdf?sfvrsn=c2cd6d20_0
- [27].Data G. Country Focus: healthcare, Regulatory and Reimbursement Landscape – belgium. 2018. Available at: https://pharma.globaldata.com/Reportsview.aspx?DocID=54645
- [28].Pauwels K, Huys I, Vogler S, et al. Managed Entry Agreements for Oncology Drugs: lessons from the European Experience to Inform the Future. 2017. Available at: https://www.researchgate.net/publication/316272818_Supplementary_Material_2. [DOI] [PMC free article] [PubMed]
- [29].KCE . 2015. “Study 2015-13 HSR”. Available at: https://kce.fgov.be/study-program/study-2015-13-hsr-the-effectiveness-of-the-belgian-reimbursement-system-for-pharmaceuticals
- [30].‘PACT FOR THE FUTURE’ . Available atwww.deblock.belgium.de.
- [31].O’Donnell P. Belgium Steps Further into Immunotherapies – highlighting Open European Questions. Applied Clinical Trials. 2017. Available at: https://www.appliedclinicaltrialsonline.com/view/belgium-steps-further-immunotherapies-highlighting-open-european-questions [Google Scholar]
- [32].Rijksoverheid . Betaalbaar houden van medicijnen. 2016. Available at: https://www.rijksoverheid.nl/onderwerpen/geneesmiddelen/betaalbaar-houden-van-geneesmiddelen.
- [33].Pauwels K, Huys I, Vogler S, et al. Managed entry agreements for oncology drugs: lessons from the european experience to inform the future. 2017. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5378787/. [DOI] [PMC free article] [PubMed]
- [34].Kamerbrief over Beëindiging ‘Sluis’ Nivolumab per 1Maart 2016. Rijksoverheid, March1, 2016. www.rijksoverheid.nl/ministeries/ministerie-van-volksgezondheid-welzijn-en-sport/documenten/kamerstukken/2016/01/28/kamerbrief-over-beeindiging-sluis-nivolumab-per-1-maart-2016.
- [35].Skipr . ‘Neem Longkankermiddel Atezolizumab Niet Op in Basispakket’. ‘Den Haag Voert Wmo Slecht Uit’ – Actueel – Skipr, www.skipr.nl/actueel/id33568-neem-longkankermiddel-atezolizumab-niet-op-in-basispakket.html.
- [36].Bos HV, Franken R. Developments pricing and reimbursement in the Netherlands. 2015. Available at: https://www.lexology.com/library/detail.aspx?g=c73eaa88-53d4-4541-a0f6-6a50c5684f46
- [37].Schippers EIMedicines Policy Plan: new drugs available to patients fast at an acceptable cost. 2016. Available at: https://www.government.nl/binaries/government/documents/letters/2016/03/07/medicines-policy-plan-new-drugs-available-to-patients-fast-at-an-acceptable-cost/medicines-policy-plan.pdf.
- [38].Volksgezondheid MVGeslaagde prijsonderhandeling borstkankermiddel pertuzumab en longkankermiddel pembrolizumab. 2017. Available at: https://www.rijksoverheid.nl/ministeries/ministerie-van-volksgezondheid-welzijn-en-sport/nieuws/2017/06/09/geslaagde-prijsonderhandeling-borstkankermiddel-pertuzumab-en-longkankermiddel-pembrolizumab.
- [39].NHS England , Appraisal and Funding of Cancer Drugs fromJuly2016. (including the new Cancer Drugs Fund). Available at: https://www.england.nhs.uk/wp-content/uploads/2013/04/cdf-sop.pdf
- [40].Charlton V. NICE and Fair? Health Technology Assessment Policy Under the UK’s National Institute for Health and Care Excellence, 1999–2018. Health Care Anal. 2019. DOI: 10.1007/s10728-019-00381-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].NICE . Cancer Drugs Fund. Available at: https://www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/nice-technology-appraisal-guidance/cancer-drugs-fund
- [42].NHS. National Cancer Drugs Fund list . Available at: https://www.england.nhs.uk/publication/national-cancer-drugs-fund-list/
- [43].Ltd M, Thomas C. The Payor Landscape in Italy. 2016. Available at: https://www.medicysltd.co.uk/articles/payor-landscape-in-italy.php.
- [44].Parlamento Italiano . Classificazione dei farmaci e regime di rimborsabilita. 2018. Available at: http://www.camera.it/leg17/561?appro=classificazione_dei_farmaci_e_regime_di_rimborsabilit_.
- [45].Money-back guarantees on cancer drugs are real in Italy, and the world is watching . 2016, January 19. Available at: https://www.fiercepharma.com/sales-and-marketing/money-back-guarantees-on-cancer-drugs-are-real-italy-and-world-watching.
- [46].Toumi MValue added medicines: rethink, reinvent & optimize medicines, improving patient health & access. 2016. Available at: http://www.medicinesforeurope.com/wp-content/uploads/2016/05/White-Paper-30-May-2016-Toumi-Value-added-medicines-Rehink-reinvent-optimize-medicines-improving-patient-health-access.pdf.
- [47].AIFA. Registri Farmaci sottoposti a monitoraggio . 2015. Available at: http://www.agenziafarmaco.gov.it/it/content/registri-farmaci-sottoposti-monitoraggio.
- [48].Jørgensen J, Hanna E, Kefalas P. Outcomes-based reimbursement for gene therapies in practice: the experience of recently launched CAR-T cell therapies in major European countries. J Mark Access Health Policy. 2020;8(1):1715536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Macaulay R, Wang G. PMU80 THE ITALIAN INNOVATION ACCELERATION? BEST PRACTICE LESSONS OF REIMBURSEMENT INCENTIVES FOR INNOVATIVE PRODUCTS. Value Health. 2020May1;23:S247. [Google Scholar]
- [50].Galeone C, Bruzzi P, Jommi C. Key drivers of innovativeness appraisal for medicines: the Italian experience after the adoption of the new ranking system. BMJ Open. 2021Jan1;11(1):e041259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Agenzia Italiana del Farmaco . Gazzetta Ufficiale, June3, 2018. www.gazzettaufficiale.it/eli/id/2018/04/20/18A02929/sg.
- [52].Hogan L. EU pricing and reimbursement – pricing and reimbursement schemes in major European countries. 2014. Available at: http://www.hoganlovells.com/files/Publication/41a7b0f3-b653-45b9-a2c8-6a8ee504274c/Presentation/PublicationAttachment/9300f058-5514-40b4-a8dd-057cc1756059/EU%20Pricing%20%20Reimbursement%20Newsletter%20-%20November%202014.pdf.
- [53].MSD MHGermany hoping for more flexibility in AMNOG procedure. 2018. Available at: https://www.apmhealtheurope.com/freestory/0/58186/msd-germany-hoping-for-more-flexibility-in-amnog-procedure.
- [54].Craddy P, Foxon G. No confidential discounts in Germany with AMNOG 2.0! 2017. Available at: http://www.remapconsulting.com/wp-content/uploads/2018/03/Article-2_German-update-1_0-Q3-17.pdf.
- [55].Flume M, Bardou M, Capri S, et al. On behalf of Payers’ Insight. Feasibility and attractiveness of indication value-based pricing in key EU countries. J Mark Access Health Policy. 2016;4(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].G-BA . Benefit assessment procedure for the active ingredient pembrolizumab (new application: renal cell carcinoma, first line, combination with axitinib). Available at: https://www.g-ba.de/bewertungsverfahren/nutzenbewertung/511/
- [57].G-BA . Benefit assessment procedure for the active ingredient pembrolizumab (new area of application: squamous cell carcinoma of the head and neck). Available at: https://www.g-ba.de/bewertungsverfahren/nutzenbewertung/513/
- [58].Towse A, Cole A, Zamora B. The debate on indication-based pricing in the US and five major European countries. OHE Consulting Report. London: Office of Health Economics. 2018May 1. [Google Scholar]
- [59].Urbinati D, Rova A, Mantuano M. The impact of managed entry agreements on drug time to market in Italy. 2017. Available at: https://www.valueinhealthjournal.com/article/S1098-3015(17)32164-2/abstract?code=jval-site
- [60].Nawrat A. The UK’s Cancer Drugs Fund: a model for access and reimbursement? Pharmaceutical Technology 2019. Available at: https://www.pharmaceutical-technology.com/features/cancer-drugs-fund-nhs-reimbursement/
- [61].PharmaPhorum . Real-world data unlocks Ibrance ok in male breast cancer. 2019. Available at: https://pharmaphorum.com/news/real-world-data-unlocks-ibrance-ok-in-male-breast-cancer/
- [62].Ivandic V. Requirements for benefit assessment in Germany and England – overview and comparison. Health Econ Rev. 2014Dec;4(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sievers H, Joos A, Hiligsmann M. Real-world evidence: perspectives on challenges, value, and alignment of regulatory and national health technology assessment data collection requirements. Int J Technol Assess Health Care. 2021 Feb 24;37:e40.. [DOI] [PubMed] [Google Scholar]
- [64].van Volksgezondheid M, Welzijn en Sport. Geslaagde prijsonderhandeling borstkankermiddel pertuzumab en longkankermiddel pembrolizumab. 2017. Available at: https://www.rijksoverheid.nl/ministeries/ministerie-van-volksgezondheid-welzijn-en-sport/nieuws/2017/06/09/geslaagde-prijsonderhandeling-borstkankermiddel-pertuzumab-en-longkankermiddel-pembrolizumab