

Abstract
The fungus Paracoccidioides lutzii is one of the species of the Paracoccidioides genus, responsible for a neglected human mycosis, endemic in Latin America, the paracoccidioidomycosis (PCM). In order to survive in the host, the fungus overcomes a hostile environment under low levels of oxygen (hypoxia) during the infectious process. The hypoxia adaptation mechanisms are variable among human pathogenic fungi and worthy to be investigated in Paracoccidoides spp. Previous proteomic results identified that P. lutzii responds to hypoxia and it has a functional homolog of the SrbA transcription factor, a well-described hypoxic regulator. However, the direct regulation of genes by SrbA and the biological processes it governs while performing protein interactions have not been revealed yet. The goal of this study was to demonstrate the potential of SrbA targets genes in P. lutzii. In addition, to show the SrbA three-dimensional aspects as well as a protein interaction map and important regions of interaction with predicted targets. The results show that SrbA-regulated genes were involved with several biological categories, such as metabolism, energy, basal processes for cell maintenance, fungal morphogenesis, defense, virulence, and signal transduction. Moreover, in order to investigate the SrbA’s role as a protein, we performed a 3D simulation and also a protein-protein network linked to this hypoxic regulator. These in silico analyses revealed relevant aspects regarding the biology of this pathogen facing hypoxia and highlight the potential of SrbA as an antifungal target in the future.
Supplementary Information
The online version contains supplementary material available at 10.1007/s42770-021-00527-x.
Keywords: Hypoxia, P. lutzii, SrbA-regulated genes, Three-dimensional structure, Protein-protein interactions
Introduction
The pathogenic fungi of the Paracoccidioides genus infect humans and cause paracoccidioidomycosis (PCM), an endemic systemic mycosis in South America and prevalent in Brazil [1, 2]. The genus comprises five distinct species: Paracoccidioides brasiliensis, Paracoccidioides lutzii, Paracoccidioides americana, Paracoccidioides venezuelensis and Paracoccidioides restrepiensis [3, 4]. The species are dimorphic presenting a mycelium phase that grows in the environment or in the culture at 22 °C and a yeast form that grows at 37 °C in culture or within the host lungs [1]. In host tissues, fungal pathogens face low levels of oxygen when compared to atmospheric levels, a condition named hypoxia. This condition is considered a key factor for the virulence of fungi such as Cryptococcus neoformans and Aspergillus fumigatus, important human pathogens [5, 6].
The hypoxia adaptation mechanisms are variable among human pathogenic fungi [7–11] and deserve to be characterized. However, a common aspect of fungal responses to hypoxia is based on oxygen sensing by a protein of the SREBP family (sterol-responsive element-binding protein): Sre1 from C. neoformans and Schizosaccharomyces pombe, SrbA from A. fumigatus and P. lutzii, and Srb1 from Histoplasma capsulatum [8, 12–15]. Data from transcriptional profiling experiments showed that the Sre1 controlled 68% of genes significantly induced (≥ 2-fold) in the fission yeast S. pombe [15]. In A. fumigatus, SrbA controls the expression of genes related to the heme group and lipids synthesis, including ergosterol [6]. The ergosterol molecule is essential, an exclusive component of fungal cell membranes. Defects in its synthesis cause abnormalities in membrane permeability and cell death [16]. The SREBP pathway activation from fungi metabolism, such as in A. fumigatus and C. neoformans, has been proposed as an excellent target for antifungal drugs, due to its requirement for the virulence of these pathogens [8, 12, 17]. Nevertheless, the SREBP proteins are not the only hypoxia regulators that exist. In the fungus Candida albicans, for example, there is no evidence of SREBP orthologs; thus, the hypoxia adaptation is mediated by Upc2 transcriptional factor [11, 18].
SREBPs of fungi contain predicted transmembrane segments and a bHLH DNA-binding domain with the unique tyrosine residue found in all SREBP orthologs [17]. The SREBPs from S. pombe and C. neoformans, named both Sre1, are synthesized as membrane-bound precursors and are cleaved in response to a decrease in ergosterol or hypoxia [8, 9]. In silico analyses showed that members of Paracoccidioides genus also contain homologs of SREBPs with bHLH (basic helix-loop-helix leucine zipper DNA-binding domain), specific to this family. The encoded protein presents transmembrane domains, responsible for associating the protein with the endoplasmic reticulum (ER). It suggests that, for all Paracoccidioides species, the srbA gene encodes an integral membrane protein which are post-translational processed releasing the N terminus portion [14]. This portion can act as a transcription factor in the nucleus, as previously described [17]. In A. fumigatus, an opportunistic pathogenic mold, a SREBP protein, also named SrbA, was characterized. The AfSrbA is relevant to hypoxia adaptation, ergosterol biosynthesis, resistance to the azole class of antifungal drugs, and to the maintenance of the cell polarity of the fungus [6]. The AfSrbA processing involves the Dsc complex, similar to that found in S. pombe, which is required for SrbA cleavage [19].
Previous proteomic results have identified that P. lutzii responds to hypoxia by altering its metabolism in order to adapt to hypoxia. In addition, the study showed that P. lutzii has a functional homolog of the SREBP protein, the SrbA transcription factor. The PbsrbA functionality could be analyze using genetic complementation in A. fumigatus SrbA mutant strains. In addition, SrbA was required for iron deprivation and azoles adaptation [14]. However, despite these findings, the direct regulation of genes by SrbA, the SrbA three-dimensional structure, and the protein interactions it performs were not determined so far. In this context, this study becomes relevant. This is the first study that shows these findings, unveiling the central role of this hypoxic regulator in P. lutzii. Moreover, this study improves the characterization of hypoxia in the Paracoccidioides genus and reinforces SrbA as a potential target for the future development of antifungal drugs.
Methods
Data from Lima et al. (2015) were used to select the differentially expressed proteins under hypoxia and were used as a database in this study. The proteins were subjected to filter criteria in order to remove those that appeared in more than one condition or in more than one time point of the experiments (12 and 24 h). The correspondent genes were obtained using the Paracoccidioides genome databank available at NCBI (National Center for Biotechnology Information: https://www.ncbi.nlm.nih.gov/genome/15356). The DNA Pattern Find tool from Sequence Manipulation Suite (http://www.bioinformatics.org/sms2/dna_pattern.html) was used in the search of consensus sequences at promoters of the selected genes. For this purpose, 1000-bp upstream the start codon of each gene was used. The transcription start sites were defined based on predictions and were represented by bent arrows in Fig. 1. The sequences used for the searches were (A/G)TCA(T/C/G)(C/G)CCAC(T/C); (A/G)(C/T)C(A/G/T)NN(C/T)(C/T/G)A(C/T) and ATC(G/A)(T/G)(A/G)(C/T)(G/C)AT, previously described as SrbA DNA binding sites [12, 15, 20]. These motifs are called sterol regulatory elements (SRE) [21]. Bioinformatics tools such as Multiple EM for Motif Elicitation (MEME) were used for this purpose [12, 15]. The EM means expectation maximization (EM), an algorithm used to estimate motifs [22, 23] but it has some limitations when compared to the MEME (Multiple EM for Motif Elicitation) [24], also implemented as a web service [25]. The number of the SrbA DNA-binding motifs is shown in Supplementary Table 1 and each motif location is shown in Supplementary Files 1, 2 and 3 (including detection from direct or reverse DNA strand). In order to analyze the predominant nucleotides of detected SrbA motifs, we used WebLogo (http://weblogo.threeplusone.com/), a web-based application designed to generate sequence logos. Logos are a graphical representation of an amino acid or nucleic acid multiple sequence alignment, consisting of stacks of symbols, one stack for each position in the sequence, indicating the relative frequency of each amino or nucleic acid at that position [26]. The result is presented in Fig. 2.
Fig. 1.

SrbA DNA binding sites in the promoter regions of P. lutzii genes. The SrbA DNA motifs are demonstrated for six randomly selected genes that presented matched with consensus sequences obtained previously. Sequence 1: 5’-(A/G)(C/T)C(A/G/T)NN(C/T)(C/T/G)A(C/T)-3’ described in Todd and co-workers (2006)—black arrows; sequence 2: 5’-(A/G)TCA(T/C/G)(C/G)CCAC(T/C)-3’ described in Chung and co-workers (2014)—gray arrows; and sequence 3: 5’-ATC(G/A)(T/G)(A/G)(C/T)(G/C)AT-3’, described in Linde and co-workers (2012)—dashed arrows. A 1-kb DNA sequence upstream to each gene was used for analysis. The motifs are shown as arrows, which also demonstrate the matched orientation. The bent arrows represent the transcription start site. The amount of SrbA binding sites for each gene is presented in Supplementary Table 1 and their sequences, strand direction, and exact position in the promotor regions are presented in Supplementary Files 1, 2 and 3. Genes ID and annotation: PAAG_00221 (acetolactate synthase); PAAG_02170 (adenylosuccinate synthetase); PAAG_00851 (6,7-dimethyl-8-ribityllumazine synthase); PAAG_00871 (30-kDa heat shock protein); PAAG_05780 (2,5-diketo-d-gluconic acid reductase A), and PAAG_02653 (acetyl-coenzyme A synthetase)
Fig. 2.
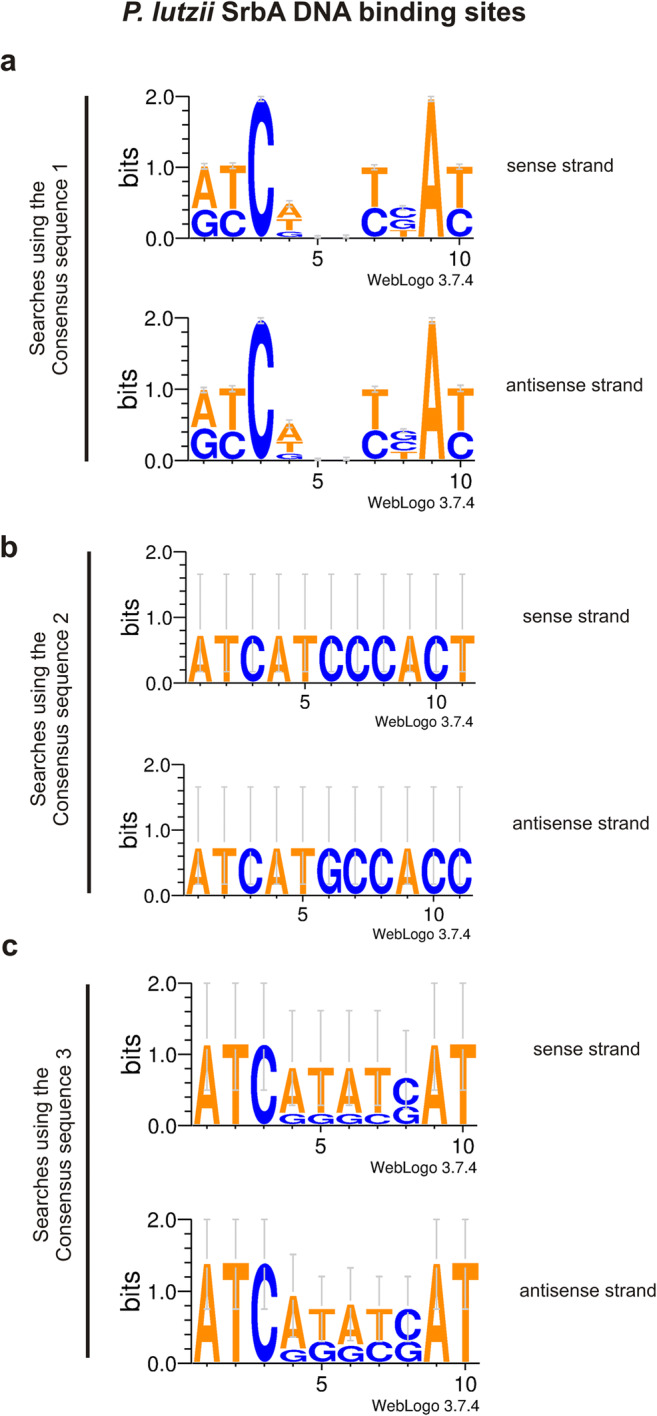
Graphical representation of SrbA DNA binding sites. From this study, all SrbA DNA binding sites found were analyzed to detect the predominate nucleotides. The software WebLogo (http://weblogo.threeplusone.com/) was used. Sequence logos depict the level of sequence conservation (show as bits of information) at each position. a Logo built with 532 and 466 sequences (motifs)—sense and reverse DNA strands, respectively—which were obtained from searches on the gene promoters using consensus sequence 1 from S. pombe defined by Todd et al. (2006). b Logo built with 2 sequences (motifs)—one for each DNA strand—which were obtained from searches on the gene promoters using consensus sequence 2 from A. fumigatus defined by Chung et al. (2014). c Logo built with 5 and 10 sequences (motifs)—sense and reverse DNA strands, respectively—which were obtained from searches on the gene promoters using consensus sequence 3 from A. fumigatus defined by Linde et al. (2012)
In the next step, orthologs from each P. lutzii selected proteins were screened using previous studies from A. fumigatus [12, 27]. These studies depicted genes and transcripts that were experimentally confirmed to be regulated by AfSrbA, even considering distinct strains such as, Af293 and Af1163 (http://www.aspgd.org/). Then, we used these studies as a database for orthologs searches (see Supplementary Table 2). Additionally, the web platform OrthoVenn [28] was employed in the identification of orthologs, as described previously [29].
The predicted SrbA amino acids were obtained (https://www.ncbi.nlm.nih.gov/protein/XP_002794199.1) and were used to search orthologs in S. cerevisiae via the BlastP tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The expectation value was set to less than or equal to 10−5. We found the ortholog HMS1 (L000004441, YOR032C) from the S288c strain. Then, the profile of protein-protein interactions (PPI) was retrieved from the BioGrid database (https://thebiogrid.org/) [30], an interaction repository that compiles data through comprehensive curation efforts. The result summary can be accessed at https://thebiogrid.org/34434/summary/saccharomyces-cerevisiae/hms1.html. Interactors with their respective amino acid sequences and their orthologs from P. lutzii databank are listed in Supplementary File 4. For orthologs searches, the BlastP tool was used (https://blast.ncbi.nlm.nih.gov/Blast.cgi); we also considered an expectation value less than or equal to 10−5, as described previously [29].
The 3-D structures used in the analyses were built using the predicted amino acids from SrbA; 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (PGM); succinate-semialdehyde dehydrogenase (SSDH); acetyl-coenzyme A transporter 1 (ACAT); and gamma-glutamyltranspeptidase (GGT). The accession numbers from NCBI database (https://www.ncbi.nlm.nih.gov/) for each protein are XP_002794199.1; XP_002790551.1; XP_002789363.1; XP_002794270.2; and XP_015700505.1, respectively. The homologs and 3D structures were obtained using the PDB (protein databank; https://www.rcsb.org/) [31] and the I-TASSER (Iterative Threading Assembly Refinement) server [32] to model the three-dimensional conformation of the proteins under study. The process is based on the homology of protein structures resolved by crystallography and deposited in the PDB (protein databank). The best conformation is determined by fold recognition via Monte Carlo simulations. The six main steps of the molecular modeling of the target protein structures consist, firstly, on the prediction of the secondary structure by PSSpred (Protein Secondary Structure Prediction) and identification of templates by LOMETS (Local Meta-Threading-Server) [33]. Secondly, assembly of ranked fragments through Monte Carlo simulations was applied in order to identify fragments with similar conformational scores [34] and following clusterization of such fragments according to conformation and energy through simulations [35]. The last step helps to select conformations of the target proteins that are similar to their correspondent native structures. Eventually, molecular dynamics allowed the refinement and the prediction of biological functions via COACH [36]. The protein docking was performed by ClusPro [37]. The hotspots in the proteins under study were identified by KFC2 [38]. We used PyMol (https://pymol.org) for the visualization of the interface of interaction and the visualization of hotspots. The hotspot prediction was based on characteristics regarding conformation specificity (K-FADE) and biochemical features such as hydrophobicity (K-CON) [39, 40]. The amino acids from each protein and those related to the hotspot interactions are shown in Supplementary File 5.
Results and discussion
SrbA as a hypoxic transcription regulator
In order to obtain a list of experimentally regulated proteins under hypoxia in P. lutzii we selected the two hundred eighty-eight (288) proteins obtained previously [14]. Then, the respective genes were obtained from Paracoccidioides spp. genomic databank (https://www.ncbi.nlm.nih.gov/genome/15356). The P. lutzii SREBP ortholog was also described in Paracoccidioides [14], called PlSrbA (Paracoccidioides lutzii SrbA). Until now, the interconnection of those regulated proteins and SrbA in P. lutzii has not been demonstrated.
In fungi, consensus sequences have been defined as DNA-binding motifs for both SrbA [12, 20] and its homolog in fission yeast, Sre1 [15]. The binding of SrbA to its own promoter reveals an autoregulatory-positive feedback loop for modulation of srbA mRNA levels [12]. In this context and using these previous data, analyses were performed here (Suppl. Files 1, 2 and 3 and Suppl. Table 1). An overview of the results is shown in Supplementary Figure 1. Only nine (9) genes did not present SrbA recognized sites, from any consensus sequences. A total of sixteen (16) genes presented the SrbA recognized sites from two consensus sequences simultaneously: those defined by Todd et al. [15] and Chung et al. [12] or those defined by Todd et al. [15] and Linde et al. [20]: 2 and 14 genes, respectively. For the remaining genes, two hundred twenty-eight (228), there were SrbA recognized DNA binding sites from the consensus sequence defined by Todd et al. [15]. The detailed results for all genes (number of the SrbA DNA binding sites with its specific sequences; position and direction on the DNA strand) are presented in Suppl. Files 1, 2 and 3 and Suppl. Table 1. We chose random genes that presented SrbA motifs matched only with consensus sequence 1 (defined by Todd et al.) [15] and others which motifs matched with at least two consensus sequences: 1 and 2 (defined by Chung et al.) [12] or 1 and 3 (defined by Linde et al.) [20] (Fig. 1).
Table 1.
SrbA-regulated proteins: biological processes, three-dimensional prediction and protein-protein interaction
| Accession numbera | Annotationa | Biological processb | 3D and protein-protein interaction predictionsc | |
|---|---|---|---|---|
| Metabolism | ||||
| Amino acid metabolism | ||||
| 1 | XP_015701561.1 | Adenylosuccinate synthetase | Aspartate biosynthesis | |
| 2 | XP_002795217.1 | Saccharopine dehydrogenase | Lysine biosynthesis | |
| 3 | XP_015703092.1 | d-3-Phosphoglycerate dehydrogenase | Serine biosynthesis/l-serine biosynthesis | |
| 4 | XP_002789912.1 | 4-Hydroxyphenylpyruvate dioxygenase | Glycine biosynthesis | |
| 5 | XP_002790306.1 | Acetolactate synthase small subunit | Valin, leucine, and isoleucine biosynthesis | |
| 6 | XP_015701886.1 | ATP phosphoribosyltransferase | l-Histidine biosynthesis | |
| 7 | XP_015701781.1 | Kynurenine-oxoglutarate transaminase | Glutamate biosynthesis | |
| 8 | XP_002795127.2 | Aspartate aminotransferase | Amino acid metabolism/aspartate biosynthetic process | |
| 9 | XP_002797285.1 | Aspartate aminotransferase | Amino acid metabolism/aspartate biosynthetic process | |
| 10 | XP_002797451.1 | 2-Oxoisovalerate dehydrogenase subunit alpha, mitochondrial | Degradation of isoleucine/conversion of alpha-keto acids to acyl-CoA and CO2 | |
| C-compound and carbohydrate metabolism | ||||
| 11 | XP_002795462.1 | Glucosamine 6-phosphate synthetase | Carbohydrate biosynthetic process/carbohydrate binding | # |
| 12 | XP_002792638.1 | Alcohol dehydrogenase zinc-binding domain-containing protein | Amino acid catabolic process to alcohol via Ehrlich pathway | |
| Nitrogen metabolism | ||||
| 13 | XP_002790569.1 | Allantoinase | Nitrogen, sulfur, and selenium metabolism | |
| Purine nucleotide/nucleoside/nucleobase metabolism | ||||
| 14 | XP_002792883.1 | Hit family protein 1 | Nucleotide metabolic process | |
| 15 | XP_002792520.1 | Inosine-5-monophosphate dehydrogenase IMD2 | Purine anabolism | |
| 16 | XP_002792838.1 | Adenylosuccinate lyase | Purine nucleotide/nucleoside/nucleobase anabolism | |
| Lipid, fatty acid, and isoprenoid metabolism | ||||
| 17 | XP_002793014.1 | ATP-citrate synthase subunit 1 | Citrate biosynthethic process | |
| 18 | XP_002793015.1 | ATP-citrate-lyase | Oxaloacetate and acetyl-CoA from citrate/ acetyl-CoA biosynthethic process | |
| 19 | XP_002790742.1 | Farnesyl pyrophosphate synthetase | Isoprenoid metabolism | |
| 20 | XP_002789106.1 | Leukotriene A-4 hydrolase | Lipid and fatty acid metabolism/leukotriene biosynthetic process | |
| 21 | XP_002795085.1 | 12-Oxophytodienoate reductase | Oxylipin biosynthetic process | |
| Energy | ||||
| Glycolysis/gluconeogenesis | ||||
| 22 | XP_002790551.1 | 2,3-Bisphosphoglycerate-independent phosphoglycerate mutase | 3-Phosphoglycerate and 2-phosphoglycerate interconversion | Δ |
| 23 | XP_002791787.1 | Glucose-6-phosphate isomerase | Interconverts glucose-6-phosphate and fructose-6-phosohate | |
| Electron transport and membrane-associated energy conservation | ||||
| 24 | XP_002796595.1 | Cytochrome b2 | l-Lactate cytochrome c oxidoreductase activity | |
| 25 | XP_002789970.2 | ATP synthase subunit beta | Aerobic respiration | |
| 26 | XP_002790709.1 | Cytochrome c oxidase* | Complex IV/ oxidative phosphorylation | |
| TCA cycle | ||||
| 27 | XP_015700403.1 | Fumarate hydratase | TCA cycle | |
| GABA shunt | ||||
| 28 | XP_002789363.1 | Succinate-semialdehyde dehydrogenase | γ-Aminobutyric acid or GABA degradation | Δ |
| Cell cycle and DNA processing | ||||
| 29 | XP_015700277.1 | Dynactin | Nuclear division | |
| 30 | XP_002791404.1 | Curved DNA-binding protein 42 kDa protein | DNA binding | |
| 31 | XP_002794643.1 | Nuclear movement protein nudC | Mitotic nuclear division | |
| 32 | XP_002796298.1 | Nuclear segregation protein Bfr1 | Mitotic cell cycle and cell cycle control | |
| 33 | XP_002789159.1 | Late histone H2B.L4 | Chromosome condensation | |
| Transcription | ||||
| 34 | XP_002793586.2 | Nascent polypeptide-associated complex subunit beta | Transcriptional control | |
| 35 | XP_002790295.1 | ATP-dependent RNA helicase DOB1 | rRNA processing | |
| 36 | XP_015699578.1 | Pirin | mRNA synthesis | |
| Translation | ||||
| 37 | XP_002791457.2 | 60S ribosomal protein L32 | Ribosome biogenesis | |
| 38 | XP_002790934.1 | Eukaryotic translation initiation factor 3 135-kDa subunit | Translate | |
| 39 | XP_002792522.1 | 40S ribosomal protein S21 | Ribosomal proteins | |
| 40 | XP_002797891.2 | 60S ribosomal protein L2* | Ribosome biogenesis | |
| 41 | XP_002796956.1 | Eukaryotic translation initiation factor 3 subunit A | Translation initiation | |
| 42 | XP_002792608.1 | 40S ribosomal protein S22 | Ribosomal proteins | |
| 43 | XP_002797846.1 | 40S ribosomal protein S23 | Ribosome biogenesis | |
| 44 | XP_002793097.2 | 60S ribosomal protein L26 | Ribosome biogenesis | |
| 45 | XP_002797549.2 | 60S ribosomal protein L3 | Ribosomal proteins | |
| 46 | XP_015700690.1 | 60S ribosomal protein L44 | Ribosomal proteins | |
| 47 | XP_002793662.1 | 60s ribosomal protein L14 | Ribosomal proteins | |
| 48 | XP_002793375.1 | ATP binding cassette sub-family F member 2 | Positive regulation of translation | |
| 49 | XP_002794622.1 | Elongation factor G1 | Translation elongation | |
| 50 | XP_015702202.1 | Prolyl-tRNA synthetase | Aminoacyl-tRNA-synthetases/translation | |
| 51 | XP_002796830.2 | ATP-dependent RNA helicase eIF4A | Protein biosynthesis | |
| 52 | XP_002795027.2 | Glutaminyl-tRNA synthetase | Aminoacyl-tRNA-synthetases/translation | |
| 53 | XP_002797837.1 | Eukaryotic translation initiation factor 3 subunit F | Translation | |
| 54 | XP_015702626.1 | Eukaryotic translation initiation factor 3 subunit M* | Protein biosynthesis | |
| Protein fate | ||||
| 55 | XP_002797245.1 | Proteasome component PUP1 | Proteasomal degradation (ubiquitin pathway) | |
| 56 | XP_015702126.1 | Proteasome component PUP2 | Proteasomal degradation (ubiquitin/proteasomal pathway) | |
| 57 | XP_002795711.1 | ATP-dependent protease La 2 | Protein/peptide degradation/ATP dependent | |
| 58 | XP_002789465.2 | 40S ribosomal protein S12 | Ribosomal proteins | |
| 59 | XP_002791336.1 | 40S ribosomal protein S24 | Ribosome biogenesis | |
| 60 | XP_002791366.1 | Ubiquitin-40S ribosomal protein S31 fusion protein | Protein degradation | # |
| 61 | XP_002795210.2 | Hsp90 co-chaperone AHA1 | Protein folding and stabilization | |
| Transport | ||||
| 62 | XP_015701282.1 | SNF7 family protein Fti1/Did2* | Protein transport | |
| 63 | XP_002794270.2 | Acetyl-coenzyme A transporter 1 | Acetyl-CoA transmembrane transporter | Δ |
| 64 | XP_002794592.1 | Vacuolar protein sorting-associated protein | Protein transport | |
| 65 | XP_015703025.1 | MFS alpha-glucoside transporter* | Alpha-glucosides and protons | |
| Cell rescue, defense, and virulence | ||||
| 66 | XP_015700505.1 | Gamma-glutamyltranspeptidase | Redox homeostasis; involved with glutathione metabolism | Δ |
| 67 | XP_015699733.1 | Hsp90 binding co-chaperone (Sba1) | Stress response/protein folding and stabilization | |
| 68 | XP_015701548.1 | Hsp70 | Stress response | |
| 69 | XP_015699872.1 | Heat shock protein | Stress response/protein folding and stabilization | # |
| 70 | XP_002797012.2 | 30-kDa heat shock protein | Heat shock response | |
| 71 | XP_002794671.1 | Mitochondrial peroxiredoxin PRX1 | Oxidative stress response | |
| Cell growth/morphogenesis | ||||
| 72 | XP_002789006.1 | TCTP family protein | Microtubule stabilization | |
| Signal transduction | ||||
| 73 | XP_002789810.2 | Calmodulin | Calcium binding | |
| Miscellaneous | ||||
| 74 | XP_015700508.1 | Short-chain dehydrogenase/reductase family | Oxidoreductase activity | |
| 75 | XP_015701087.1 | Phosphorylase family protein | - | |
| Unclassified | ||||
| 76 | XP_002794694.1 | Hypothetical protein | - | |
aP. lutzii protein accession number and annotation from NCBI database (https://www.ncbi.nlm.nih.gov/genome/15356). The annotation was obtained from homology using the BLAST tool and is indicated by asterisks (*)
bBiological categories from Uniprot (http://www.uniprot.org/)
cIn silico analysis: Δ (delta) ➔ three-dimensional protein prediction obtained from ClusPro tool (https://cluspro.bu.edu/login.php) and interaction with SrbA confirmed by the KFC2 tool (ZHU; MITCHELL, 2011); # (hashtag) ➔ SrbA’s protein-protein interactions prediction detected by the BioGrid tool (https://thebiogrid.org/) and S. cerevisiae database
The SrbA DNA-binding motifs were organized (Supplementary Table 2) in order to identify the predominant nucleotides used by P. lutzii SrbA to detected binding sequences in the promoters (Fig. 2). Using a logo, a graphical representation of a nucleic acid multiple sequence alignment consisting of stacks of symbols (one stack for each position in the sequence with the relative frequencies of each nuclei acid indicated), we detected a total of ten (10) nucleotides for all motifs, regardless of the consensus sequence used from other fungi and the DNA strand direction. However, almost all binding sequences were detected using sequence 1 (a total of 532 to sense and 466 to reverse strands) as defined by Todd and co-workers [15] (Supplementary Table 2). The predominant nucleotides detected in the motifs were ATC in positions 1–3, and AT in positions 9–10, especially to cytosine in position 3 and adenine in position 9 (Fig. 2).
Promoters are DNA sequences located upstream of the coding regions of the genes which drives RNA polymerase II to recognize where to start DNA transcription and in which direction to continue the process. RNA polymerase II itself does not have the ability to recognize specific DNA sequences including the promoter sequences. General transcription factors help the polymerase to find a promotor sequence [41–43]. Families of DNA sequence elements direct transcription via RNA polymerase II: the TATA element and the initiator motif, which are recognized by components of the transcription machinery, and the promoter-proximal and distal elements. The proximal is situated 50 to a few hundred base pairs upstream of the starting site; the distal are found up to tens of thousands of base pairs away from the transcription starting site. Both contain binding sites for proteins that modulate transcription [42, 44], also known as regulatory elements [43]. These sequences are called upstream activation sequences (UASs) in yeast and enhancers in higher eukaryotes such as humans, providing binding sites for transcriptional activators that increase the levels of gene transcription [43].
A common feature of most enhancers and promoters is the presence of multiple binding sites (enhansons) [45]. As consequence, cooperative binding of regulatory factor might occur resulting in directing protein-protein interaction between the factors, leading to a significant increase in the affinity of each factor for nucleosomal DNA, more than 2 orders of magnitude, for example [46]. In addition, multiple transcription factors during cooperative nucleosome binding require sequence-specific binding for all of them, regardless of the orientation of binding sites on the nucleosome [46].
In this context, we believe that P. lutzii SrbA transcription factor possibly interacts with other regulatory factors since many SrbA DNA binding sites were found in the promoter genes, in both DNA strands (Fig. 1; Suppl. Files 1, 2 and 3; Suppl. Table 1). P. lutzii SrbA protein has a bHLH domain and is a SREPB (sterol regulatory element-binding protein) ortholog protein [14]. It is well established that the bHLH proteins form multiple dimer combination based on the availability of binding partners, which is relevant to gene regulation [47]. In addition, SREBPs physically interact to regulate gene expression through the formation of homo and heterodimers in mammals [48, 49]. In A. fumigatus, a new transcriptional regulator of the fungal hypoxia response and virulence was identified, the SrbB. The data showed that SrbB co-regulates a sub-set of SrbA target genes in this fungus and interacts with each other in order to regulate gene expression [12].
This effect of the SrbA transcription factor has been extensively investigated in A. fumigatus during hypoxia [6, 12, 19, 27], using gene deletion. Microarray analysis revealed significant changes in transcript levels of approximately 12% of the A. fumigatus genome in absence of SrbA [27]. Furthermore, the ChIP-seq analysis revealed a total of 97 genes that are SrbA dependent. In addition, RNA-seq analysis showed that more than five hundred (500) genes had changes in transcript levels when SrbA was absent [12]. These findings highlight the amplitude of gene regulation promoted by SrbA during hypoxia adaptation.
SrbA’s role as a protein
P. lutzii orthologs from A. fumigatus
We investigated whether P. lutzii proteins share orthologs with A. fumigatus studies. We considered just the two hundred forty-four (244) genes, which presented SrbA DNA binding sites in their promoters (Supplementary Table 1). The results obtained previously [12, 27] were selected as database since they proved experimentally that SrbA regulates several genes in the fungus. The studies were performed using distinct strains (Af1163 and Af293), and then, we considered them in our analysis. From 244 proteins of P. lutzii, we did not found orthologs with Af1163 neither with Af293 for twenty-two (22) proteins (Supplementary Table S2 – green color lines). Furthermore, we found that five (5) of them presented orthologs just with one strain of A. fumigatus, Af1163 or Af293 (Supplementary Table S2 – pink color lines). On the other hand, two hundred seventeen (217) proteins shared an ortholog regulated by SrbA from both strains of A. fumigatus (Supplementary Table S2 – yellow and gray color lines). Among those, seventy-six (76) were detected from RNA-seq [12] and from microarray [27], and 3 of them were also detected by ChIP-seq data (Supplementary Table S2 – yellow color lines). The remaining, one hundred forty-one (141) proteins were detected only from RNA-seq [12] (Supplementary Table S2 – gray color lines). Thus, this cross-linked reference method has revealed that most proteins regulated in P. lutzii during hypoxia [14] have the potential to be directly regulated by PlSrbA.
Noteworthy, the two-factor authentication applied here increases the reliability of the results. Firstly, the genes encoding these proteins presented the SrbA DNA binding sites in their promoter regions, and secondly, they share orthologs which were experimentally detected as regulated by SrbA in A. fumigatus using ChiP-seq, RNA-seq and Microarray techniques [12, 27].
Biological categories of P. lutzii orthologs
The biological processes were searched for seventy-six (76) proteins (Table 1, Fig. 3). The results show that the SrbA is involved with proteins related to several biological categories, highlighting its importance to P. lutzii metabolism and hypoxia adaptation (Table 1).
Fig. 3.

Biological process of the proteins potentially regulated by SrbA from the pathogenic fungus P. lutzii. Biological categories and subcategories of the seventy-six (76) proteins listed in Table 1 are shown in the scheme (the correspondent percentages are shown). The biological process was determined using the Uniprot Knowledgebase database (https://www.uniprot.org/uniprot/). TCA, tricarboxylic cycle; GABA, aminobutyric acid degradation route
The most abundant biological processes were metabolism (27.6%, first) and energy (9.2%, third), similar to the results found in the hypoxia proteome of P. lutzii [14]. Adaptation to hypoxia is variable among fungi, for example, glycolysis is decreased in C. neoformans [5, 51] and in early times in P. lutzii [14], while it is increased and C. albicans [11, 52], Aspergillus nidulans [53], and P. brasiliensis [54]. Serious functional damage to the TCA [14] and respiratory chain are noted [14, 54, 55]. Thus, the energetic processes are deviated to alternative secondary routes, such as the GABA shunt in P. lutzii [14], A. fumigatus [7], and A. nidulans [10, 53]. Alternatively, the methylcitrate cycle was induced by P. brasiliensis in order to obtain energy [54].
Proteins involved with cell cycle and DNA processing; transcription; translation; protein fate; cell rescue/defense/virulence; transport; cell growth/ morphogenesis; and signal transduction are represented by forty-four (44) proteins or 59.2% of the processes. Interestingly, these categories have been reported, in general, to be activated in P. lutzii [14] and suppressed in P. brasiliensis [54] during the lack of oxygen. The results emphasize that SrbA possibly is the main positive/negative hypoxic regulator in Paracoccidioides and it is relevant to hypoxic stress compensation in this fungus.
Protein-protein interactions: PlSrbA network prediction
The investigation of the PlSrbA regarding its relationship with other proteins was performed. An interactome of PlSrbA was built (Supplementary Figure 2) and we performed 3D analyses involving PlSrbA and some partners (Figs. 4 and 5). The P. lutzii SrbA homolog protein was found in S. cerevisiae (a model fungus) protein databank. We found the ortholog HMS1 (L000004441, YOR032C) from the S288c strain, a bHLH protein with similarity to myc-family transcription factors. Then, a protein interaction network was constructed (Supplementary Figure 2; Supplementary File 4; https://thebiogrid.org/34434/summary/saccharomyces-cerevisiae/hms1.html). The SrbA ortholog presented 76 interactors, detected by physical or genetic pieces of evidence. All BioGrid repository proteins were previously studied by physical or genetic approaches. Physical pieces of evidence represent interactions detected, for example, by purified protein in vitro; two-hybrid system; biochemical activity; and affinity capture (identified by western blot or RNA or mass spectrometry technics). Genetic approaches represent high- or low-throughput genetic studies, such as, mutations or gene deletion, differential epistasis mapping, epistatic mini-array profile (E-MAP), phenotypic suppression (when the mutation or overexpression of one gene results in suppression of any phenotype associated with mutation or overexpression of another gene), and dosage rescue (when the overexpression or increased dosage of one gene rescues the lethality or growth defect of a strain that is mutated or deleted for another gene).
Fig. 4.
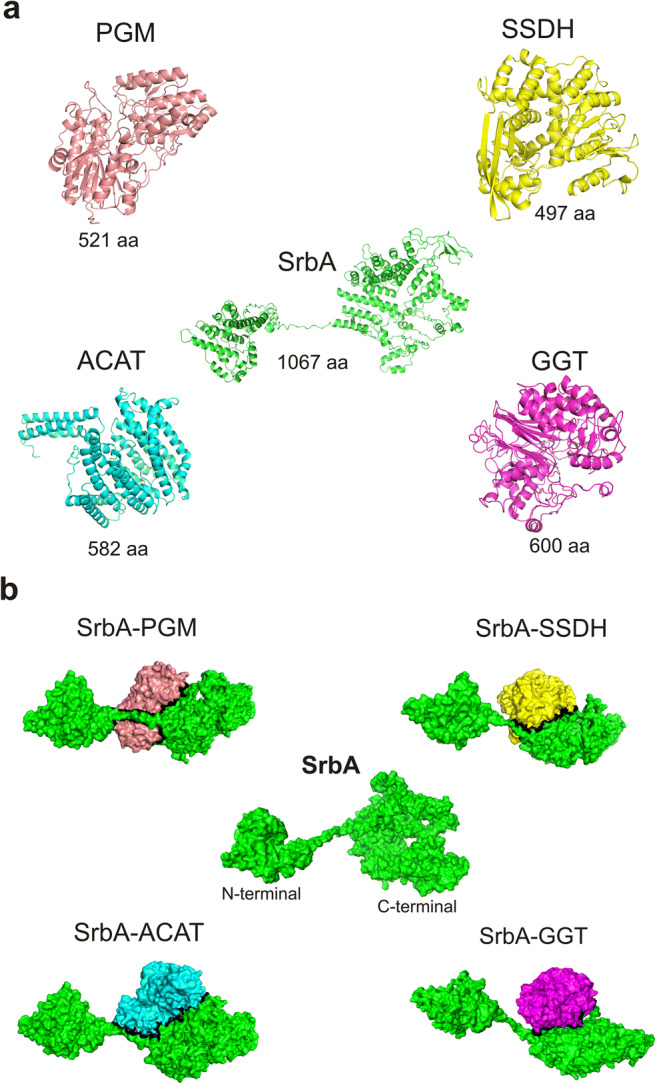
Three-dimensional structure of SrbA and its selected partners from P. lutzii. Four (4) proteins listed in Table 1 were selected for three-dimensional structure and protein-protein interactions predictions with SrbA, accession number from NCBI database (https://www.ncbi.nlm.nih.gov/): XP_002794199.1. These proteins belong to relevant metabolic pathways regulated by hypoxia in Lima and co-workers (2015). a The molecular structure of proteins 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (PGM); succinate-semialdehyde dehydrogenase (SSDH); acetyl-coenzyme A transporter 1 (ACAT), and gamma-glutamyltranspeptidase (GGT) was shown. The accession numbers from NCBI database (https://www.ncbi.nlm.nih.gov/) for each protein are XP_002790551.1; XP_002789363.1; XP_002794270.2; and XP_015700505.1, respectively. b The surface of proteins PGM, SSDH, ACAT, and GGT showing the interaction interfaces with SrbA (black line). The 3-D structures were obtained using i-TASSER server [32], in association with the PDB protein databank (https://www.rcsb.org/). The images were constructed using the PyMol software (https://pymol.org/2/). For protein-protein interactions, the ClusPro protein-protein docking software (https://cluspro.bu.edu/login.php) and the algorithm KFC2 [38] were used. The amino acids from each protein and those related to the hotspots interactions are shown in Supplementary File 5
Fig. 5.

Interaction between hotspots residues from SrbA and target proteins. a Cartoon view of a region from the interface of interaction between SrbA and PGM. The most significant interaction is between residues W691-Q094 with the lowest hydrogen bond, a major contribution to the best conformation of the complex. b Cartoon view of a region from the interface of interaction between SrbA and SSDH. Here, the most significant interaction was between residues D676-H003 via a 2.9 Å hydrogen bond. c Cartoon view of a region from the interface of interaction between SrbA and ACAT. The most significant interaction is between residues D582-R270 with the lowest hydrogen bond, a major contribution to the best conformation of the complex. d Cartoon view of a region from the interface of interaction between SrbA and GGT. Here, the most significant interaction was between residues R009-Q565 via a 2.6 Å hydrogen bond. Green = SrbA; red = PGM; yellow = SSDH; cyan = ACAT; magenta = GGT; R = arginine; D = aspartic acid; F = phenylalanine; E = glutamic acid; W = tryptophan; L = lysine; Q = glutamine; M = methionine
The major categories detected were cell cycle and growth/morphogenesis; metabolism and energy, biological processes, such as those related to amino acid precursors; fatty acid synthesis (including ergosterol biosynthesis); glycolysis; and proteins involved with the cell wall and GABA shunt pathway. Regarding cell cycle and growth/morphogenesis, the SrbA ortholog interacted with PCL1 (a cyclin involved in the regulation of polarized growth and morphogenesis through the cell cycle), CBF1 (a basic helix-loop-helix protein which forms homodimers with other transcription factors required for chromosome segregation), CLN1 (cyclin involved in the regulation of the cell cycle), and SIN3 (involved with chromosomal integrity). In addition, the SrbA ortholog interacted with IXR1 (transcriptional repressor that regulates hypoxic genes during normoxia) and NAP1 (histone chaperone that increases in response to DNA replication stress). These results are consistent with SrbA’s activity as a transcription activator presented in this study (Fig. 1; Fig. 2; Supp. Table 1; Supp. Files 1, 2 and 3), probably forming protein complexes during gene expression regulation. It is well established that the basic helix-loop-helix (bHLH) proteins form multiple dimer combinations based on the availability of binding partners being relevant to gene regulation [47], as discussed in the “SrbA as hypoxic transcription regulator” section. Interestingly, the CBF1 protein is also a bHLH protein that forms homodimers with other transcription factors, and IXR1 is a transcriptional repressor that regulates hypoxic genes during normoxia. The interactions involving SrbA/proteins related to cell cycle/morphogenesis show the influence of SrbA on the growth and morphology of the fungus to compensate for hypoxia stress.
Other interactors were related to metabolism and energy processes. The SrbA ortholog showed interaction with ASI2 (regulator of sterol biosynthesis); CEM1 (beta-keto-acyl synthase with a possible role in fatty acid synthesis); ERG27 (3-keto sterol reductase, an intermediate in ergosterol biosynthesis); HTD2 (3-hydroxyacyl-thioester dehydratase; involved in fatty acid biosynthesis); HXK2 (hexokinase that catalyzes the phosphorylation of glucose); and UGA3 (transcriptional activator for GABA-dependent induction of GABA genes). Enzymes involved in beta-oxidation and in the production of ergosterol precursor molecules, glycolysis, and GABA shunt were upregulated according to global hypoxic proteomic data in P. lutzii [14]. The metabolism of fatty acids and ergosterol are increased in C. albicans, C. neoformans, A. fumigatus, and A. nidulans in response to hypoxia; these molecules are required for stability, fluidity, and structure of the fungus plasma membrane [5, 7, 10, 51, 52, 56]. Moreover, ergosterol is known to be is an immunoactive fungal molecule [57, 58]. The dosage of the ergosterol molecule was performed in P. brasiliensis and it was increased during hypoxia responses [54].
The hexokinase protein regulates the glycolysis pathway [59] and the GABA shunt is described as an alternative route to the TCA cycle in P. lutzii, A. nidulans, and A. fumigatus under hypoxia [7, 14, 53]. More details about GABA shunt are described forward (next subtopic). A protein related to the cell wall was also detected in the network, the DAN4 protein that is expressed under anaerobic conditions and repressed during aerobic growth. Altogether, these results reinforce that the SrbA protein possibly acts as the main hypoxic regulator in P. lutzii. Conversely, all hypoxia responses detected in previous researches were not specifically attributed to SrbA.
Three-dimensional predictions
In silico predictions involving three-dimensional structures (3D predictions) were performed to PlSrbA and other four selected proteins (Figs. 4, 5, Table 1, Supplementary File 5). These proteins are representants of glycolysis, 4-aminobutyrate shunt (GABA shunt), acetyl-CoA, and glutathione metabolism processes, which were regulated by hypoxia adaptation in P. lutzii [14]. They were chosen to strengthen our suggestions related to SrbA’s regulated pathways. Interestingly, the interactions occurred at the C-terminal portion of the SrbA protein, after the transmembrane domain (Fig. 4). This might be explained because the N-terminal portion carries the bHLH domain, which is cleaved from SrbA in the endoplasmic reticulum and acts as a transcriptional activator [17, 60, 61].
The glycolysis pathway is ubiquitous throughout nature and essential for energy provision and also other physiological functions in heterotrophic cells [62]. Here, the 3D structure of the 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (PGM) protein and also its interaction with SrbA are shown (Fig. 4). The PGM enzyme catalyzes the reversible conversion of 3-phosphoglycerate and 2-phosphoglycerate as part of glycolysis and gluconeogenesis pathways [63]. The glycolysis was detected as upregulated by hypoxia in P. lutzii after 24 h of stress [14].
In addition, similar results were obtained for gamma-glutamyltranspeptidase (GGT) and succinate-semialdehyde dehydrogenase (SSDH) enzymes (Fig. 4). The GGT enzyme is involved with glutathione metabolism releasing glutamate as a product [64]. The glutathione metabolism was considered essential to Candida glabrata and also for the virulence of C. albicans, the two major human opportunistic fungal pathogens [65]. The glutamate molecule is relevant in this context because it is driven to γ-aminobutyric acid (GABA) production [66]. The GABA shunt is a metabolic pathway controlled by three enzymes and the last one, the succinate-semialdehyde dehydrogenase (SSDH), produces a succinate molecule which can enter in the tricarboxylic acid cycle (TCA) [65, 67]. When the TCA cycle is affected by stress conditions, succinate acts as an intermediate product from GABA. In fact, the GABA shunt is described as an alternative route to the TCA cycle [53] and it was detected as being utilized by A. fumigatus in response to hypoxia [7]. We predicted the best conformation of the interaction between GGT and SSDH with SrbA (Fig. 4), both enzymes were detected as upregulated by hypoxia in P. lutzii and the TCA pathway was partially downregulated [14]. Altogether, our results reinforce the relevance of GABA shunt to P. lutzii hypoxic responses and its connection to SrbA regulation.
We also determined the interaction between the acetyl-coenzyme A transporter 1 (ACAT) with SrbA (Fig. 4). ACAT is a reticulum endoplasmic localized protein involved with acetyl-CoA transportation [68]. In the P. lutzii context, it is relevant to the ergosterol biosynthesis process, because the acetyl-CoA transportation results in the production of precursor molecules. Transcripts related to ergosterol biosynthesis were upregulated during hypoxia in P. lutzii [14]. The relevance of this molecule was discussed above. The results reinforce our suggestion that SrbA might be a main hypoxic regulator in P. lutzii.
Hotspots analyses
The modes of protein-protein interaction are based on balanced coefficients of energy that includes electrostatic, hydrophobic, and Van der Waals force. Several studies have shown how electrostatic forces contribute to PPIs [69, 70] including those related to diseases development and infectious processes [71]. Amino acids categorized as hotspots are found in clusters within the interface of interaction and hydrophobic effects are the main reasons why hotspots tend to be near one another. This effect improves the free energy of the complex and helps to stabilize the complex [72]. The hotspots found to SrbA and each partner are shown in Supplementary File 5.
Figure 5 shows the interface of interaction between SrbA and PGM, SSDH, ACT, and GGT proteins. The former interaction showed a region of low free energy with a cluster of several hotspot residues binding to amino acids of the partner protein. The interface of interaction between SrbA and PGM defined a region of low free energy with a cluster of two main hotspot residues that perform several hydrogen bonds and the shortest distance was 2.9 Å between Q094 and W691 (Fig. 5a). The lowest hydrogen bond distance found in the interaction between SrbA and SSDH was 2.9 Å between D675 and H003 (Fig. 5b). This very same region presents several bonds between hotspot residues that contribute to the folding and stabilization of the binary complex conformation (W691-R050, W691-V692, R279-R050, L722-T046, D676-H003). About SrbA and ACAT, the residue R270 on the ACAT structure interacts twice with D582 of SrbA via hydrogen bonds (2.7 and 2.8 Å). Other residues interact via more distant hydrogen bonds but they contribute to stabilizing the binary complex as well (E645-F264, W726-L285, and W726-W288) (Fig. 5c). The interaction between SrbA-GGT showed a region with a cluster of hotspot residues near the residue R009 on the SrbA structure. This residue interacts with Q565 of GGT, which is 2.6 Å away. The other residues of this cluster are Q005-M591, R009-M591, R009-Q565, and M012-Q565, with hydrogen bonds ranging from 2.8–3.8 Å (Fig. 5d).
The residues R729 (Fig. 5b), R270 and R267 (Fig. 5c), and R009 (Fig. 5d) interact with residues of the partner. The arginine side chain is very amphipathic is frequently present on the surface of proteins. The hydrophilic part of arginine is able to bind to other polar residues or to the surrounding environment [73]. Arginine residues contribute enormously to the interaction between toxin proteins from pathogens and ion channel proteins from hosts through electrostatic forces [74]. These residues act as an electrostatic adhesive force among macromolecules [75]. Here, arginine hotspot residues form hydrogen bonds with neighbor residues from the same polypeptide chain and with amino acids from the polypeptide chain of the partner protein. Thus, it greatly influences the conformation stability of the binary complexes formed by SrbA.
The residues Q094 and W086 (Fig. 5a); H003, D675, and D676 (Fig. 5b); D582, W726, and W288 (Fig. 5c); and Q005 and Q565 (Fig. 5d) contribute significantly to the stability and the biological function of the SrbA complex and target proteins. Glutamine (Q) plays important role in the intermolecular association and aggregation of proteins through polar bonds [76] and is able to influence the interaction between proteins and nucleic acids [50]. Tryptophan (W) is frequently present in protein-protein interfaces. This residue has the potential to enhance the bond between proteins working as an anchor [77] and reducing the free energy of binding sites [78]. Residues of histidine present roles regarding molecular interactions due to the properties showed by its structure (Fig. 5b) and its ability to regulate electrostatic interactions of charged residues. Thus, histidine is directly related to the stability of protein complexes [79] and the design of small molecules used and testes in therapeutic approaches [80] including diseases related to the host-pathogen interaction.
Conclusions
The hypoxia responses of P. lutzii and its functional SREBP homolog were previously described [14]. However, the specific mechanisms to hypoxia adaptation linking the responses to SrbA protein have not been analyzed yet. Moreover, there is no silenced or mutant strain for the SrbA gene in P. lutzii at this time. Here, proteins previously detected as regulated by hypoxia in P. lutzii were selected and the correspondent genes were analyzed regarding elements to SrbA binding in their promotor regions. Additional in silico analyses showed that genes have the potential to be regulated directly by SrbA in this fungus. The regulation by the SrbA was also investigated at a protein level. A network with interactors was built and the three-dimensional structures of SrbA and other four proteins were determined; a protein-protein prediction was also performed for them. Although the analyses were performed in this study, the mechanisms of the hypoxia responses in P. lutzii and other species from Paracoccidioides species are largely unknown. This is the first screening that depicted the potential genes regulated by SrbA in P. lutzii. The SrbA protein is localized at the endoplasmic reticulum; under hypoxia, the protein N-terminal region is cleaved and acts as a transcription factor in the nucleus [9, 17]. It is a major transcriptional regulator in A. fumigatus that may act as both a positive and negative regulator of transcription [27]. All results obtained here highlight the relevance of the SrbA to P. lutzii hypoxia adaptation. In this sense, it has a great potential to be an antifungal drug target.
Supplementary information
{kind=link}
Schematic overview ofin silicoanalysis performed in this study. The steps of the analyzes are depicted in the scheme showing the procedures and number of proteins/ genes obtained for each step. Af1163 and Af293: A. fumigatus strains (Blatzer et al., 2011 and Chung et al., 2014); PGM: 2,3-bisphosphoglycerate independent phosphoglycerate mutase; SSDH: succinate-semialdehyde dehydrogenase; ACAT: acetyl-coenzyme A transporter 1; GGT: gamma-glutamyltranspeptidase. (PNG 284 kb)
{kind=link}
Network of interactors of the SrbA homolog fromSaccharomyces cerevisiae, Hms1. The Hms1 protein, homolog of the SrbA from P. lutzii, presented 76 unique interactors in S. cerevisiae. Some of these proteins also interact with each other. The results can be accessed in https://thebiogrid.org/34434/summary/saccharomyces-cerevisiae/hms1.html. The predicted SrbA amino acids were obtained and used to search the homolog in S. cerevisiae. The interaction network was constructed using the BioGrid tool (https://thebiogrid.org/), an interaction repository that compiles data through comprehensive curation efforts. The network shows the genes and its interactions associations related to genetic, physical and chemical evidences. Each interactor is shown in Supplementary File 4. (PNG 2342 kb)
(DOCX 69 kb)
(DOCX 33 kb)
(DOCX 33 kb)
(DOCX 159 kb)
(DOCX 22 kb)
(DOCX 75 kb)
(XLSX 21 kb)
(XLSX 155 kb)
Funding
This work was supported by grants from Instituto Nacional de Ciência e Tecnologia -Fundação de Amparo à Pesquisa do Estado de Goiás (FAPEG).
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brummer C. Restrepo. Paracoccidioidomycosis: an update. Clin Microbiol Rev. 1993;6(2):89–117. doi: 10.1128/CMR.6.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.San-Blas G, Niño-Vega G, Iturriaga T. Paracoccidioides brasiliensis and paracoccidioidomycosis: molecular approaches to morphogenesis, diagnosis, epidemiology, taxonomy and genetics. Med Mycol. 2002;40(3):225–242. doi: 10.1080/mmy.40.3.225.242. [DOI] [PubMed] [Google Scholar]
- 3.Muñoz JF, Farrer RA, Desjardins CA et al (2016) Genome diversity, recombination, and virulence across the major lineages of Paracoccidioides. Mitchell AP, ed. mSphere 1(5). 10.1128/mSphere.00213-16 [DOI] [PMC free article] [PubMed]
- 4.Turissini DA, Gomez OM, Teixeira MM, McEwen JG, Matute DR. Species boundaries in the human pathogen Paracoccidioides. Fungal Genet Biol. 2017;106:9–25. doi: 10.1016/j.fgb.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chun CD, Liu OW, Madhani HD. A link between virulence and homeostatic responses to hypoxia during infection by the human fungal pathogen Cryptococcus neoformans. PLoS Pathog. 2007;3(2):e22. doi: 10.1371/journal.ppat.0030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willger SD, Puttikamonkul S, Kim K-H, Burritt JB, Grahl N, Metzler LJ, Barbuch R, Bard M, Lawrence CB, Cramer RA. A sterol-regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus. PLoS Pathog. 2008;4(11):e1000200. doi: 10.1371/journal.ppat.1000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barker BM, Kroll K, Vödisch M, Mazurie A, Kniemeyer O, Cramer RA. Transcriptomic and proteomic analyses of the Aspergillus fumigatus hypoxia response using an oxygen-controlled fermenter. BMC Genomics. 2012;13:62. doi: 10.1186/1471-2164-13-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang YC, Bien CM, Lee H, Espenshade PJ, Kwon-Chung KJ. Sre1p, a regulator of oxygen sensing and sterol homeostasis, is required for virulence in Cryptococcus neoformans. Mol Microbiol. 2007;64(3):614–629. doi: 10.1111/j.1365-2958.2007.05676.x. [DOI] [PubMed] [Google Scholar]
- 9.Hughes AL, Todd BL, Espenshade PJ. SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell. 2005;120(6):831–842. doi: 10.1016/j.cell.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu M, Fujii T, Masuo S, Fujita K, Takaya N. Proteomic analysis of Aspergillus nidulans cultured under hypoxic conditions. Proteomics. 2009;9(1):7–19. doi: 10.1002/pmic.200701163. [DOI] [PubMed] [Google Scholar]
- 11.Synnott JM, Guida A, Mulhern-Haughey S, Higgins DG, Butler G. Regulation of the hypoxic response in Candida albicans. Eukaryot Cell. 2010;9(11):1734–1746. doi: 10.1128/EC.00159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung D, Barker BM, Carey CC, Merriman B, Werner ER, Lechner BE, Dhingra S, Cheng C, Xu W, Blosser SJ, Morohashi K, Mazurie A, Mitchell TK, Haas H, Mitchell AP, Cramer RA. ChIP-seq and in vivo transcriptome analyses of the Aspergillus fumigatus SREBP SrbA reveals a new regulator of the fungal hypoxia response and virulence. PLoS Pathog. 2014;10(11):e1004487. doi: 10.1371/journal.ppat.1004487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DuBois JC, Smulian AG. Sterol regulatory element binding protein (Srb1) is required for hypoxic adaptation and virulence in the dimorphic fungus Histoplasma capsulatum. Chaturvedi V, ed. PLoS One. 2016;11(10):e0163849. doi: 10.1371/journal.pone.0163849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lima P, de S, Chung D, Bailão AM, Cramer RA, Soares CM, de A (2015) Characterization of the Paracoccidioides hypoxia response reveals new insights into pathogenesis mechanisms of this important human pathogenic fungus. Vinetz JM, ed. PLoS Negl Trop Dis 9(12). 10.1371/journal.pntd.0004282 [DOI] [PMC free article] [PubMed]
- 15.Todd BL, Stewart EV, Burg JS, Hughes AL, Espenshade PJ. Sterol regulatory element binding protein is a principal regulator of anaerobic gene expression in fission yeast. Mol Cell Biol. 2006;26(7):2817–2831. doi: 10.1128/MCB.26.7.2817-2831.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson DL, Cox M (2014) Princípios de Bioquímica de Lehninger. 6th ed. Artmed.
- 17.Bien CM, Espenshade PJ. Sterol regulatory element binding proteins in fungi: hypoxic transcription factors linked to pathogenesis. Eukaryot Cell. 2010;9(3):352–359. doi: 10.1128/EC.00358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob Agents Chemother. 2005;49(5):1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willger SD, Cornish EJ, Chung D, Fleming BA, Lehmann MM, Puttikamonkul S, Cramer RA. Dsc orthologs are required for hypoxia adaptation, triazole drug responses, and fungal virulence in Aspergillus fumigatus. Eukaryot Cell. 2012;11(12):1557–1567. doi: 10.1128/EC.00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linde J, Hortschansky P, Fazius E, Brakhage AA, Guthke R, Haas H. Regulatory interactions for iron homeostasis in Aspergillus fumigatus inferred by a Systems Biology approach. BMC Syst Biol. 2012;6(1):6. doi: 10.1186/1752-0509-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith JR, Osborne TF, Goldstein JL, Brown MS. Identification of nucleotides responsible for enhancer activity of sterol regulatory element in low density lipoprotein receptor gene. J Biol Chem. 1990;265(4):2306–2310. doi: 10.1016/S0021-9258(19)39976-4. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence CE, Reilly AA. An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins. 1990;7(1):41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- 23.Bailey TL, Elkan C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Mach Learn. 1995;21:51–80. [Google Scholar]
- 24.Bailey TL, Williams N, Misleh C, Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Research. ;34(Web Server):W369-W373. doi:10.1093/nar/gkl198 [DOI] [PMC free article] [PubMed]
- 25.Bailey TL, Boden M, Buske FA, et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research. 37(Web Server):W202-W208. doi:10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed]
- 26.Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blatzer M, Barker BM, Willger SD, Beckmann N, Blosser SJ, Cornish EJ, Mazurie A, Grahl N, Haas H, Cramer RA. SREBP coordinates iron and ergosterol homeostasis to mediate triazole drug and hypoxia responses in the human fungal pathogen Aspergillus fumigatus. PLoS Genet. 2011;7(12):e1002374. doi: 10.1371/journal.pgen.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Coleman-Derr D, Chen G, Gu YQ. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015;43(W1):W78–W84. doi: 10.1093/nar/gkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva-Bailão MG. Potenciano da Silva KL, dos Anjos LRB, et al. Mechanisms of copper and zinc homeostasis in pathogenic black fungi. Fungal Biology. 2018;122(6):526–537. doi: 10.1016/j.funbio.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Chatr-Aryamontri A, Oughtred R, Boucher L, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45(D1):D369–D379. doi: 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu S, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35(10):3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swendsen RH, Wang J-S. Replica Monte Carlo simulation of spin-glasses. Phys Rev Lett. 1986;57(21):2607–2609. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Skolnick J. SPICKER: a clustering approach to identify near-native protein folds. J Comput Chem. 2004;25(6):865–871. doi: 10.1002/jcc.20011. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Roy A, Zhang Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29(20):2588–2595. doi: 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu X, Mitchell JC. KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins. 2011;79(9):2671–2683. doi: 10.1002/prot.23094. [DOI] [PubMed] [Google Scholar]
- 39.Darnell SJ, LeGault L, Mitchell JC (2008) KFC Server: interactive forecasting of protein interaction hot spots. Nucleic Acids Res. 36(Web Server issue):W265-269. doi:10.1093/nar/gkn346 [DOI] [PMC free article] [PubMed]
- 40.Darnell SJ, Page D, Mitchell JC. An automated decision-tree approach to predicting protein interaction hot spots. Proteins. 2007;68(4):813–823. doi: 10.1002/prot.21474. [DOI] [PubMed] [Google Scholar]
- 41.Nechaev S, Adelman K. Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2011;1809(1):34–45. doi: 10.1016/j.bbagrm.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes Dev. 1996;10(21):2657–2683. doi: 10.1101/gad.10.21.2657. [DOI] [PubMed] [Google Scholar]
- 43.Ma J. Transcriptional activators and activation mechanisms. Protein Cell. 2011;2(11):879–888. doi: 10.1007/s13238-011-1101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ptashne M. How eukaryotic transcriptional activators work. Nature. 1988;335(6192):683–689. doi: 10.1038/335683a0. [DOI] [PubMed] [Google Scholar]
- 45.Dynan WS. Modularity in promoters and enhancers. Cell. 1989;58(1):1–4. doi: 10.1016/0092-8674(89)90393-0. [DOI] [PubMed] [Google Scholar]
- 46.Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol. 1995;15(3):1405–1421. doi: 10.1128/MCB.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson KA. SURVEY AND SUMMARY: Saccharomyces cerevisiae basic helix-loop-helix proteins regulate diverse biological processes. Nucleic Acids Res 2000;28(7):1499-1505. doi:10.1093/nar/28.7.1499 [DOI] [PMC free article] [PubMed]
- 48.Datta S, Osborne TF. Activation domains from both monomers contribute to transcriptional stimulation by sterol regulatory element-binding protein dimers. J Biol Chem. 2005;280(5):3338–3345. doi: 10.1074/jbc.M411222200. [DOI] [PubMed] [Google Scholar]
- 49.Zoumi A, Datta S, Liaw L-HL, Wu CJ, Manthripragada G, Osborne TF, LaMorte VJ Spatial distribution and function of sterol regulatory element-binding protein 1a and 2 homo- and heterodimers by in vivo two-photon imaging and spectroscopy fluorescence resonance energy Transfer. MCB. 2005;25(8):2946-2956. doi:10.1128/MCB.25.8.2946-2956.2005 [DOI] [PMC free article] [PubMed]
- 50.Rhys NH, Soper AK, Dougan L. The hydrogen-bonding ability of the amino acid glutamine revealed by neutron diffraction experiments. J Phys Chem B. 2012;116(45):13308–13319. doi: 10.1021/jp307442f. [DOI] [PubMed] [Google Scholar]
- 51.Lee H, Bien CM, Hughes AL, Espenshade PJ, Kwon-Chung KJ, Chang YC. Cobalt chloride, a hypoxia-mimicking agent, targets sterol synthesis in the pathogenic fungus Cryptococcus neoformans. Mol Microbiol. 2007;65(4):1018–1033. doi: 10.1111/j.1365-2958.2007.05844.x. [DOI] [PubMed] [Google Scholar]
- 52.Askew C, Sellam A, Epp E, Hogues H, Mullick A, Nantel A, Whiteway M. Transcriptional regulation of carbohydrate metabolism in the human pathogen Candida albicans. PLoS Pathog. 2009;5(10):e1000612. doi: 10.1371/journal.ppat.1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masuo S, Terabayashi Y, Shimizu M, Fujii T, Kitazume T, Takaya N. Global gene expression analysis of Aspergillus nidulans reveals metabolic shift and transcription suppression under hypoxia. Mol Gen Genomics. 2010;284(6):415–424. doi: 10.1007/s00438-010-0576-x. [DOI] [PubMed] [Google Scholar]
- 54.Oliveira LN, Lima P de S, Araújo DS, et al. iTRAQ-based proteomic analysis of Paracoccidioides brasiliensis in response to hypoxia. Microbiol Res. 2021;247:126730. doi:10.1016/j.micres.2021.126730 [DOI] [PubMed]
- 55.Ingavale SS, Chang YC, Lee H, McClelland CM, Leong ML, Kwon-Chung KJ. Importance of mitochondria in survival of Cryptococcus neoformans under low oxygen conditions and tolerance to cobalt chloride. PLoS Pathog. 2008;4(9):e1000155. doi: 10.1371/journal.ppat.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Setiadi ER, Doedt T, Cottier F, Noffz C, Ernst JF. Transcriptional response of Candida albicans to hypoxia: linkage of oxygen sensing and Efg1p-regulatory networks. J Mol Biol. 2006;361(3):399–411. doi: 10.1016/j.jmb.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 57.Koselny K, Mutlu N, Minard AY, Kumar A, Krysan DJ, Wellington M (2018) A genome-wide screen of deletion mutants in the filamentous Saccharomyces cerevisiae background identifies ergosterol as a direct trigger of macrophage pyroptosis. mBio 9(4). 10.1128/mBio.01204-18 [DOI] [PMC free article] [PubMed]
- 58.Rodrigues ML (2018) The multifunctional fungal ergosterol. mBio. 9(5):e01755-18, /mbio/9/5/mBio.01755-18.atom. doi:10.1128/mBio.01755-18 [DOI] [PMC free article] [PubMed]
- 59.Lobo Z, Maitra PK. Physiological role of glucose-phosphorylating enzymes in Saccharomyces cerevisiae. Arch Biochem Biophys. 1977;182(2):639–645. doi: 10.1016/0003-9861(77)90544-6. [DOI] [PubMed] [Google Scholar]
- 60.Butler G. Hypoxia and gene expression in eukaryotic microbes. Annu Rev Microbiol. 2013;67:291–312. doi: 10.1146/annurev-micro-092412-155658. [DOI] [PubMed] [Google Scholar]
- 61.Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- 62.Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol. 2004;7(3):254–261. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 63.Johnsen U, Schönheit P. Characterization of cofactor-dependent and cofactor-independent phosphoglycerate mutases from Archaea. Extremophiles. 2007;11(5):647–657. doi: 10.1007/s00792-007-0094-x. [DOI] [PubMed] [Google Scholar]
- 64.Mehdi K, Thierie J, Penninckx MJ. gamma-Glutamyl transpeptidase in the yeast Saccharomyces cerevisiae and its role in the vacuolar transport and metabolism of glutathione. Biochem J. 2001;359(Pt 3):631–637. doi: 10.1042/0264-6021:3590631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yadav AK, Desai PR, Rai MN, Kaur R, Ganesan K, Bachhawat AK. Glutathione biosynthesis in the yeast pathogens Candida glabrata and Candida albicans: essential in C. glabrata, and essential for virulence in C. albicans. Microbiology (Reading, Engl) 2011;157(Pt 2):484–495. doi: 10.1099/mic.0.045054-0. [DOI] [PubMed] [Google Scholar]
- 66.Schousboe A, Waagepetersen HS (2007) GABA: homeostatic and pharmacological aspects - PubMed. Published. Accessed August 21, 2020. https://pubmed.ncbi.nlm.nih.gov/17499106/ [DOI] [PubMed]
- 67.Aoki H, Uda I, Tagami K, Furuya Y, Endo Y, Fujimoto K. The production of a new tempeh-like fermented soybean containing a high level of gamma-aminobutyric acid by anaerobic incubation with Rhizopus. Biosci Biotechnol Biochem. 2003;67(5):1018–1023. doi: 10.1271/bbb.67.1018. [DOI] [PubMed] [Google Scholar]
- 68.Kanamori A, Nakayama J, Fukuda MN, Stallcup WB, Sasaki K, Fukuda M, Hirabayashi Y. Expression cloning and characterization of a cDNA encoding a novel membrane protein required for the formation of O-acetylated ganglioside: a putative acetyl-CoA transporter. Proc Natl Acad Sci U S A. 1997;94(7):2897–2902. doi: 10.1073/pnas.94.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Z, Witham S, Alexov E. On the role of electrostatics in protein–protein interactions. Phys Biol. 2011;8(3):035001. doi: 10.1088/1478-3975/8/3/035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Norel R, Sheinerman F, Petrey D, Honig B. Electrostatic contributions to protein-protein interactions: fast energetic filters for docking and their physical basis. Protein Sci. 2001;10(11):2147–2161. doi: 10.1110/ps.12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li L, Jia Z, Peng Y, Godar S, Getov I, Teng S, Alper J, Alexov E. Forces and disease: electrostatic force differences caused by mutations in kinesin motor domains can distinguish between disease-causing and non-disease-causing mutations. Sci Rep. 2017;7(1):8237. doi: 10.1038/s41598-017-08419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y, Huang Y, Swaminathan CP, Smith-Gill SJ, Mariuzza RA. Magnitude of the hydrophobic effect at central versus peripheral sites in protein-protein interfaces. Structure. 2005;13(2):297–307. doi: 10.1016/j.str.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 73.Dong J-Y, Qin L-Q, Zhang Z, Zhao Y, Wang J, Arigoni F, Zhang W. Effect of oral l-arginine supplementation on blood pressure: a meta-analysis of randomized, double-blind, placebo-controlled trials. Am Heart J. 2011;162(6):959–965. doi: 10.1016/j.ahj.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 74.Feng J, Hu Y, Yi H, Yin S, Han S, Hu J, Chen Z, Yang W, Cao Z, de Waard M, Sabatier JM, Li W, Wu Y. Two conserved arginine residues from the SK3 potassium channel outer vestibule control selectivity of recognition by scorpion toxins. J Biol Chem. 2013;288(18):12544–12553. doi: 10.1074/jbc.M112.433888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tesei G, Vazdar M, Jensen MR, Cragnell C, Mason PE, Heyda J, Skepö M, Jungwirth P, Lund M. Self-association of a highly charged arginine-rich cell-penetrating peptide. Proc Natl Acad Sci U S A. 2017;114(43):11428–11433. doi: 10.1073/pnas.1712078114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Law MJ. The role of positively charged amino acids and electrostatic interactions in the complex of U1A protein and U1 hairpin II RNA. Nucleic Acids Res. 2006;34(1):275–285. doi: 10.1093/nar/gkj436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Jesus AJ, Allen TW. The role of tryptophan side chains in membrane protein anchoring and hydrophobic mismatch. Biochim Biophys Acta. 2013;1828(2):864–876. doi: 10.1016/j.bbamem.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 78.Samanta U, Chakrabarti P. Assessing the role of tryptophan residues in the binding site. Protein Eng Des Sel. 2001;14(1):7–15. doi: 10.1093/protein/14.1.7. [DOI] [PubMed] [Google Scholar]
- 79.Zheng P, Cao Y, Bu T, Straus SK, Li H. Single molecule force spectroscopy reveals that electrostatic interactions affect the mechanical stability of proteins. Biophys J. 2011;100(6):1534–1541. doi: 10.1016/j.bpj.2011.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu J, Luther PW, Leng Q, Mixson AJ. Synthetic histidine-rich peptides inhibit Candida species and other fungi in vitro: role of endocytosis and treatment implications. AAC. 2006;50(8):2797–2805. doi: 10.1128/AAC.00411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic overview ofin silicoanalysis performed in this study. The steps of the analyzes are depicted in the scheme showing the procedures and number of proteins/ genes obtained for each step. Af1163 and Af293: A. fumigatus strains (Blatzer et al., 2011 and Chung et al., 2014); PGM: 2,3-bisphosphoglycerate independent phosphoglycerate mutase; SSDH: succinate-semialdehyde dehydrogenase; ACAT: acetyl-coenzyme A transporter 1; GGT: gamma-glutamyltranspeptidase. (PNG 284 kb)
Network of interactors of the SrbA homolog fromSaccharomyces cerevisiae, Hms1. The Hms1 protein, homolog of the SrbA from P. lutzii, presented 76 unique interactors in S. cerevisiae. Some of these proteins also interact with each other. The results can be accessed in https://thebiogrid.org/34434/summary/saccharomyces-cerevisiae/hms1.html. The predicted SrbA amino acids were obtained and used to search the homolog in S. cerevisiae. The interaction network was constructed using the BioGrid tool (https://thebiogrid.org/), an interaction repository that compiles data through comprehensive curation efforts. The network shows the genes and its interactions associations related to genetic, physical and chemical evidences. Each interactor is shown in Supplementary File 4. (PNG 2342 kb)
(DOCX 69 kb)
(DOCX 33 kb)
(DOCX 33 kb)
(DOCX 159 kb)
(DOCX 22 kb)
(DOCX 75 kb)
(XLSX 21 kb)
(XLSX 155 kb)