

Abstract
The Conserved Oligomeric Golgi (COG) complex is an eight subunit protein complex associated with Golgi membranes. Genetic defects affecting individual COG subunits cause congenital disorders of glycosylation (CDGs), due to mislocalization of Golgi proteins involved in glycosylation mechanisms. While the resulting defects in N-and O-glycosylation have been extensively studied, no corresponding study of proteoglycan (PG) synthesis has been undertaken. We here show that glycosaminoglycan (GAG) modification of PGs is significantly reduced, regardless which COG subunit that is missing in HEK293T cells. Least reduction was observed for cells lacking COG1 and COG8 subunits, that bridge the A and B lobes of the complex. Lack of these subunits did not reduce GAG chain lengths of secreted PGs, which was reduced in cells lacking any other subunit (COG2-7). COG3 knock out (KO) cells had particularly reduced ability to polymerize GAG chains. For cell-associated GAGs, the mutant cell lines, except COG4 and COG7 KO, displayed longer GAG chains than wild-type cells, indicating that COG subunits play a role in cellular turnover of PGs. In light of the important roles PGs play in animal development, the effects KO of individual COG subunits have on GAG synthesis could explain the variable severity of COG associated CDGs.
Keywords: Congenital disorder of glycosylation (CDG), Glycosaminoglycans (GAGs), glycosylation, proteoglycans (PGs), the Conserved Oligomeric (COG) complex, the Golgi apparatus
Introduction
The Conserved Oligomeric Golgi (COG) complex consists of eight protein subunits with conserved expression from yeast to man. All COG subunits are required for normal N- and O-glycosylation of proteins in the Golgi apparatus, genetic defects in genes encoding individual subunits have been classified as congenital disorders of glycosylation (CDGs) (1-3). The COG complex helps to tether COPI vesicles to cisternal Golgi membranes; vesicles that have pinched off and moved retrogradely from a more distal Golgi region (4-5). This process is essential for recycling components of the glycosylation machinery within the secretory pathway to maintain a proper glycosylation output from the cells.
While N-glycan precursor synthesis, transfer to protein, and early processing takes place in the endoplasmic reticulum (ER), glycosaminoglycan (GAG) synthesis onto proteoglycan (PG) protein cores takes mostly place in the Golgi apparatus (6-7). In COG-deficient cells, the later processing steps occurring in the Golgi apparatus are those that are most affected in the case of N-glycan synthesis (8-9), while one would expect that GAG polymerization and modification, which occurs all together in the Golgi apparatus, would be more dramatically affected.
PGs are proteins that share the common feature of being modified with long linear GAG chains in the Golgi apparatus, underway from the ER to the cell surface. Cell surface PGs, like glypicans, that are linked to the surface by glycosyl-phosphatidyl inositol (GPI) anchors, or syndecans that are attached via their transmembrane domains, are important for normal development at both the tissue and organismal level (10,11). Other PGs are secreted and are often contributors to the structure and function of extracellular matrices in a variety of tissues. PGs are classified according to the nature of their GAG chains. Heparan sulfate (HS), heparin, chondroitin sulfate (CS), and dermatan sulfate (DS) share a common precursor, where a linker tetrasaccharide, consisting of xylose-galactose-galactose-glucuronic acid, is attached to a serine with a neighboring glycine (SG site) in a protein core translocated into the ER to pass through secretory pathway (12). The exception from this principle is keratan sulfate (KS) PGs, which carry glycans that are initiated in the same way as N- or O-linked glycoproteins, but acquire GAG chains polymerized of galactose and N-acetyl-glucosamine (GlcNAc) units in the Golgi apparatus (6). HSPGs and heparin GAGs are both polymerized from disaccharides of GlcNAc and glucuronic acid extending from the linker tetrasaccharide, but are modified differently by epimerization (to iduronic acid) and sulfation. Likewise, CSPG and DSPG GAGs are polymers of N-acetyl-galactosamine (GalNAc) and glucuronic acid, that also are differentially modified by epimerization and sulfation (6). A number of PGs are of hybrid type, carrying GAG chains of more than one type, often also in combination with other types of glycans. All the GAG variants carried by the various PG types are modified by sulfation in the Golgi apparatus. Sulfate ions are imported through the cell membrane via specific transporters (13) and are converted to 3’-phosphoadenosine-5’-phosphosulfate (PAPS) in the cytoplasm before translocation into the lumen of the Golgi apparatus through PAPS transporters, where sulfate is incorporated into the GAG chains by a number of different sulfotransferases (14). The metabolic label 35S-sulfate is predominantly incorporated into GAGs in mammalian cells, and is therefore the most specific metabolic label for PGs. By following 35S-sulfate labelled molecules by various techniques, we have undertaken a study of PG synthesis and secretion in HEK293T clonal cell lines lacking individual COG subunits (3). All cell lines displayed reduced synthesis capacity of GAG chains, both of HS and CS/DS type. There were, however, significant differences upon knock-out (KO) of individual subunits. These differences may shed light on the variable severity observed for CDGs caused by genetic defects in genes coding the different subunits of the COG complex.
Materials and methods
Culture of cell lines
Wild-type HEK293T cells and HEK293T cell lines with CRISPR/Cas9 KO of individual COG subunits (COG1-COG8) derived from the parental cell line as described (3), were grown in DMEM/F12 medium (Gibco, Life Technologies, Paisley, UK) supplemented with 10 % fetal calf serum (FCS; HyClone, GE Healthcare Life Sciences, Logan, UT, USA), and 1 % penicillum/streptomycin (PS; Lonza, Verviers, Belgium). The cell lines were kept at 37 °C and 5 % CO2 in a humidified incubator. Cells were passaged via gentle resuspension every 72 hours (h) with or without the use of 0.05 % Trypsin/EDTA. Collected cells were preserved in growth medium containing 10 % DMSO in 2 ml vials and stored in liquid nitrogen for future use.
Metabolic labelling of proteins (35S-cys/met) and proteoglycans (35S-sulfate) and isolation of macromolecules
A commonly used method to monitor the synthesis and transport of macromolecules in cell culture is to introduce an isotope labelled (radioactive) molecule that is incorporated into macromolecules during synthesis. We have previously taken advantage of the fact that sulfate ions are predominantly incorporated into the glycosaminoglycan (GAG) chains of proteoglycans (PGs), by adding 35S-sulfate to the cell culture medium. When labelling with radioactive sugars, like 3H-glucosamine, the label is incorporated both into glycoproteins and PGs, the majority in glycoproteins in most cell types. Another much used metabolic label is 35S-cysteine/methionine (cys/met), which is incorporated into all types of proteins, including PGs (15,16). In this study, we used 35S-sulfate to address metabolic labelling of PG associated GAG chains and 35S-cys/met to address total protein secretion.
For metabolic labelling experiments, HEK293T wild type and COG1-8 KO cell lines were grown to confluency in cell culture bottles (75 cm2) and transferred into and grown to confluency in 6-well plates (Costar 3516, Corning inc., New York, NY, USA) in regular growth medium (see above) before transfer to labelling conditions. Labelling with 35S-cys/met was carried out with 0.2 mCi/ml of 35S-met and 35S-cys (ARS-0110; Hartmann Analytic, Braunschweig, Germany) in 1.2 ml DMEM without cysteine, methionine, and glutamine (Sigma, St. Louis, USA), supplemented with 1% glutamine, 1% PS, and 2 % FCS for 24 h at 37 °C and 5 % CO2 in a humidified incubator. For labeling with 35S-sulfate (16-24 h), the cells were supplemented with 1.2 ml sulfate-free RPMI 1640 medium (Gibco; 041-90985M), supplemented with 2 % FCS and 0.2 mCi 35S-sulfate/ml (S-RA-1; Hartmann Analytic, Braunschweig, Germany) replacing the regular growth medium. At the end of the labeling period, the labeling medium was removed, and the cells were washed twice in ice cold phosphate buffered saline (PBS), followed by lysis in 1.2 ml of lysis buffer (10 mM Tris/HCl (pH 7.5), 0.5 % Nonidet P40, 150 mM NaCl, 0.6 mM PMSF, 3.2 μg/ml aprotinin, and 10 μl/ml phosphatase inhibitor cocktail II (Sigma–Aldrich, St. Louis, MI, USA) for 30 minutes (min) at room temperature.
Analysis of radioactive incorporation into macromolecules
Metabolic label (35S-sulfate or 35S-cys/met) not incorporated into macromolecules during the labelling time period was removed from medium samples and cell lysates by Sephadex G50 fine (GE Healthcare, Chicago, IL, USA) gel filtration, as previously described (17,), where 1 ml of each sample is applied to a 4 ml column and macromolecules are eluted in 1.5 ml PBS or H20. Aliquots of the eluate (40 – 50 μl) were added to 3 ml of Ultima Gold XR scintillation cocktail (PerkinElmer, Waltham, MA, USA) and counted in a TR 1900 Packard scintillation counter (Meridan, CT, USA).
SDS-PAGE
Samples of macromolecules (normally 15-20.000 cpm) metabolically labelled with 35S-sulfate were added SDS sample buffer (BioRad Laboratories, Hercules, CA, USA) and reducing agent (XT, BioRad), heated at 96°C for 3 min and loaded onto 4-12 % Criterion-XT gradient gels (BioRad) and run in MOPS buffer (BioRad). The gels were fixed for 30 min and treated with Amplify (GE Healthcare), dried, and exposed to PhosphorImager screens and subjected to imaging in a Typhoon 9400 (GE Healthcare), followed by quantitation with ImageQuant TL v2003.02 (GE Healthcare). Some samples were subjected to selective degradation of HS chains by combined Heparitinase (hep) I, II, and III (Grampian Enzyme, Aberdeen, Scotland, UK) or CS/DS chains by cABC (chondroitinase ABC; EC 4.2.2.4, Amsbio, Abingdon, UK) as described (18) and loaded onto the gels next to untreated control samples.
Analysis of GAG chain-length by gel filtration
GAG chain lengths were analyzed by gel filtration, using Sepharose Cl-6B (GE Healthcare) columns (19,20). The 35S-labelled GAG chains released by alkaline β-elimination treatment, by incubation for 24 h after addition of 1/10 of the sample volume of 5 M NaOH followed by neutralization with 5 M HCl. The samples were applied to gel filtration columns (1 x 40 cm) together with blue dextran and K2CrO4 as void (Vo) and total (Vt) volume markers, respectively. Fractions were eluted at 6 ml/h with 0.15 M NaCl, 0.05 M Tris-HCl buffer, pH 8.
Flow cytometry
HEK293T cells (wild type or KO) were suspended in PBS, 1 % BSA and incubated with an irrelevant control antibody or with 20 μg/ml of an anti-HS antibody (10E4, Amsbio) for 30 min at 4°C, before staining with goat anti-mouse IgM FITC, Alexa Fluor 488 (ThermoFisher, Waltham, MA, USA). Control staining was performed with second layer antibody only. Cells were analyzed with a FACSort flow cytometer (BD Biosciences), flow data collected with CellQuest 3.3 (BD Biosciences, San Jose, CA, USA), and data generated using Kaluza Flow Cytometry analysis v 1.2. At least 20,000 cells gated as viable were analyzed per sample.
Results
Synthesis and secretion of 35S-sulfate labelled and 35S-cys/met labelled macromolecules in HEK293T cells deficient in individual COG complex subunits
A complete set of HEK293T cells lacking individual COG subunits has previously been generated and characterized with respect to Golgi apparatus morphology, stability of COG subunits (in the absence of a depleted sister subunit), and N-glycan structure (3). We grew all of these cell lines to confluency in parallel in 6-well plates and shifted to medium for metabolic labelling with 35S-sulfate or 35S-cys/met. At the end of the labelling period, the media were harvested to collect the secreted macromolecules, and the cells were washed and lysed to extract the cell associated labelled macromolecules. The macromolecules were separated from unincorporated label by gel filtration of each sample. As seen in figure 1, there is a dramatic reduction in the amounts of both cell associated and secreted 35S-sulfated macromolecules in all the cell lines that are deficient in one of the COG subunits. The least reduction was observed for the cell lines lacking the COG1 or COG8 subunits. There was little variability among the parallels in each experiment, but there was some variability among the experiments performed. The reduction in 35S-labelled macromolecules was not the result of a general block in secretion of macromolecules, since changing the metabolic label to labelling the protein cores with 35S-cys/met gave only a modest reduction in protein secretion, when compared to control cells (figure 2).
Figure 1. Synthesis and secretion of sulfate labelled macromolecules in wild type and COG subunit KO HEK293T cells.

HEK293T wild type and cell lines depleted of individual COG subunits were incubated for 24 h in the presence of 0.2 mCi/ml 35S-sulfate in sulfate-depleted medium. The media and cell fractions were processed as described in materials and methods, and the content of sulfated macromolecules was quantified by scintillation counting and is expressed as % of that of wild type HEK293T cells. The figure is based on four experiments with three parallels in each.
Figure 2. Secretion of metabolically labelled proteins from wild type and COG subunit KO HEK293T cells.
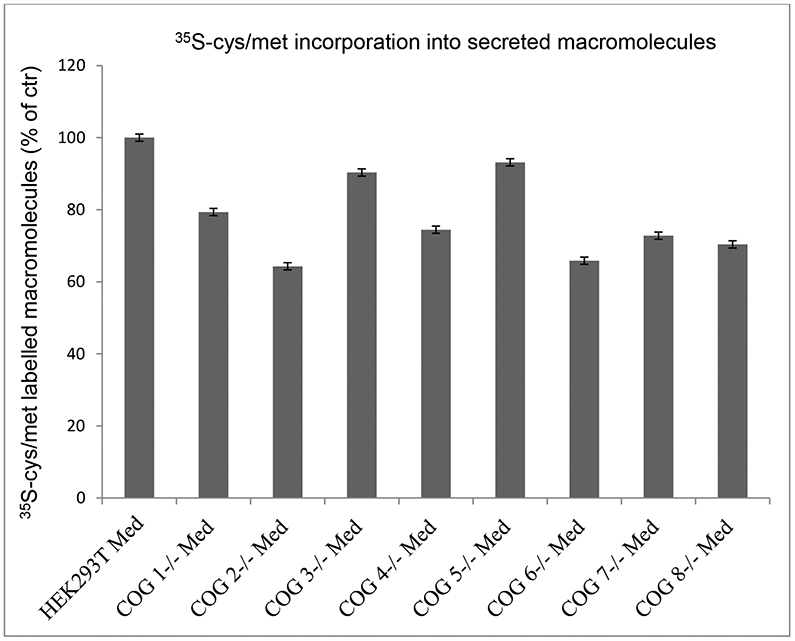
HEK293T wild type and cell lines depleted of individual COG subunits were incubated for 20 h in the presence of 0.2 mCi/ml 35S-cys/met in cys/met-depleted medium. The medium fractions were processed as described in Materials and methods, and the content of 35S-labelled macromolecules was quantified by scintillation counting and is expressed as % of that of wild type HEK293T cells. The figure shows one typical experiment of three with three parallels for each cell line.
Proteoglycan analysis by sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
Aliquots of the samples presented in figure 1 were analyzed by SDS-PAGE. The aliquots used were estimated to contain approximately the same amount of macromolecule-associated cpm of 35S-sulfate. Each sample was divided in three equal parts, where one was left untreated, one was treated with chondroitinase ABC (cABC), while one was treated with a heparitinase I, II, III (hep) mix. For medium samples from wild type HEK293T cells, PGs were distributed over a broad size range. Those of lower mass carry HS chains (degraded by hep), while those of higher mass carry CS chains (degraded by cABC). The sharper band appearing after cABC treatment are typical for high molecular mass CSPGs where the label is on the six sugar units that remain closest to the protein core (16,21). For several of the COG subunit deficient cell lines, the pattern of secreted PGs is similar to that of the wild type cells, although reduced in amount (figure 1). However, COG3 deficient cells clearly have a dramatically reduced ability to polymerize GAG chains and secrete the corresponding PGs (figure 3, panel A), when compared to the other cell lines. The major labelled bands in the 45-55 kDa range are insensitive to enzyme treatment, and may consist of PG protein cores with sulfated GAG linker regions and/or sulfated proteins. Both of these types of molecules were potentially enriched, since these samples were the most concentrated to achieve the desired level of macromolecules with 35S label. The absence of visible sulfated PGs in the medium from COG3 deficient cells is an interesting observation regarding the importance of PG synthesis for development, since no CDG patient with a COG3 defect has been reported to date (22). The same phenomenon is also partially observed for COG5, COG6, and COG7 deficient cells (figure 3, panel B) and to an even lesser extent for COG8 deficient cells (figure 3, panel E).
Figure 3. SDS-PAGE analysis of synthesis and secretion of sulfated macromolecules in wild type and COG subunit KO HEK293T cells.
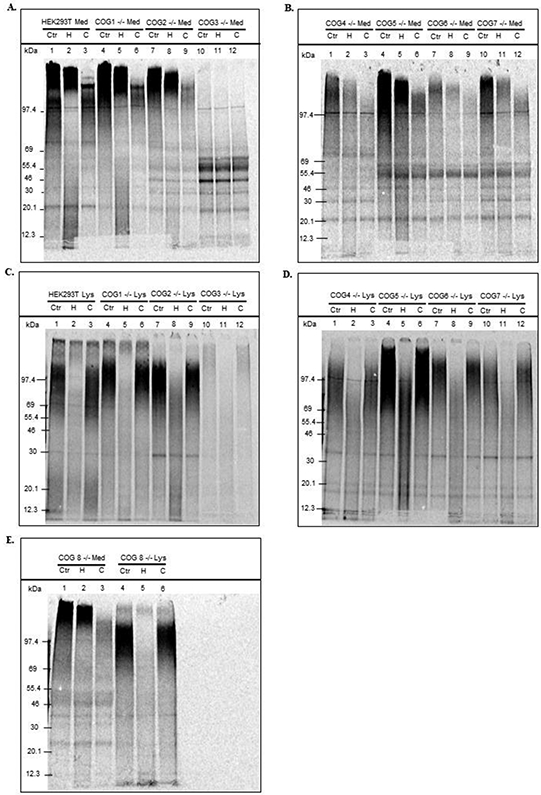
The eluates from Sephadex G-50 fine gel filtration chromatography columns were concentrated by freeze drying to contain approximately 15-20,000 cpm each in SDS-PAGE sample buffer. Parallel aliquots of each sample were subjected to no treatment (Cnt) or to treatment with cABC to degrade CS/DS (C) or hep to degrade HS and heparin (H) prior to SDS-PAGE (4-12 % gradient gels). Medium samples in panels A and B and cell lysate samples in panels C and D. Samples from COG8 deficient cells (both medium and cell lysate) in panel E.
For the cell lysate samples (figure 3, panels C-E), hep treatment has a greater effect than cABC treatment on all the samples, showing that HSPGs are the dominating cell associated PGs in HEK293T cells. While hep treatment of lysates of HEK293T wild type and COG1 and COG8 deficient cells removed almost all macromolecule-associated 35S-sulfate label from the gel lanes, the more concentrated cell lysates still displayed some activity of lower molecular mass. This could be a result of the variable extent of sample concentration to obtain similar cpm counts, where less affected minor components may become more prominent.
Another observation was that the PGs in the lysates of some of the COG deficient cell lines had a higher molecular mass than that of the PGs in the HEK293T cell lysates. The most likely explanation would be that these PGs carry longer GAG chains. These observations made by SDS-PAGE pointed to a more thorough analysis of GAG chain length by gel filtration.
Gel filtration analysis of chain lengths after GAG detachment from protein cores
Intact GAG chains can be chemically detached from the PG protein cores to which they are linked in the secretory pathway. This is mediated by alkaline treatment, inducing β-elimination of the linkage of xylose residues to serines in the protein backbone (12). The samples were then neutralized and applied to Sepharose Cl-6B columns together with Vo and Vt markers.
The GAG chains of the PGs secreted to the medium are polymerized, epimerized, and modified by sulfation in the Golgi apparatus and then secreted without further modification (6). The sizes of the GAG chains of the PGs secreted by HEK293T wild-type cells were in the 0.2 to 0.8 Kav range (figure 4). COG1 and COG8 (figure 4, panels C and J) deficient cells displayed GAG chains of similar length to the wild type cells, while PGs secreted from the cells deficient in one of the other COG subunits in addition carried shorter GAG chains, to a variable extent. This was particularly prominent for secretion from COG3 deficient cells (figure 4, panel E), and is in agreement with the observations made by SDS-PAGE (figure 3). Also the other cell lines that displayed labelled molecules of lower molecular mass (cells deficient in COG5, COG6, or COG7) secreted PGs with GAG chains of shorter length than wild type cells. These findings demonstrated that lack of some COG subunits affects GAG chain polymerization more than the lack of other subunits does.
Figure 4. Gel filtration analysis of GAG chain length of secreted PGs.
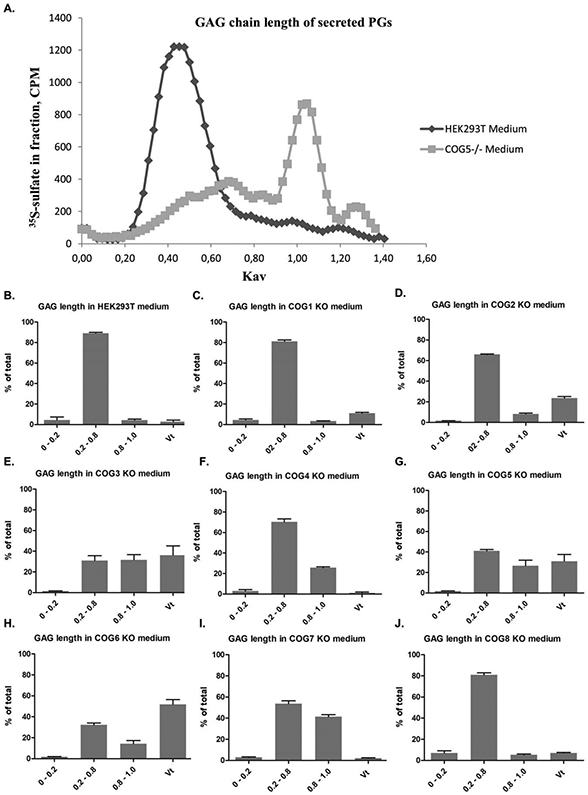
Medium samples eluted from Sephadex G50-fine columns were subjected to alkaline treatment (see Materials and methods) to detach GAG chains from their protein cores. The samples were added the Vo and Vt markers Blue Dextran and K2CrO4, respectively, for Ve and Kav calculations, and then applied to Sepharose CL-6B columns to compare the chain lengths of GAGs synthesized by HEK293T cells and COG subunit deficient cell lines. Panel A shows two examples (HEK293T control and COG5) runs. The panels show the size distribution of GAG chains in at least three samples for each cell line. The fractionation range of Sepharose CL-6B being 10,000 to 1,000,000 (for dextrans). 0-0.2 are the longer chains, 0.2-0.8 are intermediate chains, while 0.8-1.0 are shorter chains. In the Vt, are very short chains with full access to all of the column material, of less than (approximately) 20 disaccharides.
The GAG chains associated with cellular PGs of wild type HEK293T cells are shorter than those attached to secreted PGs. This may be explained by the fact that cell surface PGs turn over by endocytic internalization resulting in shortening of GAG chains, followed by either complete degradation, or recycling of PGs that acquire novel GAG chains in the Golgi apparatus (23-25). In agreement with the observations made by SDS-PAGE, several of the cell lines deficient in one of the COG subunits have PGs carrying longer GAG chains than the wild type cells (figure 5, panels B-J), the exception being COG4 and COG7 deficient cells. This indicates that in the cell lines deficient in COG1, COG2, COG3, COG5, COG6, and COG8, the endocytosis and transport deeper into the cell is significantly reduced, as observed particularly for COG1 deficient cells, where 85 % of the GAG chains are of the same length as those secreted to the medium (figure 5, panel C). In these COG subunit deficient cell lines, the relative fraction of PGs at the cell surface is most likely larger than for the wild type cells, where a significant portion of the PGs is circulating to intracellular organelles. For COG4 and COG7 deficient cells, the GAG chain length is similar to that of the wild type cells, thus in these cells the PGs might follow a normal circulation pathway.
Figure 5. Gel filtration analysis of GAG chain length of cell-assciated PGs.
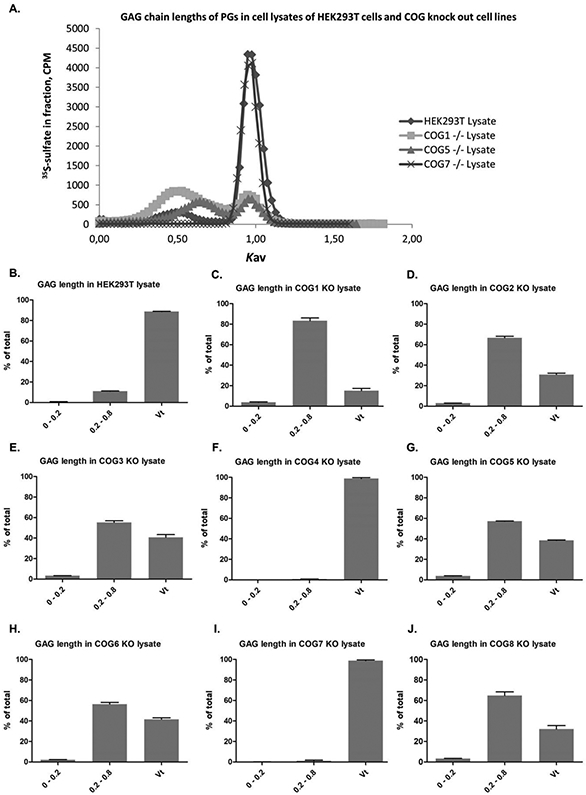
Cell lysate samples eluted from Sephadex G50-fine columns were subjected to alkaline treatment (see Materials and methods) to detach GAG chains from their protein cores. The samples were added the Vo and Vt markers Blue Dextran and K2CrO4, respectively, for Ve and Kav calculations, and then applied to Sepharose CL-6B columns to compare the chain lengths of GAGs synthesized by HEK293T cells and COG subunit deficient cell lines. Panel A shows two examples (HEK293T control and COG5) runs. The panels show the size distribution of GAG chains in at least three samples for each cell line. The fractionation range of Sepharose CL-6B being 10,000 to 1,000,000 (for dextrans). 0-0.2 are the longer chains, 0.2-0.8 are intermediate chains. In the Vt, are very short chains with full access to all of the column material, of less than (approximately) 20 disaccharides.
Flow cytometry of HEK293T wild type and COG subunit deficient cells with an anti-HS antibody.
The 10E4 antibody recognizes heparan sulfate (HS) with N-sulfated glucosamines in the epitope, thus an N-deacetylase/N-sulfotransferase (NDST) must have acted on the polymerizing HS chain. The epitope may be destroyed later by cellular metabolism (26). Prior to flow cytometry, HEK293T wild type cells, and cells deficient in a particular COG subunit, were grown to confluency and detached by mechanical means (pipetting), without any enzymatic treatment, to preserve intact HSPGs at the cell surface. The wild type cells were well recognized by the antibody. A surprising finding was that, both COG1 and COG8 deficient cells were recognized to the same extent as wild type cells (figure 6). For the other COG subunit deficient cell lines, there was only a very small fraction of cells that was recognized. The COG1 and COG8 lacking cell lines are those with the least reduction in PG synthesis, but the synthesis is still significantly reduced when compared to wild type cells. The flow cytometry result may be explained by the fact that a relatively speaking larger fraction of the HSPGs remains at the cell surface with more epitopes recognized by the anti- HS antibody per PG, as suggested based on the determination of GAG chain lengths that showed that the PGs of COG1 and COG8 deficient cells carry longer GAG chains than wild type cells. These findings could contribute to explain why CDG-IIh patients with COG8 deficiency (22, 27) and patients with COG1 deficiency (22,28,29) display milder phenotypes than patients with genetic defects in other COG subunits. This possibility requires further investigation, however, due to the limited number of patients described to date.
Figure 6. Flow cytometry with cell surface bound anti HS-antibody.
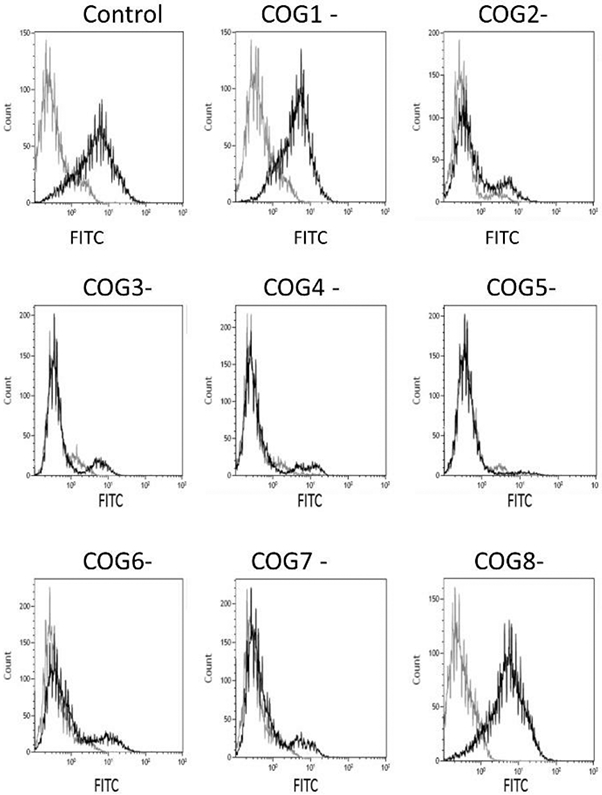
HEK293T cells and COG subunit deficient cell lines were suspended in PBS and incubated with the 10E4 anti-HS antibody for 30 min at 4°C before staining with goat anti-mouse IgM FITC. Controls are incubated with goat anti-mouse FITC, only. Cells were analyzed with a FACSort flow cytometer. Flow data collection was carried out with the program CellQuest 3.3 and the data generated using Kaluza Flow Cytometry analysis v 1.2. A total of 20,000 or more cells gated as viable were analyzed per sample. The figure shows data for wild type HEK293T cells (control) and the cell lines deficient in individual COG subunits 1-8.
Discussion
We have studied proteoglycan (PG) synthesis in the HEK293T cell line and how depletion of single subunits (COG 1-8) by CRISPR/Cas9 of the conserved oligomeric Golgi (COG) complex affects glycosaminoglycan (GAG) polymerization onto PG protein cores, as these are passing through the Golgi apparatus in transit through the secretory pathway. We show that there are significant differences in the effects, depending on which COG subunit that has been depleted. The cell lines used here have been extensively characterized with respect to Golgi morphology, subunit expression, and other glycosylation mechanisms (3).
There is consensus that the COG complex is organized in two lobes (lobe A and lobe B), where lobe A (COG1-4) and lobe B (COG5-8) are connected by COG1-COG8 interaction (2,5,30). Deficiency of a single subunit, partly or completely, has been shown to result in defects in N- and O-glycosylation (8,30,31). Until now, no systematic study of the roles the COG complex could play in GAG synthesis and modification has been undertaken, although indications of a relationship have been reported (32,). The limited focus on PG synthesis in COG deficient cells is surprising, since GAG polymerization takes place in the Golgi apparatus (6,7), and an intact COG complex has been proven essential to normal Golgi structure and function. At the same time, the essential role of PGs and their attached GAG chains in human development and physiology (33), should motivate such studies.
When monitoring synthesis of GAG chains by incorporation of 35S-sulfate as metabolic label, HEK293T cells lacking COG1 or COG8 subunits were the least affected among the COG subunit depleted lines. This, could be the contribution from the PG side to the milder phenotypes observed for patients with defects in the corresponding genes (2,5,8,30-31), but could also be related to the fact that loss of one of these two subunits has not been shown to affect the stability of other subunits of the complex that were tested (all but COG1 and COG2), to the same extent as when any other subunit (COG2-7) was depleted (3). For instance, the transient loss of the COG3 subunit in HeLa cells lead to reduction in COG1, COG2, and COG4 levels (34). It is reasonable to think that the situation is similar in HEK293T cells, although only the COG4 was addressed and found to be reduced after COG3 KO in these cells (3). Thus, with all lobe A subunits affected after COG3 KO, the cells have a dramatically reduced capacity to produce GAG chains of some length that are sensitive to HS- or CS/DS-degrading enzymes (figure 3). This could contribute to the explanation why no patient with COG3 deficiency has been identified to date (22).
Interestingly, although depletion of COG5 leads to degradation of COG7 in the cell lines we have used in these studies, and vice versa (3), the effects on PG synthesis are not identical with respect to their GAG chains. A larger fraction of the PGs secreted from COG5 depleted cells have very short GAG chains (10,000 Da or shorter based on dextran standards, figure 4). For the cell lysates, the PGs of COG7 depleted cells are of similar length to those of the wild type cells, while more than half of the GAG chains of COG5 depleted cells are longer (figure 5). A potential explanation could be that both COG5 or COG7 KO also reduce the expression of the COG6 and COG8 subunits, and that subtle expression level differences in the combination of subunits gives a variable effect (33). The differences between COG5 and COG7 KO could, however, possibly be due to mechanisms outside the Golgi apparatus (39). An interesting observation is that particularly COG4 and COG7 KO leads to accumulation of enlarged endo-lysosomal structures (EELS) in HEK293T cells (37).
A future task would be to address the COG complex subunit interaction partners (5) and how these contribute to proper PG synthesis. For instance, does the COG complex interact with Golgin-84 via the COG7 subunit (38)? KO of the Golgi tethering protein giantin alters the expression at the mRNA level of a number of enzymes involved in PG synthesis (39). Such studies of COG subunits and their interaction partners could shed more light on the synthesis and transport of PGs in mammalian cells.
In conclusion, our data show that lacking different COG complex subunits has variable effects on glycosaminoglycan (GAG) synthesis and turnover in HEK293T cells. Further PG studies of the modified cell lines used here will potentially generate more knowledge about organization of PG synthesis in the Golgi apparatus and also lead to further investigations of PG synthesis in COG related CDGs.
Synopsis:
Glycosaminoglycan (GAG) modification of proteoglycan (PG) protein cores takes place in the Golgi apparatus. The Conserved Oligomeric Golgi (COG) complex regulates the dynamic positioning and therefore also function of Golgi proteins. We report how lack of individual COG protein subunits (COG 1-8) affects GAG synthesis in HEK293T cells. GAG modification of PGs is least affected by COG1 and COG8 deficiency, the subunits bridging lobe A and B of the complex and most dramatically affected by COG3 deficiency.
Acknowledgements
The work was financed in part by a PhD fellowship from the University of Oslo to Ravi Adusumalli. V.L. was supported by NIH grant R01GM083144.
References
- 1).Pokrovskaya ID, Willett R, Smith RD, Morelle W, Kudlyk T, Lupashin VV. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011; 21(12): 1554–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Fisher P, Ungar D. Bridging the gap between glycosylation and vesicle traffic. Front Cell Dev Biol. 2016; 4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Bailey Blackburn J, Pokrovskaya I, Fisher P, Ungar D, Lupashin VV. COG complex complexities: detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Front Cell Dev Biol. 2016; 4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Miller VJ, Ungar D. Re’COG’nition at the Golgi. Traffic 2012; 13(7): 891–7. [DOI] [PubMed] [Google Scholar]
- 5).Willett R, Ungar D, Lupashin V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem Cell Biol. 2013; 140(3)271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Prydz K, Dalen KT. Synthesis and sorting of proteoglycans. J Cell Sci. 2000113(2): 193–205. [DOI] [PubMed] [Google Scholar]
- 7).Karamanos NK, Piperigkou Z, Theocharis AD, Watanabe H, Franchi M, Baud S, Brézillon S, Götte M, Passi A, Vigetti D, Ricard-Blum S, Sanderson RD, Neill T, Iozzo RV. Proteoglycan Chemical Diversity Drives Multifunctional Cell Regulation and Therapeutics. Chem Rev. 2018; 118(18): 9152–9232. [DOI] [PubMed] [Google Scholar]
- 8).Reynders E, Foulquier F, Annaert W, Matthijs G. How Golgi glycosylation meets and needs trafficking: the case of the COG complex. Glycobiology 2011; 21(7):853–63. [DOI] [PubMed] [Google Scholar]
- 9).Rosnoblet C, Peanne R, Legrand D, Foulquier F. Glycoslylation disorders and membrane trafficking. Glycoconj J. 2013; 30(1): 23–31. [DOI] [PubMed] [Google Scholar]
- 10).Fransson LÅ, Belting M, Cheng F, Jönsson M, Mani K, Sandgren S. Novel aspects of glypican biology. Cell Mol Life Sci. 2004; 61: 1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Chung H, Multhaupt HA, Oh ES, Couchman JR. Minireview: Syndecans and their crucial roles during tissue regeneration. FEBS Lett. 2016; 590(15):2408–17. [DOI] [PubMed] [Google Scholar]
- 12).Prydz K. Determinants of glycosaminoglycan (GAG) structure. Biomolecules 2015; 5(3): 2003–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Galante LL, Schwarzbauer JE. Requirements for sulfate transport and the diastrophic dysplasia sulfate transporter in fibronectin matrix assembly. J Cell Biol. 2007; 179(5): 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Dick G, Grøndahl F, Prydz K. Overexpression of the 3'-phosphoadenosine 5'-phosphosulfate (PAPS) transporter 1 increases sulfation of chondroitin sulfate in the apical pathway of MDCK II cells. Glycobiology 18(1): 53–65. [DOI] [PubMed] [Google Scholar]
- 15).Fjeldstad K, Pedersen ME, Vuong TT, Kolset SO, Nordstrand LM, Prydz K. Sulfation in the Golgi lumen of Madin-Darby canine kidney cells is inhibited by brefeldin A and depends on a factor present in the cytoplasm and on Golgi membranes. J Biol Chem. 2002; 277(39):36272–9. [DOI] [PubMed] [Google Scholar]
- 16).Tveit H, Dick G, Skibeli V, Prydz K. A proteoglycan undergoes different modifications en route to the apical and basolateral surfaces of Madin-Darby canine kidney cells. J Biol Chem. 2005; 19: 280(33):29596–603. [DOI] [PubMed] [Google Scholar]
- 17).Svennevig K, Prydz K, Kolset SO. Proteoglycans in polarized epithelial Madin-Darby canine kidney cells. Biochem J. 1995; 311(3): 881–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Grøndahl F, Tveit H, Akslen-Hoel LK, Prydz K. Easy HPLC-based separation and quantitation of chondroitin sulphate and hyaluronan disaccharides after chondroitinase ABC treatment. Carbohydr Res. 2011; 346(1):50–7. [DOI] [PubMed] [Google Scholar]
- 19).Hafte TT, Fagereng GL, Prydz K, Grøndahl F, Tveit H. Protein core-dependent glycosaminoglycan modification and glycosaminoglycan-dependent polarized sorting in epithelial Madin-Darby canine kidney cells. Glycobiology. 2011; 21(4):457–66. [DOI] [PubMed] [Google Scholar]
- 20).Vuong TT, Prydz K, Tveit H. Differences in the apical and basolateral pathways for glycosaminoglycan biosynthesis in Madin-Darby canine kidney cells. Glycobiology. 2006; 16(4):326–32. [DOI] [PubMed] [Google Scholar]
- 21).Tveit H, Akslen LK, Fagereng GL, Tranulis MA, Prydz K. A secretory Golgi bypass route to the apical domain of epithelial MDCK cells. Traffic 2009; 10(11):1685–95. [DOI] [PubMed] [Google Scholar]
- 22).Haijes HA, Jaeken J, Foulquier F, van Hasselt PM. Hypothesis: lobe A (COG1-4)-CDG causes a more severe phenotype than lobe B (COG5-8)-CDG. J Med Genet. 2018; 55(2): 137–142. [DOI] [PubMed] [Google Scholar]
- 23).Egeberg M, Kjeken R, Kolset SO, Berg T, Prydz K. internalization and stepwise degradation of heparan sulfate proteoglycans in rat hepatocytes. Biochem Biophys Acta 2001; 1541: 135–149. [DOI] [PubMed] [Google Scholar]
- 24).Mani K, Jönsson M, Edgren G, Belting M, Fransson LÅ. A novel role for nitric oxide in the endogenous degradation of heparan sulfate during recycling of glypican-1 in vascular endothelial cells. Glycobiology 2000; 10(6): 577–586. [DOI] [PubMed] [Google Scholar]
- 25).Ding K, Jönsson M, Mani K, Sandgren S, Belting M, Fransson LÅ. N-unsubstituted glucosamine in heparn sulfate of recycling glypican-1 from suramin-treated and nitrite-deprived endothelial cells. J Biol Chem. 2001; 276(6): 3885–94. [DOI] [PubMed] [Google Scholar]
- 26).Mani K, Cheng F, Sandgren S, van den Born J, Havsmark B, Ding K, Fransson LÅ. The heparan sulfate-specific epitope 10E4 is NO-sensitive and partly inaccessible in glypican-1. Glycobiology 2004; 14(7): 599–607. [DOI] [PubMed] [Google Scholar]
- 27).Kranz C, Ng BG, Sun L, Sharma V, Eklund EA, Miura Y, Ungar D, Lupashin V, Winkel RD, Cipollo JF, Costello CE, Loh E, Hong W, Freeze HH. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum Mol genet. 2007; 16: 731–41. [DOI] [PubMed] [Google Scholar]
- 28).Folquier F, Vasile E, Schollen E, Callewaert N, Raemaekers T, Quelhas D, Jaeken J, Mills P, Winchester B, Krieger M, Annaert W, Matthijs G. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci USA 2006; 103: 3764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Zeevaert R, Folquier F, Dimitrov B, Reynders E, Van damme-Lombaerts R, Simeonov E, Annaert W, Matthijs G, Jaeken J. Cerebrocostomandibular-like syndrome and a mutation in the conserved oligomeric Golgi complex, subunit 1. Human Mol genet. 2009; 18: 517–24. [DOI] [PubMed] [Google Scholar]
- 30).Foulquier F. COG defects, birth and rise! Biochem Biophys Acta (2009) 1792: 896–902. [DOI] [PubMed] [Google Scholar]
- 31).Climer LK, Dobretsov M, Lupashin V. Defects in COG complex and COG-related trafficking regulators affect neuronal Golgi function. Front Neurosci. 2015; 9: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Riblett AM, Blomen VA, Jae LT, Altamura LA, Doms RW, Brummelkamp TR, Wojcechowskyj JA. A haploid gentic screen identifies heparan sulfate proteoglycans supporting rift valley fever virus infection. J Virol. 2016; 90(3): 1414–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Townley RA, Bülow HE. Dechiphering functional glycosaminoglycan motifs in development. Curr Opin Struct Biol. 2018; 50: 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Zolov SN, Lupashin VV. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J Cell Biol. 2005; 168(5): 747–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Oka T, Vasile E, Penman M, Novina CD, Dykxhoorn DM, Ungar D, Hughson FM, Krieger M. Genetic analysis of the subunit organization and function of the conserved oligomeric Golgi (COG) complex. Studies of COG5- and COG7-deficient mammalian cells. J Biol Chem. 2005; 280(38): 32736–45. [DOI] [PubMed] [Google Scholar]
- 36).Blackburn JB, Kudlyk T, Pokrovskaya I, Lupashin VV. More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects. Traffic 2018; 19: 463–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).D’Souza Z, Blackburn JB, Kudlyk T, Pokrovskaya ID, Lupashin VV. Defects in COG-mediated Golgi trafficking alter endo-lysosomal system in human cells. Front. Cell Dev. Biol 2019; 7: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Sohda M, Misumi Y, Yamamoto A, Nakamura N, Ogota S, Sakisaka S, Hirose S, Ikehara Y, Oda K. Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic 2010; 11:1552–66. [DOI] [PubMed] [Google Scholar]
- 39).Stevenson NL, Bergen DJM, Skinner REH, Kague E, Martin-Silverstone E, Robson Brown KA, Hammond CL, Stephens DJ. Giantin-knockout models reveal a feedback loop between Golgi function and glycosyltransferase expression. J Cell Sci. 2017; 130: 4132–43. [DOI] [PMC free article] [PubMed] [Google Scholar]