

Abstract
A combination therapy using Prussian blue nanoparticles (PBNP) as photothermal therapy (PTT) agents coated with CpG oligodeoxynucleotides, an immunologic adjuvant, as a nanoimmunotherapy (CpG-PBNP-PTT) for neuroblastoma (NB) is described. NB driven by MYCN amplification confers high risk and correlates with a dismal prognosis, accounting for the majority of NB-related mortality. The efficacy of the CpG-PBNP-PTT nanoimmunotherapy in a clinically relevant, TH-MYCN murine NB model (9464D) overexpressing MYCN is tested. When administered to 9464D NB cells in vitro, CpG-PBNP-PTT triggers thermal dose-dependent immunogenic cell death and tumor cell priming for immune recognition in vitro, measured by the expression of specific costimulatory and antigen-presenting molecules. In vivo, intratumorally administered CpG-PBNP-PTT generates complete tumor regression and significantly higher long-term survival compared to controls. Furthermore, CpG-PBNP-PTT-treated mice reject tumor rechallenge. Ex vivo studies confirm these therapeutic responses result from the generation of robust T cell-mediated immunological memory. Consequently, in a synchronous 9464D tumor model, CpG-PBNP-PTT induces complete tumor regression on the treated flank and significantly slows tumor progression on the untreated flank, improving animal survival. These findings demonstrate that localized administration of the CpG-PBNP-PTT nanoimmunotherapy drives potent systemic T cell responses in solid tumors such as NB and therefore has therapeutic implications for NB.
Keywords: Prussian blue nanoparticles, photothermal therapy, CpG oligodeoxynucleotides, nanoimmunotherapy, abscopal effect, neuroblastoma, MYCN amplification
Graphical Abstract
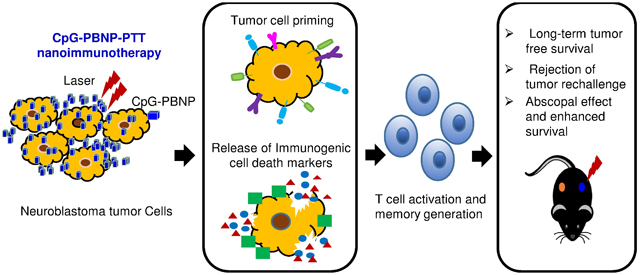
CpG-PBNP-PTT-based nanoimmunotherapy mediates thermal dose-dependent immunogenic cell death and tumor cell priming in neuroblastoma tumor cells, leading to T cell activation and the generation of T cell memory. These effects elicit long-term, tumor-free survival, and rejection of tumor rechallenge in a single tumor model of TH-MYCN neuroblastoma and a potent abscopal effect, leading to slower tumor progression of distal, untreated tumors in a synchronous tumor model.
1. Introduction
Neuroblastoma (NB) is a prevalent solid tumor of childhood with a 5-year survival rate of approximately 40% despite intense multimodal treatment regimens. The survival rates are particularly dismal for patients bearing a MYCN amplification that classifies them as high-risk NB patients.[1, 2] The therapeutic resistance observed in high-risk NB is due to many factors, including a complex tumor microenvironment (TME), which includes impaired T cell activity, reduced immune cell infiltration, along with defective antigen processing and presentation in NB cells.[3–7] Among immune cells, T cells are crucial players in antigen-specific recognition and antitumor cytotoxicity, leading to tumor regression and persistent disease remission. The success of T cell responses relies primarily on the ability of T cells to act as effectors and generate long-term antigen-specific memory. However, if tumor cells are able to evade T cell recognition for an extended period, the T cells can become exhausted and no longer effectively recognize or target the tumor. Thus, it is critical to develop therapeutic interventions that can reverse T cell exhaustion and optimize and maintain T cell activity[8, 9] for better outcomes in high-risk NB patients.
Here we expand upon our previous work using Prussian blue nanoparticles (PBNPs) as agents of photothermal therapy (PTT; PBNP-PTT)[10–16] by investigating the efficacy of PTT using PBNPs coated with unmethylated cytosine-phosphate-guanine oligodeoxynucleotides (CpG) in a syngeneic, murine model of NB.[17, 18] PBNPs are ideal candidates for use as PTT agents, as they are biodegradable with limited toxicity,[13–15] and are FDA approved.[19] We have previously reported that PBNP-PTT elicits efficient conversion of near-infrared (NIR) wavelength light into heat by biocompatible PBNPs causing local heating of the tumor and stimulating immunogenic cell death (ICD),[10, 13–16] a favorable cell death phenotype that engages an antitumor immune response.[20, 21] This triggers release of tumor-associated antigens as well as damage-related molecular patterns from the dying tumor cells which then stimulates the activation of immune cells to target nearby un-irradiated residual cancer cells in a system known as the “abscopal effect”.[22–25] We and others have previously reported that using PTT in combination with immunotherapy further improves the antitumor response of PTT compared to either therapy administered alone, by boosting immunity of the treated subjects and extending long-term durable protection against cancer recurrence.[13, 15, 16]
CpG, the immunologic adjuvant that we have attached to PBNPs, is recognized by toll-like receptor 9 (TLR9) and stimulates a cascade of innate and adaptive immune responses,[26, 27] and has recently been approved for clinical use in a hepatitis B vaccine by the FDA[28]. CpG has been previously studied in the context of NB.[13, 15, 16, 29, 30] Also, Molenkamp et al. demonstrated that intratumoral injection of CpG alone or in combination with radiotherapy increases T cell immune response in cancer patients.[31] However, CpG injected directly into a tumor is rapidly eliminated, thus restricting its immunostimulatory benefits.[32, 33] By coating CpG onto the PBNPs (CpG-PBNPs), we prevent its rapid clearance thus increasing its bioavailability in the local TME. We have previously reported that CpG-PBNP-based photothermal therapy (CpG-PBNP-PTT) stimulates antigen-presenting cells (APCs), such as dendritic cells (DCs), and helps to overcome the immunosuppressive TME by triggering a T cell response in the Neuro-2a model of NB.[15, 16]
However, most immunotherapeutic studies for NB, including our previous studies, use C1300-derived cell lines such as Neuro-2a, which lack MYCN amplification and the expression of the GD2 antigen, which is prominently expressed on the surface of high-risk human NB tumors.[18, 34] Thus, to model high-risk NB patients, in the current study we explore the potential of locally administered CpG-PBNP-PTT therapy in 9464D TH-MYCN model. The 9464D TH-MYCN model (referred to as 9464D model hereafter) generated by Weiss et al. is a transgenic model in which human MYCN is over expressed through a rat tyrosine hydroxylase (TH) promoter.[17, 18, 34] This model shows strong histological and genetic similarities with human NB including the expression of GD2, thus rendering it a more clinically relevant model.[35, 36]
Several nanoparticle-based platforms have been developed to administer treatments including photodynamic therapy, chemotherapy, or developmental photoactivated chemotherapy in combination with photothermal therapy, with encouraging results in diverse cancer models. [24, 37–42]. The specific nanoparticle platform selected for treating a particular tumor indication depends, among other factors, on the biology of tumor being treated and the ease of manufacture of the nanoparticles under consideration. To address ease of manufacture, in the current study, we employ a simple and scalable layer-by-layer coating scheme to generate stable PBNPs functionalized with CpG (CpG-PBNP) for use in PTT (CpG-PBNP-PTT) as a minimally invasive method to trigger a robust systemic antitumor immune response and potentially generate long-term antigen-specific T cell memory culminating in robust and persistent disease remission in the 9464D model of NB. The overall treatment scheme and hypothesized mechanism of action is summarized in (Figure 1). We first test whether CpG-PBNP-PTT “primes” 9464D cells for recognition by immune effector cells and elicits immunogenic cell death (ICD) as a function of thermal dose, in vitro. This thermal dose-dependent expression of costimulatory and antigen-presenting molecules has not been previously studied for PTT. Subsequently, we analyze the efficacy of CpG-PBNP-PTT to elicit complete tumor regression and robust long term memory on single tumor-bearing mice in vivo. We then conduct ex vivo co-cultures of the splenocytes from any long-term surviving, rechallenge mice with 9464D cells to confirm tumor-specific responses and immunological memory generation by the T cells. These ex vivo studies involving T cells also represent a novel aspect of our study for PTT. Finally, we assess the effects of our nanoimmunotherapy to generate a potent abscopal effect in a synchronous tumor model. Through these studies, we seek to offer CpG-PBNP-PTT as a potent anti-cancer nanoimmunotherapy leveraging the photothermal heating characteristics of the PBNPs along with the immunostimulatory properties of both PBNP-PTT and CpG in a clinically elevant 9464D NB model.
Figure 1. Schematic of the mechanism of action of the CpG-PBNP-PTT-based nanoimmunotherapy in the TH-MYCN model of neuroblastoma (NB).
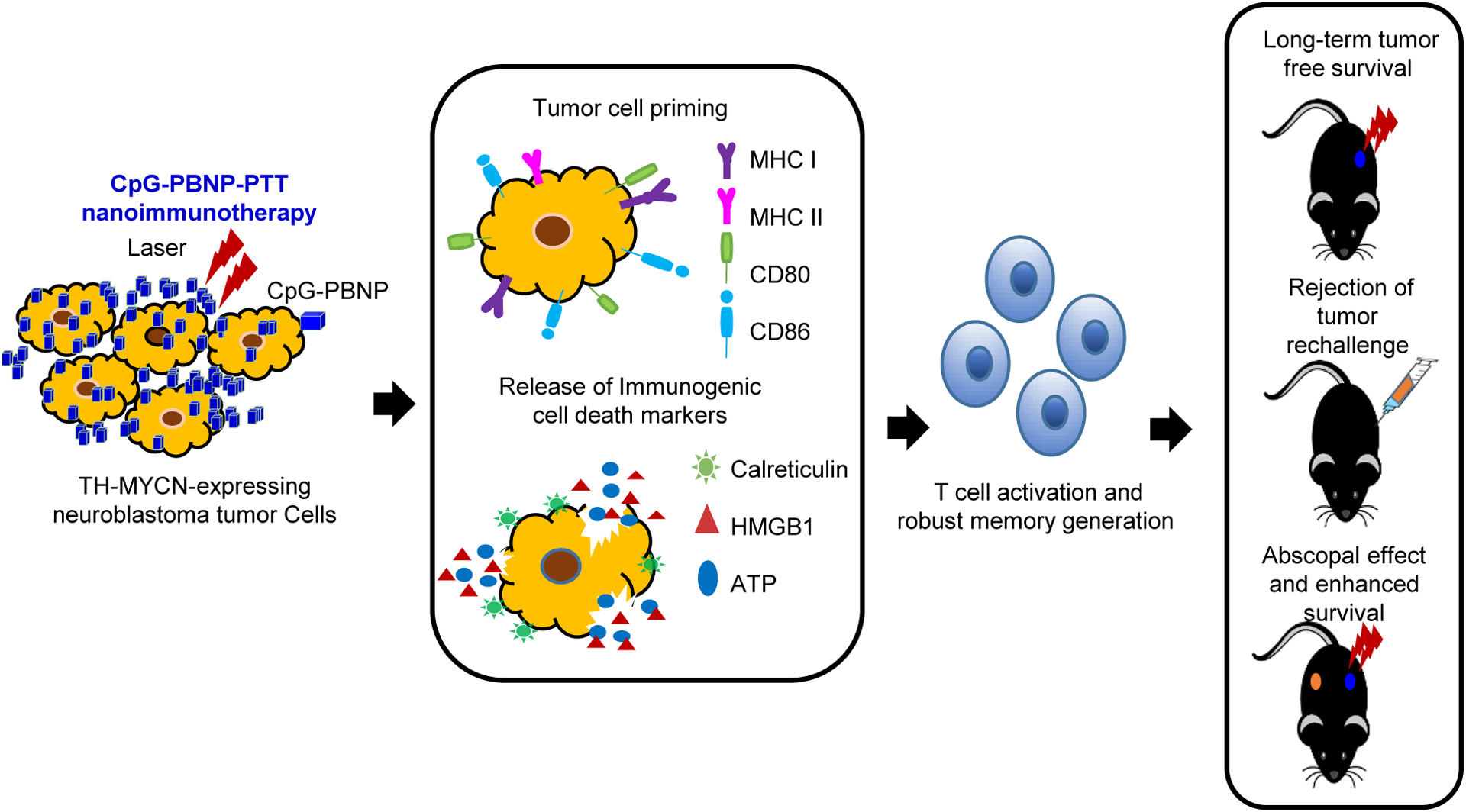
The CpG-PBNP-PTT-based nanoimmunotherapy mediates tumor cell priming along with immunogenic cell death (ICD) administered at a specific thermal dose, leading to T cell activation and generation of potent T cell memory, which can elicit long-term, tumor-free survival, and rejection of tumor rechallenge in a TH-MYCN model of NB. Further, the nanoimmunotherapy generates a potent abscopal effect, which slowers the progression of distal, untreated tumors enhancing the survival in the TH-MYCN NB model.
2. Results
2.1. CpG-PBNPs function as effective PTT agents, upregulate molecules implicated in driving T cell responses, and elicit ICD in 9464D cells in vitro
We employed a layer-by-layer coating technique[43, 44] to synthesize CpG-PBNPs and assessed the properties of the resultant CpG-PBNPs by measuring size distributions and zeta potentials using dynamic light scattering (Figure S1A, B). We analyzed the photothermal heating properties of the CpG-PBNPs in vitro which demonstrated a laser power-dependent increase (0.2 to 1.5 W) in temperature along with an increased thermal dose (expressed as cumulative equivalent minutes at 43 °C; log∑CEM43).[45, 46] We observed a maximum temperature of 77 °C (Figure S1C) and the corresponding thermal dose of 9.58 at a concentration of 0.15 mg mL−1 CpG-PBNPs and laser power of 1.5 W (Figure S1D). We further observed that PBNP-PTT- and CpG-PBNP-PTT-treated 9464D cells exhibited a laser-power dependent thermal injury, as assessed by inverted phase contrast microscope using 10× magnification 24 h post treatment. (Figure S1E).
We also analyzed a cohort of 137 pediatric patients with NB via RNA-seq data from the 2018 NCI TARGET study. We confirmed that high MYCN expression is restricted to patients with high risk tumors and observed an inverse correlation with the expression of MYCN and markers of immunogenicity, T cell infiltration and function, immune suppression, and toll-like receptors (Figure S2A, B). Thus to understand the correlation between MYCN and immune signatures, we leveraged the 9464D NB model which overexpresses MYCN to determine the baseline immunogenicity of these tumors, and assess the effect of CpG-PBNP-PTT on these immune signatures.
We first measured the induction of MHC molecules which are critical for antigen presentation to T cells.[47, 48] PBNP-PTT and CpG-PBNP-PTT at 1.5 W significantly enhanced MHC I expression on 9464D cells as compared to vehicle (Figure 2A, B; Figure S3A). MHC II expression was also significantly higher on 9464D cells treated with PBNP-PTT at 1.5 W, or CpG-PBNP-PTT at 0.5 W, and 1.5 W, as compared to vehicle (Figure 2C, D; Figure S3B). Next, we examined the effect of CpG-PBNP-PTT on the expression of costimulatory molecules CD80 and CD86, which play a pivotal role in T cell activation and proliferation, and their absence can result in T cell anergy or apoptosis.[49–52] We observed a significant increase in the expression of CD80, and CD86 post 1.5 W PBNP-PTT, or 1.5 W CpG-PBNP-PTT as compared to vehicle (Figure 2E–H; Figure S3C–D).
Figure 2. CpG-PBNP-PTT-induced immune modulation and ICD in 9464D NB cells in vitro.
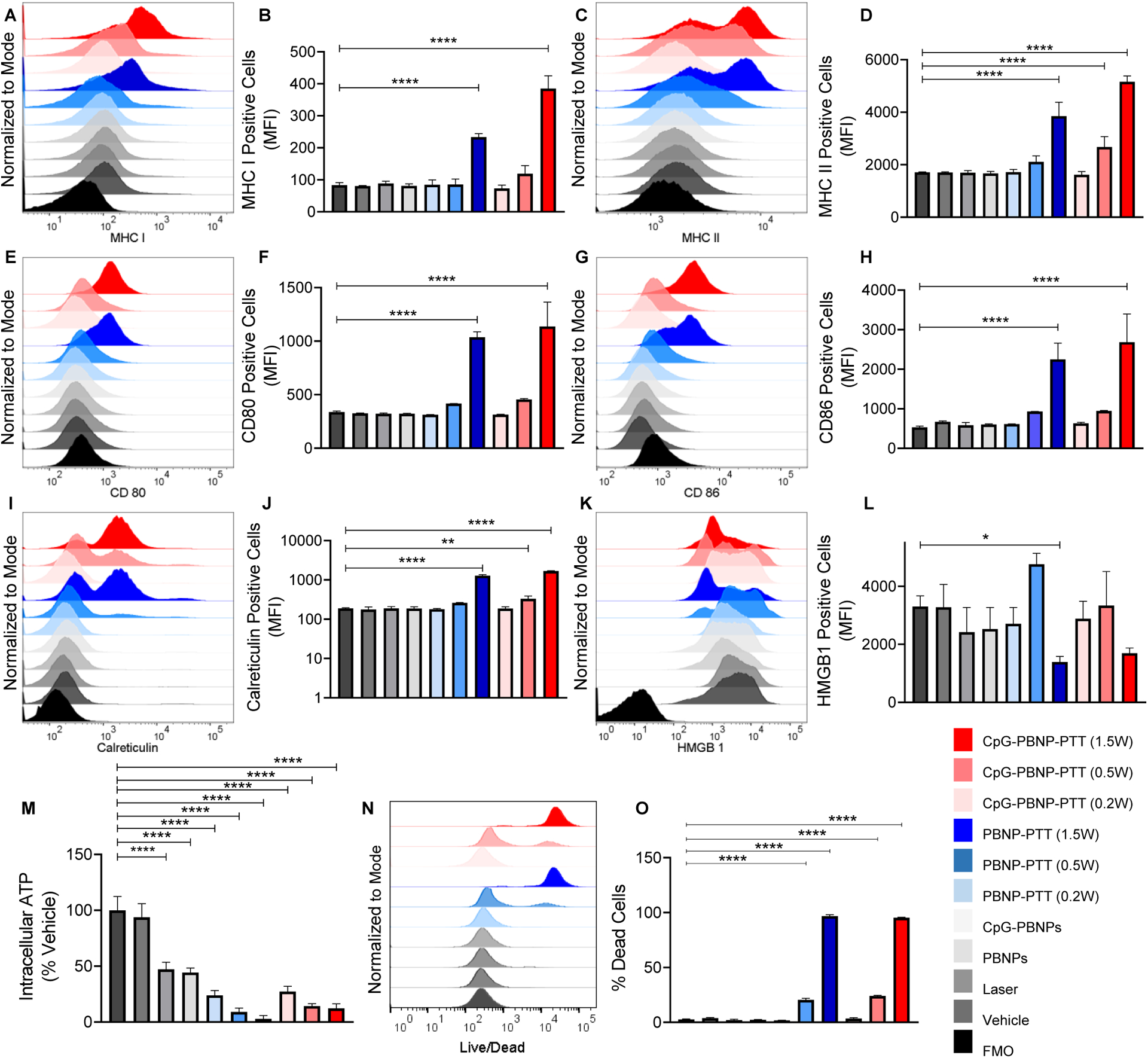
9464D NB cells cultured in vitro were treated with vehicle (PBS), laser (1.5 W), PBNPs (0.15 mg mL−1), CpG-PBNPs (0.15 mg mL−1), PBNP-PTT (0.15 mg mL−1 at 0.2, 0.5, and 1.5 W), or CpG-PBNP-PTT (0.15 mg mL−1 at 0.2, 0.5, and 1.5 W), and analyzed 24 h post PTT. (A–B) Surface MHC I expression levels in the various treatment groups analyzed by flow cytometry represented as (A) histograms, and (B) MFI (mean fluorescence intensity). (C–D) Surface MHC II expression levels in the various treatment groups analyzed by flow cytometry represented as (C) histograms, and (D) MFI. (E–F) Surface CD80 expression levels in the various treatment groups analyzed by flow cytometry represented as (E) histograms, and (F) MFI. (G–H) Surface CD86 expression levels in the various treatment groups analyzed by flow cytometry represented as (G) histograms, and (H) MFI. (I–J) Surface calreticulin expression levels in the various treatment groups analyzed by flow cytometry represented as (I) histograms, and (J) MFI. (K–L) Intracellular HMGB1 expression levels in the various treatment groups analyzed by flow cytometry represented as (K) histograms, and (L) MFI. (M) Intracellular ATP levels in the various treatment groups (as % of the vehicle-treated group) analyzed by CellTiter-Glo luminescent cell viability assay. (N–O) Quantification of cell death in the various treatment groups analyzed by flow cytometry represented as (N) histograms, and (O) as % of the vehicle-treated group. Results are expressed as mean ± std. deviation (n=3); *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (ANOVA). FMO (fluorescence minus one control).
Next, we assessed the potential of CpG-PBNP-PTT to elicit ICD, which is important to trigger tumor immunogenicity and strengthen therapeutic outcomes.[15, 16] We first investigated the status of the biochemical correlates indicating ICD, which include cell surface expression of calreticulin, along with extracellular release of ATP and high mobility group box 1 (HMGB1).[14, 20, 21] We observed a significant increase in surface calreticulin after PBNP-PTT at 1.5 W or CpG-PBNP-PTT at 0.5 W and 1.5 W relative to the vehicle (Figure 2I, J; Figure S3E). Further, a decrease in intracellular HMGB1 levels (suggestive of HMGB1 release) was observed on treatment with PBNP-PTT at 1.5 W or CpG-PBNP-PTT at 1.5 W as compared to the vehicle (Figure 2K, L; Figure S3F). Next, we observed a significant decrease in intracellular ATP levels in cells treated with PBNP-PTT at 0.2 W, 0.5 W and 1.5 W or CpG-PBNP-PTT at 0.2 W, 0.5 W and 1.5 W as compared to vehicle; as well as PBNPs or CpG-PBNPs treated groups, suggesting significant ATP release (Figure 2M; Table 1). Finally, we observed that 9464D cells exhibited significantly enhanced cell death in a laser power-dependent manner with PBNP-PTT at 0.5 W or 1.5 W or CpG-PBNP-PTT at 0.5 W and 1.5 W (Figure 2N, O). Our in vitro studies demonstrate that CpG-PBNP-PTT nanoimmunotherapy is potent in increasing expression of molecules involved in antigen presentation, costimulatory markers and triggering ICD in 9464D cells when administered at a specific thermal dose (laser power of 1.5 W).
Table 1:
List of p-values for the in vitro intracellular ATP release study (Figure 2)
| Tukey’s multiple comparisons test | Adjusted p-value |
|---|---|
| PBNPs vs. PBNP-PTT at 0.2 W | <0.0001 |
| PBNPs vs. PBNP-PTT at 0.5 W | <0.0001 |
| PBNPs vs. PBNP-PTT at 1.5 W | <0.0001 |
| PBNPs vs. CpG-PBNP-PTT at 0.2 W | <0.0001 |
| PBNPs vs. CpG-PBNP-PTT at 0.5 W | <0.0001 |
| PBNPs vs. CpG-PBNP-PTT at 1.5 W | <0.0001 |
| CpG-PBNPs vs. PBNP-PTT at 0.2 W | <0.0001 |
| CpG-PBNPs vs. PBNP-PTT at 0.5 W | <0.0001 |
| CpG-PBNPs vs. PBNP-PTT at 1.5 W | <0.0001 |
| CpG-PBNPs vs. CpG-PBNP-PTT at 0.2 W | <0.0001 |
| CpG-PBNPs vs. CpG-PBNP-PTT at 0.5 W | <0.0001 |
| CpG-PBNPs vs. CpG-PBNP-PTT at 1.5 W | <0.0001 |
2.2. CpG-PBNP-PTT triggers complete tumor regression, long-term survival, and rejection of tumor rechallenge in 9464D tumor-bearing mice
Encouraged by the induction of immune signaling markers and ICD in vitro culminating in tumor cell death, we next investigated the potential of CpG-PBNP-PTT to treat 9464D tumors in vivo. To study the effect of CpG-PBNP-PTT on the TME, we first established 9464D tumors in C57BL/6J mice and evaluated the phenotype of the resulting tumors for human NB associated genes (Figure S4A). Most importantly, for the correlation to human tumors, we observed the expression of GD2 synthase (one of the key enzymes in the production of the disialoganglioside GD2) in 9464D cells, which is not expressed in other NB mouse models.[18] Interestingly, our in vitro studies also showed significantly upregulated expression of the GD2 antigen in 9464D cells treated with PBNP-PTT (1.5 W) or CpG-PBNP-PTT (0.5 W and 1.5 W) (Figure S4B–D).
Next, we tested the therapeutic efficacy of our nanoimmunotherapy. For these studies 9464D cells were inoculated in the mice (Day 0). Tumor-bearing mice (~ 5 mm tumors; Day 18 were divided into three treatment groups (n=5/group); 1) Vehicle, 2) PBNP-PTT, and 3) CpG-PBNP-PTT, and the treatments were conducted accordingly (Figure 3A). The maximum temperature maintained during PTT was ~120 °C as measured by a thermal imaging camera (Figure 3B). The corresponding thermal dose administered was ~23.5 (log (∑CEM43)) for both the PBNP-PTT or CpG-PBNP-PTT treated groups (Figure 3C). Importantly, 100% of mice (5/5) treated with CpG-PBNP-PTT exhibited complete tumor regression and survival on Day 80 post tumor inoculation compared to 60% (3/5) PBNP-PTT-treated mice (Figure 3D). However 100% of vehicle treated mice (5/5) succumbed to high tumor burden by Day 65 (median survival 38 days) (Figure 3D, E).
Figure 3. Effect of CpG-PBNP-PTT on tumor regression and long-term survival in a primary, 9464D model of NB.
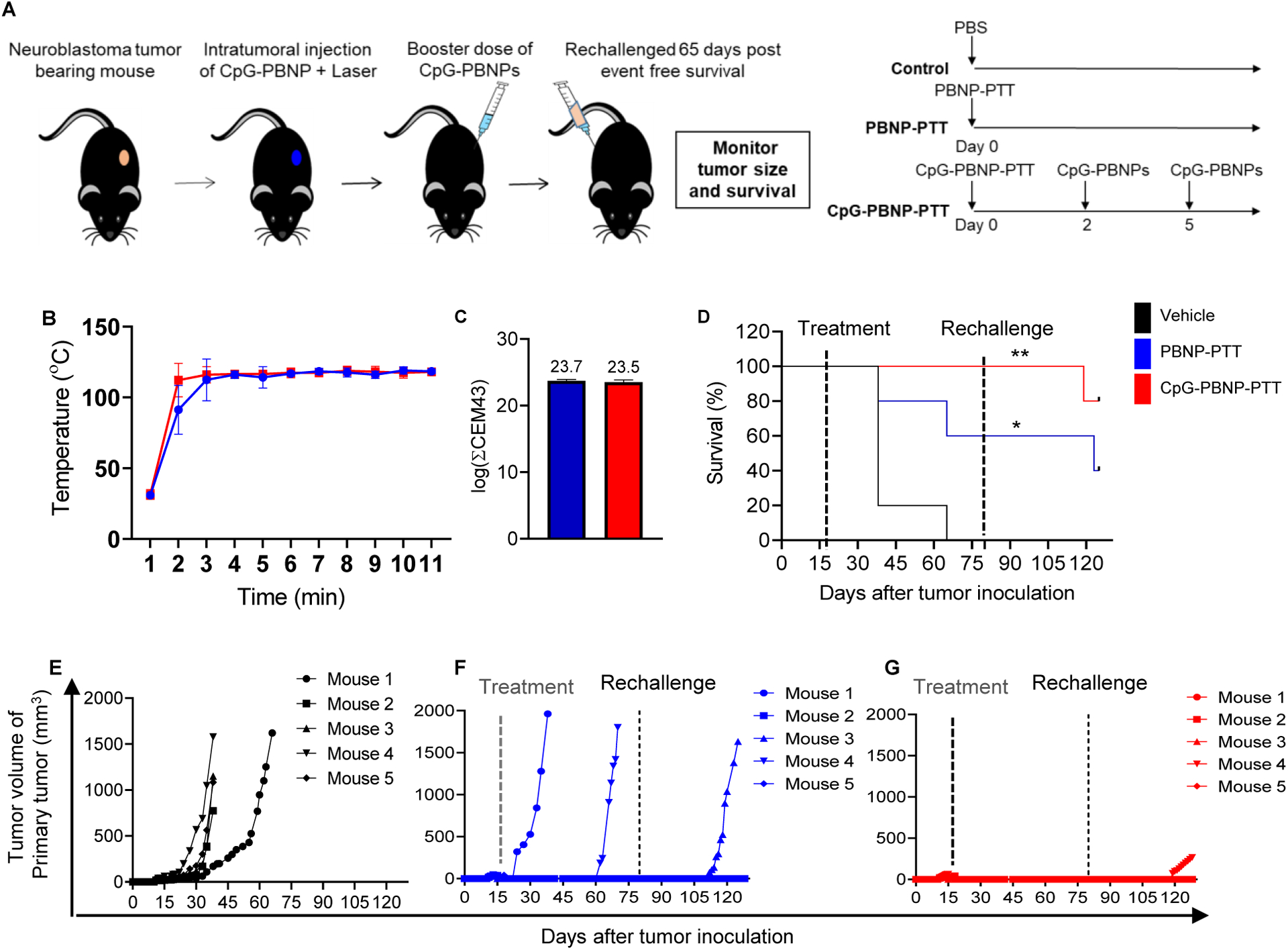
(A) Schematic of the treatments. (Left) Mice bearing ~5 mm diameter 9464D tumors were treated with vehicle (PBS), PBNP-PTT or CpG-PBNP-PTT,. (Right) The vehicle group received 50 μL of PBS intratumorally (i.t.), The PTT-treated groups received 50 μL of 1 mg mL−1 PBNPs or CpG-PBNPs i.t., and were irradiated with an 808 nm laser for 10 minutes at a temperature maintained at 120 °C. Additionally, the CpG-PBNP-PTT group received two boosters with CpG-PBNPs on days 2 and 5. The dosage of CpG for this group was 2 μg of CpG conjugated onto PBNP on days 0, 2, and 5 i.t.. (B–C) Temperature versus time profiles (B) and thermal doses administered expressed in cumulative equivalent minutes at 43 °C (log(∑CEM43)) (C) of 9464D tumor-bearing mice treated i.t. with 50 μL of 1 mg mL−1 CpG-PBNPs or PBNPs and irradiated with a NIR laser at 120°C for 10 minutes. (D) Kaplan–Meier survival plots of 9464D tumor-bearing mice that were treated with CpG-PBNP-PTT, PBNP-PTT, or vehicle (PBS) on Day 18 and rechallenged on Day 80. (E–G) Individual tumor growth curves of 9464D tumor-bearing mice receiving vehicle (PBS) (E), PBNP-PTT (F), or CpG-PBNP-PTT (G). Results are expressed as mean ± std. deviation; (n=5); *p < 0.05, **p < 0.01 (ANOVA).
In order to elucidate whether CpG-PBNP-PTT generated immunological memory and conferred protection against tumor recurrence, we rechallenged the surviving mice from PBNP-PTT or CpG-PBNP-PTT groups with one million 9464D cells on Day 80. We observed that 2 of 3 surviving mice in the PBNP-PTT-treated group survived tumor rechallenge (Figure 3F). Remarkably, 4 of 5 CpG-PBNP-PTT treated mice exhibited protection against the tumor rechallenge and continued to survive tumor-free for an additional 45 days (Day 125 in the study) at which point they were sacrificed for downstream analysis (Figure 3G). Thus, long-term survival after tumor inoculation and rechallenge at Day 125 was 80% for tumor-bearing mice treated with CpG-PBNP-PTT, two-fold higher than that observed in mice treated with PBNP-PTT (40%). Taken together, our findings suggest CpG-PBNP-PTT is a robust therapy capable of inducing tumor regression, long-term survival, and protection against tumor rechallenge in the 9464D NB model.
2.3. T cells isolated from long-term surviving, PTT-treated mice exhibit long-term memory and a tumor-specific T cell response ex vivo
In order to elucidate the antitumor effects triggered by CpG-PBNP-PTT, we analyzed the levels of T cells in the spleens of the long-term surviving, rechallenged mice on Day 125. In this study, as a treatment-naive control group (control), we used age-matched mice that were inoculated with one million 9464D cells. When tumors in these animals reached ~15 mm, we sacrificed mice from treatement naive, control group (n = 4), PBNP-PTT-treated (n = 2) and CpG-PBNP-PTT-treated (n = 4) groups on the same day (Day 125 for the rechallenged mice) for downstream analysis.
We observed a significant increase in CD3+ lymphocytes in the spleens of PBNP-PTT and CpG-PBNP-PTT rechallenged mice; compared to the spleens of mice in the control group (Figure 4A, B). However, there were no statistically significant differences in the numbers of CD4+ or CD8+ T cells between treatment groups (Figure 4C–E). Development of long-term, antigen-specific memory is a hallmark of the adaptive immune system.[53, 54] Expression of the adhesion molecule CD44 is observed on memory cells distinguishing it from the naïve T cells.[55] Further, CD69 is a key marker found on the tissue-resident memory T cells, and is prominently expressed on memory T cells both in humans and mice at various sites including, lymph nodes, liver, skin, intestines, and lungs.[56]
Figure 4. Immunostimulatory effects of CpG-PBNP-PTT on long-term surviving rechallenged mice.
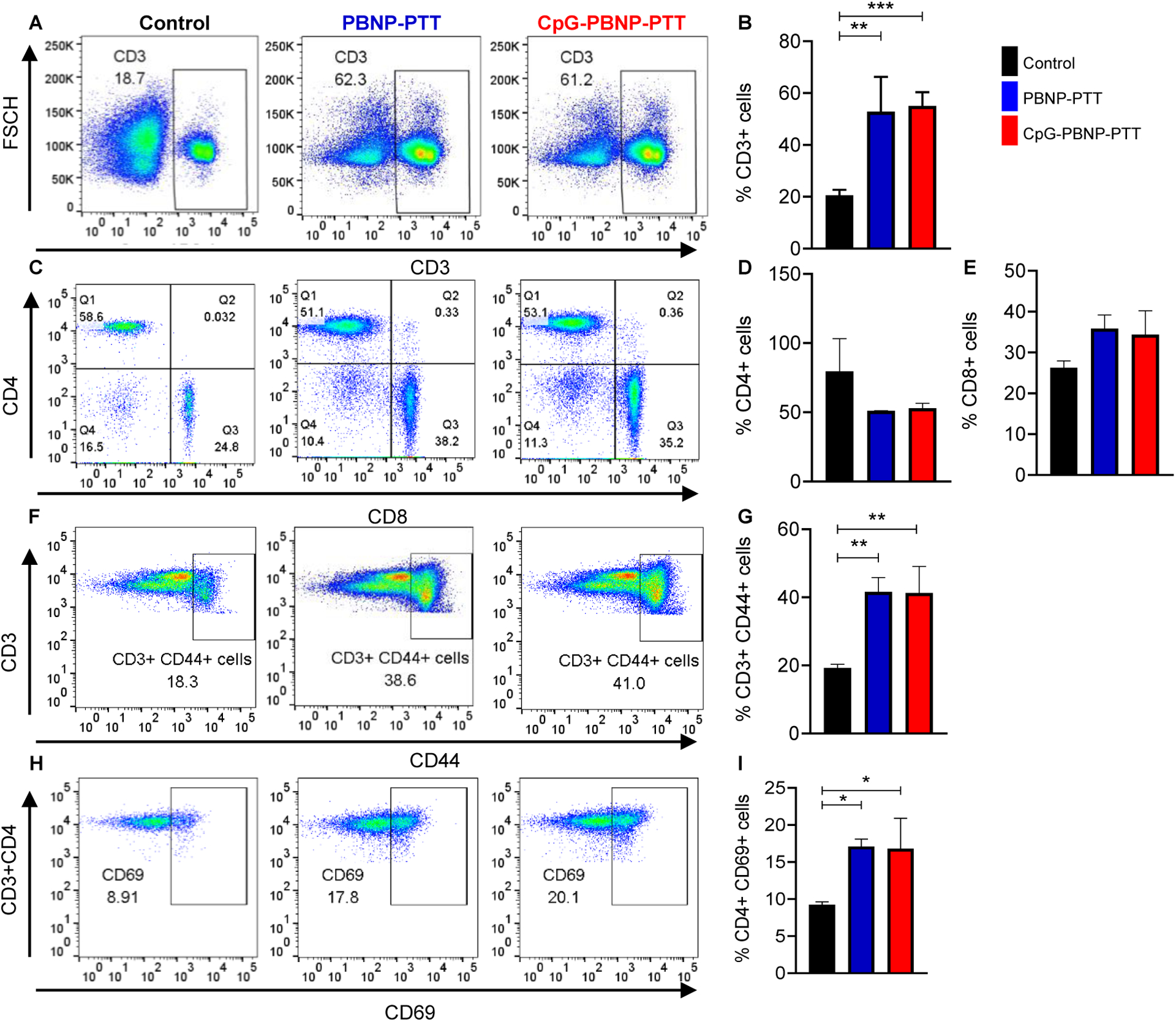
(A) Representative histograms of CD3+ cells in the spleens of PBNP-PTT and CpG-PBNP-PTT rechallenge mice compared to age-matched, treatment-naive control mice. (B) %CD3+ cells in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to mice in the control group. (C) Representative scatter plots of CD4+ and CD8+ cells (gated on CD3+ cells) in the splenocytes of rechallenged mice compared to control mice. (D) %CD4+ cells in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (E) %CD8+ cells in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (F) Representative scatter plots of CD3+ CD44+ memory cells in the splenocytes of PBNP-PTT and CpG-PBNP-PTT rechallenged mice compared to control mice. (G) %CD3+ CD44+ memory cells in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (H) Representative scatter plots of CD4+ CD69+ memory cells in the splenocytes of rechallenged mice compared to control mice. (I) %CD4+ CD69+ cells in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. Results are expressed as mean ± std. deviation; Treatment-naive control (n=4), PBNP-PTT (n=2), CpG-PBNP-PTT (n=4); *p < 0.05, **p < 0.01, ***p < 0.001 (ANOVA).
We observed a significant increase in the CD3+ CD44+ memory cells in the spleens of PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to mice in the control group (Figure 4F, G). We also observed a significant increase in the CD69 expressing CD4+ T cells in PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice (Figure 4H, I).
Memory T cells can be further classified into central memory T cells (TCM) and effector memory T cells (TEM) depending on their proliferative capacity, effector function, and migration potential. While protective memory is mediated by TEM that display immediate effector function, TCM cells home to secondary lymphoid organs and provide reactive memory by readily proliferating and differentiating to effector cells upon antigenic stimulation.[37, 57] Therefore, to understand the T cell responses driving the rejection of 9464D tumor rechallenge, we measured the proportions of both TCM (CD3+CD62L+CD44+) and TEM (CD3+CD62L-CD44+) cells for the CD4+ and CD8+ populations in the spleens of the various treatment groups. We observed that the percentage of CD4+ TCM and CD4+ TEM cells was significantly higher in PBNP-PTT or CpG-PBNP-PTT rechallenged mice as compared to mice in the control group (Figure 5A–C). We observed no significant difference in the percentage of CD8+ TCM cells, however the percentage of CD8+ TEM cells was significantly increased in the PBNP-PTT or CpG-PBNP-PTT treated rechallenged groups compared to control (Figure 5D–F), indicating that a robust immunologic memory is elicited by PTT-based treatments.
Figure 5. Long-term immune memory effects of CpG-PBNP-PTT.
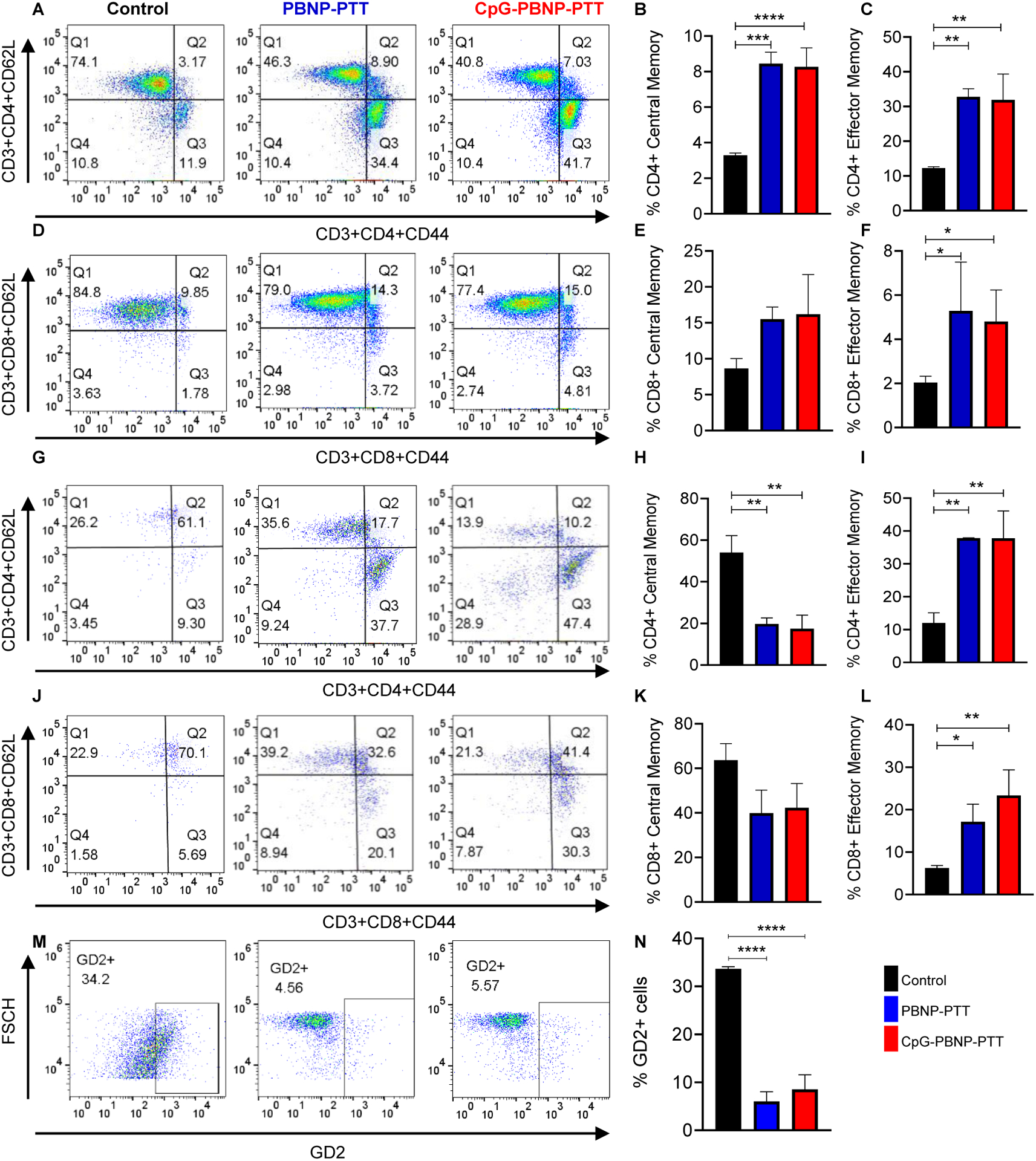
(A) Representative scatter plots of central memory (TCM) and effector memory (TEM) lymphocytes in the CD4+ subset of T cells isolated from the spleens of PBNP-PTT and CpG-PBNP-PTT rechallenged mice compared to age-matched, treatment-naive control mice. (B) %CD4+ central memory (TCM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to mice in the control group. (C) %CD4+ effector memory (TEM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (D) Representative scatter plots of central memory (TCM) and effector memory (TEM) lymphocytes in the CD8+ subset of T cells isolated from the spleen of PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (E) %CD8+ central memory (TCM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (F) %CD8+ effector memory (TEM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (G) Representative scatter plots of central memory (TCM) and effector memory (TEM) lymphocytes in the CD4+ subset of T cells isolated from the spleen of PBNP-PTT and CpG-PBNP-PTT rechallenged mice upon co-culture with 9464D cells ex vivo as compared to control mice. (H) %CD4+ central memory (TCM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice upon co-culture with 9464D cells ex vivo as compared to control mice. (I) %CD4+ effector memory (TEM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice upon co-culture with 9464D cells ex vivo as compared to control mice. (J) Representative scatter plots and histograms of central memory (TCM) and effector memory (TEM) lymphocytes in the CD8+ subset of T cells isolated from the spleen of PBNP-PTT and CpG-PBNP-PTT rechallenged mice upon co-culture with 9464D cells ex vivo as compared to control mice. (K) %CD8+ central memory (TCM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice upon co-culture with 9464D cells ex vivo as compared to control mice. (L) %CD8+ effector memory (TEM) in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice upon co-culture with 9464D cells ex vivo as compared to control imce. (M) Representative scatter plots of GD2+ live 9464D cells from co-cultures with splenic T cells from PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. (N) %GD2+ cells from co-cultures with splenic T cells from PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to control mice. Results are expressed as mean ± std. deviation; treatment-naive control (n=4), PBNP-PTT (n=2), CpG-PBNP-PTT (n=4) for spleen samples and CpG-PBNP-PTT (n=3) for co-culture samples; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (ANOVA).
Building on these observations, we performed an ex vivo study using isolated splenic T cells from the PBNP-PTT (n = 2) or CpG-PBNP-PTT (n = 4) rechallenged or treatment-naive control mice (n = 4; same animals/treatment groups used in Figure 4) and co-cultured them with 9464D cells to confirm the establishment of a 9464D cell-specific immunological memory post-rechallenge. T cells isolated from the spleens of control, PBNP-PTT, or CpG-PBNP-PTT rechallenged mice were cultured for four days in the presence of IL-2, anti-CD3, and anti-CD28 (Figure S5A), following which we co-cultured them with 9464D cells for two days. Importantly, we observed a significant decrease in the percentage of CD4+ TCM cells and a significant increase in the percentage of both CD4+ and CD8+ TEM cells in the PBNP-PTT and CpG-PBNP-PTT rechallenged mice as compared to the control mice (Figure 5G–L). These data support the idea that tumor-specific memory T cells can become reactivated upon re-exposure to 9464D cells.
We further analyzed the tumor-specific memory developed by our nanoimmunotherapy by evaluating the percentage of live GD2+ 9464D cells in the co-culture (Figure S5B). We observed a significant decrease in live GD2+ 9464D cell population in the PBNP-PTT or CpG-PBNP-PTT rechallenged mice compared to control mice (Figure 5M, N). These data suggest that our nanoimmunotherapy generated a potent tumor-specific cytotoxicity, as decreased GD2+ staining in the co-culture is suggestive of T cell-mediated 9464D cell killing. Important to note here that although we do not observe a statistically significant difference in the magnitude of responses (e.g. %CD3+CD44+ or %CD4+ effector memory cells) between the PBNP-PTT and CpG-PBNP-PTT treatments for the assays/analyses in Figures 4 and 5, which may suggest that the CpG coating does not provide any benefit over PBNP-PTT. Importantly we must take into account that the PBNP-PTT group comprised of 2 out of 5 mice that were able to survive the rechallange where as 3 of the 5 mice had already succumbed to the tumor burden. However CpG-PBNP-PTT treatment significant improved the therapeutic outcomes and comprised of 4 out of 5 mice (2-fold increase in long-term survival, Figure 3), suggesting the importance of the CpG coating for treatment outcomes.
2.4. CpG-PBNP-PTT eradicates primary tumors, slows secondary tumor growth, and improves survival in synchronous 9464D tumor-bearing mice
To determine the PTT-based T cell response in a disseminated tumor model, we established a synchronous tumor model, where one million 9464D cells each were simultaneously inoculated in both flanks of mice. Synchronous (two) tumor-bearing mice were randomly divided into five treatment groups (n=5/group): 1) Vehicle (PBS), 2) PBNP, 3) CpG-PBNP (no laser irradiation), 4) PBNP-PTT, and 5) CpG-PBNP-PTT (Figure 6A, B). For these studies, the tumor that reached a size of 5 mm (~60 mm3) first (i.e. the larger tumor) was designated as the “primary” tumor (4.5–5mm) and treated with the treatments in groups 1–5. The contralateral tumors in these groups (designated “secondary” tumor), which were generally slightly smaller(4–4.7mm), were left untreated to evaluate the abscopal effect of the treatments. A maximum tumor temperature of ~120 °C was maintained attained during PTT (Groups 4 and 5), which was measured using a thermal imaging camera (Figure 6C) and the corresponding thermal doses were ~24.4 log (∑CEM43) (Figure 6D). Both PBNP-PTT and CpG-PBNP-PTT generated complete regression of primary tumors in 100% of mice, compared to 0% of vehicle-treated, PBNP-treated, and CpG-PBNP-treated mice (Figure 6E (a-e); Figure 6G; Table 2). Additionally, mice treated with either PBNP-PTT or CpG-PBNP-PTT exhibited significantly delayed tumor progression of secondary tumors, compared to mice in the vehicle-treated and PBNP-treated groups (Figure 6F (a-e); Figure 6H; Table 2). The tumor progression in secondary tumors of the PBNP-PTT-treated or CpG-PBNP-PTT-treated mice at day 50 was significantly delayed with respect to CpG-PBNP-treated mice demonstrating the abscopal effect elicited by our PTT-based nanoimmunotherapy (Figure S6; Table 2). In line with these findings, we observed that un-irradiated PBNPs or CpG-PBNPs was unable to extend survival benefits to the tumor-bearing mice relative to vehicle-treated mice. However, all animals irradiated with PTT (PBNP-PTT or CpG-PBNP-PTT) exhibited significantly increased survival relative to vehicle-treated and PBNP-treated controls (Figure 6H; Table 3). There was also a significant increase in the survival of the CpG-PBNP-PTT treated mice relative to the CpG-PBNP-treated mice (p < 0.05). Although there was no statistically significant difference in survival between the CpG-PBNP-PTT or PBNP-PTT treated groups, the median survival was higher for the CpG-PBNP-PTT-treated group (62 days) versus the PBNP-PTT-treated group (59 days), which can likely be attributed to the presence of the CpG coating on the CpG-PBNPs used for PTT. Based on these findings, we conclude that this nanoimmunotherapy potently eradicated the primary tumors and generated an abscopal effect on contralateral tumors as evident by the significantly delayed tumor progression, and increased survival relative to vehicle-treated and PBNP-treated groups for PBNP-PTT, and all others groups for CpG-PBNP-PTT; except CpG-PBNP-PTT vs. PBNP-PTT where there was a slight increase in median survival.
Figure 6. Effect of CpG-PBNP-PTT on tumor regression and long-term survival in a synchronous, 9464D tumor-bearing mice.
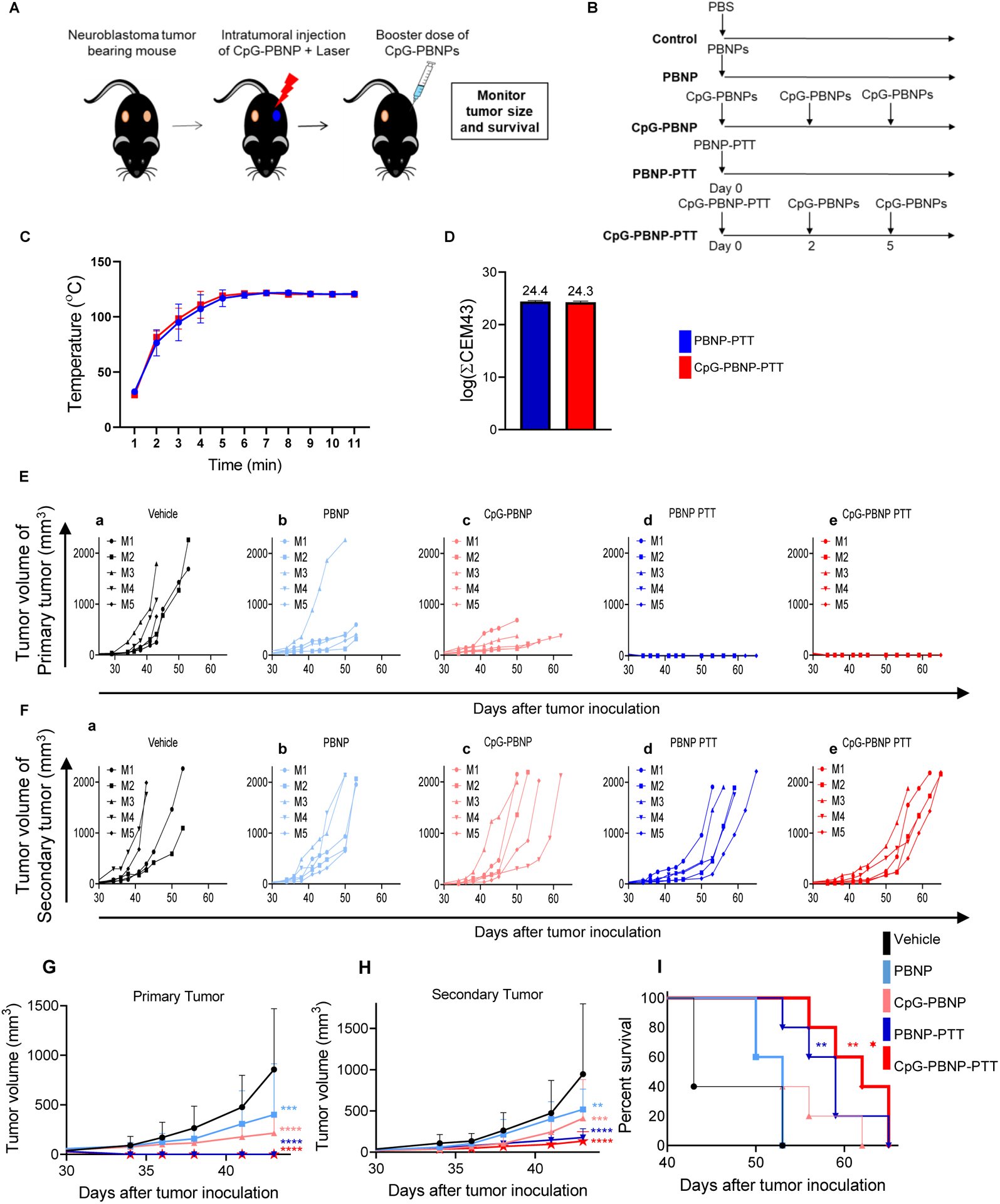
(A–B) Schematic of the treatments. (A) Mice bearing two, contralateral ~5 mm diameter 9464D tumors were treated on one tumor (designated “primary” tumor) with either vehicle (PBS), PBNP, CpG-PBNP, PBNP-PTT or CpG-PBNP-PTT and the contralateral tumor (designated “secondary” tumor) was left untreated. (B) The the vehicle group received 50 μL of PBS i.t., the PBNP-treated group received 50 μL of 1 mg mL−1 PBNPs i.t, the CpG-PBNP-treated group received 50 μL of 1 mg mL−1 CpG-PBNPs i.t, the PTT-treated groups received 50 μL of 1 mg mL−1 PBNPs or CpG-PBNPs i.t., and were irradiated with an 808 nm laser for 10 minutes at a temperature maintained at 120 °C. Additionally, the CpG-PBNP and CpG-PBNP-PTT groups received two boosters with CpG-PBNP on days 2 and 5. The dosage of CpG for these two groups was 2 μg of CpG conjugated onto PBNP on days 0, 2, and 5 i.t. (C–D) Temperature versus time profiles (C) and thermal doses administered expressed in cumulative equivalent minutes at 43 °C (log(∑CEM43)) (D) of 9464D tumor-bearing mice treated i.t. with 50 μL of 1 mg mL−1 CpG-PBNPs or PBNPs and irradiated with a NIR laser at 120°C for 10 minutes. 0.15 mg mL−1. (E) Tumor growth curves of “primary” tumors of 9464D tumor-bearing mice that were treated with (a) PBS, (b) PBNP, (c) CpG-PBNP, (d) PBNP-PTT, or (e) CpG-PBNP-PTT. (F) Tumor growth curves of “secondary” tumors of 9464D tumor-bearing mice that were treated with (a) PBS, (b) PBNP, (c) CpG-PBNP, (d) PBNP-PTT, or (e) CpG-PBNP-PTT. (G) Aggregated tumor growth curves of primary tumors of 9464D TH-MYCN murine model of NB that were treated with PBS, PBNP, CpG-PBNP, PBNP-PTT, or CpG-PBNP-PTT on the primary tumor side. (H) Aggregated tumor growth curves of secondary tumors of 9464D TH-MYCN murine model of NB that were treated with PBS, PBNP, CpG-PBNP, PBNP-PTT, or CpG-PBNP-PTT on the primary tumor side.(I) Kaplan–Meier survival plots of 9464D tumor bearing mice that were treated with CpG-PBNP-PTT, PBNP-PTT, PBNP, CpG-PBNP or vehicle (PBS). Results are expressed as mean ± std. deviation; (n=5); *p < 0.05 (with respect to CpG-PBNP), **p < 0.01, ****p < 0.0001 (ANOVA).
Table 2:
List of p-values for effect of the CpG-PBNP-PTT on tumor progression in a synchronous, 9464D TH-MYCN murine model of NB (Figure 6; Supplementary Figure. 6)
| Tukey’s multiple comparisons test | Adjusted p-value |
|---|---|
| Primary Tumor Day 43 | |
| Vehicle vs. PBNP | 0.0001 |
| Vehicle vs. CpG-PBNP | <0.0001 |
| Vehicle vs. PBNP-PTT (120) | <0.0001 |
| Vehicle vs. CpG-PBNP-PTT (120) | <0.0001 |
| PBNP vs. CpG-PBNP | 0.3497 |
| PBNP vs. PBNP-PTT (120) | 0.0010 |
| PBNP vs. CpG-PBNP-PTT (120) | 0.0010 |
| CpG-PBNP vs. PBNP-PTT (120) | 0.2159 |
| CpG-PBNP vs. CpG-PBNP-PTT (120) | 0.2159 |
| PBNP-PTT (120) vs. CpG-PBNP-PTT (120) | >0.9999 |
| Secondary Tumor Day 43 | |
| Vehicle vs. PBNP | 0.0043 |
| Vehicle vs. CpG-PBNP | 0.0001 |
| Vehicle vs. PBNP-PTT (120) | <0.0001 |
| Vehicle vs. CpG-PBNP-PTT (120) | <0.0001 |
| PBNP vs. CpG-PBNP | 0.8977 |
| PBNP vs. PBNP-PTT (120) | 0.0407 |
| PBNP vs. CpG-PBNP-PTT (120) | 0.0139 |
| CpG-PBNP vs. PBNP-PTT (120) | 0.3010 |
| CpG-PBNP vs. CpG-PBNP-PTT (120) | 0.1476 |
| PBNP-PTT (120) vs. CpG-PBNP-PTT (120) | 0.9960 |
| Secondary Tumor Day 50 | |
| CpG-PBNP vs. PBNP-PTT (120) | <0.0001 |
| CpG-PBNP vs. CpG-PBNP-PTT (120) | <0.0001 |
| PBNP-PTT (120) vs. CpG-PBNP-PTT (120) | 0.9201 |
Table 3:
List of p-values for the Kaplan–Meier survival plots of synchronous, 9464D tumor-bearing mice that were treated with CpG-PBNP-PTT, PBNP-PTT, PBNP, CpG-PBNP or vehicle (PBS).
| Log-rank (Mantel-Cox) test | Adjusted p-value |
|---|---|
| Vehicle vs. PBNP-PTT | 0.0023 |
| Vehicle vs. CpG-PBNP-PTT | 0.0023 |
| PBNP vs. PBNP-PTT | 0.0116 |
| PBNP vs. CpG-PBNP-PTT | 0.0031 |
| CpG-PBNP vs. PBNP-PTT | 0.2249 |
| CpG-PBNP vs. CpG-PBNP-PTT | 0.0490 |
3. Discussion
Here we illustrated a CpG-PBNP-PTT-mediated nanoimmunotherapy that triggered systemic antitumor immune responses, leading to long-term antigen-specific T cell memory, providing robust and persistent tumor remission in a clinically relevant syngeneic, 9464D model of NB. Our previous studies in Neuro-2a NB suggest that PBNP-PTT triggers ICD.[14, 15] Further, CpG-PBNPs were also observed to activate DCs which then culminates in T cell activation.[15, 16] To mechanistically define the role of the T cell response of our novel nanoimmunotherapy in a clinically relevant MYCN overexpressing NB model, we first focused on T cell activation. T cell activation relies on the MHC I/MHC II and T cell receptor (TCR) interaction. While MHC I molecules are known to be expressed by most nucleated cells, MHC II molecules were initially believed to be expressed only by professional antigen-presenting cells. However, recent reports suggest the expression of MHC II and components related to its pathway on a variety of human tumor cells, including many solid tumors like melanoma, breast cancer, prostate cancer, glioma, colorectal cancer, where they play a pivotal role in T cell activation.[48, 58–65] Further studies in multiple tumor types have reported an association between MHC II expression on tumor cells and a favorable prognosis.[44] Our in vitro studies revealed that PTT mediates increases in both MHC I and MHC II on 9464D cells at a specfic thermal dose (Figure 2D–G), suggestive of engaging the first step in T cell activation (antigen presentation).
The MHC:TCR crosslinking is the initial step in T cell activation, followed by the crosslinking of costimulatory molecules with this complex that initiates T cell proliferation and survival.[51] Thus, we next investigated the expression of costimulatory molecules CD80 and CD86, which are known to interact with CD28 to activate both naïve and memory T cells.[48, 51, 66] Further, the paucity of these costimulatory molecules is linked to T cell anergy,[50] and their overexpression on tumor cells enhances antitumor immune response, which has been explored in several cancer immunotherapy clinical trials.[49, 51, 67] Our findings revealed significantly higher expression of CD80 and CD86 in high thermal dose PTT-treated groups as compared to controls (Figure 2H–K), suggesting that PTT primes these cells for immune recognition at this thermal dose. Non-immunogenic “cold” tumor cells, like NB, have typically downregulated or lost their antigen-presenting capabilities, along with low expression of costimulatory molecules.[50, 68, 69] Here, we have shown that PBNP-PTT or CpG-PBNP-PTT can reverse this downregulation and elicit expression of both costimulatory molecules and markers involved in antigen presentation. Thus our in vitro findings illustrate that our nanoimmunotherapy has the potential to prime the tumor cells for immune cell recognition by upregulating MHC I, MHC II, CD80, and CD86. It can further trigger the immunogenic responses by causing ICD (Figure 2L–R), findings consistent with earlier studies by our group that show PBNP-PTT leading to cytotoxicity and ICD generation in another NB model.[14, 15] Interestingly, we also observed significantly higher GD2 expression on 9464D cells after treatment with 1.5 W PBNP-PTT or CpG-PBNP-PTT. The current standard treatment for high-risk NB involves anti-disialoganglioside GD2 monoclonal antibody that targets GD2 on neuroblasts.[70, 71] Therefore3, the PTT-mediated enhanced GD2 expression observed here may offer an improved response to this current treatment modality (Figure S4B, C).
Another critical feature of a robust T cell response is the generation of long-term immunological memory. To investigate whether PBNP-PTT or CpG-PBNP-PTT could confer immunological protection against relapse, we rechallenged the long-term surviving PBNP-PTT or CpG-PBNP-PTT treated mice with one million 9464D cells on Day 80. 80% of mice in the CpG-PBNP-PTT treatment group rejected rechallenge, compared to 40% in the PBNP-PTT treatment group, indicating the development of a robust antitumor immunological memory generated by CpG-PBNP-PTT based nanoimmunotherapy (Figure 3D–G).
To further illustrate a robust T cell memory suggested by the rechallenge studies, we first analyzed the infiltration of T cells in the spleens of rechallenged mice. T cells play a critical role in eliciting immune responses and influencing the clinical outcome against various diseases, including autoimmune diseases, infection, allergic diseases, and cancer.[52, 72] We found significantly higher infiltration of CD3+ cells (Figure 4A–B) along with a significant increase in CD3+ CD44+ memory cells in the spleens of PBNP-PTT and CpG-PBNP-PTT treated mice that survived rechallenge, compared to treatment-naive, control mice (Figure 4F, G). Further there was a significant increase in the CD69 expression on CD4+ T cells, which is a key marker for tissue resident memory T cells both in humans and mice[56] ( Figure 4H, I).
We also found an increase in CD4+ central and effector memory along with CD8+ effector memory T cells in the spleens of the surviving CpG-PBNP-PTT and PBNP-PTT rechallenged mice compared to the treatment-naive, control mice (Figure 5A–F). Central memory T cells are responsible for long-term memory and usually reside in the T cell niche of secondary lymphoid organs, however on antigenic stimulation they readily proliferate and differentiate into effector memory T cells which then elicit immediate effector function.[73, 74] Building upon our in vivo study, we validated the generation of a long-term antigen-specific T cell memory response in the ex vivo co-culture analysis. Importantly, we observed a decrease in the CD4+ central memory and an increase in the CD4+ and CD8+ effector memory upon co-culturing the T cells with 9464D cells, suggesting the generation of antigen-specific memory in the T cells, critical to the rejection of tumor rechallenge (Figure 5G–L). Further, we observed a T cell mediated 9464D tumor cells killing upon co-culture, suggestive of tumor-specific memory (Figure 5M, N), a key criteria for therapeutic success.
Finally, we evaluated the potential of CpG-PBNP-PTT to elicit an abscopal effect in a synchronous tumor model (Figure 6). This aspect is particularly important as approximately 70% of NB cases present with metastatic disease at initial diagnosis.[75] Thus, an effective robust treatment should be able to extend its effects to more distant sites of tumor dissemination. Induction of abscopal effect is shown to involve activation of a systemic antitumor immune response against tumor antigens.[24, 25] Our results indicated complete tumor regression on the PTT-treated flank (Figure 6E (a-e)) and a significantly slower tumor progression on the untreated flank of PBNP-PTT- and CpG-PBNP-PTT-treated mice compared to vehicle-treated nice(Figure 6F (a-e)), with significantly enhanced long-term survival in the PBNP-PTT- and CpG-PBNP-PTT-treated mice compared to vehicle treated mice thus extending its abscopal effect to the secondary tumor site. Although we were unable to observe a statistically significant difference in survival between PBNP-PTT and CpG-PBNP-PTT in the synchronous tumor model with CpG-PBNP-PTT yielding only a modest increase in median survival compared to PBNP-PTT, we are investigating combinations of CpG-PBNP-PTT with other immunotherapies checkpoint inhibitors or immune effector cell therapies to better exploit any therapeutic advantage offered by the CpG coating (as observed in the single tumor model).
Overall, we observed that CpG-PBNP-PTT nanoimmunotherapy potently triggered strong antitumor immune responses with abscopal effects driven by T cell activation and long-term robust tumor-specific T cell memory. We elucidated that the antitumor effects CpG-PBNP-PTT are mediated by direct killing of cancer cells by PTT, along with generation of ICD and upregulation of immunostimulatory molecules, which ultimately leads to T cell activation and generation of tumor-specific T cell memory that prevents tumor relapse. As we tested our nanoimmunotherapy in a syngeneic preclinical NB model, there is potential to build upon these promising results for clinical translation.
4. Experimental Section
4.1. Materials
Nanoparticles were synthesized using DI water, Potassium hexacyanoferrate (II) trihydrate (MW 422.39; K4[Fe(CN)6]·3H2O), iron (III) chloride hexahydrate (MW 270.3; Fe(Cl)3·6H2O), Poly (ethylenimine) (PEI, MW 2,000, Mn 1,800, 50% w/v in H2O), acetate buffer (pH 5.2), citric acid, acetone, and ethanol obtained from Sigma-Aldrich (St. Louis, MO, USA). Murine CpG oligodeoxynucleotide (CpG) TLR9 ligand (ODN 1585; Class A) was purchased from InVivoGen (San Diego, CA, USA). Fluorescent antibodies against HMGB1 (EPR3507) and calreticulin (FMC75) were purchased from Abcam (Cambridge, UK). Dulbecco’s Modified Eagle Media, non-essential amino acids, antibiotic/antimitotic, and β-Mercaptoethanol were purchased from Thermo Fisher (Waltham, Massachusetts). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). CD28 (37.51) and CD3e (145–2C11) antibodies were purchased from eBioscience (San Diego, CA). TexMACS Medium and mouse Pan T Cell Isolation Kit II were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany).
4.2. Pediatric NB analyses
The results published here are based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET, https://ocg.cancer.gov/programs/target) initiative, (NB, phs000467). The data used for this analysis are available at https://portal.gdc.cancer.gov/projects. Data from the NB (TARGET, 2018) study was obtained through Pediatric cBioPortal, downloaded in June 2020[73–75]. MYCN expression in NB risk groups was compared using one-way ANOVA. The heat map was generated through Pediatric cBioPortal. 139 patients annotated with RNA-seq data from TARGET 2018 study were chosen for this analysis, but patients TARGET-30-PASSWW, TARGET-30-PARGKK were excluded as their transcriptomes were not complete.
4.3. Nanoparticle synthesis and characterization
PBNPs were synthesized using a one-pot synthesis system as previously described,[15] where an aqueous solution of 1.0 mM FeCl3·6H2O and 0.5 mM citric acid in 20 mL of DI water was mixed with 20 mL aqueous solution containing 1 mM K4Fe(CN)6·3H2O and 0.5 mM citric acid with vigorous stirring. The resulting precipitate containing PBNPs was isolated by adding equal volumes of acetone and centrifuging at (10,400 ×g for 10 minutes) at room temperature (RT). The collected PBNPs were rinsed three times and resuspended in Milli-Q water by sonication for 5 s using a Q500 sonicator (QSonica LLC, Newton, CT, USA) at high power (amplitude = 40 %).
CpG-PBNPs were synthesized using a layer-by-layer coating approach[15], where PBNPs (3 mg mL−1) were contacted with equal volumes of PEI (12 mg mL−1) in acetate buffer (pH 5.2) at RT for 1 h on an orbital shaker. PEI-coated PBNPs (PEI-PBNPs) were washed four times in 50 mL ethanol and centrifuged at 10,400 ×g for 10 minutes and resuspended by sonication in Milli-Q water. We further coated 500 μL PEI-PBNPs (2 mg mL−1) with 300 μL of an aqueous solution of CpG (containing 100 μg CpG in endotoxin-free water) under stirring at RT for 15 minutes. The resultant CpG-coated PEI-PBNPs (CpG-PBNPs) were collected by centrifuging the mixture at 21,000 ×g for 15 minutes. Dynamic light scattering on a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) was used to measure the stability of the nanoparticles by measuring the size (hydrodynamic diameter) and charge distributions (zeta potential) of the PBNPs, PEI-PBNPs, and CpG-PBNPs.
4.4. In vitro cell culture
The transgenic murine NB cell line 9464D, derived from spontaneous tumors from TH-MYCN transgenic mice on C57BL/6J background, were provided by Dr. Carol Thiele (Pediatric Oncology Branch, NIH, Bethesda). 9464D cells were cultured in DMEM containing 10% FBS, 1% non-essential amino acids, 0.5% antibiotic/antimycotic, and 0.05% β-mercaptoethanol. All cells and were tested negative for mycoplasma contamination.
4.5. In vitro PTT experiments
Five million 9464D cells were exposed to varying degrees of laser power (0.2 W cm−2 to 1.5 W cm−2) for 10 minutes with a fixed concentration (0.15 mg mL−1) of PBNPs or CpG-PBNPs using an 808 nm NIR continuous wave, collimated diode laser (Laserglow Technologies, Toronto, ON, Canada) in 1.5 mL tube. A power meter (Thorlabs, Newton, NJ) was used to confirm the laser power administered in each study. Time-based temperature measurements were captured using an i7 thermal imaging camera (FLIR, Arlington, VA, USA) at 1 minute interval for 10 minutes. 9464D cells were plated in 6-well plate and incubated at 37 °C under 5% CO2 post treatment, and visualized under the inverted phase contrast microscope (Leica DMi1 Inverted Microscope, Leica Microsystems) post 24 h for morphological changes using 10X magnification. The cells were further analyzed for viability using the CellTiter-Glo assay (Promega Corporation, Madison, WI) according to manufacturer’s instruction. Briefly, CellTiter-Glo reagent was added to the wells (50 μL and 50 μL media) and incubated at room temperature for 10 min in the dark and then the luminescence was recorded using SpectraMax i3x (Molecular Devices, LLC. San Jose, CA). Blank wells (media alone) were measured for luminescence and subtracted from the values in experimental wells. Results were expressed as a percentage of vehicle treated control cells. For flow cytometric analysis 24 h post treatment, cells were blocked using TruStain FcX (anti-mouse CD16/32) Antibody (101320) and then stained with PE/Cy7 anti GD2 (357308), APC/Fire 750 anti MHC I (114618), Alexa Fluor 488 anti MHC II (116410), Brilliant Violet 605 anti CD80 (104729), Pacific Blue anti CD86 (105022), and Zombie Violet Fixable Viability Dye (423114) purchased from BioLegend (San Diego, CA). Alexa Fluor 647 anti HMGB1 (ab195011) and PE anti calreticulin (ab83220) were purchased from Abcam (Cambridge, UK); all antibodies were used at 1:100 dilution. Stained cells were visualized on the BD Celesta Cell Analyzer (BD Biosciences Franklin Lakes, NJ) and analyzed using FlowJo (Ashland, OR) software and only live single cells were gated for analysis (Figure S7). All in vitro experiments were conducted in triplicate.
4.6. In vivo studies
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the George Washington University, Washington, DC, USA (Protocol # A396). Humane care of the animals was ensured in accord with IACUC guidelines. For all in vivo studies, five-week-old female C57BL/6J mice purchased from Jackson Laboratory (Bar Harbor, ME, USA) and acclimated for a week. The tumor sizes were measured twice a week using a digital caliper, and the tumor volume was estimated by calculation as Volume (mm3) = (length × width2) / 2. Animals were euthanized through cervical dislocation after CO2 narcosis when tumor sizes reached 15 mm diameter in any dimension or ulcerations occurred. If the animals displayed any signs of distress they were immediately euthanized.
4.7. In vivo NB model
The 9464D model was established by subcutaneously inoculating one million 9464D cells in 100 μL PBS in 5–6 week old female C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME), as described by Kroesen et al.[18] Mice were treated when tumors reached a diameter of at least 5 mm (~60 mm3). For the synchronous tumor model, one million 9464D cells were inoculated in both flanks of mice simultaneously. The tumor that reached a size of 5 mm (~60 mm3) first (i.e. the larger tumor) was designated as the “primary” tumor and treated. The contralateral tumors (generally slightly smaller; designated as the “secondary” tumor ) were left untreated to evaluate the abscopal effect of the treatments.
To confirm the establishment of the model, total RNA was extracted from the mice tumor cells using the RNeasy Mini Kit (Cat# 74104) following the manufacturer’s instructions (Qiagen, Hilden, Germany) and quantified using ND-1000 NanoDrop Spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE). The cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, 1708891), and mRNA was quantified using MyIQ single-color real-time PCR detection system (Bio-Rad, Hercules, CA) and iQ SYBR green Supermix (Bio-Rad, 1708882). Primers (Table 4) were customized from Invitrogen (Waltham, Massachusetts, USA) and checked for correct amplification and dissociation of the PCR products. mRNA expression was determined relative to PBGD expression using the method described by Kroesen et al.[18]
Table 4:
Primer sequences used in this study
| Gene name | Species | Primer sequence |
|---|---|---|
| PBGD | Mouse | CCTACCATACTACCTCCTGGCTTTAC TTTGGGTGAAAGACAACAGCAT |
| survivin | Mouse | CTGCTTTAAGGAATTGGAAGGCT CTGACGGGTAGTCTTTGCAGT |
| S100A6 | Mouse | TGGCTCCAAGCTGCAGG CCCAGGAAGGCGACATACTC |
| ODC | Mouse | GCACATCCAAAGGCAAAGTTG CAAACTTAACACTGAGGCGACAGA |
| hMYCN | Mouse | CGACCACAAGGCCCTCAGTA CAGCCTTGGTGTTGGAGGAG |
| GD2-synthase | Mouse | CCAAGGAGCCGAGTACAACAT GTAGGGTAAAAGCGTCGGATG |
4.8. In vivo PTT
For single tumor studies, mice were randomly divided into 3 groups when tumors reached a diameter of at approximatley 5 mm (~60 mm3) (n=5/group): 1) Vehicle (intratumoral (i.t.) injection of 50 μL PBS on day 0), 2) PBNP-PTT (i.t. injection of 50 μL of 1 mg mL−1 PBNPs and irradiated with NIR laser to temperature of 120°C for 10 minutes), and 3) CpG-PBNP-PTT nanoimmunotherapy (i.t. injection of 50 μL of 1 mg mL−1 CpG-PBNP with 2 μg bound CpG, irradiated with NIR laser to reach a maximum temperature of 120°C for 10 minutes, 50 μL CpG-PBNP boosts were administered at the initial site of tumor (without PTT) on days 2 and 5 ). Surviving mice were rechallenged with one million 9464D cells ~80 days post tumor-free survival to assess the immunological memory.
Synchronous tumor-bearing mice were randomly divided into five groups (n=5/group): 1) Vehicle (injected i.t. with 50 μl PBS on day 0), 2) PBNP (i.t. injection of 50 μL of 1 mg mL-1 PBNP), 3) CpG-PBNP (i.t. injection of 50 μL of 1 mg mL-1 CpG-PBNP with 2 μg bound CpG (without PTT) on days 0, 2, and 5), 4) PBNP-PTT (i.t. injection of 50 μL of 1 mg mL-1 PBNPs and irradiated with NIR laser to temperature of 120°C for 10 minutes), and 5) CpG-PBNP-PTT (i.t. injection of 50 μL of 1 mg mL-1 CpG-PBNPs with 2 μg bound CpG, irradiated with NIR laser to temperature of 120°C for 10 minutes, CpG-PBNPs boosts were administered at the initial site of tumor (without PTT) on days 2 and 5). The tumor that reached a size of 5 mm (~60 mm3) first (i.e. the larger tumor) was designated as the “primary” tumor and treated. The contralateral tumors (generally slightly smaller; designated as the “secondary” tumor) were left untreated to evaluate the abscopal effect of the treatments.
Mice were anesthetized during the procedure using 2–5% isoflurane. Temperatures attained during PTT treatment were monitored using the i7 FLIR camera. Tumor growth of the mice was monitored following inoculation and treatments by routine caliper measurements.
4.9. Ex vivo T cell studies
T cells were isolated from the spleens of either treatement naive, control group or PBNP-PTT- /CpG-PBNP-PTT-treated mice (125 days post tumor inoculation and rechallenge for these PTT-treated groups) using a Pan T Cell Isolation Kit II (Miltenyi Biotec). For the treatement naive, control group we used age-matched mice that were inoculated with one million 9464D cells and euthanized when their tumors reached ~15 mm diameter. This was scheduled so that we were able to acquire spleens of mice from the treatement naive, control group, PBNP-PTT- and CpG-PBNP-PTT-treated groups on the same day for downstream analysis. T cells were expanded in vitro for 4 days using TexMACS medium supplemented with 100 U mouse Recombinant IL-2 (STEMCELL Technologies) in the presence of anti-CD3 and anti-CD28 antibodies. Subsequently, 9464D cells were co-cultured with the T cells for 2 days, after which the cells were blocked using TruStain FcX (anti-mouse CD16/32) antibody and then stained with fluorescent antibodies APC anti CD3, Brilliant Violet 421 anti CD4, Alexa Fluor 488 anti CD8, APC/Cy7 anti CD44, Brilliant Violet 650 anti CD62L, and PE/Cy7 anti GD2 and Zombie Violet Fixable Viability Dye; all antibodies were used at 1:100 dilution. Stained cells were visualized on the BD Celesta Cell Analyzer (BD Biosciences Franklin Lakes, NJ). Flow cytometry results were analyzed using FlowJo (Ashland, OR) software and only live single cells were gated for analysis.
4.10. Statistical analysis
1. Pre-processing of data: Normalization was used in this study to represent flow cytometry count data (normalized to mode) and the expression of intracellular ATP levels in the various treatment groups (as a percentage of the vehicle-treated group). All animal studies were performed in a non-blinded fashion, and mice that attained similar average volumes of the primary tumors were randomized into the various treatment groups. All other data acquired in this study were presented as acquired without any transformation or exclusion of outliers. 2. Data presentation: results obtained in this study are expressed as mean ± standard deviation. 3. Sample size (n) for each statistical analysis: All in vitro studies had a sample size of n = 3. All in vivo studies had a sample size of n = 5. For the ex vivo studies, sample sizes were as follows; treatment-naive group (n = 4), PBNP-PTT (n = 2), CpG-PBNP-PTT (n = 4) for spleen samples and treatment-naive group (n = 4), PBNP-PTT (n = 2), CpG-PBNP-PTT (n = 3) for co-culture samples. 4. Statistical methods: To assess statistically significant differences between treatment groups, a one-way or two-way ANOVA followed by Tukey’s multiple comparison tests were performed. To assess statistical differences in animal survival post-treatment, log-rank (Mantel-Cox) tests were performed. Data were approximated as normally distributed assuming variance was similar between the treatment groups. Statistical significance throughout the study is indicated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. 5. Software used for statistical analysis: analysis was performed using the Prism 8.1.1 software (GraphPad).
Supplementary Material
5. Acknowledgements
This work was supported by Alex’s Lemonade Stand Foundation for Childhood Cancer’s ‘A’ Award and the George Washington University Cancer Center. Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health under Award Number R37CA226171. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Elizabeth E. Sweeney, The George Washington Cancer Center, The George Washington University, 800 22nd St NW, Science and Engineering Hall 8th Floor, Washington, DC 20052, USA.
Rohan Fernandes, The George Washington Cancer Center, The George Washington University, 800 22nd St NW, Science and Engineering Hall 8th Floor, Washington, DC 20052, USA.; The Institute for Biomedical Sciences, The George Washington University,2300 Eye Street NW, Ross Hall Room 561, Washington, DC 20037, USA. Department of Medicine, The George Washington University, 2150 Pennsylvania Avenue, NW, Suite 8-416, Washington, DC 20037, USA.
References
- 1.Vanichapol T; Chutipongtanate S; Anurathapan U; Hongeng S, Biomed Res Int 2018, 2018, 1812535. DOI 10.1155/2018/1812535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casey DL; Cheung NV, Cancer Immunol Res 2020, 8 (2), 161–166. DOI 10.1158/2326-6066.CIR-19-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borriello L; Seeger RC; Asgharzadeh S; DeClerck YA, Cancer Lett 2016, 380 (1), 304–14. DOI 10.1016/j.canlet.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galland S; Vuille J; Martin P; Letovanec I; Caignard A; Fregni G; Stamenkovic I, Cell Rep 2017, 20 (12), 2891–2905. DOI 10.1016/j.celrep.2017.08.089. [DOI] [PubMed] [Google Scholar]
- 5.Pelizzo G; Veschi V; Mantelli M; Croce S; Di Benedetto V; D’Angelo P; Maltese A; Catenacci L; Apuzzo T; Scavo E; Moretta A; Todaro M; Stassi G; Avanzini MA; Calcaterra V, BMC Cancer 2018, 18 (1), 1176. DOI 10.1186/s12885-018-5082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang P; Wu X; Basu M; Dong C; Zheng P; Liu Y; Sandler AD, Front Immunol 2017, 8, 1473. DOI 10.3389/fimmu.2017.01473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Layer JP; Kronmuller MT; Quast T; van den Boorn-Konijnenberg D; Effern M; Hinze D; Althoff K; Schramm A; Westermann F; Peifer M; Hartmann G; Tuting T; Kolanus W; Fischer M; Schulte J; Holzel M, Oncoimmunology 2017, 6 (6), e1320626. DOI 10.1080/2162402X.2017.1320626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagni F; Guerini-Rocco E; Schultheis AM; Grazia G; Rijavec E; Ghidini M; Lopez G; Venetis K; Croci GA; Malapelle U; Fusco N, Int J Mol Sci 2019, 20 (21). DOI 10.3390/ijms20215452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegde PS; Chen DS, Immunity 2020, 52 (1), 17–35. DOI 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Dumont MF; Hoffman HA; Yoon PR; Conklin LS; Saha SR; Paglione J; Sze RW; Fernandes R, Bioconjug Chem 2014, 25 (1), 129–37. DOI 10.1021/bc4004266. [DOI] [PubMed] [Google Scholar]
- 11.Sweeney EE; Burga RA; Li C; Zhu Y; Fernandes R, Sci Rep 2016, 6, 37035. DOI 10.1038/srep37035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kale SS; Burga RA; Sweeney EE; Zun Z; Sze RW; Tuesca A; Subramony JA; Fernandes R, Int J Nanomedicine 2017, 12, 6413–6424. DOI 10.2147/IJN.S144515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cano-Mejia J; Burga RA; Sweeney EE; Fisher JP; Bollard CM; Sandler AD; Cruz CRY; Fernandes R, Nanomedicine 2017, 13 (2), 771–781. DOI 10.1016/j.nano.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweeney EE; Cano-Mejia J; Fernandes R, Small 2018, 14 (20), e1800678. DOI 10.1002/smll.201800678. [DOI] [PubMed] [Google Scholar]
- 15.Cano-Mejia J; Bookstaver ML; Sweeney EE; Jewell CM; Fernandes R, Biomater Sci 2019, 7 (5), 1875–1887. DOI 10.1039/c8bm01553h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cano-Mejia J; Shukla A; Ledezma DK; Palmer E; Villagra A; Fernandes R, Transl Oncol 2020, 13 (10), 100823. DOI 10.1016/j.tranon.2020.100823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss WA; Aldape K; Mohapatra G; Feuerstein BG; Bishop JM, EMBO J 1997, 16 (11), 2985–95. DOI 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kroesen M; Nierkens S; Ansems M; Wassink M; Orentas RJ; Boon L; den Brok MH; Hoogerbrugge PM; Adema GJ, Int J Cancer 2014, 134 (6), 1335–45. DOI 10.1002/ijc.28463. [DOI] [PubMed] [Google Scholar]
- 19.https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021626s007lbl.pdf2008.
- 20.Kroemer G; Galluzzi L; Kepp O; Zitvogel L, Annu Rev Immunol 2013, 31, 51–72. DOI 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 21.Galluzzi L; Buque A; Kepp O; Zitvogel L; Kroemer G, Nat Rev Immunol 2017, 17 (2), 97–111. DOI 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 22.Mole RH, Br J Radiol 1953, 26 (305), 234–41. DOI 10.1259/0007-1285-26-305-234. [DOI] [PubMed] [Google Scholar]
- 23.Bezu L; Gomes-de-Silva LC; Dewitte H; Breckpot K; Fucikova J; Spisek R; Galluzzi L; Kepp O; Kroemer G, Front Immunol 2015, 6, 187. DOI 10.3389/fimmu.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y; Dong Y; Kong L; Shi F; Zhu H; Yu J, J Hematol Oncol 2018, 11 (1), 104. DOI 10.1186/s13045-018-0647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farias VA; Tovar I; Del Moral R; O’Valle F; Exposito J; Oliver FJ; Ruiz de Almodovar JM, Front Oncol 2019, 9, 1381. DOI 10.3389/fonc.2019.01381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gursel M; Gursel I, Methods Mol Biol 2016, 1404, 289–298. DOI 10.1007/978-1-4939-3389-1_20. [DOI] [PubMed] [Google Scholar]
- 27.Lai CY; Yu GY; Luo Y; Xiang R; Chuang TH, Front Immunol 2019, 10, 179. DOI 10.3389/fimmu.2019.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.https://www.fda.gov/media/108745/download2017.
- 29.Carpentier AF; Chen L; Maltonti F; Delattre JY, Cancer Res 1999, 59 (21), 5429–32. [PubMed] [Google Scholar]
- 30.Guo LH; Schluesener HJ, Oligonucleotides 2004, 14 (4), 287–98. DOI 10.1089/oli.2004.14.287. [DOI] [PubMed] [Google Scholar]
- 31.Crittenden M; Kohrt H; Levy R; Jones J; Camphausen K; Dicker A; Demaria S; Formenti S, Semin Radiat Oncol 2015, 25 (1), 54–64. DOI 10.1016/j.semradonc.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourquin C; Anz D; Zwiorek K; Lanz AL; Fuchs S; Weigel S; Wurzenberger C; von der Borch P; Golic M; Moder S; Winter G; Coester C; Endres S, J Immunol 2008, 181 (5), 2990–8. DOI 10.4049/jimmunol.181.5.2990. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X; Wu F; Men K; Huang R; Zhou B; Zhang R; Zou R; Yang L, Nanoscale Res Lett 2018, 13 (1), 240. DOI 10.1186/s11671-018-2661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ash S; Stein J; Askenasy N; Yaniv I, Br J Cancer 2010, 103 (10), 1597–605. DOI 10.1038/sj.bjc.6605924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore HC; Wood KM; Jackson MS; Lastowska MA; Hall D; Imrie H; Redfern CP; Lovat PE; Ponthan F; O’Toole K; Lunec J; Tweddle DA, J Clin Pathol 2008, 61 (10), 1098–103. DOI 10.1136/jcp.2007.054627. [DOI] [PubMed] [Google Scholar]
- 36.Terrile M; Bryan K; Vaughan L; Hallsworth A; Webber H; Chesler L; Stallings RL, PLoS One 2011, 6 (12), e28356. DOI 10.1371/journal.pone.0028356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Q; Xu L; Liang C; Wang C; Peng R; Liu Z, Nat Commun 2016, 7, 13193. DOI 10.1038/ncomms13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wansong Chen KZ, Hong Liu, Jiang Ouyang, Liqiang Wang, Ying Liu, Hao Wang, Liu Deng,* and Liu You-Nian, Advanced Functional Material 2017, 27, 1605795. [Google Scholar]
- 39.Liu J; Liu K; Feng L; Liu Z; Xu L, Biomater Sci 2017, 5 (2), 331–340. DOI 10.1039/c6bm00526h. [DOI] [PubMed] [Google Scholar]
- 40.Wen M; Ouyang J; Wei C; Li H; Chen W; Liu YN, Angew Chem Int Ed Engl 2019, 58 (48), 17425–17432. DOI 10.1002/anie.201909729. [DOI] [PubMed] [Google Scholar]
- 41.Liu B; Wang W; Fan J; Long Y; Xiao F; Daniyal M; Tong C; Xie Q; Jian Y; Li B; Ma X; Wang W, Biomaterials 2019, 217, 119301. DOI 10.1016/j.biomaterials.2019.119301. [DOI] [PubMed] [Google Scholar]
- 42.Shi H; Sadler PJ, Br J Cancer 2020, 123 (6), 871–873. DOI 10.1038/s41416-020-0926-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang P; Chiu YC; Tostanoski LH; Jewell CM, ACS Nano 2015, 9 (6), 6465–77. DOI 10.1021/acsnano.5b02153. [DOI] [PubMed] [Google Scholar]
- 44.Chiu YC; Gammon JM; Andorko JI; Tostanoski LH; Jewell CM, ACS Biomater Sci Eng 2015, 1 (12), 1200–1205. DOI 10.1021/acsbiomaterials.5b00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sapareto SA; Dewey WC, Int J Radiat Oncol Biol Phys 1984, 10 (6), 787–800. DOI 10.1016/0360-3016(84)90379-1. [DOI] [PubMed] [Google Scholar]
- 46.van Rhoon GC; Samaras T; Yarmolenko PS; Dewhirst MW; Neufeld E; Kuster N, Eur Radiol 2013, 23 (8), 2215–27. DOI 10.1007/s00330-013-2825-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neefjes J; Jongsma ML; Paul P; Bakke O, Nat Rev Immunol 2011, 11 (12), 823–36. DOI 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 48.Axelrod ML; Cook RS; Johnson DB; Balko JM, Clin Cancer Res 2019, 25 (8), 2392–2402. DOI 10.1158/1078-0432.CCR-18-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harding FA; McArthur JG; Gross JA; Raulet DH; Allison JP, Nature 1992, 356 (6370), 607–9. DOI 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 50.Chen L; McGowan P; Ashe S; Johnston JV; Hellstrom I; Hellstrom KE, Cancer Res 1994, 54 (20), 5420–3. [PubMed] [Google Scholar]
- 51.Chen L; Flies DB, Nat Rev Immunol 2013, 13 (4), 227–42. DOI 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia A; Zhang Y; Xu J; Yin T; Lu XJ, Front Immunol 2019, 10, 1719. DOI 10.3389/fimmu.2019.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pennock ND; White JT; Cross EW; Cheney EE; Tamburini BA; Kedl RM, Adv Physiol Educ 2013, 37 (4), 273–83. DOI 10.1152/advan.00066.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin H; Iwasaki A, Immunol Rev 2013, 255 (1), 165–81. DOI 10.1111/imr.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schumann J; Stanko K; Schliesser U; Appelt C; Sawitzki B, PLoS One 2015, 10 (7), e0132479. DOI 10.1371/journal.pone.0132479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar BV; Ma W; Miron M; Granot T; Guyer RS; Carpenter DJ; Senda T; Sun X; Ho SH; Lerner H; Friedman AL; Shen Y; Farber DL, Cell Rep 2017, 20 (12), 2921–2934. DOI 10.1016/j.celrep.2017.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang L; Li Y; Du Y; Zhang Y; Wang X; Ding Y; Yang X; Meng F; Tu J; Luo L; Sun C, Nat Commun 2019, 10 (1), 4871. DOI 10.1038/s41467-019-12771-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feinmesser M; Sulkes A; Morgenstern S; Sulkes J; Stern S; Okon E, J Clin Pathol 2000, 53 (4), 286–91. DOI 10.1136/jcp.53.4.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soos JM; Krieger JI; Stuve O; King CL; Patarroyo JC; Aldape K; Wosik K; Slavin AJ; Nelson PA; Antel JP; Zamvil SS, Glia 2001, 36 (3), 391–405. DOI 10.1002/glia.1125. [DOI] [PubMed] [Google Scholar]
- 60.Bustin SA; Li SR; Phillips S; Dorudi S, Tumour Biol 2001, 22 (5), 294–8. DOI 10.1159/000050630. [DOI] [PubMed] [Google Scholar]
- 61.Younger AR; Amria S; Jeffrey WA; Mahdy AE; Goldstein OG; Norris JS; Haque A, Prostate Cancer Prostatic Dis 2008, 11 (4), 334–41. DOI 10.1038/sj.pcan.4501021. [DOI] [PubMed] [Google Scholar]
- 62.Loi S; Dushyanthen S; Beavis PA; Salgado R; Denkert C; Savas P; Combs S; Rimm DL; Giltnane JM; Estrada MV; Sanchez V; Sanders ME; Cook RS; Pilkinton MA; Mallal SA; Wang K; Miller VA; Stephens PJ; Yelensky R; Doimi FD; Gomez H; Ryzhov SV; Darcy PK; Arteaga CL; Balko JM, Clin Cancer Res 2016, 22 (6), 1499–509. DOI 10.1158/1078-0432.CCR-15-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laidlaw BJ; Craft JE; Kaech SM, Nat Rev Immunol 2016, 16 (2), 102–11. DOI 10.1038/nri.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spitzer MH; Carmi Y; Reticker-Flynn NE; Kwek SS; Madhireddy D; Martins MM; Gherardini PF; Prestwood TR; Chabon J; Bendall SC; Fong L; Nolan GP; Engleman EG, Cell 2017, 168 (3), 487–502 e15. DOI 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson DB; Bordeaux J; Kim JY; Vaupel C; Rimm DL; Ho TH; Joseph RW; Daud AI; Conry RM; Gaughan EM; Hernandez-Aya LF; Dimou A; Funchain P; Smithy J; Witte JS; McKee SB; Ko J; Wrangle JM; Dabbas B; Tangri S; Lameh J; Hall J; Markowitz J; Balko JM; Dakappagari N, Clin Cancer Res 2018, 24 (21), 5250–5260. DOI 10.1158/1078-0432.CCR-18-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharpe AH, Immunol Rev 2009, 229 (1), 5–11. DOI 10.1111/j.1600-065X.2009.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antonia SJ; Seigne J; Diaz J; Muro-Cacho C; Extermann M; Farmelo MJ; Friberg M; Alsarraj M; Mahany JJ; Pow-Sang J; Cantor A; Janssen W, J Urol 2002, 167 (5), 1995–2000. [PubMed] [Google Scholar]
- 68.Cabrera T; Ruiz-Cabello F; Garrido F, Scand J Immunol 1995, 41 (4), 398–406. DOI 10.1111/j.1365-3083.1995.tb03584.x. [DOI] [PubMed] [Google Scholar]
- 69.Ostroumov D; Fekete-Drimusz N; Saborowski M; Kuhnel F; Woller N, Cell Mol Life Sci 2018, 75 (4), 689–713. DOI 10.1007/s00018-017-2686-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu AL; Gilman AL; Ozkaynak MF; London WB; Kreissman SG; Chen HX; Smith M; Anderson B; Villablanca JG; Matthay KK; Shimada H; Grupp SA; Seeger R; Reynolds CP; Buxton A; Reisfeld RA; Gillies SD; Cohn SL; Maris JM; Sondel PM; Children’s Oncology G, N Engl J Med 2010, 363 (14), 1324–34. DOI 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Voeller J; Erbe AK; Slowinski J; Rasmussen K; Carlson PM; Hoefges A; VandenHeuvel S; Stuckwisch A; Wang X; Gillies SD; Patel RB; Farrel A; Rokita JL; Maris J; Hank JA; Morris ZS; Rakhmilevich AL; Sondel PM, J Immunother Cancer 2019, 7 (1), 344. DOI 10.1186/s40425-019-0823-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Borst J; Ahrends T; Babala N; Melief CJM; Kastenmuller W, Nat Rev Immunol 2018, 18 (10), 635–647. DOI 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 73.Gattinoni L; Lugli E; Ji Y; Pos Z; Paulos CM; Quigley MF; Almeida JR; Gostick E; Yu Z; Carpenito C; Wang E; Douek DC; Price DA; June CH; Marincola FM; Roederer M; Restifo NP, Nat Med 2011, 17 (10), 1290–7. DOI 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reiser J; Banerjee A, J Immunol Res 2016, 2016, 8941260. DOI 10.1155/2016/8941260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khan FH; Pandian V; Ramraj S; Aravindan S; Herman TS; Aravindan N, BMC Genomics 2015, 16, 501. DOI 10.1186/s12864-015-1642-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.