

Abstract
Current fuel-cell catalysts for oxygen reduction reaction (ORR) and H2 oxidation use precious metals and, for ORR, require high overpotentials. In contrast, metalloenzymes perform their respective reaction at low overpotentials using earth-abundant metals, making metalloenzymes ideal candidates for inspiring electrocatalytic design. Critical to the success of these enzymes are redox-active metal centers surrounding the enzyme active sites that ensure fast electron transfer (ET) to or away from the active site, by tuning the catalytic potential of the reaction as observed in multicopper oxidases but also in dictating the catalytic bias of the reaction as realized in hydrogenases. This review summarizes recent advances in studying these ET centers in multicopper oxidases and heme-copper oxidases that perform ORR and hydrogenases in carrying out H2 oxidation. Insights gained from understanding how the reduction potential of the ET centers effects reactivity at the active site in both the enzymes and their models are provided.
Introduction
As the global demand for energy increases, research into other electrical energy sources like fuel cells has gained significant interest due to fuel cells’ ability to generate electricity through the coupling of oxidation of a fuel and reduction of an oxidant. Because energy is generated from the coupling of these two half reactions, more sustainable fuel sources can be used in the generation of electrical energy. A primary example of a shift towards more sustainable fuel sources in fuel cell catalysis is the combination of the O2 reduction reaction (ORR) and H2 oxidation, which not only produces clean byproducts like water but also produces a maximum voltage of the cell of over 1 V due to the large potential difference between these two half reactions, potentially giving the cell access to a large amount of energy.1 However, issues arise with current fuel catalysts with actualizing this source of energy due to high energy input and slow or inefficient catalysis. For ORR, current fuel cell catalysts employ Pt based catalysts which not only require large overpotentials for the reaction but are also sluggish in catalytic rates and inefficient in the four-electron reduction of O2 to H2O. The methods that are often employed to speed up the ORR rate require increasing the Pt catalyst concentration which is expensive due to the high intrinsic cost of Pt materials.2 Pt-based catalyst are also inefficient in the selective 4 electron reduction of O2 to H2O. These catalysts produce other detrimental byproducts such as peroxide, which can lead to Pt catalyst degradation as well as nanofilm degradation between the two half cells.2 Other issues also arise with current catalysts for H2 oxidation, arising mainly from the economic cost of Pt metal .
Enzymes perform both of these reactions with high selectivity and high turnover rates using earth abundant metals instead of precious metals that are currently being employed.3 Therefore, enzymes not only provide key insights for developing electrocatalysts but also can be employed in the development of bio fuel cells, which use enzymes as the electrocatalysts.4 Enzymes also operate very close to the potentials required for the biochemical reactions and thus perform the catalysis at low overpotentials. In contrast to other Pt based catalysts for ORR, multicopper oxidases (MCO) and heme-copper oxidases (HCO) use copper and iron to carry out ORR. In particular , MCO’s perform ORR with much lower overpotentials than observed in Pt. 5,6 For H2 oxidation, hydrogenases perform the reaction using nickel and iron with nearly no overpotential. Therefore, learning how these enzymes succeeded in using these earth-abundant metals to carry out the ORR and H2 oxidation will not only help us design better electrocatalysts for fuel cells but these enzymes can also be implemented in bio fuel cell designs. Specifically, the efficiency of these enzymes relies on the rapid supply of electrons from protein surfaces to an often-buried catalytic site. Nature has exploited multiple metal-based and organic cofactors for these electron transfer (ET) steps and extensive research has focused on understanding or modulating the driving force and ET properties of these centers.7–9 These ET centers are not only important in providing fast ET to the active site but can also have significant impact on the catalytic overpotential and the catalytic bias of the chemical reaction, both of which are important in tuning these enzymes for efficient electrobiocatalysis.1,10 Therefore, instead of discussing the catalytic centers of these enzymes, this review will focus on the recent advances in understanding the redox properties of the ET centers around the catalytic sites in MCOs, HCOs and hydrogenases and how tuning the ET properties of these centers affects catalysis.
Oxygen reduction reaction (ORR)
Two classes of enzymes, multicopper oxidases (MCO) and heme-copper oxidases (HCO) can perform efficient 4e− reduction of O2 to H2O. Both enzymes not only contain earth-abundant metal cofactors to perform ORR but also contain ET centers to ensure fast ET to the active site for catalysis. MCOs are well established examples of fuel cell biocatalysts that perform ORR at low overpotentials comparable to Pt-based catalyts.11 Four copper ions are present in MCOs, which are involved in substrate oxidation and O2 reduction (Figure 1a). Intermolecular electron transfer from a reduced substrate occurs at the Type 1 copper (T1Cu) center located near the surface of the protein. Four electrons are liberated from the oxidation of four substrates at the T1Cu center and transferred 13 Å away through a Cys-His superexchange pathway to the trinuclear copper center (TNC) where O2 binding and reduction occur. The TNC contains a mononuclear Type 2 (T2) center and a coupled binuclear Type 3 (T3) center. The ligation of the TNC center is fairly conserved among the family of MCOs whereas differences in ligation arise at the T1Cu center. The T1Cu centers of MCOs are coordinated by two His residues and one Cys residue but either exhibit a weakly bound axial Met or a non-coordinating residue such as a Phe (Figure 1b).
Figure 1.
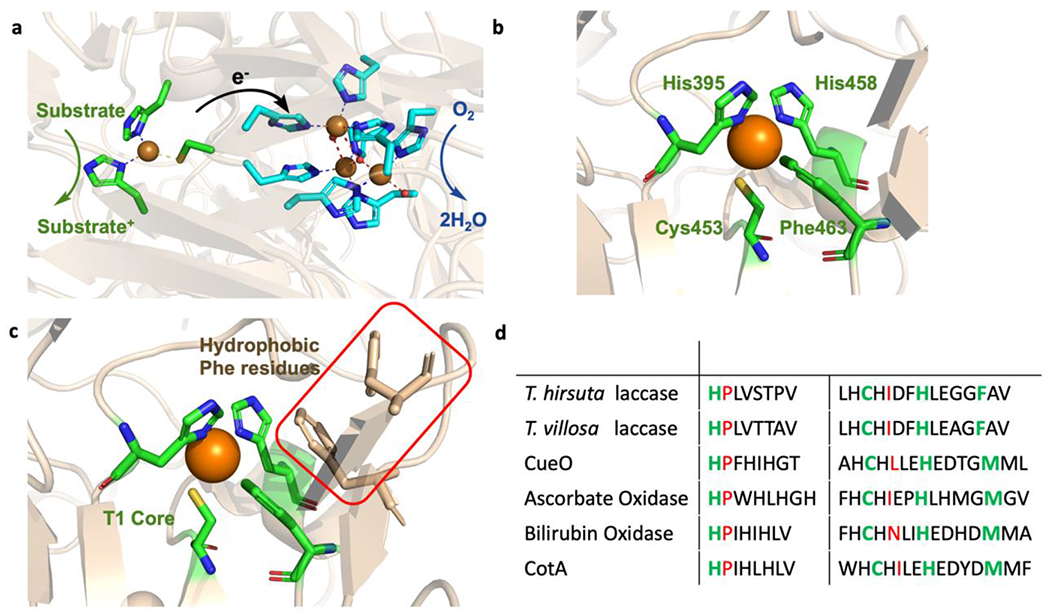
(a) The active site of fungal laccase from Trametes hirsuta (PDB:3fpx) showing the electron transfer from the T1Cu center to the site of O2 reduction at the trinuclear copper center in MCOs. (b) The T1Cu center of the fungal laccase from T. hirsuta. (c) The hydrophobic Phe residues in the SCS of fungal laccase are shown in tan and the T1Cu center is highlighted in green. (d) Sequence alignment of MCOs showing the PCS in green of the T1Cu center and the analogous sites for hydrogen bonding to the S(Cys) residue shown in red. MCOs have a conserved Pro residue in the analogous azurin Asn47 position indicating one hydrogen bond to the S(Cys) rather than two.
MCOs exhibit a wide range of T1Cu center E°′ from 300 mV up to 800 mV as observed in the fungal laccase from Trametes hirsuta12 Previous studies have shown a correlation between the low overpotentials of ORR with the E°′ of the T1Cu center.5 Therefore, MCOs with high T1Cu E°′ such as fungal laccases are attractive for fuel cell catalysis as these enzymes are able to reduce O2 at higher potentials than lower potential MCOs. However, the utility of fungal laccases in fuel cell catalysis is difficult due to the level of protein glycosylation and the presence of halide ions (F− and Cl−) both of which have been shown to affect ORR activity.13 Therefore, research has focused on altering the potential of other MCO T1Cu centers to achieve lower overpotentials for ORR.14,15
Tuning the E°′ of the T1Cu center in mononuclear cupredoxins has been extensively studied16–18 and similar approaches have been applied to E°′ tuning of the T1Cu center of MCOs. Differences in axial ligand coordination, electrostatic interactions and hydrophobicity all play a role in the wide range of E°′ exhibited by the T1Cu center of Cu proteins.18 Previous studies of mononuclear cupredoxin proteins have shown that the reduction potential of the T1Cu center is influenced by the nature of the axial ligand in the primary coordination sphere (PCS).18 The relatively high reduction potential of the T1Cu center in fungal laccase has been attributed to the presence of a noncoordinating hydrophobic ligand in the axial position, whereas the relatively low reduction potential of stellacyanin has been attributed to a stronger field O(Glu) residue. Solomon and coworkers demonstrated a 100 mV decrease in E°′ in the fungal laccase T. villosa when the axial Phe was mutated to a Met residue.19 Likewise, substitution of the axial Met with a Leu or Phe in B. subtilis CoA laccase resulted in an increase in the E°′ of T1Cu center by 100 mV.20 Hydrophobicity of the residues in the secondary coordination sphere (SCS) of the T1Cu center also has an effect on the E°′ where inclusion of Phe residues in the SCS of the T1Cu center in the mononuclear Cu protein azurin resulted in an increase of approximately 30 mV per Phe residue.21 Examination of the crystal structures of multiple high potential fungal laccases indicate there are a number of hydrophobic Phe residues in the vicinity of the T1Cu center, where two of the Phe residues can form a π stacking interaction with the axially bound Phe (Figure 1c). The presence of these hydrophobic residues in the SCS could also contribute to the high T1Cu E°′ of these enzymes.8
The hydrogen bonding interactions to the coordinated S(Cys) of the T1Cu center also have significant effect on tuning E°′,22–24 Studies in azurin have shown two important hydrogen bonding interactions to the S(Cys) thiolate from the backbone amides of both Phe114 and Asn47. Elimination of the Phe114 hydrogen bonding interaction through a Phe114Pro mutation in azurin resulted in a shorter Cu-S(Met) bond length and increased Cu-S(Cys) covalency consistent with a decrease in E°′ 22,25 The effect of tuning the electron-donating abilities of the S(Cys) through hydrogen bonding interactions was also investigated in MCOs. Sakurai and coworkers explored the effects of the hydrogen bonding interactions to the Cu-S(Cys) of the T1Cu center in the copper efflux oxidase (CuEO), which contains an axially bound Met residue. This enzyme contains only one hydrogen bond to the S(Cys) from a backbone amide of Leu502 in the analogous Phe114 position in azurin. The second potential hydrogen bonding interaction in the analogous Asn47 position in azurin is a proline residue, thus eliminating any additional hydrogen bonding interaction to the thiolate. Mutations of Pro444 to Ala, Leu or Ile to include the second hydrogen bonding interaction to the S(Cys) from a backbone amide resulted in an increase of ~ 40-70 mV with no substantial changes in the UV/vis absorption or EPR spectra.26 Sequence alignment with multiple axial Met containing MCOs indicates that the position of the Pro residue in CuEO is highly conserved among MCOs (Figure 1d).27 The lack of the additional hydrogen bond to the S(Cys) in MCOs would generate a more covalent Cu-S(Cys) bond. This increased interaction results in larger orbital overlap between donor and acceptor through the Cys-His pathway to the TNC promoting faster intramolecular ET, 8,18 thus, suggesting that MCOs are tuned to perform efficient intramolecular ET to the TNC through this increased Cu-S(Cys) interaction.
Model systems and their analogous mutations in MCOs have demonstrated the ability to modulate the E°′ of the T1Cu center. However, changes in the E°′ of the T1Cu center of MCOs can produce changes in ET properties and subsequently ORR activity. For example, the increase in 100 mV of the T1Cu center of CoA laccase from B. subtilis coincided with a decline in activity associated with unfavorable ET from the T1Cu to the TNC.28 Similarly a high potential triple mutant variant from CuEO also lacked ORR activity due to the significant increase in E°′ and its inability to transfer electrons to the TNC.29 Thus, there appears to be a balance between the two sites and a limit to the E°′ tuning of the T1Cu center. Solomon and coworkers sought to investigate the effect of T1Cu E°′ on ORR activity. The authors studied the effect of a 350 mV increase in the T1Cu E° on the mechanism of ORR by comparing a low E° plant laccase from Toxiconedron vernivifluum (430 mV) to a high E°′ fungal laccase from Trametes versicolor (780 mV).30 The studies indicated that both plant and fungal laccases exhibit similar mechanisms of ORR but vary in the rate determining step (RDS). The increased E°′ of the T1Cu center in fungal laccase results in a decrease in driving force for the first intramolecular electron step and thus a slower rate. Alternatively, the lower E°′ of the T1Cu center of plant laccase decreases the driving force for the first intermolecular ET step making the T1Cu reduction step rate limiting. However, the difference between fungal and plant laccases in the driving force for the first intramolecular ET step cannot be solely attributed to their differences in the E°′ of the T1Cu center. Instead, the difference in the driving force for the first intramolecular ET also arises from differences in the E°′ of the first intermediate at the TNC. The authors indicate that even though both TNC centers exhibit similar PCS of the T2 and binuclear T3 centers, there are multiple differences between the two enzymes in the SCS of the TNC. The TNC in fungal laccase exhibits a more positively charged protein environment, which would favor an increased E°′ of the TNC. Therefore, the authors suggest that the decrease in driving force for the first intramolecular ET step is also due to an increased E°′ of the first TNC intermediate in fungal laccase. This study demonstrates the importance of not only considering the E°′ of the T1Cu center but also its redox relationship with the TNC. This work also suggests a remedy for the inactivity observed in the engineered high E°′ T1Cu MCOs through E°′ tuning of not only the T1Cu center but also considering E°′ tuning of the TNC.
Heme-copper oxidases (HCO) also perform ORR with earth abundant metals and high catalytic efficiency. HCOs are a superfamily of terminal oxygen reductases in the respiratory chain. As the name suggests, HCO contains a heme-copper binuclear center (BNC) consisting of a Cu-His3 moiety (CuB), a five-coordinate heme and a tyrosine residue covalently linked to one of the His ligands (Figure 2a). Upon binding and cleavage of O2, the BNC is fully oxidized to form a ferryl species and a tyrosyl radical (Figure 2b). Electrons are delivered to the buried BNC by multiple metal cofactors. The CuA center in cytochrome oxidase (COX), a major class of HCO, mediates electron transfer from its redox partner reduced cytochrome c to the heme a site before ultimately reaching the BNC. At the end of electron transfer, the tyrosyl radical in the BNC receives the first of the four electrons, followed by intramolecular ET between the Cu(II) and heme, to complete the ORR cycle.
Figure 2.
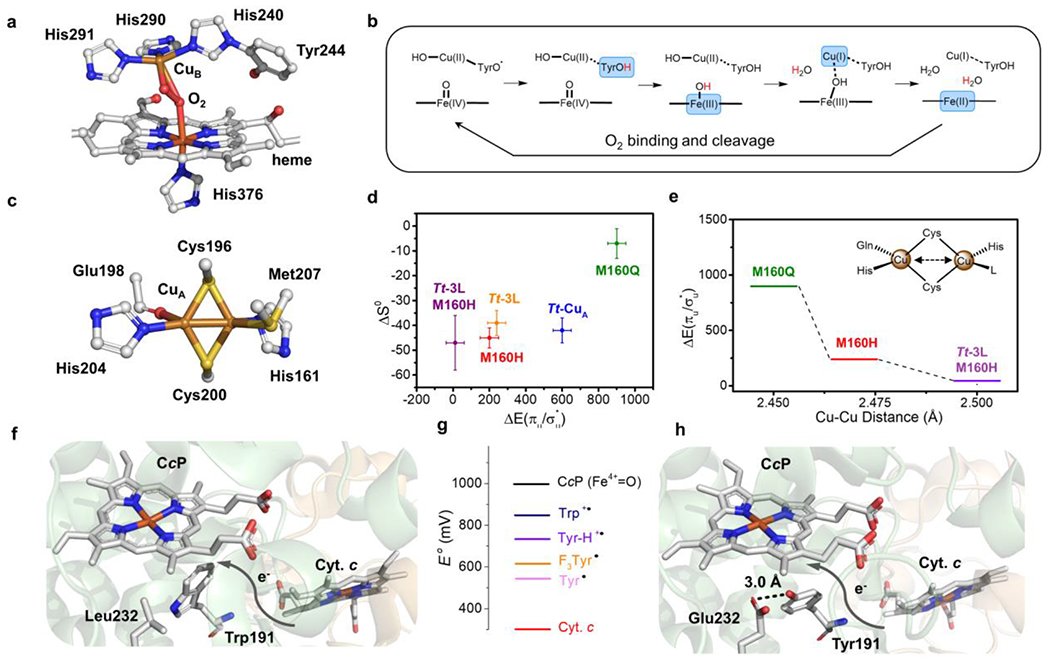
(a) Heme-CuB site (BNC) of bovine heart HCO (PDB 2OCC) with O2 bound. (b) Proposed mechanism for HCO which shows the four-electron reduction from fully oxidized BNC to the rest state. Each arrow on the top represents an one-proton-one-electron reduction. The reduced moieties in each step are showed in blue blocks and the coupled protons are labelled in red. (c) Active site structure of Tt-CuA (PDB 2CUA). (d) Scatter plot of the Cu(II)-Cu(I) entropy change (ΔS0, y-axis) of Tt-CuA mutants versus the energy gap between two thermally accessible ground states (ΔEσu*/πu), x-axis). (e) Change of (ΔEσu*/πu) (y-axis) with different Cu-Cu distances in Tt-CuA mutants. (f) Structural representation of wild-type yeast CcP (co-crystallized with Cyt. c, PDB 2PCC). (g) Scheme of reduction potentials of different amino acid residue radicals and iron species. (h) Structural representation of W191Y/L232E CcP (PDB 6P41). Die hydrogen bond between Glu232 and Tyr191 is shown as a black dash.
Similar to MCOs, there has been extensive research on modulating the E°′ of the ET centers in HCOs. Due to the complexity of HCO, these sites were mostly studied in model proteins. For example, the CuA site found in COX has been incorporated into various homologous β-barrel cupredoxins31. de novo designed α -helical peptides32 and recently into an α-helical cytochrome c peroxidase (CcP) scaffold.33 The CuA site is a binuclear Cu center, where the two copper ions are bridged by two S(Cys) ligands and each Cu is coordinated by one histidine residue (Figure 2c). This PCS configuration is conserved among most native CuA proteins In typical CuA centers, there is one axial methionine weakly bound to one copper and one backbone carbonyl bound to the other copper.34 A rare type of CuA was recently discovered with the carbonyl replaced by another methionine.35 The electronic structure of the oxidized CuA has been extensively studied and is best described as two thermally accessible ground states σu and πu of different orbital symmetry arising from the Cu2S2 core.36 In the CuA site, the σu is at slightly lower energy than the πu state.37 The small energy gap (ΔEσu/πu) has been associated with differences in Cu-Cu distances, where the σu is associated with shorter Cu-Cu distances and πu is associated with longer Cu-Cu distances.
Previous studies have shown that both the E°′ and ΔEσu*/πu could be tuned through axial Met mutations, where a mutation of the axial Met to a Leu or Ile resulted in the largest increase of ~120 mV/38–40 Recently, Murgida and co-workers expanded on this work to understand the correlation between structural changes at the CuA center and ΔEσu*/πu. The authors used axial Met mutations and loop-replacement to generate variants with ΔEσu*/πu ranging from 900 to 13 cm−1. Thus, demonstrating the combined effects of PCS and SCS tuning to produce a spectrum of variants ranging from purely σu* ground state to equal populations of both σu* and πu ground states. The Thermus thermophilus (Tt) Met160His variant which exhibited near degenerate σu* and πu states was further charactered by Extended X-Ray Absorption Fine Structure (EXAFS) to understand the connection between ΔEσu*/πu and the geometric changes at the CuA in particular the Cu-Cu distance.41 Along with its change in electronic structure, this variant exhibited a decrease in E°′ by 182 mV relative to the wild-type. However, structural characterization by EXAFS only resulted in a mere 0.06 Å change in Cu-Cu bond. Therefore, substantial changes in the geometric structure of the site are not required to generate dramatic changes in electronic structure nor in E°′. The authors’ work suggests that minor structural changes to the CuA site such as those arising after a redox protein partner binds could result in major changes in electronic structure and E°′. The binding of a redox partner near the CuA center could favor one ground state over another, suggesting a way that biology could control ET through allosteric binding.38,42 Therefore, these studies demonstrate that even minimal changes in PCS of CuA has a significant impact on the electronic structure, including redox potential, and thereby effects the electron transfer properties.
Another important ET site in HCO’s is a conserved tyrosine residue. As shown in Figure 2b,43–45 the tyrosyl radical of the BNC forms upon O2 bond cleavage and is first reduced by the electrons transferred by the CuA center.46 Interestingly, another type of terminal oxidase, known as cytochrome bd oxidase, has a tryptophan instead of tyrosine in proximity of the oxygen reduction site.47 Therefore it renders the question as to whether tyrosine and tryptophan are functionally interchangeable for ORR. More importantly, DFT calculation on the HCO active site implies that the protonation state of tyrosine is important for the ORR cycle. The residue must be protonated before O2 binding to form an activated intermediate and maintain the function of HCO.46
These two questions were addressed by Crane and co-workers in a recent study on yeast cytochrome c peroxidase (CcP), an enzyme accepting electron from cytochrome c to reduce the peroxide substrates. 48 The electron transfer between the two heme cofactors of cytochrome c and CcP is mediated by the Trp191 residue (Figure 3f). Replacing Trp191 with tyrosine (W191Y) effectively unpaired the electron transfer from cytochrome c to the active site heme of CcP, where the deprotonated tyrosyl radical was found to have a significantly lower reduction potential (E°′) than tryptophan and lacks the driving force to accept electrons from cytochrome c. Introduction of a hydrogen bond donor of either His232 or Glu232 to Tyr191 (Figure 2h) resulted in an increase of the protonated tyrosyl radical E°′ by 200 mV. The tuning effect made the E°′ of the tyrosyl radical comparable to the native tryptophan (Figure 2g), and thereby rescued the electron transfer at a 30-fold greater rate. Another mutation to a fluorinated unnatural tyrosine (F3Y191) showed a higher electron transfer rate than the deprotonated Tyr191 mutant in accordance with its higher E°′. However, due to the higher acidity, F3Y191 formed a weaker hydrogen bond with the Glu232 residue, which caused slower electron transfer than the combination of Tyr191 and Glu232. This study suggests that well-positioned hydrogen bonding can significantly affect the reduction potential of tyrosine that enable it to replace tryptophan in the electron transfer pathway. This opens up new strategies to tune the reduction potential of tyrosyl radicals and furthermore the activities of ORR enzymes employing aromatic residues for electron transfer 49
Figure 3.
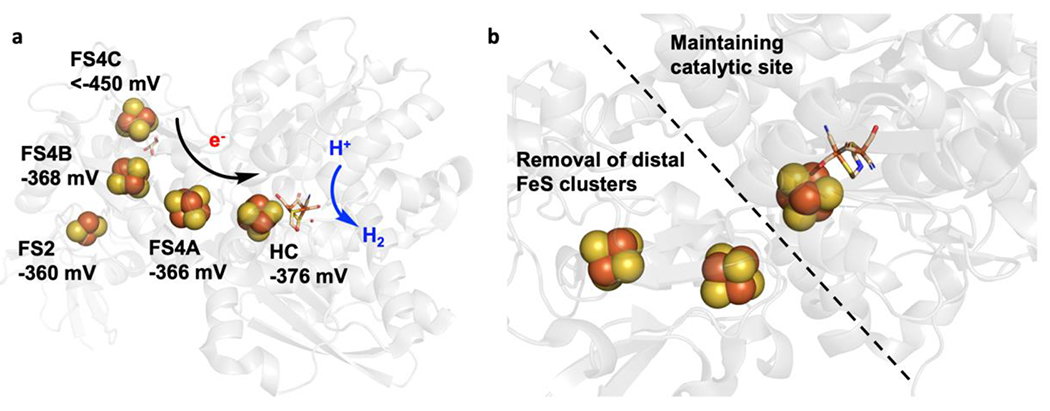
(a) The active site and accessory [FeS] cluster array in CPI [FeFe] hydrogenase (PDB 3C8Y). The reduction potential of each [FeS] cluster was determined and labeled. The proposed electron transfer array from distal [4Fe-4S] cluster to the active site was depicted favoring proton reduction. (b) [FeFe] Hydrogenase from Megasphaera elsdenii (MeHydA) and its N-terminal truncated form (MeH-HydA) which keeps the catalytic site but lacks the distal accessory [4Fe-4S] cluster.
Hydrogenases
Hydrogenases are enzymes that perform the reversible proton reduction/H2 oxidation reaction using nickel and iron and are comparable in efficiency to fuel cell Pt electrodes.9,50,51 Hydrogenases can host either an [FeFe] or [NiFe] cluster active site along with an array of accessory iron-sulfur clusters for fast intramolecular ET between the buried active site and the surface of the protein.52,53 The catalytic bias or inherent preference of H2 oxidation or H2 production varies across different hydrogenases. Typically [NiFe] hydrogenases perform H2 oxidation and are O2 tolerant, thus making these enzymes more applicable for fuel cell catalysis, whereas the [FeFe] hydrogenases typically perform H2 production. Recently, modifications in the iron-sulfur (FeS) clusters of the ET chain of a [NiFe] hydrogenase biased the reactivity towards H2 production rather than H2 oxidation. Therefore, extensive research has focused on understanding the contributions of the intramolecular ET pathway on biasing catalysis given that there is variation in the composition of iron-sulfur clusters throughout different hydrogenases.9,54
Iron-sulfur (FeS) containing proteins offer a wide range of reduction potentials (E°′) for numerous functions in biology. Typically, these clusters are coordinated to the Fe atom through cysteine thiolate ligation in varying numbers of Fe or S atoms to generate either [2Fe2S], [3Fe4S] or [4Fe4S] clusters.55 The ET chain of hydrogenase contain different combinations of these clusters, where the [NiFe] hydrogenases typically contain two [4Fe4S] clusters and one [3Fe4S] cluster and [FeFe] hydrogenases contain two [4Fe4S] and two [2Fe2S] clusters.56 The E°′ of these FeS clusters can be tuned through mutations of the coordinating Cys residues and through modulation of H-bonding interactions to the (S)Cys of the FeS cluster. The correlation between the variation in FeS cluster chains of these hydrogenases and the catalytic bias of the hydrogenase’s reactivity have alluded many researchers due to the challenges of overlapping spectroscopic signals from multiple metal cofactors. Recently, Peters and coworkers employed an [FeFe]-hydrogenase from Clostridium pasteurianum (CpI) as a model system to study the functions of the accessory FeS clusters on biasing proton reduction catalysis.57 CpI [FeFe]-hydrogenase contains a total of four accessory [FeS] clusters called F-clusters which transfer electrons to the [FeFe] active site (Figure 3a).58 The active site of [FeFe] hydrogenase is the H-cluster which consists of a [4Fe4S] cluster covalently attached to a bridging cysteine thiolate to the [2Fe] subcluster ([2Fe]H). Each Fe in the [2Fe] subcluster is bound to a CN− and a CO ligand and the two metal centers are bridged by an azadithiolate (ADT) ligand and a CO ligand. Replacement of the native active site ligand with the synthetic analogue propanedithiolate (PDT) suppresses proton reduction, which allowed the authors to characterize the FeS ET chain at different potentials. Electron Paramagnetic Resonance (EPR) spectroscopy was used to measure the reduction potential of the four accessory [FeS] clusters. The E°′ of distal or the [4Fe4S] cluster closest to the protein surface was determined to be the lowest (~ < −450 mV) with others having similar reduction potentials of about −360 mV, which makes the subsequent electron transfer through the additional accessory [FeS] clusters to the active site H-cluster thermodynamically favorable. The lower reduction potential exhibited by the distal [4Fe4S] cluster is suggested to arise from its coordination environment where the cluster is coordinated to three Cys and one His residue rather than the typically observed four-Cys coordination. This E array and ET flow of [FeS] thermodynamically biases the reaction direction to proton reduction over H2 oxidation.
Although the fuel cell electrode reaction is H2 oxidation instead of proton reduction, nature’s strategy of using distal [4Fe4S] cluster to bias the reaction direction could guide the design of fuel cell electrodes for H2 oxidation that could prevent the reverse reaction or further favor H2 oxidation. The importance of the distal [4Fe4S] cluster in biasing the reaction direction was further supported by Fontecave and coworkers’ work on [FeFe]-hydrogenase from Megasphaera elsdenii (MeHydA)59 where a N-terminal truncated enzyme (MeH-HydA) was generated that lacks the distal accessory [4Fe4S] cluster (Figure 3b). The removal of this distal [4Fe4S] cluster resulted in an increased bias toward proton reduction than what was observed in the native MeHydA. As the catalytic bias is the ratio between the maximal rates of the forward and reverse reactions, the difference in bias between the holo and truncated enzyme relates to differences in the rate determining step (RDS) when the accessory clusters are removed. In the holo enzyme, the slow intramolecular electron transfer between [FeS] cluster and the active site determines the catalytic rates in both directions (i.e., H2 production/H2 oxidation). Removal of the accessory [FeS] clusters change the RDS to a chemical step at the active site and thus biases the reaction towards purely H2 production rather than the reversible reaction observed in the holo enzyme. If the RDS is not based on the active site chemistry but rather ET, then the accessory [FeS] clusters dictate the catalytic direction. Therefore, these results highlight the importance of understanding the mechanism in order to tune the bias towards H2 oxidation and if the RDS is ET based, through engineering [FeS] clusters.
Conclusions
This review highlights the recent advances in understanding the factors effecting both the E°′ of the ET sites and the reactivity of the H2 oxidation/production or ORR. These ET centers not only are important for efficient ET but also can bias the reactivity and/or determine the catalytic potential. Therefore, attention must also be drawn to these ET centers when looking towards biofuel design. Protein engineering in simple model systems have widened our knowledge on how both the PCS and the SCS interact with these ET sites to modulate its E°′60,61 Dovetailing the knowledge gained through these model systems with more complex enzymatic systems will illuminate not only the factors that contribute to a higher E°′ in the ET center but also how these principles apply to the enzymes activity. Merging these E°′ tuning factors gathered in model systems with the cofactors of more complex systems is the next step in engineering fuel cell catalysts with lower overpotentials and higher catalytic rates.
Acknowledgement
The Lu group research described in this review has been supported by the US National Science Foundation (under award CHE 17-10241), National Institute of Health (under award GM06211), as well as by the Chemical Transformation Initiative at Pacific Northwest National Laboratory (PNNL), conducted under the Laboratory Directed Research and Development Program at PNNL, a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest Statement
The authors declare no conflict of interests to the subjective matters written in this review.
References
- 1.Cracknell JA, Vincent KA & Armstrong FA Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 108, 2439–2461 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Gewirth AA & Thorum MS Electroreduction of dioxygen for fuel-cell applications: Materials and challenges, Inorg. Chem 49, 3557–3566 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Wang X et al. Review of Metal Catalysts for Oxygen Reduction Reaction: From Nanoscale Engineering to Atomic Design. Chem. vol.51486–1511 (2019). [Google Scholar]
- 4.Bullen RA, Arnot TC, Lakeman JB & Walsh FC Biofuel cells and their development. Biosens. Bioelectron. 21, 2015–2045 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Shleev S et al. Direct electron transfer reactions of laccases from different origins on carbon electrodes. Bioelectrochemistry 67, 115–124 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Dagys M et al. Oxygen electroreduction catalysed by laccase wired to gold nanoparticles via the trinuclear copper cluster. Energy Environ. Sci. 10, 498–502 (2017). [Google Scholar]
- 7.Liu J et al. Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers. Chemical Reviews vol. 114 4366–4369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solomon EI, Sundaram UM & Machonkin TE Multicopper Oxidases and Oxygenases. (1996) doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 9.Vincent KA, Parkin A & Armstrong FA Investigating and exploiting the electrocatalytic properties of hydrogenases. Chemical Reviews vol. 107 4366–4413 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Chumillas S, Maestro B, Feliu JM & Climent V Comprehensive study of the enzymatic catalysis of the electrochemical Oxygen Reduction Reaction (ORR) by Immobilized Copper efflux oxidase (CueO) from Escherichia coli. Front. Chem 6, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giardina P et al. Laccases: A never-ending story. Cell. Mol. Life Sci. 67, 369–385 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodgers CJ et al. Designer laccases: a vogue for high-potential fungal enzymes? Trends Biotechnol. 28, 63–72 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Vite-Vallejo O et al. The role of N-glycosylation on the enzymatic activity of a Pycnoporus sanguineus laccase. Enzyme Microb. Technol. 45, 233–239 (2009). [Google Scholar]
- 14.Hong G, Ivnitski DM, Johnson GR, Atanassov P & Pachter R Design parameters for tuning the type 1 Cu multicopper oxidase redox potential: Insight from a combination of first principles and empirical molecular dynamics simulations. J. Am. Chem. Soc. 133, 4802–4809 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Mateljak I et al. Increasing Redox Potential, Redox Mediator Activity, and Stability in a Fungal Laccase by Computer-Guided Mutagenesis and Directed Evolution. ACS Catal. 9, 4561–4572 (2019). [Google Scholar]
- 16.Warren JJ, Lancaster KM, Richards JH & Gray HB Inner- and outer-sphere metal coordination in blue copper proteins ☆. J. Inorg. Biochem. 115, 119–126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lancaster KM Biological outer-sphere coordination. Struct. Bond. 142, 119–154 (2012). [Google Scholar]
- 18.Solomon EI, Szilagyi RK, DeBeer George S & Basumallick L Electronic Structures of Metal Sites in Proteins and Models: Contributions to Function in Blue Copper Proteins. Chem. Rev. 104,419–458 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Xu F et al. Targeted mutations in a Trametes villosa laccase: Axial perturbations of the T1 copper. J. Biol. Chem. 274, 12372–12375 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Pardo I & Camarero S Laccase engineering by rational and evolutionary design. Cell. Mol. Life Sci. 72, 897–910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berry SM, Baker MH & Reardon NJ Reduction potential variations in azurin through secondary coordination sphere phenylalanine incorporations. J. Inorg. Biochem. 104, 1071–1078 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Hadt RG et al. Spectroscopic and DFT studies of second-sphere variants of the type 1 copper site in azurin: Covalent and nonlocal electrostatic contributions to reduction potentials. J. Am. Chem. Soc. 134, 16701–16716 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Machczynski MC, Gray HB & Richards JH An outer-sphere hydrogen-bond network constrains copper coordination in blue proteins. 88, 375–380 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Warren JJ, Lancaster KM, Richards JH & Gray HB Inner- and outer-sphere metal coordination in blue copper proteins. J. Inorg. Biochem. 115, 119–126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanagisawa S, Banfield MJ & Dennison C The role of hydrogen bonding at the active site of a cupredoxin: The Phe114Pro azurin variant. Biochemistry 45, 8812–8822 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Kamitaka Y et al. Effects of axial ligand mutation of the type I copper site in bilirubin oxidase on direct electron transfer-type bioelectrocatalytic reduction of dioxygen. J. Electroanal. Chem. 601, 119–124 (2007). [Google Scholar]
- *27.Glazunova OA, Polyakov KM, Moiseenko KV, Kurzeev SA & Fedorova TV Structure-function study of two new middle-redox potential laccases from basidiomycetes Antrodiella faginea and Steccherinum murashkinskyi. Int. J. Biol. Macromol. 118, 406–418 (2018). [DOI] [PubMed] [Google Scholar]; The authors compared the structure around the T1Cu center of two middle redox potential laccases with other high and low potential laccases. The authors also correlated the total solvent accessible surface area of the conserved regions in the T1Cu center with the reduction potential of the T1Cu.
- 28.Sakurai T & Kataoka K Structure and function of type I copper in multicopper oxidases. Cell. Mol. Life Sci. 64, 2642–2656 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kajikawa T, Kataoka K & Sakurai T Modifications on the hydrogen bond network by mutations of Escherichia coli copper efflux oxidase affect the process of proton transfer to dioxygen leading to alterations of enzymatic activities. Biochem. Biophys. Res. Commun. 422, 152–156(2012). [DOI] [PubMed] [Google Scholar]
- *30.Sekretaryova A, Jones SM & Solomon EI O2 Reduction to Water by High Potential Multicopper Oxidases: Contributions of the T1 Copper Site Potential and the Local Environment of the Trinuclear Copper Cluster. J. Am. Chem. Soc. 141, 11304–11314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors compare the ORR mechanism of a high potential laccase with a low reduction potenial laccase and demonstrate that both display similar mechanisms of ORR but differ in the rate determining step of the ORR reaction.
- 31.Wilson TD, Yu Y & Lu Y Understanding copper-thiolate containing electron transfer centers by incorporation of unnatural amino acids and the Cu A center into the type 1 copper protein azurin. Coordination Chemistry Reviews vol. 257 260–276 (2013). [Google Scholar]
- 32.Shiga D et al. Creation of a binuclear purple copper site within a de novo coiled-coil protein. Biochemistry 51, 7901–7907 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Mirts EN, Dikanov SA, Jose A, Solomon EI & Lu Y A Binuclear CuACenter Designed in an All α-Helical Protein Scaffold. J. Am. Chem. Soc. 142, 13779–13794 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solomon EI et al. Copper Active Sites in Biology. Chem. Rev. 114, 3659–3853 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross MO et al. Formation and Electronic Structure of an Atypical CuA Site. J. Am. Chem. Soc. 141,4678–4686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abriata LA, Ledesma GN, Pierattelli R & Vila AJ Electronic structure of the ground and excited states of the CuA site by NMR spectroscopy. J. Am. Chem. Soc. 131, 1939–1945 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorelsky SI, Xie X, Chen Y, Fee JA & Solomon EI The two-state issue in the mixed-valence binuclear CuA center in cytochrome c oxidase and N2O reductase. J. Am. Chem. Soc. 128, 16452–16453 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Abriata LA et al. Alternative ground states enable pathway switching in biological electron transfer. 109, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ledesma GN et al. The met axial ligand determines the redox potential in CuA sites. J. Am. Chem. Soc. 129, 11884–11885 (2007). [DOI] [PubMed] [Google Scholar]
- *40.Alvarez-Paggi D et al. Tuning of Enthalpic/Entropic Parameters of a Protein Redox Center through Manipulation of the Electronic Partition Function. J. Am. Chem. Soc. 139, 9803–9806 (2017). [DOI] [PubMed] [Google Scholar]; The reduction potentials of CuA in different PCS were measured and analyzed by enthalpic/entropic contributions, which were correlated with ligand hydrophobicity and ground state electronic structure changes, respectively.
- *41.Leguto AJ et al. Dramatic Electronic Perturbations of Cu A Centers via Subtle Geometric Changes. J. Am. Chem. Soc. 141, 1373–1381 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors measured the energy gap between two accessible ground states of different CuA mutants and validated that the tuning of CuA potential originated from a small perturbation of Cu-Cu distance by 0.06 Å.
- 42.Leguto AJ et al. Dramatic Electronic Perturbations of CuA Centers via Subtle Geometric Changes. J. Am. Chem. Soc. 141, 1373–1381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams PA et al. The Cu(A) domain of Thermus thermophilus ba3-type cytochrome C oxidase at 1.6 Å resolution. Nat. Struct. Biol. 6, 509–516 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Yoshikawa S et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science (80-. ). 280, 1723–1729 (1998). [DOI] [PubMed] [Google Scholar]
- 45.Pelletier H & Kraut J Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science (80-. ). 258, 1748–1755 (1992). [DOI] [PubMed] [Google Scholar]
- 46.Blomberg MRA Mechanism of Oxygen Reduction in Cytochrome c Oxidase and the Role of the Active Site Tyrosine. Biochemistry 55, 489–500 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Safarian S et al. Structure of a bd oxidase indicates similar mechanisms for membraneintegrated oxygen reductases. Science (80-. ). 352, 583–586 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- *48.Yee EF, Dzikovski B & Crane BR Tuning Radical Relay Residues by Proton Management Rescues Protein Electron Hopping. J. Am. Chem. Soc. 141, 17571–17587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; The electron transfer to active site in CcP was impaired by substituting Trp191 with Tyr, and was restored by introducing electron donor Glu232 to protonate the Tyr191 radical and significantly increase its potential.
- 49.Yu Y et al. Significant improvement of oxidase activity through the genetic incorporation of a redox-active unnatural amino acid. Chem. Sci. 6, 3881–3885 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones AK, Sillery E, Albracht SPJ & Armstrong FA Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum catalyst. Chem. Commun. 2, 866–867 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Armstrong FA & Hirst J Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc. Natl. Acad. Sci. 108, 14049–14054 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rumpel S et al. Enhancing hydrogen production of microalgae by redirecting electrons from photosystem i to hydrogenase. Energy Environ. Sci. 7, 3296–3301 (2014). [Google Scholar]
- 53.Mulder DW et al. Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure vol. 19 1038–1052 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Abou Hamdan A et al. Understanding and Tuning the Catalytic Bias of Hydrogenase. J. Am. Chem. Soc 134, (2012). [DOI] [PubMed] [Google Scholar]
- 55.Johnson DC, Dean DR, Smith AD & Johnson MK Structure, function, and formation of biological iron-sulfur clusters. Annual Review of Biochemistry vol. 74 247–281 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Lubitz W, Ogata H, Rü O & Reijerse E Hydrogenases. (2014) doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- *57.Artz JH et al. Reduction Potentials of [FeFe]-Hydrogenase Accessory Iron-Sulfur Clusters Provide Insights into the Energetics of Proton Reduction Catalysis. J. Am. Chem. Soc. 139, 9544–9550(2017). [DOI] [PubMed] [Google Scholar]; Four accessory [Fe4-S4] in an [FeFe]-hydrogenase from Clostridium pasteurianum (CpI) forms electron array to faciliate electron transfer through the additional accessory [FeS] clusters to the active site H-cluster favorable, thus biases the reaction direction to proton reduction over H2 oxidation.
- 58.Peters JW X-ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom Resolution. Science (80-.). 282, 1853–1858 (1998). [DOI] [PubMed] [Google Scholar]
- *59.Caserta G et al. Engineering an [FeFe]-Hydrogenase: Do Accessory Clusters Influence O2 Resistance and Catalytic Bias? J. Am. Chem. Soc. 140, 5516–5526 (2018). [DOI] [PubMed] [Google Scholar]; Truncated Hydrogenase (MeH-HydA), lacking the N-terminal domain with distal accessory [4Fe-4S] cluster, has significant effect on catalysis to show lower reactivity compared to the native form MeHydA, but more biased toward proton reduction.
- 60.Marshall NM et al. Rationally tuning the reduction potential of a single cupredoxin beyond the natural range. Nature 462, 113–116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hosseinzadeh P et al. Design of a single protein that spans the entire 2-V range of physiological redox potentials. Proc. Natl. Acad. Sci. 113, 262 LP – 267 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]