

Abstract
Termination of antidepressant therapy often has negative consequences. Although symptoms of antidepressant withdrawal are widely recognized, the molecular processes that underlie them are not well characterized. We show that certain aspects of Gαs signaling remain suppressed after antidepressant withdrawal, even after others have reverted to baseline. Antidepressant treatment causes translocation of Gαs protein from lipid rafts to nonraft membrane regions. This results in augmented Gαs signaling, including facilitated activation of adenylyl cyclase and increased cAMP accumulation. Using CC6 or SK-N-SH cells and a lipid raft–localized cAMP sensor, we show that Gαs signaling is reduced in lipid rafts, even while signaling is enhanced elsewhere in the cell. These signaling changes mirror the changes in Gαs localization observed after antidepressant treatment. Furthermore, we show that suppression of Gαs signaling in lipid rafts persists at least 24 hours after cessation of antidepressant treatment. Gαs localization was quantified after membrane isolation and sequential detergent extraction. We show that suppression of lipid raft Gαs signaling persists for an extended time period after antidepressant withdrawal, whereas increased nonraft membrane Gαs signaling reverts partially or fully upon cessation of antidepressant treatment. Translocation of Gαs out of lipid rafts is also persistent. These events may reflect cellular adaptations to antidepressant treatment that contribute to antidepressant discontinuation syndromes and may aid in the discovery of new treatments and strategies to mitigate the symptoms of depression and antidepressant withdrawal.
SIGNIFICANCE STATEMENT
This work explores, for the first time, the effects of antidepressants on Gαs signaling after drug withdrawal. This provides novel insight into the cellular and molecular processes affected by antidepressant drugs and their persistence after discontinuation of treatment.
Introduction
Major depressive disorder is currently the leading cause of disability worldwide, and its impact is expected to continue growing (World Health Organization, 2017). No single treatment is fully effective in all people, with as many as two out of three individuals failing to remit after initial treatment (Rush et al., 2006a). Follow-up treatments are also often ineffective, with many patients failing to remit even after second- and third-line treatment options have been exhausted (Insel and Wang 2009). These failures are exacerbated by the long delay between initiation of treatment and subsequent antidepressant response, with some drugs taking 8 weeks before effects can be evaluated (Rush et al., 2006b). This means many individuals will undergo months or even years of failed therapy before finding relief or will drop out of treatment altogether (Sharma et al., 2019). These challenges necessitate a deeper understanding of the processes leading to a positive antidepressant response and the specific factors that distinguish individuals who will not respond to antidepressant treatment.
After cessation of long-term antidepressant treatment, a constellation of symptoms known as antidepressant discontinuation syndrome can occur (Gabriel and Sharma, 2017). These symptoms may include sleep disturbances, anxiety, flu-like symptoms, and sensory abnormalities, including electric shock–like experiences (Baldwin et al., 2006). Specific symptoms depend on the drug used and vary between individuals (Fava et al., 2015). Tricyclic antidepressants such as desipramine tend to produce more severe symptoms, including akathisia and parkinsonian reactions (Charney et al., 1982; Garner et al., 1993; Haddad, 2001). Nonetheless, antidepressant drugs with diverse primary mechanisms of action, including the serotonin and norepinephrine reuptake inhibitor venlafaxine, the selective serotonin reuptake inhibitor paroxetine, and the monoamine oxidase inhibitor phenelzine, among others, can all produce an antidepressant withdrawal syndrome (Ogle and Akkerman, 2013). Symptom severity tends to increase with longer treatment duration and higher dosage (Haddad, 1997; Warner et al., 2006). Antidepressant discontinuation syndromes can be debilitating and last for over 2 weeks, and not all patients are fully informed of this risk before treatment initiation (Bull et al., 2002; Warner et al., 2006). Although there has been some research into the mechanistic basis of this syndrome, its etiology remains poorly understood (Blier and Tremblay, 2006; Murata et al., 2010; Zabegalov et al., 2018). A better understanding of the residual effects that persist after antidepressant withdrawal is sorely needed and will facilitate improved patient care after antidepressant treatment.
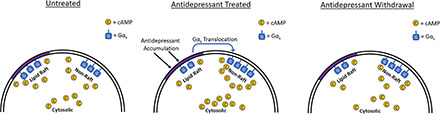
Lipid rafts are a subcellular membrane microdomain high in cholesterol content and with distinct signaling characteristics compared with other membrane regions (Simons and Toomre, 2000; Allen et al., 2007). A wide variety of antidepressant drugs are known to cause translocation of Gαs out of lipid rafts (Senese et al., 2018). This action is specific to Gαs, as other Gα proteins are not similarly affected (Donati and Rasenick, 2005). Antidepressants with distinct primary targets cause these effects and can do so even in model systems lacking that target. For example, the selective serotonin reuptake inhibitor (SSRI) escitalopram causes Gαs translocation in cells lacking the serotonin transporter, and the rapid-acting antidepressant ketamine causes translocation even after N-methyl-d-aspartate receptor knockdown (Eisensamer et al., 2005; Wray et al., 2018). In the case of escitalopram, this effect is stereospecific, as the inactive stereoisomer r-citalopram fails to affect Gαs localization or signaling (Czysz et al., 2015).
This translocation produces a distinct change in Gαs signaling, notably increased coupling to adenylyl cyclase, and resultant potentiation of downstream signaling (Chen and Rasenick 1995a,b). Potentiation of cAMP generation by antidepressants has been observed in both preclinical and clinical studies (Mooney et al., 2013; Singh et al., 2018). These changes likely represent cellular adaptations to extended antidepressant treatment. Multiday treatments are required before these effects manifest, and these changes are still observed at least 30 minutes after antidepressants have been withdrawn (Zhang and Rasenick, 2010). These signaling changes may provide novel insight into the cellular adaptations occurring in response to antidepressants with diverse mechanisms of action.
The cAMP signaling cascade has been linked to depression and antidepressant action in various contexts (Dwivedi and Pandey, 2008). Suicide completers have reduced central adenylyl cyclase activity, cAMP, and downstream signaling via protein kinase A (Cowburn et al., 1994; Pandey et al., 2005; Fujita et al., 2017). Furthermore, in post-mortem cortical tissue from individuals diagnosed with unipolar depression, Gαs is localized in microenvironments with impaired adenylyl cyclase coupling (Donati et al., 2008). Conversely, 8 weeks of treatment with an SSRI restored cAMP generation in the brains of subjects with depression (Fujita et al., 2017).
We sought to determine, in a cellular model, whether previously observed antidepressant effects on Gαs signaling and translocation persist after drug withdrawal. Furthermore, we differentiated antidepressant-induced signaling changes in lipid rafts and nonraft regions using fluorescent cAMP sensors. The lipid raft–targeted cAMP sensor is expressed specifically in rafts because of the addition of a dually myristoylated/palmitoylated peptide sequence. This sequence restricts fluorophore expression to high-density TX-100 insoluble regions (i.e., lipid rafts) and engenders significant caveolin colocalization (Zacharias et al., 2002). This raft localization is reduced by the raft disruptor methyl-β-cyclodextrin (Zacharias et al., 2002), confirming the lipid raft specificity of this targeting sequence. We determined that cellular hallmarks of antidepressant action, including translocation of Gαs from lipid rafts, persist after drug withdrawal in this model system. Further, we demonstrated that in contrast to antidepressant-induced increases in whole-cell cAMP, lipid raft cAMP signaling was suppressed after antidepressant withdrawal. The ability to model antidepressant discontinuation in vitro may facilitate the development of effective antidepressant compounds without the risk of discontinuation syndromes.
Materials and Methods
Cell Culture
HEK-293 and C6 cells were obtained from the American Type Culture Collection (VA). SK-N-SH cells were a generous donation from the laboratory of Dr. Ankur Saxena at University of Illinois at Chicago. HEK-293 and C6 cells were cultured in DMEM with 4.5 g/l glucose and l-glutamine without sodium pyruvate (Corning, NY) supplemented with 10% Gibco newborn calf serum (Thermo Fisher Scientific, MA). SK-N-SH cells were cultured in Minimum Essential Media with Earle’s salts and l-glutamine (Corning, NY) supplemented with 10% heat-inactivated fetal bovine serum (Corning, NY).
Drug Treatments
Stock solutions (10 mM) of all drugs were made as follows. Phenelzine sulfate (Sigma-Aldrich, MO), fluoxetine HCl (Sigma-Aldrich, MO), venlafaxine HCl (Sigma-Aldrich, MO), escitalopram oxalate (Sigma-Aldrich, MO), and desipramine HCl (Santa Cruz Biotechnology Inc., TX) were all dissolved in dd H2O. r-Citalopram oxalate was a generous gift from Lundbeck (Denmark) and was dissolved in ddH2O. Because of the low aqueous solubility, paroxetine HCl (Sigma-Aldrich, MO) and MDL 12330A (Tocris Bioscience, UK) were dissolved in DMSO. Ketamine HCl 100 mg/ml solution (Hospira Inc., IL) was diluted with ddH2O. Drugs were diluted from 10 mM to 10× final assay concentration in appropriate culture media for each cell type.
All antidepressants were administered for 3 days, except for ketamine, which requires only a 15-minute treatment (Wray et al., 2018). Antidepressants were removed from culture flasks or plates either 30 minutes before start of experiment or 24 hours before start of experiment, as indicated. An antidepressant is defined as having a full reversal if the measured effect was significantly different between 3 days of treatment and 24 hours after withdrawal but not different from vehicle-treated cells. A partial reversal is designated when 24-hour withdrawal is not different from either 3 days of treatment or vehicle treatment.
“No reversal” indicates the 24-hour withdrawal effect remained significantly different from vehicle-treated cells but not different from the 3-day treatment.
Lipid Raft Extraction
After drug treatment, two T150 culture flasks (Thermo Fisher Scientific, MA) containing approximately 25 million cells total were washed twice with TME buffer (10 mM Tris-HCl, 1 mM MgCl, 1 mM EDTA, pH 7.5) to remove residual media, scraped in TME buffer containing protease inhibitor cocktail II (MilliporeSigma, MA), and homogenized using a glass Dounce and a benchtop drill. Homogenized cells sat on ice for 30 minutes.
Homogenized cells were centrifuged at 60,000g for 60 minutes at 4°C. After aspirating supernatant, each pellet was resuspended in 0.5 ml of TME containing protease inhibitor and 1.0% TX-100 (Thermo Fisher Scientific, MA) and again homogenized. Homogenized samples sat on ice for 30 minutes.
Samples were again centrifuged at 60,000g for 60 minutes at 4°C, generating the TX-100 soluble fraction in the supernatant (nonraft membrane fraction). After aspirating supernatant, each pellet was resuspended in 0.5 ml of TME containing protease inhibitor and 1.4% Triton X-114 (TX-114; Thermo Fisher Scientific, MA) and again homogenized. Homogenized samples then sat on ice for 30 minutes. Finally, samples were centrifuged at 60,000g for 60 minutes at 4°C, generating the TX-114 soluble fraction in the supernatant (lipid raft membrane fraction).
This protocol was modified for membranes prepared for AlphaScreen cAMP experiments (Fig. 2E). The presence of TX-114 interferes with cAMP measurement using this assay (unpublished observation). Therefore, after extraction of nonraft membranes with TX-100, the TX-100 insoluble pellet containing lipid raft membranes was resuspended in AlphaScreen stimulation buffer. Per 20 ml, this buffer consists of 19.81 ml Hanks’ balanced salt solution, 100 μl HEPES (pH 7.5), 20 μl 500 mM 3-isobutyl-1-methylxanthine, and 66.7 μl 30% bovine serum albumin solution dissolved in ddH2O. This buffer was supplemented with (final concentration) 25 mM MgCl, 375 mM NaCl, 250 μM ATP, 2.5 μM GDP, and 2.5 nM GTP.
Fig. 2.

Desipramine and escitalopram reduce baseline and isoproterenol-stimulated cAMP in lipid rafts. A C6 cell expressing the lipid raft–localized fMP Green Downward cADDis cAMP sensor (A). Three days of treatment of C6 cells with 10 μM desipramine does not affect sensor localization or expression (B). This treatment increases baseline fluorescence in C6 cells (C). Desipramine pretreatment increases baseline and isoproterenol-stimulated fluorescence in lipid rafts (D). Isoproterenol-stimulated (1 μM) cAMP generation after desipramine pretreatment in lipid raft membranes prepared from C6 cells is reduced, as measured by the AlphaScreen assay (E). ****P < 0.0001, compared with top end of vehicle control curve. ***P < 0.001 compared with vehicle baseline fluorescence. #P < 0.05, compared with bottom end of vehicle control curve. ++P < 0.01, compared with vehicle-treated membranes. WCL = Whole Cell Lysate. Veh = Vehicle. Iso = Isoproterenol. Frsk = Forskolin. n = 6–12.
Protein Quantification
Fractions were diluted and normalized for total protein content after determination of protein concentration using the Pierce Bicinchoninic Acid assay kit (Thermo Fisher Scientific, MA). Gαs, Cav-1, and cADDis sensor content were quantified for each sample using the Wes instrument (ProteinSimple, Inc., CA).
Primary antibody for Gαs detection is anti-Gs protein, α-subunit N192/12 (Antibodies Incorporated, CA) diluted 1:300 from starting concentration. Primary antibody for Cav-1 is rabbit anti–caveolin-1 antibody (Abcam, UK) diluted 1:250 from starting concentration. Primary antibody for fMP cADDis cAMP sensor (i.e., lipid raft cAMP sensor) is mNeon tag antibody 53061 (Cell Signaling Technology, MA) diluted 1:50 from starting concentration.
For detection of Gαs and Cav-1, samples were heated to 98°C for 5 minutes immediately before quantification. For detection of the cADDis cAMP sensor, samples were heated to 37°C for 30 minutes, with agitation every 10 minutes to reduce high-molecular-weight aggregates. Cav-1 and cADDis cAMP sensor were quantified from the same samples, and as such, Cav-1 serves as the protein loading control for the cADDis cAMP sensor expression data.
Representative readouts showing protein signal detected with each antibody are provided (Supplemental Figs. 2 and 3). Area under the curve analysis is performed for each peak corresponding to target protein, and these values are normalized to the mean area under the curve (AUC) value for the vehicle-treated control fractions for each run.
cADDis cAMP Assay
cAMP accumulation in C6 cells was determined as described previously, with minor modification (Wray et al., 2018). Briefly, C6 cells were cultured in T75 tissue culture flasks (Thermo Fisher Scientific, MA) such that they would reach ∼80%–90% confluence 24 hours before cAMP measurement. Cells were treated with antidepressant or vehicle in culture flasks, with treatments starting at least 24 hours after plating.
Cells were dissociated from tissue culture flask using Cell-Stripper nonenzymatic cell dissociation reagent (Corning Inc., NY) 24 hours before cAMP measurement and were plated into Costar black-sided clear-bottom sterile tissue culture–treated 96-well plates (Corning Inc., NY). For SK-N-SH and C6 cells, ∼48,000 cells were plated into each well. As HEK-293 cells divide more rapidly, only ∼30,000 cells were plated per well. Immediately after plating cells, 20 μl of baculovirus expressing either the cytoplasmic Green Upward cADDis sensor (Montana Molecular, MT) or the lipid raft–restricted fMP Green Downward cADDis sensor (Montana Molecular, MT) was added to each well. Sodium butyrate (Sigma-Aldrich, MO) was added to each well to promote sensor expression, using a final concentration of 7.5 mM for C6 cells and 2.5 mM for HEK-293 and SK-N-SH cells.
Culture media were replaced with warmed, serum-free Fluorobrite DMEM (Thermo Fisher Scientific, MA) 30 minutes before cAMP measurement to reduce background fluorescence and remove antidepressant. Plate was incubated for 30 minutes at room temperature and protected from light to prevent sensor bleaching. Green fluorescent protein signal intensity was quantified using either a Synergy H4 plate reader, a Synergy Neo2 plate reader (Biotek Instruments Inc., VT), or a SpectraMax i3x plate reader (Molecular Devices, CA). After average baseline fluorescence was determined for each well, either isoproterenol or vehicle was added, and fluorescence intensity was measured again.
For time course experiments, a fluorescence measurement was taken every 30 seconds after isoproterenol addition. For isoproterenol dose-response experiments, fluorescence was quantified 5 minutes after isoproterenol addition in HEK-293 cells and 30 minutes after isoproterenol addition in C6 and SK-N-SH cells, as these time points produced the most robust signal (see time courses in Figs. 3, 5, 7, 8, 9, and 10).
Fig. 3.

Reduction of lipid raft cAMP by desipramine persists for over 24 hours but reverses 72 hours after withdrawal. The increased fluorescence after 3 days of 10 μM desipramine treatment persists 24 hours after desipramine withdrawal in C6 cells (A and D) but returns to untreated levels 3 days after desipramine withdrawal (B). Isoproterenol (1 μM) maximally reduces fluorescence intensity after 3 minutes in C6 cells, and this response is maintained for at least 30 minutes (C and D). The increase in fluorescence after desipramine pretreatment in C6 cells is apparent over the entire 30-minute isoproterenol time course (C), as well as 24 hours after desipramine withdrawal (D). **P < 0.01, compared with top end of vehicle control curve. #P < 0.05, compared with maximal isoproterenol effect in vehicle pretreated wells. n = 6.
Fig. 5.

Desipramine increases maximal isoproterenol-stimulated whole-cell cAMP A C6 cell expressing the cytoplasmic Green Upward cADDis cAMP sensor (A). Baseline fluorescence intensity is not affected by desipramine treatment in C6 cells expressing this sensor (B). Three days of treatment of C6 cells with 10 μM desipramine significantly increases maximal isoproterenol-stimulated cAMP production (C and E). This effect is no longer significantly different from vehicle-treated cells 24 hours after desipramine withdrawal (D and F). Isoproterenol (1 μM) maximally increases fluorescence intensity in C6 cells after 3 minutes, and this response is maintained over the entire 30-minute isoproterenol time course (E and F). #P < 0.05, compared with top end of vehicle control curve. n = 4.
Fig. 7.

Desipramine does not affect lipid raft cAMP in SK-N-SH cells. Three days of 10 μM desipramine treatment has no effect on baseline lipid raft cAMP levels immediately after drug withdrawal (A) or 24 hours later (B) in the neuronal SK-N-SH cell line. This cell line has no quantifiable change in lipid raft–localized cAMP levels after isoproterenol treatment, and this is not modified by desipramine treatment (A and B). Forskolin produces a concentration-dependent response in SK-N-SH cells expressing this sensor (C). SK-N-SH cells have virtually no response to 1 μM isoproterenol at any point along the 30-minute time course (D and E).
Fig. 8.

Desipramine potentiates isoproterenol-stimulated whole-cell cAMP in SK-N-SH cells Desipramine treatment potentiates isoproterenol-stimulated cAMP in SK-N-SH cells expressing the cytoplasmic cAMP sensor (A and C), but this effect reverses to vehicle-treated levels 24 hours after desipramine withdrawal (B and D). Response to 1 μM isoproterenol peaks after 3 minutes and then declines over the 30-minute time course (C and D). #P < 0.05 compared with top end of vehicle control curve. n = 4.
Fig. 9.

Desipramine does not affect lipid raft cAMP signaling in HEK-293 cells. HEK-293 cells expressing the lipid raft cAMP sensor respond to isoproterenol (A and B), but this response is not affected by desipramine treatment (A and C), and signaling remains unchanged 24 hours after desipramine withdrawal (B and D). Response to 1 μM isoproterenol peaks after 2 minutes and then declines over the 30-minute time course (C and D). n = 4.
Fig. 10.

Desipramine does not affect whole-cell cAMP signaling in HEK-293 cells. HEK-293 cells expressing the cytoplasmic cAMP sensor respond to isoproterenol (A and B), but this response is not affected by desipramine treatment (A and C), and signaling remains unchanged 24 hours after desipramine withdrawal (B and D). Response to 1 μM isoproterenol peaks after 1 minute and then declines over the 30-minute time course (C and D). n = 4.
For colchicine and methyl-β-cyclodextrin (MβCD) experiments, either 10 μM colchicine or 10 mM MβCD was added to the assay plate 15 minutes before isoproterenol addition. MβCD powder (Sigma-Aldrich, MO) was dissolved in ddH2O to 4× final assay concentration (40 mM), whereas colchicine (Sigma-Aldrich, MO) stock solution was diluted in ddH2O to 10× final assay concentration (100 μM). After 15 minutes of MβCD or colchicine exposure, isoproterenol was added and allowed to incubate for 30 minutes before final fluorescence measurement. Because of the poor aqueous solubility, MDL 12330A was added at 2× final concentration (2 mM), and fluorescence output was measured for 30 minutes after addition.
AlphaScreen cAMP Assay
AlphaScreen cAMP experiments were performed per the manufacturer’s instructions (Perkin Elmer, MA), with slight modification. These experiments used the AlphaScreen cAMP detection kit with lipid raft containing membranes prepared as described above. Briefly, cAMP standard curve, stimulation buffer, bovine serum albumin solution, isoproterenol, and forskolin stock solutions were prepared fresh on the day of the assay. In total, 20 μg of lipid raft membrane homogenates were added to each well of a white 384-well PerkinElmer Optiplate. Agonist and acceptor beads were added to wells containing membrane homogenates and allowed to incubate for 30 minutes. Final isoproterenol concentration was 1 μM, and final forskolin concentration was 10 μM. Signaling was terminated by addition of donor beads prepared in lysis buffer. Plate was then incubated for 1 hour at room temperature and protected from light before measurement using the SpectraMax i3x plate reader with Alpha module (Molecular Devices, CA). Detected fluorescence signal for each well was fitted to a cAMP standard curve prepared with each experiment to determine sample cAMP concentrations.
Live-Cell Imaging
High-resolution images of cells expressing fluorescent cAMP sensors were obtained using the LSM 880 confocal microscope (ZEISS, Germany) in Airyscan detection mode with a 40× objective. C6 cells were plated on glass microscopy dishes 48 hours before imaging. Cells were infected with either Green Upward cADDis sensor (cytoplasmic) or fMP Green Downward cADDis sensor (lipid raft) expressing baculovirus (Montana Molecular, MT). Infection took place 24 hours before live-cell imaging. Culture media were replaced with serum-free Fluorobrite DMEM (Thermo Fisher Scientific, MA) 30 minutes before imaging to decrease background fluorescence.
Statistics
AUC was determined for chemiluminescent peaks automatically identified by Protein Simple’s Compass for SW software (version 4.0.0). These AUC values were compared to assess changes in Gαs protein expression levels. Concentration-effect results from cAMP assays were fitted to sigmoidal concentration-effect curves (Hill slope = 1) in GraphPad Prism (version 8.2.1). These curves were used to determine EC50, maximal effect, and baseline values. The 3-day antidepressant treatments and their corresponding 24-hour withdrawal conditions were always run on the same plate and, as such, share the same vehicle control curve, but they are presented as separate subfigures for clarity. Venlafaxine and paroxetine (cytoplasmic sensor), escitalopram and desipramine (cytoplasmic sensor), as well as escitalopram and r-citalopram (lipid raft sensor) were also tested together and share vehicle control data. In time course experiments, signal intensity was typically measured once every 30 seconds for the stated experiment run time. For experiments using the upward cytoplasmic cADDis cAMP sensor, raw fluorescence output is corrected using the baseline fluorescence of each well (ΔF/F0). Because of the drug-induced changes in baseline fluorescence intensity using the downward fMP cADDis cAMP sensor, raw fluorescence output is corrected using the mean fluorescence of vehicle-treated wells (ΔF/Fveh). Data are presented as ±S.E.M. with between three and six replicates, as indicated, and compared using unpaired t test, except when stated otherwise.
Results
Desipramine and Escitalopram Translocate Gαs from C6 Glioma Lipid Rafts; Desipramine’s Effect Persists for over 24 Hours after Drug Withdrawal
We first sought to determine the effects of desipramine and escitalopram on Gαs distribution in C6 cells immediately after treatment and 24 hours after drug withdrawal. Three days of treatment of C6 cells with 10 μM desipramine significantly reduces Gαs distribution in lipid rafts (72.67% ± 1.54% of control; Fig. 1A). This reduction persists at least 24 hours after desipramine withdrawal (71.25% ± 3.01% of control; Fig. 1A). Three days of 10 μM desipramine treatment has no significant effect on Gαs expression in nonraft membrane regions (90.97% ± 2.72% of control; Fig. 1B); however, 24 hours after desipramine withdrawal, nonraft Gαs content is decreased compared with vehicle-treated control cells (80.53% ± 3.04% of control; Fig. 1B).
Fig. 1.

Desipramine and escitalopram translocate Gαs from lipid rafts. Treatment of C6 cells with 10 μM desipramine for 3 days reduces Gαs presence in lipid rafts, and this effect persists for over 24 hours after drug withdrawal (A). Desipramine has no immediate effect on Gαs in nonraft membrane regions; however, there is a significant decrease 24 hours after drug withdrawal (B). Treatment with 10 μM escitalopram for 3 days also reduces lipid raft Gαs; however, this effect is reversed 24 hours after drug withdrawal (C). There is no change in nonraft membrane regions immediately after escitalopram treatment or 24 hours after withdrawal (D). r-Citalopram has no effect on lipid raft Gαs (E) or in nonraft membranes (F). *P < 0.05 and **P < 0.01, compared with vehicle control. #P < 0.05, compared with antidepressant treated cells. n = 3 to 4.
Three days of 10 μM escitalopram treatment of C6 cells also reduces Gαs in lipid rafts (77.32% ± 4.33% of control; Fig. 1C). In contrast to desipramine-treated cells, this returns to baseline levels 24 hours after escitalopram withdrawal (95.94% ± 3.24% of control; Fig. 1C). Three days of 10 μM escitalopram treatment does not affect Gαs in nonraft membrane regions (110.74% ± 4.07% of control; Fig. 1D), and there is no change 24 hours after escitalopram withdrawal (111.99% ± 4.86% of control; Fig. 1D).
Three days of treatment of C6 cells with 10 μM r-citalopram did not affect Gαs distribution in lipid rafts (Fig. 1E) or in nonraft membranes (Fig. 1F). Both measures remained unchanged 24 hours after r-citalopram withdrawal (Fig. 1, E and F).
Desipramine and Escitalopram Treatments Reduce Baseline and Isoproterenol-Stimulated cAMP in Lipid Rafts
We next tested whether changes in Gαs localization were reflected by changes in lipid raft cAMP signaling. The fMP cADDis cAMP sensor (Montana Molecular, MT) is highly expressed in lipid raft membranes as a result of the addition of a dually myristoylated/palmitoylated peptide sequence (Fig. 2A). Fluorophores tagged with this sequence generally colocalize with the lipid raft marker caveolin and are highly expressed in TX-100 insoluble (i.e., lipid raft) membrane fractions (Zacharias et al., 2002). In C6 cells expressing this lipid raft–localized cAMP sensor (Fig. 2, A and B), baseline fluorescence is significantly increased after 3 days of 10 μM desipramine treatment (118.46% ± 2.59% of control; Fig. 2C) and 24 hours after desipramine withdrawal (115.99% ± 2.56% of control; Fig. 2C). As the lipid raft cAMP sensor gains fluorescence intensity in response to decreasing cAMP, this increased baseline fluorescence after desipramine treatment suggests lower baseline cAMP concentrations proximal to the lipid raft sensor. Considering that changes in sensor expression can also affect this baseline reading, we also quantified sensor expression after desipramine treatment. Desipramine did not affect overall sensor expression in C6 cells or its distribution between raft/nonraft membranes (Fig. 2B). Therefore, the desipramine-induced alterations in baseline fluorescence cannot be explained by altered sensor expression or localization and likely reflect reduced baseline cAMP proximal to the lipid raft sensor.
If desipramine alters sensor expression or affects fluorescence output nonspecifically, then a difference in maximal fluorescence should be apparent when cAMP levels are reduced to a minimum level. cAMP can be reduced below baseline levels using chemical inhibitors of adenylyl cyclase, and this strategy has been used previously to determine the fluorescence range of cytoplasmic and lipid raft–localized fluorescent cAMP sensors (Agarwal et al., 2018).
We tested the effects of a saturating concentration of the adenylyl cyclase inhibitor MDL 12330A on fluorescence output of C6 cells expressing the lipid raft cAMP sensor. In cells with no antidepressant treatment, MDL 12330A increased fluorescence over the 30-minute treatment window (Supplemental Fig. 5A). This demonstrates that reductions in baseline cAMP result in increased fluorescence with this sensor.
We also compared the maximal fluorescence of C6 cells expressing this sensor, treated initially with either vehicle or desipramine, and then treated with MDL 12330A. The maximal fluorescence was not different between vehicle- and desipramine-pretreated cells after 30 minutes (Supplemental Fig. 5B). This shows that desipramine does not affect the maximal fluorescence of this lipid raft cAMP sensor and that reduced lipid raft cAMP is the most likely explanation for the increased baseline fluorescence after desipramine treatment.
Results using this sensor are reported using the final fluorescence reading from vehicle-treated wells as a baseline (ΔF/Fveh), and all drug-induced fluorescence changes are reported as percent change compared with the baseline fluorescence of vehicle control wells. Three days of 10 μM desipramine treatment reduced both baseline cAMP in range of the lipid raft sensor (27.61% ± 3.01% increase over vehicle; Fig. 2D) and the maximal isoproterenol response (46.20% ± 2.72% decrease from baseline with vehicle vs. 34.87% ± 3.09% decrease from baseline with desipramine; Fig. 2D). The reduction in maximal isoproterenol response is not due to limited sensor range, as forskolin treatment reduces fluorescent intensity to a greater degree than the maximal change observed with isoproterenol (Supplemental Fig. 4), in line with higher maximal cAMP concentrations typically observed with forskolin compared with isoproterenol.
Isoproterenol-stimulated cAMP peaks rapidly in C6 cells and remains near peak value for at least 30 minutes (Fig. 3C). The desipramine-induced suppression of C6 cell lipid raft cAMP is evident immediately after addition of 1 μM isoproterenol and remains separated over the entire 30-minute testing duration (Fig. 3C).
Both effects on lipid raft cAMP produced by 3 days of 10 μM desipramine treatment persist 24 hours after drug withdrawal. Baseline cAMP remains suppressed (19.24% ± 2.87% increase over vehicle; Fig. 3A), and maximal isoproterenol stimulation is still decreased (35.49% ± 3.11% decrease from baseline after desipramine withdrawal; Fig. 3A). The suppression of lipid raft cAMP observed 24 hours after desipramine withdrawal is apparent over the entire 1 μM isoproterenol time course (Fig. 3D).
As these effects were not attenuated 24 hours after desipramine withdrawal, we next tested whether lipid raft cAMP effects persist 3 days after desipramine withdrawal. In C6 cells that have been pretreated for 3 days with 10 μM desipramine, neither baseline fluorescence (1.88% ± 2.82% decrease from vehicle) nor maximal isoproterenol stimulation (48.72% ± 2.53% decrease from baseline with vehicle vs. 46.43% ± 2.93% decrease from baseline after desipramine withdrawal) were significantly changed 3 days after withdrawal compared with vehicle-treated cells (Fig. 3B).
Three days of 10 μM escitalopram treatment produces effects similar to desipramine on lipid raft cAMP. This treatment reduces baseline lipid raft cAMP (25.55% ± 6.73% increase over vehicle; Fig. 4A), as well as maximal isoproterenol-stimulated lipid raft cAMP (46.13% ± 6.06% decrease from baseline with vehicle vs. 18.96 ± 4.43% decrease from baseline with escitalopram; Fig. 4B). Escitalopram treatment had a strong trend (P = 0.0588) toward increased isoproterenol potency for cAMP generation (1.77 ± 1.76 nM isoproterenol EC50 with vehicle vs. 107.40 ± 91.58 pM isoproterenol EC50 with escitalopram; Fig. 4A), a trend not observed after desipramine treatment.
Fig. 4.

Reduction of lipid raft cAMP by escitalopram reverses 24 hours after withdrawal. Three days of treatment with 10 μM escitalopram significantly reduces baseline and isoproterenol-stimulated cAMP in C6 lipid rafts; (A) however, this is no longer observed 24 hours after escitalopram withdrawal (B). Three days of treatment with 10 μM r-citalopram does not affect baseline or isoproterenol-stimulated cAMP in C6 lipid rafts, either immediately after drug removal (C) or 24 hours after drug withdrawal (D). **P < 0.01, compared with top end of vehicle control curve. ##P < 0.01, compared with maximal isoproterenol effect in vehicle-pretreated wells. n = 6.
At 24 hours after drug withdrawal, escitalopram treatment produced no significant effects on lipid raft cAMP in C6 cells, with baseline (16.91% ± 7.82% over vehicle), maximal isoproterenol stimulation (39.15% ± 8.64% decrease from baseline) and isoproterenol EC50 (1.83% ± 1.95 nM) not significantly different from vehicle-treated cells (Fig. 4B).
Lipid raft cAMP signaling was also probed in C6 cells after 3 days of 10 μM r-citalopram treatment. r-Citalopram did not affect baseline lipid raft cAMP, maximal isoproterenol stimulation, or isoproterenol EC50 (Fig. 4C). These measures were still unaffected 24 hours after r-citalopram withdrawal (Fig. 4D).
Whole-Cell cAMP Increases Subsequent to Escitalopram or Desipramine Treatment
We compared antidepressant effects on lipid raft cAMP with effects on whole-cell cAMP concentration in C6 cells expressing a cytoplasmic cAMP sensor (Fig. 5A). As drug treatments did not affect baseline fluorescence generated by the cytoplasmic sensor, fluorescence output is normalized to the pretreatment fluorescence output of each well (ΔF/F0). The maximal whole-cell cAMP concentration produced by isoproterenol in C6 cells is increased after 3 days of treatment with 10 μM desipramine (110.01% ± 4.41% increase over baseline with vehicle vs. 133.60% ± 5.63% with desipramine; Fig. 5C). This effect is noticeable almost immediately after addition of 1 μM isoproterenol and remains elevated over 30 minutes after isoproterenol addition (Fig. 5E).
This is in contrast to 3 days of treatment with 10 μM escitalopram, which increased isoproterenol potency (1.17 ± 0.39 nM isoproterenol EC50 with vehicle treatment vs. 231.8 ± 73.52 pM isoproterenol EC50 with escitalopram treatment; Fig. 6A) with no change in maximal accumulation (110.01% ± 4.41% increase over baseline with vehicle vs. 114.53% ± 3.84% with escitalopram; Fig. 6A).
Fig. 6.

Escitalopram increases potency of isoproterenol-stimulated whole-cell cAMP. Three days of treatment with 10 μM escitalopram increases the potency of isoproterenol-stimulated cAMP production but has no effect on maximal accumulation in C6 cells expressing the cytoplasmic cAMP sensor (A). This is reversed 24 hours after escitalopram withdrawal (B). r-Citalopram has no effect on isoproterenol-stimulated cytoplasmic cAMP in C6 cells (C and D). #P < 0.05, compared with top end of vehicle control curve. n = 4.
This effect of escitalopram treatment is dose-dependent (Supplemental Fig. 1A), with 3 days of 1 μM escitalopram shifting isoproterenol EC50 by 2.3-fold compared with vehicle and 10 μM escitalopram producing a 6.1-fold EC50 shift (Supplemental Fig. 1C). The 100 nM and 10 nM escitalopram treatments were ineffective.
Unlike Gαs translocation from lipid rafts, desipramine effects on whole-cell cAMP accumulation are not sustained after drug withdrawal (110.01% ± 4.41% increase over baseline with vehicle vs. 120.46% ± 4.83% after desipramine withdrawal; Fig. 5, D and F). The increase in isoproterenol potency induced by 3 days of treatment with 10 μM escitalopram is also reversed 24 hours after withdrawal (1.17 ± 0.39 nM isoproterenol EC50 with vehicle vs. 1.03 ± 0.24 nM isoproterenol EC50 after escitalopram withdrawal; Fig. 6B). Lower escitalopram concentrations also fail to affect whole-cell cAMP 24 hours after withdrawal (Supplemental Fig. 1B).
Three days of 10 μM r-citalopram treatment in C6 cells did not affect whole-cell cAMP response, with both maximal isoproterenol stimulation and isoproterenol potency remaining unchanged after r-citalopram treatment (Fig. 6C) and 24 hours after r-citalopram withdrawal (Fig. 6D).
Sustained Antidepressant Effects on cAMP Accumulation Vary with Drug
In addition to escitalopram and desipramine, we also tested the effects of fluoxetine, phenelzine, venlafaxine, ketamine, and paroxetine on whole-cell cAMP. The effects of these drugs are summarized in Table 1. Fluoxetine treatment (3 days, 10 μM) increased maximal isoproterenol stimulation, and this was partially reversed 24 hours after fluoxetine withdrawal (118.84% ± 8.04% increase over baseline with vehicle vs. 146.77% ± 4.30% with fluoxetine vs. 130.75% ± 5.72% after fluoxetine withdrawal). Phenelzine treatment (3 days, 10 μM) increased maximal isoproterenol stimulation, and this effect was fully persistent 24 hours after phenelzine withdrawal (122.40% ± 10.17% increase over baseline with vehicle vs. 167.78% ± 10.28% with phenelzine vs. 151.21% ± 3.80% after phenelzine withdrawal). Venlafaxine treatment (3 days, 10 μM) increased maximal isoproterenol stimulation, and this was partially reversed 24 hours after venlafaxine withdrawal (122.00% ± 3.62% increase over baseline with vehicle vs. 137.77% ± 4.54% with venlafaxine vs. 125.04% ± 11.06% after venlafaxine withdrawal). Ketamine treatment (15 minutes, 10 μM) increased maximal isoproterenol stimulation, and this was fully reversed 24 hours after ketamine withdrawal (134.80% ± 7.83% increase over baseline with vehicle vs. 159.58% ± 6.67% with ketamine vs. 124.11% ± 6.28% after ketamine withdrawal). Paroxetine treatment (3 days, 10 μM) did not increase maximal cAMP accumulation, and there was still no effect 24 hours after paroxetine withdrawal (122.00% ± 3.62% increase over baseline with vehicle vs. 113.04% ± 5.48% with paroxetine vs. 115.45% ± 6.06% after paroxetine withdrawal). Aside from escitalopram (Fig. 4B), none of the tested antidepressants alter the potency of isoproterenol for cytoplasmic cAMP generation (Table 1).
TABLE 1.
Effects of antidepressant treatment and withdrawal on C6 whole-cell cAMP
Antidepressants were applied for 3 days at 10 μM (desipramine, escitalopram, fluoxetine, phenelzine, venlafaxine, paroxetine, or r-citalopram) or for 30 min at 10 μM (ketamine). Desipramine, fluoxetine, venlafaxine, and ketamine all increased maximal whole-cell isoproterenol response. Phenelzine’s effect was fully persistent 24 h after withdrawal, whereas desipramine, fluoxetine, and venlafaxine reversed partially, and ketamine reversed fully. In contrast, escitalopram increased isoproterenol potency but not maximal response, an effect that reversed fully 24 h after withdrawal. Paroxetine and r-citalopram had no effect.
| Isoproterenol Stimulated cAMP Accumulation |
|||
|---|---|---|---|
| Treatment | Efficacy | Potency | 24 h Reversal |
| Desipramine | * | Partial | |
| Escitalopram | * | Full | |
| Fluoxetine | * | Partial | |
| Phenelzine | * | No reversal | |
| Venlafaxine | * | Partial | |
| Ketamine | * | Full | |
| Paroxetine | N/A | ||
| r-Citalopram | N/A | ||
*P < 0.05, compared with vehicle treatment. n = 3 to 4.
Desipramine Potentiates Isoproterenol-Stimulated Cytoplasmic cAMP in SK-N-SH Cells without Affecting Lipid Raft cAMP
The effects of antidepressants on lipid raft cAMP in C6 cells were next compared with effects in the neuronal SK-N-SH cell line. SK-N-SH cells expressing a lipid raft–restricted cAMP sensor did not have a measurable response to isoproterenol (Fig. 7A) at any point during the 30 minutes after isoproterenol addition (Fig. 7D). In contrast, forskolin produced a large lipid raft cAMP response in SK-N-SH cells expressing this sensor, demonstrating that the lipid raft sensor, and adenylyl cyclase, functions normally in this cell line (Fig. 7C).
Three days of 10 μM desipramine treatment did not affect baseline lipid raft cAMP in these cells and did not facilitate an isoproterenol response (Fig. 7A). No changes occurred on either measure 24 hours after desipramine withdrawal (Fig. 7, B and E).
In contrast, SK-N-SH cells expressing a cytoplasmic cAMP sensor produced a robust isoproterenol response (104.44% ± 4.59% increase over baseline; Fig. 8A). This response peaked within 5 minutes of 1 μM isoproterenol addition and then decayed slightly over 30 minutes (Fig. 8C). The maximal isoproterenol effect was significantly increased by 3 days of 10 μM desipramine treatment (151.22% ± 1.25% increase over baseline; Fig. 8A). This effect is evident within 5 minutes of isoproterenol addition and persists for the 30-minute testing period (Fig. 8C).
At 24 hours after desipramine withdrawal, these measures reverted to vehicle control levels, with both baseline cytoplasmic cAMP (10.17% ± 2.25% over vehicle) and maximal isoproterenol-stimulated cAMP (99.60% ± 3.61% over baseline) not significantly different from vehicle-treated cells (Fig. 8, B and D).
The lack of a measurable isoproterenol response in SK-N-SH cells expressing the lipid raft–targeted sensor (Fig. 7A) suggests that this sensor measures local lipid raft cAMP concentrations rather specifically. The robust whole-cell cAMP response (Fig. 8A) did not bleed over into a detectable response with the lipid raft sensor.
Desipramine Does Not Affect Lipid Raft or Whole-Cell cAMP Signaling in HEK-293 Cells
In addition to glial C6 and neuronal SK-N-SH cells, we also sought to determine the effects of antidepressants on lipid raft signaling in human kidney–derived HEK-293 cells. HEK-293 cells expressing a lipid raft–restricted cAMP sensor, like C6 cells, produce a measurable increase in cAMP after isoproterenol stimulation (25.87% ± 6.40% decrease from baseline; Fig. 9A). This effect peaks within 3–5 minutes of 1 μM isoproterenol addition and then decays over the 30-minute testing period (Fig. 9C).
Three days of 10 μM desipramine treatment did not affect baseline lipid raft cAMP in these cells (5.21% ± 2.96% increase over vehicle) or maximal isoproterenol effect (29.97% ± 3.39% decrease from baseline; Fig. 9A), despite affecting both of these measures in C6 cells expressing this sensor (Fig. 2D). At 24 hours after desipramine withdrawal, these measures remained unchanged (Fig. 9B). There is no measurable effect of desipramine at any time point after 1 μM isoproterenol addition (Fig. 9, C and D).
Desipramine also failed to affect cAMP signaling in HEK-293 cells expressing a cytoplasmic cAMP sensor, with baseline and maximal isoproterenol-stimulated cAMP unchanged compared with vehicle, both after 3 days of 10 μM desipramine treatment (Fig. 10A) and 24 hours after desipramine withdrawal (Fig. 10B).
C6 cells reach a maximal lipid raft cAMP response approximately 3 minutes after isoproterenol addition and maintain this response over the 30-minute testing period (Fig. 3, C and D). SK-N-SH cells have essentially no response to isoproterenol regardless of treatment conditions (Fig. 7, D and E). The response to isoproterenol in HEK-293 cells peaks 1 to 2 minute after isoproterenol addition but then decays over the 30-minute testing period (Fig. 9, C and D). At the 30-minute time point, HEK-293 cells treated with vehicle and 1 μM isoproterenol have similar cAMP levels (Fig. 9, C and D).
Within 3–5 minutes of 1 μM isoproterenol addition, HEK-293 cells produced a more rapid cytoplasmic cAMP response (Fig. 10C) compared with C6 cells (Fig. 5E). This peak in HEK-293 cells is relatively transient, such that 15 minutes after isoproterenol addition, HEK-293 and C6 cells have comparable responses. Nonetheless, desipramine treatment fails to affect HEK-293 cytoplasmic cAMP response at any point during the 30-minute testing period, either after 3 days of treatment (Fig. 10C) or 24 hours after desipramine withdrawal (Fig. 10D).
The Cholesterol Chelator MβCD and the Microtubule Disruptor Colchicine Produce Effects Similar to Desipramine on cAMP Signaling in C6 Cells
Microtubule disruption with colchicine affects lipid raft organization and induces translocation of Gαs out of lipid rafts (Donati and Rasenick, 2005). In C6 cells expressing either the lipid raft cAMP sensor (Fig. 11A) or the cytoplasmic cAMP sensor (Fig. 11B), response to colchicine was similar to the effects observed after desipramine treatment. Treatment for 15 minutes with 10 μM colchicine suppressed baseline lipid raft cAMP (27.73% ± 8.48% increase over vehicle) but did not significantly affect maximal isoproterenol response (56.08% ± 6.28% decrease from baseline with vehicle vs. 31.71% ± 16.87% with colchicine), despite trending toward a reduced effect (Fig. 11A). Colchicine treatment did not affect baseline fluorescence detected by the cytoplasmic sensor (Fig. 11B) but increased the maximal isoproterenol response compared with vehicle-treated cells (80.02% ± 13.31% increase from baseline with vehicle vs. 132.60% ± 6.40% with colchicine).
Fig. 11.

Lipid raft disruptors MβCD and colchicine reduce baseline cAMP in lipid rafts, whereas colchicine potentiates isoproterenol-stimulated whole-cell cAMP in C6 cells. Colchicine reduces baseline lipid raft cAMP in C6 cells (A) while also potentiating maximal isoproterenol-induced whole-cell cAMP (B). Treatment with 10 mM MβCD for 15 minutes reduces baseline and isoproterenol-stimulated lipid raft cAMP (C). *P < 0.05, compared with top end of vehicle control curve. #P < 0.05, compared with top end of vehicle control curve. n = 4.
The cholesterol chelator MβCD also disrupts lipid raft organization (Zidovetzki and Levitan, 2007) and reduces raft-localized Gαs (Allen et al., 2009). Similar to desipramine, MβCD trends toward reduced baseline cAMP detected by the lipid raft sensor in C6 cells (32.57% ± 14.65% increase over vehicle; Fig. 11C) and also reduces the maximal isoproterenol response (48.47% ± 4.81% decrease from baseline with vehicle vs. 14.61% ± 10.34% with MβCD; Fig. 11C).
Together with the desipramine data presented above, these results suggest that treatments that induce translocation of Gαs out of lipid rafts will generally decrease cAMP detected by a lipid raft–localized sensor while simultaneously increasing total cellular cAMP response.
Discussion
Results from this study suggest that, for some antidepressants, effects on Gαs signaling and localization persist after drug withdrawal. Desipramine-induced reduction of lipid raft Gαs persists for over 24 hours after drug withdrawal in C6 cells (Fig. 1A). In contrast, escitalopram’s effects on lipid raft Gαs localization revert to baseline within 24 hours of escitalopram withdrawal (Fig. 1C). This effect is stereospecific, as the inactive stereoisomer r-citalopram has no effect on Gαs localization (Fig. 1, E and F). Although the ratio of nonraft/raft Gαs localization returns to baseline 24 hours after withdrawal of either drug, both nonraft and raft Gαs are reduced after withdrawal of desipramine but not escitalopram (Fig. 1). Therefore, this cellular model of antidepressant action reveals a persistent desipramine effect not observed with escitalopram.
The sustained reduction of lipid raft Gαs after desipramine withdrawal corresponds with a persistent increase in baseline fluorescence emitted by a lipid raft–localized cAMP biosensor (Fig. 2C). These changes in baseline fluorescence do not result from changes in overall sensor expression or localization, as desipramine treatment did not significantly affect expression or raft localization (Fig. 2B).
In addition to the desipramine-induced increase in baseline fluorescence, desipramine treatment also reduced the maximal change in fluorescence after isoproterenol challenge in C6 cells (Fig. 2D). This suggests that desipramine treatment inhibits the maximal effect of isoproterenol on cAMP generated proximal to the lipid raft cAMP sensor. Consistent with this interpretation, lipid raft membranes purified from desipramine-treated C6 cells produce less cAMP after isoproterenol challenge (Fig. 2E). Together, these data suggest that the increased fluorescence emitted by the lipid raft cAMP sensor after desipramine pretreatment corresponds with a reduction in baseline cAMP proximal to lipid rafts, as well as a reduction in maximal isoproterenol-stimulated cAMP in this region.
The cholesterol chelator MβCD and the microtubule disruptor colchicine increase baseline fluorescence emitted by the lipid raft cAMP sensor and attenuate maximal isoproterenol response (Fig. 11). Both of these compounds liberate Gαs from lipid rafts, albeit through divergent mechanisms (Donati and Rasenick, 2005; Zidovetzki and Levitan, 2007; (Zacharias et al., 2002) Allen et al., 2009). As such, reduction of lipid raft–localized Gαs appears to be a sufficient strategy to reduce lipid raft cAMP. This also suggests that the cAMP detected by the lipid raft sensor is primarily produced after activation of raft-localized Gαs.
The reduction of lipid raft cAMP after 3 days of desipramine treatment persists for over 24 hours after desipramine withdrawal in C6 cells but reverts to baseline 72 hours after withdrawal (Fig. 3, A and B). In contrast, escitalopram effects do not persist after escitalopram withdrawal (Fig. 4, A and B). Translocation of lipid raft Gαs after desipramine and escitalopram treatments (Fig. 1, A and C) adheres to a similar time course. This reveals a strong correlation between the reduction in lipid raft Gαs and the reduction of lipid raft cAMP after treatment with these antidepressants.
Desipramine increases the maximal effect of isoproterenol-stimulated whole-cell cAMP in C6 cells (Fig. 3B); however, this effect begins to revert to baseline within 24 hours (Fig. 4A). Desipramine has no effect on the potency of the β-agonist isoproterenol, nor does any other antidepressant tested, save escitalopram (Table 1). This represents an initial demonstration that during the process of removing Gαs from lipid rafts, antidepressants inhibit Gαs evoked signaling in those rafts while contemporaneously enhancing cAMP signaling in the whole cell.
The reduction in lipid raft cAMP observed here is consistent with predictions from earlier work showing that many antidepressants reduce Gαs distribution in lipid rafts (Toki et al., 1999; Donati and Rasenick, 2005; Zhang and Rasenick, 2010) without altering cellular content of this protein. This process is specific to antidepressants, as non-antidepressants, including olanzapine, haloperidol, lithium, and diazepam, lack these effects (Czysz et al., 2015; Donati et al., 2015). Antidepressant drugs also accumulate in lipid raft membranes of C6 cells (Erb et al., 2016). As the C6 cell line lacks monoamine transporters (Eshleman et al., 1997), the lipid raft binding target for these antidepressants remains unknown. Nonetheless, the signaling effects of antidepressant accumulation in lipid rafts are becoming more well understood–namely, reduced cAMP proximal to lipid rafts, facilitated Gαs signaling elsewhere in the cell, and cAMP-dependent increases in signaling factors including phosph-cAMP Response Element-Binding Protein and BDNF (Singh et al., 2018) as well as the BDNF target Tropomyosin receptor kinase B (Casarotto et al., 2021). Note that lipid raft translocation of Gαs and the resultant sustained increases in cAMP and BDNF are hallmarks of antidepressant action that unfold over the course of extended drug treatment. The identified pharmacologic target of many of these drugs (e.g., monoamine transporters or catabolizing enzymes) are affected immediately by antidepressants, and the clinical effects show a hysteresis of up to 2 months.
Escitalopram’s effect on isoproterenol potency was surprising, and it differed from all other antidepressants tested (Table 1). This effect was dose-dependent (Supplemental Fig. 1A) and stereospecific (Fig. 4). Furthermore, escitalopram had a strong trend (P = 0.0588) toward increasing isoproterenol potency in lipid rafts while simultaneously decreasing isoproterenol’s maximal effect in raft domains (Fig. 4A). This suggests a dual action of escitalopram on signaling downstream of Gαs in lipid rafts, both driving Gαs out of this region while at the same time facilitating β-adrenergic receptor signaling.
Similar to the effect observed in C6 cells, desipramine treatment increased the maximal isoproterenol effect on whole-cell cAMP in the neuronal SK-N-SH cell line (Fig. 8A), an effect that did not persist after desipramine withdrawal (Fig. 8B). Unlike C6 cells, SK-N-SH had no measurable response to isoproterenol in lipid rafts and did not respond to desipramine treatment in rafts (Fig. 7A). This suggests that SK-N-SH lack functional β-adrenergic receptors in lipid rafts, and an intact forskolin response demonstrates these cells have the capacity to generate cAMP detectable by the lipid raft sensor (Fig. 7C). In contrast, HEK-293 cells respond robustly to isoproterenol stimulation, when measuring both whole-cell cAMP (Fig. 10A) and lipid raft cAMP (Fig. 9A). Nonetheless, neither of these measures is affected by desipramine treatment, consistent with the lack of antidepressant effect on Gαs signaling in rat kidney (Menkes et al., 1983).
The inverse effect of desipramine pretreatment on whole-cell and lipid raft cAMP in C6 cells was surprising because of the assumption that cAMP would generally diffuse freely and rapidly throughout the cell. If this were true, lipid raft sensors in SK-N-SH would detect overall cellular increases in isoproterenol-stimulated cAMP. They do not (Fig. 7). In fact, several reports provide evidence that cAMP concentration is regulated independently across subcellular domains and that cAMP diffuses at a rate significantly lower than that predicted for free diffusion (Saucerman et al., 2006; Agarwal et al., 2016). In fact, cAMP concentration gradients have been observed between nanometer-sized subcellular domains in HEK-293 cells (Bock et al., 2020), confirming that cAMP does not diffuse freely throughout the cytoplasm.
In HEK-293 cells, prostaglandin E1 increases cAMP, gradually, to a steady state in cytoplasmic regions distant from the plasma membrane (Rich et al., 2001a,b). The rapid reduction in membrane cAMP was prevented by pretreatment with a phosphodiesterase inhibitor, suggesting that differences in the rate of cAMP breakdown contribute to differential concentrations between various cellular subdomains (Rich et al., 2001a,b; Oliveira et al., 2010). The rate of cAMP production by adenylyl cyclase is also affected by minute changes in local pH, particularly in caveolae (Willoughby et al., 2005). Distinct expression of proteins that form complexes with adenylyl cyclase between membrane subdomains (e.g., calmodulin, protein kinase A, A-kinase-anchoring protein) may also contribute to subregion-specific cAMP regulation (Simpson et al., 2006; Di Benedetto et al., 2008; Zaccolo and Pozzan, 2002). Together these localized differences in cAMP breakdown and production allow for distinct, compartmentalized regulation of cellular cAMP.
Cell-specific variation in whole-cell versus lipid raft cAMP among C6, SK-N-SH, and HEK-293 cells demonstrates cell type specificity for desipramine’s effects. Both the neuronal SK-N-SH and glial C6 cells generated higher maximal whole-cell cAMP levels after desipramine treatment, whereas HEK-293 cells were unaffected. Only C6 cells displayed a reduction in lipid raft cAMP after desipramine treatment, suggesting these cells (and, perhaps, glia generally) have somewhat unique antidepressant-responsive elements in this membrane region. This is consistent with the observation that both C6 cells and primary astrocytes, but not HEK-293 cells, display magnified cAMP responses after treatment with the antidepressant ketamine (Wray et al., 2018).
Although antidepressant efficacy has traditionally been assumed to involve neuronal targets, glial cells have also been heavily implicated in antidepressant action and discontinuation syndromes. SSRIs such as citalopram evoke a calcium response in astrocytes, an effect that persists for longer time periods than other neurotransmitter-induced calcium signaling (Schipke et al., 2011). Antidepressants also induce gliogenesis in mouse embryonic stem cells (Kusakawa et al., 2010), an effect consistent with increased expression of neurotrophic factors after antidepressant treatment in C6 cells (Singh et al., 2018). Furthermore, astrocytes derived from neural stem cells of subjects with depression responded to n-3 polyunsaturated fatty acid with cellular responses similar to those described in this study (Yu et al., 2020). Finally, altered choline-to-creatine ratio observed in the anterior cingulate of individuals experiencing antidepressant discontinuation syndrome has been attributed to disrupted astrocytic function (Kaufman et al., 2003), affirming the involvement of glial cells not only in antidepressant action but also in the residual effects after antidepressant discontinuation.
The use of baseline fluorescence output to determine changes in baseline cAMP has the potential to be confounded by changes in sensor expression. We control for this possibility by quantifying sensor expression directly after vehicle and desipramine pretreatments (Fig. 2B) and show that sensor expression and localization are unaltered. Using a chemical inhibitor of adenylyl cyclase to reduce baseline cAMP, we also show that desipramine does not affect the maximal fluorescent output of this sensor (Supplemental Fig. 5).
Furthermore, fluorescence alterations detected with lipid raft cAMP biosensors are consistent with reduced lipid raft cAMP generation detected with the AlphaScreen cAMP assay (PerkinElmer, MA). Although we did not detect a baseline cAMP difference between vehicle and desipramine-treated lipid raft membranes using the AlphaScreen assay, this is likely due to the lipid raft purification process (Fig. 2E). The cytoplasmic contents are removed during membrane purification, so baseline measurements reflect cAMP that is generated during the AlphaScreen assay rather than resting cAMP levels in intact and alive cells with the fluorescent sensor. Desipramine reduced isoproterenol-stimulated cAMP to a greater degree using AlphaScreen detection compared with the fluorescent lipid raft sensor. This result was surprising, but it may indicate that some cAMP generated during isoproterenol stimulation in a live C6 cell diffuses from nonraft membrane regions such that it is detected by the lipid raft sensor.
This work underscores the importance of measuring G-protein signaling in relevant subcellular locales. The lipid raft specific signaling events described here complement recent studies exploring the importance of G-protein signaling from endosomes (Lyga et al., 2016; Thomsen et al., 2016), in recycling tubules (Bowman et al., 2016), and the trans-Golgi network (Godbole et al., 2017). Persistent endosomal cAMP signaling is a consistent finding after receptor internalization (Ferrandon et al., 2009; Tsvetanova and von Zastrow, 2014; Jean-Alphonse et al., 2017). Persistent antidepressant-induced changes in cAMP signaling are likely independent from this endosomal signaling, as the antidepressant-induced changes are evident within minutes of isoproterenol addition.
Dysregulated cAMP signaling is a hallmark feature of depression. Depressed individuals have widespread reductions in cAMP levels throughout the brain (Fujita et al., 2017), and suicidal subjects with depression have reduced adenylyl cyclase IV expression and activity in post-mortem temporal cortex (Reiach et al., 1999). The reduced cAMP level observed in unmedicated patients with depression is corrected to healthy control levels after effective SSRI treatment (Fujita et al., 2017), and the rapid-acting antidepressant ketamine facilitates cAMP signaling in glial cells (Wray et al., 2018). In animal models, chronic but not acute antidepressant treatments increase brain cAMP levels (Menkes et al., 1983; Ozawa and Rasenick, 1989) and downstream signaling factors including cAMP response element-binding protein (Nibuya et al., 1996). Furthermore, phosphodiesterase 4B inhibitors specifically increase cAMP concentrations and produce antidepressant-like effects in preclinical models (Zhang et al., 2006, 2017).
Together, these studies indicate that antidepressant-induced increases in cAMP are an important determinant of treatment effectiveness and that reductions are generally indicative of unmedicated depression. Our results complement these findings, showing for the first time that antidepressant-induced increases in cAMP reverse relatively quickly after antidepressant withdrawal in both glial and neuronal cells. They also validate Gαs translocation from lipid rafts and the sequelae of cAMP signaling events accompanying this as consistent biomarkers of antidepressant action. The residual depression of lipid raft cAMP signaling may represent an as of yet unrecognized cellular mechanism underlying antidepressant discontinuation syndromes. In future studies, we hope to determine how a wider spectrum of antidepressants affect lipid raft signaling and whether this action correlates with severity of discontinuation symptoms.
Acknowledgments
We thank Dr. Ankur Saxena and colleagues for donating the SK-N-SH cells used in these experiments. We also thank Montana Molecular for donating the lipid raft cAMP sensor used for preliminary experiments and Lundbeck for their donation of r-citalopram.
Abbreviations
- AUC
area under the curve
- BDNF
Brain Derived Neurotrophic Factor
- Cav-1
Caveolin-1
- ddH2O
double-distilled H2O
- DMEM
Dulbecco’s modified Eagle’s medium
- fMP
fluorescent Myr/Palm
- MβCD
methyl-β-cyclodextrin
- SSRI
selective serotonin reuptake inhibitor
- TX-100
Triton X-100
- TX-114
Triton X-114
Authorship Contributions
Participated in research design: Senese, Rasenick.
Conducted experiments: Senese.
Performed data analysis: Senese, Rasenick.
Wrote or contributed to the writing of the manuscript: Senese, Rasenick.
Footnotes
This work was supported by the Department of Veterans Affairs [Merit Award BX001149], National Institutes of Health (NIH) National Institute of Mental Health [Grant T32-MH067631], and National Institute of Health (NIH) National Center for Complementary & Alternative Medicine [Grant R01-AT009169].
M.M.R. has received research support from Eli Lilly and Lundbeck, Inc., and is a consultant to Otsuka Pharmaceuticals. M.M.R. has ownership in Pax Neuroscience, Inc. N.B.S. has no conflicts to declare.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Allen JA, Halverson-Tamboli RA, Rasenick MM (2007) Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci 8:128–140 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Allen JA, Yu JZ, Dave RH, Bhatnagar A, Roth BL, Rasenick MM (2009) Caveolin-1 and lipid microdomains regulate Gs trafficking and attenuate Gs/adenylyl cyclase signaling. Mol Pharmacol 76:1082–1093 10.1124/mol.109.060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Clancy CE, Harvey RD (2016) Mechanisms restricting diffusion of intracellular cAMP. Sci Rep 6:19577 10.1038/srep19577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Gratwohl J, Cozad M, Yang PC, Clancy CE, Harvey RD (2018) Compartmentalized cAMP signaling associated with lipid raft and non-raft membrane domains in adult ventricular myocytes. Front Pharmacol 9:332 10.3389/fphar.2018.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin DS, Cooper JA, Huusom AKT, Hindmarch I (2006) A double-blind, randomized, parallel-group, flexible-dose study to evaluate the tolerability, efficacy and effects of treatment discontinuation with escitalopram and paroxetine in patients with major depressive disorder. Int Clin Psychopharmacol 21:159–169 10.1097/01.yic.0000194377.88330.1d. [DOI] [PubMed] [Google Scholar]
- Blier P, Tremblay P (2006) Physiologic mechanisms underlying the antidepressant discontinuation syndrome. J Clin Psychiatry 67 (Suppl 4):8–13. [PubMed] [Google Scholar]
- Bock A, Annibale P, Konrad C, Hannawacker A, Anton SE, Maiellaro I, Zabel U, Sivaramakrishnan S, Falcke M, Lohse MJ (2020) Optical mapping of cAMP signaling at the nanometer scale. Cell 182:1519–1530.e17 10.1016/j.cell.2020.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman SL, Shiwarski DJ, Puthenveedu MA (2016) Distinct G protein-coupled receptor recycling pathways allow spatial control of downstream G protein signaling. J Cell Biol 214:797–806 10.1083/jcb.201512068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull SA, Hu XH, Hunkeler EM, Lee JY, Ming EE, Markson LE, Fireman B (2002) Discontinuation of use and switching of antidepressants: influence of patient-physician communication. JAMA 288:1403–1409 10.1001/jama.288.11.1403. [DOI] [PubMed] [Google Scholar]
- Casarotto PCGirych MFred SMKovaleva VMoliner REnkavi GBiojone CCannarozzo CSahu MPKaurinkoski K, et al. (2021) Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184:1299–1313.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney DS, Heninger GR, Sternberg DE, Landis H (1982) Abrupt discontinuation of tricyclic antidepressant drugs: evidence for noradrenergic hyperactivity. Br J Psychiatry 141:377–386 10.1192/bjp.141.4.377. [DOI] [PubMed] [Google Scholar]
- Chen J, Rasenick MM (1995a) Chronic antidepressant treatment facilitates G protein activation of adenylyl cyclase without altering G protein content. J Pharmacol Exp Ther 275:509–517. [PubMed] [Google Scholar]
- Chen J, Rasenick MM (1995b) Chronic treatment of C6 glioma cells with antidepressant drugs increases functional coupling between a G protein (Gs) and adenylyl cyclase. J Neurochem 64:724–732 10.1046/j.1471-4159.1995.64020724.x. [DOI] [PubMed] [Google Scholar]
- Cowburn RF, Marcusson JO, Eriksson A, Wiehager B, O’Neill C (1994) Adenylyl cyclase activity and G-protein subunit levels in postmortem frontal cortex of suicide victims. Brain Res 633:297–304 10.1016/0006-8993(94)91552-0. [DOI] [PubMed] [Google Scholar]
- Czysz AH, Schappi JM, Rasenick MM (2015) Lateral diffusion of Gαs in the plasma membrane is decreased after chronic but not acute antidepressant treatment: role of lipid raft and non-raft membrane microdomains. Neuropsychopharmacology 40:766–773 10.1038/npp.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Benedetto G, Zoccarato A, Lissandron V, Terrin A, Li X, Houslay MD, Baillie GS, Zaccolo M (2008) Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ Res 103:836–844 10.1161/CIRCRESAHA.108.174813. [DOI] [PubMed] [Google Scholar]
- Donati RJ, Dwivedi Y, Roberts RC, Conley RR, Pandey GN, Rasenick MM (2008) Postmortem brain tissue of depressed suicides reveals increased Gs α localization in lipid raft domains where it is less likely to activate adenylyl cyclase. J Neurosci 28:3042–3050 10.1523/JNEUROSCI.5713-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati RJ, Rasenick MM (2005) Chronic antidepressant treatment prevents accumulation of gsalpha in cholesterol-rich, cytoskeletal-associated, plasma membrane domains (lipid rafts). Neuropsychopharmacology 30:1238–1245 10.1038/sj.npp.1300697. [DOI] [PubMed] [Google Scholar]
- Donati RJ, Schappi J, Czysz AH, Jackson A, Rasenick MM (2015) Differential effects of antidepressants escitalopram versus lithium on Gs alpha membrane relocalization. BMC Neurosci 16:40 10.1186/s12868-015-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Pandey GN (2008) Adenylyl cyclase-cyclicAMP signaling in mood disorders: role of the crucial phosphorylating enzyme protein kinase A. Neuropsychiatr Dis Treat 4:161–176 10.2147/ndt.s2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisensamer B, Uhr M, Meyr S, Gimpl G, Deiml T, Rammes G, Lambert JJ, Zieglgänsberger W, Holsboer F, Rupprecht R (2005) Antidepressants and antipsychotic drugs colocalize with 5-HT3 receptors in raft-like domains. J Neurosci 25:10198–10206 10.1523/JNEUROSCI.2460-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb SJ, Schappi JM, Rasenick MM (2016) Antidepressants accumulate in lipid rafts independent of monoamine transporters to modulate redistribution of the G protein, Goαs. J Biol Chem 291:19725–19733 10.1074/jbc.M116.727263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman AJ, Stewart E, Evenson AK, Mason JN, Blakely RD, Janowsky A, Neve KA (1997) Metabolism of catecholamines by catechol-O-methyltransferase in cells expressing recombinant catecholamine transporters. J Neurochem 69:1459–1466 10.1046/j.1471- 4159.1997.69041459.x. [DOI] [PubMed] [Google Scholar]
- Fava GA, Gatti A, Belaise C, Guidi J, Offidani E (2015) Withdrawal symptoms after selective serotonin reuptake inhibitor discontinuation: A systematic review. Psychother Psychosom 84:72–81 10.1159/000370338. [DOI] [PubMed] [Google Scholar]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP (2009) Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 5:734–742 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita MRichards EMNiciu MJIonescu DFZoghbi SSHong JTelu SHines CSPike VWZarate CA, et al. (2017) cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol Psychiatry 22:754–759 10.1038/mp.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel M, Sharma V (2017) Antidepressant discontinuation syndrome. CMAJ 189:E747 10.1503/cmaj.160991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner EM, Kelly MW, Thompson DF (1993) Tricyclic antidepressant withdrawal syndrome. Ann Pharmacother 27:1068–1072 10.1177/106002809302700912. [DOI] [PubMed] [Google Scholar]
- Godbole A, Lyga S, Lohse MJ, Calebiro D (2017) Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat Commun 8:443 10.1038/s41467-017-00357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad P (1997) Newer antidepressants and the discontinuation syndrome. J Clin Psychiatry 58 (Suppl 7):17–21, discussion 22. [PubMed] [Google Scholar]
- Haddad PM (2001) Antidepressant discontinuation syndromes. Drug Saf 24:183–197 10.2165/00002018-200124030-00003. [DOI] [PubMed] [Google Scholar]
- Insel TR, Wang PS (2009) The STAR*D trial: revealing the need for better treatments. Psychiatr Serv 60:1466–1467 10.1176/ps.2009.60.11.1466. [DOI] [PubMed] [Google Scholar]
- Jean-Alphonse FG, Wehbi VL, Chen J, Noda M, Taboas JM, Xiao K, Vilardaga JP (2017) β2-adrenergic receptor control of endosomal PTH receptor signaling via Gβγ. Nat Chem Biol 13:259–261 10.1038/nchembio.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman MJ, Henry ME, Frederick Bd, Hennen J, Villafuerte RA, Stoddard EP, Schmidt ME, Cohen BM, Renshaw PF (2003) Selective serotonin reuptake inhibitor discontinuation syndrome is associated with a rostral anterior cingulate choline metabolite decrease: a proton magnetic resonance spectroscopic imaging study. Biol Psychiatry 54:534–539 10.1016/s0006- 3223(02)01828-0. [DOI] [PubMed] [Google Scholar]
- Kusakawa S, Nakamura K, Miyamoto Y, Sanbe A, Torii T, Yamauchi J, Tanoue A (2010) Fluoxetine promotes gliogenesis during neural differentiation in mouse embryonic stem cells. J Neurosci Res 88:3479–3487 10.1002/jnr.22509. [DOI] [PubMed] [Google Scholar]
- Lyga S, Volpe S, Werthmann RC, Götz K, Sungkaworn T, Lohse MJ, Calebiro D (2016) Persistent cAMP signaling by internalized LH receptors in ovarian follicles. Endocrinology 157:1613–1621 10.1210/en.2015-1945. [DOI] [PubMed] [Google Scholar]
- Menkes DB, Rasenick MM, Wheeler MA, Bitensky MW (1983) Guanosine triphosphate activation of brain adenylate cyclase: enhancement by long-term antidepressant treatment. Science 219:65–67 10.1126/science.6849117. [DOI] [PubMed] [Google Scholar]
- Mooney JJ, Samson JA, McHale NL, Pappalarado KM, Alpert JE, Schildkraut JJ (2013) Increased Gsα within blood cell membrane lipid microdomains in some depressive disorders: an exploratory study. J Psychiatr Res 47:706–711 10.1016/j.jpsychires.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Kobayashi D, Imuta N, Haraguchi K, Ieiri I, Nishimura R, Koyama S, Mine K (2010) Effects of the serotonin 1A, 2A, 2C, 3A, and 3B and serotonin transporter gene polymorphisms on the occurrence of paroxetine discontinuation syndrome. J Clin Psychopharmacol 30:11–17 10.1097/JCP.0b013e3181c8ae80. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Nestler EJ, Duman RS (1996) Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 16:2365–2372 10.1523/jneurosci.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogle NR, Akkerman SR (2013) Guidance for the discontinuation or switching of antidepressant therapies in adults. J Pharm Pract 26:389–396 10.1177/0897190012467210. [DOI] [PubMed] [Google Scholar]
- Oliveira RF, Terrin A, Di Benedetto G, Cannon RC, Koh W, Kim M, Zaccolo M, Blackwell KT (2010) The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PLoS One 5:e11725 10.1371/journal.pone.0011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa H, Rasenick MM (1989) Coupling of the stimulatory GTP-binding protein Gs to rat synaptic membrane adenylate cyclase is enhanced subsequent to chronic antidepressant treatment. Mol Pharmacol 36:803–808. [PubMed] [Google Scholar]
- Pandey GN, Dwivedi Y, Ren X, Rizavi HS, Mondal AC, Shukla PK, Conley RR (2005) Brain region specific alterations in the protein and mRNA levels of protein kinase A subunits in the post-mortem brain of teenage suicide victims. Neuropsychopharmacology 30:1548–1556 10.1038/sj.npp.1300765. [DOI] [PubMed] [Google Scholar]
- Reiach JS, Li PP, Warsh JJ, Kish SJ, Young LT (1999) Reduced adenylyl cyclase immunolabeling and activity in postmortem temporal cortex of depressed suicide victims. J Affect Disord 56:141–151 10.1016/s0165-0327(99)00048-8. [DOI] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW (2001a) A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci USA 98:13049–13054 10.1073/pnas.221381398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Tse TE, Rohan JG, Schaack J, Karpen JW (2001b) In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J Gen Physiol 118:63–78 10.1085/jgp.118.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AJTrivedi MHWisniewski SRNierenberg AAStewart JWWarden DNiederehe GThase MELavori PWLebowitz BD, et al. (2006a) Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163:1905–1917 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Rush AJTrivedi MHWisniewski SRStewart JWNierenberg AAThase MERitz LBiggs MMWarden DLuther JF, et al. ; STAR*D Study Team (2006b) Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 354:1231–1242 10.1056/NEJMoa052963. [DOI] [PubMed] [Google Scholar]
- Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY, McCulloch AD (2006) Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci USA 103:12923–12928 10.1073/pnas.0600137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipke CG, Heuser I, Peters O (2011) Antidepressants act on glial cells: SSRIs and serotonin elicit astrocyte calcium signaling in the mouse prefrontal cortex. J Psychiatr Res 45:242–248 10.1016/j.jpsychires.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Senese NB, Rasenick MM, Traynor JR (2018) The role of G-proteins and G-protein regulating proteins in depressive disorders. Front Pharmacol 9:1289 10.3389/fphar.2018.01289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma T, Guski LS, Freund N, Meng DM, Gøtzsche PC (2019) Drop-out rates in placebo-controlled trials of antidepressant drugs: A systematic review and meta-analysis based on clinical study reports. Int J Risk Saf Med 30:217–232 10.3233/JRS-195041. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31–39 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simpson RE, Ciruela A, Cooper DMF (2006) The role of calmodulin recruitment in Ca2+ stimulation of adenylyl cyclase type 8. J Biol Chem 281:17379–17389 10.1074/jbc.M510992200. [DOI] [PubMed] [Google Scholar]
- Singh H, Wray N, Schappi JM, Rasenick MM (2018) Disruption of lipid-raft localized Gαs/tubulin complexes by antidepressants: a unique feature of HDAC6 inhibitors, SSRI and tricyclic compounds. Neuropsychopharmacology 43:1481–1491 10.1038/s41386-018-0016-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Thomsen ARBPlouffe BCahill TJ 3rdShukla AKTarrasch JTDosey AMKahsai AWStrachan RTPani BMahoney JP, et al. (2016) GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling. Cell 166:907–919 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toki S, Donati RJ, Rasenick MM (1999) Treatment of C6 glioma cells and rats with antidepressant drugs increases the detergent extraction of G(s α) from plasma membrane. J Neurochem 73:1114–1120 10.1046/j.1471-4159.1999.0731114.x. [DOI] [PubMed] [Google Scholar]
- Tsvetanova NG, von Zastrow M (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10:1061–1065 10.1038/nchembio.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner CH, Bobo W, Warner C, Reid S, Rachal J (2006) Antidepressant discontinuation syndrome. Am Fam Physician 74:449–456 10.1016/s1097-8690(11)70382-1. [PubMed] [Google Scholar]
- Willoughby D, Masada N, Crossthwaite AJ, Ciruela A, Cooper DMF (2005) Localized Na+/H+ exchanger 1 expression protects Ca2+-regulated adenylyl cyclases from changes in intracellular pH. J Biol Chem 280:30864–30872 10.1074/jbc.M414355200. [DOI] [PubMed] [Google Scholar]
- World Health Organization; Depression and other common mental disorders: global health estimates WHO World Heal Organ; [Internet]. 2017;1–24. Available from: http://apps.who.int/iris/bitstream/10665/254610/1/WHO-MSD-MER-2017.2-eng.pdf
- Wray NH, Schappi JM, Singh H, Senese NB, Rasenick MM (2018) NMDAR-independent, cAMP dependent antidepressant actions of ketamine. Mol Psychiatry 24:1–11 10.1038/s41380-018-0083-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J-Z, Wang J, Sheridan SD, Perlis RH, and Rasenick MM (2020) N-3 polyunsaturated fatty acids promote astrocyte differentiation and neurotrophin production independent of camp in patient-derived neural stem cells. Mol Psychiatry. [DOI] [PMC free article] [PubMed]
- Zabegalov KNKolesnikova TOKhatsko SLVolgin ADYakovlev OAAmstislavskaya TGAlekseeva PAMeshalkina DAFriend AJBao W, et al. (2018) Understanding antidepressant discontinuation syndrome (ADS) through preclinical experimental models. Eur J Pharmacol 829:129–140 10.1016/j.ejphar.2018.04.003. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T (2002) Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295:1711–1715 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- Zhang L, Rasenick MM (2010) Chronic treatment with escitalopram but not R-citalopram translocates Galpha(s) from lipid raft domains and potentiates adenylyl cyclase: a 5-hydroxytryptamine transporter-independent action of this antidepressant compound. J Pharmacol Exp Ther 332:977–984 10.1124/jpet.109.162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HT, Zhao Y, Huang Y, Deng C, Hopper AT, De Vivo M, Rose GM, O’Donnell JM (2006) Antidepressant-like effects of PDE4 inhibitors mediated by the high-affinity rolipram binding state (HARBS) of the phosphodiesterase-4 enzyme (PDE4) in rats. Psychopharmacology (Berl) 186:209–217 10.1007/s00213-006-0369-4. [DOI] [PubMed] [Google Scholar]
- Zhang C, Xu Y, Zhang HT, Gurney ME, O’Donnell JM (2017) Comparison of the pharmacological profiles of selective PDE4B and PDE4D inhibitors in the central nervous system. Sci Rep 7:40115 10.1038/srep40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R, and Levitan I (2007) Use of cyclodextrins to manipulate plasma membrane cholesterol content:evidence, misconceptions and control strategies. Biochim Biophys Acta1768:1311–24. [DOI] [PMC free article] [PubMed]