

Abstract
The impeccable control over chemo-, site-, and stereoselectivity possible in enzymatic reactions has led to a surge in the development of new biocatalytic methods. Despite carbon-carbon (C–C) bonds providing the central framework for organic molecules, development of biocatalytic methods for their formation has been largely confined to the use of a select few lyases over the last several decades, limiting the types of C–C bond-forming transformations possible through biocatalytic methods. This Review provides an update on the suite of enzymes available for highly selective biocatalytic C–C bond formation. Examples will be discussed in reference to the (1) native activity of enzymes, (2) alteration of activity through protein or substrate engineering for broader applicability, and (3) utility of the biocatalyst for abiotic synthesis.
Graphical Abstract
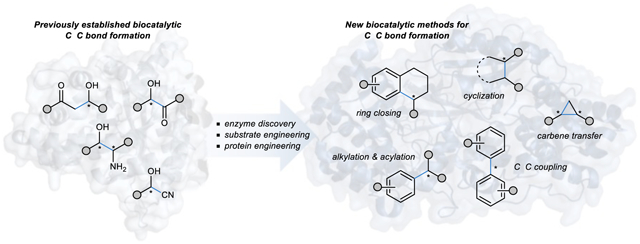
Discovery and application of enzymes that carry out new reactions are essential for the broader implementation of biocatalysts in organic synthesis. This Review highlights the most recent developments in biocatalytic methods for carbon-carbon bond formation.
Introduction
In nature, enzymes catalyse a myriad of highly selective transformations in the biosynthesis of primary and secondary metabolites in nature.1–3 Major advances in enzyme discovery and engineering over the last several decades have brought with them a surge in the development and implementation of biocatalytic reactions in organic syntheses executed in both academic and industrial laboratories.4–6 Enzymes are attractive catalysts in organic synthesis due to (1) the control they exhibit over the chemo-, site-, and stereoselectivity in the reactions they catalyse, (2) the new retrosynthetic disconnections they can offer over small molecule-mediated transformations, and (3) their sustainable footprint, especially in industrial-scale reactions.6–9
The major barrier in the use of biocatalysts in organic synthesis is the identification and development of a biocatalyst for a given reaction.10 This can be challenging because, while the three-dimensional control of a protein active site lends itself toward high levels of selectivity in biocatalytic reactions, this same tight regulation can limit the versatility of a biocatalyst through a restricted substrate scope.11 To overcome this limitation, enzymes can be engineered for the desired chemistry through rational design or random mutagenesis.12 The power of iterative rounds of random mutagenesis and screening was recently recognized with the 2018 Nobel Prize in Chemistry, awarded for the directed evolution of enzymes. In this approach, evolutionary pressure inherent to the development of enzymes in nature is expedited and applied artificially in a laboratory setting to evolve an enzyme for a target transformation or process (Fig. 1a).13–15 Additionally, advances in bioinformatic tools and machine learning are progressively merging the use of random and rational protein design for increasingly rapid protein engineering.16–20
Figure 1 |. Engineering enzymes for organic synthesis.
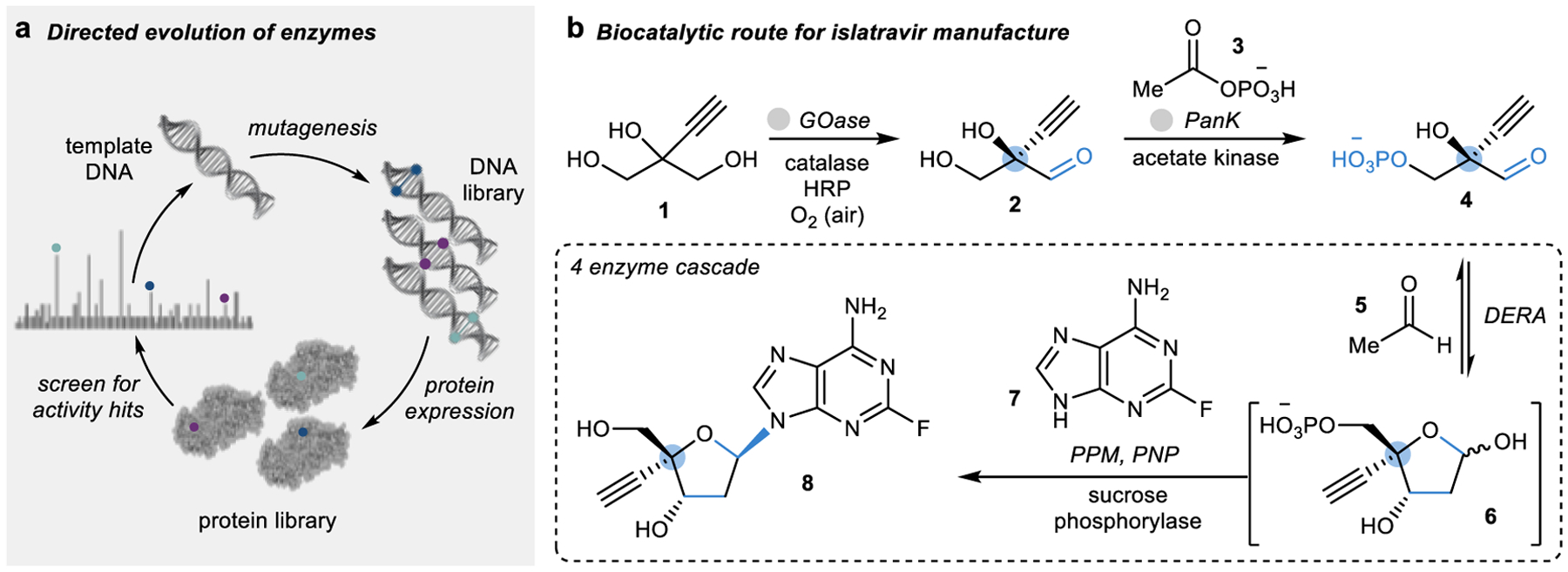
a | Typical iterative workflow of the directed evolution of enzymes for the development of new biocatalysts. b | Five enzymes were evolved by Merck and Codexis for the manufacture of islatravir (8), including galactose oxidase (GOase), pantothenate kinase (PanK), deoxyribose 5-phosphate aldolase (DERA), phosphopentomutase (PPM), and purine nucleoside phosphorylase (PNP).21
The biocatalytic route developed by Merck and Codexis for the manufacture of an anti-HIV drug, islatravir (8), demonstrates the tunability of enzymes for a desired transformation achievable with protein engineering.21 To overcome the inefficient toggling of protecting groups required to successfully set stereocenters present in non-natural nucleoside 8,22 five enzymes were selected based on the nucleoside salvage pathway present in bacteria in a new retrosynthetic approach (Fig. 1b).21 Each of the selected enzymes was engineered for a distinct purpose; for example, the oxidase (GOase) was engineered for improved activity and stereoselectivity whereas the aldolase (DERA) was engineered specifically for acetaldehyde (5) tolerance. Using these five engineered enzymes along with four auxiliary enzymes, islatravir (8) could be synthesized in a three-step biocatalytic sequence with a yield triple that of the original chemical synthesis (Fig. 1b).21,22
In addition to biocatalytic cascades, such as the islatravir (8) synthesis,21,23 biocatalysts can be used to catalyse key transformations in synthetic routes that draw on both biocatalytic methods and traditional small molecule reagents.24 The power of using enzymes to address synthetic challenges has been demonstrated over the last several decades with the increased implementation of biocatalytic processes in the synthesis of pharmaceutical compounds.5,6 However, a major limitation to the broader use of biocatalysts in both academic and industrial labs is the relatively narrow set of biocatalysts that can be reliably designed into a synthesis.
Carbon-carbon (C–C) bonds provide the central framework for all organic molecules and thus there are countless enzymes involved in their formation in primary and secondary metabolite pathways. Yet, the commonly employed biocatalysts for C–C bond formation are primarily restricted to a small subset of lyases, thereby limiting the scope of biocatalytic C–C bond formation to aldol reactions, acyloin condensations, and cyanohydrin formation in organic synthesis.25 Aldolases are by far the most commonly employed lyases for biocatalytic C–C bond formation and have been extensively developed into a versatile class of biocatalysts for site- and stereoselective aldol reactions.26–28 Thiamine diphosphate (ThDP)-dependent enzymes perform acyloin condensation reactions and have been used as valuable biocatalysts in the synthesis of enantioenriched building blocks.29–31 Lastly, biocatalytic cyanohydrin formation is achievable through several different mechanisms: (1) the addition of a cyanide ion to a ketone or aldehyde catalysed by a hydroxynitrile lyase32–34 or (2) the opening of an epoxide catalysed halohydrin dehalogenases.35 The breadth of research on these biocatalysts for C–C bond formation is captured in previous reviews on this topic.28,36,37
Discovery and application of enzymes that carry out new reactions are essential for the broader implementation of biocatalysts in organic synthesis, including new catalysts for C–C bond formation. Over the last several years, there have been numerous advances in biocatalytic C–C bond formation, using both wild-type enzymes and enzymes engineered for non-natural functions. This Review provides an update on the exciting new advances in biocatalytic C–C bond formation reactions including alkylation, acylation, oxidative C–C coupling, cyclisation, and carbene transfer reactions.
Alkylation and acylation reactions
Alkylation reactions are ubiquitous in nature and are one of the key C–C bond-forming events in natural product biosynthesis. These reactions can occur through various mechanisms, however one of the most common is an SN2-type reaction utilizing an activated electrophilic cofactor and a nucleophilic substrate (Fig. 2)
Figure 2 |. Biocatalytic alkylation reaction using activated electrophilic cofactors or substrates as alkyl donors.
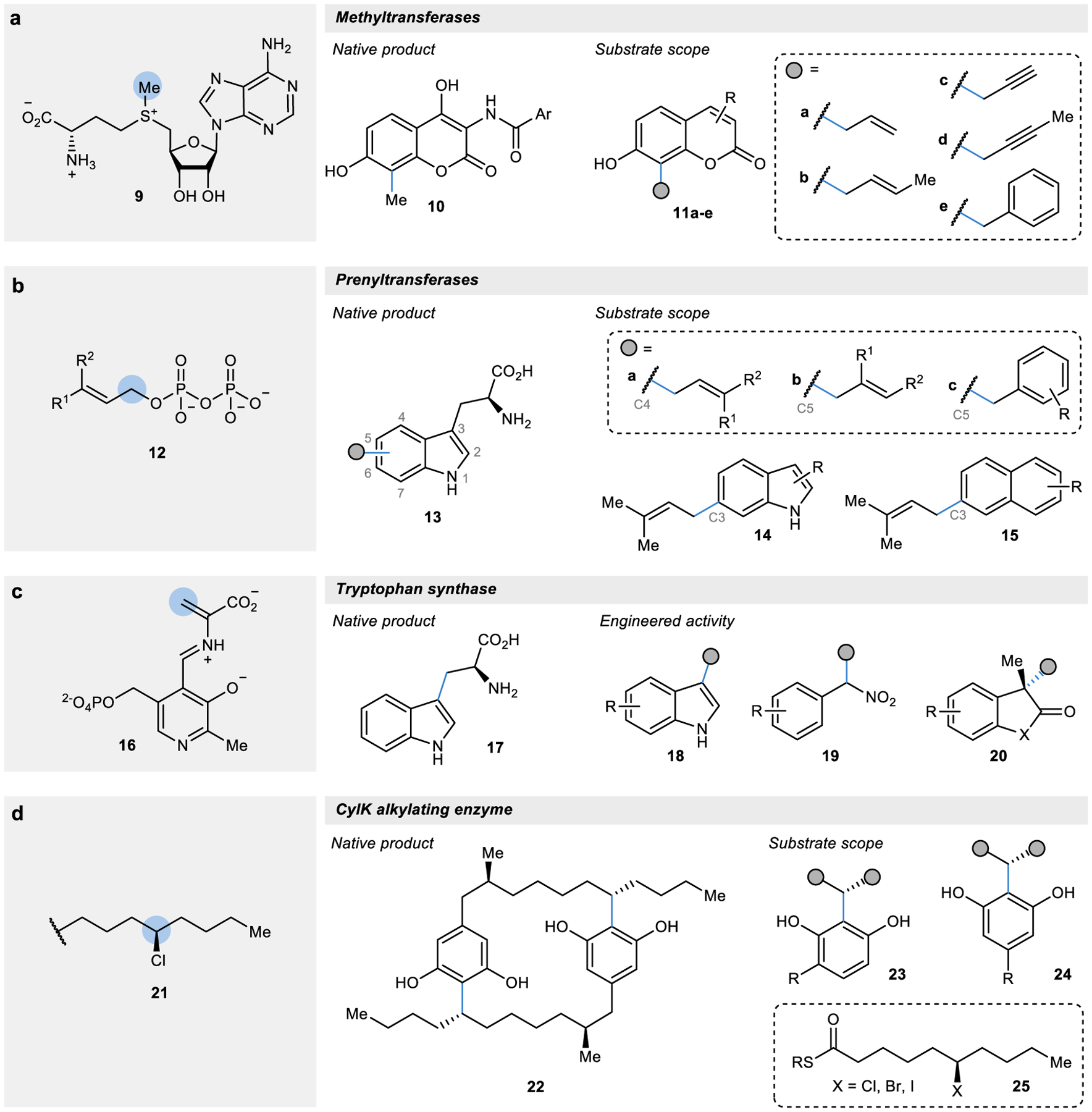
a | Methylation reactions catalysed by SAM-dependent enzymes can be diversified with the use of synthetic SAM analogues for broader C–C alkylation reactions.50 b | The diphosphate prenyl donors and acceptors in prenyltransferase-catalysed reactions can be modified for catalyst- and substrate-controlled site-selective alkylation reactions.54,55 c | Tryptophan synthase, a PLP-dependent enzyme, has been engineered to catalyse alkylation reactions on substituted indoles, nitro alkanes, and oxindoles.63–68 d | CylK utilizes an alkyl halide in the Friedel-Crafts alkylation of resorcinol derivatives.69,70
S-adenosyl-L-methionine (SAM)-dependent enzymes.
SAM (9) is the major methyl donor in biological systems and the second most common enzymatic cofactor after ATP.38,39 The most common reaction catalysed by enzymes harbouring a SAM cofactor is the transfer of the electrophilic methyl group bound to the sulfonium centre of SAM through an SN2 reaction (Fig. 2a). However, SAM-dependent enzymes can also perform radical reactions for the transfer of methyl or adenosyl groups,40,41 including those for C–C bond formation.42 Methyltransferases utilizing SAM cofactors are incredibly important in biological systems for applications including the biosynthesis of small molecules, protein repair, gene silencing, and chromatin remodelling.43
Unsurprisingly, given the crucial biological ramifications of these methylation events, the ability of these methyltransferases to selectively methylate their substrates is critical. This selective alkylation of such a diverse range of biomolecules has motivated the use of SAM-dependent methyltransferases as tools for biorthogonal labelling in biotechnology.44–48 In these systems, synthetic analogues of SAM are used to label nucleic acids or proteins by replacing the sulfonium centre with an aziridine ring or larger alkyl or allyl group.49 Upon nucleophilic attack of the target molecule on the SAM analogues, the resulting product will either be trapped by the aziridine analogue or alkylated with the alternative sulfonium substituent.49
Although methyltransferases have been used extensively for the modification and labelling of nucleic acids and proteins, methyltransferases using SAM analogues have only sparingly been explored for the biocatalytic C–C alkylation of small molecules in abiotic synthesis. Currently, the only example of biocatalytic C–C alkylation using SAM analogues is for a site-selective Friedel-Crafts alkylation at the unactivated C–H bond of a coumarin substrate to form 10 in Streptomyces secondary metabolite pathways (Fig. 2a).50 In these reactions, SAM analogues synthesized with bulkier substituents at the sulfonium centre were used as cofactors for the methyltransferase-catalysed Friedel-Crafts alkylation of several different coumarin derivatives to form coumarin 11 modified with allyl (a-b), propargyl (c-d), and benzyl (e) groups with excellent site-selectivity.50
Prenyltransferases.
Using a similar strategy to SAM-dependent methyltransferases, prenyltransferases take advantage of the excellent leaving group capability of the diphosphate group on allyl diphosphate substrates (12) for the transfer of an allylic group to a second more electron-rich substrate.51,52 The catalyst- and substrate-controlled site-selectivity exhibited by these enzymes have made them attractive biocatalysts for late-stage functionalization of natural products, especially due to the diverse biological effects the addition of a hydrophobic prenyl group can provide to a molecule.53
The investigation of the substrate scopes of several tryptophan prenyltransferases have shown that these enzymes are often promiscuous in their ability to catalyse the site-selective carbon-prenylation of a broad scope of aromatic tryptophan derivatives.52 Like SAM-dependent methyltransferases, both the alkyl donating and accepting substrates can be modified to diversify these reactions (Fig. 2b). A recent report demonstrated that a single wild-type prenyltransferase (FgaPT2) could donate alkyl groups from 34 different diphosphate derivatives, each forming a distinct C–C bond to give an alkylated product (13a-c).54 Interestingly, these FgaPT2-catalysed reactions exhibited substrate-dependent site-selectivity, with changes to the electronics and bulkiness of the alkyl group being donated conferring a switch in selectivity from the native C4 site of alkylation to a non-natural C5 alkylation. The divergent site-selectivity demonstrated by these reactions adds to the rapidly growing toolbox of site-selective alkylations, such as the C3 alkylations performed by the bacterial prenyltransferase PriB.55 This recently discovered enzyme is highly promiscuous, possessing the ability to alkylate a broad range of indole (14) and naphthalene (15) derivatives.55
Overall, the relative promiscuity of these enzymes enables rapid substrate engineering through perturbation of the alkyl group being transferred and the substrate being alkylated, making these enzymes an attractive class of potentially robust biocatalysts.
Pyridoxal 5-phosphate (PLP)-dependent enzymes.
PLP is a versatile cofactor present in enzymes that catalyse many transformations including transamination, epimerization, and decarboxylation. Despite this diversity, the shared driving force in all PLP-dependent mechanisms is the “electron sink” character of the PLP cofactor as it uses its π-system to stabilize the negative charge that develops at its Cα position.56 Several PLP-dependent enzymes have been identified to catalyse the formation of C–C bond forming reactions, including (1) aldol reactions catalysed by threonine aldolase,57 (2) the formation of alpha-amino ketones by a polyketide-like synthase,58,59 and (3) the formation of tryptophan and tryptophan derivatives by tryptophan synthase.60,61
In the catalytic mechanism of tryptophan synthase, a serine amino acid condenses onto the PLP cofactor and, upon dehydration, forms the highly electrophilic aminoacrylate intermediate species (16). Activation of this electrophilic cofactor allows for the nucleophilic attack by an indole to forge a new C–C bond, ultimately forming tryptophan (17). Tryptophan synthase is composed of an α subunit (TrpA) that acts as a regulatory subunit to the catalytic β subunit (TrpB), which binds PLP and catalyses the C–C bond formation event.62 Through directed evolution of the TrpB subunit, it was identified that the allosteric regulation provided by the TrpA subunit could be circumvented through a few key mutations in TrpB, increasing the biocatalytic utility of this enzyme.63 Since this first discovery, TrpB has been engineered extensively for broader C–C bond forming reactions on substituted indoles (18), nitro alkanes (19), and oxindoles (20) to form quaternary bonds (Fig. 2c).63–68
Halogenated electrophiles.
Unlike the highly activated electrophilic cofactors described so far, an alkylating enzyme (CylK) was recently discovered that solely relies on a halogenated alkyl chain (21) as the electrophilic alkyl group in an intermolecular Friedel-Crafts alkylation.69 The native reaction catalysed by this alkylating enzyme is a dimerization reaction between two halogenated resorcinol derivatives to form 22. In addition to this native reaction, CylK can catalyse the alkylation of a panel of resorcinol derivates (such as 23 and 24) using truncated alkyl halides (25; Fig. 2d).70
Intramolecular Friedel-Crafts alkylations.
Intermolecular Friedel-Crafts alkylations occur routinely in primary and secondary metabolite pathways and can be catalysed by some of the enzymes described so far; however, C–C bonds can also be formed in intramolecular Friedel-Crafts alkylations to forge a new ring, such as in the biosynthesis of many isoquinoline alkaloid and lignan natural products (Fig. 3a,c).71,72
Figure 3 |. Biosynthetic and biocatalytic ring-closing reactions.
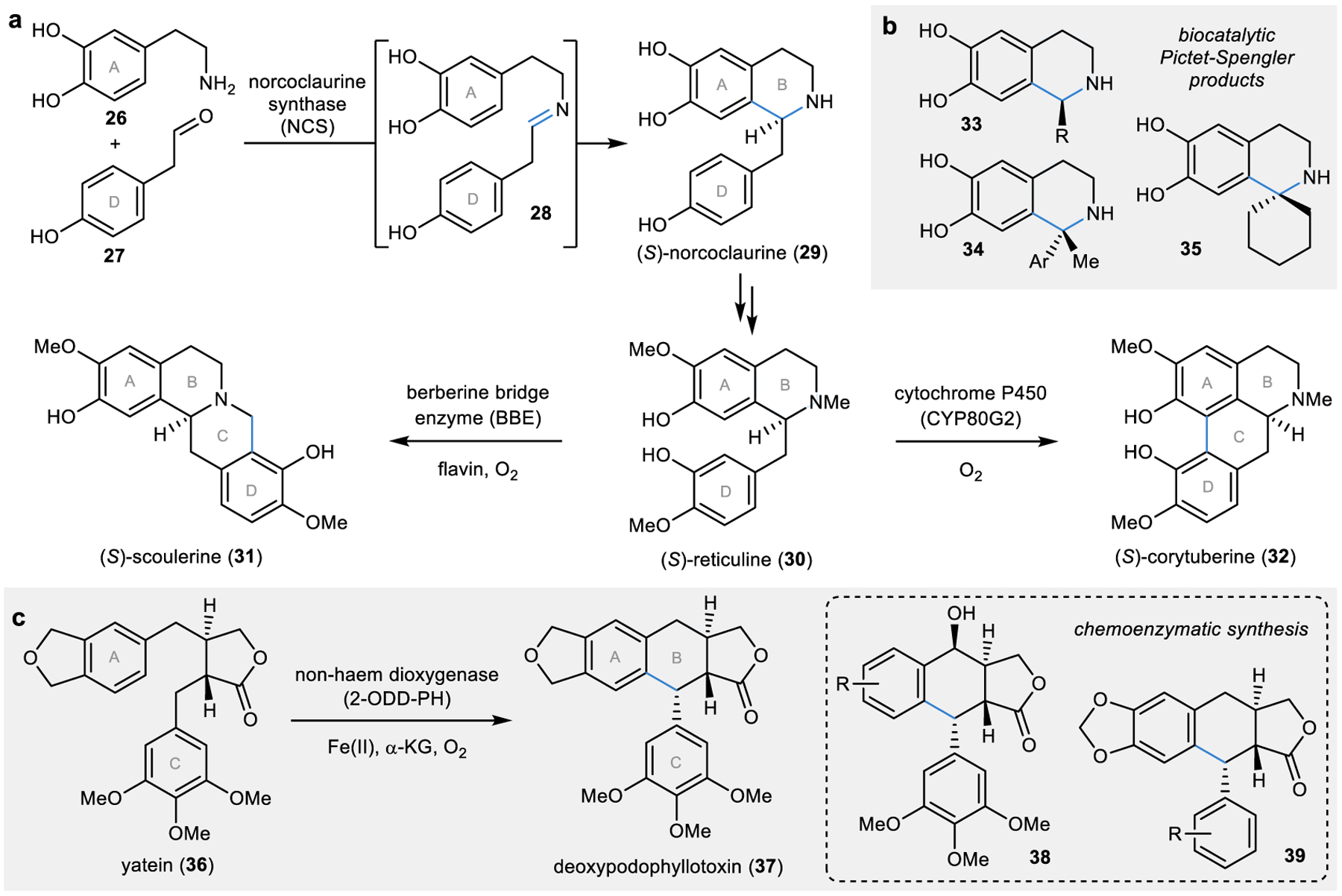
a | Several different enzymes catalyse intramolecular alkylation or oxidative coupling reactions to form a new ring in the biosynthesis of isoquinoline alkaloids.71 b | Noroclaurine synthases (NCSs) have been utilized as biocatalytic Pictet-Spenglerases for the formation of a panel of products.74,75 c | A non-haem dioxygenase catalyses an intramolecular alkylation reaction to form a new ring in the biosynthesis of podophyllotoxin lignan products.85 This biocatalytic reaction has been applied to the chemoenzymatic synthesis of several natural product analogues.88,89
One critical enzyme in the biosynthesis of isoquinoline alkaloid natural products is norcoclaurine synthase (NCS; Fig. 3a). NCS is a Pictet-Spenglerase that catalyses the condensation of an aldehyde 27 onto an amine 26 to form the iminium intermediate 28. This electrophilic intermediate (28) is then poised for an intramolecular Friedel-Crafts alkylation by nucleophilic attack to forge a C–C bond with the phenolic ring A to form (S)-norcoclaurine (29).73 Through the exploration NCSs for their broader biocatalytic utility for Pictet-Spengler reactions, the substrate scopes of these enzymes have been found to be relatively broad with the ability to accept a wide range of aldehyde derivates to form various tetrahydroisoquinoline products (33).74 As such, these enzymes have been applied to the chemoenzymatic syntheses of a range of alkaloid natural products and natural product derivatives.74 Additionally, a recent report demonstrated the ability of these enzymes to perform this ring closure with unactivated ketone coupling partners, thereby overcoming the limitation of coupling only to aldehydes.75 Using ketone substrates, quaternary centres can be formed in chiral disubstituted- and spiro-tetrahydroisoquinolines (see 34 and 35, respectively; Fig. 3b).75
Modification of the NCS product, (S)-norcoclaurine (29), by several methyltransferases and a cytochrome P450 yields (S)-reticuline (30), a branchpoint in the biosynthesis of many isoquinoline alkaloids.71,76 Isoquinoline alkaloids encompass many pharmacologically valuable natural products such as morphine and berberine. As such, extensive research has focused on the metabolic engineering of microorganisms for the scalable production of these molecules.77–81 The in vivo production of valuable molecules can be beneficial, especially when the chemical syntheses are not amenable to large-scale synthesis or the enzymes involved in the pathway are challenging to work with in vitro.82
As a branchpoint in the biosynthesis of isoquinoline alkaloids, several different C–C bond-forming reactions can be performed on (S)-reticuline (30) for further diversification. In particular, a fourth ring can be formed either through another intramolecular Friedel-Crafts alkylation to form (S)-scoulerine (31) or through an oxidative coupling reaction (described in more detail in the next section of this Review) to form (S)-corytuberine (32; Fig. 3a). The enzyme responsible for the Friedel-Crafts alkylation is the flavin-dependent berberine bridge enzyme (BBE). In all the examples described thus far, the first step before an alkylation event is the activation of an electrophilic carbon source; however, in the case of the BBE-catalysed alkylation, the unactivated methyl group is directly attacked by the phenol in 30 (ring D).83 This is made possible by the presence of a flavin cofactor in the active site of BBE which accepts a hydride from the methyl group being attacked in a single concerted step.83 This enzyme has not been explored for use as a biocatalyst as extensively as NCS; however, it has been identified that the substituents on ring D in 30 can be modified with successful C–C bond formation. Additionally, the site-selectivity of the bond formation can be biased in cases where a C–F bond is present at a potential alkylation site.84
Beyond alkaloids, another intramolecular Friedel-Crafts alkylation was recently identified in the biosynthesis of podophyllotoxin lignans (Fig. 3c).85 In particular, a non-haem dioxygenase was identified to catalyse the formation of the final ring in this biosynthetic pathway to form deoxypodophyllotoxin (37).85,86 Enzymes containing a non-haem iron are most commonly involved in hydroxylation events; however, this class of enzymes can also perform a myriad of other oxidative transformations.87 Evaluation of the substrate scope of the C–C coupling non-haem dioxygenase, 2-ODD-PH, demonstrated its ability to perform oxidative C–C alkylations on a variety of derivatives of 36, particularly with modifications to the substituents around rings A and C (Fig. 3c).88 This chemistry was used to perform late-stage C–C coupling reactions for the chemoenzymatic synthesis of products such as 38 and 39.89
Friedel-Crafts acylations.
Typically, acyl transfers in nature are carrier protein-dependent processes that proceed through SNAc-type reactions.90 However, similarly to the Friedel-Crafts alkylations described in this section so far, one unique bacterial acyltransferase has been identified to catalyse a Friedel-Crafts acylation on resorcinol substrates (Fig. 4).91 This acyltransferase natively transfers an acetyl group from 43 to the resorcinol substrate (40), either by directly forming the C–C bond in the acetylated product (41), or through an ester intermediate (42).92 In the latter mechanism, the acyltransferase can catalyse the biocatalytic equivalent of a Fries-rearrangement with an intramolecular acetyl transfer to form 41.
Figure 4 |. Site-selective acetylation of resorcinol derivatives catalysed by an acyltransferase.
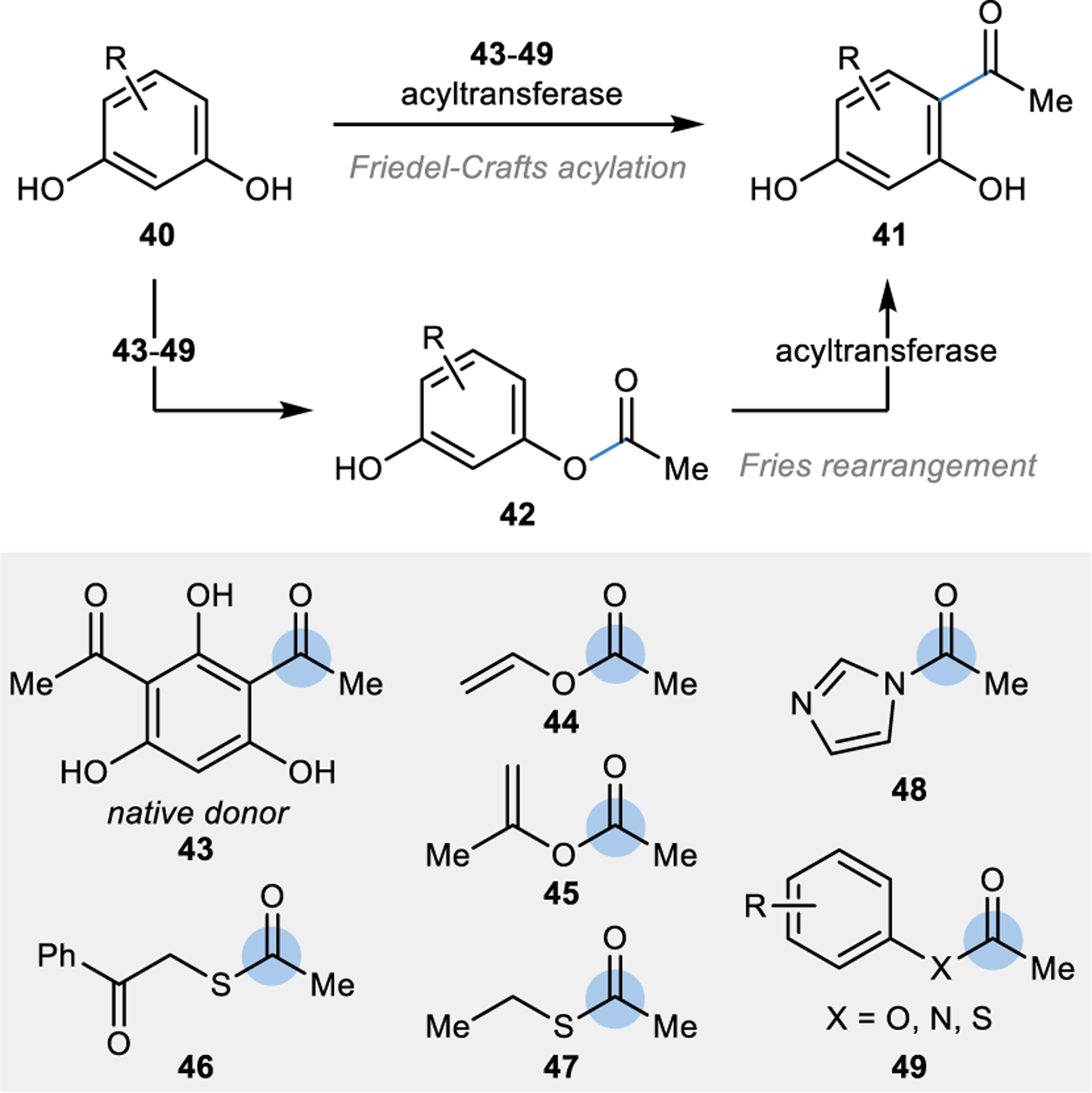
In addition to the native acyl donor (43), various acyl donors (44-49) were successfully employed in a biocatalytic Friedel-Crafts acylation or Fries rearrangement-like reaction to acetylate resorcinol derivates (40).92–94
Like the previously described intermolecular Friedel-Crafts reactions, both the acyl donor and acceptor could be modified to increase the versatility of these biocatalytic reactions. Specifically, a broad range of commercially available acetyl donors were productively utilized, including activated esters (44-45), thioesters (46-47), and amides (48-49) to the selectively acetylate various resorcinol derivates (40).92–94
Oxidative C–C coupling reactions
Oxidative coupling reactions perhaps offer the most efficient approach to C–C bond formation as two building blocks are directly stitched together.95 However, a major limitation in current synthetic methods for oxidative coupling lies in the control over the chemo-, site-, and stereochemical outcome of the reaction, particularly in the case of intermolecular coupling reactions.96,97 Nature has evolved enzymes that can control the oxidative dimerization of phenolic substrates with precise control of the site- and stereoselectivity of bond formation. These enzymes include cytochromes P450, laccases, and peroxidases. These three classes of enzymes share the ability to initiate radical mechanisms on phenolic substrates through the abstraction of a hydrogen atom from the phenol. However, the mechanism of hydrogen abstraction and extent of control exerted over the bond formation event in intermolecular and intramolecular reactions vary among these enzymes.
Intramolecular C–C coupling.
Cytochromes P450 catalyse a wide range of oxidative transformations leveraging their iron-haem cofactor.98 Often, these oxidative transformations involve a hydrogen atom abstraction from the substrate, followed by oxygenation through a rebound hydroxylation.99 However, the mechanism of these oxidative transformations can diverge following initial hydrogen atom abstraction, including C–C bond formation.98 The C–C bond formed through an oxidative coupling event in a number of natural products have been attributed to P450-catalysed reactions.100
Many of these P450-catalysed C–C coupling reactions occur intramolecularly, such as in the biosynthesis of (S)-corytuberine (32).101 In this reaction, the cytochrome P450 CYP80G2 initiates the oxidative mechanism on the substrate 30 with a hydrogen atom abstraction from one of the phenols. Instead of the canonical rebound hydroxylation typically performed by these enzymes, a C–C bond is formed either through a radical-radical coupling or by radical addition to a second phenolic group to form 32 (Fig. 3a). Another key intramolecular C–C coupling catalysed by a P450, is in the biosynthesis of the glycopeptide antibiotic vancomycin.102 Upon identification of the P450 OxyC responsible for catalysing this reaction, it was applied for the in vitro chemoenzymatic synthesis of vancomycin derivatives.103
Intermolecular C–C coupling.
In addition to intramolecular C–C coupling, oxidative coupling reactions can occur in nature between two phenolic substrates in an intermolecular fashion. These reactions are key steps in the biosynthesis of many dimeric biaryl secondary metabolites and can be catalysed by P450s, laccases, or peroxidases (Fig. 5).
Figure 5 |. Intermolecular oxidative coupling reactions in nature.
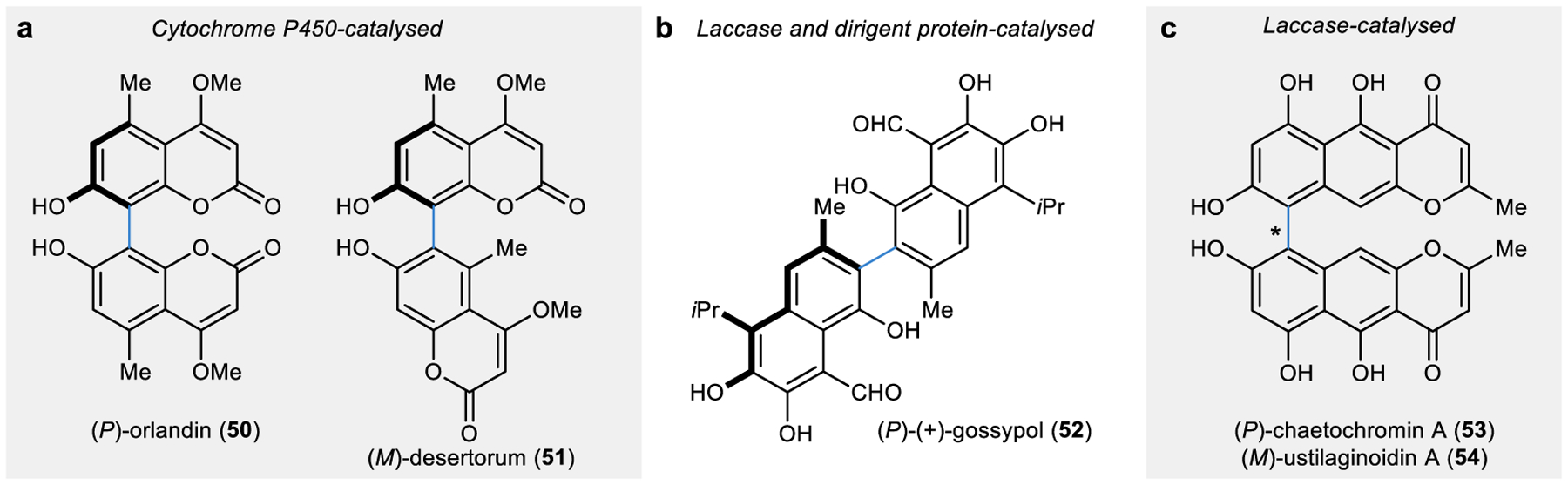
a | Cytochrome P450-catalysed site-selective oxidative coupling of coumarin substrates to form bicoumarin isomers.105 b | Laccase-catalysed oxidation of polyphenol substrates followed by dirigent protein-mediated radical coupling to yield enantioenriched gossypol.115 c | Laccase-catalysed atroposelective oxidative coupling of naphthopyrone substrates to form axially chiral biaryl products.116
P450s have been identified as the catalysts for intermolecular C–C coupling reactions for site-selective biaryl bond formation in several secondary metabolite pathways. For example, a group of Aspergillus P450s have been identified that catalyse the dimerization of a coumarin substrate to form up to six distinct axially chiral bicoumarins with catalyst-controlled site-selectivity.104 Two of these enzymes, KtnC and DesC, were characterized to selectively catalyse the site- and atroposelective C–C coupling to form axially chiral (P)-orlandin (50) and (M)-desertorum (51), respectively (Fig. 5a).105 This divergent site-selectivity among P450 homologues appears in several other bacterial and fungal secondary metabolite pathways, suggesting high levels of control exerted over the C–C bond formation event in P450 active sites.106–108
While P450s have only recently demonstrated an ability to catalyse site- and atroposelective intermolecular C–C coupling reactions, laccases and peroxidases have long been used in biotechnological applications for their oxidative coupling capabilities.109,110 Until recently, however, these enzymes were not known to impart site- or enantioselectivity in their oxidative coupling reactions limiting their utility for selective biocatalytic reactions. At a mechanistic level, these oxidative enzymes are responsible for the generation of radical intermediates but were not implicated in templating the substrates to exert control over the bond-forming event.111 In contrast, select radical substrates formed by laccases or peroxidases can rapidly be captured by a second class of protein catalysts, dirigent proteins.112,113 Dirigent proteins contain two distinct substrate binding sites and are uniquely poised to rapidly bind radical species in an aqueous solution and orient two of the substrates in a productive position with control over the site- and enantioselectivity of the C–C bond formation.113,114 Dirigent proteins have been identified in plant secondary metabolite pathways, including in the formation of lignans (such as 37) and the biaryl molecule gossypol (52; Fig. 5b).113,115
Recently, however, the first report of laccases that demonstrated control over the bond-forming event independently of an auxiliary dirigent protein was reported in the biosynthesis of axially chiral fungal ustilaginoidin natural products (Fig. 5c).116 Interestingly, while three of the characterized laccases selectively catalysed the formation of the (P)-atropisomer (53), one of the laccases, UstL, exhibited a catalyst-loading dependence on the atroposelectivity of the product, where the authors noted increasing concentrations of the catalyst gave an increased preference for the (M)-atropisomer (54).116 Although the mechanism behind the selectivity switch observed with UstL is unclear at this point, this unique catalyst-controlled atroposelectivity is unprecedented among laccases and holds promise for the development of stereodivergent biocatalysts.
This growing subset of P450s and laccases that perform oxidative coupling reactions to afford biaryl products with complementary site- and atroposelectivity has been vastly underexplored as biocatalysts despite the promise they offer for these challenging transformations.
Cyclisation reactions
Nature has mastered the construction of diverse scaffolds from common precursors by evolving catalysts capable of controlling the outcome of cyclisation reactions. These reactions vary in complexity with some catalysing the formation of a single C–C bond, whereas others can catalyse the stereoselective formation of a series of new C–C bonds in cascade cyclisation reactions.
Terpene cyclases.
Terpenes constitute the largest natural product family with over 80,000 members.117 Despite the magnitude and structural diversity of this family, all these natural products stem from a similar five-carbon precursor unit. Most of the structural complexity of the products are installed in cascade cyclisation reactions wherein multiple fused rings and stereocenters are installed by a single enzyme (such as in Fig. 6a). The enzymes that catalyse these reactions are referred to as terpene cyclases (or terpene synthases). These enzymatic reactions are considered the most complex in nature, with a typical cyclisation reaction breaking or making new bonds at the majority of the carbon atoms comprising the backbone of the terpene precursor substrate.117 The complexity-generating nature of terpene cyclase-catalysed reactions have influenced the biomimetic syntheses of terpenoid natural products, such as the use of Lewis acid-mediated opening of vinyl epoxides for the cascade polycyclisations of terpenoids, however the stereocontrol of the cyclisation is still lacking in chemical methods.118,119
Figure 6 |. Cyclisation reactions catalysed by natural and engineered enzymes.
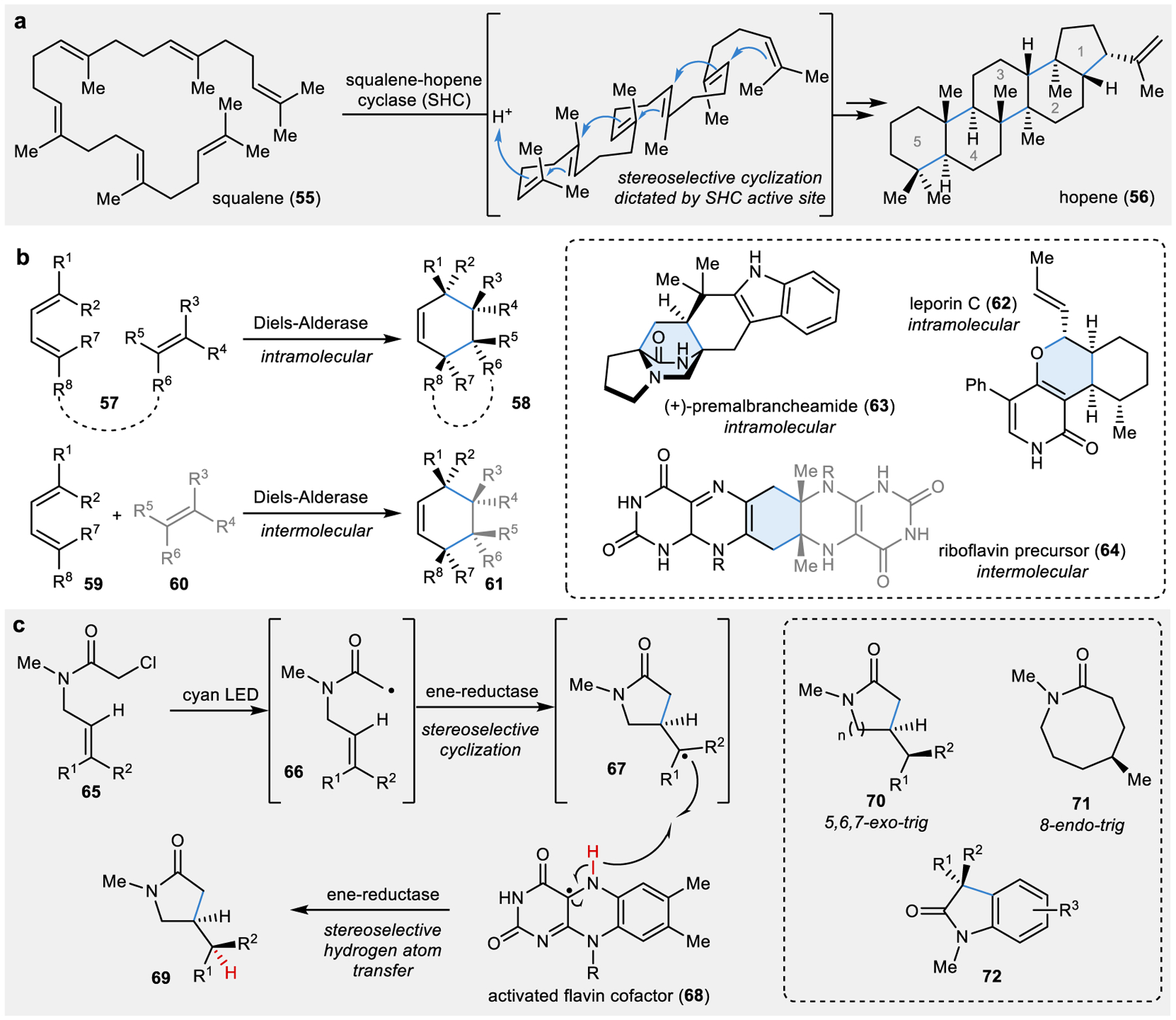
a | Terpene cyclases catalyse stereoselective cascade cyclisation reactions for the generation of polycyclic products.121 b | Diels-Alderases have been identified to catalyse intramolecular Diels-Alder reactions in secondary metabolite pathways and are hypothesized to be involved in intermolecular cycloaddition reactions.131,135 c | Engineered flavin-dependent ene-reductases can catalyse non-natural photobiocatalytic radical cyclisation reactions.141,142
Triterpene cyclases, such as the squalene-hopene cyclase (SHC), have been studied extensively for their structure and mechanism.120 In the native SHC-catalysed cascade cyclisation of squalene (55) to form hopene (56), five new C–C bonds are forged to form five fused rings and ten new stereocenters (Fig. 6a). It has been found that SHCs can catalyse a much wider variety of cyclisation reactions;121 examples of these include (1) stereoselective cascade cyclisations with smaller linear terpenes to form molecules with two or three fused rings,122,123 (2) Friedel-Crafts alkylations,124 (3) Prins-type reactions,125–127 and (4) isomerisation reactions.121 Additionally, these enzymes have demonstrated their amenability to evolution campaigns, often with a single amino acid change in the binding pocket of the enzyme resulting in a dramatic increase in activity with a non-native reaction.121 However, this promiscuity can also lead to the formation of multiple product isomers from a single linear substrate due to the ability of the active site to accommodate multiple conformations of the carbocation skeleton during cyclisation.128 Despite this potential limitation of some terpene cyclases, engineered SHC variants have successfully been employed in chemoenzymatic syntheses, such as in the synthesis of (+)-ambrein, a high-commodity natural product that is difficult to isolate from natural sources.129
Diels-Alderases.
In the list of classic, complexity-generating C–C bond forming reactions, the Diels-Alder reaction comes close to the top. This transformation has been central to the synthesis of six-membered rings in complex natural products and simple structures alike. This bread-and-butter reaction is embedded in the retrosynthetic logic chemists use to develop strategies toward target molecules, which has driven the development of variants of the Diels-Alder reaction that allow this cycloaddition to be applied intra- or intermolecularly and on substrates with a broad range of electronic properties (see 57 for intramolecular cycloaddition and 59-60 for intermolecular cycloaddition in Fig. 6b). Despite the common occurrence of the cyclohexene motif in natural product cores, the first enzymatic Diels-Alder reactions were carried out with proteins engineered to this non-native reactivity by leveraging the rate enhancement possible through templating of a given diene and dienophile and the acid-base type activation possible in an active site.130
While a number of artificial Diels-Alderases have been developed, natural Diels-Alderases have proven to be a much more elusive target and have garnered a great deal of attention over the last several decades.131 Elements of this controversy arise from the dual action of many “Diels-Alderases” as many enzymes given this label generate one of the Diels-Alder partners in addition to potentially catalysing the cyclization event. This controversy has also been fuelled by debate over what mechanistically constitutes a Diels-Alder cycloaddition, with strict definitions calling for concerted formation of two new C–C bonds and more liberal labelling of Diels-Alderases catalysing these bond formations in a step-wise fashion.
Recently, crystal structures of several bifunctional enzymes were reported that catalyse intramolecular Diels-Alder reactions: (1) SAM-dependent O-methyltransferase LepI in leporin C (62) biosynthesis132,133 and (2) short-chain dehydrogenases MalC and PhqE in (+)-premalbrancheamide (63) biosynthesis.134 Although these enzymes are homologous to functionally diverse enzyme classes, it appears that enzymes have evolved away from their canonical functions to act as a scaffold for stereoselective Diels-Alder reactions. This possesses a challenge in assigning cyclase function based on sequence. Although, several intermolecular [4+2] cycloadditions have been proposed to be catalysed by a Diels-Alderase, these proposals have not been experimentally confirmed.135 One example of these is riboflavin synthase, which catalyses the putative intermolecular cycloaddition of two molecules to form a key intermediate 64 in the riboflavin biosynthetic pathway that has been isolated and characterized.136 Ultimately, both characterization of the chemistry mediated by putative cyclases and mechanistic investigations are required to accurately label a Diels-Alderase.
Ene-reductases.
Ene-reductases are a class of enzymes that natively catalyse the asymmetric reduction of activated alkenes. These flavin-dependent enzymes deliver a hydride to one face of the alkene substrate. Subsequent protonation of the reduced C–C bond by a conserved tyrosine residue reduces the alkene in a stereo-controlled fashion.137 Ene-reductases are robust enzymes, typically with broad substrate scopes, high yields, and excellent selectivity, garnering them a great deal of attention as industrial biocatalysts.138,139
Beyond their widely studied and utilized canonical function, several ene-reductases were recently employed in the reduction of other functional groups. Ene-reductase variants lacking the conserved tyrosine residue responsible for the protonation event can transfer a hydride to α,β-unsaturated aldehydes and ketones triggering a reductive cyclisation event to form cyclopropanes.140 Although these reactions only exhibit moderate stereoselectivity, these experiments represent an important proof-of-concept that the reactivity of these transformations could be controlled through protein and substrate engineering beyond the native reactivity observed for ene-reductases.
As well as the non-natural cyclopropanation reactions, several recent reports demonstrated the ability for ene-reductases to catalyse the radical cyclisation of five- to eight-membered rings in photobiocatalytic reactions (Fig. 6c).141,142 In contrast to the typical mechanism of ene-reductases, where hydride transfer from the flavin cofactor to the substrate initiates the reaction, the delivery of a hydrogen atom quenches a radical species such as 66 to form a cyclised product (69; Fig. 6c).143 In the first report of this photobiocatalytic reaction, a panel of chloroacetamide derivatives (65) were exposed to cyan LEDs, causing the formation of a radical and subsequent radical cyclisation and hydride transfer to form products such as 70 and 71.141 A second report demonstrated the effectiveness of this reaction on a panel of chloroacetamide substrates to form diverse oxindole products (72).142
In addition to engineering ene-reductases for a completely new type of reaction when combined with photocatalysis, engineered haemoproteins for carbene transfer reactions serve as an excellent case study in engineering enzymes for completely new-to-nature types of transformations.
Carbene transfer reactions
The diversity of transformations achievable through carbene transfer including cyclopropanations, heteroatom-hydrogen (X–H) bond insertions, and rearrangements make carbene transformations valuable in synthetic chemistry.144–149 Although carbene transfer is not known to occur in nature, oxene transfer is commonly performed by cytochromes P450.99 These enzymes harbour a haem cofactor (73) in their active sites, which upon reduction binds molecular oxygen to form a highly reactive iron-oxene (74) intermediate species.99 As was previously described, this reactive iron-oxene species (74) typically performs a hydrogen atom abstraction, after which it can perform a myriad of oxidative transformations such as a rebound hydroxylation or insertion of oxygen into a double bond to form an epoxide (Fig. 7a).98,150
Figure 7 |. Engineering non-natural carbene transferases for biocatalytic C–C bond formation.
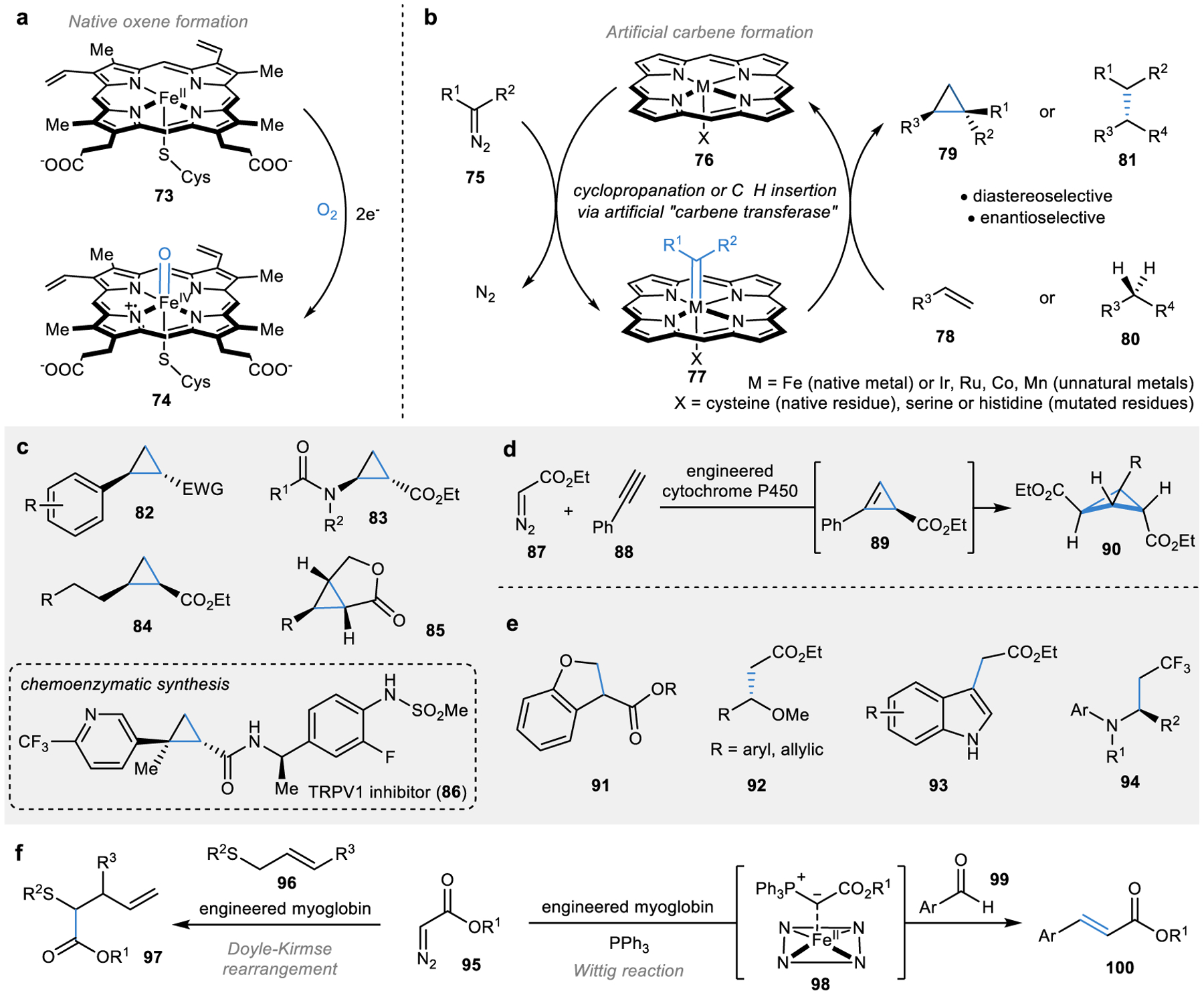
a | Natural iron-oxene formation in the haem cofactor of haemoproteins. b | Engineering artificial carbene-transferases through iron-carbene formation.151,168–170 c | Scope of cyclopropanation reactions catalysed by carbene transferases158–160,162–164 and representative use in chemoenzymatic synthesis.166 d | Carbene transfer into alkyne forms highly strained cyclopropene and bicyclobutane products.165 e | Carbene insertion into C–H bonds forms alkylated products.168,169,171–174 f | Carbene transferases have catalysed C–C bond formation through rearrangement reactions and Wittig reactions.175–177
In 2013, Arnold and co-workers published the first of a series of reports describing the utilization of the bacterial P450 BM3 for carbene transfer.151 Arnold discovered that replacement of molecular oxygen with a diazoacetate reagent (75) allowed for the formation of an iron-carbene species (77) in the P450 haem cofactor. In the presence of an olefin (78), carbene transfer could occur to form a cyclopropane (79) with the stereo-control provided by an enzyme active site, marking the first instance of a non-natural “carbene transferase” (Fig. 7b).
Engineering carbene transferases.
Following Arnold’s initial report, several research groups have led the way in engineering P450 BM3 and other haemoproteins into carbene transferases for cyclopropanation of olefins, alkylation through insertion into C–H bonds, and C–C bond formation through rearrangement reactions.152 Engineering efforts have focused on mutagenesis and alterations to the haem cofactor and coordinating metal ion to modulate oxidation potential and overcome obstacles including (1) limited substrate scope, (2) access to a single isomer of the desired product, and (3) competition with the native iron-oxene formation.152 The pursuit of this new-to-nature biocatalyst serves as an excellent example for how enzymes can be engineered into refined biocatalysts for a given reaction.
In 2013, Arnold’s group identified that mutagenesis of the cysteine residue, which serves as the axial ligand to the P450 haem cofactor, to a serine increased the reduction potential of the resting state enzyme thereby abolishing monooxygenase activity and giving a preference to the formation of a carbene over an oxene.153 This point mutation was then applied to this highly conserved residue in other P450s, forming a suite of enzymes dubbed “P411s” due to the change in their absorbance from 450 nm to 411 nm.154 Additionally, nucleophilic residues in the active site causing mechanism-based inhibition were identified and removed to further promote carbene transfer to a desired substrate.155 The Fasan group ventured beyond P450s to other haemoproteins and successfully engineered the first myoglobin capable of carbene transfer.156 For the myoglobin-derived carbene transferases, modification of the haem cofactor through installation of alternative substitution patterns increased the efficiency of iron-carbene formation and enabled reactions to be performed aerobically without inhibition of carbene transfer by oxygen.157
Olefin cyclopropanation.
Over the last five years, haemoproteins have been engineered for modified selectivity and substrate scope of olefin cyclopropanations performed by carbene transferases through directed evolution and rational mutagenesis of the active site.152 These engineering efforts have provided access to stereo-divergent cyclopropane products with control over both the diastereo- and enantioselectivity in carbene transfer reactions.
The substrate scope of these reactions has moved beyond phenyl-substituted olefins (forming 82) to form cyclopropanes substituted with heteroatoms (83) or aliphatic chains (84) to access more diverse cyclopropanes (Fig. 7c).158–161 Furthermore, the electron withdrawing group on the diazo reagent could be modified from an ester moiety to a nitrile or trifluoromethyl substituent to form the corresponding cyclopropanes (82).162,163 Additionally, these cyclopropanations can occur intramolecularly to form products such as 85.164 Recently, this cyclopropanation chemistry was extended beyond alkenes to alkynes (88), in which a strained cyclopropene (89) was generated.165 Cyclopropene products (89) are primed to undergo a second carbene transfer to form a bicyclobutane product (90) or be derivatized with chemical methods (Fig. 7d).165
These reactions are generally performed in whole E. coli cells, requiring no purification of the active catalysts. Additionally, these reactions can typically be performed on gram-scale and have been applied to the syntheses of several cyclopropane-containing natural products and pharmacologically-relevant molecules, such as the TRPV1 inhibitor (86).166,167
Carbene insertion into C–H bonds.
Beyond cyclopropanation, these engineered haemoproteins can perform insertion into C–H bonds to form various types of products (Fig. 7e). In 2016, Hartwig and co-workers found that replacing the iron in the P450 or myoglobin haem with iridium promoted carbene insertion into C–H bonds to form products such as 91.168,169 This demonstrated the ability to alter the reactivity of an enzyme by altering the catalytic metal ion without the need for extensive evolution. Shortly after these initial findings, the Fasan group demonstrated that several first-row transitions metals such as cobalt and manganese could inflict similar changes in reactivity to enable C–H insertions.170
More recently, several groups have demonstrated the ability to perform sp3 C–H alkylations on unactivated substrates using evolved P411 or myoglobin variants containing the native iron-haem cofactor.171 These C–H insertion reactions can be performed on a wide range of substrates (92). Further evolution broadened the substrate scope even more to alkylate unprotected indoles (93)172,173 and N-aryl pyrrolidine substrates (94).174 For the latter substrates, trifluoroethyl groups were installed through insertion into α-amino C–H bonds.174 These reactions can either be performed with purified enzyme or in whole E. coli cells expressing the active catalyst as described for the cyclopropanation reactions. Additionally, the scalability and relevance of haemoprotein-catalysed C–H insertions were demonstrated in several chemoenzymatic syntheses, such as in the synthesis of the anti-inflammatory drug indomethacin172 and enantio-complementary alkaloids.171
Other reactions catalysed by carbene transferases.
Although cyclopropanations and C–H insertions have been the most well-established C–C bond-forming reactions catalysed by engineered carbene transferases, extensive substrate and protein engineering have resulted in the discovery of other reactivity (Fig. 7f). For example, exposure of the diazoester reagent (95) and an allylic sulfide (96) to an engineered myoglobin led to the formation of a sulfur ylide intermediate that then underwent a [2,3]-sigmatropic rearrangement to form 97.175 Alternatively, exposure of the diazoester reagent (95) and an engineered myoglobin to triphenylphosphine promoted the formation of the proposed intermediate 98, which, when in the presence of an aldehyde (99) could undergo a Wittig reaction to form olefin 100.176,177
Conclusion and outlook
Enzymes offer an attractive alternative to many small molecule-mediated transformations due to the high levels of chemo-, site-, and stereoselectivity they impart in their reactions. However, a major obstacle to the routine incorporation of biocatalysts in organic synthesis currently lies in the relative lack of diversity and availability of biocatalysts for a given transformation. Despite the surge in the development of new biocatalytic methods over the last several decades, additional biocatalysts need to be developed to increase the library of biocatalysts, particularly for C–C bond formation. Until recently, biocatalytic C–C bond formation has primarily been limited to the use of a small subset of enzymes, limiting the versatility of biocatalytic C–C bond formation. This Review highlights the major advances over the last several years in enzyme discovery and the development of biocatalysts for more versatile C–C bond-forming reactions including alkylation, acylation, oxidative coupling, cyclisation, and carbene transfer reactions. Technological advances have accelerated the discovery of novel biocatalysts, and as more synthetic chemist embrace biocatalytic methods, this area is ripe for development of transformations that will build molecular scaffolds. We anticipate the C–C bond forming transformations outlined in this Review will be enabling for synthetic chemists brave enough to incorporate biocatalysts into their syntheses and direct chemists to additional resources that bridge the educational gap between organic chemistry and biocatalysis.7,178–180
Acknowledgements
The authors are grateful for support from the National Institutes of Health (R35 GM124880 and T32 GM008353).
Glossary terms
- Lyase
A class of enzymes characterized by their ability to break or form a new chemical bond using mechanisms other than hydrolysis or oxidation.
- Atroposelective
A preference for the formation of a specific stereoisomer (or atropisomer) containing an axis of chirality caused by hindered rotation around a single bond that results in only one of two possible stable conformations.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Drew SW & Demain AL Effect of primary metabolites on secondary metabolism. Annu. Rev. Microbiol 31, 343–356 (1977). [DOI] [PubMed] [Google Scholar]
- 2.Williams DH, Stone MJ, Hauck PR & Rahman SK Why are secondary metabolites (natural products) biosynthesized? J. Nat. Prod 52, 1189–1208 (1989). [DOI] [PubMed] [Google Scholar]
- 3.Maplestone RA, Stone MJ & Williams DH The evolutionary role of secondary metabolites: A review. Gene 115, 151–157 (1992). [DOI] [PubMed] [Google Scholar]
- 4.Devine PN et al. Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem 2, 409–421 (2018). [Google Scholar]
- 5.Campos KR et al. The importance of synthetic chemistry in the pharmaceutical industry. Science 363, eaat0805 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Bornscheuer UT et al. Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Turner NJ & O’Reilly E Biocatalytic retrosynthesis. Nat. Chem. Biol 9, 285–288 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Sheldon RA & Woodley JM Role of biocatalysis in sustainable chemistry. Chem. Rev 118, 801–838 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Hönig M, Sondermann P, Turner NJ & Carreira EM Enantioselective chemo- and biocatalysis: Partners in retrosynthesis. Angew. Chem. Int. Ed 56, 8942–8973 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Schoemaker HE, Mink D & Wubbolts MG Dispelling the myths: Biocatalysis in industrial synthesis. Science 299, 1694–1697 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Hult K & Berglund P Enzyme promiscuity: Mechanism and applications. Trends Biotechnol 25, 231–238 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Brustad EM & Arnold FH Optimizing non-natural protein function with directed evolution. Curr. Opin. Chem. Biol 15, 201–210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Packer MS & Liu DR Methods for the directed evolution of proteins. Nat. Rev. Genet 16, 379–394 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Renata H, Wang ZJ & Arnold FH Expanding the enzyme universe: Accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed 54, 3351–3367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner NJ Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol 5, 567–573 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Fox RJ et al. Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol 25, 338–344 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Fox RJ & Huisman GW Enzyme optimization: Moving from blind evolution to statistical exploration of sequence-function space. Trends Biotechnol 26, 132–138 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Lutz S Beyond directed evolution: Semi-rational protein engineering and design. Curr. Opin. Biotechnol 21, 734–743 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Currin A, Swainston N, Day PJ & Kell DB Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem. Soc. Rev 44, 1172–1239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang KK, Wu Z & Arnold FH Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Huffman MA et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 366, 1255–1259 (2019). [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin M et al. Enantioselective synthesis of 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) via enzymatic desymmetrization. Org. Lett 19, 926–929 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Schrittwieser JH, Velikogne S, Hall M & Kroutil W Artificial biocatalytic linear cascades for preparation of organic molecules. Chemical Reviews 118, 270–348 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Rudroff F et al. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal 1, 12–22 (2018). [Google Scholar]
- 25.Brovetto M, Gamenara D, Saenz Méndez P & Seoane GA C–C bond-forming lyases in organic synthesis. Chem. Rev 111, 4346–4403 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Fessner W-D Enzyme mediated C–C bond formation. Curr. Opin. Chem. Biol 2, 85–97 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Machajewski TD & Wong C-H The catalytic asymmetric aldol reaction. Angew. Chem. Int. Ed 39, 1352–1375 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Schmidt NG, Eger E & Kroutil W Building bridges: Biocatalytic C–C-bond formation toward multifunctional products. ACS Catal 6, 4286–4311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pohl M, Sprenger GA & Müller M A new perspective on thiamine catalysis. Curr. Opin. Biotechnol 15, 335–342 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Müller M, Gocke D & Pohl M Thiamin diphosphate in biological chemistry: Exploitation of diverse thiamin diphosphate-dependent enzymes for asymmetric chemoenzymatic synthesis. FEBS J 276, 2894–2904 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Pohl M, Lingen B & Müller M Thiamin diphosphate-dependent enzymes: New aspects of asymmetric C–C bond formation. Chem. Eur. J 8, 5288–5295 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Effenberger F, Förster S & Wajant H Hydroxynitrile lyases in stereoselective catalysis. Curr. Opin. Biotechnol 11, 532–539 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Griengl H, Schwab H & Fechter M The synthesis of chiral cyanohydrins by oxynitrilases. Trends Biotechnol 18, 252–256 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Sukumaran J & Hanefeld U Enantioselective C–C bond synthesis catalysed by enzymes. Chem. Soc. Rev 34, 530–542 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Schallmey A & Schallmey M Recent advances on halohydrin dehalogenases-from enzyme identification to novel biocatalytic applications. Appl. Microbiol. Biotechnol 100, 7827–7839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miao Y, Rahimi M, Geertsema EM & Poelarends GJ Recent developments in enzyme promiscuity for carbon-carbon bond-forming reactions. Curr. Opin. Chem. Biol 25, 115–123 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Resch V, Schrittwieser JH, Siirola E & Kroutil W Novel carbon-carbon bond formations for biocatalysis. Curr. Opin. Biotechnol 22, 793–799 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cantoni GL Biological methylation: selected aspects. Annu. Rev. Biochem 44, 435–451 (1975). [DOI] [PubMed] [Google Scholar]
- 39.Chiang PK et al. S-Adenosylmethionine and methylation. FASEB J 10, 471–480 (1996). [PubMed] [Google Scholar]
- 40.Fontecave M, Atta M & Mulliez E S-adenosylmethionine: Nothing goes to waste. Trends Biochem. Sci 29, 243–249 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Lin H S-adenosylmethionine-dependent alkylation reactions: When are radical reactions used? Bioorg. Chem 39, 161–170 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yokoyama K & Lilla EA C–C bond forming radical SAM enzymes involved in the construction of carbon skeletons of cofactors and natural products. Nat. Prod. Rep 35, 660–694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schubert HL, Blumenthal RM & Cheng X Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci 28, 329–335 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Struck AW, Thompson ML, Wong LS & Micklefield J S-adenosyl-methionine-dependent methyltransferases: Highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem 13, 2642–2655 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Klimasauskas S & Weinhold E A new tool for biotechnology: AdoMet-dependent methyltransferases. Trends Biotechnol 25, 99–104 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Dalhoff C, Lukinavičius G, Klimasăuskas S & Weinhold E Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat. Chem. Biol 2, 31–32 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Motorin Y et al. Expanding the chemical scope of RNA: Methyltransferases to site-specific alkynylation of RNA for click labeling. Nucleic Acids Res 39, 1943–1952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peters W et al. Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angew. Chem. Int. Ed 49, 5170–5173 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Deen J et al. Methyltransferase-directed labeling of biomolecules and its applications. Angew. Chem. Int. Ed 56, 5182–5200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stecher H et al. Biocatalytic Friedel-Crafts alkylation using non-natural cofactors. Angew. Chem. Int. Ed 48, 9546–9548 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Liang PH, Ko TP & Wang AH Structure, mechanism and function of prenyltransferases. Eur. J. Biochem 269, 3339–3354 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Tanner ME Mechanistic studies on the indole prenyltransferases. Nat. Prod. Rep 32, 88–101 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Epifano F, Genovese S, Menghini L & Curini M Chemistry and pharmacology of oxyprenylated secondary plant metabolites. Phytochem 68, 939–953 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Bandari C et al. FgaPT2, a biocatalytic tool for alkyl-diversification of indole natural products. MedChemComm 10, 1465–1475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elshahawi SI et al. Structure and specificity of a permissive bacterial C-prenyltransferase. Nat. Chem. Biol 13, 366–368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eliot AC & Kirsch JF Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem 73, 383–415 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Fesko K Threonine aldolases: Perspectives in engineering and screening the enzymes with enhanced substrate and stereo specificities. Appl. Microbiol. Biotechnol 100, 2579–2590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chun SW & Narayan ARH Biocatalytic synthesis of α-amino ketones. Synlett 30, 1269–1274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chun SW, Hinze ME, Skiba MA & Narayan ARH Chemistry of a unique polyketide-like synthase. J. Am. Chem. Soc 140, 2430–2433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miles EW Tryptophan synthase: A multienzyme complex with an intramolecular tunnel. Chem. Rec 1, 140–151 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Francis D, Winn M, Latham J, Greaney MF & Micklefield J An engineered tryptophan synthase opens new enzymatic pathways to β-methyltryptophan and derivatives. ChemBioChem 18, 382–386 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Dunn MF Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch. Biochem. Biophys 519, 154–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buller AR et al. Directed evolution of the tryptophan synthase beta-subunit for stand-alone function recapitulates allosteric activation. Proc. Natl. Acad. Sci. U.S.A 112, 14599–14604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boville CE et al. Engineered biosynthesis of beta-alkyl tryptophan analogues. Angew. Chem. Int. Ed 57, 14764–14768 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Boville CE, Romney DK, Almhjell PJ, Sieben M & Arnold FH Improved synthesis of 4-cyanotryptophan and other tryptophan analogues in aqueous solvent using variants of TrpB from Thermotoga maritima. J. Org. Chem 83, 7447–7452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murciano-Calles J, Romney DK, Brinkmann-Chen S, Buller AR & Arnold FH A panel of TrpB biocatalysts derived from tryptophan synthase through the transfer of mutations that mimic allosteric activation. Angew. Chem. Int. Ed 55, 11577–11581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dick M, Sarai NS, Martynowycz MW, Gonen T & Arnold FH Tailoring tryptophan synthase TrpB for selective quaternary carbon bond formation. J. Am. Chem. Soc, 19817–19822 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Romney DK, Murciano-Calles J, Wehrmüller JE & Arnold FH Unlocking reactivity of TrpB: A general biocatalytic platform for synthesis of tryptophan analogues. J. Am. Chem. Soc 139, 10769–10776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakamura H, Schultz EE & Balskus EP A new strategy for aromatic ring alkylation in cylindrocyclophane biosynthesis. Nat. Chem. Biol 13, 916–921 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Schultz EE, Braffman NR, Luescher MU, Hager HH & Balskus EP Biocatalytic Friedel–Crafts alkylation using a promiscuous biosynthetic enzyme. Angew. Chem. Int. Ed 58, 3151–3155 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Facchini PJ Alkaloid biosynthesis in plants: Biochemistry, cell biology, molecular regulation, and metabolic engineering applications. Annu. Rev. Plant Physiol. Plant Mol. Biol 52, 29–66 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Suzuki S & Umezawa T Biosynthesis of lignans and norlignans. J. Wood Sci 53, 273–284 (2007). [Google Scholar]
- 73.Sheng X & Himo F Enzymatic Pictet-Spengler reaction: Computational study of the mechanism and enantioselectivity of norcoclaurine synthase. J. Am. Chem. Soc 141, 11230–11238 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Patil MD, Grogan G & Yun H Biocatalyzed C–C bond formation for the production of alkaloids. ChemCatChem 10, 4783–4804 (2018). [Google Scholar]
- 75.Lichman BR, Zhao J, Hailes HC & Ward JM Enzyme catalysed Pictet-Spengler formation of chiral 1,1’-disubstituted- and spiro-tetrahydroisoquinolines. Nat. Comm 8, 14883 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sato F, Inai K & Hashimoto T in Applications of Plant Metabolic Engineering (eds Verpoorte R, Alfermann AW, & Johnson TS) 145–173 (Springer; Netherlands, 2007). [Google Scholar]
- 77.Leonard E, Runguphan W, O’Connor S & Prather KJ Opportunities in metabolic engineering to facilitate scalable alkaloid production. Nat. Chem. Biol 5, 292–300 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Galanie S, Thodey K, Trenchard IJ, Filsinger Interrante M & Smolke CD Complete biosynthesis of opioids in yeast. Science 349, 1095–1103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Diamond A & Desgagne-Penix I Metabolic engineering for the production of plant isoquinoline alkaloids. Plant Biotechnol. J 14, 1319–1328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thodey K, Galanie S & Smolke CD A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol 10, 837–844 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marienhagen J & Bott M Metabolic engineering of microorganisms for the synthesis of plant natural products. J. Biotechnol 163, 166–178 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Du J, Shao Z & Zhao H Engineering microbial factories for synthesis of value-added products. J. Ind. Microbiol. Biotechnol 38, 873–890 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Winkler A et al. A concerted mechanism for berberine bridge enzyme. Nat. Chem. Biol 4, 739–741 (2008). [DOI] [PubMed] [Google Scholar]
- 84.Resch V et al. Inverting the regioselectivity of the berberine bridge enzyme by employing customized fluorine-containing substrates. Chemistry 18, 13173–13179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lau W & Sattely ES Six enzymes from mayapple that complete the biosynthetic pathway to the etoposide aglycone. Science 349, 1224–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chang W-C, Yang Z-J, Tu Y-H & Chien T-C Reaction mechanism of a non-heme iron enzyme catalyzed oxidative cyclization via C–C bond formation. Org. Lett 21, 228–232 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Martinez S & Hausinger RP Catalytic mechanisms of Fe(II)- and 2-oxoglutarate-dependent oxygenases. J. Biol. Chem 290, 20702–20711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lazzarotto M et al. Chemoenzymatic total synthesis of deoxy-, epi-, and podophyllotoxin and a biocatalytic kinetic resolution of dibenzylbutyrolactones. Angew. Chem. Int. Ed 58, 8226–8230 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li J, Zhang X & Renata H Asymmetric chemoenzymatic synthesis of (−)-podophyllotoxin and related aryltetralin lignans. Angew. Chem. Int. Ed 58, 11657–11660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Austin MB & Noel JP The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep 20, 79–110 (2003). [DOI] [PubMed] [Google Scholar]
- 91.Hayashi A et al. Molecular and catalytic properties of monoacetylphloroglucinol acetyltransferase from Pseudomonas sp. YGJ3. Biosci Biotechnol Biochem 76, 559–566 (2012). [DOI] [PubMed] [Google Scholar]
- 92.Schmidt NG et al. Biocatalytic Friedel-Crafts acylation and Fries Reaction. Angew. Chem. Int. Ed 56, 7615–7619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmidt NG & Kroutil W Acyl donors and additives for the biocatalytic Friedel-Crafts acylation. Eur. J. Org. Chem 2017, 5865–5871 (2017). [Google Scholar]
- 94.Żądło-Dobrowolska A, Schmidt NG & Kroutil W Thioesters as acyl donors in biocatalytic Friedel-Crafts-type acylation catalyzed by acyltransferase from Pseudomonas protegens. ChemCatChem 11, 1064–1068 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kozlowski MC Oxidative coupling in complexity building transforms. Acc. Chem. Res 50, 638–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kozlowski MC, Morgan BJ & Linton EC Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem. Soc. Rev 38, 3193–3207 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bringmann G, Gulder T, Gulder TAM & Breuning M Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev 111, 563–639 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Isin EM & Guengerich FP Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta 1770, 314–329 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Denisov IG, Makris TM, Sligar SG & Schlichting I Structure and chemistry of cytochrome P450. Chem. Rev 105, 2253–2278 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Guengerich FP & Yoshimoto FK Formation and cleavage of C–C bonds by enzymatic oxidation–reduction reactions. Chem. Rev 118, 6573–6655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ikezawa N, Iwasa K & Sato F Molecular cloning and characterization of CYP80G2, a cytochrome P450 that catalyzes an intramolecular C–C phenol coupling of (S)-reticuline in magnoflorine biosynthesis, from cultured Coptis japonica cells. J. Biol. Chem 283, 8810–8821 (2008). [DOI] [PubMed] [Google Scholar]
- 102.Pylypenko O, Vitali F, Zerbe K, Robinson JA & Schlichting I Crystal structure of OxyC, a cytochrome P450 implicated in an oxidative C–C coupling reaction during vancomycin biosynthesis. J. Biol. Chem 278, 46727–46733 (2003). [DOI] [PubMed] [Google Scholar]
- 103.Forneris CC & Seyedsayamdost MR In vitro reconstitution of OxyC activity enables total chemoenzymatic syntheses of vancomycin aglycone variants. Angew. Chem. Int. Ed 57, 8048–8052 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Gil Girol C et al. Regio- and stereoselective oxidative phenol coupling in Aspergillus niger. Angew. Chem. Int. Ed 51, 9788–9791 (2012). [DOI] [PubMed] [Google Scholar]
- 105.Mazzaferro LS, Hüttel W, Fries A & Müller M Cytochrome P450-catalyzed regio- and stereoselective phenol coupling of fungal natural products. J. Am. Chem. Soc 137, 12289–12295 (2015). [DOI] [PubMed] [Google Scholar]
- 106.Präg A et al. Regio- and stereoselective intermolecular oxidative phenol coupling in Streptomyces. J. Am. Chem. Soc 136, 6195–6198 (2014). [DOI] [PubMed] [Google Scholar]
- 107.Obermaier S & Muller M Biaryl-forming enzymes from Aspergilli exhibit substrate-dependent stereoselectivity. Biochemistry 58, 2589–2593 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Zhao B et al. Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J. Biol. Chem 280, 11599–11607 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Mate DM & Alcalde M Laccase: A multi-purpose biocatalyst at the forefront of biotechnology. Microb. Biotechnol 10, 1457–1467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Azevedo AM et al. Horseradish peroxidase: A valuable tool in biotechnology. Biotechnol. Annu. Rev 9, 199–247 (2003). [DOI] [PubMed] [Google Scholar]
- 111.Jones SM & Solomon EI Electron transfer and reaction mechanism of laccases. Cell Mol. Life Sci 72, 869–883 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Davin LB et al. Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center. Science 275, 362–366 (1997). [DOI] [PubMed] [Google Scholar]
- 113.Pickel B & Schaller A Dirigent proteins: Molecular characteristics and potential biotechnological applications. Appl. Microbiol. Biotechnol 97, 8427–8438 (2013). [DOI] [PubMed] [Google Scholar]
- 114.Gasper R et al. Dirigent protein mode of action revealed by the crystal structure of AtDIR6. Plant Physiol 172, 2165–2175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu J, Stipanovic RD, Bell AA, Puckhaber LS & Magill CW Stereoselective coupling of hemigossypol to form (+)-gossypol in moco cotton is mediated by a dirigent protein. Phytochemistry 69, 3038–3042 (2008). [DOI] [PubMed] [Google Scholar]
- 116.Obermaier S, Thiele W, Fürtges L & Müller M Enantioselective phenol coupling by laccases in the biosynthesis of fungal dimeric naphthopyrones. Angew. Chem. Int. Ed 58, 9125–9128 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Christianson DW Structural and chemical biology of terpenoid cyclases. Chem. Rev 117, 11570–11648 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baunach M, Franke J & Hertweck C Terpenoid biosynthesis off the beaten track: Unconventional cyclases and their impact on biomimetic synthesis. Angewandte Chemie International Edition 54, 2604–2626 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Pronin SV & Shenvi RA Synthesis of highly strained terpenes by non-stop tail-to-head polycyclization. Nature Chemistry 4, 915–920 (2012). [DOI] [PubMed] [Google Scholar]
- 120.Siedenburg G & Jendrossek D Squalene-hopene cyclases. Appl. Environ. Microbiol 77, 3905–3915 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Syren PO, Henche S, Eichler A, Nestl BM & Hauer B Squalene-hopene cyclases-evolution, dynamics and catalytic scope. Curr. Opin. Struct. Biol 41, 73–82 (2016). [DOI] [PubMed] [Google Scholar]
- 122.Hoshino T, Kumai Y, Kudo I, Nakano S & Ohashi S Enzymatic cyclization reactions of geraniol, farnesol and geranylgeraniol, and those of truncated squalene analogs having C20 and C25 by recombinant squalene cyclase. Org. Biomol. Chem 2, 2650–2657 (2004). [DOI] [PubMed] [Google Scholar]
- 123.Seitz M et al. Synthesis of heterocyclic terpenoids by promiscuous squalene-hopene cyclases. Chembiochem 14, 436–439 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Hammer SC, Dominicus JM, Syrén P-O, Nestl BM & Hauer B Stereoselective Friedel–Crafts alkylation catalyzed by squalene hopene cyclases. Tetrahedron 68, 7624–7629 (2012). [Google Scholar]
- 125.Seitz M et al. Substrate specificity of a novel squalene-hopene cyclase from Zymomonas mobilis. J. Mol. Catal. B Enzym 84, 72–77 (2012). [Google Scholar]
- 126.Siedenburg G et al. Activation-independent cyclization of monoterpenoids. Appl. Environ. Microbiol 78, 1055–1062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hammer SC, Marjanovic A, Dominicus JM, Nestl BM & Hauer B Squalene hopene cyclases are protonases for stereoselective Brønsted acid catalysis. Nat. Chem. Biol 11, 121–126 (2015). [DOI] [PubMed] [Google Scholar]
- 128.Degenhardt J, Kollner TG & Gershenzon J Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70, 1621–1637 (2009). [DOI] [PubMed] [Google Scholar]
- 129.Ueda D, Hoshino T & Sato T Cyclization of squalene from both termini: Identification of an onoceroid synthase and enzymatic synthesis of ambrein. J. Am. Chem. Soc 135, 18335–18338 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Siegel JB et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 329, 309–313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Klas K, Tsukamoto S, Sherman DH & Williams RM Natural Diels-Alderases: Elusive and irresistable. J. Org. Chem 80, 11672–11685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ohashi M et al. SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature 549, 502–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cai Y et al. Structural basis for stereoselective dehydration and hydrogen-bonding catalysis by the SAM-dependent pericyclase LepI. Nat. Chem 11, 812–820 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dan Q et al. Fungal indole alkaloid biogenesis through evolution of a bifunctional reductase/Diels-Alderase. Nat. Chem 11, 972–980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Minami A & Oikawa H Recent advances of Diels-Alderases involved in natural product biosynthesis. J. Antibiot 69, 500–506 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Kim R-R et al. Mechanistic insights on riboflavin synthase inspired by selective binding of the 6,7-dimethyl-8-ribityllumazine exomethylene anion. J. Am. Chem. Soc 132, 2983–2990 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kohli RM & Massey V The oxidative half-reaction of Old Yellow Enzyme. The role of tyrosine 196. J. Biol. Chem 273, 32763–32770 (1998). [DOI] [PubMed] [Google Scholar]
- 138.Winkler CK, Tasnádi G, Clay D, Hall M & Faber K Asymmetric bioreduction of activated alkenes to industrially relevant optically active compounds. J. Biotechnol 162, 381–389 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Toogood HS & Scrutton NS Discovery, characterisation, engineering and applications of ene reductases for industrial biocatalysis. ACS Catal 8, 3532–3549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heckenbichler K et al. Asymmetric reductive carbocyclization using engineered ene reductases. Angew. Chem. Int. Ed 57, 7240–7244 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Biegasiewicz KF et al. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 364, 1166–1169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Black MJ et al. Asymmetric redox-neutral radical cyclization catalysed by flavin-dependent ‘ene’-reductases. Nat. Chem, 71–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sandoval BA, Meichan AJ & Hyster TK Enantioselective hydrogen atom transfer: Discovery of catalytic promiscuity in flavin-dependent ‘ene’-reductases. J. Am. Chem. Soc 139, 11313–11316 (2017). [DOI] [PubMed] [Google Scholar]
- 144.Ye T & McKervey MA Organic synthesis with alpha-diazo carbonyl compounds. Chem. Rev 94, 1091–1160 (1994). [Google Scholar]
- 145.Doyle MP & Forbes DC Recent advances in asymmetric catalytic metal carbene transformations. Chem. Rev 98, 911–936 (1998). [DOI] [PubMed] [Google Scholar]
- 146.Davies HM & Manning JR Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 451, 417–424 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang Y & Wang J Recent development of reactions with α-diazocarbonyl compounds as nucleophiles. Chem. Comm, 5350–5361 (2009). [DOI] [PubMed] [Google Scholar]
- 148.Ford A et al. Modern organic synthesis with α-diazocarbonyl compounds. Chem. Rev 115, 9981–10080 (2015). [DOI] [PubMed] [Google Scholar]
- 149.Franssen NMG, Walters AJC, Reek JNH & de Bruin B Carbene insertion into transition metal–carbon bonds: A new tool for catalytic C–C bond formation. Catal. Sci. Technol 1, 153–165 (2011). [Google Scholar]
- 150.Vaz ADN, McGinnity DF & Coon MJ Epoxidation of olefins by cytochrome P450: Evidence from site-specific mutagenesis for hydroperoxo-iron as an electrophilic oxidant. Proc. Natl. Acad. Sci. U.S.A 95, 3555–3560 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Coelho PS, Brustad EM, Kannan A & Arnold FH Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–310 (2013). [DOI] [PubMed] [Google Scholar]
- 152.Brandenberg OF, Fasan R & Arnold FH Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol 47, 102–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Coelho PS et al. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol 9, 485–487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Heel T, McIntosh JA, Dodani SC, Meyerowitz JT & Arnold FH Non-natural olefin cyclopropanation catalyzed by diverse cytochrome P450s and other hemoproteins. Chembiochem 15, 2556–2562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Renata H et al. Identification of mechanism-based inactivation in P450-catalyzed cyclopropanation facilitates engineering of improved enzymes. J. Am. Chem. Soc 138, 12527–12533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bordeaux M, Tyagi V & Fasan R Highly diastereoselective and enantioselective olefin cyclopropanation using engineered myoglobin-based catalysts. Angew. Chem 127, 1764–1768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sreenilayam G, Moore EJ, Steck V & Fasan R Stereoselective olefin cyclopropanation under aerobic conditions with an artificial enzyme incorporating an iron-chlorin e6 cofactor. ACS Catal 7, 7629–7633 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Key HM et al. Beyond iron: Iridium-containing P450 enzymes for selective cyclopropanations of structurally diverse alkenes. ACS Cent. Sci 3, 302–308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brandenberg OF et al. Stereoselective enzymatic synthesis of heteroatom-substituted cyclopropanes. ACS Catal 8, 2629–2634 (2018). [Google Scholar]
- 160.Knight AM et al. Diverse engineered heme proteins enable stereodivergent cyclopropanation of unactivated alkenes. ACS Cent. Sci 4, 372–377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Carminati DM & Fasan R Stereoselective cyclopropanation of electron-deficient olefins with a cofactor redesigned carbene transferase featuring radical reactivity. ACS Catal 9, 9683–9697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tinoco A, Steck V, Tyagi V & Fasan R Highly diastereo- and enantioselective synthesis of trifluoromethyl-substituted cyclopropanes via myoglobin-catalyzed transfer of trifluoromethylcarbene. J. Am. Chem. Soc 139, 5293–5296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chandgude AL & Fasan R Highly diastereo- and enantioselective synthesis of nitrile-substituted cyclopropanes by myoglobin-mediated carbene transfer catalysis. Angew. Chem. Int. Ed 57, 15852–15856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chandgude AL, Ren X & Fasan R Stereodivergent intramolecular cyclopropanation enabled by engineered carbene transferases. J. Am. Chem. Soc 141, 9145–9150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen K, Huang X, Kan SBJ, Zhang RK & Arnold FH Enzymatic construction of highly strained carbocycles. Science 360, 71–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bajaj P, Sreenilayam G, Tyagi V & Fasan R Gram-scale synthesis of chiral cyclopropane-containing drugs and drug precursors with engineered myoglobin catalysts featuring complementary stereoselectivity. Angew. Chem. Int. Ed 55, 16110–16114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang ZJ et al. Improved cyclopropanation activity of histidine-ligated cytochrome P450 enables the enantioselective formal synthesis of levomilnacipran. Angew. Chem. Int. Ed 53, 6810–6813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Key HM, Dydio P, Clark DS & Hartwig JF Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 534, 534–537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dydio P et al. An artificial metalloenzyme with the kinetics of native enzymes. Science 354, 102–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sreenilayam G, Moore EJ, Steck V & Fasan R Metal substitution modulates the reactivity and extends the reaction scope of myoglobin carbene transfer catalysts. Adv. Synth. Catal 359, 2076–2089 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang RK et al. Enzymatic assembly of carbon-carbon bonds via iron-catalysed sp3 C–H functionalization. Nature 565, 67–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vargas DA, Tinoco A, Tyagi V & Fasan R Myoglobin-catalyzed C–H functionalization of unprotected indoles. Angew. Chem. Int. Ed 57, 9911–9915 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Brandenberg OF, Chen K & Arnold FH Directed evolution of a cytochrome P450 carbene transferase for selective functionalization of cyclic compounds. J. Am. Chem. Soc 141, 8989–8995 (2019). [DOI] [PubMed] [Google Scholar]
- 174.Zhang J, Huang X, Zhang RK & Arnold FH Enantiodivergent α-amino C–H fluoroalkylation catalyzed by engineered cytochrome P450s. J. Am. Chem. Soc 141, 9798–9802 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tyagi V, Sreenilayam G, Bajaj P, Tinoco A & Fasan R Biocatalytic synthesis of allylic and allenyl sulfides through a myoglobin-catalyzed Doyle-Kirmse reaction. Angew. Chem. Int. Ed 55, 13562–13566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tyagi V & Fasan R Myoglobin-catalyzed olefination of aldehydes. Angew. Chem. Int. Ed 55, 2512–2516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Weissenborn MJ et al. Enzyme-catalyzed carbonyl olefination by the E. coli protein YfeX in the absence of phosphines. ChemCatChem 8, 1636–1640 (2016). [Google Scholar]
- 178.Clouthier CM & Pelletier JN Expanding the organic toolbox: A guide to integrating biocatalysis in synthesis. Chem. Soc. Rev 41, 1585–1605 (2012). [DOI] [PubMed] [Google Scholar]
- 179.Sheldon RA & Brady D Broadening the scope of biocatalysis in sustainable organic synthesis. ChemSusChem 12, 2859–2881 (2019). [DOI] [PubMed] [Google Scholar]
- 180.Sheldon RA, Brady D & Bode ML The hitchhiker’s guide to biocatalysis: Recent advances in the use of enzymes in organic synthesis. Chem. Sci 11, 2587–2605 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]