

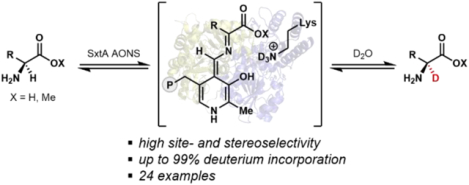
Abstract
α-2H amino acids are valuable precursors toward labeled pharmaceutical agents and tools for studying biological systems; however, these molecules are costly to purchase and challenging to synthesize in a site- and stereoselective manner. Here, we show that an α-oxo-amine synthase that evolved for saxitoxin biosynthesis, SxtA AONS, is capable of producing a range of α-2H amino acids and esters site- and stereoselectively using D2O as the deuterium source. Additionally, we demonstrate the utility of this operationally simple reaction on preparative scale in the stereoselective chemoenzymatic synthesis of a deuterated analog of safinamide, a drug used to treat Parkinson’s disease.
Keywords: biocatalysis, α-οxοamine synthase, pyridoxal phosphate, deuterium labeling, amino acid, chemoenzymatic synthesis
Graphical Abstract
Deuterated compounds are important tools in fundamental research and commercial endeavors including drug discovery and development.1–4 Deuterated drugs are often metabolized slower than the analogous protio-compounds due to the kinetic isotope effect, affording longer half-lives and allowing for lower doses.5 The FDA has recognized the potential benefits of deuterated pharmaceutical agents, approving the first analog of an existing drug, deutetrabenazine, in 2017.6
The value of deuterated compounds creates a demand for methods to selectively introduce isotopic labels into available building blocks. For example, α-2H amino acids are potential pharmaceutical precursors,7 can be incorporated into proteins to improve NMR signal,8 and are tools to probe protein and natural product metabolism.9,10 Despite the value of these molecules, synthetic access is challenging, with difficulties in achieving site- and stereoselectivity in the deuterium incorporation step. Established methods toward α-2H -amino acids follow multi step routes where protecting groups and chiral auxiliaries are required (see 1 to 2, Figure 1A), accounting for the hundred-fold higher cost for enantioenriched α-2H-amino acids relative to their protio counterparts.11–13 Recent approaches offer more efficient access by targeting deuterated compounds directly from the corresponding unlabeled acids (see 3 to 2, Figure 1A). Although these methods provide high-levels of deuterium incorporation, they often fall short in achieving site- and stereoselectivity on a wide array of substrates.14,15
Figure 1.
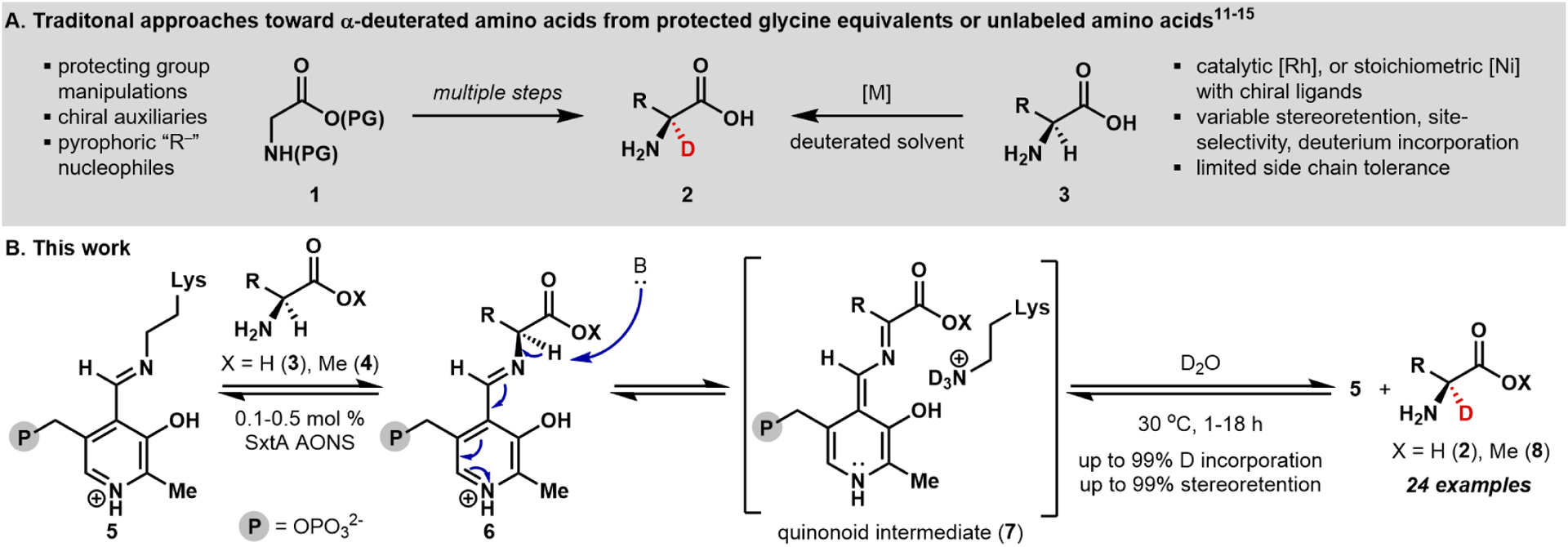
Preparations of deuterated compounds and amino acids. (A) Classic methods for synthesizing α-deuterated amino acids are lengthy (left). Recent chemical routes starting from the corresponding protio-amino acids vary in activity (right). (B) SxtA AONS mediates α-deuterium incorporation on a wide range of substrates.
A biocatalytic approach to the transformation of protio-amino acids to α-deuterated analogs offers several potential advantages, including precise stereoselectivity, the elimination of protecting groups, and mild, sustainable reaction conditions.16 Deuterium sources such as D2 and D2O are economical and available in bulk.17 Pyridoxal phosphate (PLP)-dependent enzymes are one class of enzymes that reversibly deprotonate α-amino acids,18,19 initiated through deprotonation of external aldimine intermediate 6, (Figure 1B).20,21 The corresponding quinonoid species 7 thus generated is deuterated by D2O or a deuterated catalytic lysine residue before release from the active site (see 7 to 8). In contrast to PLP-dependent transaminases and racemases, which catalyze deuterated amino acid formation with some substrate flexibility but rarely have control over site- and stereoselectivity,22,23 α-oxoamine synthases (AOS) have been shown to install a deuterium atom at the α-position of select amino acids stereoselectively. For example, in mechanistic studies of AOS proteins BioF and SPT, which naturally mediate the net decar-boxylative condensation of an amino acid with a thioester,24 incubation in D2O led to α-deuteration of their native substrates l-Ala and l-Ser, respectively.20,21
We recently reported the native activity of one AOS, SxtA AONS (8-amino-7-oxononanoate synthase), a domain within a multi-domain polyketide-like synthase.25 During saxitoxin biosynthesis, SxtA AONS generates ketone 11 from l-Arg (Figure 2). We now envision leveraging AOS enzymes as biocatalysts to produce enantioenriched α-2H amino acids. Here, we show that SxtA AONS is indeed an effective catalyst for α-deuteration of select l-amino acids and a broad panel of α-amino ester substrates (Figure 1B). These deuterated building blocks can be generated on preparative-scale and employed in multi-step synthesis as we have demonstrated in the synthesis of a deuterated form of the Parkinson’s drug, safinamide.26 To test the feasibility of AOS-mediated deuterium labeling, we first identified a robust enzyme, selecting the Microseira wollei SxtA AONS among four recombinant cyanobacterial homologs for its superior expression levels.27–29 SxtA AONS successfully catalyzed deuterium incorporation into its native amino acid substrate l-Arg (l-9) at modest levels (Figure 3A). After incubating SxtA AONS with 4 mM l-Arg overnight in D2O, approximately 20% of the acid had one deuterium atom incorporated as determined by LC-MS analysis (Figure 3C, black line). We then assayed the deuteration activity of SxtA AONS with a panel of related substrates: ketone 11 (Figure 3C, light blue line), d-Arg (d-9, green line), and both enantiomers of Arg-OMe (13, red lines).30 Whereas deuterium labeling was moderate for l-Arg, both ketone 11 and its structural mimic l-Arg-OMe reached maximum theoretical deuteration within 15 minutes. Interestingly, substrates deuterated at a low level by SxtA AONS were readily transformed into compounds that were efficiently deuterated through esterification to the corresponding methyl ester (see d-Arg vs. d-Arg-OMe). Minor deuterium incorporation into the acids and esters was observed in controls lacking active enzyme, while the lower pKa of ketone 11 led to 15% nonenzymatic deuteration (see Table S3).20
Figure 2.
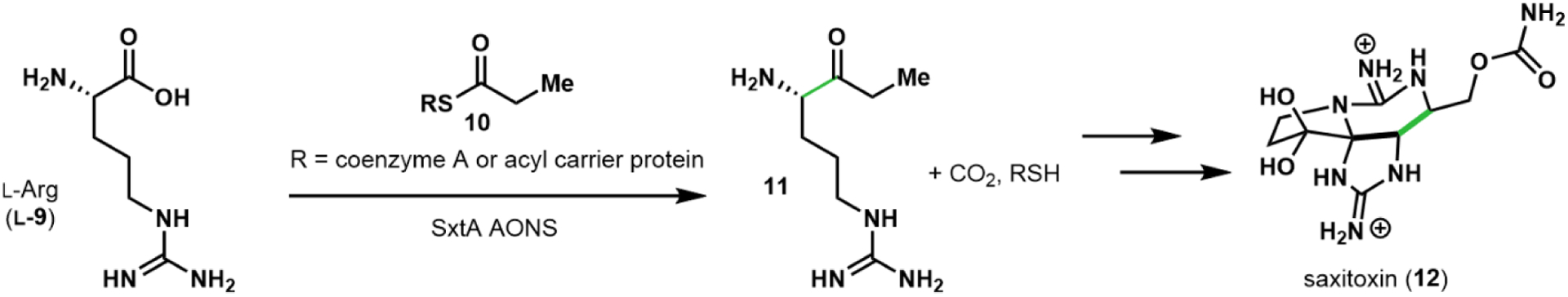
In saxitoxin biosynthesis, SxtA AONS produces ketone 11 from l-Arg and propionyl thioesters.
Figure 3.
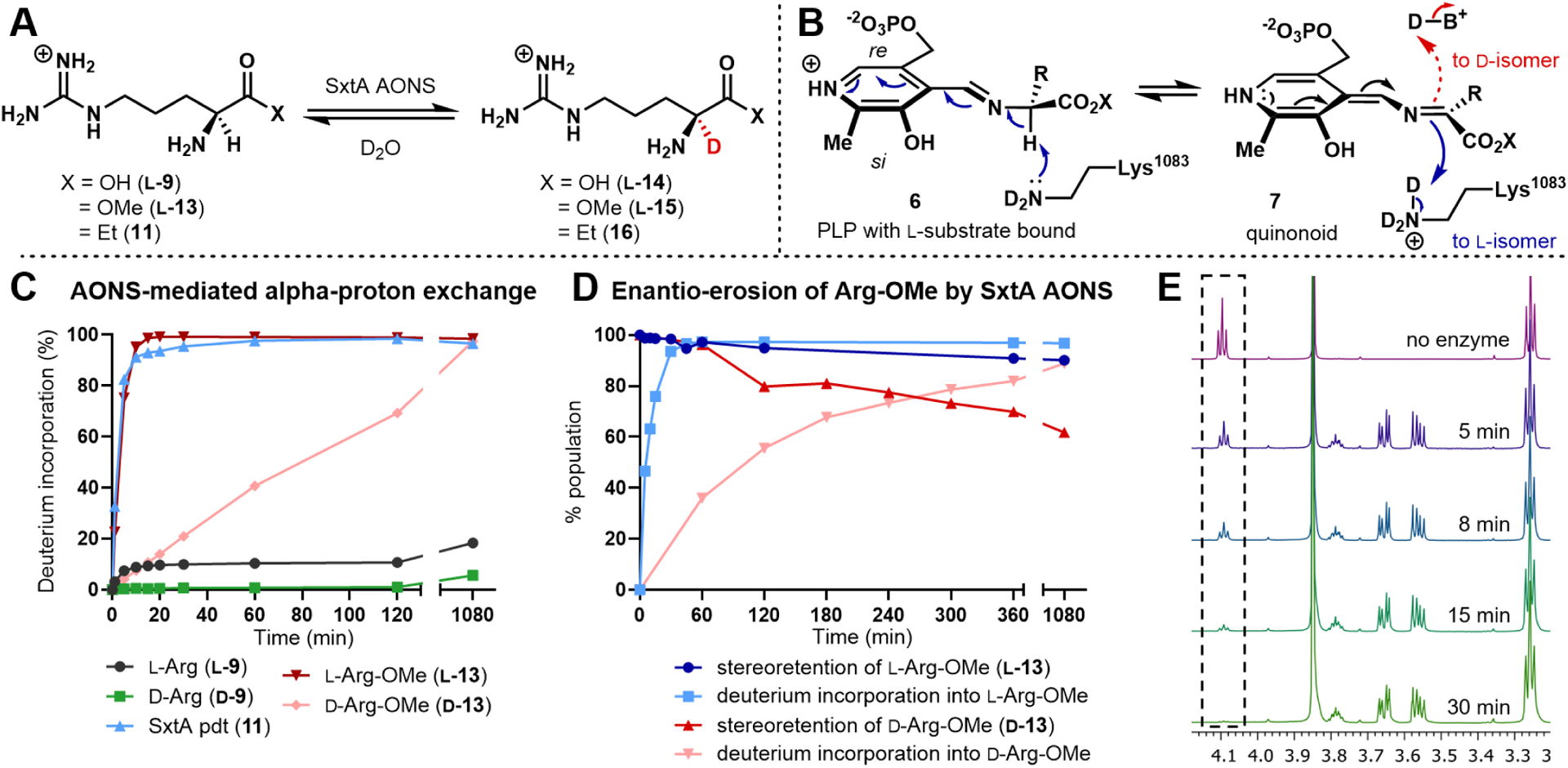
Deuteration of arginine-related substrates. (A) α-deuterated compound formation. (B) Potential mechanisms of proton/deuterium transfers in SxtA AONS. (C) Timecourse of α-deuteration by mass spectrometry. (D) Comparison of deuterium incorporation and enantiomeric composition when starting with l-Arg-OMe (blue) or d-Arg-OMe (red). (E) 1H NMR of l-Arg-OMe (l-13) incubated with AONS over 30 min, confirming exchange of the α-proton to deuterium (see SI for conditions). Peaks present between 3.7−3.5 ppm are attributed to glycerol from the enzyme storage buffer.
The site of deuterium incorporation was confirmed to be the α-carbon of l-Arg-OMe (l-13). In a reaction monitored by 1H NMR, the α-proton triplet signal at 4.09 ppm disappeared over 45 min, demonstrating complete replacement by deuterium (Figure 3E). No decrease in the β-proton signals was observed, even at extended timepoints (see Figure S6), indicating a higher level of site-selectivity for SxtA AONS compared with deuteration mediated by other PLP-dependent enzymes.22,31,32 The exception to this site-selectivity was observed only with ketone 11, which possesses additional acidic protons alpha to the carbonyl group.20 No more than a single deuterium atom was incorporated into non-glycine-based amino acid and methyl ester substrates.
As SxtA AONS deuterates both enantiomers of Arg-OMe, we next investigated the configuration of the deuterated products (15). In prior studies of AOS proteins, the catalytic lysine residue (Lysl083 in M. wollei SxtA) is proposed to stereospecifically mediate both the initial deprotonation of the l-amino acid substrate and the subsequent reprotonation of the α-carbon to generate l-products (Figure 3B, blue arrows).33,34 Notably, this residue is located on the si face of PLP in all published AOS crystal structures.35–39 On the basis of this general mechanism, the deprotonation and deuteration of d-Arg-OMe (d-13) is unexpected. Ongoing structural studies aim to distinguish if SxtA AONS accommodates different binding modes40 for each Arg-OMe enantiomer or if an alternative active site residue or water can serve as a base41 to account for activity on d-configured substrates (see Figure 3B, red arrows, and Figure S29). To determine the degree of stereo control exhibited by SxtA AONS, 20 mM of each Arg-OMe enantiomer was separately incubated with 20 μM SxtA AONS and assessed for deuterium labeling and enantiopurity at various timepoints. Analysis of Fmoc-derivatized compounds by chiral supercritical fluid chromatography (SFC) provided the enantiopurity of each product (see Table S6). For l-Arg-OMe, complete deuterium incorporation and a high degree of retention of the l configuration was observed at 45 min, with slight enantio-erosion to 90:10 l to d occurring at extended timepoints (Figure 3D, blue lines). At a higher substrate concentration (20 mM), d-Arg-OMe reached 89% deuteration (red lines). The rate of enantio-erosion was faster than that of the l-enantiomer, ending at a 38:62 l to d enantiomeric ratio after 18 h. For both enantiomers of ester 13, the retention of the original configuration does not track directly with the deuteration level, nor is the reprotonation step observed to be completely stereoselective. The reprotonation step for l-Arg-OMe is generally stereoretentive to afford primarily the deuterated l-isomer. The putative reprotonation step with a d-Arg-OMe substrate is less selective, producing significant amount of l-product. The mechanism of deprotonation and reprotonation remains unclear in the absence of a SxtA AONS crystal structure.
To evaluate the substrate promiscuity of SxtA AONS for deuterium incorporation, we first screened twenty-three additional l-amino acids. These substrates consisted of the remaining proteinogenic amino acids and other Arg-related compounds. In line with our initial studies with l-Arg (l-9), most of these amino acids, including the structurally similar substrates l-ornithine, l-Lys and l-homoarginine, showed no or low levels of deuteration (Figure 4A, blue). Although some polar substrates demonstrated modest levels of deuterium incorporation, only two substrates were >90% deuterated, l-His and Arg-mimic l-canavanine. Based on our previous observation that esterification of Arg free acids to the corresponding methyl esters significantly increased both the rate and levels of deuterium incorporation (Figure 3C), we next assessed the corresponding methyl esters of the entire substrate panel (Figure 4A, gray). Gratifyingly, every l-methyl ester tested was labeled to some extent with deuterium, and a majority (twenty side chains) were >90% labeled upon overnight incubation. The lowest-converting substrates were those with acidic side chains, as well as the polar ester l-Cys-OMe, and the aliphatic substrate l-Leu-OMe. Esterification slightly decreased the deuterium incorporation of Asp-based compounds, whereas l-Cys-OMe had significantly lower deuterium incorporation, likely due to the high levels of disulfide formation. Thus, for SxtA AONS, a significantly greater breadth of methyl esters was deuterated compared to the analogous panel of amino acid substrates. The increased activity on methyl ester substrates may be due to the lower pKa of the α-proton,33,42,43 higher structural similarity to the ethyl ketone product 11, or both.
Figure 4.
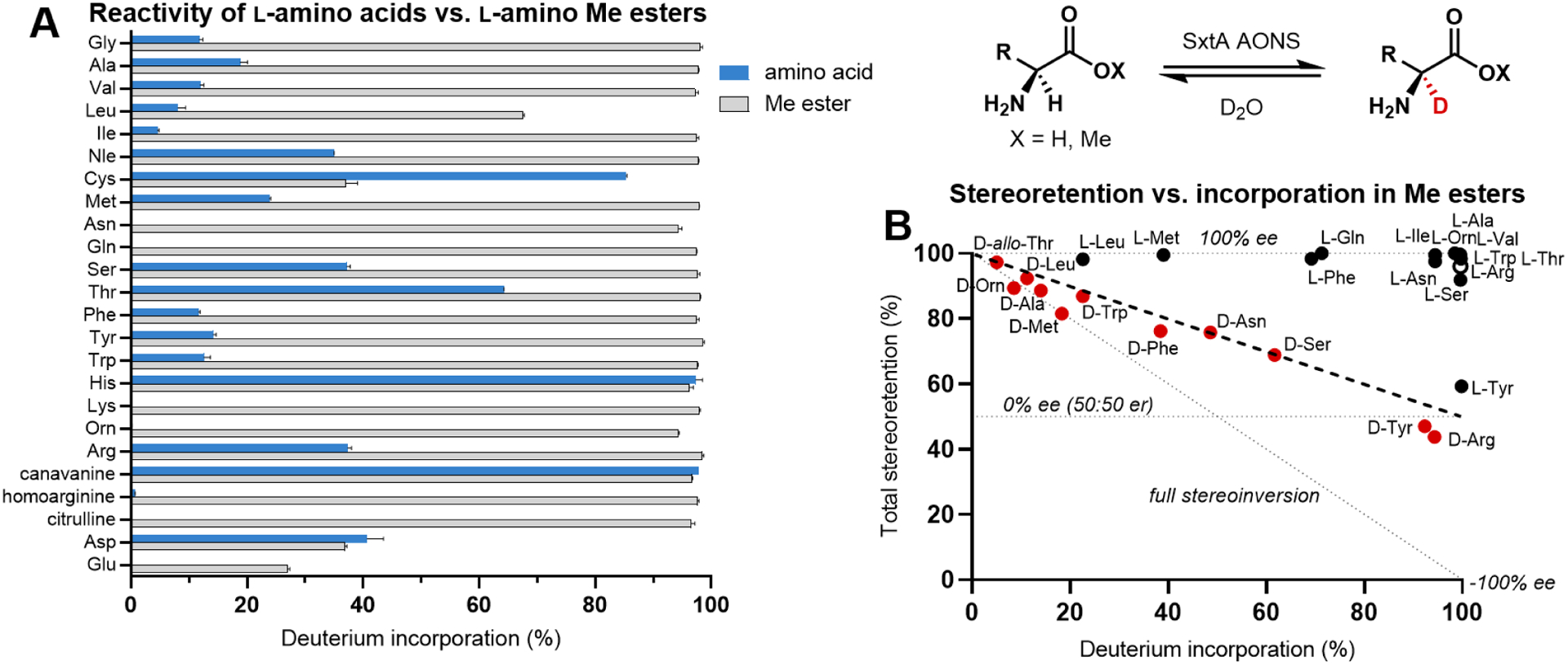
Deuterium incorporation at the α-carbon of amino acids and their methyl esters. (A) Deuterium incorporation into 4 mM of l-substrates after overnight incubation with SxtA AONS in D2O. (B) Total stereoretention of 20 mM of the original methyl ester substrate configuration at the time of highest deuterium incorporation (see Table S8 for full data set).
To assess the stereocontrol in SxtA AONS-catalyzed deuteration of non-arginine esters, we resolved the stereoisomers of compounds possessing fifteen different side chains by chiral SFC (Figure 4B). In reactions with l-methyl esters (black dots), the l configuration was retained, with many substrates affording both high deuterium incorporation and retention of the original configuration at the α-position. l-Tyr-OMe was the only l-substrate observed to have relatively low stereoretention (41:59 d/l er). There is no significant correlation between the steric bulk or class of side chain and level of deuterium incorporation. For d-esters (red dots), deuterium incorporation varied widely from <1% in select aliphatic side chains (e.g., d-Val-OMe, see Table S8 for full data set) to 94% for d-Arg-OMe. The mixtures of stereoisomers resulting from d-substrates are approximately halfway between full stereoretention and stereoinversion to l for every molecule processed. Thus, after deprotonation of a d-substrate and formation of a putative quinonoid intermediate, the reprotonation step appears to be non-selective (see Figure 3B). A racemic mixture of deuterated ester was also observed nonenzymatically when SxtA AONS was replaced with free PLP; however, only low amounts of deuteration occur in the absence of enzyme (see Table S2).44,45 For the achiral ester Gly-OMe, approximately 5% of the product mixture showed incorporation of two deuterium atoms, but the singly deuterated isotopomers could not be separated in our hands by chiral SFC analysis.
With evidence that SxtA AONS performs over a thousand turnovers, we sought to test the scalability of this transformation. As an initial target, we selected the Parkinson’s drug, safinamide. Preparative-scale reactions on hundreds of milligrams were fruitful. For example, a 200 mg-scale reaction with l-Ala-OMe was carried out to afford deuterated l-Ala-OMe (17) in a 60% recovered yield with >99% deuterium incorporation and 96% ee. This deuterated building block was elaborated over three steps according to an established route to safinamide to deliver deutero-safinamide (19) with 96% final deuterium incorporation (Figure 5).46 Ester 17 was also easily saponified with LiOH to acid 20, maintaining 97% of the α-deuterium label, an example of a building block useful in protein synthesis or other coupling reactions.47,48
Figure 5.
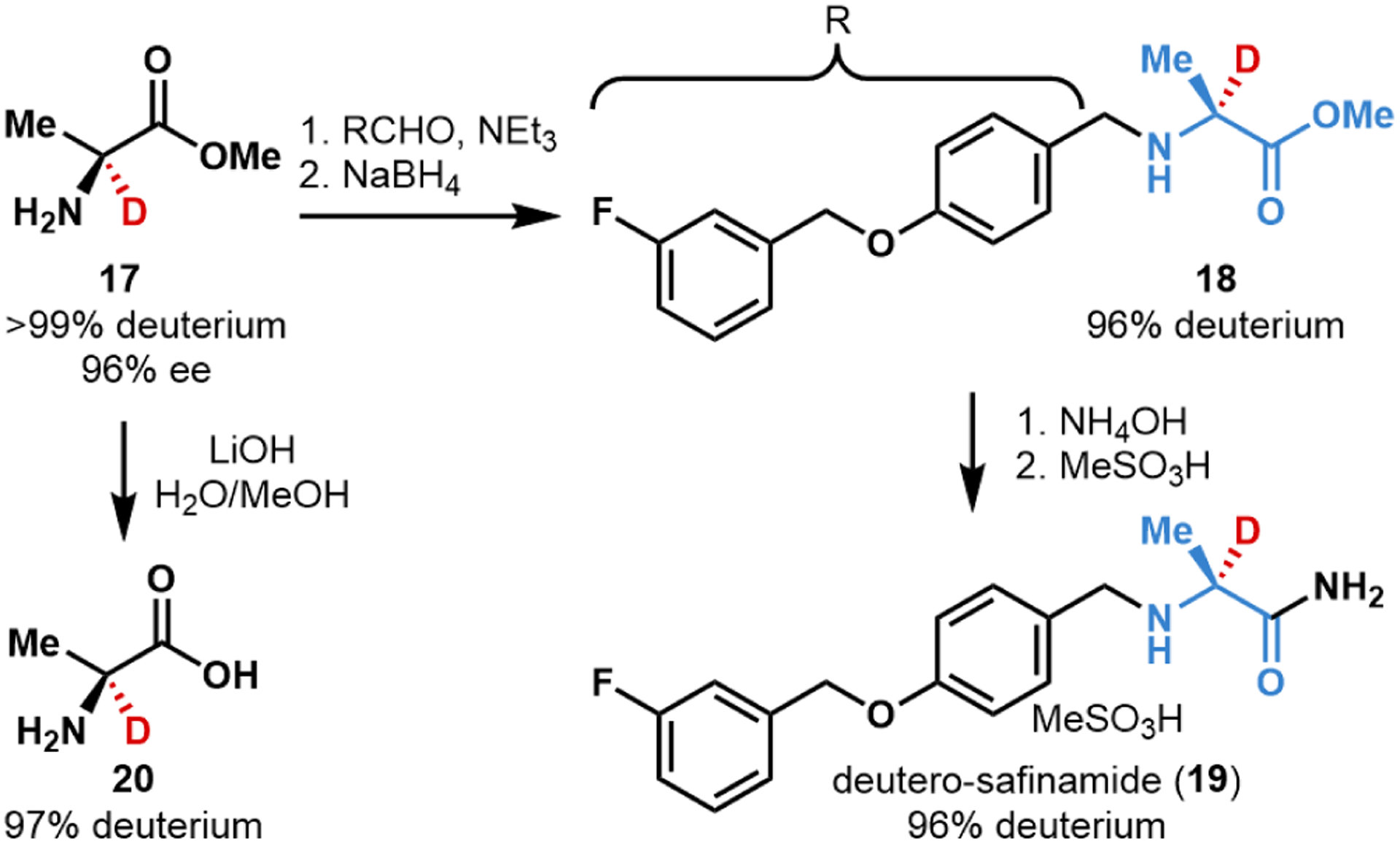
Synthesis of deutero-safinamide (19) and l-[2-2H]alanine (20). See SI for full conditions.
We have demonstrated that SxtA AONS, a PLP-dependent enzyme that natively transforms an amino acid into an α-amino ketone, can be repurposed to install deuterium atoms site-selectively at the α-carbon of amino acids and amino esters. The method is particularly robust on a broad range of methyl ester substrates affording α-deuterated products under mild conditions. Even among promiscuous PLP-dependent amino acid-derivatizing enzymes, such as the broad-specificity racemases, the ability to accommodate such a wide range of substrates that vary in structure and stereochemical configuration like SxtA AONS has not been previously demonstrated. The overall stereoselectivity of the C–D bond-forming step varies according to the side chain of α-amino methyl esters but in general, SxtA AONS exhibits high stereoselectivity with l-substrates, affording deuterated products with high levels of stereocontrol to generate products with the l configuration. Finally, the utility and scalability of this one-step, protecting group-free, biocatalytic method was applied to the chemoenzymatic synthesis of deuterium-labeled safinamide. We anticipate that this method can easily be extended to the syntheses of other deuterated or tritiated products.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by funds from the University of Michigan Life Sciences Institute, University of Michigan Department of Chemistry, and the National Institutes of Health R35 GM124880.
Footnotes
The authors declare no competing financial interest.
Supporting Information.
The Supporting Information is available free of charge at http://pubs.acs.org.
Materials and methods; NMR spectra; mass spectra; SFC traces (PDF)
REFERENCES
- (1).Harbeson SL; Tung RD Deuterium in Drug Discovery and Development. Annu. Rep. Med. Chem 2011, 46, 403–417. [Google Scholar]
- (2).Westheimer FH The Magnitude of the Primary Kinetic Isotope Effect for Compounds of Hydrogen and Deuterium. Chem. Rev 1961, 61, 265–273. [Google Scholar]
- (3).Pony Yu R; Hesk D; Rivera N; Pelczer I; Chirik PJ Iron-Catalysed Tritiation of Pharmaceuticals. Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]
- (4).Loh YY; Nagao K; Hoover AJ; Hesk D; Rivera NR; Colletti SL; Davies IW; MacMillan DWC Photoredox-Catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017,358, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Timmins GS Deuterated Drugs; Updates and Obviousness Analysis. Expert Opin. Ther. Pat 2017, 27, 1353–1361. [DOI] [PubMed] [Google Scholar]
- (6).Dean M; Sung VW Review of Deutetrabenazine: A Novel Treatment for Chorea Associated with Huntington’s Disease. Drug Des. Devel. Ther 2018,12, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).de la Torre BG; Albericio F The Pharmaceutical Industry in 2017. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2018, 23, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sheppard D; Li DW; Brüschweiler R; Tugarinov V Deuterium Spin Probes of Backbone Order in Proteins: 2H NMR Relaxation Study of Deuterated Carbon α Sites. J. Am. Chem. Soc 2009, 131, 15853–15865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bornø A; Van Hall G Quantitative Amino Acid Profiling and Stable Isotopically Labeled Amino Acid Tracer Enrichment Used for In Vivo Human Systemic and Tissue Kinetics Measurements. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2014, 951–952, 69–77. [DOI] [PubMed] [Google Scholar]
- (10).Bode HB; Brachmann AO; Jadhav KB; Seyfarth L; Dauth C; Fuchs SW; Kaiser M; Waterfield NR; Sack H; Heinemann SH; Arndt H-D Structure Elucidation and Activity of Kolossin A, the d-/l-Pentadecapeptide Product of a Giant Nonribosomal Peptide Synthetase. Angew. Chem. Int. Ed 2015, 54, 10352–10355. [DOI] [PubMed] [Google Scholar]
- (11).Seebach D; Dziadulewiczl E; Behrendt L; Cantoreggi S; Fitzi R Synthesis of Nonproteinogenic (R)- or (S)-Amino Acids Analogues of Phenylalanine, Isotopically Labeled and Cyclic Amino Acids from tert-Butyl 2-(Tert-Butyl)-3-Methyl-4-Oxo-l-Imidazolinecarboxylate (Boc-BMl). Liebigs Ann. der Chemie 1989, 1215–1232. [Google Scholar]
- (12).Hoppe D; Ludger B Selective Mono- and Dialkylation of N-[Bis(Alkylthio)Methylene] Glycine Ethyl Ester for Synthesis of Higher and α-Branched α-Amino Acids. Liebigs Ann. der Chemie 1979, 2066–2075. [Google Scholar]
- (13).Elemes Y; Ragnarsson U Synthesis of Enantiopure α-Deuteriated Boc-l-Amino Acids. J. Chem. Soc. Perkin Trans 11996, 537–540. [Google Scholar]
- (14).Takeda R; Abe H; Shibata N; Moriwaki H; Izawa K; Soloshonok VA Asymmetric Synthesis of α-Deuterated α-Amino Acids. Org. Biomol. Chem 2017,15, 6978–6983. [DOI] [PubMed] [Google Scholar]
- (15).Chatterjee B; Krishnakumar V; Gunanathan C Selective α-Deuteration of Amines and Amino Acids Using D2O. Org. Lett 2016, 18, 5892–5895. [DOI] [PubMed] [Google Scholar]
- (16).Truppo MD Biocatalysis in the Pharmaceutical Industry : The Need for Speed. ACS Med. Chem. Lett 2017, 8, 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).$15.80 and $1,165 per Liter of D2 and D2O, respectively on Sigma-Aldrich in April 2020.
- (18).Dunathan HC Conformation and Reaction Specificity in Pyridoxal Phosphate Enzymes. Proc. Natl. Acad. Sci 1966, 55, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Toney MD Controlling Reaction Specificity in Pyridoxal Phosphate Enzymes. Biochim. Biophys. Acta - Proteins Proteomics 2011, 1814, 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ploux O; Marquet A Mechanistic Studies on the 8-Amino-7-Oxopelargonate Synthase, a Pyridoxal-5’-Phosphate-Dependent Enzyme Involved in Biotin Biosynthesis. Eur. J. Biochem 1996, 236, 301–308. [DOI] [PubMed] [Google Scholar]
- (21).Ikushiro H; Fujii S; Shiraiwa Y; Hayashi H Acceleration of the Substrate Cα Deprotonation by an Analogue of the Second Substrate Palmitoyl-CoA in Serine Palmitoyltransferase. J. Biol. Chem 2008, 283, 7542–7553. [DOI] [PubMed] [Google Scholar]
- (22).Babu UM; Johnston RB D2O-Alanine Exchange Reactions Catalyzed by Alanine Racemase and Glutamic Pyruvic Transaminase. Biochem. Biophys. Res. Commun 1974, 58, 460–466. [DOI] [PubMed] [Google Scholar]
- (23).Lim Y-H; Yoshimura T; Soda K; Esaki N Stereospecific Labeling at α-Position of Phenylalanine Phenylglycine with Amino Acid Racemase. J. Ferment. Bioeng 1998, 86,400–402. [Google Scholar]
- (24).Webster SP; Alexeev D; Campopiano DJ; Watt RM; Alexeeva M; Sawyer L; Baxter RL Mechanism of 8-Amino-7-Oxononanoate Synthase: Spectroscopic, Kinetic, and Crystallographic Studies. Biochemistry 2000, 39, 516–528. [DOI] [PubMed] [Google Scholar]
- (25).Chun SW; Hinze ME; Skiba MA; Narayan ARH Chemistry of a Unique Polyketide-like Synthase. J. Am. Chem. Soc 2018, 140, 2430–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Salvati P; Maj R; Caccia C; Cervini MA; Fornaretto MG; Lamberti E; Pevarello P; Skeen GA; White HS; Wolf HH; Faravelli L; Mazzanti M; Mancinelli E; Varasi M; Fariello RG Biochemical and Electrophysiological Studies on the Mechanism of Action of PNU-151774E, a Novel Antiepileptic Compound. J. Pharmacol. Exp. Ther 1999, 288, 1151–1159. [PubMed] [Google Scholar]
- (27).Kellmann R; Mihali TK; Young JJ; Pickford R; Pomati F; Neilan BA Biosynthetic Intermediate Analysis and Functional Homology Reveal a Saxitoxin Gene Cluster in Cyanobacteria. Appl. Environ. Microbiol 2008, 74, 4044–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mihali TK; Kellmann R; Neilan BA Characterisation of the Paralytic Shellfish Toxin Biosynthesis Gene Clusters in Anabaena circinalis AWQC131C and Aphanizomenon sp. NH-5. BMC Biochem 2009, 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mihali TK; Carmichael WW; Neilan BA A Putative Gene Cluster from a Lyngbya wollei Bloom that Encodes Paralytic Shellfish Toxin Biosynthesis. PLoS ONE 2011, 6, el4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kerbarh O; Campopiano DJ; Baxter RL Mechanism of α-Oxoamine Synthases: Identification of the Intermediate Claisen Product in the 8-Amino-7-Oxononanoate Synthase Reaction. Chem. Commun 2006, 18, 60–62. [DOI] [PubMed] [Google Scholar]
- (31).Cooper AJL Proton Magnetic Resonance Studies of Glutamate-Alanine Deuterium Exchange. J. Biol. Chem 1976, 251, 1088–1097. [PubMed] [Google Scholar]
- (32).Golichowski A; Harruff RC; Jenkins WT The Effects of pH on the Rates of Isotope Exchange Alanine Aminotransferase. Arch. Biochem. Biophys 1977, 178, 459–467. [DOI] [PubMed] [Google Scholar]
- (33).Hunter GA; Ferreira GC Lysine-313 of 5-Aminolevulinate Synthase Acts as a General Base during Formation of the Quinonoid Reaction Intermediates. Biochemistry 1999, 38, 3711–3718. [DOI] [PubMed] [Google Scholar]
- (34).Raman MCC; Johnson KA; Yard BA; Lowther J; Carter LG; Naismith JH; Campopiano DJ The External Aldimine Form of Serine Palmitoyltransferase. J. Biol. Chem 2009, 284, 17328–17339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Alexeev D; Alexeeva M; Baxter RL; Campopiano DJ; Webster SP; Sawyer L The Crystal Structure of 8-Amino-7-Oxononanoate Synthase: A Bacterial PLP-Dependent, Acyl-CoA-Condensing Enzyme. J. Mol. Biol 1998, 284, 401–419. [DOI] [PubMed] [Google Scholar]
- (36).Schmidt A; Sivaraman J; Li Y; Larocque R; Barbosa JARG; Smith C; Matte A; Schrag JD; Cygler M Three-Dimensional Structure of 2-Amino-3-Ketobutyrate CoA Ligase from Escherichia coli Complexed with a PLP–Substrate Intermediate: Inferred Reaction Mechanism. Biochemistry 2001, 40, 5151–5160. [DOI] [PubMed] [Google Scholar]
- (37).Astner I; Schulze JO; van den Heuvel J; Jahn D; Schubert W-D; Heinz DW Crystal Structure of 5-Aminolevulinate Synthase, the First Enzyme of Heme Biosynthesis, and Its Link to XLSA in Humans. EMBO J. 2005, 24, 3166–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yard BA; Carter LG; Johnson KA; Overton IM; Dorward M; Liu H; McMahon SA; Oke M; Puech D; Barton GJ; Naismith JH; Campopiano DJ The Structure of Serine Palmitoyltransferase; Gateway to Sphingolipid Biosynthesis. J. Mol. Biol 2007, 370, 870–886. [DOI] [PubMed] [Google Scholar]
- (39).Jahan N; Potter JA; Sheikh MA; Botting CH; Shirran SL; Westwood NJ; Taylor GL Insights into the Biosynthesis of the Vibrio cholerae Major Autoinducer CAI-1 from the Crystal Structure of the PLP-Dependent Enzyme CqsA. J. Mol. Biol 2009, 392, 763–773. [DOI] [PubMed] [Google Scholar]
- (40).Walton CJW; Thiebaut F; Brunzelle JS; Couture J-F; Chica RA Structural Determinants of the Stereoinverting Activity of Pseudomonas Stutzeri d-Phenylglycine Aminotransferase. Biochemistry 2018, 57, 5437–5446. [DOI] [PubMed] [Google Scholar]
- (41).Spies MA; Toney MD Multiple Hydrogen Kinetic Isotope Effects for Enzymes Catalyzing Exchange with Solvent: Application to Alanine Racemase. Biochemistry 2003, 42, 5099–5107. [DOI] [PubMed] [Google Scholar]
- (42).Richard JP; Amyes TL; Crugeiras J; Rios A Pyridoxal 5′-Phosphate: Electrophilic Catalyst Extraordinaire. Curr. Opin. Chem. Biol 2009, 13, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rios A; Amyes TL; Richard JP Formation and Stability of Organic Zwitterions in Aqueous Solution: Enolates of the Amino Acid Glycine and Its Derivatives. J. Am. Chem. Soc 2000,122,9373–9385. [Google Scholar]
- (44).Pugnière M; Commeyras A; Previero A Racemization of Amino Acid Esters Catalysed by Pyridoxal 5′ Phosphate as a Step in the Production of L-Amino Acids. Biotechnol. Lett 1983, 5, 447–452. [Google Scholar]
- (45).Zabinski RF; Toney MD Metal Ion Inhibition of Nonenzymatic Pyridoxal Phosphate Catalyzed Decarboxylation and Transamination. J. Am. Chem. Soc 2001, 123, 193–198. [DOI] [PubMed] [Google Scholar]
- (46).Cao X; Chen H; Du Q; Shen X High-Purity Safinamide Preparing Method. CN105061245A, 2015. [Google Scholar]
- (47).El-Faham A; Albericio F Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev 2011, 111, 6557–6602. [DOI] [PubMed] [Google Scholar]
- (48).Kazemi M; Shiri L Thioesters Synthesis : Recent Adventures in the Esterification of Thiols. J. Sulfur Chem 2015, 35, 613–623. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.