

Abstract
People are exposed to wide range of redox-active environmental pollutants. Air pollution, heavy metals, pesticides, and endocrine disrupting chemicals can disrupt cellular redox status. Redox-active pollutants in our environment all trigger their own sets of specific cellular responses, but they also activate a common set of general stress responses that buffer the cell against homeostatic insults. These cellular defense system (CDS) pathways include the heat shock response, the oxidative stress response, the hypoxia response, the unfolded protein response, the DNA damage response, and the general stress response mediated by the stress-activated p38 mitogen-activated protein kinase. Over the past two decades, the field of environmental epigenetics has investigated epigenetic responses to environmental pollutants, including redox-active pollutants. Studies of these responses highlight the role of chromatin modifications in controlling the transcriptional response to pollutants and the role of transcriptional memory, often referred to as “epigenetic reprogramming”, in predisposing previously exposed individuals to more potent transcriptional responses on secondary challenge. My central thesis in this review is that high dose or chronic exposure to redox-active pollutants leads to transcriptional memories at CDS target genes that influence the cell’s ability to mount protective responses. To support this thesis, I will: (1) summarize the known chromatin features required for inducible gene activation; (2) review the known forms of transcriptional memory; (3) discuss the roles of inducible chromatin and transcriptional memory in CDS responses that are activated by redox-active environmental pollutants; and (4) propose a conceptual framework for CDS pathway responsiveness as a readout of total cellular exposure to redox-active pollutants.
Keywords: Oxidative stress, heat shock, hypoxia, DNA damage response, unfolded protein response, cellular stress
Graphical Abstract
1. Introduction
People are exposed to wide range of redox-active environmental pollutants [1, 2]. Air pollution, heavy metals, pesticides, and endocrine disrupting chemicals can disrupt cellular redox status [3, 4]. Over the past two decades, the field of environmental epigenetics has investigated epigenetic responses to environmental pollutants, including redox-active pollutants [5]. Epigenetic phenomena are responsible for changes in phenotype without changes in genotype [6, 7]. The majority of these phenotypic changes are achieved by differential regulation of gene expression by chemical modifications to DNA and its associated proteins (collectively, chromatin), as well as the three-dimensional structure of chromatin [7, 8]. The original definition of “epigenetics” required these modifications to be heritable across cell division [7], although the common usage has expanded to include non-heritable, trans-acting transcriptional regulators, including non-coding RNA [9]. The field of environmental epigenetics has focused on two primary areas: the role of chromatin modifications in controlling the transcriptional response to pollutants and the role of transcriptional memory, often referred to as “epigenetic reprogramming”, in predisposing previously exposed individuals to more potent transcriptional responses on secondary challenge [10]. Past and current work in this field primarily characterizes the epigenetic effects of individual pollutants, with the goal of identifying specific signatures of exposure and effect that can be used to evaluate past exposures and predict future disease [11]. However, individuals are rarely exposed to a single pollutant. Most people are exposed to multiple mixtures of pollutants that may act additively or synergistically to affect health [12]. Therefore, an individual’s total burden of pollutant exposure and their combined effects is likely the best predictor of disease.
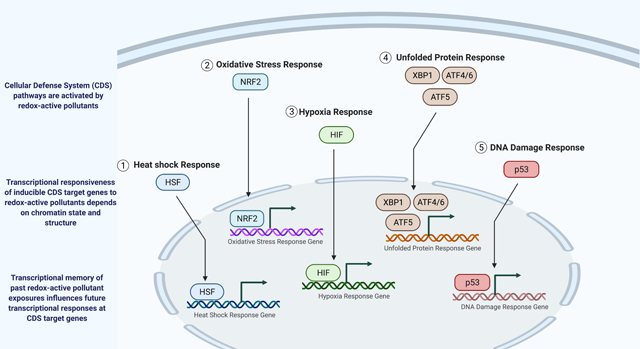
Redox-active pollutants in our environment all trigger their own sets of specific cellular responses, but they also activate a common set of general stress responses that buffer the cell against homeostatic insults, including oxidative damage and protein misfolding [13]. These general stress responses occur at the cellular level, rather than the organismal level [13, 14]. In order to avoid confusion with organismal effects of psychosocial stressors (e.g., through the hypothalamic-pituitary-adrenal axis), in this review, I will refer to these pathways as cellular defense system (CDS) pathways. CDS pathways include the heat shock response, the oxidative stress response, the hypoxia response, the unfolded protein response, and the DNA damage response, as well as the general stress response mediated by the stress-activated p38 mitogen-activated protein kinase (p38-MAPK) [13, 14]. Disease can result when these systems are overwhelmed, develop resistance (e.g., fail to respond) over time, or if their sustained activation is harmful to the cell [13, 14]. Therefore, an individual’s ability to mount protective CDS responses by turning on CDS genes is a critically important protection against disease. In addition, because many pollutants activate CDS responses, transcriptional memory at CDS target genes likely reflects an individual’s total redox-active pollutant burden.
My central thesis in this review is that high dose or chronic exposure to redox-active pollutants leads to transcriptional memories at CDS target genes that influence the cell’s ability to mount protective responses. This work builds directly on prior work by Simmons and Ramabhadran that leveraged transcriptional readout of CDS pathways as biomarkers for high-throughput toxicity screening of environmental pollutants [13]. To support this thesis, I will: (1) summarize the known chromatin features required for inducible gene activation; (2) review the known forms of transcriptional memory; (3) discuss the roles of inducible chromatin and transcriptional memory in CDS responses that are activated by redox-active environmental pollutants; and (4) propose a conceptual framework for CDS pathway responsiveness as a readout of total cellular exposure to redox-active pollutants. This review is not intended to be a comprehensive review of the literature. I have included selected examples to illustrate discussed concepts.
2. Chromatin features of inducible genes
CDS pathways sense cellular stress and transmit that information through intracellular signaling pathways that terminate in activation of specific transcription factors [13, 14]. CDS-activated transcription factors translocate to the nucleus and bind their respective transcription factor binding sites, or response elements, to activate target genes [13, 14]. CDS target genes are inducible genes that are newly transcribed or transcribed to a greater degree in response to a stimulus [15, 16]. Inducible genes can be contrasted with constitutively transcribed “housekeeping” genes that are responsible for the baseline functioning of the cell [17, 18]. Inducible genes require flexible and responsive chromatin states that can prevent inappropriate transcription at baseline but can respond rapidly and transiently to stimulus by significantly increasing or decreasing transcription [17, 18]. Target gene sets must remain inducible across cell division, implying mitotic heritability of some component(s) of inducible chromatin at these loci [19]. In this section, I will briefly review the sequence of events required for transcriptional activation and then discuss the evidence for chromatin features that confer inducibility.
2.1. Two-dimensional and three-dimensional control of gene transcription
Gene expression is controlled at the level of gene transcription by chromatin modifications and protein binding to the two-dimensional (2D) genome (reviewed in [20]) and by looping dynamics of the three-dimensional (3D) genome [8, 21]. The eukaryotic 2D genome, also called the linear genome, refers to the DNA double-helix wound around protein complexes called nucleosomes, in a configuration that resembles “beads on a string” (Figure 1.1) [20]. Nucleosomes are protein octamers comprising two heterodimers of histones H2A and H2B and two heterodimers of histones H3 and H4 [20]. Chromatin that is reversibly repressed is called facultative heterochromatin and is formed when nucleosomes are densely packed together, blocking transcription factors from binding DNA and turning on genes [20]. (In contrast, constitutive heterochromatin is not dynamic; it stably silences chromatin near centromeres, telomeres and some repetitive elements [22].) In accessible chromatin, or euchromatin, nucleosomes are sparser and can be easily shifted or removed from DNA to allow binding of transcription factors [20]. Regulatory regions, or elements, within DNA (e.g., promoters, enhancers, silencers, and insulators) contain clusters of transcription factor binding sites that can be exposed to transcription factor binding in euchromatin or restricted from transcription factor binding in facultative heterochromatin [20]. Both DNA and histone proteins can be modified with chemical groups that affect chromatin accessibility [23]. Histone proteins can be modified at both their globular domains and their amino (N-) and carboxy (C-) terminal tails (most commonly at lysine, arginine, or serine residues) with a wide variety of chemical groups, most commonly acetyl and methyl groups [23]. Histone acetylation (e.g., acetylation of lysine 27 on histone H3, or H3K27ac) is generally permissive of transcription (with the notable exception of H4K20ac [24]) and functions both to relax the affinity of DNA for nucleosomes [23], which makes DNA more accessible for binding of proteins required for gene transcription, as well as to recruit chromatin remodeling proteins that further promote DNA accessibility [23]. Histone methylation can enable or repress transcription, depending on the residue that is modified and the protein complexes that are recruited by that specific modification [23]. For example, trimethylation of lysine 4 on the N-terminal tail of histone H3 (H3K4me3) is permissive of transcription, but trimethylation of lysine 9 or lysine 27 on the N-terminal tail of histone H3 (H3K9me3 or H3K27me3) are repressive [25, 26]. Specific nucleotides within DNA can be methylated, too [27]. Methylation of cytosine residues within CG dinucleotides is the most common form of DNA methylation in mammalian genomes but other dinucleotide or trinucleotide sequences can be methylated, as well [28].) DNA methylation within regulatory sequences can repress transcription both by directly blocking binding of some transcription factors [29] and by recruiting other proteins, including those that modify histones, to promote a transcriptionally repressive state [30]. Histone modifying enzymes include histone methyltransferases (HMTs), histone acetyltransferases (HATs), histone demethylases (HDMs), and histone deacetylases (HDACs) [23]. DNA modifying enzymes including DNA methyltransferases (DNMTs) and ten-eleven-translocation (TET) proteins, which promote DNA demethylation [31]. Active regulatory elements are enriched for permissive histone modifications and deficient in DNA methylation and repressed or silenced regulatory elements are enriched in repressive histone modifications and DNA methylation [32]. Elements that contain both permissive and repressive histone modifications are termed “bivalent”; these regulatory regions can pivot quickly to either a more repressive (by losing permissive modifications) or a more permissive (by losing repressive modifications) state [33].
Figure 1. Chromatin characteristics of inducible genes.
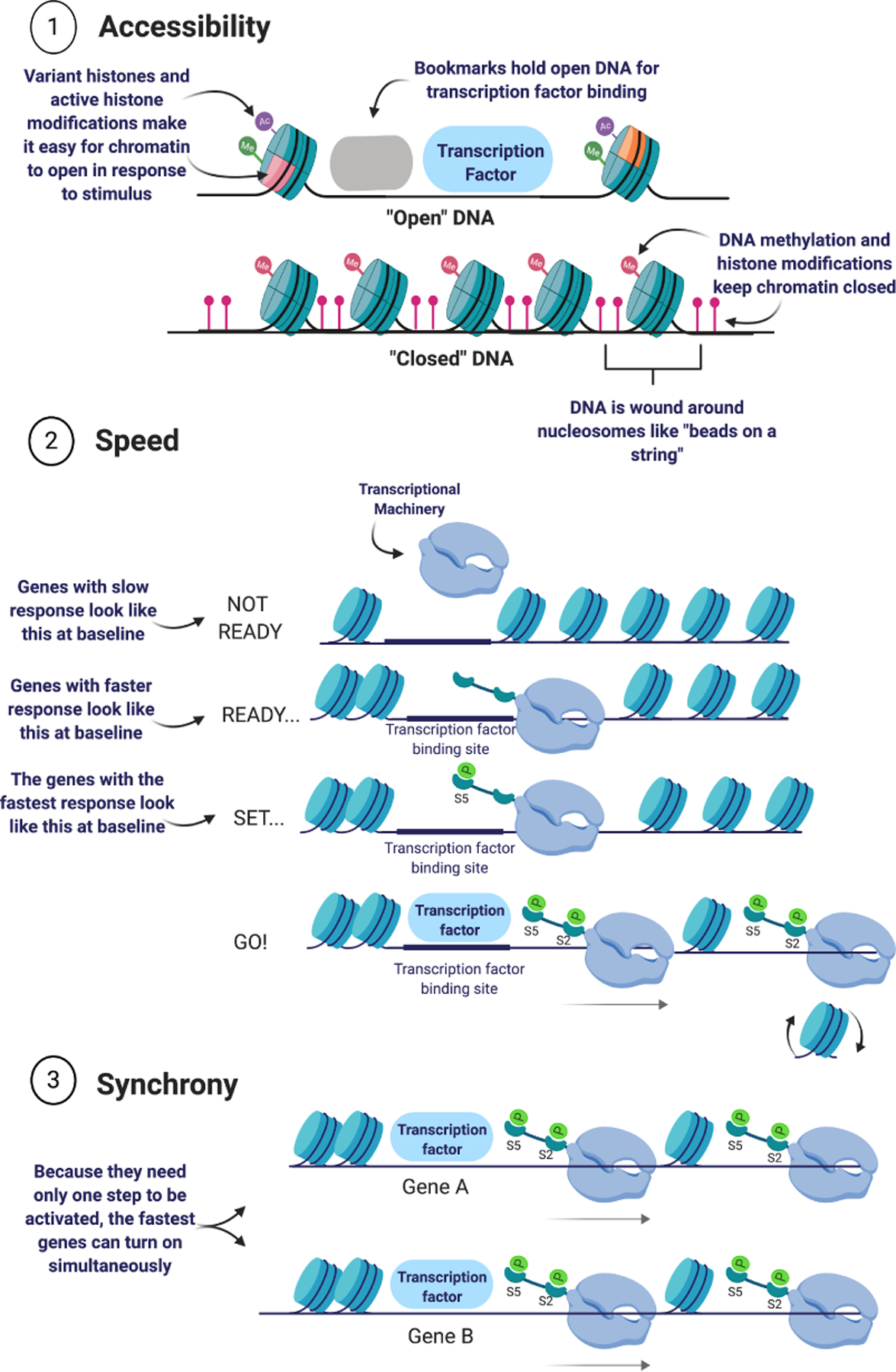
Inducible genes use chromatin strategies to regulate their responsiveness to stimuli. Inducible genes maintain accessible chromatin at transcription factor binding sites that enable transcription factors to bind and initiate a sequence of events, including remodeling of inducible chromatin to promote accessibility along the gene body, which is required for transcription. Inducible genes with poised RNA polymerase II can be induced very rapidly, since transcription factor binding triggers pause-release of the polymerase directly into elongation, rather than triggering initial assembly of the transcriptional machinery. Inducible genes with poised RNA polymerase II can be induced synchronously, as well.
The first step in transcription of an inducible gene is the binding of a stimulus-responsive transcription factor to an accessible promoter or enhancer [18]. When a transcription factor binds to a regulatory element that is relatively near the target gene’s transcription start site, it recruits a chromatin remodeling complex that increases chromatin accessibility at the core promoter [18]. Chromatin remodeling proteins or complexes (e.g., the SWI/SNF family, the ISWI family, the CHD family and the INO80 family) [34] are multi-subunit complexes that contain catalytic units that require ATP for activity and translocase subunits that regulate chromatin accessibility by controlling nucleosomes’ histone composition, position along the chromatin, and partial or total eviction from or incorporation into chromatin [35–41]. Chromatin remodeling complexes often contain subunits that can modify histone modifications [20].
Chromatin remodeling of the core promoter enables RNA polymerase II (RNAPII) binding and formation of the pre-initiation complex (PIC) [42, 43]. In addition to RNAPII, the PIC contains general transcriptional factors, as well as the positive elongation factor P-TEFb, the HAT complex SAGA and the transcriptional co-activator Mediator [42, 43]. If a transcription factor binds an accessible distal enhancer, then the chromatin must form a loop to bring the enhancer in close proximity to the core promoter of the target gene [44]. These enhancer-promoter contact loops represent 3D control of gene transcription [20, 44]. Enhancer-promoter contact loops form within larger chromatin loops called topologically active domains (TADS) [8, 21]. TADS are anchored at the loop base by the presence of two CCTC-binding factor (CCTF) insulator proteins, which binding orientation at the base of the loop are critical for loop formation, as well as cohesin complexes through which DNA is threaded to form the loop [8, 21]. TADS form genomic “neighborhoods” of gene regulation, and enhancers can only contact promoters that are contained within the same TAD [21, 45].
Transcription factor binding and PIC formation is not sufficient for a gene to be transcribed. First, RNAPII must be released into active elongation to transcribe the gene (reviewed in [42, 43]). RNAPII contains a C-terminal domain (CTD) that can be modified to regulate transcription [42, 43]. Once RNAPII is bound to the core promoter, its CTD is phosphorylated at serine 5 by the Cdk7 kinase, a subunit of the PIC [42, 43]. This initial phosphorylation triggers RNAPII poising: RNAPII escapes the PIC, transcribes a short stretch of DNA (20–50bp), and then pauses [42, 43]. Poised RNAPII is bound by negative elongation factors that stabilize the paused state [42, 43]. When the CTD of RNAPII is phosphorylated additionally at serine 2, positive elongation factor P-TEFb phosphorylates negative elongation factors DSIF and NELF which then dissociate from chromatin, and RNAPII is released into productive elongation, in a process termed “pause-release” [42, 43]. The chromatin remodeling complex bound at the promoter then shifts, modifies (either via chemical modification or by removal of a single dimer to form less stable hexamer nucleosomes), or evicts nucleosomes in the target gene body to enable RNAPII to process along the full length of the gene [42, 46].
All of the steps described above are required for any gene to be transcribed. At constitutively transcribed genes, the core promoter is maintained in an accessible state due to continuous, sequential binding of RNAPII [18, 20]. However, inducible genes respond to specific cellular cues or stressors [18]. Their responsiveness relies on three key characteristics. First, an initial regulatory element must be accessible to transcription factor binding [47], followed by a subsequent increase in chromatin accessibility at the target gene, both at the core promoter to enable PIC binding and along the gene body to enable RNAPII processivity [18] (Figure 1.1). Second, the speed at which these proteins bind and turn on genes is critical for a timely transcription [48] (Figure 1.2). Third, the ability for multiple genes to be expressed in synchrony is a critical factor for an effective cellular response [49] (Figure 1.3).
2.2. Chromatin accessibility at inducible genes
Transcription factors that are activated by environmental stressors or cues can only bind chromatin at accessible transcription factor binding sites [47]. Current evidence supports a model in which the full repertoire of regulatory elements with accessible transcription factor binding sites is established and maintained within the cell during differentiation [50]. Regulatory element accessibility is stably maintained through cell division by pioneer transcription factors, which bind to these regions and hold them open (i.e., serve as “bookmarks”) (Figure 1.1) [50]. A subset of pioneer transcription factors called lineage-determining transcription factors establish and maintain cellular identity through differential chromatin accessibility at transcription factor binding sites, which enables cell type-specific transcriptional programs [50].
In addition to baseline chromatin accessibility of transcription factor binding sites, chromatin at inducible target genes must be responsive. In order to facilitate rapid increases in chromatin accessibility in response to stimuli, inducible genes often contain unstable nucleosomes [51]. Unstable nucleosomes can be more easily shifted or evicted by chromatin remodeling proteins to increase chromatin accessibility [51]. A common form of an unstable nucleosome is a hexamer nucleosome that contain three, rather than four, protein dimers [52]. Hexamer nucleosomes are formed after eviction of a single histone dimer, rather than an entire nucleosome, by a chromatin remodeling complex [52]. Unstable nucleosomes are more likely to form in CG-rich DNA sequences [53]. Therefore, nucleosomes within CG-rich sequence are inherently less stable and more easily evicted from chromatin stochastically [53]. Almost 40% of mammalian coding genes contain proximal promoter elements with regions of high CpG site density, termed CpG islands [54, 55]. Genes with CpG islands in their promoters may spontaneously activate without the assistance of chromatin remodeling proteins [53]. It is possible that these genes can also be easily induced to higher transcriptional levels in the presence of active chromatin remodeling.
Non-canonical nucleosomes containing certain histone variants are often components of inducible chromatin [56]. For example, the baseline presence of H2A.z (a variant of the canonical histone H2A that forms an unstable nucleosome) at gene promoters is essential for induction target genes of the nuclear hormone receptor estrogen receptor-α (ER-α) [57–60] and H2A.z promoter enrichment increases with the strength of the estrogen receptor binding site [61]. H2A.z is lost from enhancers during induction [57, 62–64], likely due to full nucleosomal eviction or partial eviction of only H2A–H2B dimers during the chromatin remodeling that accompanies activation [65]. However, H2A.z is likely re-incorporated into nucleosome octamers after transcription is completed, in order to maintain baseline inducibility [64]. In another example, the presence of H3.3 (a variant of the canonical histone H3) in gene bodies potentiates transcriptional elongation. The N-terminus of H3.3 contains a serine 31 residue that is absent in the canonical H3 which is phosphorylated (H3.3S31ph) in response to stimulation in nucleosomes within gene bodies of rapidly induced genes [66]. H3.3S31ph binds the histone methyltransferase SETD2 and evicts the elongation co-repressor ZMYND11, which promotes RNAPII processivity and rapid elongation [66]. H3.3 is incorporated into genes immediately after induction and requires active transcription for incorporation [67]. H3.3 turns over continuously at constitutively active ribosomal DNA genes in Drosophila but is stably incorporated at induced Hsp70 genes after they have returned to baseline states [67].
Chromatin remodeling complexes can be blocked from remodeling nucleosomes by the presence of histone modifications [68]. Specifically, mono-ubiquitination of lysine 120 on the C-terminal tail of histone H2B (mono-ubiquitination of H2BK120, or H2Bub1) can block eviction of H2A.z-containing nucleosomes or histone heterodimers by blocking INO80 binding [68]. The H2B de-ubiquitinase USP22 is required for transcription initiation of inducible genes [69]. Overexpression of the E3 ubiquitin ligases RNF20 and RNF40 that deposit H2Bub1 is sufficient to block induction of ER-α target genes in response to hormone stimulus and knockdown of RNF20/40 is sufficient to promote increased induction in response to stimulus [70]. H2Bub1 may also function to define the boundaries of new chromatin opening in response to stimulus. For example, in MCF-7 breast cancer cells, H2Bub1 levels are high overall at ER-α inducible enhancers, except at transcription factor binding sites, to a degree proportional to the strength of the ER binding site [68]. Immediately after induction with estradiol, a “valley” of H2Bub1 enrichment forms at the centers of strong enhancers and these valleys gain other histone modifications of active enhancers [68]. The “peaks” of H2Bub1 on either side of these valleys may prevent the spread of accessible chromatin [68]. In these cells, chromatin rich in H2Bub1 overlaps chromatin that is rich in H2A.z, but H2A.z levels drop when H2Bub1 valleys form and impaired H2Bub1 leads to a decrease in H2A.z both before and after induction [68]. These data suggest that H2Bub1 stabilizes H2A.z in nucleosomes and that H2A.z-containing nucleosomes can be evicted when H2Bub1 is removed, ostensibly by USP22, in response to stimulus [68]. Disruption of H2Bub1 is also associated with aberrant gene activation in primary human cancers [61]. H2Bub1 levels are low in primary tumor samples of late-stage breast cancer, and estrogen-dependent breast cancer cells can proliferate in the absence of estrogen when RNF20/40 is knocked down [61]. However, RNF20/40 knockdown has only a moderate effect on constitutive gene expression [70, 71], supporting a specific role for H2Bub1 in regulating inducible, rather than constitutively active, genes. H2Bub1 may only block chromatin remodeling when it is present in regulatory elements and have different functions in different genetic contexts. For example, H2Bub1 is required for RNAPII elongation within gene bodies in yeast [72–74]. H2Bub1 is also enriched in actively transcribed regions of human genes [75] and likely plays a similar role in transcriptional elongation in human cells [72, 75]. In support of this model, H2Bub1 increases across the gene bodies of ER-α target genes GREB1 and TFF1 in human cancer cell lines on induction with estradiol [61, 76–78]. Overall, current evidence supports H2Bub1 as a critical component of active transcription within gene bodies but as a potent repressor of initiation in promoters and enhancers [79, 80].
2.3. Speed and synchrony of gene induction
Although all transcribed genes must go through all of the same steps to achieve successful transcription, genes differ in their transcriptional speed. Transcriptional speed is partly determined by the state of RNAPII at baseline, which can be described using the phrase “ready, set, go!”[42, 43, 81] At many genes, RNAPII is unbound at baseline (“not ready”), and must be recruited to the core promoter as part of the PIC [42, 43, 81]. However, RNAPII may also be bound to the core promoter at baseline but unmodified (i.e., a “ready” state) [42, 43, 81]. Alternatively, RNAPII may be bound in the poised state, in which serine 5 of the RNAPII CTD is phosphorylated and transcription has been primed (i.e., a “set” state) [42, 43, 81]. In this third instance, binding of a transcription factor does not trigger initial PIC assembly but rather results in phosphorylation of serine 2 on the RNAPII CTD, which triggers release of RNAPII into productive elongation (“go!”) [42, 43, 81]. Genes in the unready state at baseline are the slowest to respond, and genes in the set state are the fastest [82]. In addition, a target gene set in which all genes are all maintained in the set state at baseline can be activated with a high degree of synchrony [49], enabling a specific rapid response at the cellular level.
The genes that respond fastest to an initial stimulus are termed primary response genes (PRGs) [83]. PRGs can be induced without any new protein synthesis, so any required transcription factors or cofactors are already present in the cell at baseline [83]. PRGs can be subdivided into those genes with immediate response, termed immediate-early genes (IEGs), and those with delayed response but that still do not require new protein synthesis, termed delayed PRGs [48, 84]. PRGs often encode transcription factors and signaling molecules that, once translated, trigger transcription of the second wave of target genes (secondary response genes, or SRGs) [48]. IEGs have the fastest induction kinetics of known inducible genes, which is accomplished by pre-setting genes with poised RNAPII that is then released into elongation by the positive elongation factor P-TEFb which phosphorylates serine 2 on the RNAPII CTD [48, 85]. P-TEFb is recruited to IEG promoters by the bromodomain protein 4 (BRD4), which is itself recruited by the acetylation of histone H4 at lysine 16 (H4K16ac) [86]. H4K16ac is deposited by the histone acetyltransferase MOF, which is recruited by phosphorylation of histone H3 at serine 10 (H3S10ph) [86]. Therefore, the H3S10ph modification kicks off a sequence of events that results in pause-release of RNAPII from IEG promoters [86]. However, this modification is not present at IEGs at baseline [86]. Rather, IEGs are generally bivalent at baseline and contain the repressive modification H3K27me3 and the activating modification H3K4me2/3, as well as activating acetylated histones [87]. IEGs generally produce short transcripts and their promoters are enriched for specific transcription factors, including serum response factor (SRF), nuclear factor-κB (NF-κB), cyclic AMP response element-binding protein (CREB), and Zeste-like factor [84] Therefore, a likely sequence of events entails activation of one of the above transcription factors by a stimulus, binding of that transcription factor to the IEG promoter, recruitment by that transcription factor of histone modifying enzymes that remove H3K27me2/3 and deposit H3S10ph [86]. H3S10ph then recruits MOF, which deposits H4K16ac [86]. H4K16ac recruits BRD4 and P-TEFb, which triggers RNAPII pause-release and transcriptional induction [86]. Additional chromatin modifications, including H3S28ph [86], and the enzyme PARP-1 [88] have been linked to IEG activation, although their roles are unclear.
2.4. Chromatin looping controls the speed and intensity of transcription at inducible genes
Inducible genes and their respective enhancers are enclosed within distinct chromatin loops (<200kb) that are anchored by CTCF, within which smaller enhancer-promoter contact loops can form [89]. These larger chromatin loops support efficient induction, by bringing enhancers and promoters of inducible genes closer to one another in physical space, and they also restrict the enclosed inducible genes to activation by enhancers that are also present within the CTCF loop [89]. They also insulate inducible genes from neighboring housekeeping genes which are constitutively expressed and therefore constantly recruiting RNAPII and other transcriptional activators [89]. These CTCF loops enclose dynamic sets of inducible genes that are required for cellular function [89]. In primary cells, these loops contain genes involved in transcriptional regulation, cell motility and stem cell differentiation, but in immortalized cells, the genes responsible for these functions are not contained within distinct CTCF loops [89], which tracks with these genes’ differential regulation in immortalized cells [89]. CTCF is required for these loops and when CTCF is not present, inducible enhancers lose chromatin modifications indicative of active (H3K4me1 and H3K27ac) or poised (H3K4me1) enhancers [89, 90], indicating that these inducible enhancers are inactive in the absence of CTCF. This is an important result in light of general acceptance that knockdown of CTCF has little effect on global gene expression [91]. Although this is true in unstressed cells, the phenotypic effects of CTCF knockdown are apparent when the appropriate stimuli are applied. In fact, published case reports on three unrelated patients with mutations in CTCF showed reduced expression of enhancer-dependent inducible genes [92]. These case reports support a model in which inducible gene transcription can be initiated by promoters but not amplified by contact with enhancers in the absence of CTCF-mediated loops [92]. Notably, some inducible genes also show enhancer-promoter loops at baseline [93, 94]. This pre-setting of enhancer-promoter contacts further increases speed of target gene induction [93–95]. Loop pre-setting occurs both in undifferentiated cells during embryonic development [95] and in fully differentiated cells [96]. In both mouse and Drosophila embryonic stem cells, genes that are set to be activated at later stages of differentiation are pre-looped and associated with chromatin modifications characteristic of poised enhancers [95]. In differentiated human cells, TNF-α-responsive genes show pre-looped enhancers and promoters [96]. One of these target genes shows a unique pre-loop that connects the gene’s 5’ and 3’ ends and requires CTCF; this pre-loop is required for induction of this particular gene [96]. Since TNF-α is one of the transcription factors with motif enrichment in IEG promoters [48], these data suggest the possibility that pre-looping may be a general characteristic of IEGs. Chromatin loops can be actively disassembled to repress inducible genes [97]. For example, an enhancer-promoter loop that induces the Kit gene during development is later actively removed by replacing the loop-inducing transcription factor GATA-1 with the non-loop-inducing GATA-2 at the distal enhancer, which functions to repress Kit [97].
3. Transcriptional memory
Transcriptional memory describes a stable shift in a gene’s inducibility at the chromatin level [98, 99]. Cells can develop transcriptional memories of prior stress responses that alter either active transcriptional profiles or secondary inducibility profiles [98, 99]. Transcriptional memory suggests that an individual’s baseline chromatin state at CDS genes is determined by past exposures and will influence response to future exposures. Transcriptional memory can take four forms: (1) sustained activation of a gene even after exposure ceases (Figure 2); (2) sustained repression or silencing after an exposure ceases (Figure 2); (3) positive priming, in which a gene that responds to an initial hit of a stressor will respond either more quickly or more strongly to a second hit (Figure 2); or (4) negative priming, in which a gene that responds to an initial hit of a stressor will respond less strongly or not at all to a second hit (Figure 2).
Figure 2. Transcriptional memory takes several forms.
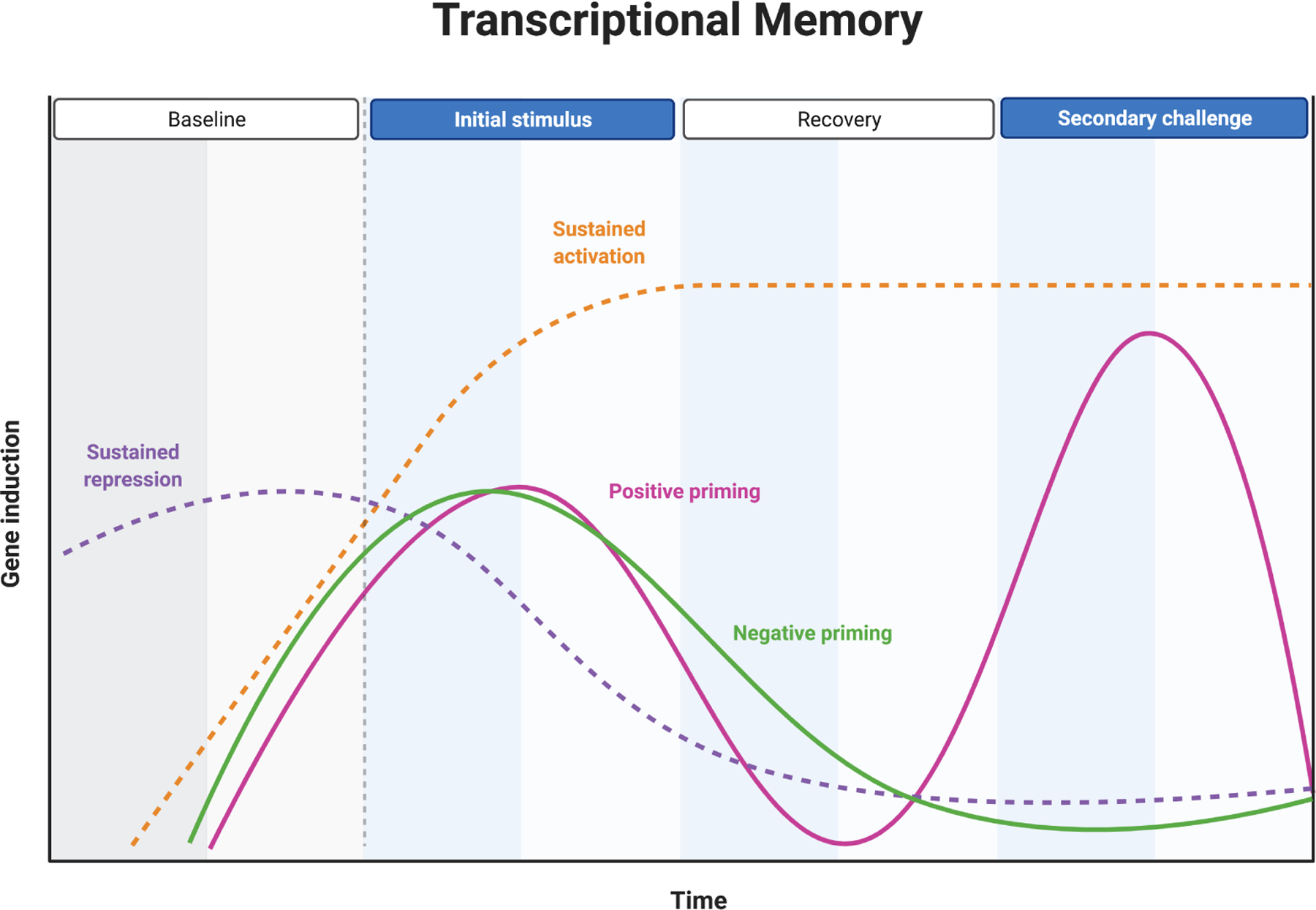
Inducible genes that are activated (or repressed) by a stressor respond rapidly but transiently to stressor. However, in some cases transcriptional memory of stress exposure forms at stress-responsive genes. Transcriptional memory can take one of four forms. In sustained activation, genes that are induced by a stressor can remain at high levels of transcription instead of returning to a baseline state. In sustained repression, genes repressed by a stressor remain repressed, instead of returning to a baseline state. In positive priming, genes induced by a stressor return to baseline after stress ceases, but induction is faster or stronger on re-stimulation. In negative priming, genes induced by a stressor return to baseline after stress ceases, but induction is decreased or absent on re-stimulation.
All four of these forms of memory can occur in transient or relatively stable forms [100–105], although the mechanisms governing which genes within relevant target gene sets develop memories and how long those memories last are unclear. Stable memories are not necessarily epigenetic, as they may not be inherited across cell division [106, 107]. However, there is some evidence that chromatin modifications can be mitotically inherited [108], providing a mechanism for epigenetic memory. Similar to inducible genes’ dependence on DNA sequence motifs, transcriptional memory formation is also influenced by underlying DNA sequence [99, 109, 110]. Importantly, transcriptional memory can be accomplished through non-chromatin mechanisms. For example, transcription factors can be post-translationally modified to increase their residence time on chromatin to increase transcription level and duration [111] and sequestration proteins can be overexpressed or their associations with transcription factors can be stabilized to inhibit a transcription factor’s ability to translocate to the nucleus or bind DNA [112]. In addition, transcriptional regulators can be deposited by a parent cell into daughter cells during mitosis to confer parental transcriptional states on daughter cell genomes [113].
Genes that are newly expressed by a stimulus usually return to baseline after removal of the stimulus [18]. In some cases, however, induced genes show sustained activation [100, 114, 115]. Sustained activation likely depends on the strength of the initial activation [100] and may vary even among cells within a tissue [100]. For example, individual cells that showed particularly strong initial transcriptional responses to doxycycline, hypoxia and DNA damaging agents showed sustained changes in gene expression, as well as growth rates and viability, for multiple cellular generations after the initial stimulus, but cells with lower initial transcriptional responses did not [100]. Sustained gene activation can be a result of persistent, aberrant de-repression of gene sets, as well. For example, loss of repressive marks H3K27me and H3K9me3 in Caenorhabditis elegans associated with developmental bisphenol A exposure is inherited for multiple generations [114]. In another example, loss of DNA methylation at the retroelement viable yellow Agouti associated with developmental bisphenol A (BPA) [116] or genistein [115, 116] exposure persists well into adulthood. However, it is not clear that these two examples represent memories that are triggered by an initial transcriptional event.
Genes that are newly repressed by a stimulus can show sustained repression after removal of the stimulus [101]. Sustained repression does not imply lack of responsiveness on secondary challenge, only that repression induced by an initial stressor is persistent. Over time, transient repression, which is characterized by the loss of activating histone modifications (e.g., H3K4me3, H3K9ac, H3K14ac) and the presence of repressive histone modifications (e.g., H3K27me3), can be stabilized into a fully heterochromatic silenced state, which is characterized by the addition of H3K9me3 and DNA methylation [117], which trigger 3D chromatin compaction [101]. Heterochromatin that is artificially induced via recruitment of the transcription factor heterochromatic protein-1 (HP-1) [118] or the repressive histone modification H3K9me3 [119] is epigenetically maintained, in that it can be inherited across cell division [101, 118, 119]. Sustained repression may even be inherited across generations. In C. elegans, the repressive modification H3K27me3 can be meiotically inherited [120] and newly repressed genes may remain repressed for many generations [120]. In Drosophila, repressive Polycomb group proteins (including the histone methyltransferase EZH2, which deposits H3K27me3) form repressive loops between an endogenous homeotic gene Abdominal-B and a cis-regulatory Polycomb response element elsewhere in the genome [121]. This repressive loop can be reconstituted between the response element and an artificial transgene and abolishment of the loop is sufficient to de-repress the gene [121]. De-repression of Abdominal-B is inherited by several Drosophila progeny, suggesting that loss (or gain) of sustained repressive memory may be meiotically heritable in metazoans [121]. Polycomb group proteins form similar repressive loops in humans [122]. Until recently, it was thought that humans did not have Polycomb response elements to serve as loop anchors; however, a recent study reported four human Polycomb response elements [123]. DNA methylation is often deposited at H3K27me3-modified heterochromatin, as well [124]. DNA methylation is a particularly attractive candidate for this form of memory, since there is a clear mechanism for its propagation during DNA replication [125]. DNA methyltransferase 1 (DNMT1) associates with the DNA replication machinery and adds new methyl groups to newly synthesized strands at locations that are methylated in the parent template strand [125].
Inducible genes that show positive priming are characterized on the 2D level by baseline increases in active histone modifications (e.g., H3K4me1/2/3) [102, 103] RNAPII occupancy and/or RNAPII poising [85, 103]. An increase in active histone modifications and RNAPII occupancy or poising generally increases the rate and intensity of transcriptional activation on secondary stimulus challenge [102, 103, 126]. Positive chromatin priming has been observed in repeated studies of immune system cells (reviewed in [127]). For example, human macrophages exposed to Candida albicans show increased H3K4me3 at pro-inflammatory genes and signal transducers of the immune response that lasts for a week after exposure and is associated with faster transcriptional response of those genes on secondary exposure [128]. H3K4me3 is newly deposited after activation of inducible immune genes that have no basal activity [129, 130]. However, limited studies in other cell types suggest that priming mechanisms are common across cell types in response to multiple stimuli. H3K4me3 is newly deposited at genes associated with prostate cancer in prostate tissue of rats following postnatal exposure to the endocrine disrupting chemical bisphenol A [131]. Subsequent to this initial postnatal exposure, these prostate cancer genes show elevated expression at baseline, as well as increased expression intensity in response to hormone in adulthood [131]. A series of studies in yeast provides more detail on a potential mechanism. Yeast develop positive priming at the inositol-1-phosphate synthase (INO1) gene by recruiting a remodeled form of the Set1/COMPASS histone methyltransferase complex that deposits only H3K4me2 and cannot deposit H3K4me3 [132]. The H3K4me2 modification recruits Set3, a component of the SET3C HDAC complex, which is in turn required for both RNAPII recruitment and H3K4me2 maintenance at INO1 [103]. In addition, a modified form of the transcriptional co-activator Mediator complex is required for the recruitment and poising of RNAPII at INO1 [103]. A Mediator complex that contains the Cdk8 kinase controls RNAPII recruitment and poising at the INO1 gene under memory conditions, but not under active conditions, suggesting a highly specific role for this remodeled Mediator complex [103]. In rare cases (or, at least, infrequently reported cases), positively primed enhancers that were previously not readily accessible to transcription factor binding can become newly and stably accessible, even in differentiated cells with established enhancer repertoires [133]. Fully differentiated human macrophages show sequential binding of stimulus-activated transcription factors (STAT1 and STAT6) and a lineage-determining transcription factor (Pu.1), followed by acquisition of active enhancer modifications, including H3K4me1 and H3K27ac, to previously inactive but not silenced enhancers in response to stimulus [133]. These new enhancers lose Pu.1 binding after stimulus ceases, likely because of a low affinity Pu.1 binding site that mandates cooperative binding of Pu.1 with stimulus-responsive transcription factors [133]. However, H3K4me1 is retained at ~30% of these enhancers and mediates a faster and stronger response on re-stimulation [133]. In addition, some regulatory elements that are active during embryonic development retain a hypomethylated state (low levels of DNA methylation) in fully differentiated cells, despite those elements’ inactivity in differentiated cells, which suggests a mechanism for reactivating developmental programs, given the right stimulus [134]. How does H3K4me1/2/3 accomplish positive priming? H3K4me may recruit histone acetyltransferases that accelerate transcriptional response [135]. Alternatively, H3K4me may not be responsible for priming, but rather the enzyme complexes that deposit or maintain it. In the above yeast example, Set3 recruits RNAPII to the INO1 gene under memory conditions [103]. In mouse embryonic stem cells, the MLL2 subunit of the MLL2/COMPASS complex can bind developmental genes and prevent their repression by Polycomb repressive complexes (PRCs) and DNA methyltransferases [136]. Although acetylated histones are generally lost when inducible genes are repressed following stimulus removal, there are some examples of retained histone acetylation at genes with positive priming. RNAPII and the HAT p300, as well as acetylated histones, are newly bound at baseline in some IEGs in T-cells after stimulation, and their binding correlates with faster re-activation of those genes [85]. Retained histone modifications can also increase the sensitivity of regulatory elements to a stimulus. For example, after initial stimulation, T-cells with newly bound RNAPII and the HAT p300 are able to respond to stimuli that previously were unable to induce a T-cell response [85].
Positive priming events can also occur on the 3D level, in the form of new chromatin loops or stabilized chromatin loops, and/or by association with the nuclear pore complex (reviewed in [127, 137]. In yeast, when inducible genes are activated by specific transcription factors, those genes form loops that link their 5’ and 3’ ends, and these loops move to the nuclear periphery and interact with proteins contained within the nuclear pore complex [98]. Heterochromatic regions containing silenced or repressed genes are often found on the nuclear periphery of eukaryotic cells and euchromatin is localized to the center of the nucleus [138]. Therefore, the nuclear pore complex represents an exception to this nuclear organization rule [139]. These induced genes generally return to the nucleoplasm when gene activation ceases, but in cases of positive priming, they remain looped and associated with the nuclear pore complex and more rapidly recruit RNAPII to their promoters, yielding faster and stronger gene transcription on secondary stimulus challenge [98]. Similar nuclear pore complex association is required for the induction of the Drosophila heat shock protein gene hsp70 [140], as well as in mammalian genes modified with acetylated histones [141], suggesting that localization to the nuclear pore complex is a common step in gene induction. An additional study in Drosophila demonstrate that the nuclear pore protein Nup98 has a distinct architectural role in forming enhancer-promoter loops at target genes [126] and a conserved role for Nup98 is apparent in humans, as well, where its presence is associated with H3K4 methylation and poised RNAPII [126]. Histone variants may play a role in 3D positive priming, as well. In yeast, the histone variant H2A.z is incorporated into the promoters of two genes, GAL1 and INO1, just after transcriptional activation [142]. These genes localize to the nuclear pore complex during activation, and H2A.z is required to retain them there after repression [142]. This continued association with the nuclear pore is required for faster reactivation of these genes on secondary challenge [142]. However, the roles of H2A.z and nuclear pore complex proteins in transcriptional memory are not supported by all evidence. At least one report shows that these chromatin factors do not control memory, but rather mitotically inherited transcription factors and a classic signaling feedback loop are responsible for apparent memory at the yeast GAL gene cluster [143]. The controversy about the relative roles of feedback signaling loops and structural chromatin features in transcriptional regulation is not a new one [107]. It may be possible that both phenomena are at play and must be carefully dissected mechanistically. An additional study in yeast showed that mild salt stress led to later resistance to hydrogen peroxide that persisted for four to five generations [144]. This resistance phenotype was entirely due to long-lived catalase that was synthesized during the initial salt stress and distributed to daughter cells [144]. However, two forms of memory may operate simultaneously; in a separate effect from hydrogen peroxide resistance, these cells also showed faster gene expression response at stress-induced genes, which response required the nuclear pore protein Nup42p [144].
Inducible genes that show negative priming show baseline loss of active histone modifications [104, 105]. Negative priming is sometimes referred to as transient silencing, although this alternate term does not capture the lack of responsiveness to secondary challenge. A classic example of negative priming is endotoxin tolerance, in which target genes have a refractory period after an initial exposure to a pathogen or pathogen-like (i.e., lipopolysaccharides, or LPS, present on pathogen cell membranes that stimulate immune responses) [104, 105, 145]. Negative priming likely avoids adverse consequence of exaggerated, chronic or repeated activation of target gene sets, but it can also present serious consequences. For example, human monocytes in people recovering from sepsis or other immune trauma cannot respond to a subsequent pathogen challenge for a substantial period of time, often several days, placing these patients at risk for mortality due to immunosuppression [104, 145]. Endotoxin tolerance is associated with suppression of inflammatory cytokines, including IL-1β, and upregulation of negative regulators of the inflammatory response, including the IL-1R-associated kinase-M (IRAK-M) which regulates TLR4 signaling, soluble triggering receptor expressed on myeloid cells-1 (TREM-1), and suppressor of cytokine signaling 3 (SOCS3), among others, which temporarily block an inflammatory reaction to LPS and other immune stimuli (reviewed in [145]). Endotoxin-tolerant human monocytes that suppress IL-1β do not show the increase in induction-associated H3S10ph and decrease in repressive H3K9me3 at this gene that endotoxin-sensitive cells do in the presence of LPS [146]. This negative priming effect extends to a full set of inflammatory cytokine genes [105]. Specifically, the SWI/SNF remodeler BRG1 was not recruited to these genes in response to LPS in tolerant cells and these genes show decreased baseline activating histone modifications (total H4 acetylation, H3K4m3) and decreased chromatin accessibility; however, these genes do not carry histone modifications that indicate gene silencing (H3K9me3, H3K27me3) [105]. Instead, repression is mediated by the high mobility group box 1 protein (HMGB1) and the linker histone H1 [147]. Specifically, HMGB1 binds the TNF-α promoter and promotes assembly of the RelB repressor complex [147]. H1, which displaces HMGB1 from chromatin under baseline conditions, co-binds with HMGB1 to the repressed promoter [147]. HMGB1 and H1 are recruited independently to the promoter and their binding requires H3K9me2 [147]. This mechanism is gene-specific; a similar repression is seen at the pro-inflammatory IL-1β promoter, but not at the anti-inflammatory IκBα promoter [147]. In addition, IEGs, delayed PRGs and SRGs showed differences in chromatin remodeling requirements during endotoxin tolerance [148]. Under normal conditions, chromatin remodeling by SWI/SNF is required for activation of SRGs and delayed PRGs, but not IEGs [149]. The Mi-2β remodeling protein co-binds with SWI/SNF at some of these genes to repress their activation, providing an alternate mechanism for negative priming in endotoxin tolerance [149]. It is interesting to note that endotoxin tolerance also involves positive priming of antimicrobial genes, which were characterized by higher baseline and inducible histone acetylation and faster BRG1 recruitment [105], highlighting that multiple types of transcriptional memory may regulate a biological response.
4. CDS pathways respond to redox-active pollutants
CDS pathways sense shifts in cellular homeostasis caused by redox-active pollutants and response by activating inducible CDS target genes [13]. In this section, I will briefly review each CDS pathway, how it senses cellular stress, the sequence of events required for induction of its target genes and evidence for transcriptional memory formation in response to pathway stimulation. Although I have not reviewed them here, redox-active pollutants can trigger induction and transcriptional memory at genes not within CDS pathways (reviewed in [150]) and redox-active pollutants can also affect chromatin state through general mechanisms that do not specifically target CDS genes [151].
4.1. Stress-activated p38 MAP kinase signaling
In addition to specific CDS responses, mammals have a general stress response pathway governed by p38 mitogen-activated protein kinases (MAPKs) that can respond to a wide range of stressors (reviewed in [152]). (P38-MAPKs also have many functions in the cell that are unrelated to CDS pathways, including involvement in immune and inflammatory responses [152].) Much of our understanding of the mammalian general stress response is based on research on the yeast general stress response [18]. Unlike humans, yeast only have this general stress response system (in yeast, called the Environmental Stress Response or ESR [153].) The ESR is activated by a single stress-activated MAPK, Hog1, that regulates expression of a general set of ESR target genes (~300 genes are induced and ~600 are repressed, although increasing stress leads to induction of a larger gene set) [154]. The classic Hog1 inducer is osmotic stress, although the ESR responds to a variety of stressors (reviewed in [15]). Hog1 is rapidly activated by phosphorylation after osmotic stress, after which it translocates to the nucleus [153]. Hog1 affects transcription both through transient phosphorylation and subsequent activation of transcription factors and by physically associating with target gene promoters [153, 155]. Hog1 is tethered to chromatin by its interaction with transcription factors, which enables Hog1 to phosphorylate other transcriptional co-activators [153]. For example, Hog1 can activate the Sko1p-Cyc8p-Tup1p complex to recruit the HAT complex SAGA, the HDAC Rpd3, SWI/SNF chromatin remodelers, and RNAPII [155–157]. However, chromatin remodeling may not be required for stress gene induction [158], suggesting that Hog1 binding to gene promoters may only be necessary under some conditions. When H3K4 methylation, particularly H3K4me1, is present at stress gene promoters, chromatin remodeling is required for induction [158]. When this modification is removed, SWI/SNF is no longer required for target gene induction and alternative remodeling complexes, including the Swr1 complex that deposits H2A.z, can bind to these genes [158]. These data suggest that yeast stress response genes are controlled by two remodeling mechanisms: SWI/SNF remodels gene promoters in the presence of H3K4 methylation and Swr1 deposits the unstable histone H2A.z in its absence [158], which may promote accessible chromatin in the absence of active remodeling.
The mammalian general stress response is much more restricted than the yeast response [159]. In mammals, ~100–150 genes are rapidly upregulated on heat stress, osmotic stress and oxidative stress but the common response is restricted to ~30% of them [152]. Human p38 MAPK has four isoforms (p38α-δ, or MAPK11–14, sometimes called stress-activated kinases or SAPKs); p38α is the isoform most commonly activated in mammalian stress response [152]. Outside of the common response, p38α interacts with specific CDS responses [152]; these interactions are highlighted in later sections of this review. A significant fraction of specific mammalian CDS responses rely on p38α [159]. For example, p38α is recruited to target genes in response to osmotic shock, anisomycin, or TNF-α activation by their respectively activated transcription factors, and p38α is then required for RNAPII recruitment [157]. In another example, exposure of mammalian cells to heat shock and oxidative stress for 30 minutes induced around 100 genes [160]; exposure of mammalian cells to osmotic stress, the inflammatory cytokine TNF-α and the protein synthesis inhibitor anisomycin for 45 minutes induced more than 120 genes; the majority of both gene sets were dependent on p38α, including a set of common response genes (~20% of total target genes) activated by different stimuli [159]. Like Hog1, p38α can be recruited to target gene promoters and recruit SWI/SNF complexes [161]. P38α phosphorylates mitogen- and stress-activated protein kinases MSK1/2, which in turn phosphorylate both the chromatin architectural protein high mobility group protein 14 (HMG-14) and serine 10 on histone H3 (H3S10ph), as well as the IEG transcription factors CREB and NF-κB to trigger stress gene transcription [162]. MSKs themselves can be recruited to IEG promoters, as well, to initiate transcription [163]. P38α also phosphorylates MAP kinase-activated protein kinase 2 and 3 (MK2/3), which phosphorylate the IEG transcription factors CREB and SRF [164]. Last, p38α directly phosphorylates the transcription factors p53 and ATF6, which have distinct roles in CDS pathways (DNA damage response and unfolded protein response, respectively) (reviewed in [152]). P38α associates both with regulatory elements and along the entire length of transcribed gene bodies [165, 166], suggesting that it also plays a role in transcriptional elongation of stress response genes.
4.2. Heat shock signaling
The heat shock response is an evolutionarily conserved cellular defense that is triggered by proteotoxic stress caused by protein misfolding or denaturation and subsequent protein aggregation [167]. The heat shock response was first described in 1962 by Ferrucio Ritossa, who discovered that increased temperature induced chromosomal puffs to form in the large chromosomes of the salivary glands of Drosophila busckii [168]. These puffs were later identified as regions of actively transcribed genes encoding heat shock proteins, which are protein chaperones that support protein folding and clear protein aggregates [168]. Current data show that the heat shock response is characterized by more complex transcriptional changes than the upregulation of a small number of heat shock proteins [82]. The mammalian heat shock response triggers the rapid induction of several hundred genes and repression of several thousand genes [169]. Genes that are upregulated are required for maintaining cellular homeostasis, including heat shock proteins and cytoskeletal proteins required to maintain cellular integrity and cellular transport [170].The cell downregulates all other transcription globally, including constitutively expressed genes that control cell cycle, general transcription and metabolism, to prevent misfolding of newly synthesized proteins and an exacerbation of proteotoxicity [169]. Under heat shock conditions, ubiquitination of misfolded nascently translated proteins is detected by p38α which then translocates to the nucleus and directly binds the promoters of repressed genes [169]. In addition to sensing denatured proteins, increased temperature is sensed directly via thermosensory structures in DNA, RNA, proteins and lipids. DNA topology can be altered, RNA hairpins can melt, heat shock proteins can shift conformations, and ion channels are activated in mammalian cell membranes [171–173]. A sustained heat shock response can be harmful to the cell. In Drosophila, induction of the transcription factor heat shock factor (HSF) is restricted within minutes of heat stress, and when this restriction is lost (during HSF overexpression or overexpression of HSF targets), cells show reduced fitness and compromised viability [174].
4.2.1. Redox-active pollutants activate the heat shock response
Proteotoxicity can be triggered by stimuli other than heat. For example, heavy metals and metalloids (e.g., lead, mercury, cadmium, arsenite) can bind thiol groups within proteins, disrupting their structures and function, causing proteotoxicity [150, 175]. In addition, any pollutant that increases reactive oxygen species in the cell can cause damage to proteins, triggering a heat shock response [176] (see Oxidative Stress Response.) Heat shock signaling occurs in response to misfolding of nascently translated proteins, rather than existing proteins present in the cell [169]. This suggests that the heat shock response is triggered partly by misfolding of proteins upregulated by pollutant exposure, rather than by inactivation of proteins present in the cell at the time of exposure.
4.2.2. Heat shock response in the 2D genome
At the 2D genome level, several heat shock transcription factors orchestrate a rapid change in global transcription in the cell in response to proteotoxic stress [14, 169]. The majority of upregulated heat shock response genes are activated by increased release of poised RNAPII from target gene promoters [14, 169]. This release is directly triggered by the positive elongation factor P-TEFb, which phosphorylates and releases negative elongation factors NELF and DSIF, as well as phosphorylating serine 2 on the CTD of RNAPII [169]. P-TEFb is recruited to target gene promoters when transcription factors bind [169]. This regulation implies that the initial steps of transcriptional activation (increased chromatin accessibility at the core promoter, assembly of the PIC, poising of RNAPII) all occur at baseline, prior to proteotoxic stress exposure. Unlike heat shock gene promoters, heat shock enhancers are generally unbound by RNAPII at baseline, and their chromatin accessibility is bookmarked by pioneer transcription factors (GAGA-associated factor, or GAF, in Drosophila, and GATA-2 and TAL1 in humans) [14, 18]
A recent time-course study in mouse embryonic fibroblasts demonstrates that three groups of genes are activated, and two groups of genes are repressed, in response to heat shock, but they differ in their transcription factor control and timing [82]. Heat shock factor 1 (HSF1), originally considered the “master regulator” of the heat shock response [177–179], controls only a small number of heat shock-induced genes, primarily heat shock proteins and other chaperones, by increasing release of poised RNAPII from heat shock promoters [82]. These genes are upregulated rapidly (within 2 hours of heat shock) and maintained at a high transcriptional level for at least four days (60 hours) [82]. The transcription factor SRF induces IEGs, primarily cytoskeletal genes, also by triggering release of poised RNAPII from target gene promoters [82]. A subset of these genes is activated rapidly (within 2.5 hours of heat shock), and another subset is activated more slowly (within 12 hours of heat shock) [82]. Both subsets of genes are activated only transiently; all genes return to baseline by the 60-hour mark [82]. A third group of genes, primarily apopotic genes, is upregulated, likely by the transcription factor NRF2 [82]. This group of genes is also induced by increased release of poised RNAPII, but their induction occurs late after heat shock [82]. Although all three of these groups of induced genes are primarily regulated by increased release of poised RNAPII, there is some evidence that all three are also regulated to a lesser degree by an increase in new RNAPII recruitment and PIC formation [82]. For example, HSF1 can recruit the chromatin remodeling complex SWI/SNF to mammalian target gene promoters to enable PIC binding (reviewed in [14]). The majority of the heat shock response involves global transcriptional repression of active genes, classified into two groups: those involved in cell cycle and metabolism (repressed early, within 2.5 hours of heat shock) and those involved in mRNA processing (repressed late, within 12 hours of heat shock) [82]. Both groups show sustained repression for at least four days (60 hours) [82]. It was previously thought that HSF1 was responsible for transcriptional repression, too, by binding gene bodies and creating a physical obstacle to transcript elongation [180], but current evidence shows that HSF1 bound at intragenic (and intergenic) sites is more likely binding to enhancers [180]. Transcriptional repression is accomplished by reduced release of poised RNAPII, mediated by increased recruitment of the negative elongation factor NELF to target gene promoters [82], in some cases by p38α [169]. Heat shock also activates transcription of non-coding transposable elements (Alu in humans, SINE B2 in mice) that prevent initiation of transcription on some repressed genes by interfering with PIC formation [181–183].
Histone modification by the chemical groups PAR (PARylation) and SUMO can promote heat shock transcriptional responses [184–186]. PARylation is a stress-inducible histone modification deposited by the poly[ADP-ribose] polymerase 1 (PARP-1) in the promoter and gene body of the target heat shock protein gene HSPA1A on heat shock [148, 184]. PARylation promotes increased chromatin accessibility and allows HSF1 binding to the gene promoter [184, 187]. SUMO is a complex of small ubiquitin-like modifier proteins that is increased at the promoters of highly transcribed genes and enhancers during heat shock [185, 186, 188]. SUMO restricts the activity of these genes and promotes their repression [185]. Both positive (P-TEFb) and negative (NELF) elongation factors are also modified with SUMO groups, which targets them for proteasomal inhibition [185]. This result suggests that SUMOylation of elongation factors can affect release of poised RNAPII, although not in a single clear direction. Heat shock triggers a decrease in SUMOylation of regions containing CTCF-binding sites, which suggests a role for SUMO in TAD shifts during heat shock (discussed below) [185].
In addition to programmed gene responses, heat shock (among other forms of stress, including oxidative and osmotic stress), leads to failed transcriptional termination in several genes [189, 190]. The resulting transcripts are called “downstream of gene-containing transcripts” (DoGs), because they include readthrough transcription that can span thousands of base pairs downstream of target genes’ ends [189, 190]. The function of DoGs is unknown, although they may be needed for nuclear scaffold maintenance during stress [189, 190].
4.2.3. Heat shock response in the 3D genome
At the 3D genome level, both TAD reconfiguration and formation of chromatin loops that bring enhancers in contact with heat shock target gene promoters are important parts of the heat shock response [191, 192]. The involvement of enhancers in the heat shock response provides a model for tissue-specific heat shock transcriptional programs, either through differential target gene sets, or differential speed, intensity or stability of target gene responses. Pluripotency factors OCT4, NANOG and KLF4 are involved in loop formation between heat shock enhancers and target genes, as well as target gene regulation, in human embryonic stem cells [191], suggesting that developmental timing of proteotoxic stress may influence the transcriptional response, as well. Higher order TADs are reorganized during the heat shock response by re-localization of CTCF to new sites and formation of stress-induced TADs in human embryonic stem cells [191]. New TADs enable new enhancer-promoter contacts that were not possible in baseline TAD conformations; new TADs also disrupt baseline enhancer-promoter contacts, which may result in gene repression [89, 137]. In contrast to the human embryonic stem cell data, Drosophila cells exposed to heat shock do not disrupt baseline loops and form new ones, but rather re-localize CTCF and cohesin from TAD boundaries to enhancers and promoters contained within the original TADs, which weakens TAD boundaries and increases inter-TAD interactions [193]. These inter-TAD interactions function to repress certain genes during heat shock, by enabling new interactions between re-localized cohesin and the repressive Polycomb complex [193]. In addition to TAD dynamics, heat shock protein genes located on different chromosomes in yeast can physically interact with one another through HSF1-mediated contacts, in a function independent from its role as a transcription factor [192]. These inter-chromosomal contacts may support formation of transcriptionally active foci, or transcription factories, to drive synchronous induction of heat shock target genes [192]. These foci suggest that heat shock response requires phase separation [194–196], or formation of biochemically distinct nuclear sub-compartments, to accomplish a rapid, synchronous response.
4.2.4. Transcriptional memory in heat shock response
Heat shock signaling can lead to transcriptional memory in plants and animals [173, 197, 198]. In plants, temperature shock leads to H3K4me2/3 marking of induced heat shock genes that is associated with hyper-induction on secondary heat shock [25]. This memory requires the transcription factor HSFA2, which appears to be dedicated to memory formation, since it has no role in initial induction [25]. Heat shock leads to transcriptional memory in rodents and chicks [197, 198]. Chick exposure to heat stress three to five days after hatching leads to long-lasting transcriptional changes at the heat shock protein hsp70 in chick anterior hypothalamus [197]. Harsh heat stress triggered both a sustained induction response and a positive priming response. Specifically, harsh heat yielded an immediate 13.7-fold increase in hsp70 transcription which increased to 16.5-fold induction at 6 hours and remained high (4.8-fold over baseline) for the next 18 hours [197]. In contrast, mild heat stress triggered a negative priming response. Specifically, mild heat yielded only a 2.3-fold initial increase in hsp70 transcript levels that remained stable through 24 hours and then returned to baseline levels [197]. On secondary challenge with mild heat, chicks conditioned with harsh heat showed a stronger hsp70 expression response as compared to mild heat-conditioned chicks (3.1-fold vs 1.7-fold increase after 6 hours), indicating a positive priming response in the harsh heat group, but the mild heat group showed a blunted transcriptional response, as compared to its initial response to a heat stress of comparable intensity [197]. These data suggest that mild heat triggered conditioning, in which the chick acclimatized to a higher heat environment, but that harsh heat triggered a stress response. The harsh heat-conditioned group showed increased DNA methylation at repressive elements within the hsp70 promoter; this increased DNA methylation decreased binding of repressive transcription factors, as well as a chromatin remodeling complex containing a histone deacetylase [197]. Therefore, this increased promoter DNA methylation led to increased promoter histone acetylation at baseline, which is responsible for higher inducible hsp70 transcription in high heat-stressed chicks [197]. In rodents, mild heat stress triggered a positive priming response. Specifically, mild heat stress in rats led to transiently increased H3S10ph but sustained increase in histone H4 acetylation in the promoters of heat shock elements of heat shock genes hsp70 and hsp90 [198]. In this case, H4 acetylation levels predicted faster transcription of these genes on secondary challenge [198]. The authors hypothesize that the initial H3S10ph is required for deposition but not maintenance of histone acetylation [198]. This response extends to physiological increases in heat that occur during physical exertion. Specifically, monocytes isolated from female mice 30 days after exertional heat stress showed changes in promoter DNA methylation and increased intensity of transcription of heat shock genes following secondary heat shock [160]. Together, these data underscore the complexity of cellular responses to differential exposure duration and stressor intensity, as well as the importance of standardized stress exposure designs to clarify response patterns.
4.3. Oxidative stress signaling
The oxidative stress response is caused by reactive oxygen species (ROS) that disturb cellular redox status by oxidizing proteins, lipids, and nucleic acids (reviewed in [199]). A majority of endogenous cellular ROS is generated as a byproduct of mitochondrial oxidative phosphorylation (OXPHOS), but ROS is also generated by NADPH oxidases and during protein folding in the endoplasmic reticulum (ER) [199, 200]. ROS include hydrogen peroxide, superoxide radicals and hydroxyl radicals [199, 200]. ROS can serve as signaling molecules and also as undesirable cytotoxic byproducts of oxidative phosphorylation in mitochondria and other metabolic pathways in the peroxisome [199]. The mammalian cell’s oxidative stress response is primarily regulated by the NRF transcription factors, NRF1, NRF2, and NRF3 [201], although most research has focused on NRF2 [200]. The classic regulatory mechanism for NRF2 involves the Kelch-like ECH-associated protein (Keap1) [202]. At baseline, NRF2 is sequestered in the cytoplasm through association with Keap1, which makes NRF2 accessible to ubiquitination by the E3 ubiquitin ligase Cullin 3-Ring-box protein 1 (CUL3) and subsequent proteasomal degradation [112, 202–204]. When the cellular environment becomes more oxidative, cysteine residues on Keap1 are oxidized, which prevents NRF2 ubiquitination, resulting it NRF2 accumulation, nuclear translocation and chromatin binding to antioxidant response elements (AREs) in target gene promoters and enhancers [112, 202, 203, 205]. These target genes include antioxidant enzymes and enzymes required for biosynthesis of glutathione, the cell’s primary ROS scavenger (reviewed in [14]). Knockout of NRF2 increases susceptibility to many chemical pollutants and worsens diseases associated with oxidative stress (reviewed in [200]). Conversely, biochemical or genetic increases in NRF2 protects the cell against oxidative damage [200]. However, NRF2 activation can harm the cell, as well (reviewed in [206]). NRF2 signaling is associated with increased cisplatin resistance in certain cancers (reviewed in [150]). Oxidative stress signaling requires additional transcription factors, specifically members of the forkhead box O (FOXO) transcription factor family: FOXO1, FOXO3, FOXO4 [14]. FOXOs are activated by post-translational phosphorylation by upstream signaling molecules (JNK and MST1) and translocate to the nucleus, where they trigger transcription of specific antioxidant genes [14]. Other cellular signaling pathways, including NF-kB and JUN, may play a role in regulation of certain oxidative stress target genes (reviewed in [14]). Oxidative stress can also cause indirect effects by modulating levels of epigenetic substrates and mitochondrial metabolites/second messengers that themselves affect gene expression profiles (reviewed in [151, 207]). Importantly, ROS do not always cause oxidative stress and have independent functions as redox signaling molecules in cellular homeostasis and organismal development (reviewed in [199, 208]).
4.3.1. Redox-active pollutants activate the oxidative stress response
A large number of chemical pollutants can increase cellular ROS during their metabolism by cytochrome P450 proteins, by binding reactive thiol groups on certain proteins and inactivating them, by redox cycling, or by triggering mitochondrial dysfunction (reviewed in [150]). For example, non-dioxin-like polychlorinated biphenyls (PCBs) generate ROS as byproducts of metabolism by cellular cytochrome P450 enzymes [209]. Other pollutants (e.g., lead, cadmium, mercury) bind thiol groups within antioxidant defense proteins that protect the cell from free radicals like ROS or reactive nitrogen species (RNS), deactivating these proteins and causing free radical levels to rise passively in the cell (reviewed in [210]). In addition, certain toxicants (e.g., iron, cobalt, copper, and chromium) can redox cycle, or alternately function as electron acceptors and donors, which disrupts the stability of cellular proteins and lipids [210]. Last, select toxicants (e.g., lead, mercury) also accumulate in mitochondria, which have high ROS levels due to OXPHOS [210]. It is unclear whether metals directly trigger mitochondrial dysfunction, leading to ROS overproduction and leakage into the cytosol, or simply amplify their effects in an organelle rich in ROS scavengers, like glutathione, and antioxidant defense enzymes, including glutathione peroxidase and superoxide dismutase [210]. Some pesticides are known to cause mitochondrial dysfunction by inhibiting one or more complexes in the electron transport chain, which leads to electron leakage and ROS formation, with an eventual increase in cellular ROS [150].
4.3.2. Oxidative stress response in the 2D genome
During oxidative stress, NRF2 target genes are induced by new chromatin accessibility and PIC formation at the core promoters [211]. However, a subset of target genes show poised RNAPII at baseline and these genes are induced through increased release of poised RNAPII [211]. Unlike HSF1, which recruits the positive elongation factor P-TEFb to release RNAPII, during oxidative stress, the negative elongation factor NELF dissociates from chromatin and enables RNAPII release even in the presence of a P-TEFb inhibitor, suggesting that mechanisms of RNAPII pause-release may vary by stressor [211]. Chromatin remodelers can be recruited to oxidative stress target genes by DNA structures. For example, the NRF2 target gene heme-oxygenase 1 (HO-1) forms Z-DNA in response to NRF2 binding, which in turn assists in recruiting the SWI/SNF remodeler BRG1, which is required for activation of this particular gene [212]. However, chromatin remodeling complexes can also restrict oxidative stress gene expression. Decreased expression of SWI/SNF family remodelers BRG1 and BRM is correlated with increased expression of some NRF2 target genes [213]. This result indicates that SWI/SNF restricts NRF2 binding at some sites via local nucleosome remodeling that restricts accessibility. Higher order chromatin structure may also function in repressing a NRF2 response. For example, the high-mobility group box 1 (HMGB1), a chromatin architectural protein, suppresses NRF2 transcription, as well as transcription of NRF2 target genes, in response to hydrogen peroxide in human melanocytes [214]. Although the majority of the oxidative stress response is regulated by NRF2 directed to AREs, some target genes have alternate mechanisms of activation. At least two oxidative stress target genes contain DNA sequences within their proximal promoters that serve as oxidative damage sensors that trigger the genes’ activation [215]. Oxidative damage to DNA forms 8-oxo-7,8-dihydroguanine residues [215]. When these damaged residues occur within a G-quadruplex sequence in the proximal promoter elements of the vascular endothelial growth factor (VEGF) and the endonuclease III-like protein 1 (NTHL1) genes, a component of the DNA repair machinery, OGG1, is recruited to the damaged promoter elements [215] where it directly recruits transcription factors and RNAPII to the promoter and stimulates transcription of the genes [216]. The heat shock response transcription factor HSF1 is also activated in an oxidative cellular environment [150]. At baseline, HSF1 is sequestered in the cytoplasm by the heat shock proteins HSP90 and HSP70 [150]. Both of these heat shock proteins contain reactive cysteines that can be oxidized during a shift in cellular redox status [150]. When oxidized, HSP90 and HSP70 dissociate from HSF1, and HSF1 translocates to the nucleus and triggers gene expression, although it is unclear whether HSF1 mounts separate transcriptional programs during oxidative stress and heat shock responses [150].
Oxidative stress can directly damage the genome and the 2D epigenome (reviewed in [151, 217]). Peroxynitrite can nitrate tyrosines on histones, which increase their thermostability and possibly protects DNA from oxidative damage [218–220]. ROS promotes the production of reactive carbonyls that form adducts with reactive cysteines, arginines, lysines and histidines on proteins, including abundant histones; these adducts can alternately stabilize [221] or destabilize [222, 223] histones, cause histone loss from chromatin [224], or prevent programmed modifications at residues blocked by adducts [225, 226]. Adduct formation occurs more easily in free histones, which cannot be reincorporated into chromatin if they cannot be modified by chromatin remodeling enzymes [225]. Histones can also be modified with glutathione during oxidative stress [227]. Guanine residues within DNA can be oxidized, which can block binding of methyl binding proteins [228], and methyl groups bound to DNA (5-methylcytosine) can be directly oxidized to 5-hydroxymethylcytosine, which is an intermediate in the active DNA demethylation pathway and can lead to global loss of DNA methylation [229]. The methionine adenosyltransferase that catalyzes formation of the methyl donor S-adenosylmethionine required for methylation of cytosines in DNA can itself be oxidized and deactivated, leading to a passive decrease in DNA methylation through a second pathway [230, 231]. In contrast, TET DNA demethylases can also be oxidized and inactivated, which can lead to global increases in DNA methylation [232]. The DNA methyltransferase DNMT1 and the HMT EZH2 is recruited to sites of oxidized DNA damage, resulting in localized increases in DNA methylation and H3K27me3 [233, 234]. HMTs and HDACs [235, 236] can also be inhibited by oxidative stress [237] and DNA and histone demethylases can be inhibited by lack of methyl donor availability [151, 238]. These responses may cause specific effects on target gene sets or global, non-programmed effects that are a function of enzyme kinetics and substrate specificity, rather than gene functionality. One example of a specific effect is inhibition of histone deacetylases that are components of co-repressor complexes bound at baseline to promoters of redox-sensitive transcription factors [236]. Release of this repression by inhibition of these enzymes permits transcription of these transcription factors [236].
4.3.3. Oxidative stress response in the 3D genome
The chromatin remodeler Cockayne syndrome group B protein (CSB), a SWI/SNF family member, interacts with CTCF bound to chromatin under oxidative stress conditions and forms long-range chromatin loops in which CSB is bound to the regulatory element and CTCF to its sequence motif [239]. CSB deficiency increases cells’ sensitivity to ROS, suggesting that this remodeler is particularly important in the oxidative stress response [239]. CTCF deficiency also increases cells’ sensitivity to ROS [240], highlighting the importance of long-range chromatin loop formation or maintenance in mounting an effective oxidative stress response. Expression of the insulator and chromatin architectural protein CTCF is decreased in oxidative stress conditions (via NF-κB signaling, not NRF2), which leads to loss of insulator function and appropriate mono-allelic expression at the imprinted locus H19/IGF2 [241].
4.3.4. Transcriptional memory in oxidative stress response
Transcriptional memory in the oxidative stress response occurs in yeast, worms, fish and rodents [144, 154, 242–245]. Yeast cells conditioned with salt stress show positive priming at 77 genes induced by challenge with oxidative stress [144, 154, 244]. This memory persists for four generations and requires the nuclear pore protein Nup42 [144, 154, 244]. Stochastic increases in physiological levels of endogenous ROS during early development decrease global H3K4me3 and increase stress resistance and lifespan in adult C. elegans [245]. This study indicates that mild environmental exposures may be sufficient to trigger transcriptional memory. This finding is particularly interesting in light of another example that involves the transcription factor aryl hydrocarbon receptor (AhR). AhR activates its target gene cytochrome P450 1A (CYP1A) in response to PAH or dioxin [243]. In mice with a transient adult dioxin exposure, CYP1A shows positive priming that persists for forty days post-exposure [243]. However, in Atlantic killifish chronically exposed for multiple generations to PAH pollution, CYP1A loses its ability to be induced by AhR, but regains responsiveness within one generation of clean-water rearing [242]. This result suggests either negative priming or sustained repression that is lost within one generation [242]. In the AhR-CYP1A example, positive priming is the initial protective response to stress in mice and negative priming appears to be the later response to chronic stress in killifish. However, in the low-level ROS study in C. elegans, ROS levels are mild and insufficient to induce stress. The decrease in H3K4me3, if it confers negative priming, suggests differential memory formation pathways in mild, non-stressful exposures that may be protective and adaptive.
4.4. Hypoxia signaling
Healthy mammalian cells require oxygen to generate energy through OXPHOS [246]. Normal tissue oxygen levels are referred to as a state of normoxia [246]. Hypoxia refers to a state of decreased cellular oxygen levels, which leads to diminished energy production [246]. In order to survive this oxygen reduction, cells activate transcription of genes that restore oxygen levels, as well as downregulating genes related to energy production [246]. Transcriptional activation triggered by hypoxia is controlled by transcription factors called hypoxia inducible factors (HIFs), including HIF-1α, HIF-2α, HIF-3α, and their common dimerization partner, HIF-1β [246]. HIF-1β is constitutively expressed, and the levels of the other three HIFs are regulated by cellular oxygen levels [247]. HIF-1α is the best studied of the HIFs [248]. HIF-1α is prolyl hydroxylated during normoxia, which triggers ubiquitination by the E3 ubiquitin ligase von Hippel-Lindau protein (pVHL) and lysosomal degradation [248]. Under hypoxic conditions, this hydroxylation is inhibited, and HIF-1α dimerizes with HIF-1β, translocates to the nucleus, and binds to hypoxia response elements (HREs) in HIF-1α target genes [248]. HIF-1α is stabilized by PARylation deposited by PARP-1, which facilitates HIF-1α binding to chromatin [249]. HIF-2α and HIF-3α are similarly regulated by pVHL and activate separate transcriptional programs, although there is some overlap in target genes [246]. Although hypoxia does induce a subset of target genes, hypoxia primarily represses genes to reduce energy demanding processes and redirect limited resources to housekeeping functions in the cell [247]. At least ten different transcriptional repressors have bene reported for HIFs, including Repressor Element-1 Silencing Transcription Factor (REST) [246]. About 20% of genes repressed in hypoxia are downregulated by REST [250].
4.4.1. Redox-active pollutants activate the hypoxia response
Mercury and cadmium can exert toxicity by inhibiting protective hypoxia signaling even under normoxic conditions [251–255]. One class of HMTs that contain Jumonji C domains require oxygen for optimal function and can be inhibited by heavy metals/metalloids (e.g., cadmium, arsenic, chromium, nickel), so exposure to metals in the presence of cellular hypoxia likely decreases histone methylation levels (reviewed in [256, 257]). In some cases, toxicant action is potentiated by existing dysregulation of hypoxia signaling. For example, endocrine disrupting chemicals (EDCs), including BPA, benzyl butyl phthalate (BBP), and di(2-ethylhexyl) phthalate (DEHP) decreased activation of ER-α mediated transcription in the presence of hypoxia in human breast cancer cells [258]. This suggests that EDCs can potentiate cancer pathology associated with hypoxia [258].
4.4.2. Hypoxia response in the 2D genome
Genes induced by hypoxia generally display basal expression levels that are enhanced under low oxygen conditions [248] and therefore are bound by activating histone marks, like H3K4me3, at baseline [and by high levels RNAPII in core promoters and moderate levels of RNAPII along gene bodies [259], indicating active transcription. HIFs may increase pause-release of RNAPII at target genes, based on more equal levels of RNAPII binding at promoters and along gene bodies seen under hypoxic conditions [259] and based on evidence that HIFs recruit CDK8 and TIP60, which promote elongation [260, 261]. The evidence for widespread changes in chromatin accessibility in hypoxia is mixed: some studies report globally decreased [262, 263] chromatin accessibility and others report no changes or changes limited to specific target gene promoters [264].
Hypoxia leads to changes in DNA methylation but not in one clear direction. For example, in some reports, DNA methylation is globally increased in cancer cells under hypoxic conditions (by inhibiting TET activity) [265], but in others, DNA methylation is globally decreased [266]. These discrepancies may be due to experimental techniques that do not account for DNA hydroxymethylation, which is an intermediate in the DNA demethylation pathway that also serves as a regulatory modification [267]. However, there is mixed evidence for hypoxia effects on TET enzymes, which catalyze oxidation of DNA methylation to DNA hydroxmethylation [267, 268]. One study reports that DNA hydroxymethylation was reduced under normoxic conditions via TET degradation through the pVHL pathway [268], which is consistent with reports of increased TET activity under hypoxic conditions [267, 269]. However, at least two studies report TET inhibition under hypoxic conditions [265, 270]. However, these studies were all performed in cancer cell lines and the results may be specific to cancer subtypes.
The hypoxia response is also limited by pre-existing DNA methylation. DNA methylation of the HRE inhibited HIF activation of target gene Epo [271]. In malignant cancer cells, DNA methylation inhibited HIF binding to the EGLN3/PHD3 gene, the protein product of which hydroxylates HIF [272, 273], suggesting that DNA methylation can inhibit this negative feedback loop in disease states.
Hypoxia leads to changes in histone acetylation. Hypoxia triggers global loss of histone acetylation, as well as specific loss of acetylation at hypoxia-repressed genes [274, 275]. Histone deacetylase 1 (HDAC1) is induced by hypoxia and represses the tumor suppressor genes VHL and p53 to enable angiogenesis (which tumor suppressor genes inhibit) [276]. The HATs CBP, p300, and SRC-1 directly interact with HIF transcription factors to promote induction of the genes activated by hypoxia (reviewed in [247, 277]). CBP and p300 bind HIF-1α under hypoxic conditions; when HIF-1α binds a target gene, these HATs acetylate the gene promoter to promote chromatin remodeling and gene induction [278–281]. Specifically, these HATs are required for induction of the target genes EPO, VEGF and lactate dehydrogenase-A (LDH-A) [282]. Inactivation of p300 abolishes HIF-1α gene induction [280, 281] and overexpression of the HIF-1α domain that binds CBP and p300 potentiates induction [283]. However, CBP or p300 HAT function is required for only 35–50% of HIF-1α-induced genes [279]. SRC-1 is an important third HAT at some target genes that functions cooperatively with CBP and p300 but may not require CBP and p300 for gene activation [282]. CBP and p300 form a complex with SRC-1 and support colocalization of SRC-1 and HIF-1α [282]. SRC-1 may form a physical bridge between HIF-1α and the PIC [284] and its presence at gene promoters enhances activation of HIF-1α genes [284, 285].
The negative feedback loop for rapid cessation of a hypoxia signaling response includes disruption of HIF-1α’s association with HATs. Factor Inhibiting HIF-1 (FIH-1) requires molecular oxygen as a cofactor and disrupts the interactions between HIF-1α and p300/CBP in the presence of sufficient cellular oxygen [286–290].
Hypoxia leads to changes in histone methylation at both repressed and induced genes. Many histone demethylases require oxygen as a cofactor, and these enzymes are inhibited under hypoxic conditions (reviewed in [256]). For example, hypoxia triggered increased H3K4me3 via inhibition of the HDM JARID1A’s demethylation capacity [291]. However, not all histone demethylases appear sensitive to oxygen. For example, hypoxia downregulates the DNA repair genes BRCA1 and RAD51 by removal of activating H3K4 methylation by LSD1 [292]. In another example, the HDM JMJD1A decreased repressive H3K9 methylation at hypoxia target genes, enabling their induction [293].
HIFs recruit SWI/SNF chromatin remodeling proteins to activate target genes. The BRM and BRG1 catalytic subunits of human SWI/SNF promote transcription of a subset of HIF-1α and HIF-2α target genes and induce transcription of the HIFs themselves [294]. BRM and BRG1 are recruited to the Epo promoter by HIF-1α during hypoxia and both are necessary for Epo transcription [295]. Both chromatin remodeling proteins are required for the overall hypoxia response, but in many cases, they are targeted to distinct promoters by different transcription factors [296, 297]. In some cases, BRM and BRG1 are recruited but are not required for induction. For example, both proteins are recruited to the VEGF promoter, but VEGF transcription does not require either protein [295], suggesting that this gene does not require chromatin remodeling for induction. Both BRM and BRG1 contain bromodomains that bind acetylated lysines, and the VEGF promoter is highly acetylated [295, 298]. Therefore, these chromatin remodelers may be recruited to the VEGF promoter based on the promoter’s chromatin structure, even if the promoter does not require chromatin remodeling for activation.
4.4.3. Hypoxia response in the 3D genome
HIF-binding sites and their target genes interact over long ranges [299] through chromatin looping [300]. These loops are constitutively established at baseline, even in the absence of transcriptional activation by HIFs [300], and hypoxia does not appear to substantially alter looping patterns on a global scale [300], which is supported by gene-specific data for the target gene PAG1 [301]. These pre-established loops may function to increase the speed of the hypoxia response. However, there is some evidence a CTCF-mediated loop may be altered at another target gene, PAX6 [302]. Specifically, CTCF binds a repressor element upstream of the PAX6 gene to repress the gene [302]. At baseline, CTCF is post-translationally modified with SUMO; SUMOylated CTCF cannot bind the PAX6 repressor element [302]. Although this study does not report on 3D looping, CTCF often exerts its transcriptional regulation through chromatin loops [89], suggesting that a new loop is involved in PAX6 repression.
4.4.4. Transcriptional memory in the hypoxia response
Cells that are pre-conditioned to hypoxia retain a cross-reactive memory of that conditioning [303]. Intra-tumoral cells reside in a hypoxic microenvironment and these cells develop a ROS-resistant phenotype that provides a survival advantage during metastasis [303]. These cells show a sustained increase in the expression of a subset of hypoxia-inducible genes after they metastasize [303]. A similar memory is reported in chick embryos with neuroblastomas [304]. Live imaging of these embryos shows that exposure of neuroblastoma cells to 1% oxygen for three days leads to these cells acquiring metastatic characteristics that are retained by these cells after hypoxia ceases; this memory effect was not seen in milder hypoxia (8% oxygen) or after shorter exposure to 1% oxygen [304]. In addition, non-hypoxic cells are influenced by neighboring cells that were pre-conditioned by hypoxia; these non-hypoxic cells can acquire the same metastatic characteristics, suggesting that regulatory molecules are passed from hypoxic to non-hypoxic cells [304], possibly via extracellular vesicles [305–307], or that hypoxic cells create a path for normoxic cell invasion by promoting matrix degradation or cell migration [247]. This phenotype requires HIF-dependent transcription of genes involved in metastasis and can be blocked by HIF inhibition [304].
Transcriptional memory of hypoxia signaling can cross-react with immune functioning, as well. HIF-1α is required for trained immunity, or the epigenetic reprogramming of myeloid cells that protects them from secondary pathogen challenge [308]. Mice with a myeloid defect in HIF-1α were unable to mount this response during bacterial sepsis [308]. Neonatal mice transiently exposed to hypoxia and challenged with bacterial LPS at post-natal day 9 or at 6–8 weeks showed sex-specific augmentation of basal expression of microglial anti-inflammatory cytokines and attenuated microglial responses to LPS stimulation [309], indicating that these memories can be stably maintained into adulthood.
4.5. Unfolded protein response
The unfolded protein response is activated when proteins misfold or aggregate in either the endoplasmic reticulum, where nascent proteins are folded, or mitochondria, where ROS levels are high and proteotoxicity more likely [310, 311]. There are unfolded protein responses attributed to each of these organelles: the endoplasmic reticular unfolded protein response (UPRER) [311] and the mitochondrial unfolded protein response (UPRmt) [312].
ER stress results from accumulation of unfolded or misfolded proteins in the ER lumen, which activates transmembrane sensors on the ER: inositol-requiring protein 1 (IRE1, also known as ERN1), protein kinase RNA-like ER kinase (PERK, also known as EIF2AK3), and activating transcription factor 6 (ATF6) (reviewed in [311]). At baseline, these sensors are bound and kept inactive by the ER luminal chaperone binding immunoglobulin protein (BiP) [311]. When unfolded proteins accumulate in the ER lumen, BiP is sequestered away from the lumen, enabling sensor activation [311]. IRE1 dimerizes and removes an intron from the X-box binding protein 1 (XBP1) mRNA, which is then translated to an active transcription factor [311]. PERK phosphorylates the eukaryotic translation initiation factor-2α (eIF2α) to inhibit translation, with the notable exception of activating transcription factor 4 (ATF4), which is translated at a higher rate [311]. ATF6 localizes to the Golgi complex and is cleaved to produce an active cytosolic transcription factor [311]. The resulting three active transcription factors (XBP1, ATF4, ATF6) bind ER stress response elements (ERSEs) and unfolded protein response elements (UPREs) [313] to initiate transcription of genes encoding protein chaperones (including BiP), which increase assisted protein folding capacity, and components of the ER-associated protein degradation (ERAD) pathway, in which misfolded proteins are ubiquitinated, translocated to the cytosol and degraded by lysosomes [311]. These transcription factors can also activate the transcription factors include CCAAT/enhancer-binding protein beta (C/EBPβ) and C/EBP homology protein (CHOP, also known as DDIT3) to induce apoptosis [311]. Some UPRER genes are upregulated by other transcription factors, including JUN, TFEB, TFE3, NFAT, and TCF/LEF, but their activation and roles are unclear [311]. Even in the absence of any external stressor, ER homeostasis is disturbed during cell growth and during cellular differentiation, when the folding capacity of the ER is exceeded by the high protein synthesis rates [311]. Sustained ER stress is seen in diseases like cystic fibrosis and neurodegenerative diseases due to UPR dysregulation (reviewed in [14]).
Protein homeostasis in the mitochondria is sensed and regulated by the UPRmt [310]. In the model organism C. elegans, the primary transcription factor that regulates the UPRmt is ATFS-1, which has a functional mammalian ortholog, ATF5 [310]. ATF5 is present in mitochondria under normal conditions, but under conditions of mitochondrial proteotoxicity, it translocates to the nucleus and activates transcription of mitochondrial chaperones [310]. UPRmt transcription factors are regulated by C/EBPβ and CHOP (reviewed in [14].) In pancreatic β cells, the pancreas/duodenum homeobox protein 1 (Pdx1) regulates cell survival and ER stress susceptibility through direct transcriptional regulation of Atf4, the UPRER transcription factor, and of Atf5, the UPRmt transcription factor [314]. Pdx1 is required for Atf4 and Atf5 transcription, suggesting tissue-specific regulation of UPRs is controlled by tissue-specific transcription factors [314].
Cytoplasmic sensors of proteotoxicity are important in UPRs, too. The nascent polypeptide-associated complex α subunit (αNAC) binds to growing nascent polypeptide chains and inhibits signal recognition particles from transporting these nascent chains to the ER lumen [315]. γ-taxilin, which is involved in intracellular vesicle trafficking, binds αNAC [315]. Both of these proteins appear to function as extra-ER chaperones [315]. The αNAC/γ-taxilin pathway can be downregulated by glycogen synthase kinase 3β (GSK3β), which is activated by the kinase AKT, which is, in turn, activated via dephosphorylation by protein phosphatase 2A [315]. This downregulation triggers UPRs and leads to mitochondria-dependent apoptosis [315].
Overactivation of the UPRER can clearly be harmful to the cell. UPRER overactivation in astrocytes (both in vitro in response to ER stressors and in vivo in mice with neurodegenerative disease) caused a loss of synaptogenic function by modifying the astrocytic secretome [316]. In these astrocytes, the secretome was depleted of compounds important for synapse formation and maintenance, including extracellular matrix components, like collagen, fibronectin and filamen [316]. In addition, overactivation of the UPRER is an important acquired characteristic in cancer cells, particularly for drug resistance phenotypes, and its loss inhibits tumor growth [317].
4.5.1. Redox-active pollutants activate the unfolded protein response
UPRs are activated by pollutants that cause proteotoxicity of fully translated proteins that are located within the ER or mitochondria [318]. Although the sensing mechanism is different for UPRs and the heat shock response (which senses ubiquitination of nascently translated proteins in the cytosol), a similar set of pollutants activate both responses. For example, heavy metals that bind thiol groups in proteins, including lead, mercury and cadmium, activate UPRs [319]. In addition, DNA damaging agents, including ionizing radiation and topoisomerase inhibitors like etoposide, promote the proteasomal degradation of BiP to trigger UPRER activation [315]. Inhibition of topoisomerases, which relieve DNA torsion stress during replication and transcription, increases DNA double-strand breaks, as well as ROS, which then oxidizes DNA bases and enhances the DNA cleaving function of uninhibited topoisomerases [320]. Although synthetic topoisomerase inhibitors are used in clinical therapies, several naturally occurring compounds possess topoisomerase inhibition activity, including polyphenols, like EGCG found in green tea, genistein in soy, and resveratrol in red wine and various plants [321–323]. However, UPRER activation also triggers repression of the gene encoding topoisomerase IIα [324], for reasons that are unclear, highlighting the complexity of UPR response to cellular stress.
4.5.2. Unfolded protein response in the 2D genome
Histone modifications and transcription factor binding partners appear to be highly target-specific in the UPR, which likely confers granularity and specificity in responses. For example, in C. elegans, heterochromatin protein like-2 (HPL-2), a homolog of the human heterochromatin protein 1 (HP1), downregulates the UPRER in the intestine, perhaps by repressing XBP1 expression [325]. HPL-2 and HP1 are important chromatin binding proteins that are recruited by the repressive mark H3K9me to promote chromatin compaction and gene silencing [325]. Inactivation of HPL-2 results in enhanced resistance to UPRER signaling through increased basal XBP1 activation in the absence of stress [325]. H3K9 methylation regulates the UPRER independently of HPL-2; mutant worms lacking both hpl-2 and the histone methyltransferase met-2 (but not the histone methyltransferase set-25) showed synergistic survival phenotypes [325]. In yeast, histone acetylation is the critical factor; deletion of the histone acetyltransferase Gcn5 is sufficient to block a UPR transcriptional program [326]. In mammals, ER stress enhances chromatin accessibility in activated genes mediated by increased histone acetylation and activating histone methylation [327] but not decreased nucleosome occupancy [312]; these data indicate that the DNA relaxes to allow transcription factor binding without nucleosome eviction [40]. ATF6 is recruited to UPR target gene promoter elements by a constitutively bound transcription factor CBF/NF-Y (CBF) [328]. CBF is also required for recruitment of the TATA-binding protein to the promoter, but it is not required for H3K9 acetylation or H3K4 methylation that occur prior to target gene transcription [328]. Another constitutively expressed transcription factor, YY1, is required for expression of BiP, but YY1 binds the BiP promoter only under ER stress in a complex with ATF6 [329] YY1 recruits the arginine methyltransferase PRMT1, which methylates arginine 3 on histone H4 [329]. Histone H4 acetylation is deposited at the BiP promoter by p300, although it is unclear how p300 is recruited to the gene [329]. It is possible that ATF6 recruits p300, in a manner similar to ATF4 [330]. Chromatin regulatory mechanisms in the UPR may be specific to cell type, differentiation state, or disease state. For example, the HMT DOT1L prevents induction of key UPR genes in neural stem cells via the histone modification H3K79me; this effect is absent in mouse embryonic fibroblasts and the opposite effect is observed in human leukemic MOLM-13 cells [331].
4.5.3. Unfolded protein response in the 3D genome
Currently, there is limited, gene-specific evidence in yeast and humans for 3D control of the UPR. In yeast, inositol starvation triggers a UPR response, including activation of the UPR target gene INO1 (highlighted earlier in this review in the section on positive priming) [132]. INO1 is activated when the transcription factor Hac1 (a homolog of XBP1) triggers dissociation of a repressor upstream of the gene, causing INO1 to be physically recruited to the nuclear membrane for activation [132]. In humans, four UPRER genes (CACFD1, GTF3C5, SARDH, and FAM163B) are differentially expressed in skeletal muscle in individuals carrying a SNP in intron 9 of the G-protein signaling modulator 1 (GPSM1) gene [332]. This SNP disrupts CTCF binding to the GPSM1 allele and decreases transcription of the four UPRER target genes, ostensibly via disruption of chromatin looping [332].
4.5.4. Transcriptional memory in the unfolded protein response
UPR activation during early development can trigger transcriptional memory in adulthood. Specifically, mild UPRmt stress during early development increases lifespan in C. elegans, possibly through ROS resistance in adulthood [312]. In these worms, the gene lin-65 is essential for UPRmt response in mitochondrial stress (although lifespan extension does occur in the absence of this gene, highlighting that mitochondrial stress triggers multiple pathways that contribute to the lifespan effect) [312]. The LIN-65 protein translocates to the nucleus during stress and is also required for the translocation of a UPRmt transcription factor DVE-1 [312]. After nuclear translocation, DVE-1 binds to nuclear chromatin at accessible sites [312]. Prior to DVE-1 translocation, the UPRmt triggers the chromatin incorporation of new histones modified by repressive H3K9me2, possibly to restrict DVE-1 binding sites to a specific gene set during stress [312]. Overexpression of the histone demethylase genes jmjd-1.2 and jmjd-3.1, the protein products of which remove repressive H3K27me3, are sufficient to trigger mild UPRmt activation in early life (likely due to repression of mitochondrial genes necessary for function) and recapitulate the lifespan extension effect [333]. Although these results do not specifically demonstrate transcriptional memory, they do report an initial phenotype that requires a chromatin-based response and a secondary phenotype that is suggestive of memory.
Activation of UPRER in fully developed yeast and rodents can also trigger transcriptional memory. As outlined in the section on positive priming, yeast exposed to inositol starvation mount a UPR response that involves activating the INO1 gene by recruiting it to the nuclear membrane [132]. Yeast develop a highly specific, programmed memory at this gene by initial binding of the transcription factor Sfl1 to a Memory Recognition Sequence, followed by recruiting a modified Set1/COMPASS complex that deposits only H3K4me2 [99, 103, 132, 142]. The H3K4me2 modification recruits Set3, which in turn recruits RNAPII more quickly to the gene promoter than in the unprimed state [99, 103, 132, 142]. A UPR response in mice also yields gene-specific memories, although it is unclear if these memories are programmed or if they differ based on stochastic factors. In a mouse model of post-traumatic stress disorder, adult mice exposed to a single, prolonged stress showed transient activation of the UPRER, except for upregulation of IRE1α, which was sustained for seven days post-stress (peaking on days 1, 4, and 7), before returning to baseline [334]. Other target genes showed different memory patterns. The pro-apoptotic UPRER gene Bcl-2 showed a peak in transcription on day 1 post-stress and then returned to baseline, but the pro-apoptotic gene Bax showed sustained upregulation on days 1–3 post-stress, with a subsequent increase on day 4 that was maintained for several days [334].
4.6. DNA damage response
The DNA Damage Response (DDR) is a cellular sensor program for detecting DNA damage, activating appropriate cell cycle checkpoints to slow cell cycle progression, and activating DNA repair pathways to repair DNA lesions [335, 336]. The general DDR program is activated for all DNA damage types, although the specific responses differ by damage type [335, 336]. The regulation of these DNA repair pathways, including induction of DNA repair proteins and chromatin remodeling at sites of DNA damage to facilitate repair, is highly complex and is described elsewhere. In this review, I focus on the transcription regulation of the primary response genes that initially respond to the stress of DNA damage in healthy (non-cancerous) cells. The protein kinases ataxia-telangiectasia mutated (ATM), ATM- and Rad3-Related (ATR), and DNA-dependent protein kinases (DNA-PKs) are the primary kinases active in the DDR (reviewed in [335, 336]). ATM and ATR target the protein kinases CHK1 and CHK2 to reduce cyclin-dependent kinase (CDK) activity, partly through the action of the p53 transcription factor [335, 336]. CDK inhibition slows or arrests the cell cycle to allow for DNA repair prior to DNA replication [335, 336]. ATM and ATR also activate new transcription of DNA repair proteins, in addition to modifying these proteins post-translationally and recruiting them to sites of damage [335, 336]. If the damage is repaired, DDR signaling ceases; if the damage is unrepaired, chronic DDR signaling triggers cell death via apoptosis or cellular senescence [337].
4.6.1. Redox-active pollutants activate the DNA damage response
A large number of redox-active genotoxic pollutants trigger the DDR. These include ultraviolet light, ionizing radiation, environmental carcinogens including PAH, all of which cause specific forms of DNA damage, including single strand breaks, double strand breaks, and bulky DNA adducts, among many more [338–340]. In addition, many pollutants that are not considered genotoxic can induce ROS, including many heavy metals, endocrine disrupting compounds and pesticides [150]; ROS itself can cause oxidative DNA damage [3]. This topic has been extensively reviewed elsewhere [338–340].
4.6.2. DNA damage response in the 2D genome
The primary transcriptional regulators of the initial DDR transcriptional response include the transcription factor p53 and the kinases, ATM, ATR and p38α [341]. P53 is ubiquitinated and targeted for degradation by the E3 ubiquitin ligase Mouse double minute 2 (MDM2) under non-stress conditions [341]. Under stress, ATM disrupts the P53-MDM2 interaction by phosphorylating p53 in its MDM2-interaction domain, which stabilizes p53 [341]. P53 then translocates to the nucleus and mounts a transcriptional program to accomplish one of three outcomes: transient cell cycle arrest to enable DNA repair, permanent cell cycle arrest (senescence), or apoptosis [341]. Many transcriptional co-regulators modify p53 activity and refine its transcriptional program [341].
P53 binds DNA with the HAT p300, and p300 acetylates chromatin to recruit chromatin remodelers at activated genes [342]. P53 can also be acetylated by p300, which promotes p53’s association with other co-activators and HATs [342] and increases its residence time bound to regulatory elements to increase transcription of target genes [111]. At p53-activated target genes, chromatin is enriched with active chromatin marks (e.g., H3K4me3) [343] and p53 associates with the chromatin remodelers SWI/SNF or Remodeling and spacing factor 1 (RSF) [344], which remodel promoter chromatin and induce transcription [345]. The CSB chromatin remodeling protein can also bind to p53 directly, but in a reciprocally regulated manner [346]. CSB enables p53 binding to chromatin when p53 levels are low but at high p53 concentrations, p53 occludes CSB’s nucleosome interaction surface and prevents CSB from binding DNA [346]. P53 can also activate genes that are enriched with the silencing histone modification H3K9me3 at baseline [347]. P53 downregulates the HMT SUV39H1 that deposits H3K9me3, after which these promoters are modified with the activating mark H3K4me3 by the HMT MLL1 [347]. A subunit of the COMPASS/MLL1 complex, Ash2L, is required for p53 to activate transcription of these genes [347]. Ash2L deficiency results in decreased poising of RNAPII, reduced general transcription factor binding, and reduced gene induction, even though p53 and RNAPII binding are unaffected [347]. P53 can repress target genes in cooperation with the histone demethylase LSD1; P53 and LSD1 co-bind a regulatory element and LSD1 demethylates H3K4 to repress a classic target gene, α-fetoprotein (AFP) [348].
P53 activity is repressed by the linker histone H1.2 and by co-binding with MDM2 [349] Linker histones bind DNA between nucleosomes and are often considered transcriptional repressors because they prevent transcription factor binding [349]. The linker histone H1 directly interacts with regulatory factors to repress transcription [349]. The linker variant H1.2 is recruited to p53 target genes where it associates with p53 to repress p300-mediated chromatin acetylation [349]. Acetylation of p53 by p300 and H1.2 phosphorylation by DNA-PK disrupt the p53-H1.2 association and release p53 to activate transcription [349]. P53 bound to DNA can also be blocked from activating transcription by co-binding of MDM2 [350, 351]. MDM2 is itself a p53 target gene, which suggests both a negative feedback loop and crosstalk with the oxidative stress pathway [342, 352]. MDM2 is recruited to chromatin independently of p53 via tethering by the transcription factors ATF3/4 to regulate cellular redox state during oxidative stress [352]. Depletion of MDM2 in p53-deficient cells impairs serine/glycine metabolism, glutathione recycling, and the ratio of oxidized to reduced nicotinamide adenine dinucleotide (NAD+/NADH) [352].
Interestingly, p53 plays a role in limiting lineage conversion, in which one cell type switches cellular identity via epigenetic remodeling with lineage-specific transcription factors [353]. In mouse fibroblasts treated with liver-specific lineage-determining transcription factors, ATM-p53 was strongly activated and triggered proliferation arrest and cell death [353]. Most interestingly, ATM’s activation was not triggered by DNA damage, but rather responded to regions of chromatin that were newly opened by the lineage-determining transcription factors [353]. Baf60b, a subunit of SWI/SNF, recruits ATM to these newly opened sites to initiate a cell death response [353].
P38α also plays an important role in chromatin responses in the DDR. Ultraviolet radiation activates p38α to phosphorylate MK2, which, in turn, phosphorylates negative elongation factor polypeptide E (NELF-E) [164]. Phosphorylated NELF-E dissociates from chromatin, enabling release of poised RNAPII at more than 2,000 genes involved the DDR [164]. In contrast, P-TEFb is recruited to target gene promoters by RNA-binding motif protein 7 (RBM7) in response to DNA damage; this recruitment is mediated by p38α [354].
4.6.3. DNA damage response in the 3D genome
Chromatin looping is an important regulatory component of the p53 transcriptional program, as evidenced by the altered chromatin looping and differential transcription program activated by mutant p53, as compared to wild-type p53 [355]. In one example in healthy cells, p53 uses a loop to repress its target gene CDKN1A, and abolishing the loop is sufficient to activate the gene [356]. Human cells exposed to the DNA damaging agent doxorubicin showed eviction of cohesin from the CDKN1A gene body, which correlated with transcriptional induction of that gene [356]. Cohesin is evicted by RNAPII as it moves along the p53 target gene CDKN1A, and loss of cohesin disrupts a chromatin loop that connects a downstream repressor to an alternative promoter within the gene, permitting gene activation [356]. More globally, cohesin is evicted from gene bodies of multiple p53 target genes [356]. In addition, p53 target genes cluster in chromatin looped hubs at baseline, which allows faster transcription when p53 binds enhancers that are already in contact with gene promoters [356]. P53 can also regulate multiple target genes from a single distal enhancer, a process that requires physical bridging from enhancer RNA, which are transcribed from active enhancers [357].
4.6.4. Transcriptional memory in the DNA damage response
Although we have known for some time that DNA lesions can be inherited mitotically [358], it has only recently been reported that DDR signaling can be inherited across cellular generations. Transcriptional memory of DDR signaling determines whether daughter cells will immediately enter the next cell cycle or will remain in a quiescent state. Proliferative human breast cancer cells directly transmit p53 protein and mitogen-induced cyclin D1 (CCND1) mRNA to daughter cells [113]. [113]. The cyclin-dependent kinase inhibitor p21, which is induced by p53 to promote cell cycle exit, and the newly translated cyclin D1 protein are variably expressed in daughter cells and compete to determine cell cycle commitment [113]. This is a remarkable example of epigenetic (mitotically heritable) transcriptional memory directly involving key transcriptional regulators. It is unclear whether this memory is chromatin-based, since it is unknown whether mitotically inherited p53 is bound to chromatin, and, if so, whether the bound sites represent sites of transcriptional memory.
5. Crosstalk and cross-reactivity in CDS pathways
Although I have presented CDS pathways as functionally independent in this review, in reality they crosstalk extensively. I list a few examples here to illustrate these concepts. One form of crosstalk involves a key regulator of one CDS pathway subsequently activating a second CDS pathway. For example, in oxidative stress caused by arsenite or peroxide, NRF2 activates the HSF1 promoter to trigger a heat shock response [359]. Another form of crosstalk involves one CDS pathway repressing another. For example, the DNA damage response and hypoxia compete for activation. In hypoxic conditions, the DNA repair genes BRCA1 [292] and MLH1 [360] are silenced. Alternately, p53 represses the transcription of HIF-1α via competition for binding with the HATs p300/CBP [361–366]. In a third form of crosstalk, the protective response of one CDS pathway creates secondary stress that triggers a second CDS pathway. For example, hypoxia triggers the UPRER due to protective inhibition of protein translation, which is one way that the cell conserves energy in low oxygen conditions [255, 315, 329]. In addition to crosstalk, CDS pathways are cross-reactive. For example, early life exposure to ROS causes a more robust response to adult exposure to heat shock [245].
6. A conceptual framework for CDS responsiveness as a readout of redox-active pollutant exposure
I propose a conceptual framework in which an individual’s cumulative redox-active pollutant exposures can alter chromatin inducibility and induce transcriptional memories at CDS target genes. These memories can either be protective (in response to mild pollutant exposure) or they can cause an exaggerated subsequent response to stress or a dampened ability to mount a protective response (in response to severe or chronic pollutant exposure). This framework is supported by existing evidence that exaggerated and dampened CDS responses can harm the cell and are associated with disease. For example, comparative studies of yeast and human cells show that stress responses must be temporally restricted because their continuous induction is detrimental to cell growth [244, 367]. This slowed cell growth may be due to chronic activation of cell cycle checkpoint systems and subsequent cell cycle delays or to energy diversion into stress response [368, 369]. This effect is apparent in whole organisms, too. Loss of Nrf2 in mice increases susceptibility to cancer [150] and defects in the heat shock response disrupt mouse development [370]. Human diseases, including cardiovascular disease [371], cancer [304], diabetes [372], Alzheimer’s disease [373], and Parkinson’s disease [374], are also associated with CDS dysfunction.
I hypothesize that transcriptional memories at CDS genes are tissue specific. First, tissue specificity in toxicological responses can be due to a higher relative dose in a specific target tissue where a pollutant may accumulate [1]. Higher doses of exposures elicit stronger induction and subsequent memory formation at target genes [160]. In addition, the landscape of accessible transcription factor binding sites differs among cell types [47]. Different cell types and cells at different stages of differentiation show different CDS transcriptional programs in response to stress [154], suggesting that CDS gene set bookmarking may diverge during lineage-determination in early development. This suggests the possibility of alternative enhancer bookmarking in early development by a developmental redox-active pollutant exposure, which is supported by evidence that NRF2 regulates expression of the lineage-determining factor PU.1 in differentiated cells [375], which suggests that similar regulation may occur at earlier stages of development. Disruption of CDS bookmarking during development (perhaps by activation of genes critical for cellular protection in response to pollutant exposure) could permanently alter cells’ abilities to mount CDS responses, as well as the observed cell type differences in CDS gene set activation.
This framework highlights several open questions. First, the mechanisms of transcriptional memory formation are still poorly understood. Second, it is not known why some target genes form memories and others do not. Specifically, are transcriptional memories programmed, as suggested by the binding of transcription factor Sfl1 to a Memory Recognition Sequence to promote highly specific memory at the INO1 gene in yeast, or are they stochastically established [132], as suggested by evidence showing that memories preferentially form within cells that show stochastically greater initial transcriptional responses at target genes [100]? Both possibilities are supported by current evidence. Third, why are some memories retained for long periods, or through cell division, and others are not? Fourth, what factors determine positive vs. negative priming responses? Fifth, given evidence that genetic variation impacts inter-individual differences in CDS gene expression [376], does genetic variation also play a role in inter-individual differences in chromatin inducibility and transcriptional memories at CDS genes?
Within this framework, environmental stressors that exert effects in other ways can still impact CDS responses. The CDS comprises master pathways that are highly interconnected with others in the cell, many of which cannot be turned on at the same time; sustained activation of CDS pathways can potentially block other cellular responses, and vice versa (e.g., the transcription factor AhR competes for a heterodimerization partner with HIF-1α [377], so activation of one pathway blocks activation of the other).
This framework offers a new way of conceptualizing biological impacts of multiple or chronic exposures to redox-active pollutants. I note that it is possible that CDS transcriptional memories will scale in proportion to total cellular burden of redox-active pollutant exposures, which would suggest these loci as cumulative biomarkers of effect.
7. Conclusion
In summary, redox-active pollutants can activate a common set of cellular defense system pathways, in addition to those pathways activated specifically by individual pollutants. These cellular defense pathways are activated in response to an individual’s cumulative redox-active pollutant exposure and may be more likely than other pathways to develop dysfunction. CDS pathway dysfunction is a common hallmark of many chronic and complex diseases, suggesting that redox-active pollutant exposure contributes to complex disease development through activation or dysfunction of CDS pathways. Therefore, increasing our understanding of CDS pathway function is an important step in preventing disease. The majority of current evidence for redox-active pollutant effects on these pathways focuses on pollutant sensing and cellular signaling. However, cellular signaling pathways trigger different gene expression patterns in different cell types and in response to different stimuli. I suggest that part of this specificity in CDS response is due to chromatin inducibility at target genes in different cell types and that chromatin inducibility can change (i.e., transcriptional memories form) with repeated exposures. A clearer understanding of chromatin determinants of CDS responses will significantly clarify sources of inter-individual differences in cellular responses to redox-active pollutants and will support better prediction of individual disease risk in response to these pollutants.
Highlights.
Redox-active pollutants activate cellular defense system pathways
Inducibility of CDS genes requires a responsive chromatin state and structure
Transcriptional memories at CDS genes influence responses to future exposure
Acknowledgements.
I thank Drs. Kent Anger, Charles Allen, Stephen Lloyd, Mitchell Turker, Matthew Butler and Leslie Hammer for critical reviews of a prior version of this manuscript. All figures were generated using BioRender.
Funding.
This work is supported by the Oregon Institute of Occupational Health Sciences and NIH grant K01ES032044-01.
Abbreviations:
- 2D
two-dimensional
- 3D
three-dimensional
- AFP
α-fetoprotein
- AhR
aryl hydrocarbon receptor
- AKT
AKT serine/threonine kinase
- αNAC
nascent polypeptide-associated complex α subunit
- ARE
antioxidant response element
- Ash2L
ASH2-like histone lysine methyltransferase complex subunit
- ATF4
activating transcription factor 4
- ATF5
activating transcription factor 5
- ATF6
activating transcription factor 6
- ATFS-1
activating transcription factor associated with stress
- ATM
ataxia-telangiectasia mutated
- ATR
ATM- and Rad3-Related
- Baf60b
BRG1-associated factor 60B
- Bax
BCL2-associated X
- BBP
benzyl butyl phthalate
- Bcl-2
B-cell CLL/Lymphoma 2
- BiP
binding immunoglobulin protein
- BPA
bisphenol A
- BRCA1
breast cancer type 1 susceptibility protein
- BRD4
bromodomain-containing protein 4
- BRG1
Brahma-related gene 1
- BRM
Brahma
- C/EBPbeta
CCAAT-enhancer binding protein betea
- CACFD1
calcium channel flower domain containing 1
- CBF
core binding factor
- CCND1
mitogen-induced cyclin D1
- CDK
cyclin-dependent kinase
- Cdk7
cyclin-dependent kinase 7
- Cdk8
cyclin-dependent kinase 8
- CDKN1A
cyclin-dependent kinase inhibitor 1A
- CDS
cellular defense system
- CHD
chromodomain helicase DNA-binding
- CHK1
checkpoint kinase 1
- CHK2
checkpoint kinase 2
- CHOP
C/EBP homology protein
- COMPASS
complex proteins associated with Set1
- CPB
CREB-binding protein
- CREB
c-AMP response element binding protein
- CSB
Cockayne syndrome group B protein
- CTD
C-terminal domain
- CUL3
Cullin 3-RING-box E3 ubiquitin ligase
- CYP1A
cytochrome P450 1A
- DDIT3
DNA damage inducible transcript 3
- DDR
DNA Damage Response
- DEHP
di(2-ethylhexyl) phthalate
- DNA
deoxyribonucleic acid
- DNA-PK
DNA-dependent protein kinase
- DNMT
DNA methyltransferase
- DoG
downstream-of-gene-containing transcript
- DOT1L
DOT1-like histone lysine methyltransferase
- DSIF
DRB sensitivity inducing factor
- DVE-1
defective proventriculus in Drosophila homolog 1
- EDC
endocrine disrupting chemical
- EGCG
epigallocatechin gallate
- EGLN3/PHD3
Egl-9 family hypoxia inducible factor 2/prolyl hydroxylase domain 3
- EIF2AK3
eukaryotic translation initiation factor 2-alpha kinase 3
- eIF2α
eukaryotic translation initiation factor 2-α
- Epo
erythropoietin
- ER
endoplasmic reticulum
- ER-α
estrogen receptor-α
- ERAD
endoplasmic reticulum-associated protein degradation pathway
- ERN1
endoribonuclease 1
- ERSE
endoplasmic reticulum stress response element
- ESR
environmental stress response
- EZH2
Enhancer of Zeste homology 2
- FAM163B
family with sequence similarity 163 member B
- FIH
factor inhibiting HIF-1
- FOXO
Forkhead box O
- FOXO1
Forkhead box O1
- FOXO3
Forkhead box O3
- FOXO4
Forkhead box O4
- GAF
GAGA-associated factor
- GAL
galanin and GMAP prepropeptide
- GATA-1
GATA-binding factor 1
- GATA-2
GATA-binding factor 2
- Gcn5
general control non-repressed 5
- GPSM1
G-protein signaling modulator 1
- GREB1
growth regulating estrogen receptor binding 1
- GSK3β
glycogen synthase kinase 3β
- GTF3C5
general transcription factor IIIC subunit 5
- H1
histone H1
- H1.2
histone H1.2
- H19/IGF2
h19 transcript/insulin like growth factor 2
- H2A
histone 2A
- H2B
histone 2B
- H2Bub1
histone 2B mono-ubiquitinated
- H3
histone 3
- H3.3S31ph
histone 3.3 serine 31 phosphorylated
- H3K14ac
histone 3 lysine 14 acetylated
- H3K27ac
histone 2 lysine 27 acetylated
- H3K27me3
histone 3 lysine 27 tri-methylated
- H3K4me1
histone 3 lysine 4 mono-methylated
- H3K4me3
histone 3 lysine 4 tri-methylated
- H3K9me3
histone 3 lysine 9 tri-methylated
- H3S10ph
histone 3 serine 10 phosphorylated
- H4
histone 4
- H4K16ac
histone 4 lysine 16 acetylated
- H4K20ac
histone 4 lysine 20 acetylated
- Hac1
hyperpolarization-activated cation channel 1
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HDAC1
histone deacetylase 1
- HDM
histone demethylase
- HIF
hypoxia inducible factor
- HIF-1α
hypoxia inducible factor 1α
- HIF-1β
hypoxia inducible factor 1β
- HIF-2α
hypoxia inducible factor 2α
- HIF-3α
hypoxia inducible factor 3α
- HMG-14
high mobility group protein 14
- HMGB1
high mobility group protein B1
- HMT
histone methyltransferase
- HO-1
heme oxygenase-1
- Hog1
high-osmolarity glycerol 1
- HP-1
heterochromatin protein-1
- HPL-2
heterochromatin protein like-2
- HRE
hypoxia response element
- HSF
heat shock factor
- HSF1
heat shock factor 1
- HSFA2
heat shock factor A2
- Hsp70
heat shock protein 70
- Hsp90
heat shock protein 90
- HSPA1A
heat shock protein A1A
- IEG
immediate early gene
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor α
- IL-1 β
interleukin-1β
- INO1
inositol-3-phosphate synthase
- INO80
inositol-requiring mutant 80
- IRAK-M
interleukin-1 receptor-associated kinase M
- IRE1
inositol-requiring protein 1
- ISWI
imitation SWI of Drosophila melanogaster
- JARID1A
JumonjiC and ARID-domain containing histone lysine demethylase 1A
- Jmjd-1.2
Jumonji domain protein 1.2
- Jmjd-3.1
Jumonji domain protein 3.1
- JNK
c-JUN N-terminal kinase
- JUN
jun proto-oncogene
- Keap1
Kelch-like ECH-associated protein 1
- Kit
tyrosine protein kinase KIT
- KLF4
Kruppel-like factor 4
- LDH-A
lactate dehydrogenase-A (gene)
- Lin-65
abnormal cell lineage 65
- LPS
lipopolysaccharide
- LSD1
lysine-specific histone demethylase 1
- MAPK
mitogen-activated protein kinase
- MCF-7
Michigan Cancer Foundatio-7 cell line
- MDM2
Mouse double minute 2
- Mi-2β
chromodomain helicase DNA-binding protein Mi-2
- MK2/3
MAPK-activated protein kinase 2/3
- MLH1
mutL homolog 1
- MLL1
mixed-lineage leukemia histone methyltransferase 1
- MLL2
mixed-lineage leukemia histone methyltransferase 2
- MOF
Ortholog of Drosophila Males Absent on the First
- MOLM-13
human leukemia cell line with MLL gene rearrangement
- MSK1/2
mitogen- and stress-activated kinase 1/2
- MST1
macrophage stimulating 1
- NAD+
oxidized nicotinamide adenine dinucleotide
- NADH
reduced nicotinamide adenine dinucleotide
- NANOG
Homeobox transcription factor NANOG (derived from Celtic ‘Tir nan Og’)
- NELF-E
negative elongation factor polypeptide E
- NF-κB
nuclear factor kappa-light chain-enhancer of activated B cells
- NFAT
nuclear factor of activated T-cells
- NRF1
nuclear respiratory factor 1
- NRF2
nuclear respiratory factor 2
- NRF3
nuclear respiratory factor 3
- NTHL1
Nth like DNA glycosylase 1
- Nup98
nuclear pore protein 98
- OCT4
octamer transcription factor 4
- OGG1
8-oxoguanine glycosylase
- OXPHOS
oxidative phosphorylation
- P-TEFb
positive transcription elongation factor b
- P300
adenovirus E1A-associated cellular p300 transcriptional co-activator protein
- P53
tumor protein 53
- PAG1
phosphoprotein membrane anchor with glycosphingolipid microdomains
- PAH
polycyclic aromatic hydrocarbon
- PAR
poly[ADP-ribose]
- PARP-1
poy[ADP-ribose]polymerase-1
- PAX6
paired box protein 6
- PCB
polychlorinated biphenyl
- Pdx1
pancreas/duodenum homeobox protein 1
- PERK
protein kinase RNA-like ER kinase
- PIC
pre-initiation complex
- PRG
primary response gene
- Pu.1
PU-box binding protein 1
- pVHL
von Hippel-Lindau protein
- RAD51
recA-like protein 51
- RBM7
RNA-binding motif protein 7
- RelB
RELB proto-oncogene
- REST
repressor element-1 silencing transcription factor
- RNA
ribonucleic acid
- RNAPII
RNA polymerase II
- RNF20
ring finger protein 20
- RNF40
ring finger protein 40
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- Rpd3
reduced potassium dependency 1 histone deacetylase
- RSF
Remodeling and spacing factor 1
- SAGA
Spt5-Ada-Gcn5 histone acetyltransferase complex
- SAPK
stress-activated protein kinase
- SARDH
sarcosine dehydrogenase
- Set1
Set domain-containing 1 histone methyltransferase
- SET3
Set domain-containing 3 histone methyltransferase
- Sfl1
suppressor gene for flocculation 1
- SINE
short interspersed nuclear element
- Sko1p-Cyc8p-Tup1p
Suppressor of Kinase Overexpression 1 - Cytochrome C 8 - dTMP-Uptake 1 yeast co-repressor complex
- SNP
single nucleotide polymorphism
- SOCS3
Suppressor of Cytokine Signaling 3
- SRC-1
Steroid Receptor Coactivator-1
- SRF
serum response factor
- SRG
secondary response gene
- STAT1
Signal transducer and activator of transcription 1
- STAT6
Signal transducer and activator of transcription 6
- SUMO
Small ubiquitin-like modifier
- SUV39H1
Suppressor of variegation 3–9 homolog 1
- SWI/SNF
Switch/Sucrose Non-Fermentable chromatin remodeling complex
- Swr1
SWi/Snf2-Related 1
- TAD
topologically-associated domain
- TAL1
TAL BHLH Transcription Factor 1
- TCF/LEF
transcription factor/lymphoid enhancer binding factor
- TET
ten-eleven translocase
- TFE3
transcription factor binding to IGHM enhancer
- TFEB
transcription factor EB
- TFF1
Trefoil factor 1
- TIP60
Tat interactive protein 60-kDa histone acetyltransferase
- TLR4
Toll-like receptor 4
- TNF-α
Tumor Necrosis Factor-α
- TREM-1
Triggering Receptor Expressed on Myeloid Cells 1
- UPR
unfolded protein response
- UPRE
unfolded protein response element
- UPRER
unfolded protein response - endoplasmic reticulum pathway
- UPRmt
unfolded protein response - mitochondrial pathway
- USP22
Ubiquitin Specific Peptidase 22
- VEGF
Vascular endothelial growth factor
- XBP1
X-box binding protein 1
- YY1
Yin Yang 1
- ZMYND11
Zinc Finger MYND-Type Containing 11
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest. I have no conflicts of interest.
References
- 1.Vermeulen R, et al. , The exposome and health: Where chemistry meets biology. Science, 2020. 367(6476): p. 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niedzwiecki MM, et al. , The Exposome: Molecules to Populations. Annu Rev Pharmacol Toxicol, 2019. 59: p. 107–127. [DOI] [PubMed] [Google Scholar]
- 3.Go YM and Jones DP, Redox biology: interface of the exposome with the proteome, epigenome and genome. Redox Biol, 2014. 2: p. 358–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dennis KK, Go YM, and Jones DP, Redox Systems Biology of Nutrition and Oxidative Stress. J Nutr, 2019. 149(4): p. 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baccarelli A and Bollati V, Epigenetics and environmental chemicals. Curr Opin Pediatr, 2009. 21(2): p. 243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harvey ZH, Chen Y, and Jarosz DF, Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol Cell, 2018. 69(2): p. 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henikoff S and Greally JM, Epigenetics, cellular memory and gene regulation. Curr Biol, 2016. 26(14): p. R644–8. [DOI] [PubMed] [Google Scholar]
- 8.Rao SS, et al. , A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 2014. 159(7): p. 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, Notani D, and Rosenfeld MG, Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet, 2016. 17(4): p. 207–23. [DOI] [PubMed] [Google Scholar]
- 10.Feil R and Fraga MF, Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet, 2012. 13(2): p. 97–109. [DOI] [PubMed] [Google Scholar]
- 11.Wang T, et al. , The NIEHS TaRGET II Consortium and environmental epigenomics. Nat Biotechnol, 2018. 36(3): p. 225–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drakvik E, et al. , Statement on advancing the assessment of chemical mixtures and their risks for human health and the environment. Environ Int, 2020. 134: p. 105267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simmons SO, Fan CY, and Ramabhadran R, Cellular stress response pathway system as a sentinel ensemble in toxicological screening. Toxicol Sci, 2009. 111(2): p. 202–25. [DOI] [PubMed] [Google Scholar]
- 14.Himanen SV and Sistonen L, New insights into transcriptional reprogramming during cellular stress. J Cell Sci, 2019. 132(21). [DOI] [PubMed] [Google Scholar]
- 15.de Nadal E, Ammerer G, and Posas F, Controlling gene expression in response to stress. Nat Rev Genet, 2011. 12(12): p. 833–45. [DOI] [PubMed] [Google Scholar]
- 16.Vihervaara A, Duarte FM, and Lis JT, Molecular mechanisms driving transcriptional stress responses. Nat Rev Genet, 2018. 19(6): p. 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim PS, et al. , Defining the chromatin signature of inducible genes in T cells. Genome Biol, 2009. 10(10): p. R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weake VM and Workman JL, Inducible gene expression: diverse regulatory mechanisms. Nat Rev Genet, 2010. 11(6): p. 426–37. [DOI] [PubMed] [Google Scholar]
- 19.Festuccia N, et al. , Mitotic bookmarking in development and stem cells. Development, 2017. 144(20): p. 3633–3645. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Carey M, and Workman JL, The role of chromatin during transcription. Cell, 2007. 128(4): p. 707–19. [DOI] [PubMed] [Google Scholar]
- 21.Lieberman-Aiden E, et al. , Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 2009. 326(5950): p. 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsano RM, et al. , A New Portrait of Constitutive Heterochromatin: Lessons from Drosophila melanogaster. Trends Genet, 2019. 35(9): p. 615–631. [DOI] [PubMed] [Google Scholar]
- 23.Zentner GE and Henikoff S, Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol, 2013. 20(3): p. 259–66. [DOI] [PubMed] [Google Scholar]
- 24.Kaimori JY, et al. , Histone H4 lysine 20 acetylation is associated with gene repression in human cells. Sci Rep, 2016. 6: p. 24318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamke J, et al. , A hit-and-run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J, 2016. 35(2): p. 162–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fabrizio P, Garvis S, and Palladino F, Histone Methylation and Memory of Environmental Stress. Cells, 2019. 8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zilberman D and Henikoff S, Genome-wide analysis of DNA methylation patterns. Development, 2007. 134(22): p. 3959–65. [DOI] [PubMed] [Google Scholar]
- 28.He Y and Ecker JR, Non-CG Methylation in the Human Genome. Annu Rev Genomics Hum Genet, 2015. 16: p. 55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heberle E and Bardet AF, Sensitivity of transcription factors to DNA methylation. Essays Biochem, 2019. 63(6): p. 727–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashimoto H, Vertino PM, and Cheng X, Molecular coupling of DNA methylation and histone methylation. Epigenomics, 2010. 2(5): p. 657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng H, et al. , DNA methylation, its mediators and genome integrity. Int J Biol Sci, 2015. 11(5): p. 604–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allis CD and Muir TW, Spreading chromatin into chemical biology. Chembiochem, 2011. 12(2): p. 264–79. [DOI] [PubMed] [Google Scholar]
- 33.Xiang Y, et al. , Epigenomic analysis of gastrulation identifies a unique chromatin state for primed pluripotency. Nat Genet, 2020. 52(1): p. 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clapier CR and Cairns BR, The biology of chromatin remodeling complexes. Annu Rev Biochem, 2009. 78: p. 273–304. [DOI] [PubMed] [Google Scholar]
- 35.Venkatesh S and Workman JL, Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol, 2015. 16(3): p. 178–89. [DOI] [PubMed] [Google Scholar]
- 36.Hassan AH, Neely KE, and Workman JL, Histone acetyltransferase complexes stabilize swi/snf binding to promoter nucleosomes. Cell, 2001. 104(6): p. 817–27. [DOI] [PubMed] [Google Scholar]
- 37.Schwabish MA and Struhl K, The Swi/Snf complex is important for histone eviction during transcriptional activation and RNA polymerase II elongation in vivo. Mol Cell Biol, 2007. 27(20): p. 6987–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shivaswamy S and Iyer VR, Stress-dependent dynamics of global chromatin remodeling in yeast: dual role for SWI/SNF in the heat shock stress response. Mol Cell Biol, 2008. 28(7): p. 2221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schick S, et al. , Dynamics of chromatin accessibility and epigenetic state in response to UV damage. J Cell Sci, 2015. 128(23): p. 4380–94. [DOI] [PubMed] [Google Scholar]
- 40.Mueller B, et al. , Widespread changes in nucleosome accessibility without changes in nucleosome occupancy during a rapid transcriptional induction. Genes Dev, 2017. 31(5): p. 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vihervaara A, et al. , Transcriptional response to stress is pre-wired by promoter and enhancer architecture. Nat Commun, 2017. 8(1): p. 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Core L and Adelman K, Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev, 2019. 33(15–16): p. 960–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adelman K and Lis JT, Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet, 2012. 13(10): p. 720–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pombo A and Dillon N, Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol, 2015. 16(4): p. 245–57. [DOI] [PubMed] [Google Scholar]
- 45.Dixon JR, et al. , Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 2012. 485(7398): p. 376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ehara H, et al. , Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science, 2019. 363(6428): p. 744–747. [DOI] [PubMed] [Google Scholar]
- 47.Thurman RE, et al. , The accessible chromatin landscape of the human genome. Nature, 2012. 489(7414): p. 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tullai JW, et al. , Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J Biol Chem, 2007. 282(33): p. 23981–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zobeck KL, et al. , Recruitment timing and dynamics of transcription factors at the Hsp70 loci in living cells. Mol Cell, 2010. 40(6): p. 965–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mayran A and Drouin J, Pioneer transcription factors shape the epigenetic landscape. J Biol Chem, 2018. 293(36): p. 13795–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou M, et al. , Structural basis of nucleosome dynamics modulation by histone variants H2A.B and H2A.Z.2.2. EMBO J, 2021. 40(1): p. e105907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kulaeva OI, Hsieh FK, and Studitsky VM, RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc Natl Acad Sci USA, 2010. 107(25): p. 11325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramirez-Carrozzi VR, et al. , A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell, 2009. 138(1): p. 114–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deaton AM and Bird A, CpG islands and the regulation of transcription. Genes Dev, 2011. 25(10): p. 1010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fatemi M, et al. , Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res, 2005. 33(20): p. e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin C, et al. , H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet, 2009. 41(8): p. 941–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gevry N, et al. , Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev, 2009. 23(13): p. 1522–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brunelle M, et al. , The histone variant H2A.Z is an important regulator of enhancer activity. Nucleic Acids Res, 2015. 43(20): p. 9742–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weake VM and Workman JL, Histone ubiquitination: triggering gene activity. Mol Cell, 2008. 29(6): p. 653–63. [DOI] [PubMed] [Google Scholar]
- 60.Watanabe S, et al. , A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science, 2013. 340(6129): p. 195–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prenzel T, et al. , Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res, 2011. 71(17): p. 5739–53. [DOI] [PubMed] [Google Scholar]
- 62.Chauhan S and Boyd DD, Regulation of u-PAR gene expression by H2A.Z is modulated by the MEK-ERK/AP-1 pathway. Nucleic Acids Res, 2012. 40(2): p. 600–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Papamichos-Chronakis M, et al. , Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell, 2011. 144(2): p. 200–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yen K, Vinayachandran V, and Pugh BF, SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell, 2013. 154(6): p. 1246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weber CM, Ramachandran S, and Henikoff S, Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol Cell, 2014. 53(5): p. 819–30. [DOI] [PubMed] [Google Scholar]
- 66.Armache A, et al. , Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature, 2020. 583(7818): p. 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwartz BE and Ahmad K, Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev, 2005. 19(7): p. 804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Segala G, et al. , Monoubiquitination of Histone H2B Blocks Eviction of Histone Variant H2A.Z from Inducible Enhancers. Mol Cell, 2016. 64(2): p. 334–346. [DOI] [PubMed] [Google Scholar]
- 69.Zhang XY, et al. , The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell, 2008. 29(1): p. 102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennesch MA and Picard D, Minireview: Tipping the balance: ligand-independent activation of steroid receptors. Mol Endocrinol, 2015. 29(3): p. 349–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shema E, et al. , The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev, 2008. 22(19): p. 2664–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pavri R, et al. , Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell, 2006. 125(4): p. 703–17. [DOI] [PubMed] [Google Scholar]
- 73.Fleming AB, et al. , H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol Cell, 2008. 31(1): p. 57–66. [DOI] [PubMed] [Google Scholar]
- 74.Fuchs G, et al. , Cotranscriptional histone H2B monoubiquitylation is tightly coupled with RNA polymerase II elongation rate. Genome Res, 2014. 24(10): p. 1572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minsky N, et al. , Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol, 2008. 10(4): p. 483–8. [DOI] [PubMed] [Google Scholar]
- 76.He HH, et al. , Differential DNase I hypersensitivity reveals factor-dependent chromatin dynamics. Genome Res, 2012. 22(6): p. 1015–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Z, et al. , Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell, 2014. 159(2): p. 358–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nagarajan S, et al. , Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep, 2014. 8(2): p. 460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Batta K, et al. , Genome-wide function of H2B ubiquitylation in promoter and genic regions. Genes Dev, 2011. 25(21): p. 2254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Materne P, et al. , Histone H2B ubiquitylation represses gametogenesis by opposing RSC-dependent chromatin remodeling at the ste11 master regulator locus. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Price DH, Poised polymerases: on your mark…get set…go! Mol Cell, 2008. 30(1): p. 7–10. [DOI] [PubMed] [Google Scholar]
- 82.Mahat DB, et al. , Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol Cell, 2016. 62(1): p. 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fowler T, Sen R, and Roy AL, Regulation of primary response genes. Mol Cell, 2011. 44(3): p. 348–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bahrami S and Drablos F, Gene regulation in the immediate-early response process. Adv Biol Regul, 2016. 62: p. 37–49. [DOI] [PubMed] [Google Scholar]
- 85.Byun JS, et al. , Dynamic bookmarking of primary response genes by p300 and RNA polymerase II complexes. Proc Natl Acad Sci USA, 2009. 106(46): p. 19286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Drobic B, et al. , Promoter chromatin remodeling of immediate-early genes is mediated through H3 phosphorylation at either serine 28 or 10 by the MSK1 multi-protein complex. Nucleic Acids Res, 2010. 38(10): p. 3196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Healy S, et al. , Histone H3 phosphorylation, immediate-early gene expression, and the nucleosomal response: a historical perspective. Biochem Cell Biol, 2012. 90(1): p. 39–54. [DOI] [PubMed] [Google Scholar]
- 88.Mostocotto C, et al. , Poly(ADP-ribosyl)ation is required to modulate chromatin changes at c-MYC promoter during emergence from quiescence. PLoS One, 2014. 9(7): p. e102575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oti M, et al. , CTCF-mediated chromatin loops enclose inducible gene regulatory domains. BMC Genomics, 2016. 17: p. 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heintzman ND, et al. , Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet, 2007. 39(3): p. 311–8. [DOI] [PubMed] [Google Scholar]
- 91.Zuin J, et al. , Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci USA, 2014. 111(3): p. 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gregor A, et al. , De novo mutations in the genome organizer CTCF cause intellectual disability. Am J Hum Genet, 2013. 93(1): p. 124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jin F, et al. , A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature, 2013. 503(7475): p. 290–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li G, et al. , Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell, 2012. 148(1–2): p. 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghavi-Helm Y, et al. , Enhancer loops appear stable during development and are associated with paused polymerase. Nature, 2014. 512(7512): p. 96–100. [DOI] [PubMed] [Google Scholar]
- 96.Larkin JD, Cook PR, and Papantonis A, Dynamic reconfiguration of long human genes during one transcription cycle. Mol Cell Biol, 2012. 32(14): p. 2738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jing H, et al. , Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol Cell, 2008. 29(2): p. 232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tan-Wong SM, Wijayatilake HD, and Proudfoot NJ, Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev, 2009. 23(22): p. 2610–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Light WH, et al. , Interaction of a DNA zip code with the nuclear pore complex promotes H2A.Z incorporation and INO1 transcriptional memory. Mol Cell, 2010. 40(1): p. 112–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burrill DR, et al. , Synthetic memory circuits for tracking human cell fate. Genes Dev, 2012. 26(13): p. 1486–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Oyer JA, et al. , Aberrant epigenetic silencing is triggered by a transient reduction in gene expression. PLoS One, 2009. 4(3): p. e4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ng HH, et al. , Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell, 2003. 11(3): p. 709–19. [DOI] [PubMed] [Google Scholar]
- 103.D’Urso A, et al. , Set1/COMPASS and Mediator are repurposed to promote epigenetic transcriptional memory. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.del Fresno C, et al. , Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol, 2009. 182(10): p. 6494–507. [DOI] [PubMed] [Google Scholar]
- 105.Foster SL, Hargreaves DC, and Medzhitov R, Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature, 2007. 447(7147): p. 972–8. [DOI] [PubMed] [Google Scholar]
- 106.Moazed D, Mechanisms for the inheritance of chromatin states. Cell, 2011. 146(4): p. 510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ptashne M, Epigenetics: core misconcept. Proc Natl Acad Sci USA, 2013. 110(18): p. 7101–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stewart-Morgan KR, Petryk N, and Groth A, Chromatin replication and epigenetic cell memory. Nat Cell Biol, 2020. 22(4): p. 361–371. [DOI] [PubMed] [Google Scholar]
- 109.Wang X and Moazed D, DNA sequence-dependent epigenetic inheritance of gene silencing and histone H3K9 methylation. Science, 2017. 356(6333): p. 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ahmed S, et al. , DNA zip codes control an ancient mechanism for gene targeting to the nuclear periphery. Nat Cell Biol, 2010. 12(2): p. 111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Loffreda A, et al. , Live-cell p53 single-molecule binding is modulated by C-terminal acetylation and correlates with transcriptional activity. Nat Commun, 2017. 8(1): p. 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Itoh K, et al. , Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev, 1999. 13(1): p. 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang HW, et al. , Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature, 2017. 549(7672): p. 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Camacho J, et al. , The Memory of Environmental Chemical Exposure in C. elegans Is Dependent on the Jumonji Demethylases jmjd-2 and jmjd-3/utx-1. Cell Rep, 2018. 23(8): p. 2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dolinoy DC, et al. , Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect, 2006. 114(4): p. 567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dolinoy DC, Huang D, and Jirtle RL, Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA, 2007. 104(32): p. 13056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Oksuz O, et al. , Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol Cell, 2018. 70(6): p. 1149–1162 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hathaway NA, et al. , Dynamics and memory of heterochromatin in living cells. Cell, 2012. 149(7): p. 1447–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ragunathan K, Jih G, and Moazed D, Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science, 2015. 348(6230): p. 1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gaydos LJ, Wang W, and Strome S, Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science, 2014. 345(6203): p. 1515–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bantignies F, et al. , Inheritance of Polycomb-dependent chromosomal interactions in Drosophila. Genes Dev, 2003. 17(19): p. 2406–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tiwari VK, et al. , PcG proteins, DNA methylation, and gene repression by chromatin looping. PLoS Biol, 2008. 6(12): p. 2911–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Du J, et al. , Three classes of response elements for human PRC2 and MLL1/2-Trithorax complexes. Nucleic Acids Res, 2018. 46(17): p. 8848–8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Laugesen A, Hojfeldt JW, and Helin K, Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol Cell, 2019. 74(1): p. 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bashtrykov P, et al. , Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem Biol, 2012. 19(5): p. 572–8. [DOI] [PubMed] [Google Scholar]
- 126.Pascual-Garcia P, et al. , Metazoan Nuclear Pores Provide a Scaffold for Poised Genes and Mediate Induced Enhancer-Promoter Contacts. Mol Cell, 2017. 66(1): p. 63–76 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Monticelli S and Natoli G, Short-term memory of danger signals and environmental stimuli in immune cells. Nat Immunol, 2013. 14(8): p. 777–84. [DOI] [PubMed] [Google Scholar]
- 128.Quintin J, et al. , Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe, 2012. 12(2): p. 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hargreaves DC, Horng T, and Medzhitov R, Control of inducible gene expression by signal-dependent transcriptional elongation. Cell, 2009. 138(1): p. 129–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.De Santa F, et al. , Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J, 2009. 28(21): p. 3341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Q, et al. , Reprogramming of the Epigenome by MLL1 Links Early-Life Environmental Exposures to Prostate Cancer Risk. Mol Endocrinol, 2016. 30(8): p. 856–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brickner JH and Walter P, Gene recruitment of the activated INO1 locus to the nuclear membrane. PLoS Biol, 2004. 2(11): p. e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ostuni R, et al. , Latent enhancers activated by stimulation in differentiated cells. Cell, 2013. 152(1–2): p. 157–71. [DOI] [PubMed] [Google Scholar]
- 134.Jadhav U, et al. , Extensive Recovery of Embryonic Enhancer and Gene Memory Stored in Hypomethylated Enhancer DNA. Mol Cell, 2019. 74(3): p. 542–554 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jeong KW, et al. , Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat Struct Mol Biol, 2011. 18(12): p. 1358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Douillet D, et al. , Uncoupling histone H3K4 trimethylation from developmental gene expression via an equilibrium of COMPASS, Polycomb and DNA methylation. Nat Genet, 2020. 52(6): p. 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Deng W and Blobel GA, Do chromatin loops provide epigenetic gene expression states? Curr Opin Genet Dev, 2010. 20(5): p. 548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang S, et al. , Spatial organization of chromatin domains and compartments in single chromosomes. Science, 2016. 353(6299): p. 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fernandez-Martinez J and Rout MP, One Ring to Rule them All? Structural and Functional Diversity in the Nuclear Pore Complex. Trends Biochem Sci, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kurshakova MM, et al. , SAGA and a novel Drosophila export complex anchor efficient transcription and mRNA export to NPC. EMBO J, 2007. 26(24): p. 4956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brown CR, et al. , Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev, 2008. 22(5): p. 627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brickner DG, et al. , H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol, 2007. 5(4): p. e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kundu S and Peterson CL, Dominant role for signal transduction in the transcriptional memory of yeast GAL genes. Mol Cell Biol, 2010. 30(10): p. 2330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guan Q, et al. , Cellular memory of acquired stress resistance in Saccharomyces cerevisiae. Genetics, 2012. 192(2): p. 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Biswas SK and Lopez-Collazo E, Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol, 2009. 30(10): p. 475–87. [DOI] [PubMed] [Google Scholar]
- 146.Chan C, et al. , Endotoxin tolerance disrupts chromatin remodeling and NF-kappaB transactivation at the IL-1beta promoter. J Immunol, 2005. 175(1): p. 461–8. [DOI] [PubMed] [Google Scholar]
- 147.El Gazzar M, et al. , Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol, 2009. 29(7): p. 1959–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Murawska M, et al. , Stress-induced PARP activation mediates recruitment of Drosophila Mi-2 to promote heat shock gene expression. PLoS Genet, 2011. 7(7): p. e1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ramirez-Carrozzi VR, et al. , Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev, 2006. 20(3): p. 282–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zheng F, et al. , Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol, 2020. 34: p. 101475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cyr AR and Domann FE, The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal, 2011. 15(2): p. 551–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cuadrado A and Nebreda AR, Mechanisms and functions of p38 MAPK signalling. Biochem J, 2010. 429(3): p. 403–17. [DOI] [PubMed] [Google Scholar]
- 153.Alepuz PM, et al. , Stress-induced map kinase Hog1 is part of transcription activation complexes. Mol Cell, 2001. 7(4): p. 767–77. [DOI] [PubMed] [Google Scholar]
- 154.Gasch AP, et al. , Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell, 2000. 11(12): p. 4241–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Proft M and Struhl K, Hog1 kinase converts the Sko1-Cyc8-Tup1 repressor complex into an activator that recruits SAGA and SWI/SNF in response to osmotic stress. Mol Cell, 2002. 9(6): p. 1307–17. [DOI] [PubMed] [Google Scholar]
- 156.De Nadal E, et al. , The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature, 2004. 427(6972): p. 370–4. [DOI] [PubMed] [Google Scholar]
- 157.Ferreiro I, et al. , The p38 SAPK is recruited to chromatin via its interaction with transcription factors. J Biol Chem, 2010. 285(41): p. 31819–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nadal-Ribelles M, et al. , H3K4 monomethylation dictates nucleosome dynamics and chromatin remodeling at stress-responsive genes. Nucleic Acids Res, 2015. 43(10): p. 4937–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ferreiro I, et al. , Whole genome analysis of p38 SAPK-mediated gene expression upon stress. BMC Genomics, 2010. 11: p. 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Murray KO, et al. , Exertional heat stroke leads to concurrent long-term epigenetic memory, immunosuppression and altered heat shock response in female mice. J Physiol, 2020. [DOI] [PubMed] [Google Scholar]
- 161.Simone C, et al. , p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet, 2004. 36(7): p. 738–43. [DOI] [PubMed] [Google Scholar]
- 162.Soloaga A, et al. , MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J, 2003. 22(11): p. 2788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vermeulen L, et al. , The versatile role of MSKs in transcriptional regulation. Trends Biochem Sci, 2009. 34(6): p. 311–8. [DOI] [PubMed] [Google Scholar]
- 164.Borisova ME, et al. , p38-MK2 signaling axis regulates RNA metabolism after UV-light-induced DNA damage. Nat Commun, 2018. 9(1): p. 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pokholok DK, et al. , Activated signal transduction kinases frequently occupy target genes. Science, 2006. 313(5786): p. 533–6. [DOI] [PubMed] [Google Scholar]
- 166.Proft M, et al. , The stress-activated Hog1 kinase is a selective transcriptional elongation factor for genes responding to osmotic stress. Mol Cell, 2006. 23(2): p. 241–50. [DOI] [PubMed] [Google Scholar]
- 167.Joutsen J and Sistonen L, Tailoring of Proteostasis Networks with Heat Shock Factors. Cold Spring Harb Perspect Biol, 2019. 11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ritossa F, Discovery of the heat shock response. Cell Stress Chaperones, 1996. 1(2): p. 97–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Aprile-Garcia F, et al. , Nascent-protein ubiquitination is required for heat shock-induced gene downregulation in human cells. Nat Struct Mol Biol, 2019. 26(2): p. 137–146. [DOI] [PubMed] [Google Scholar]
- 170.Baird NA, et al. , HSF-1-mediated cytoskeletal integrity determines thermotolerance and life span. Science, 2014. 346(6207): p. 360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Franzmann TM, et al. , Activation of the chaperone Hsp26 is controlled by the rearrangement of its thermosensor domain. Mol Cell, 2008. 29(2): p. 207–16. [DOI] [PubMed] [Google Scholar]
- 172.Hamada FN, et al. , An internal thermal sensor controlling temperature preference in Drosophila. Nature, 2008. 454(7201): p. 217–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.McClung CR and Davis SJ, Ambient thermometers in plants: from physiological outputs towards mechanisms of thermal sensing. Curr Biol, 2010. 20(24): p. R1086–92. [DOI] [PubMed] [Google Scholar]
- 174.Nollen EA and Morimoto RI, Chaperoning signaling pathways: molecular chaperones as stress-sensing ‘heat shock’ proteins. J Cell Sci, 2002. 115(Pt 14): p. 2809–16. [DOI] [PubMed] [Google Scholar]
- 175.Papaconstantinou AD, et al. , Mercury, cadmium, and arsenite enhance heat shock protein synthesis in chick embryos prior to embryotoxicity. Birth Defects Res B Dev Reprod Toxicol, 2003. 68(6): p. 456–64. [DOI] [PubMed] [Google Scholar]
- 176.Bao A, et al. , Inducible expression of heat shock protein 20 protects airway epithelial cells against oxidative injury involving the Nrf2-NQO-1 pathway. Cell Biosci, 2020. 10: p. 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rabindran SK, et al. , Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science, 1993. 259(5092): p. 230–4. [DOI] [PubMed] [Google Scholar]
- 178.Xu YM, et al. , Post-translational modification of human heat shock factors and their functions: a recent update by proteomic approach. J Proteome Res, 2012. 11(5): p. 2625–34. [DOI] [PubMed] [Google Scholar]
- 179.Jaeger AM, et al. , Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J Biol Chem, 2014. 289(44): p. 30459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guertin MJ and Lis JT, Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet, 2010. 6(9): p. e1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Allen TA, et al. , The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat Struct Mol Biol, 2004. 11(9): p. 816–21. [DOI] [PubMed] [Google Scholar]
- 182.Mariner PD, et al. , Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol Cell, 2008. 29(4): p. 499–509. [DOI] [PubMed] [Google Scholar]
- 183.Yakovchuk P, Goodrich JA, and Kugel JF, B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc Natl Acad Sci USA, 2009. 106(14): p. 5569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fujimoto M, et al. , Poly(ADP-Ribose) Polymerase 1 Promotes the Human Heat Shock Response by Facilitating Heat Shock Transcription Factor 1 Binding to DNA. Mol Cell Biol, 2018. 38(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Niskanen EA, et al. , Global SUMOylation on active chromatin is an acute heat stress response restricting transcription. Genome Biol, 2015. 16: p. 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Seifert A, et al. , Proteotoxic stress reprograms the chromatin landscape of SUMO modification. Sci Signal, 2015. 8(384): p. rs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Petesch SJ and Lis JT, Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell, 2008. 134(1): p. 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cossec JC, et al. , SUMO Safeguards Somatic and Pluripotent Cell Identities by Enforcing Distinct Chromatin States. Cell Stem Cell, 2018. 23(5): p. 742–757 e8. [DOI] [PubMed] [Google Scholar]
- 189.Vilborg A, et al. , Comparative analysis reveals genomic features of stress-induced transcriptional readthrough. Proc Natl Acad Sci USA, 2017. 114(40): p. E8362–E8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vilborg A, et al. , Widespread Inducible Transcription Downstream of Human Genes. Mol Cell, 2015. 59(3): p. 449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lyu X, Rowley MJ, and Corces VG, Architectural Proteins and Pluripotency Factors Cooperate to Orchestrate the Transcriptional Response of hESCs to Temperature Stress. Mol Cell, 2018. 71(6): p. 940–955 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chowdhary S, et al. , Heat Shock Factor 1 Drives Intergenic Association of Its Target Gene Loci upon Heat Shock. Cell Rep, 2019. 26(1): p. 18–28 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Li L, et al. , Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol Cell, 2015. 58(2): p. 216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hnisz D, et al. , A Phase Separation Model for Transcriptional Control. Cell, 2017. 169(1): p. 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Strom AR, et al. , Phase separation drives heterochromatin domain formation. Nature, 2017. 547(7662): p. 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Erdel F and Rippe K, Formation of Chromatin Subcompartments by Phase Separation. Biophys J, 2018. 114(10): p. 2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kisliouk T, Cramer T, and Meiri N, Methyl CpG level at distal part of heat-shock protein promoter HSP70 exhibits epigenetic memory for heat stress by modulating recruitment of POU2F1-associated nucleosome-remodeling deacetylase (NuRD) complex. J Neurochem, 2017. 141(3): p. 358–372. [DOI] [PubMed] [Google Scholar]
- 198.Tetievsky A and Horowitz M, Posttranslational modifications in histones underlie heat acclimation-mediated cytoprotective memory. J Appl Physiol (1985), 2010. 109(5): p. 1552–61. [DOI] [PubMed] [Google Scholar]
- 199.Sies H, Berndt C, and Jones DP, Oxidative Stress. Annu Rev Biochem, 2017. 86: p. 715–748. [DOI] [PubMed] [Google Scholar]
- 200.Schmidlin CJ, et al. , Redox regulation by NRF2 in aging and disease. Free Radic Biol Med, 2019. 134: p. 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Liu P, et al. , Differential and overlapping targets of the transcriptional regulators NRF1, NRF2, and NRF3 in human cells. J Biol Chem, 2019. 294(48): p. 18131–18149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhang DD and Hannink M, Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol, 2003. 23(22): p. 8137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liu P, et al. , Non-covalent NRF2 Activation Confers Greater Cellular Protection than Covalent Activation. Cell Chem Biol, 2019. 26(10): p. 1427–1435 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tian W, et al. , Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2-related factors 1 and 2 (NRF1 and NRF2). J Biol Chem, 2018. 293(6): p. 2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tao S, et al. , p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Mol Cell Biol, 2017. 37(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dodson M, et al. , Modulating NRF2 in Disease: Timing Is Everything. Annu Rev Pharmacol Toxicol, 2019. 59: p. 555–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Weinhouse C, Mitochondrial-epigenetic crosstalk in environmental toxicology. Toxicology, 2017. 391: p. 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hansen JM, Jones DP, and Harris C, The Redox Theory of Development. Antioxid Redox Signal, 2020. 32(10): p. 715–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Petriello MC, et al. , PCB 126 toxicity is modulated by cross-talk between caveolae and Nrf2 signaling. Toxicol Appl Pharmacol, 2014. 277(2): p. 192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Valko M, Morris H, and Cronin MT, Metals, toxicity and oxidative stress. Curr Med Chem, 2005. 12(10): p. 1161–208. [DOI] [PubMed] [Google Scholar]
- 211.Nilson KA, et al. , Oxidative stress rapidly stabilizes promoter-proximal paused Pol II across the human genome. Nucleic Acids Res, 2017. 45(19): p. 11088–11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Maruyama A, et al. , Nrf2 activation is associated with Z-DNA formation in the human HO-1 promoter. Nucleic Acids Res, 2013. 41(10): p. 5223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Song S, et al. , Loss of SWI/SNF Chromatin Remodeling Alters NRF2 Signaling in Non-Small Cell Lung Carcinoma. Mol Cancer Res, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mou K, et al. , HMGB1 deficiency reduces H2 O2 -induced oxidative damage in human melanocytes via the Nrf2 pathway. J Cell Mol Med, 2018. 22(12): p. 6148–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fleming AM, Ding Y, and Burrows CJ, Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc Natl Acad Sci USA, 2017. 114(10): p. 2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ba X, et al. , 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J Immunol, 2014. 192(5): p. 2384–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kreuz S and Fischle W, Oxidative stress signaling to chromatin in health and disease. Epigenomics, 2016. 8(6): p. 843–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dixit K, et al. , Physicochemical studies on peroxynitrite-modified H3 histone. Int J Biol Macromol, 2010. 46(1): p. 20–6. [DOI] [PubMed] [Google Scholar]
- 219.Khan MA, et al. , Studies on peroxynitrite-modified H1 histone: implications in systemic lupus erythematosus. Biochimie, 2014. 97: p. 104–13. [DOI] [PubMed] [Google Scholar]
- 220.Ashraf JM, et al. , 3-Deoxyglucosone: a potential glycating agent accountable for structural alteration in H3 histone protein through generation of different AGEs. PLoS One, 2015. 10(2): p. e0116804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mir AR, et al. , Methylglyoxal mediated conformational changes in histone H2A-generation of carboxyethylated advanced glycation end products. Int J Biol Macromol, 2014. 69: p. 260–6. [DOI] [PubMed] [Google Scholar]
- 222.Ashraf JM, et al. , Physicochemical analysis of structural alteration and advanced glycation end products generation during glycation of H2A histone by 3-deoxyglucosone. IUBMB Life, 2014. 66(10): p. 686–93. [DOI] [PubMed] [Google Scholar]
- 223.Ashraf JM, et al. , Glycation of H1 Histone by 3-Deoxyglucosone: Effects on Protein Structure and Generation of Different Advanced Glycation End Products. PLoS One, 2015. 10(6): p. e0130630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rahmanpour R and Bathaie SZ, Histone H1 structural changes and its interaction with DNA in the presence of high glucose concentration in vivo and in vitro. J Biomol Struct Dyn, 2011. 28(4): p. 575–86. [DOI] [PubMed] [Google Scholar]
- 225.Chen D, et al. , Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J Biol Chem, 2013. 288(30): p. 21678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Cervantes-Laurean D, et al. , Nuclear proteasome activation and degradation of carboxymethylated histones in human keratinocytes following glyoxal treatment. Free Radic Biol Med, 2005. 38(6): p. 786–95. [DOI] [PubMed] [Google Scholar]
- 227.de Luca A, et al. , Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem J, 2011. 440(2): p. 175–83. [DOI] [PubMed] [Google Scholar]
- 228.Valinluck V, et al. , Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res, 2004. 32(14): p. 4100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Valinluck V and Sowers LC, Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res, 2007. 67(3): p. 946–50. [DOI] [PubMed] [Google Scholar]
- 230.Avila MA, et al. , Specific interaction of methionine adenosyltransferase with free radicals. Biofactors, 1998. 8(1–2): p. 27–32. [DOI] [PubMed] [Google Scholar]
- 231.Pajares MA, et al. , Modulation of rat liver S-adenosylmethionine synthetase activity by glutathione. J Biol Chem, 1992. 267(25): p. 17598–605. [PubMed] [Google Scholar]
- 232.Niu Y, et al. , Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med, 2015. 82: p. 22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.O’Hagan HM, et al. , Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell, 2011. 20(5): p. 606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ding N, et al. , Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J Mol Cell Biol, 2016. 8(3): p. 244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Moodie FM, et al. , Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J, 2004. 18(15): p. 1897–9. [DOI] [PubMed] [Google Scholar]
- 236.Doyle K and Fitzpatrick FA, Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem, 2010. 285(23): p. 17417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Bannister AJ and Kouzarides T, Regulation of chromatin by histone modifications. Cell Res, 2011. 21(3): p. 381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hitchler MJ and Domann FE, Redox regulation of the epigenetic landscape in cancer: a role for metabolic reprogramming in remodeling the epigenome. Free Radic Biol Med, 2012. 53(11): p. 2178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lake RJ, et al. , The CSB chromatin remodeler and CTCF architectural protein cooperate in response to oxidative stress. Nucleic Acids Res, 2016. 44(5): p. 2125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Roy AR, et al. , The transcriptional regulator CCCTC-binding factor limits oxidative stress in endothelial cells. J Biol Chem, 2018. 293(22): p. 8449–8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yang B, et al. , A novel pathway links oxidative stress to loss of insulin growth factor-2 (IGF2) imprinting through NF-kappaB activation. PLoS One, 2014. 9(2): p. e88052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Meyer JN, Nacci DE, and Di Giulio RT, Cytochrome P4501A (CYP1A) in killifish (Fundulus heteroclitus): heritability of altered expression and relationship to survival in contaminated sediments. Toxicol Sci, 2002. 68(1): p. 69–81. [DOI] [PubMed] [Google Scholar]
- 243.Amenya HZ, Tohyama C, and Ohsako S, Dioxin induces Ahr-dependent robust DNA demethylation of the Cyp1a1 promoter via Tdg in the mouse liver. Sci Rep, 2016. 6: p. 34989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Berry DB and Gasch AP, Stress-activated genomic expression changes serve a preparative role for impending stress in yeast. Mol Biol Cell, 2008. 19(11): p. 4580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bazopoulou D, et al. , Developmental ROS individualizes organismal stress resistance and lifespan. Nature, 2019. 576(7786): p. 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Cavadas MAS, Cheong A, and Taylor CT, The regulation of transcriptional repression in hypoxia. Exp Cell Res, 2017. 356(2): p. 173–181. [DOI] [PubMed] [Google Scholar]
- 247.Johnson AB and Barton MC, Hypoxia-induced and stress-specific changes in chromatin structure and function. Mutat Res, 2007. 618(1–2): p. 149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Xia X and Kung AL, Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol, 2009. 10(10): p. R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Marti JM, et al. , Selective modulation by PARP-1 of HIF-1alpha-recruitment to chromatin during hypoxia is required for tumor adaptation to hypoxic conditions. Redox Biol, 2021. 41: p. 101885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Cavadas MA, et al. , REST is a hypoxia-responsive transcriptional repressor. Sci Rep, 2016. 6: p. 31355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Chun YS, et al. , Cadmium blocks hypoxia-inducible factor (HIF)-1-mediated response to hypoxia by stimulating the proteasome-dependent degradation of HIF-1alpha. Eur J Biochem, 2000. 267(13): p. 4198–204. [DOI] [PubMed] [Google Scholar]
- 252.Rocco SA, et al. , Cadmium Exposure Inhibits Branching Morphogenesis and Causes Alterations Consistent With HIF-1alpha Inhibition in Human Primary Breast Organoids. Toxicol Sci, 2018. 164(2): p. 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ion R and Bernal AL, Smoking and Preterm Birth. Reprod Sci, 2015. 22(8): p. 918–26. [DOI] [PubMed] [Google Scholar]
- 254.Chang J, et al. , Acute Methylmercury Exposure and the Hypoxia-Inducible Factor-1alpha Signaling Pathway under Normoxic Conditions in the Rat Brain and Astrocytes in Vitro. Environ Health Perspect, 2019. 127(12): p. 127006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Liu L, et al. , Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell, 2006. 21(4): p. 521–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Chervona Y and Costa M, The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic Biol Med, 2012. 53(5): p. 1041–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Costa M, et al. , Nickel carcinogenesis: epigenetics and hypoxia signaling. Mutat Res, 2005. 592(1–2): p. 79–88. [DOI] [PubMed] [Google Scholar]
- 258.Park C, et al. , The effects of bisphenol A, benzyl butyl phthalate, and di(2-ethylhexyl) phthalate on estrogen receptor alpha in estrogen receptor-positive cells under hypoxia. Environ Pollut, 2019. 248: p. 774–781. [DOI] [PubMed] [Google Scholar]
- 259.Choudhry H, et al. , Extensive regulation of the non-coding transcriptome by hypoxia: role of HIF in releasing paused RNApol2. EMBO Rep, 2014. 15(1): p. 70–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Galbraith MD, et al. , HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell, 2013. 153(6): p. 1327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Perez-Perri JI, et al. , The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep, 2016. 16(1): p. 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kirmes I, et al. , A transient ischemic environment induces reversible compaction of chromatin. Genome Biol, 2015. 16: p. 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Li Y, et al. , Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis, 2020. 11(2): p. 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Suzuki N, et al. , HIF-dependent and reversible nucleosome disassembly in hypoxia-inducible gene promoters. Exp Cell Res, 2018. 366(2): p. 181–191. [DOI] [PubMed] [Google Scholar]
- 265.Thienpont B, et al. , Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature, 2016. 537(7618): p. 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Shahrzad S, et al. , Induction of DNA hypomethylation by tumor hypoxia. Epigenetics, 2007. 2(2): p. 119–25. [DOI] [PubMed] [Google Scholar]
- 267.Mariani CJ, et al. , TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep, 2014. 7(5): p. 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Fan S, et al. , TET is targeted for proteasomal degradation by the PHD-pVHL pathway to reduce DNA hydroxymethylation. J Biol Chem, 2020. 295(48): p. 16299–16313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lin G, et al. , Hypoxia induces the expression of TET enzymes in HepG2 cells. Oncol Lett, 2017. 14(6): p. 6457–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Laukka T, et al. , Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J Biol Chem, 2016. 291(8): p. 4256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wenger RH, et al. , Oxygen-regulated erythropoietin gene expression is dependent on a CpG methylation-free hypoxia-inducible factor-1 DNA-binding site. Eur J Biochem, 1998. 253(3): p. 771–7. [DOI] [PubMed] [Google Scholar]
- 272.Place TL, et al. , Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PLoS One, 2011. 6(1): p. e14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Huang KT, et al. , DNA methylation analysis of the HIF-1alpha prolyl hydroxylase domain genes PHD1, PHD2, PHD3 and the factor inhibiting HIF gene FIH in invasive breast carcinomas. Histopathology, 2010. 57(3): p. 451–60. [DOI] [PubMed] [Google Scholar]
- 274.Murai M, et al. , Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br J Cancer, 2005. 92(6): p. 1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Koumenis C, et al. , Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol Cell Biol, 2001. 21(4): p. 1297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kim MS, et al. , Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med, 2001. 7(4): p. 437–43. [DOI] [PubMed] [Google Scholar]
- 277.Melvin A and Rocha S, Chromatin as an oxygen sensor and active player in the hypoxia response. Cell Signal, 2012. 24(1): p. 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kallio PJ, et al. , Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J, 1998. 17(22): p. 6573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kasper LH, et al. , Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J, 2005. 24(22): p. 3846–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Arany Z, et al. , An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA, 1996. 93(23): p. 12969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Ebert BL and Bunn HF, Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol, 1998. 18(7): p. 4089–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ruas JL, Poellinger L, and Pereira T, Role of CBP in regulating HIF-1-mediated activation of transcription. J Cell Sci, 2005. 118(Pt 2): p. 301–11. [DOI] [PubMed] [Google Scholar]
- 283.Kung AL, et al. , Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med, 2000. 6(12): p. 1335–40. [DOI] [PubMed] [Google Scholar]
- 284.Carrero P, et al. , Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol, 2000. 20(1): p. 402–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Ruas JL, Poellinger L, and Pereira T, Functional analysis of hypoxia-inducible factor-1 alpha-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J Biol Chem, 2002. 277(41): p. 38723–30. [DOI] [PubMed] [Google Scholar]
- 286.Dann CE 3rd, Bruick RK, and Deisenhofer J, Structure of factor-inhibiting hypoxia-inducible factor 1: An asparaginyl hydroxylase involved in the hypoxic response pathway. Proc Natl Acad Sci USA, 2002. 99(24): p. 15351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Mahon PC, Hirota K, and Semenza GL, FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev, 2001. 15(20): p. 2675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Lando D, et al. , FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev, 2002. 16(12): p. 1466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Lando D, et al. , Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science, 2002. 295(5556): p. 858–61. [DOI] [PubMed] [Google Scholar]
- 290.Hewitson KS, et al. , Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem, 2002. 277(29): p. 26351–5. [DOI] [PubMed] [Google Scholar]
- 291.Zhou X, et al. , Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res, 2010. 70(10): p. 4214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Lu Y, et al. , Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol Cell Biol, 2011. 31(16): p. 3339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Krieg AJ, et al. , Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol, 2010. 30(1): p. 344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Sena JA, Wang L, and Hu CJ, BRG1 and BRM chromatin-remodeling complexes regulate the hypoxia response by acting as coactivators for a subset of hypoxia-inducible transcription factor target genes. Mol Cell Biol, 2013. 33(19): p. 3849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Wang F, et al. , Roles of coactivators in hypoxic induction of the erythropoietin gene. PLoS One, 2010. 5(4): p. e10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Kadam S, et al. , Functional selectivity of recombinant mammalian SWI/SNF subunits. Genes Dev, 2000. 14(19): p. 2441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Kadam S and Emerson BM, Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol Cell, 2003. 11(2): p. 377–89. [DOI] [PubMed] [Google Scholar]
- 298.Jung JE, et al. , STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J, 2005. 19(10): p. 1296–8. [DOI] [PubMed] [Google Scholar]
- 299.Schodel J, et al. , High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood, 2011. 117(23): p. e207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Platt JL, et al. , Capture-C reveals preformed chromatin interactions between HIF-binding sites and distant promoters. EMBO Rep, 2016. 17(10): p. 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Schorg A, et al. , Destruction of a distal hypoxia response element abolishes trans-activation of the PAG1 gene mediated by HIF-independent chromatin looping. Nucleic Acids Res, 2015. 43(12): p. 5810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Wang J, Wang Y, and Lu L, De-SUMOylation of CCCTC binding factor (CTCF) in hypoxic stress-induced human corneal epithelial cells. J Biol Chem, 2012. 287(15): p. 12469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Godet I, et al. , Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat Commun, 2019. 10(1): p. 4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Herrmann A, et al. , Cellular memory of hypoxia elicits neuroblastoma metastasis and enables invasion by non-aggressive neighbouring cells. Oncogenesis, 2015. 4: p. e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Peinado H, et al. , Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med, 2012. 18(6): p. 883–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.King HW, Michael MZ, and Gleadle JM, Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer, 2012. 12: p. 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Wysoczynski M and Ratajczak MZ, Lung cancer secreted microvesicles: underappreciated modulators of microenvironment in expanding tumors. Int J Cancer, 2009. 125(7): p. 1595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Cheng SC, et al. , mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science, 2014. 345(6204): p. 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Kiernan EA, et al. , Neonatal Intermittent Hypoxia Induces Lasting Sex-Specific Augmentation of Rat Microglial Cytokine Expression. Front Immunol, 2019. 10: p. 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Wang S, Gao K, and Liu Y, UPR(mt) coordinates immunity to maintain mitochondrial homeostasis and animal fitness. Mitochondrion, 2018. 41: p. 9–13. [DOI] [PubMed] [Google Scholar]
- 311.Almanza A, et al. , Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J, 2019. 286(2): p. 241–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Tian Y, et al. , Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell, 2016. 165(5): p. 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Takayanagi S, et al. , Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperones, 2013. 18(1): p. 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Juliana CA, et al. , ATF5 regulates beta-cell survival during stress. Proc Natl Acad Sci USA, 2017. 114(6): p. 1341–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Hotokezaka Y, Katayama I, and Nakamura T, ATM-associated signalling triggers the unfolded protein response and cell death in response to stress. Commun Biol, 2020. 3(1): p. 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Smith HL, et al. , Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron, 2020. 105(5): p. 855–866 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Dong D, et al. , Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res, 2008. 68(2): p. 498–505. [DOI] [PubMed] [Google Scholar]
- 318.Lee Y, et al. , General anesthesia activates the mitochondrial unfolded protein response and induces age-dependent, long-lasting changes in mitochondrial function in the developing brain. Neurotoxicology, 2021. 82: p. 1–8. [DOI] [PubMed] [Google Scholar]
- 319.Ge J, et al. , Cadmium exposure triggers mitochondrial dysfunction and oxidative stress in chicken (Gallus gallus) kidney via mitochondrial UPR inhibition and Nrf2-mediated antioxidant defense activation. Sci Total Environ, 2019. 689: p. 1160–1171. [DOI] [PubMed] [Google Scholar]
- 320.Ganguly A, et al. , Betulinic acid, a catalytic inhibitor of topoisomerase I, inhibits reactive oxygen species-mediated apoptotic topoisomerase I-DNA cleavable complex formation in prostate cancer cells but does not affect the process of cell death. Cancer Res, 2007. 67(24): p. 11848–58. [DOI] [PubMed] [Google Scholar]
- 321.Lee JH, Wendorff TJ, and Berger JM, Resveratrol: A novel type of topoisomerase II inhibitor. J Biol Chem, 2017. 292(51): p. 21011–21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Vann KR, et al. , Effects of Olive Metabolites on DNA Cleavage Mediated by Human Type II Topoisomerases. Biochemistry, 2015. 54(29): p. 4531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Bandele OJ, Clawson SJ, and Osheroff N, Dietary polyphenols as topoisomerase II poisons: B ring and C ring substituents determine the mechanism of enzyme-mediated DNA cleavage enhancement. Chem Res Toxicol, 2008. 21(6): p. 1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Mann MJ, et al. , UPR-induced resistance to etoposide is downstream of PERK and independent of changes in topoisomerase IIalpha levels. PLoS One, 2012. 7(10): p. e47931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Kozlowski L, et al. , The Caenorhabditis elegans HP1 family protein HPL-2 maintains ER homeostasis through the UPR and hormesis. Proc Natl Acad Sci USA, 2014. 111(16): p. 5956–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Welihinda AA, Tirasophon W, and Kaufman RJ, The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr, 1999. 7(4–6): p. 293–300. [PMC free article] [PubMed] [Google Scholar]
- 327.Schram AW, et al. , A dual role for SAGA-associated factor 29 (SGF29) in ER stress survival by coordination of both histone H3 acetylation and histone H3 lysine-4 trimethylation. PLoS One, 2013. 8(7): p. e70035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Luo R, et al. , CBF/NF-Y controls endoplasmic reticulum stress induced transcription through recruitment of both ATF6(N) and TBP. J Cell Biochem, 2008. 104(5): p. 1708–23. [DOI] [PubMed] [Google Scholar]
- 329.Baumeister P, et al. , Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol, 2005. 25(11): p. 4529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Cherasse Y, et al. , The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res, 2007. 35(17): p. 5954–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Roidl D, et al. , DOT1L Activity Promotes Proliferation and Protects Cortical Neural Stem Cells from Activation of ATF4-DDIT3-Mediated ER Stress In Vitro. Stem Cells, 2016. 34(1): p. 233–45. [DOI] [PubMed] [Google Scholar]
- 332.Ding Q, et al. , Genome-wide meta-analysis associates GPSM1 with type 2 diabetes, a plausible gene involved in skeletal muscle function. J Hum Genet, 2020. 65(4): p. 411–420. [DOI] [PubMed] [Google Scholar]
- 333.Merkwirth C, et al. , Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell, 2016. 165(5): p. 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Kong F, et al. , Molecular Mechanisms of IRE1alpha-ASK1 Pathway Reactions to Unfolded Protein Response in DRN Neurons of Post-Traumatic Stress Disorder Rats. J Mol Neurosci, 2017. 61(4): p. 531–541. [DOI] [PubMed] [Google Scholar]
- 335.Blackford AN and Jackson SP, ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell, 2017. 66(6): p. 801–817. [DOI] [PubMed] [Google Scholar]
- 336.Marechal A and Zou L, DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol, 2013. 5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Jackson SP and Bartek J, The DNA-damage response in human biology and disease. Nature, 2009. 461(7267): p. 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Bhagat J, Combinations of genotoxic tests for the evaluation of group 1 IARC carcinogens. J Appl Toxicol, 2018. 38(1): p. 81–99. [DOI] [PubMed] [Google Scholar]
- 339.Hartwig A, et al. , Mode of action-based risk assessment of genotoxic carcinogens. Arch Toxicol, 2020. 94(6): p. 1787–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Benigni R, Predicting the carcinogenicity of chemicals with alternative approaches: recent advances. Expert Opin Drug Metab Toxicol, 2014. 10(9): p. 1199–208. [DOI] [PubMed] [Google Scholar]
- 341.Sammons MA, Zhu J, and Berger SL, A Chromatin-Focused siRNA Screen for Regulators of p53-Dependent Transcription. G3 (Bethesda), 2016. 6(8): p. 2671–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Allison SJ and Milner J, Remodelling chromatin on a global scale: a novel protective function of p53. Carcinogenesis, 2004. 25(9): p. 1551–7. [DOI] [PubMed] [Google Scholar]
- 343.Bao F, et al. , p53 binding sites in normal and cancer cells are characterized by distinct chromatin context. Cell Cycle, 2017. 16(21): p. 2073–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Min S, et al. , Chromatin-remodeling factor, RSF1, controls p53-mediated transcription in apoptosis upon DNA strand breaks. Cell Death Dis, 2018. 9(11): p. 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Lee D, et al. , SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J Biol Chem, 2002. 277(25): p. 22330–7. [DOI] [PubMed] [Google Scholar]
- 346.Lake RJ, Basheer A, and Fan HY, Reciprocally regulated chromatin association of Cockayne syndrome protein B and p53 protein. J Biol Chem, 2011. 286(40): p. 34951–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Mungamuri SK, et al. , Ash2L enables P53-dependent apoptosis by favoring stable transcription pre-initiation complex formation on its pro-apoptotic target promoters. Oncogene, 2015. 34(19): p. 2461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Tsai WW, et al. , p53-targeted LSD1 functions in repression of chromatin structure and transcription in vivo. Mol Cell Biol, 2008. 28(17): p. 5139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Kim K, et al. , Functional interplay between p53 acetylation and H1.2 phosphorylation in p53-regulated transcription. Oncogene, 2012. 31(39): p. 4290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Arva NC, et al. , A chromatin-associated and transcriptionally inactive p53-Mdm2 complex occurs in mdm2 SNP309 homozygous cells. J Biol Chem, 2005. 280(29): p. 26776–87. [DOI] [PubMed] [Google Scholar]
- 351.White DE, et al. , Mouse double minute 2 associates with chromatin in the presence of p53 and is released to facilitate activation of transcription. Cancer Res, 2006. 66(7): p. 3463–70. [DOI] [PubMed] [Google Scholar]
- 352.Riscal R, et al. , Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol Cell, 2016. 62(6): p. 890–902. [DOI] [PubMed] [Google Scholar]
- 353.Ji S, et al. , Baf60b-mediated ATM-p53 activation blocks cell identity conversion by sensing chromatin opening. Cell Res, 2017. 27(5): p. 642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Bugai A, et al. , P-TEFb Activation by RBM7 Shapes a Pro-survival Transcriptional Response to Genotoxic Stress. Mol Cell, 2019. 74(2): p. 254–267 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Qu H, et al. , Wild-type p53 regulates OTOP2 transcription through DNA loop alteration of the promoter in colorectal cancer. FEBS Open Bio, 2019. 9(1): p. 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Millau JF, Wijchers P, and Gaudreau L, High-Resolution 4C Reveals Rapid p53-Dependent Chromatin Reorganization of the CDKN1A Locus in Response to Stress. PLoS One, 2016. 11(10): p. e0163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Melo CA, et al. , eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell, 2013. 49(3): p. 524–35. [DOI] [PubMed] [Google Scholar]
- 358.Lukas C, et al. , 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol, 2011. 13(3): p. 243–53. [DOI] [PubMed] [Google Scholar]
- 359.Paul S, et al. , NRF2 transcriptionally activates the heat shock factor 1 promoter under oxidative stress and affects survival and migration potential of MCF7 cells. J Biol Chem, 2018. 293(50): p. 19303–19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Lu Y, et al. , Silencing of the DNA mismatch repair gene MLH1 induced by hypoxic stress in a pathway dependent on the histone demethylase LSD1. Cell Rep, 2014. 8(2): p. 501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Schmid T, et al. , p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem J, 2004. 380(Pt 1): p. 289–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Freedman SJ, et al. , Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat Struct Biol, 2003. 10(7): p. 504–12. [DOI] [PubMed] [Google Scholar]
- 363.Freedman SJ, et al. , Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA, 2002. 99(8): p. 5367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Fox SB, et al. , CITED4 inhibits hypoxia-activated transcription in cancer cells, and its cytoplasmic location in breast cancer is associated with elevated expression of tumor cell hypoxia-inducible factor 1alpha. Cancer Res, 2004. 64(17): p. 6075–81. [DOI] [PubMed] [Google Scholar]
- 365.Yin Z, et al. , The essential role of Cited2, a negative regulator for HIF-1alpha, in heart development and neurulation. Proc Natl Acad Sci USA, 2002. 99(16): p. 10488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Bhattacharya S, et al. , Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev, 1999. 13(1): p. 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Lamech LT and Haynes CM, The unpredictability of prolonged activation of stress response pathways. J Cell Biol, 2015. 209(6): p. 781–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Yaakov G, et al. , Combination of two activating mutations in one HOG1 gene forms hyperactive enzymes that induce growth arrest. Mol Cell Biol, 2003. 23(14): p. 4826–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Vendrell A, et al. , Sir2 histone deacetylase prevents programmed cell death caused by sustained activation of the Hog1 stress-activated protein kinase. EMBO Rep, 2011. 12(10): p. 1062–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Xiao X, et al. , HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J, 1999. 18(21): p. 5943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Pockley AG, Heat shock proteins, inflammation, and cardiovascular disease. Circulation, 2002. 105(8): p. 1012–7. [DOI] [PubMed] [Google Scholar]
- 372.Oyadomari S, Araki E, and Mori M, Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis, 2002. 7(4): p. 335–45. [DOI] [PubMed] [Google Scholar]
- 373.Terro F, et al. , Neurons overexpressing mutant presenilin-1 are more sensitive to apoptosis induced by endoplasmic reticulum-Golgi stress. J Neurosci Res, 2002. 69(4): p. 530–9. [DOI] [PubMed] [Google Scholar]
- 374.Imai Y, Soda M, and Takahashi R, Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem, 2000. 275(46): p. 35661–4. [DOI] [PubMed] [Google Scholar]
- 375.Staitieh BS, et al. , Nrf2 regulates PU.1 expression and activity in the alveolar macrophage. Am J Physiol Lung Cell Mol Physiol, 2015. 308(10): p. L1086–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Ward MC, et al. , Dynamic effects of genetic variation on gene expression revealed following hypoxic stress in cardiomyocytes. Elife, 2021. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Vorrink SU and Domann FE, Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1alpha signaling node. Chem Biol Interact, 2014. 218: p. 82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]