

SUMMARY
Scn2a encodes the voltage-gated sodium channel NaV1.2, a main mediator of neuronal action potential firing. The current paradigm suggests that NaV1.2 gain-of-function variants enhance neuronal excitability, resulting in epilepsy, whereas NaV1.2 deficiency impairs neuronal excitability, contributing to autism. However, this paradigm does not explain why ~20%–30% of individuals with NaV1.2 deficiency still develop seizures. Here, we report the counterintuitive finding that severe NaV1.2 deficiency results in increased neuronal excitability. Using a NaV1.2-deficient mouse model, we show enhanced intrinsic excitability of principal neurons in the prefrontal cortex and striatum, brain regions known to be involved in Scn2a-related seizures. This increased excitability is autonomous and reversible by genetic restoration of Scn2a expression in adult mice. RNA sequencing reveals downregulation of multiple potassium channels, including KV1.1. Correspondingly, KV channel openers alleviate the hyperexcitability of NaV1.2-deficient neurons. This unexpected neuronal hyperexcitability may serve as a cellular basis underlying NaV1.2 deficiency-related seizures.
Graphical Abstract
In brief
Conventional wisdom suggests that the inadequacy of voltage-gated sodium channels impairs intrinsic neuronal excitability. Zhang et al. report that a severe deficiency of the NaV1.2 channel unexpectedly results in hyperexcitability of striatal medium spiny neurons with an elevated voltage threshold.
INTRODUCTION
The NaV1.2 channel, encoded by Scn2a, is a major voltage-gated sodium channel in the central nervous system (CNS) supporting action potential (AP) firing (Gazina et al., 2015; Sanders et al., 2018). NaV1.2 is strongly expressed in the principal neurons of the corticostriatal circuit, including pyramidal neurons in the medial prefrontal cortex (mPFC) and medium spiny neurons (MSNs) of the caudate nucleus and the putamen (CPu) in the striatum (Miyazaki et al., 2014; Tian et al., 2014; Yamagata et al., 2017). Gain-of-function (GoF) variants of SCN2A are closely associated with unprovoked seizures and epilepsy, whereas loss-of-function (LoF) or protein-truncating variants of SCN2A (collectively referred to as NaV1.2 deficiency) are leading genetic causes of autism spectrum disorder (ASD) and intellectual disability (ID) (Hoischen et al., 2014; Johnson et al., 2016; Sanders et al., 2012; Satterstrom et al., 2020; Wang et al., 2016a). The conventional paradigm suggests that GoF variants of SCN2A increase the excitability of principal neurons, resulting in spontaneous seizures and epilepsy, whereas NaV1.2 deficiency impairs the excitability of principal neurons, leading to ASD (Sanders et al., 2018). However, clinical studies found that a portion of individuals with NaV1.2 deficiency develop “late-onset” drug-resistant seizures (Berg et al., 2021; Wolff et al., 2017, 2019). Because hyperexcitability and hypersynchronization of neuronal firing are associated with seizure generation (Tóth et al., 2018), it is intriguing how NaV1.2 deficiency, predicted to reduce neuronal excitability, contributes to seizures and epilepsy.
To understand NaV1.2 deficiency-related pathophysiology, mouse models were generated. Homozygous Scn2a−/− knockout mice die perinatally (Planells-Cases et al., 2000; Shin et al., 2019), whereas heterozygous Scn2a+/− mice (with ~50% NaV1.2 expression level) survive to adulthood. However, the earlier study did not find notable abnormalities in Scn2a+/− mice (Planells-Cases et al., 2000). More recently, absence-like seizures were reported in adult male Scn2a+/− mice (Ogiwara et al., 2018). The CPu of the striatum and the mPFC of the cortex are suggested to be key brain regions in which absence seizure-like spike-wave discharges (SWDs) were identified (Miyamoto et al., 2019; Ogiwara et al., 2018). Indeed, the corticostriatal circuit is highly involved in ASD as well as in generation of spontaneous seizures, and the excitability of principal neurons in this circuit could strongly influence seizure susceptibility (Aupy et al., 2019; Fuccillo, 2016). Despite these in vivo findings, recordings in brain slices revealed unchanged AP firing and reduced excitatory postsynaptic current in pyramidal neurons of adult Scn2a+/− mice (Ogiwara et al., 2018; Spratt et al., 2019), which are unable to explain the in vivo findings or model the clinical phenotypes.
It is not uncommon that heterozygous knockout with a close to 50% reduction in protein level is not sufficient to render major phenotypes in mice (Fallah and Eubanks, 2020). A substantial reduction of gene expression may be essential to produce robust phenotypes in the mouse model of Scn2a. Because Scn2a null (100% knockout) is lethal, we generated a NaV1.2-deficient mouse model via a gene trap strategy (Eaton et al., 2021). Using this mouse model, we investigated how severe NaV1.2 deficiency affects neuronal excitabilities in principal neurons of the corticostriatal circuit.
RESULTS
Neurons expressing a substantially low level of NaV1.2 channel have elevated excitability
To understand the consequences of severe NaV1.2 deficiency in neurons, we utilized a NaV1.2-deficient mouse model generated using a gene trap (gt) approach (Eaton et al., 2021). Homozygous (HOM) Scn2agt/gt mice can survive to adulthood and have a substantial reduction of NaV1.2 expression (~25% of the wild-type [WT] level) (Eaton et al., 2021). Because the gt cassette contains a LacZ element that is driven by the native NaV1.2 promoter (Figure S1A; Skarnes et al., 2004, 2011), we used LacZ staining as a surrogate to determine the expression and distribution of NaV1.2 in the brain. Our data show that Scn2a is widely expressed in the mouse brain, including in the cortex and striatum (Figure S1B), which is consistent with previous studies of Scn2a distribution (Miyazaki et al., 2014; Tian et al., 2014; Yamagata et al., 2017). To confirm these results, we performed a western blot analysis. We found that heterozygous (HET) Scn2aWT/gt mice have ~60% of WT NaV1.2 protein level in homogenate from CPu tissues, whereas HOM Scn2agt/gt mice have a much lower level at ~32% (Figure S1C). This result is largely consistent with our initial characterization of this mouse model using whole-brain homogenates (Eaton et al., 2021).
The CPu is a common node for ASD and seizures and is the main brain region involved in Scn2a-related absence-like seizures (Fuccillo, 2016; Miyamoto et al., 2019; Ogiwara et al., 2018). To determine how severe deficiency of NaV1.2 affects neuronal excitability, we performed ex vivo patch-clamp recordings in brain slices. Unexpectedly, we found that striatal principal MSNs from Scn2agt/gt mice were drastically more excitable (Figures 1A–1C). The AP firing triggered by current injection were elevated significantly in MSNs from Scn2agt/gt mice compared with WT neurons. We also observed depolarized resting membrane potential (RMP) and increased input resistance of these MSNs (Figures 1D and 1E), which were in line with increased neuronal excitability. Phase-plane plot analysis showed that the AP waveform in Scn2agt/gt mice was clearly altered (Figures 1F and 1G). Although rheobase was reduced, we detected a higher voltage threshold, reduced AP amplitude, elevated fast after-hyperpolarization (AHP), and increased half-width values in MSNs from Scn2agt/gt mice (Figures 1H–1L). Voltage-dependent conductance can affect neuronal RMP (Hu and Bean, 2018), and RMP is known to influence neuronal excitability (Huang et al., 2009). Therefore, we performed recordings at a fixed membrane potential (MP) to determine whether the altered RMP is a major factor contributing to the observed hyperexcitability of MSNs. Interestingly, even at the fixed MP, we were still able to detect enhanced excitability along with altered AP waveforms in MSNs from Scn2agt/gt mice (Figures S1D–S1M), suggesting that, besides RMP, other factors may play essential roles contributing to neuronal hyperexcitability. Our data reveal the counterintuitive finding that severe deficiency of NaV1.2 results in increased (rather than conventionally suggested decreased) neuronal excitability.
Figure 1. Elevated neuronal firing of striatal medium spiny neurons (MSNs) in adult NaV1.2-deficient mice.
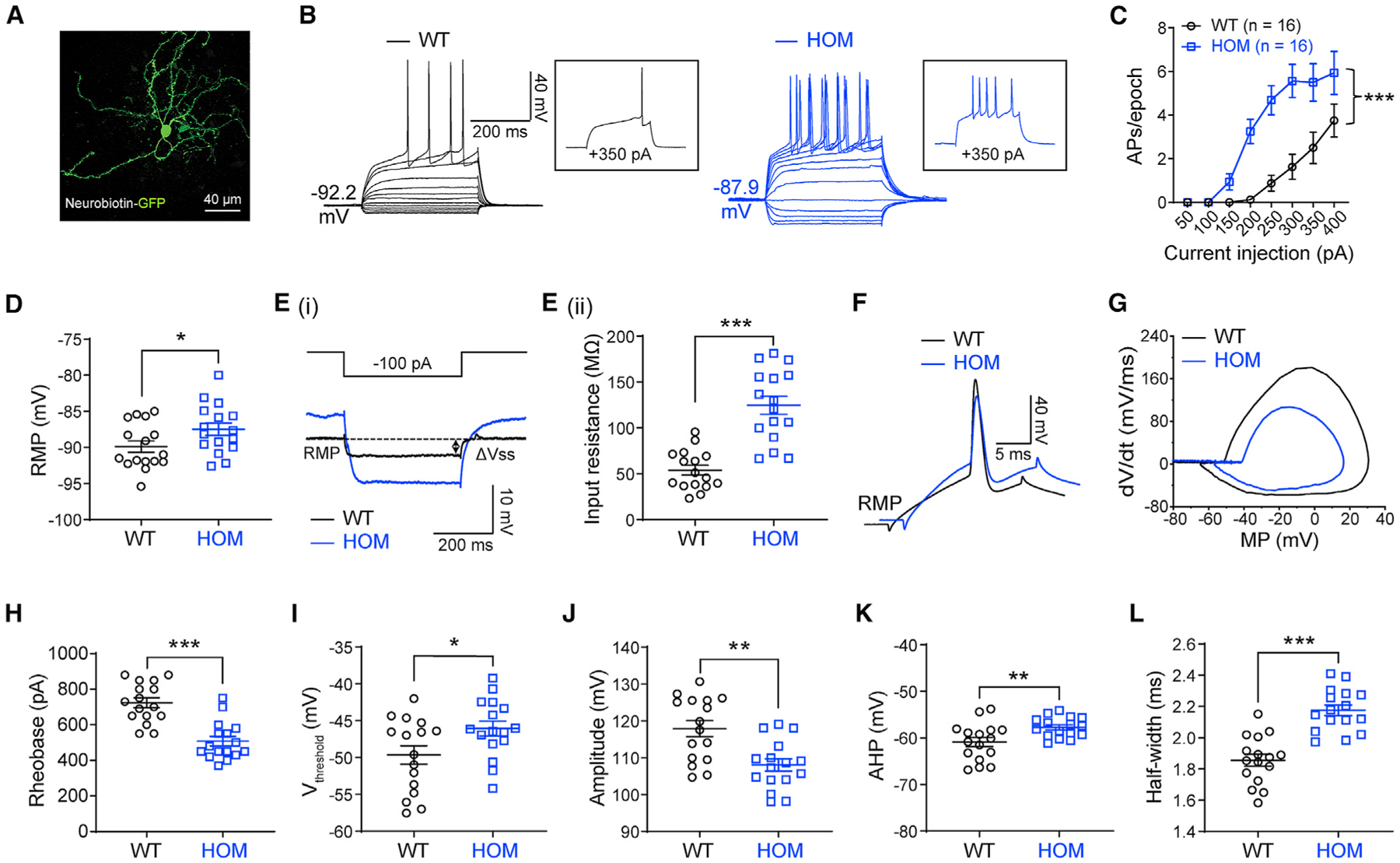
(A) A typical MSN labeled by neurobiotin. Scale bar, 40 μm.
(B) Representative current-clamp recordings of MSNs from wild-type (WT, black) and homozygous (HOM) Scn2agt/gt (blue) mice were obtained at the RMP. Inset: representative trace in response to +350 pA injection.
(C) The average number of action potentials (APs) generated in response to depolarizing current pulses. Unpaired two-tailed non-parametric Mann-Whitney U test for each current pulse.
(D) Individuals and mean RMP values. Unpaired t test.
(Ei) Representative traces in response to −100 pA injection. Vsteady-state (Vss) is the voltage recorded 0–10 ms before the end of the stimulus.
(Eii) Individuals and mean input resistance values at the RMP. Unpaired t test.
(F) Typical spikes of MSNs from WT (black) and HOM (blue) mice were obtained at the normal RMP.
(G) Associated phase-plane plots.
(H–L) Individual and mean spike rheobase, voltage threshold, amplitude, fast after-hyperpolarization (AHP), and half-width values. Unpaired t test. Data are shown as mean ± SEM.
Enhanced excitability is reversible in adult NaV1.2-deficient mice with restoration of Scn2a expression and is autonomous
Scn2agt/gt mice, generated via a gt strategy, has a built-in genetic “rescue” element for manipulation (Skarnes et al., 2011; Testa et al., 2004). The inserted “tm1a” trapping cassette is flanked with Frt sites that can be removed via a flippase recombinase (Flp) to achieve a “tm1c” allele in a temporally and spatially controlled manner (Skarnes et al., 2011; Figure S1A). This “tm1c” allele could be used as a “rescue” allele to restore the expression of the target gene. We performed experiments to restore Scn2a expression by adeno-associated virus (AAV) delivery of codon-optimized Flp (FlpO) with the goal to determine the reversibility of this enhanced neuronal firing in adult mice. Using a PHP.eB.AAV vector that can be administered via systemic delivery (Figure 2A) to transduce neurons across the brain (Chan et al., 2017), we studied the LacZ signals (Figure 2B) and protein expression level of Scn2a (Figure 2C). AAV-FlpO transduction markedly reduced the LacZ signal in brain slices of Scn2agt/gt mice (Figure 2B). We further found that AAV-FlpO transduction resulted in partial but significant elevation of NaV1.2 protein expression in adult Scn2agt/gt mice compared with control PHP.eB.AAV transduction (Figure 2C). Remarkably, this partial restoration of Scn2a expression in adult mice translated into changes in neuronal excitability. We found that adult Scn2agt/gt mice transduced with AAV-FlpO displayed decreased excitability of striatal MSNs (Figures 2D and 2E). In the FlpO-treated group, triggered AP firing of MSNs in Scn2agt/gt mice was reduced to the WT range, along with correction of other parameters, including RMP and AP waveform (Figures 2D–2J). Our data show that, even with partial restoration of Scn2a expression to ~50%–60% of the WT level, we were able to achieve close to full rescue of neuronal excitability in adult mice.
Figure 2. Elevated neuronal firing is reversible by FlpO-mediated restoration of NaV1.2 expression in adult NaV1.2-deficient mice.
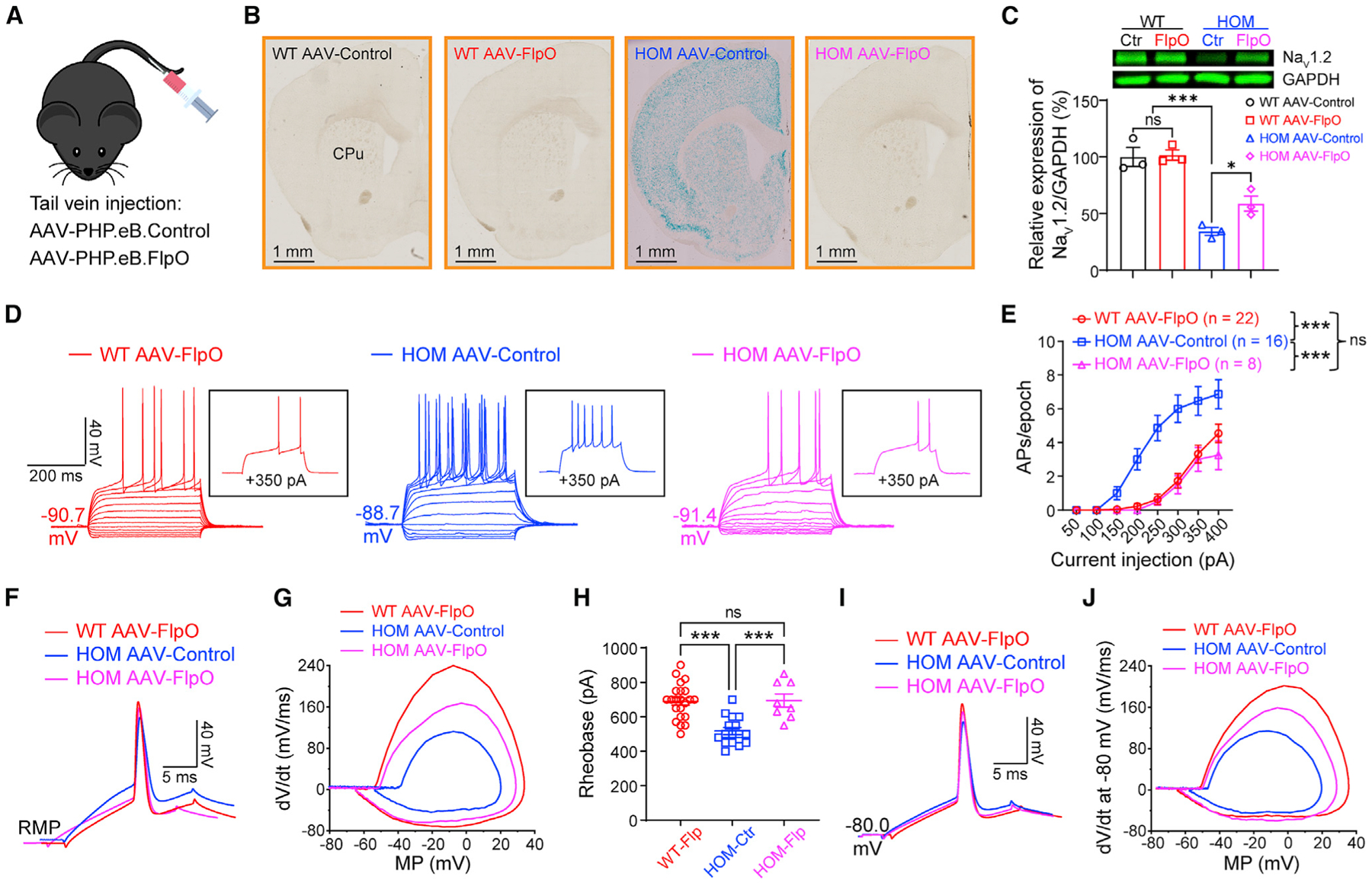
(A) Cartoon illustration of mice systemically administered PHP.eB.AAV-control or PHP.eB.AAV-FlpO via tail vein injection.
(B) Coronal views of LacZ staining of the striatum from WT and Scn2agt/gt (HOM) mice injected with AAV-control or AAV-FlpO. Blue staining of HOM mice largely disappeared in the AAV-FlpO group. CPu, caudate nucleus and the putamen (dorsal striatum). Scale bar, 1 mm.
(C) Western blot analysis showing NaV1.2 protein levels in whole-brain homogenates from HOM mice in the AAV-control or AAV-FlpO group. One-way ANOVA with multiple comparisons.
(D) Representative current-clamp recordings of MSNs from WT mice transduced with AAV-FlpO (red), HOM mice transduced with AAV-Control (blue), and HOM mice transduced with AAV-Control (magenta) obtained at the RMP. Inset: representative trace in response to +350 pA injection.
(E) The average number of APs generated in response to depolarizing current pulses at the RMP. Unpaired Mann-Whitney U test for each current pulse.
(F) Typical spikes of MSNs were obtained at the normal RMP.
(G) Associated phase-plane plots.
(H) Individuals and average spike rheobase. Unpaired t test.
(I) Typical spikes of MSNs at a fixed MP of −80 mV.
(J) Associated phase-plane plots at −80 mV. Data were shown as mean ± SEM.
In the corticostriatal circuit, principal pyramidal neurons of the mPFC project to the striatum and are suggested to be involved in seizure initiation. Because the mPFC is also implicated in absence-like seizures of Scn2a+/− mice (Miyamoto et al., 2019; Ogiwara et al., 2018), we studied the excitability of layer V pyramidal neurons of the mPFC. We found that the excitability of these NaV1.2-deficient neurons was increased significantly compared with that of the WT neurons and was reversed by FlpO-mediated partial restoration of Scn2a expression (Figure S2). Our data suggest that NaV1.2 deficiency-related hyperexcitability exists along the corticostriatal circuit, manifested in the principal neurons of the cortex and striatum.
The hyperexcitability observed in neurons with NaV1.2 deficiency could come from the altered intrinsic properties independent of other neurons (autonomous) or be a result of a disrupted circuit. To distinguish these possibilities, we performed AAV injections of FlpO-mCherry to sparsely transduce only a few MSNs in the CPu. We then performed patch-clamp recordings on adjacent neurons with or without fluorescence (AAV-negative/non-transduced neurons versus AAV-positive/transduced neurons) (Figure 3A). Strikingly, our data showed that transduced neurons (showing fluorescence) had greatly decreased neuronal excitability compared with non-transduced neurons (showing non-fluorescence) in the same brain slices of the NaV1.2-deficient mouse. In particular, we found that RMP, input resistance, and the altered AP waveform were reversed in FlpO-transduced neurons of Scn2agt/gt mice (Figures 3B–3L). Moreover, when we performed the recordings at a fixed MP of −80 mV, similar findings were obtained (Figure S3). On the other hand, non-transduced neurons displayed hyperexcitability similarly to neurons from Scn2agt/gt mice without virus transduction. Our data indicate that the hyperexcitability of each MSN can be modulated autonomously by the expression level of Scn2a and that NaV1.2 deficiency-related hyperexcitability is an intrinsic property of a particular neuron independent of its surrounding neurons or circuit.
Figure 3. Elevated neuronal excitability is autonomous in adult NaV1.2-deficient mice.
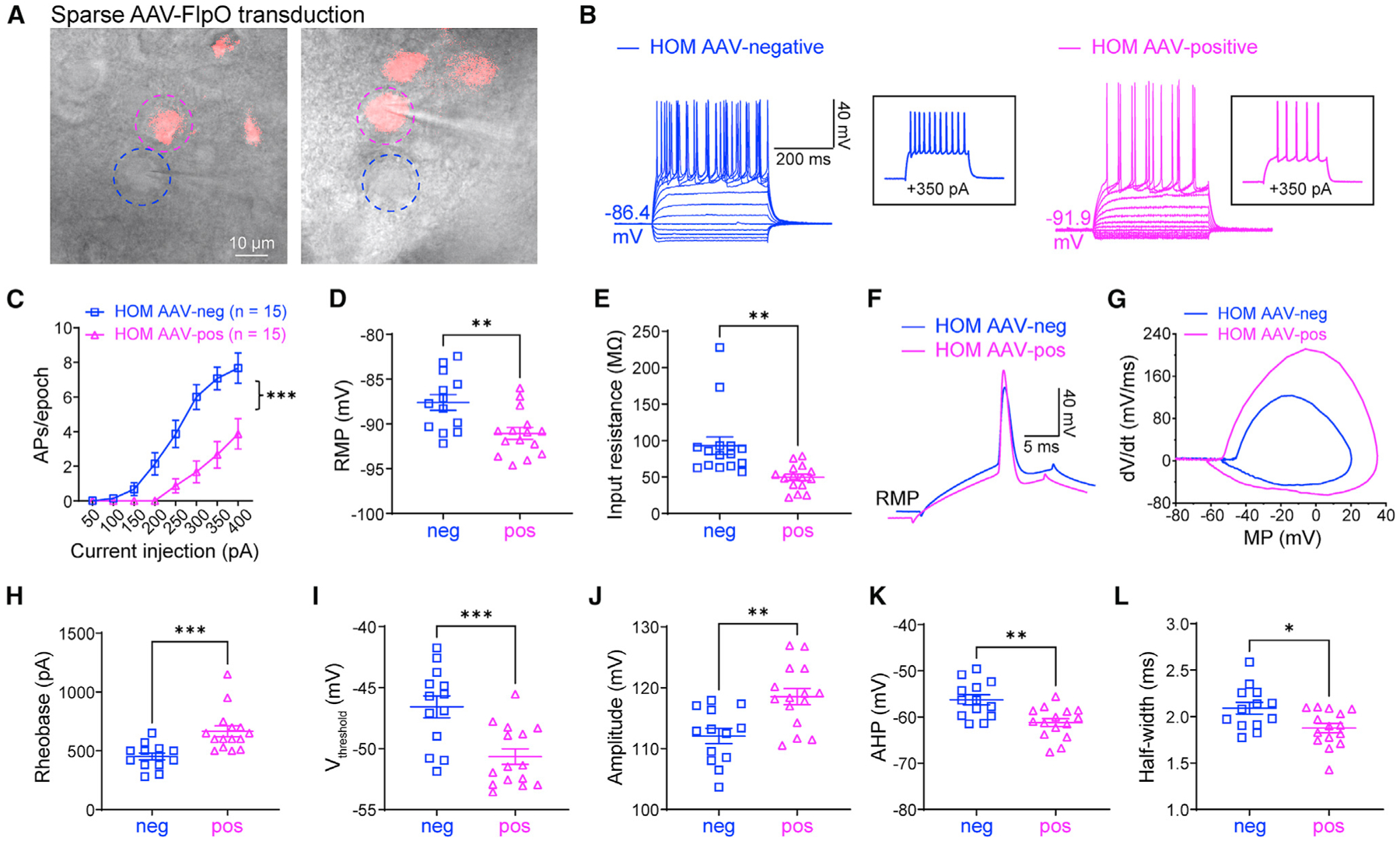
(A) Scn2agt/gt (HOM) mice were injected with a dilute FlpO virus, sparsely transducing a subset of neurons in the striatum. Dashed circles highlight two neighboring AAV-negative (blue circle) and AAV-FlpO-positive (magenta circle) neurons. Scale bar, 10 μm.
(B) Representative current-clamp recordings of AAV-negative (blue) and AAV-FlpO-positive (magenta) MSNs in the CPu of HOM mice were obtained at the RMP. Inset: representative trace in response to +350 pA injection.
(C) The average number of APs generated in response to depolarizing current pulses. Unpaired Mann-Whitney U test for each current pulse.
(D) Individuals and average RMP values. Unpaired t test.
(E) Individuals and average input resistance values at the RMP. Unpaired t test.
(F) Typical spikes were obtained at the RMP.
(G) Associated phase-plane plots.
(H–L) Individual and average spike rheobase, voltage threshold, amplitude, AHP, and half-width values. Unpaired t test. Data are shown as mean ± SEM.
Downregulation of potassium channels contributes to elevated AP firing
To identify the molecular basis underlying the enhanced neuronal excitability of Scn2agt/gt mice, we studied the gene expression profile using RNA sequencing (RNA-seq). We identified around 900 genes that were significantly up- or downregulated in Scn2agt/gt mice compared with WT littermates (Figure 4A). Scn2a expression was 29.6% of the WT value (Figure 4B), consistent with our qPCR (Figure S4A) and western blot studies (Figure 2C; Figure S1C). NaV1.6 and NaV1.2 are two major sodium channels often working in a coordinated fashion in principal neurons in the CNS, and dysfunction of NaV1.6 is involved in seizures (Bunton-Stasyshyn et al., 2019; Lopez-Santiago et al., 2017; Makinson et al., 2017; Meisler, 2019). In NaV1.6-deficient mouse models, NaV1.2 is upregulated, suggesting a compensatory relationship (Katz et al., 2018; Vega et al., 2008). Interestingly, we detected slightly reduced (rather than increased) expression of NaV1.6 in Scn2agt/gt mice in our RNA-seq analysis. This reduction of NaV1.6 did not reach statistical significance (91.4% ± 2.3% of WT, n = 4, p = 0.39) by qPCR validation (Figure S4A), indicating that our observed neuronal hyperexcitability is unlikely to result from a compensatory change of NaV1.6 channel expression.
Figure 4. Activation of KV channels reverses elevated neuronal firing in adult NaV1.2-deficient mice.
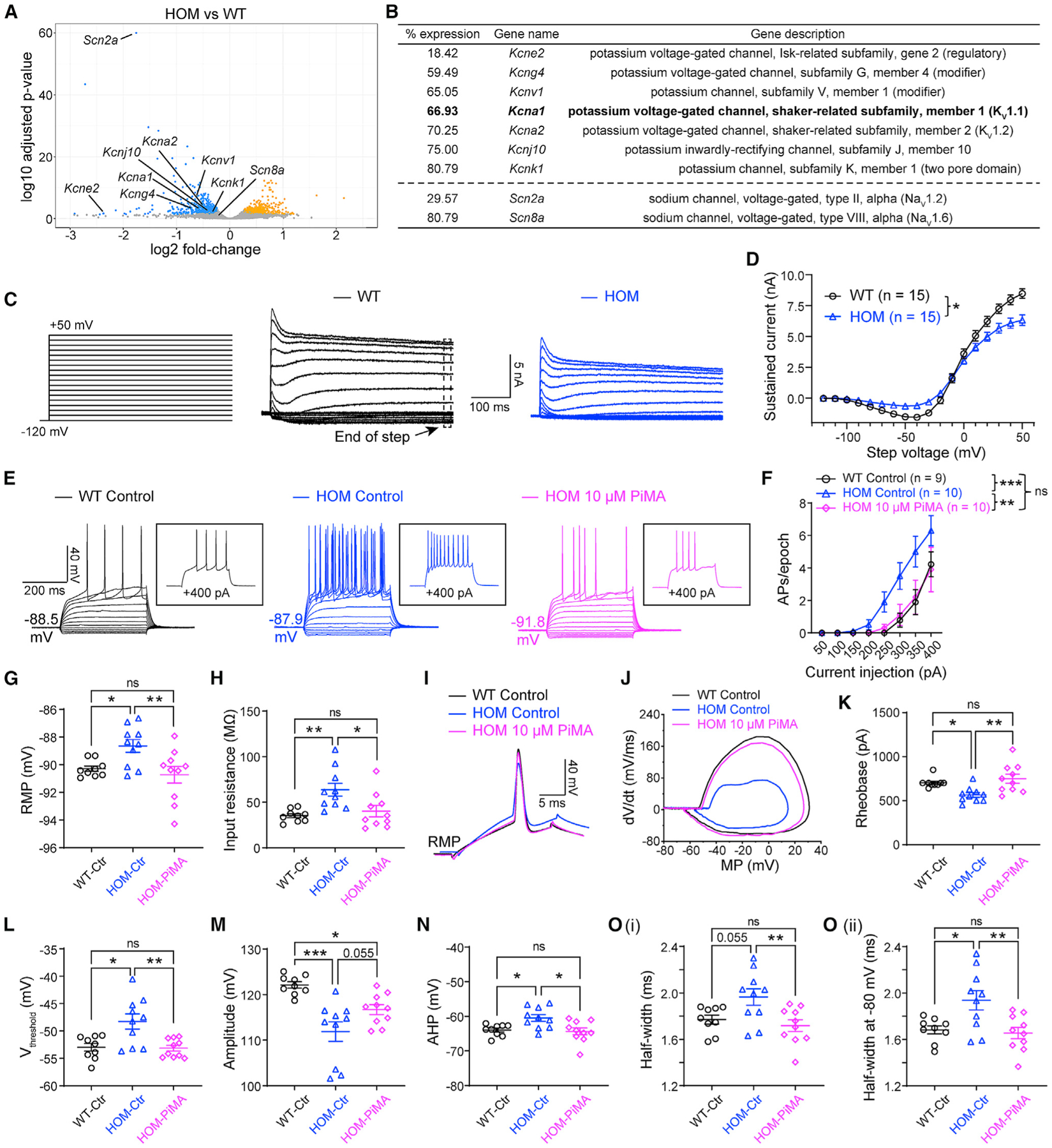
(A) Volcano plot displaying Scn2a and Scn8a as well as potassium channels that are statistically downregulated in Scn2agt/gt (HOM) mice compared with WT mice identified by RNA-seq. Significantly upregulated genes are shown in yellow, and downregulated genes are shown in blue.
(B) List of potassium channels that are significantly downregulated in HOM mice compared with the WT. Hits were identified from both DESeq2 and edgeR differential expression analysis with a false discovery rate of less than 0.05. “% expression” of WT level (100%). n = 4 mice for each group.
(C) Representative whole-cell voltage-clamp recordings of MSNs in brain slices from WT and HOM mice, showing potassium currents at voltage steps from −120 mV to +50 mV. Voltage-gated Ca2+ channels were not blocked to allow activation of Ca2+-dependent K+ channels.
(D) Summary of the total sustained current (measured at the end of steps). Two-way ANOVA with repeated measures.
(E) Representative current-clamp recordings of MSNs from WT slices perfused with 0.1% DMSO in artificial cerebrospinal fluid (aCSF) (WT control, black), HOM slices perfused with 0.1% DMSO in aCSF (HOM control, blue), and HOM slices perfused with 0.1% DMSO in aCSF containing PiMA (HOM 10 μM PiMA, magenta) at the RMP. Inset: representative trace in response to +400 pA injection.
(F) The average number of APs generated in response to depolarizing current pulses at the RMP. Unpaired Mann-Whitney U test for each current pulse.
(G) Individuals and average RMP values. Unpaired t test.
(H) Individuals and average input resistance values at the RMP. Unpaired t test.
(I) Typical spikes of MSNs were obtained at the RMP.
(J) Associated phase-plane plots.
(K–O) Individuals and average spike rheobase, voltage threshold, amplitude, AHP, and half-width values. Unpaired t test. Data are shown as mean ± SEM.
Besides NaV channels, potassium channels are also known to be major mediators setting neuronal excitability (Guan et al., 2006; Niday and Tzingounis, 2018) and are often co-localized with NaV along the axon in high density to regulate neuronal excitability (Duménieu et al., 2017; Lorincz and Nusser, 2008). Indeed, because the AP waveform was altered markedly in neurons with severe Nav1.2 deficiency, it is likely that the function or expression of potassium channels, which are responsible for many aspects of an AP waveform, was disrupted in these neurons. Thus, we expanded our survey to include potassium channels. We found multiple potassium channel genes to be downregulated significantly (Kcne2, Kcng4, Kcnv1, Kcna1, Kcna2, Kcnj10, and Kcnk1) in our RNA-seq analysis (Figure 4B). Our qPCR experiment validated significant downregulation of KV1.1 and KV1.2 channels (Figure S4B) as well as the other potassium channels (Figure S4C) identified by our RNA-seq analysis. Importantly, the reduction of mRNA levels of multiple potassium channels was accompanied by a change of total potassium current. By whole-cell voltage-clamp recording, we found that total sustained potassium currents were reduced in NaV1.2-deficient MSNs (Figures 4C and 4D).
Downregulation of KV1.1 (Kcna1) and KV1.2 (Kcna2) channels, known to be involved in neuronal excitability and seizures(Trosclair et al., 2020), motivated us to test the effect of KV channel openers. Pimaric acid (PiMA) is a relatively general K channel opener but with demonstrated properties as a KV1.1-KV2.1 opener (Sakamoto et al., 2017). We tested PiMA (10 μM) on MSNs in brain slices of Scn2agt/gt mice. Although it might not be surprising that PiMA can affect neurons from WT mice, it was quite remarkable that PiMA almost completely rescued the excitability of MSNs of Scn2agt/gt mice to the WT range without “overshooting” (Figures 4E–4O). Strikingly, most of the parameters, including RMP, input resistance, AP rheobase, voltage threshold, amplitude, AHP, and half-width values, were reversed toward WT values in the presence of PiMA as well (Figures 4G–4O).
Using 4-trifluoromethyl-L-phenylglycine (4TFMPG; 100 μM), recently reported to be a selective KV1.1 opener (Manville and Abbott, 2020), we investigated the specific role of KV1.1 in MSNs of Scn2agt/gt mice. We found that the pre-incubation with 100 μM 4TFMPG for 10 min or more could significantly reverse the hyperexcitability of MSNs as well as the RMP and rheobase values of Scn2agt/gt mice (Figures S4D–S4F, S4J, S4O–S4Q, and S4U). Interestingly, different from the relatively broad potassium channel opener PiMA, 4TFMPG was not able to rescue the input resistance, AP voltage threshold, amplitude, AHP, or half-width (Figures S4G–S4I, S4K–S4N, S4R–S4T, and S4V–S4Y).
To understand whether the change of expression in these KV channels could be influenced by the NaV1.2 expression level, we performed a qPCR experiment with striatal tissue from mice injected with AAV-FlpO. Our data revealed that, after partial restoration of Scn2a expression by FlpO, expression of KV1.1 and KV1.2 as well as other potassium channel genes was increased (Figures S4B and S4C). Our data suggest that neurons have a dynamic adaptation mechanism to regulate gene expression in response to the change of Scn2a expression level.
Remarkably, the dynamic change of potassium channel expression is accompanied by a change in potassium currents (Figures S5A–S5C). Compared with WT neurons, NaV1.2-deficient neurons displayed a smaller total step onset current, end of step current, transient current, as well as tail current, which can be reversed partially (or almost completely) by restoration of NaV1.2 via AAV-FlpO transduction (Figures S5D–S5G). To understand the contribution of KV channels, we studied α-dendrotoxin (α-DTX), a relatively selective blocker that can inhibit KV1.1/KV1.2 (Glazebrook et al., 2002; Lahiri and Bevan, 2020). We found that α-DTX (100 nM) can block ~30% of the total potassium current in WT neurons but only ~10% of that in NaV1.2-deficient neurons. With restoration of NaV1.2 channel expression via AAV-FlpO transduction, we again observed an ~30% α-DTX sensitivity current (Figures S5H–S5K), supporting our findings obtained with RNA-seq and qPCR analyses.
DISCUSSION
In this study, we report the counterintuitive finding that severe NaV1.2 deficiency results in hyperexcitability of principal MSNs in the striatum and pyramidal neurons in the mPFC, challenging the conventional paradigm. We demonstrated that this hyperexcitability is reversible even in adult mice, supporting a dynamic adaptive ability of neurons. We provided evidence to suggest that the compensatory reduction in potassium channel expression/current might be a mechanism underlying this unexpected hyperexcitability of neurons in NaV1.2-deficient mice.
The NaV1.2 channel plays a variety of roles in initiation, propagation, and backpropagation of APs in the developing and adult brain (Hu et al., 2009; Rush et al., 2005; Spratt et al., 2019; Wang et al., 2017; Westenbroek et al., 1992). In the early stage of brain development, NaV1.2 is suggested to be the main sodium channel expressed in the axon initial segment (AIS) (Gazina et al., 2015; Gazina et al., 2010; Workman et al., 2013). Later in development, NaV1.6 becomes the dominating channel in the axon and distal AIS, whereas expression of NaV1.2 is re-distributed to other parts of the neurons, including proximal AIS and dendrites (Bender and Trussell, 2012; Hu and Bean, 2018; Hu et al., 2009; Spratt et al., 2019). Here we revealed that severe NaV1.2 deficiency in neurons leads to hyperexcitability, an intrinsic property of neurons that can be modulated in adulthood. However, how severe NaV1.2 deficiency changes neuronal excitability during early brain development remains to be determined.
Increased NaV1.2 channel activity leads to enhanced excitability of principal neurons and aggravates seizures (Hedrich et al., 2019; Sanders et al., 2018; Wolff et al., 2019). NaV1.2 deficiency, on the other hand, is mainly found in ASD/ID cases and conventionally expected to impair neuronal excitability (Ben-Shalom et al., 2017; Sanders et al., 2012; Satterstrom et al., 2020). Intriguingly, an estimated 20%–30% of individuals with NaV1.2 deficiency-associated ASD/ID develop “late-onset” seizures (Sanders et al., 2018; Wolff et al., 2017). Treating unprovoked seizures in individuals with NaV1.2 deficiency is extremely difficult, and sodium channel blockers have been shown to exacerbate rather than alleviate the seizure phenotype (Wolff et al., 2017). Our current data, together with a studies of Scn2a+/− mice, could suggest a revised paradigm. A moderate deficiency in NaV1.2 (e.g., ~50% reduction) impairs neuronal excitability contributing to ASD and ID, whereas severe deficiency in NaV1.2 tips the balance, resulting in neuronal hyperexcitability and increased seizure susceptibility. An independent study (Spratt et al., 2021 [this issue of Cell Reports]) found that 100% conditional knockout of Scn2a in a subset of pyramidal neurons of the mPFC results in hyperexcitability as well.
Potassium channels are known to play major roles in neuronal excitability, spontaneous seizures, and epilepsy (Millichap et al., 2016; Niday et al., 2017; Quraishi et al., 2019, 2020; Soh et al., 2018). The AP waveform, which is highly influenced by orchestration of a variety of potassium channels, is disrupted drastically in neurons with severe NaV1.2 deficiency. Our RNA-seq results identified multiple potassium channels to be downregulated significantly in Scn2agt/gt mice, including KV1.1 and KV1.2. Additionally, the large downregulation of Kcne2 may imply a change in KV7.2/KV7.3 function (Tinel et al., 2000), which could contribute to the difference observed in a component of the tail current (Soh and Tzingounis, 2010). Based on our recording protocol, the potassium current we recorded could involve the KV2.1 channel (Park et al., 2006), which may be modulated by Kcne2 as well (David et al., 2015; McCrossan et al., 2009). Besides elevated AP firing, neurons from Scn2agt/gt mice have reduced AP amplitude, higher voltage threshold, increased input resistance, and enhanced AHP, which could be modulated by additional potassium channels to eventually enhance neuronal excitability (Johnston et al., 2010; Wulff et al., 2009). We are particularly interested in the KV1.1 channel because it is expressed abundantly in principal neurons of the CNS and contributes to the threshold as well as inter-spike intervals during repetitive firing (Guan et al., 2006). KV1.1 can form heteromultimeric channels with KV1.2 (Guan et al., 2006), which was identified to be downregulated in Scn2agt/gt mice by our RNA-seq analysis as well. Targeting KV1.1 is of significance because AAV-KV1.1 has been suggested as a promising gene therapy to reduce seizures (Snowball et al., 2019; Wykes et al., 2012).
Although our data revealed that (1) NaV1.2 deficiency results in a compensatory reduction in the expression/current of many potassium channels and (2) KV channel openers could normalize neuronal hyperexcitability, our findings do not directly establish a casual role of the KV channel in the hyperexcitability phenotype. That aberrant hyperexcitability of neurons can, in part, be rescued by pharmacological activation of one type of potassium channel does not necessarily suggest that a decrease in the same channel is the cause of the hyperexcitability. Different combinations of ion channels could produce almost identical firing patterns in neurons (Goldman et al., 2000). While a causal link might be difficult to establish, our data nevertheless provide compelling evidence that targeting the KV channel might be a viable strategy to alleviate neuronal hyperexcitability related to NaV1.2 deficiency.
Our results reveal an unexpected hyperexcitability phenotype in NaV1.2-deficient neurons with compensatory reduction of potassium channel expression/current that is reversible by restoration of NaV1.2 channel expression. Our findings may explain the puzzling clinical observation that a portion of individuals with NaV1.2 deficiency still develop unprovoked seizures and guide further development of interventions to treat NaV1.2 deficiency-related disorders.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for reagents and resources should be directed to the Lead Contact, Yang Yang (yangyang@purdue.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw and processed RNA sequencing data are publicly available at GEO through accession number GEO: GSE179818.
Custom scripts are publicly available at https://zenodo.org/record/5098353#.YO48C_lKg4s. DOI 10.5281/zenodo.5098353.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Strains
C57BL/6N-Scn2a1tm1aNarl/Narl (referred to as Scn2aWT/gt) mice were generated from the National Laboratory Animal Center Rodent Model Resource Center based on a modified gene-trap design (Skarnes et al., 2004, 2011). The generation and basic characterization of this mouse model are available in our recent article (Eaton et al., 2021). The targeting construct (tm1a trapping cassette) was electroporated into C57BL/6N embryonic stem cells, and founders in a pure C57BL/6N background were obtained to produce mice for experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC). Mice were same-sex housed in mixed-genotype groups (3–5 mice per cage) on vented cage racks with 1/8” Bed-o-cobb bedding (Anderson, Maumee, OH, USA) and > 8 g of nesting material as enrichment (shredded paper, crinkle-cut paper, and/or cotton nestles) on a 12hr light cycle. Food (2018S Teklad from Envigo) and reverse osmosis water were given ad-lib. Heterozygous (HET, Scn2aWT/gt) mice were used as breeding pairs to obtain homozygous (HOM, Scn2agt/gt) mice and WT littermates for study. Adult mice (2–5 months old) of both sexes were used. Whenever possible, investigators were blind to the genotype of the mice.
METHOD DETAILS
Reagents
Stock solutions were made for the following chemicals at the 1000 × of final concentrations and stored at −20°C: tetrodotoxin citrate (TTX, sodium channel blocker, 500 μM in pure water); pimaric acid (PiMA, KV channel opener, 10 mM in DMSO); 4-(Trifluoromethyl)-L-phenylglycine (4TFMPG, KV1.1 specific opener, 100 mM in 1 M hydrochloric acid); AP5 (NMDA receptor antagonist, 50 mM in pure water); CNQX disodium salt (AMPA/kainate receptor antagonist, 10 mM in pure water), picrotoxin (GABAA receptor antagonist, 50 mM in DMSO); CGP 55845hydrochloride (GABAB receptor antagonist, 2 mM in pure water); ZD7288 [hyperpolarization-activated, cyclic nucleotide-gated cation (HCN) channel blocker, 15 mM in pure water]; α-Dendrotoxin [α-DTX, KV1.1/KV1.2/KV1.6 blocker, 100 μM in water containing 0.1% BSA (w/v)]. All stocks were freshly diluted with normal aCSF (or aCSF containing 0.1% BSA for α-DTX) before use.
Antibodies
Primary antibodies used were: Rabbit anti-SCN2A (NaV1.2) (1: 1000, Alomone Labs, ASC-002), β-Actin (8H10D10) Mouse mAb (1:2000, Cell Signaling Technology, 3700S), and GAPDH (D16H11) XP® Rabbit mAb (1:2000, Cell Signaling Technology, 5174S). Secondary antibodies were: IRDye® 680RD Goat anti-Rabbit IgG Secondary Antibody (1:5000, LI-COR Biosciences, AB_10956166) and IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody (1:5000, LI-COR Biosciences, AB_10956588).
Genotyping
Mice were labeled and genotyped via ear punch at weaning (21–28 days old). Genotyping for the tm1a cassette was performed using gene-specific polymerase chain reaction (PCR) on DNA extracted from ear tissues with a tissue DNA extraction kit (Macherey-Nagel, Bethlehem, PA, USA) with primers (forward 5′ to 3′: GAGGCAAAGAATCTGTACTGTGGGG, reverse: GACGCCTGTGAATAAAACCA AGGAA). The wild-type allele’s PCR product is 240 base pairs (bp) and the tm1a (gt) allele’s PCR product is 340 bp.
Adeno-Associated Virus (AAV) Production
pAAV-EF1a-mCherry-IRES-Flpo was a gift from Karl Deisseroth (Addgene plasmid # 55634 and viral prep # 55634-AAVrg; http://addgene.org/55634; RRID: Addgene_55634) (Fenno et al., 2014), PHP.eB-EF1a-mCherry-IRES-Flpo-WPRE-hGH with the titer of 2.56 × 1013 GC/mL was packed by Penn Vector Core (https://pennvectorcore.med.upenn.edu/); pUCmini-iCAP-PHP.eB was a gift from Viviana Gradinaru (Addgene plasmid # 103005; http://addgene.org/103005; RRID:Addgene_103005) (Chan et al., 2017), pAAV-Ef1a-DO-mCherry-WPRE-pA was a gift from Bernardo Sabatini (Addgene plasmid # 37119; http://addgene.org/37119; RRID: Addgene_37119) (Saunders et al., 2012), control virus, PHP.eB-Ef1a-DO-mCherry-WPRE-pA with the titer of 1.2 × 1013 GC/mL was packed by Bio-Detail Corporation.
Surgical Procedures
For all surgeries (except as noted), mice were systemically anesthetized with ketamine and xylazine, and received analgesic buprenorphine to help postoperative recovery.
AAV Injections
For systemic delivery of virus, each adult mouse received 2 × 1011 infections of FlpO or control AAV virus via tail vein injection. For viral injection into the brain to label neurons sparsely, mice were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.) and secured in a stereotaxic apparatus with ear-bars (RWD Ltd, China). After exposing the skull via a small incision, small holes for each hemisphere were drilled for injection based on coordinates to bregma. Mice were bilaterally injected with AAV virus (diluted into ~5 × 1010 infections units per mL with PBS) into the caudate nucleus and the putamen (CPu, dorsal striatum) (coordinates of the injection sites relative to bregma: AP +1.30 mm, ML ± 1.25 mm, DV −3.30 mm; AP +0.50 mm, ML ± 2.00 mm, DV −3.25 mm, 0.5–1 μL per point) and the nucleus accumbens (NAc, ventral striatum) (coordinates of the injection sites relative to bregma: AP +1.30 mm, ML ± 1.25 mm, DV −4.50 mm, 0.5–1 μL per point) with sharpened glass pipettes (Sutter Instrument), self-made to have a bevel of 35° and an opening of 20-μm diameter at the tip (Liu et al., 2020), attached to syringe needles (200-μm diameter). The pipette was filled from the back end with mineral oil and attached to a syringe needle mounted in a microinjection syringe pump (World Precision Instruments, UMP3T-2). Before injection, the viral suspension was suctioned through the tip of the pipette. The skull over the target coordinates was thinned with a drill and punctured with the tip of the pipette. The pipette was inserted slowly (120 μm/min) to the desired depth. The virus was slowly (~100–150 nL/min) injected to the desired location. Before being retracted out of the brain, the pipette was left at the same place for 10 min when the injection was finished. The virus was allowed to express for at least three weeks before electrophysiological recordings. Animals were allowed to recover from surgery for one week and their body weight and health conditions were closely monitored during recovery. The accurate location of injection sites and viral infectivity were confirmed in mice post hoc by imaging sections (50 μm in thickness) containing the relevant brain regions.
Perfusions and Tissue Processing
For immunostaining, mice were administered an overdose of anesthesia and transcardiacally perfused with ice-cold PBS followed by 4% paraformaldehyde (PFA) (For LacZ staining, 4% PFA was replaced by 2% formaldehyde + 0.2% glutaraldehyde in PBS, hereinafter inclusive). After perfusion, brain slices were dissected out and post-fixed in 4% PFA overnight at 4°C. Tissues were cryoprotected by sinking in gradient sucrose (10%, 20%, and 30%) with 0.01 M PBS at 4°C and subsequently frozen in 20% sucrose and 30% sucrose in 1 × phosphate-buffered saline (PBS) for 24–48 hr. Samples were frozen in Optimal Cutting Temperature compound using dry ice and stored at −80°C. Tissue sections of 20 μm in thickness were taken on a cryostat (Leica CM1950) and allowed to air dry on slides, followed by analysis on a confocal microscope (Zeiss LSM 900 or Nikon A1R-MP).
LacZ (β-galactosidase) Staining
Both Scn2agt/gt and WT mice were processed at the same time under the same condition to minimize variation. Cryosections were fixed with 2% formaldehyde + 0.2% glutaraldehyde in PBS for 5 min. Then sections were washed at least 5 min in PBS (with 0.02% Triton X-100 for optimal reduction of unspecific binding of antibodies). Tissues were covered with a volume of freshly prepared staining solution [X-Gal solution added into Iron Buffer (1/19, v/v) and mixed thoroughly for 10 min], sufficient to fully cover the specimen (e.g., 50 μL) and incubate for 15–30 min at 37°C in a humid chamber until cells were stained blue. Color development was checked under a microscope and incubation time was continued if necessary. Specimen were washed three times with PBS and mounted in glycerol before storage after removing PBS. Images were analyzed under an upright light microscope.
Immunostaining and Imaging Analysis
Cryosections (20 μm in thickness) were permeabilized, incubated in blocking buffer (0.5% Triton X-100 and 5% normal goatserum in PBS) for one hour at room temperature, and overlaid with primary antibodies overnight at 4°C. Then, the corresponding Alexa Fluor 488-, 594-or 647-conjugated secondary antibodies were applied. Stained sections were mounted with DAPI-containing mounting solution and sealed with glass coverslips. Immunofluorescence-labeled images were acquired using a confocal microscope (Wang et al., 2020).
RNA Sequencing
RNA Extraction
Four Scn2agt/gt (HOM) and four WT littermate mice were used to extract RNA. Mice were given an overdose of anesthesia and transcardiacally perfused with ice-cold PBS. Acute coronal brain slices (300-μm in thickness) were cut using a vibratome (Leica VT1200S, Germany). Tissues were rapidly microdissected, immersed into liquid nitrogen, and stored at −80°C until use (same procedures for Western Blotting and qPCR). Based on the manufacturer’s instructions, total RNAs were extracted with TRIzol reagent (Thermo Fisher Scientific, 15596018) from mouse cerebral tissues.
Library Preparation and Sequencing
Novogene prepared libraries using the TruSeq Stranded kit (Illumina, San Diego, CA) and RNA quality was assessed using an Agilent Nano RNA ChIP. Paired-end 150 bp reads were sequenced using the NovaSeq 6000.
Analysis
Reads were quality trimmed and Illumina TruSeq adaptor sequences were removed using Trimmomatic v.0.36 (Bolger et al., 2014). A sliding window approach to trimming was performed, using a window size of 5 and a required average Phred (quality) score of 16. Bases falling below a Phred score of 10 at the start and end of reads were trimmed and reads shorter than 20 bases in length after trimming were removed. FastQC v. 0.11.7 (Andrews, 2010) was run to observe data quality before and after trimming/adaptor removal. STAR v. 2.5.4b (Dobin et al., 2013) was used to align reads to the Ensembl Mus musculus genome database version GRCm38.p6. The htseq-count script in HTSeq v.0.7.0 (Anders et al., 2015) was run to count the number of reads mapping to each gene. HTSeq used Biopython v.2.7.3 in the analysis. HTSeq was run utilizing the GTF file on “intersection-nonempty” mode. The HTSeq feature was set to “exon” and the attribute parameter was set to “gene_id” and the–stranded = reverse option was set. The Bioconductor packages DESeq2 v.1.22.2 (Love et al., 2014) and edgeR 3.24.3 (Robinson et al., 2010) were used for differential expression analysis. Genes that were identified as differentially expressed in both packages were used as high confidence differentially expressed genes and were used in subsequent pathway analysis. The Benjamini-Hochberg false discovery rate correction was used to correct p values for multiple testing. To improve power, low expression transcripts were filtered out of the data before performing differential expression analysis. Genes expressed at lower than 0.5 counts per million (CPM) in all samples combined were filtered out prior to performing the differential expression analysis. After filtering, 18,134 genes were remaining. The expression of genes between WT and HOM was deemed significant if the adjusted p value < 0.05. The Bioconductor package biomaRt v. 2.38.0 was used to perform annotation of genes (Durinck et al., 2005, 2009). ClusterProfiler v. 3.10.1 was used to perform pathway and gene ontology enrichment analysis (Yu et al., 2012).
Western Blotting
Brain tissues were homogenized in ice-cold RIPA lysis and extraction buffer (Thermo Fisher Scientific, 89901) supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific, A32953), sonicated, and cleared by centrifugation (10,000 × g, 10 min, at 4°C). Protein concentration in the supernatant was determined by Nanodrop (Thermo Scientific). Proteins in 1 × sample buffer [62.5 mM Tris-HCl (pH 6.8), 2% (w/v) SDS, 5% glycerol, 0.05% (w/v) bromophenol blue] were denatured by boiling at 95°C for 5 min. For each sample, 40 μg total proteins were loaded to the 8% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gels and transferred onto PVDF membrane (Millipore, IPFL00010) by electrophoresis. Blots were blocked in 5% nonfat milk in Tris-buffered saline and Tween 20 (TBST) for 1 h at room temperature and probed with the primary antibody in 5% milk-TSBT overnight at 4°C. After overnight incubation, the blots were washed three times in TBST for 15 min, followed by incubation with corresponding IRDye® 680RD secondary antibodies in TBST for 2h at room temperature. Following three cycles of 15 min washes with TBST, the immuno-reactive bands were scanned and captured by the Odyssey® CLx Imaging System (LI-COR Biosciences) and quantitatively analyzed by densitometry with Image Studio Lite 5.2 (LI-COR Biosciences) or ImageJ software (NIH). Each sample was normalized to its β-actin or GAPDH, then normalized with the corresponding WT littermates.
RNA Isolation, Reverse Transcription, and qPCR Analysis
Total RNAs were extracted with TRIzol reagent (Thermo Fisher Scientific, 15596018) from mouse cerebral tissues according to the manufacturer’s instructions. 4 μg RNA was subjected to reverse transcription (RT) with a Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, K1672). The resulting cDNAs were subjected to quantitative PCR analysis using the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25777) and specific primers in a C1000 Touch PCR thermal cycler (Bio-Rad). Gapdh and β-actin mRNA levels were used as an endogenous control for normalization using the ΔCt method (Livak and Schmittgen, 2001). In brief, test (T):ΔCtT = [CtT (target gene) - CtT (internal control)]; Amount of the target = 2−ΔCt.
Patch-clamp Recordings
Acute slice preparations
Electrophysiology was performed in slices prepared from 2–5 months old Scn2agt/gt and corresponding control mice. Mice were deeply anesthetized with ketamine/xylazine (100/10 mg/kg, i.p., 0.1 mL per 10 g of body weight), transcardially perfused, and decapitated to dissect brains into ice-cold slicing solution containing the following (in mM): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 25 glucose, 1 sodium ascorbate, 3.1 sodium pyruvate (bubbled with 95% O2 and 5% CO2, pH 7.4, 305–315 mOsm). Acute coronal slices containing PFC and/or striatum (300-μm in thickness) were cut by using a vibratome (Leica VT1200S, Germany), and incubated in the same solution for 10 min at 33°C. Then, slices were transferred to normal artificial cerebrospinal fluid (aCSF) (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, 10 glucose (bubbled with 95% O2 and 5% CO2, pH 7.4, 305–315 mOsm) at 33°C for 10–20 min and at room temperature for at least 30 min before use. Slices were visualized under IR-DIC (infrared-differential interference contrast) using a BX-51WI microscope (Olympus) with an IR-2000 camera (Dage-MTI).
Ex vivo electrophysiological whole-cell recordings
All somatic whole-cell patch-clamp recordings were performed from identified striatal MSNs or mPFC layer V pyramidal neurons. The selection criteria for MSNs were based on morphological characteristics with medium-sized cell body presenting polygon or diamond viewed with a microscope equipped with IR-DIC optics (BX-51WI, Olympus), and numerous dendritic spines and their hyperpolarized RMP (lower than −80 mV) based on published method (Torres-García et al., 2012). Layer V pyramidal cells with a prominent apical dendrite were visually identified mainly by location, shape, and pClampex online membrane test parameters (Mei et al., 2016). Putative pyramidal cells in layer 5b were identified based on regular spiking characteristics (Clarkson et al., 2017; Dembrow et al., 2010; Spratt et al., 2019). To minimize variability, recordings were made on cells with low or high HCN expression levels, corresponding to intratelencephalic (IT) or pyramidal tract (PT) neurons, respectively. The selection criterion for PT pyramidal cells followed the published approach (Alessi et al., 2016). Briefly, the selection was based on their firing properties and shape of the AP (i.e., cells’ intrinsic ability to generate significant membrane-potential sags induced by both hyperpolarizing and depolarizing current injection at the soma). Only the recordings from putative PT neurons were used for further analysis.
For whole-cell current-clamp recordings, the internal solution contained (in mM): 122 KMeSO4, 4 KCl, 2 MgCl2, 0.2 EGTA, 10 HEPES, 4 Na2ATP, 0.3 Tris-GTP, 14 Tris-phosphocreatine, adjusted to pH 7.25 with KOH, 295–305 mOsm. The sag ratio, input resistance, and firing number were obtained in response to a series of 400-ms hyperpolarizing and depolarizing current steps from −200 pA to +400 pA in increments of 50-pA, each sweep duration of 5 s with cells held at the normal RMP or a fixed potential of −80 mV. The sag ratio was calculated with the equation:
Where Vbaseline is the resting membrane potential or −80 mV, Vmin is the minimum voltage reached soon after the hyperpolarizing current pulse, and Vsteady-state (Vss) is the voltage recorded at 0–10 ms before the end of the −200 pA stimulus.
The input resistance was calculated with the equation:
Where Vbaseline is the resting membrane potential or −80 mV, and Vsteady-state (Vss) is the voltage recorded at 0–10 ms before the end ofthe −100 pA stimulus.
The RMP, AP threshold, amplitude, fast afterhyperpolarization (AHP), and half-width values were obtained in response to a 20 ms current step of the smallest current to obtain an intact AP, each sweep duration of 1.5 s and start-to-start intervals of 10 s with cells held at the normal RMP or a fixed potential of −80 mV. The RMP, AP threshold, amplitude, AHP, and half-width values were analyzed using the Clampfit 11.1 inbuilt statistics measurements program (Criteria included the baseline, peak amplitude, antipeak amplitude, and half-width). The threshold was defined as the Vm when dV/dt measurements first exceeded 15 V/s.
We used thin-wall borosilicate pipettes (BF150-110-10) with open-tip resistances of 3–5 MΩ. All current-clamp recordings were started at least 1 min after breakin to stabilize the contact between the glass electrode and the cell membrane, and finished within 10 min to avoid large voltage changes due to the internal solution exchange equilibrium. Recordings were performed with an Axon MultiClamp 700B amplifier (Molecular Devices) and data were acquired using pClamp 11.1 software at the normal RMP or a fixed potential of −80 mV, filtered at 2 kHz and sampling rate at 20 kHz with an Axon Digidata 1550B plus HumSilencer digitizer (Molecular Devices). Slices were maintained under continuous perfusion of aCSF at 32–33°C with a 2–3 mL/min flow. In the whole-cell configuration, recordings with stable series resistance (Rs) 15–30 MΩ were used, and recordings with unstable Rs or a change of Rs > 20% were aborted.
To study the effect of KV channel openers, the 1000 × stocks were freshly diluted with aCSF. After 10 min perfusion of each opener or the corresponding vehicle control (0.1% DMSO in aCSF for PiMA, and aCSF for 4TFMPG), the target neurons were studied with the continuous perfusion of the chemicals. One or two neurons were patched for each brain slice, and recordings were discarded if a slice was perfused with KV channel openers for more than 30 min.
Whole-cell potassium currents were recorded according to the protocols reported in previous studies (Bekkers, 2000; Brew et al., 2003; Itri et al., 2005; Wang et al., 2016b; Yang et al., 2010). The internal solution was the same as above. The whole-cell capacitance transients were compensated, and the residual fast transients that emerged after setting series resistance (Rs) were compensated again. Data were recorded with a sampling rate at 50 kHz and filtered at 2 kHz. Rs compensation of 80%–90% was used. Membrane voltages were adjusted for the liquid junction potential (10 mV). Potassium currents were activated with 500-ms voltage steps from −120 mV to +50 mV, in 10 mV increments, followed by 100-ms pulse back to −120 mV. Leak currents were auto-subtracted using P/N leak subtraction after each waveform sweep by execution of 6 subsweeps in the same polarity as the waveform. 2–3 trials were averaged per voltage step. Current amplitudes were calculated from the transient peak, sustained components (average of the last 50 ms of each step), and tail peak. Experiments were performed in 500 nM TTX, 50 μM AP5, 10 μM CNQX, 50 μM picrotoxin, 2 μM CGP 55845, and 15 μM ZD7288. Voltage-gated Ca2+ channels were not blocked to allow for activation of Ca2+-dependent K+ channels. Delayed onset currents were isolated with a 50-ms pre-pulse to −40 mV. Transient K+ currents were isolated by subtracting delayed currents from total currents.
For cell labeling, the internal solution contains 0.1%–0.2% (w/v) neurobiotin tracer. At the end of the electrophysiological recording (about 30 min), slices were treated as previously described (Fogarty et al., 2013). Briefly, sections were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 20–30 min at room temperature, and subsequently washed 3–4 times for 30 min in 0.1 M phosphate-buffered saline (PBS, pH 7.4) at 4°C. Sections were then incubated in Alexa 488-conjugated streptavidin (overnight at 4°C, 1:250 in blocking solution) to visualize neurobiotin.
QUANTIFICATION AND STATISTICAL ANALYSIS
Normality and variance similarity was measured by GraphPad Prism before we applied any parametric tests. Two-tailed Student’s t test (parametric) or unpaired two-tailed Mann-Whitney U-test (non-parametric) was used for single comparisons between two groups. Other data were analyzed using one-way or two-way ANOVA with Tukey correction (parametric) or Kruskal-Wallis with Dunn’s multi comparison correction (non-parametric) depending on the appropriate design. Post hoc comparisons were carried out only when the primary measure showed statistical significance. All data were expressed as mean ± SEM, with statistical significance determined at p values < 0.05. In details, p ≥ 0.05 is indicated as ns (no significance), p < 0.05 is indicated as one asterisk (*), p < 0.01 is indicated as two asterisks (**), and p < 0.001 is indicated as three asterisks (***) in all figures. Mice with different litters, body weights, and sexes were randomized and assigned to different treatment groups, and no other specific randomization was used for the animal studies.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-SCN2A (NaV1.2) | Alomone Labs | Cat#ASC-002; RRID: AB_2040005 |
| GAPDH (D16H11)XP® Rabbit mAb | Cell Signaling Technology | Cat#5174S; RRID: RRID: AB_10622025 |
| β-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | Cat#3700S; RRID: RRID: AB_2242334 |
| IRDye® 680RD Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | P/N: 926–68071; RRID: AB_10956166 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | P/N: 926–68070; RRID: AB_10956588 |
| IRDye® 680RD Goat anti-Rat IgG Secondary Antibody | LI-COR Biosciences | P/N: 926–68076; RRID: AB_10956590 |
| Bacterial and virus strains | ||
| pAAV-EF1a-mCherry-IRES-Flpo | Fenno et al., 2014 | Addgene 55634-AAVrg |
| PHP.eB-EF1a-mCherry-IRES-Flpo-WPRE-hGH | This paper; Penn Vector Core | N/A |
| PHP.eB-Ef1a-DO-mCherry-WPRE-pA | This paper; Bio-Detail Corporation | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| NEUROBIOTIN Tracer | Vector Laboratories | SP-1120 |
| Streptavidin, Alexa Fluor 488 Conjugate | Thermo Fisher Scientific | S32354 |
| 4% paraformaldehyde | Thermo Fisher Scientific | AAJ19943K2 |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | 89901 |
| Protease Inhibitor Mini Tablets | Thermo Fisher Scientific | A32953 |
| Pierce Rapid Gold BCA Protein Assay Kit | Thermo Fisher Scientific | A53226 |
| NuPAGE LDS Sample Buffer (4 ×) | Thermo Fisher Scientific | NP0007 |
| NuPAGE Sample Reducing Agent (10 ×) | Thermo Fisher Scientific | NP0009 |
| PageRuler Plus Prestained Protein Ladder, 10 to 250 kDa | Thermo Fisher Scientific | 26619 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | A25777 |
| SYBR Safe DNA Gel Stain | Invitrogen by Thermo Fisher Scientific | S33102 |
| Tween 20 | Fisher Scientific | AAJ20605AP |
| Formaldehyde solution | Sigma-Aldrich | F8775 |
| Glutaraldehyde solution | Sigma-Aldrich | 354400 |
| 4-(Trifluoromethyl)-L-phenylglycine | Sigma-Aldrich | 16599 |
| Choline chloride | Sigma-Aldrich | C7527 |
| Bovine Serum Albumin | Sigma-Aldrich | A9418 |
| 1 kb DNA Ladder | Sigma-Aldrich | D0428–1VL |
| Agarose | Sigma-Aldrich | A9539 |
| Tetrodotoxin citrate | Abcam | ab120055 |
| CGP 55845 hydrochloride | Abcam | ab120337 |
| D-AP5 | Abcam | ab120003 |
| CNQX disodium salt | Abcam | ab120044 |
| ZD7288 | Abcam | ab120102 |
| Pimaric acid | MP Biomedicals | 219895105 |
| (2-Fluorophenyl) glycine | VWR | 101795–980 |
| Odyssey® Blocking Buffer (PBS) | LI-COR Biosciences | 927–40000 |
| Intercept® T20 (TBS) Antibody Diluent | LI-COR Biosciences | 927–65001 |
| 10 × Tris Buffered Saline | Bio-Rad | 1706435 |
| 10 × Tris/Glycine Buffer | Bio-Rad | 1610771 |
| 10 × Tris/Glycine/SDS Electrophoresis Buffer | Bio-Rad | 1610772 |
| Immobilon®-FL PVDF membrane | Millipore | IPFL00010 |
| 50 × pH 8,3 tris-acetate EDTA buffer | Millipore | 1.06174 |
| α-Dendrotoxin | Alomone Labs | D-350 |
| Tetrodotoxin citrate | Alomone Labs | T-550 |
| Picrotoxin | Alomone Labs | P-325 |
| Optimal Cutting Temperature compound | Tissue Tek | 4583 |
| NucleoSpin® DNA RapidLyse | Macherey-Nagel | 740100.250 |
| Liquid Proteinase K | Macherey-Nagel | 740396 |
| Critical commercial assays | ||
| β-Gal Staining Set | Sigma-Aldrich | 11828673001 |
| Maxima First Strand cDNA Synthesis Kit for RT-qPCR, with dsDNase | Thermo Fisher Scientific | K1672 |
| Deposited data | ||
| Raw and processed RNA sequencing data | This paper | GEO: GSE179818 |
| Custom scripts | This paper | Zenodo Repository https://zenodo.org/record/5098353 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6N-Scn2a1tm1aNarl/Narl | The National Laboratory Animal Center Rodent Model Resource Center | Scn2a WT/gt |
| Oligonucleotides | ||
| See Table S1 | N/A | |
| Recombinant DNA | ||
| pAAV-EF1a-mCherry-IRES-Flpo | Fenno et al., 2014 | Addgene Plasmid # 55634 |
| pUCmini-iCAP-PHP.eB | Chan et al., 2017 | Addgene plasmid # 103005 |
| pAAV-Ef1a-DO-mCherry-WPRE-pA | Saunders et al., 2012 | Addgene plasmid # 37119 |
| Software and algorithms | ||
| ImageJ (Fiji) | Eliceiri/LOCI group | https://fiji.sc/ |
| Image Studio Lite 5.2 | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite/d5 |
| pClamp 11.1 | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system |
| OriginPro version 2020 | OriginLab Corporation | https://www.originlab.com/ |
| GraphPad Prism version 8.0.0 for Windows | GraphPad Software | https://www.graphpad.com/ |
| Adobe Illustrator 2020 | Adobe | https://www.adobe.com/ |
| Other | ||
| Odyssey® CLx Imaging System | LI-COR Biosciences | https://www.licor.com/bio/odyssey-clx/ |
| NanoDrop OneC Microvolume UV-Vis Spectrophotometer | Thermo Fisher Scientific | N/A |
| C1000 Touch PCR thermal cycler | Bio-Rad | N/A |
| MultiPhoton confocal microscopy | Nikon | A1R-MP |
| LSM 900 scanning confocal microscope | Zeiss | N/A |
| Cryostat | Leica Biosystems | CM1950 |
| Vibrating blade microtome | Leica Biosystems | VT1200 S |
| Micropipette Puller | Sutter Instrument | P-1000 |
| Borosilicate glass with filament | Sutter Instrument | BF150-110-10 |
| Axon MultiClamp 700B Microelectrode Amplifier | Molecular Devices | N/A |
| Axon Digidata 1550B Plus HumSilencer | Molecular Devices | Digidata 1550B4 |
| BX-51WI microscope | Olympus | N/A |
| IR-2000 Infrared Monochrome Video Camera | Dage-MTI | N/A |
| Stereotaxic for Mouse, Dual M, Digital, with Adaptor (68055) | RWD Life Science Inc. | 68046 |
Highlights.
Severe NaV1.2 deficiency results in enhanced neuronal excitability in adult mice
Increased excitability is accompanied by a higher voltage threshold in striatal MSNs
Hyperexcitability related to NaV1.2 deficiency is reversible and autonomous
Multiple K+ channels, including KV1.1, have a compensatory reduction in expression
ACKNOWLEDGMENTS
We thank Dr. Amy L. Brewster for critical reading of this manuscript. We appreciate helpful input from Dr. Mark Estacion regarding the recording of K+ currents. We thank Dr. Lan Chen from the Chemical Genomics Facility of PIDD for assay discussions. This work is partially supported by the Showalter Research Trust, and the Purdue Big Idea Challenge 2.0 on Autism (to Y.Y.). The research reported in this publication was also supported by the NINDS of the NIH (R01NS117585 and R01NS123154 to Y.Y.). The authors gratefully acknowledge support from the FamilieSCN2A Foundation for Action Potential Grant support and PIDD and PIIN for additional funding support. M.E. is supported by the NSF GRFP fellowship (DGE-1842166). This project was supported in part by the Indiana Spinal Cord & Brain Injury Research Fund and the Indiana CTSI, funded in part by UL1TR002529 from the NIH. The Yang lab appreciates bioinformatics support from the Collaborative Core for Cancer Bioinformatics (C3B) of the IU Simon Comprehensive Cancer Center (P30CA082709), PCCR (P30CA023168), and the Walther Cancer Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109495.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
REFERENCES
- Alessi C, Raspanti A, and Magistretti J (2016). Two distinct types of depolarizing afterpotentials are differentially expressed in stellate and pyramidal-like neurons of entorhinal-cortex layer II. Hippocampus 26, 380–404. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Aupy J, Wendling F, Taylor K, Bulacio J, Gonzalez-Martinez J, and Chauvel P (2019). Cortico-striatal synchronization in human focal seizures. Brain 142, 1282–1295. [DOI] [PubMed] [Google Scholar]
- Bekkers JM (2000). Properties of voltage-gated potassium currents in nucleated patches from large layer 5 cortical pyramidal neurons of the rat. J. Physiol 525, 593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, and Bender KJ (2017). Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry 82, 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, and Trussell LO (2012). The physiology of the axon initial segment. Annu. Rev. Neurosci 35, 249–265. [DOI] [PubMed] [Google Scholar]
- Berg AT, Palac H, Wilkening G, Zelko F, and Schust Meyer L (2021). SCN2A-Developmental and Epileptic Encephalopathies: Challenges to trialreadiness for non-seizure outcomes. Epilepsia 62, 258–268. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew HM, Hallows JL, and Tempel BL (2003). Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J. Physiol 548, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunton-Stasyshyn RKA, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, Bhatia K, Jones JM, Maniaci MR, Parent JM, et al. (2019). Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 142, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, and Gradinaru V (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson RL, Liptak AT, Gee SM, Sohal VS, and Bender KJ (2017). D3 Receptors Regulate Excitability in a Unique Class of Prefrontal Pyramidal Cells. J. Neurosci 37, 5846–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David J-P, Stas JI, Schmitt N, and Bocksteins E (2015). Auxiliary KCNE subunits modulate both homotetrameric Kv2.1 and heterotetrameric Kv2.1/Kv6.4 channels. Sci. Rep 5, 12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, and Johnston D (2010). Projection-specific neuromodulation of medial prefrontal cortex neurons. J. Neurosci 30, 16922–16937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duménieu M, Oulé M, Kreutz MR, and Lopez-Rojas J (2017). The Segregated Expression of Voltage-Gated Potassium and Sodium Channels in Neuronal Membranes: Functional Implications and Regulatory Mechanisms. Front. Cell. Neurosci 11, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, and Huber W (2005). BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton M, Zhang J, Ma Z, Park AC, Lietzke E, Romero CM, Liu Y, Coleman ER, Chen X, Xiao T, et al. (2021). Generation and basic characterization of a gene-trap knockout mouse model of Scn2a with a substantial reduction of voltage-gated sodium channel Nav 1.2 expression. Genes Brain Behav. 20, e12725. [DOI] [PubMed] [Google Scholar]
- Fallah MS, and Eubanks JH (2020). Seizures in Mouse Models of Rare Neurodevelopmental Disorders. Neuroscience 445, 50–68. [DOI] [PubMed] [Google Scholar]
- Fenno LE, Mattis J, Ramakrishnan C, Hyun M, Lee SY, He M, Tucciarone J, Selimbeyoglu A, Berndt A, Grosenick L, et al. (2014). Targeting cells with single vectors using multiple-feature Boolean logic. Nat. Methods 11, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty MJ, Hammond LA, Kanjhan R, Bellingham MC, and Noakes PG (2013). A method for the three-dimensional reconstruction of Neurobiotin™-filled neurons and the location of their synaptic inputs. Front. Neural Circuits 7, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo MV (2016). Striatal Circuits as a Common Node for Autism Pathophysiology. Front. Neurosci 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazina EV, Richards KL, Mokhtar MB, Thomas EA, Reid CA, and Petrou S (2010). Differential expression of exon 5 splice variants of sodium channel alpha subunit mRNAs in the developing mouse brain. Neuroscience 166, 195–200. [DOI] [PubMed] [Google Scholar]
- Gazina EV, Leaw BT, Richards KL, Wimmer VC, Kim TH, Aumann TD, Featherby TJ, Churilov L, Hammond VE, Reid CA, and Petrou S (2015). ‘Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum. Mol. Genet 24, 1457–1468. [DOI] [PubMed] [Google Scholar]
- Glazebrook PA, Ramirez AN, Schild JH, Shieh C-C, Doan T, Wible BA, and Kunze DL (2002). Potassium channels Kv1.1, Kv1.2 and Kv1.6 influence excitability of rat visceral sensory neurons. J. Physiol 541, 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MS, Golowasch J, Marder E, and Abbott L (2000). Dependence of firing pattern on intrinsic ionic conductances: sensitive and insensitive combinations. Neurocomputing 32, 141–146. [Google Scholar]
- Guan D, Lee JC, Tkatch T, Surmeier DJ, Armstrong WE, and Foehring RC (2006). Expression and biophysical properties of Kv1 channels in supra-granular neocortical pyramidal neurones. J. Physiol 571, 371–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich UBS, Lauxmann S, and Lerche H (2019). SCN2A channelopathies: Mechanisms and models. Epilepsia 60 (Suppl 3), S68–S76. [DOI] [PubMed] [Google Scholar]
- Hoischen A, Krumm N, and Eichler EE (2014). Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat. Neurosci 17, 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, and Bean BP (2018). Differential Control of Axonal and Somatic Resting Potential by Voltage-Dependent Conductances in Cortical Layer 5 Pyramidal Neurons. Neuron 99, 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, and Shu Y (2009). Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat. Neurosci 12, 996–1002. [DOI] [PubMed] [Google Scholar]
- Huang Z, Walker MC, and Shah MM (2009). Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J. Neurosci 29, 10979–10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itri JN, Michel S, Vansteensel MJ, Meijer JH, and Colwell CS (2005). Fast delayed rectifier potassium current is required for circadian neural activity. Nat. Neurosci 8, 650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MR, Shkura K, Langley SR, Delahaye-Duriez A, Srivastava P, Hill WD, Rackham OJ, Davies G, Harris SE, Moreno-Moral A, et al. (2016). Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nat. Neurosci 19, 223–232. [DOI] [PubMed] [Google Scholar]
- Johnston J, Forsythe ID, and Kopp-Scheinpflug C (2010). Going native: voltage-gated potassium channels controlling neuronal excitability. J. Physiol 588, 3187–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Stoler O, Scheller A, Khrapunsky Y, Goebbels S, Kirchhoff F, Gutnick MJ, Wolf F, and Fleidervish IA (2018). Role of sodium channel subtype in action potential generation by neocortical pyramidal neurons. Proc. Natl. Acad. Sci. USA 115, E7184–E7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri AK, and Bevan MD (2020). Dopaminergic Transmission Rapidly and Persistently Enhances Excitability of D1 Receptor-Expressing Striatal Projection Neurons. Neuron 106, 277–290.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LD, Chen S, Economo MN, Li N, and Svoboda K (2020). Accurate localization of linear probe electrodes across multiple brains. bioRxiv, 2020.2002.2025.965210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Yuan Y, Wagnon JL, Hull JM, Frasier CR, O’Malley HA, Meisler MH, and Isom LL (2017). Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc. Natl. Acad. Sci. USA 114, 2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, and Nusser Z (2008). Cell-type-dependent molecular composition of the axon initial segment. J. Neurosci 28, 14329–14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Sorokin JM, Wong JC, Christian CA, Goldin AL, Escayg A, and Huguenard JR (2017). Regulation of Thalamic and Cortical Network Synchrony by Scn8a. Neuron 93, 1165–1179.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, and Abbott GW (2020). Isoform-Selective KCNA1 Potassium Channel Openers Built from Glycine. J. Pharmacol. Exp. Ther 373, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrossan ZA, Roepke TK, Lewis A, Panaghie G, and Abbott GW (2009). Regulation of the Kv2.1 potassium channel by MinK and MiRP1. J. Membr. Biol 228, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, and Feng G (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH (2019). SCN8A encephalopathy: Mechanisms and models. Epilepsia 60 (Suppl 3), S86–S91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millichap JJ, Park KL, Tsuchida T, Ben-Zeev B, Carmant L, Flamini R, Joshi N, Levisohn PM, Marsh E, Nangia S, et al. (2016). KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet 2, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto H, Tatsukawa T, Shimohata A, Yamagata T, Suzuki T, Amano K, Mazaki E, Raveau M, Ogiwara I, Oba-Asaka A, et al. (2019). Impaired cortico-striatal excitatory transmission triggers epilepsy. Nat. Commun 10, 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki H, Oyama F, Inoue R, Aosaki T, Abe T, Kiyonari H, Kino Y, Kurosawa M, Shimizu J, Ogiwara I, et al. (2014). Singular localization of sodium channel β4 subunit in unmyelinated fibres and its role in the striatum. Nat. Commun 5, 5525. [DOI] [PubMed] [Google Scholar]
- Niday Z, and Tzingounis AV (2018). Potassium Channel Gain of Function in Epilepsy: An Unresolved Paradox. Neuroscientist 24, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niday Z, Hawkins VE, Soh H, Mulkey DK, and Tzingounis AV (2017). Epilepsy-Associated KCNQ2 Channels Regulate Multiple Intrinsic Properties of Layer 2/3 Pyramidal Neurons. J. Neurosci 37, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Tatsukawa T, Yamagata T, Nakayama T, Ata-pour N, Miura E, Mazaki E, Ernst SJ, Cao D, et al. (2018). Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun. Biol 1, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K-S, Mohapatra DP, Misonou H, and Trimmer JS (2006). Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 313, 976–979. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, Masliah E, and Montal M (2000). Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys. J 78, 2878–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quraishi IH, Stern S, Mangan KP, Zhang Y, Ali SR, Mercier MR, Marchetto MC, McLachlan MJ, Jones EM, Gage FH, and Kaczmarek LK (2019). An Epilepsy-Associated KCNT1 Mutation Enhances Excitability of Human iPSC-Derived Neurons by Increasing Slack KNa Currents. J. Neurosci 39, 7438–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quraishi IH, Mercier MR, McClure H, Couture RL, Schwartz ML, Lu-kowski R, Ruth P, and Kaczmarek LK (2020). Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding Slack (KNa1.1) Na+-activated K+ channels. Sci. Rep 10, 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, and Waxman SG (2005). Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J. Physiol 564, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Suzuki Y, Yamamura H, Ohya S, Muraki K, and Imaizumi Y (2017). Molecular mechanisms underlying pimaric acid-induced modulation of voltage-gated K+ channels. J. Pharmacol. Sci 133, 223–231. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Will-sey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, Bernier RA, Catterall WA, Chung WK, Empfield JR, et al. (2018). Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 41, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al. ; Autism Sequencing Consortium; iPSYCH-Broad Consortium (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Johnson CA, and Sabatini BL (2012). Novel recombinant adeno-associated viruses for Cre activated and inactivated transgene expression in neurons. Front. Neural Circuits 6, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W, Kweon H, Kang R, Kim D, Kim K, Kang M, Kim SY, Hwang SN, Kim JY, Yang E, et al. (2019). Scn2a Haploinsufficiency in Mice Suppresses Hippocampal Neuronal Excitability, Excitatory Synaptic Drive, and Long-Term Potentiation, and Spatial Learning and Memory. Front. Mol. Neurosci 12, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, von Melchner H, Wurst W, Hicks G, Nord AS, Cox T, Young SG, Ruiz P, Soriano P, Tessier-Lavigne M, et al. ; International Gene Trap Consortium (2004). A public gene trap resource for mouse functional genomics. Nat. Genet 36, 543–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowball A, Chabrol E, Wykes RC, Shekh-Ahmad T, Cornford JH, Lieb A, Hughes MP, Massaro G, Rahim AA, Hashemi KS, et al. (2019). Epilepsy Gene Therapy Using an Engineered Potassium Channel. J. Neurosci 39, 3159–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, and Tzingounis AV (2010). The specific slow afterhyperpolarization inhibitor UCL2077 is a subtype-selective blocker of the epilepsy associated KCNQ channels. Mol. Pharmacol 78, 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Park S, Ryan K, Springer K, Maheshwari A, and Tzingounis AV (2018). Deletion of KCNQ2/3 potassium channels from PV+ interneurons leads to homeostatic potentiation of excitatory transmission. eLife 7, e38617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ Jr., Clarkson RL, Sanders SJ, and Bender KJ (2019). The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 103, 673–685.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt PWE, Alexander RPD, Ben-Shalom R, Sahagun A, Kyoung H, Keeshen CM, Sanders SJ, and Bender KJ (2021). Paradoxical hyperexcitability from NaV1.2 sodium channel loss in neocortical pyramidal cells. Cell Rep. 36, 109483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa G, Schaft J, van der Hoeven F, Glaser S, Anastassiadis K, Zhang Y, Hermann T, Stremmel W, and Stewart AF (2004). A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles. Genesis 38, 151–158. [DOI] [PubMed] [Google Scholar]
- Tian C, Wang K, Ke W, Guo H, and Shu Y (2014). Molecular identity of axonal sodium channels in human cortical pyramidal cells. Front. Cell. Neurosci 8, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel N, Diochot S, Lauritzen I, Barhanin J, Lazdunski M, and Borsotto M (2000). M-type KCNQ2-KCNQ3 potassium channels are modulated by the KCNE2 subunit. FEBS Lett. 480, 137–141. [DOI] [PubMed] [Google Scholar]
- Torres-García ME, Solis O, Patricio A, Rodríguez-Moreno A, Camacho-Abrego I, Limón ID, and Flores G (2012). Dendritic morphology changes in neurons from the prefrontal cortex, hippocampus and nucleus accumbens in rats after lesion of the thalamic reticular nucleus. Neuroscience 223, 429–438. [DOI] [PubMed] [Google Scholar]
- Tóth K, Hofer KT, Kandrács Á, Entz L, Bagó A, Erőss L, Jordán Z, Nagy G, Sólyom A, Fabó D, et al. (2018). Hyperexcitability of the network contributes to synchronization processes in the human epileptic neocortex. J. Physiol 596, 317–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosclair K, Dhaibar HA, Gautier NM, Mishra V, and Glasscock E (2020). Neuron-specific Kv1.1 deficiency is sufficient to cause epilepsy, premature death, and cardiorespiratory dysregulation. Neurobiol. Dis 137, 104759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega AV, Henry DL, and Matthews G (2008). Reduced expression of Na(v) 1.6 sodium channels and compensation by Na(v)1.2 channels in mice heterozygous for a null mutation in Scn8a. Neurosci. Lett 442, 69–73. [DOI] [PubMed] [Google Scholar]
- Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, et al. (2016a). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun 7, 13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X-C, Wang S, Zhang M, Gao F, Yin C, Li H, Zhang Y, Hu S-J, and Duan J-H (2016b). A-Dendrotoxin-sensitive Kv1 channels contribute to conduction failure of polymodal nociceptive C-fibers from rat coccygeal nerve. J. Neurophysiol 115, 947–957. [DOI] [PubMed] [Google Scholar]
- Wang J, Ou SW, and Wang YJ (2017). Distribution and function of voltage-gated sodium channels in the nervous system. Channels (Austin) 11, 534–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, He X, Meng H, Li Y, Dmitriev P, Tian F, Page JC, Lu QR, and He Z (2020). Robust Myelination of Regenerated Axons Induced by Combined Manipulations of GPR17 and Microglia. Neuron 108, 876–886.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Noebels JL, and Catterall WA (1992). Elevated expression of type II Na+ channels in hypomyelinated axons of shiverer mouse brain. J. Neurosci 12, 2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, Lesca G, Ville D, Milh M, Villard L, et al. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336. [DOI] [PubMed] [Google Scholar]
- Wolff M, Brunklaus A, and Zuberi SM (2019). Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia 60 (Suppl 3), S59–S67. [DOI] [PubMed] [Google Scholar]
- Workman AD, Charvet CJ, Clancy B, Darlington RB, and Finlay BL (2013). Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci 33, 7368–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Castle NA, and Pardo LA (2009). Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov 8, 982–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S, and Kullmann DM (2012). Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci. Transl. Med 4, 161ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata T, Ogiwara I, Mazaki E, Yanagawa Y, and Yamakawa K (2017). Nav1.2 is expressed in caudal ganglionic eminence-derived disinhibitory interneurons: Mutually exclusive distributions of Nav1.1 and Nav1.2. Bio-chem. Biophys. Res. Commun 491, 1070–1076. [DOI] [PubMed] [Google Scholar]
- Yang J-J, Tian Y-T, Yang Z, and Zhang T (2010). Effect of melamine on potassium currents in rat hippocampal CA1 neurons. Toxicol. In Vitro 24, 397–403. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed RNA sequencing data are publicly available at GEO through accession number GEO: GSE179818.
Custom scripts are publicly available at https://zenodo.org/record/5098353#.YO48C_lKg4s. DOI 10.5281/zenodo.5098353.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.