

Abstract
Objective
To determine the efficacy and safety of the treatment with prolonged-release 4-aminopyridine (fampridine) and acetazolamide for patients with episodic ataxia type 2 (EA2), patients with EA2 were treated with a random sequence of fampridine, acetazolamide, and placebo in a 3-period crossover trial.
Methods
A total of 30 patients with EA2 (8 female; aged 20–71 years; 18 genetically confirmed, 4 with a positive family history, 8 with the clinical diagnosis) were enrolled in this phase III, randomized, double-blind, placebo-controlled, 3-period crossover trial. Each period lasted 12 weeks with a 4-week washout period. Each patient received a random sequence of 20 mg/d fampridine, 750 mg/d acetazolamide, and placebo. The primary end point was the number of attacks during the last 30 days within the 12-week treatment period. Participants, caregivers, and those assessing the outcomes were blinded to the intervention.
Results
Compared with placebo, fampridine reduced the number of attacks to 63% (95% CI 54%–74%) and acetazolamide to 52% (95% CI 46%–60%). A total of 39 (26.5%) adverse events were observed under treatment with fampridine (mostly tingling paresthesia and fatigue), 66 (44.9%) happened under acetazolamide (mostly taste disturbance and gastrointestinal complaints), and 42 (28.6%) under placebo (mostly gastrointestinal complaints).
Conclusion
Both fampridine and acetazolamide significantly reduce the number of attacks in patients with EA2 and related EA in comparison to placebo. Fampridine 10 mg twice daily had fewer side effects than acetazolamide 250 mg 3 times daily. The trial was registered with DRKS.de (DRKS00005258) and EudraCT (2013-000107-17). This study was supported by the Federal Ministry of Education and Research (BMBF) (grant number 01EO0901). Fampridine (study medication) was provided by Biogen Idec.
Classification of Evidence
Class II evidence.
Episodic ataxia type 2 (EA2) is characterized by paroxysmal attacks, defined by ataxia, vertigo, and dysarthria.1–3 EA2 is an autosomal dominant hereditary disorder caused by heterozygous mutations of the CACNA1A gene.4,5
So far, there are 2 recommended treatment options: acetazolamide and 4-aminopyridine (4-AP). The carbonic anhydrase inhibitor acetazolamide was serendipitously discovered to prevent attacks in patients with EA2, but its efficacy has never been proven in a randomized controlled trial.6 Clinical experience has shown that treatment with acetazolamide 250–1,000 mg/d prevents or attenuates the attacks.7 However, many patients discontinue the treatment due to adverse events (AEs) or loss of response during the long course of the treatment.2,7,8
The treatment option with the potassium channel blocker 4-AP was first described in an observational study.9 These findings were confirmed in a randomized, double-blind, placebo-controlled, crossover trial.10 Ten patients with EA2 (7 with a confirmed CACNA1A mutation) were treated with 4-AP (3 × 5 mg/d). The median monthly attack frequency decreased significantly under 4-AP compared with placebo (1.65 under 4-AP; 6.5 under placebo).10 4-AP was well tolerated.
Thus, the American Academy of Neurology recommended treatment with 4-AP 15 mg/d for patients with EA2.11 This formulation of 4-AP has no approval for any indication, in contrast to the prolonged-release form of 4-AP (fampridine, Fampyra, Biogen Idec, Ltd. Maidenhead, Berkshire, SL6 4AY, UK), which is approved for the symptomatic treatment of gait disorders in multiple sclerosis.12,13
However, randomized, placebo-controlled trials investigating the efficacy of acetazolamide and fampridine are lacking. Therefore, we performed a randomized, placebo-controlled, 3-period crossover trial for treatment of EA2 with fampridine and acetazolamide.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
Ethical approval was obtained from the local ethics committee (application number 163-13; May 15, 2013). The trial adhered to the ethical principles of the Declaration of Helsinki and Good Clinical Practice guidelines. Written informed consent was obtained from each patient. The trial was registered with DRKS.de (DRKS00005258) and EudraCT (2013-000107-17).
Study Design and Patients
Episodic Ataxia Type 2 TREAtment Trial (EAT2TREAT, fampridine and acetazolamide in EA2 and related familial episodic ataxias) was a randomized, double-blind, placebo-controlled, single-site, phase III, 3-period crossover trial. Because of the low prevalence of EA2, a crossover design was chosen. Based on a direct comparison of all treatments conducted on each single patient, a lower sample size results from the lower variability observed within a patient. Patients were recruited between July 26, 2013, and November 5, 2015.
Eligible patients were aged ≥18 years, had genetically confirmed EA2 or related familial episodic ataxias, and were able to follow the study instructions and likely to complete all required study visits. Women of childbearing potential had to use an acceptable method of contraception before randomization and throughout study participation. Exclusion criteria were low body weight ≤40 kg; female subjects who were pregnant, breastfeeding, or contemplating pregnancy during study duration; subjects, who were taking organic cation transporter 2 inhibitors; known hypersensitivity to APs and/or acetazolamide/sulfonamides; acute myocardial infarction (within the last 3 months); prolonged QTc interval >500 ms; atrial fibrillation; AV block ≥ II; unstable angina pectoris; severe heart failure (NYHA IV); arterial hypertension (grade III according to the guidelines of the German Association for Cardiology 2008); stroke within the last 3 months; history of seizures or known epilepsy; asthma (severity ≥ III); obstructive pulmonary disease; hepatic insufficiency (defined as aspartate aminotransferase/alanine aminotransferase/total bilirubin > 3× upper limit); mild or severe renal insufficiency (creatinine clearance ≤80 mL/min); adrenocortical insufficiency; not adjusted thyroid disease; acute gastric/intestinal ulcer; hyperchloremic acidosis; decreased sodium and/or potassium blood serum level; chronic angle-closure glaucoma; hypercalcemia; gout; known sickle cell anemia; diabetes mellitus type I/II; acute, severe disease; subject unable to understand the extent, meaning and consequences of the trial and to follow the study schedule; subject previously participated in this clinical trial or was treated with another investigational drug 30 days before study participation.
Randomization and Masking
There are 6 different sequences (sequences in detail: fampridine followed by acetazolamide followed by placebo; fampridine followed by placebo followed by acetazolamide; acetazolamide followed by fampridine followed by placebo; acetazolamide followed by placebo followed by fampridine; placebo followed by fampridine followed by acetazolamide; and placebo followed by acetazolamide followed by fampridine) containing each of the 3 treatment options (fampridine, acetazolamide, and placebo). Each of the 30 patients was equally randomized to 1 of these 6 possible treatment sequences. The randomization was web based using our in-house tool Randoulette (wwwapp.ibe.med.uni-muenchen.de/randoulette/). The study center received the packaged study medication, which was marked with a number. Randoulette informed the doctor which package should be handed out to the patient. Masking used the double dummy principle (ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf). The assignment of a patient to the treatment sequence is double blind: neither the treating doctor nor the patients know the sequence. The packing of medication was performed by the clinical pharmacy at the medical center of the Ludwig Maximilians University, Munich.
Study Procedures
Patients consecutively received in a 3-period crossover design fampridine 10 mg twice daily, acetazolamide 250 mg 3 times daily, and placebo for 12 weeks each. The first and second treatment periods were followed by a 4-week washout period to avoid carryover effects. A follow-up assessment was performed for each patient 4 weeks after the last study medication intake. Based on Strupp et al.,10 the durations of 12 weeks for each treatment period and 4 weeks for the washout periods were chosen.
Study visits took place at the beginning of a treatment period, 4 weeks after the beginning of a treatment period, at the end of a 12-week treatment period, and 4 weeks after the last intake of study medication. In total, 10 study visits and a study duration of 48 weeks were planned for each patient. A screening visit could be performed within 4 weeks before randomization. Screening and the first study visit were conducted at the same time point. Patients under treatment with 4-AP, fampridine, or acetazolamide before study participation had to stop the medication at least 1 week before randomization. As per the protocol, patients were allowed to discontinue single phases, for example, due to side effects, and after a 4-week washout period continue with the next treatment period.
At screening, patients underwent a neurologic and neuro-ophthalmologic examination, an electrocardiogram was performed, in particular to determine the QT and QTc intervals, and laboratory examinations of venous blood, including a pregnancy test in women of childbearing potential, and urine were conducted.
Patients received the drugs in standardized packages with written instructions on how to take the medication at the beginning of each treatment period. The investigators monitored drug compliance and adherence at each visit.
Patients documented the frequency, duration, and severity of the episodic attacks in structured patients' diaries. At each study visit, the investigators monitored the patients' diaries. Patients were instructed to inform the investigators about any side effects.
Laboratory examinations of venous blood and urine were performed at the beginning of each treatment period, 4 weeks after the beginning, and at the end of the treatment period. A pregnancy test using a blood sample had to be performed for participating women of childbearing potential at screening, at the beginning of each treatment period, and 4 weeks after beginning. The participants themselves conducted pregnancy tests using a urine sample 8 weeks after beginning a treatment period. The results were documented in a telephone interview. Furthermore, pregnancy tests using a urine sample were performed at the end of each treatment period.
At the beginning of each treatment period, at the end of each treatment period, and at follow-up, the Scale for the Assessment and Rating of Ataxia (SARA) and gait analysis using a pressure-sensitive carpet (GAITRite) were performed.
Quality of life was measured by using the Vestibular Disorders Activities of Daily Living Scale (VDADL) and EuroQol in five dimensions with five-level scale (EQ-5D-5L) at beginning of each treatment period, 4 weeks after the beginning of the treatment, at the end of the treatment periods, and at the follow-up.
Outcome Measures
The primary outcome measure was the number of attacks during the last 30 days within a 12-week treatment period based on the patients' diaries.
Secondary outcome measures were the median duration and strength of attacks during the last 30 days within a 12-week treatment period. Other secondary outcome measures were measured as changes after 12 weeks of treatment compared with the beginning of a 12-week treatment period and 4 weeks after the last administration of study medication: changes in the coefficient of variability of maximal walking speed (relative change [log scale])14; changes in the quality of life (measured by the questionnaires EQ-5D-5L and VDADL); and changes in the SARA score.
AEs and serious AEs (SAEs) were documented between the beginning of the trial and the end of the follow-up period, regardless of the relationship to the study medication.
Statistical Analysis
The effect assumptions for the sample size calculation are taken from Strupp et al.10 The results reported in this article allow an assumption of a 45% reduction of attack rates under treatment. It also provides data to set the standard error of the log-transformed attack rate reduction to 1.2. To adjust for the multiple testing (placebo vs fampridine and placebo vs acetazolamide), we applied a Bonferroni correction and used a 2.5% 2-sided significance level for each of the tests. To detect the effect of 45% (log(0.45) = −0.8) between placebo and treatment (given the corresponding standard error of 1.2) on the 2.5% level with power of 80%, a total of 24 patients were needed, who were distributed over the 6 sequences. To adjust for a 20% dropout rate, 5 patients per group (a total of 30 patients) were recruited.
The main analysis used a random effects Poisson regression model on the total number of attacks. Each patient had an individual random effect. Three models were studied: a model with period by treatment interaction, a model in which period and treatment are the main effects, and a model with treatment as the single main effect. All 3 models were compared by a likelihood ratio test to see whether treatment effects, period effects, or crossover effects were present.
The analysis of secondary end points also used a linear random-effects model to assess effects in score-based data following the same steps as described for the primary end point.
Descriptive statistics used percentages in case of categorical variables or in case of numeric variables mean, SD, and range. No subgroup analyses were performed.
The analysis used the statistical software R using the package lme4 for the primary analysis (Version 3.5.1, the R Foundation for Statistical Computing).
Data Availability
Data cannot be shared publicly because participants did not explicitly consent to the sharing of their data as per the European Union's General Data Protection Regulation and the corresponding German privacy laws. Data are available through the Research Ethics Board of the Ludwig Maximilian University, Munich, for researchers who meet the criteria for access to confidential data. Please address your request to ethikkommission@med.uni-muenchen.de.
Results
Thirty-six patients were screened for eligibility, 30 of whom were enrolled and randomly and equally assigned to 1 of the 6 treatment sequences (figure 1). The main reason for patients not being enrolled was the inability to attend visits for administrative reasons. Patients were recruited between July 26, 2013, and November 5, 2015.
Figure 1. Trial Profile.

AE = adverse event; SAE = serious AE.
Of the 30 patients, 22 patients were male (73.3%) and 8 female (26.7%). The mean age of the patient cohort was 43.7 ± 13.4 years (SD; range 20–71 years). Table 1 contains information on baseline characteristics of the patients.
Table 1.
Baseline Characteristics
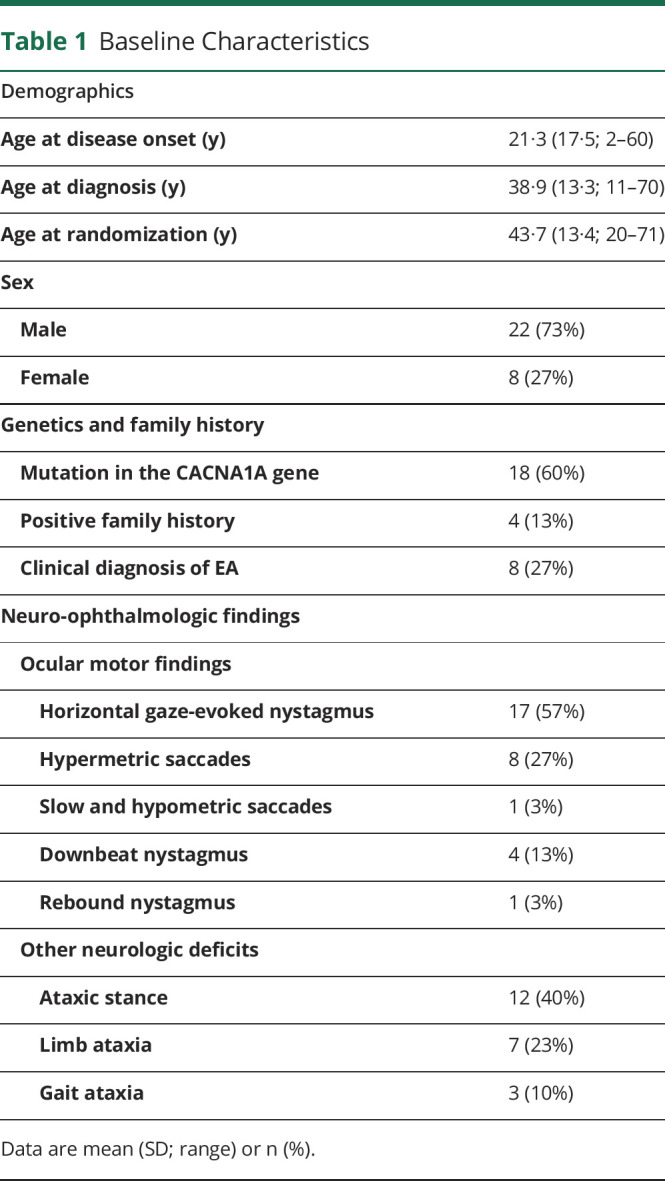
Of the 30 patients, 18 had a genetically defined CACNA1A mutation, 7 tested negative for a CACNA1A mutation, whereas 5 were not tested. Of the latter 2 groups, 4 patients presented with a positive family history and 8 with the typical clinical diagnosis (table 1). Five so far undescribed, novel CACNA1A mutations were identified: 1. c.4095_4096delTG; p.fsX heterozygous, 2. c.2192A>C; p.Glu731Ala heterozygous, 3. c.533T>C; p.Leu178Pro heterozygous, 4. c.5419-1G>A heterozygous, and 5. c.539+1 G>T heterozygous.
Patients affected by EA2 often exhibit central ocular motor disturbances. In this study, at baseline, 17 (56.7%) patients showed horizontal gaze-evoked nystagmus, 4 showed downbeat nystagmus (13.3%), and 9 pathologic saccades (30.0%) (table 1). Further neurologic disturbances were observed: 12 patients presented with ataxic stance (40.0%), 3 with gait ataxia (10.0%), and 7 with limb ataxia (23.0%) (table 1).
Twenty-two of the participating patients finished all of the 3 treatment periods completely. Eight patients discontinued treatment: 2 of them completed 2 of 3 treatment periods, 3 only 1 period, and 3 completed none of the treatment periods (figure 1). Two patients discontinued during fampridine treatment due to noncompliance. In contrast, 5 patients discontinued during acetazolamide treatment, 1 due to an SAE, 2 due to AEs, and 2 due to noncompliance. Of the theoretical 90 measurements (30 patients × 3 measurements = 90), 64 (71.1%) data points were actually collected. For further information, see figure e-1 (links.lww.com/CPJ/A233). At baseline, the median number of attacks per month was 11.0 (IQR 4.0–27.5).
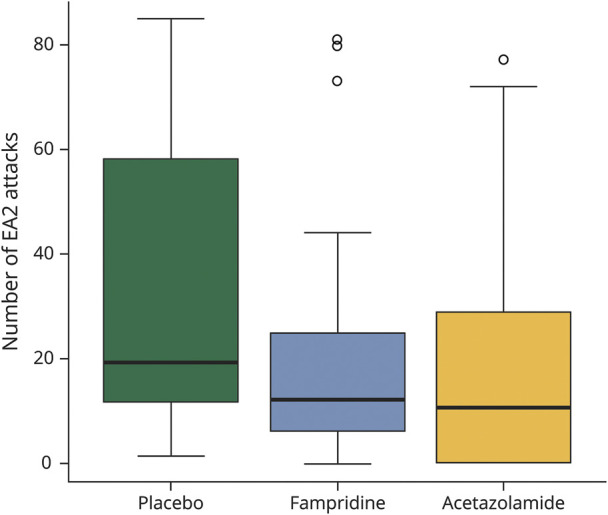
The primary outcome measurement of this study was defined as the number of attacks within the last 30 days of each treatment period, lasting 12 weeks. In the placebo treatment period, patients had a median of 19.0 (IQR 11.5–58.0) attacks. In contrast, in the fampridine treatment period, subjects had a median of 12.0 (IQR 6.0–25.0) attacks. Finally, during the acetazolamide treatment period, a median of 10.5 (IQR 0–28.0) attacks was observed (figure 2).
Figure 2. Number of EA2 Attacks Within the Last 30 Days of a 12-Week Treatment Period (Primary Efficacy Outcome Measure).
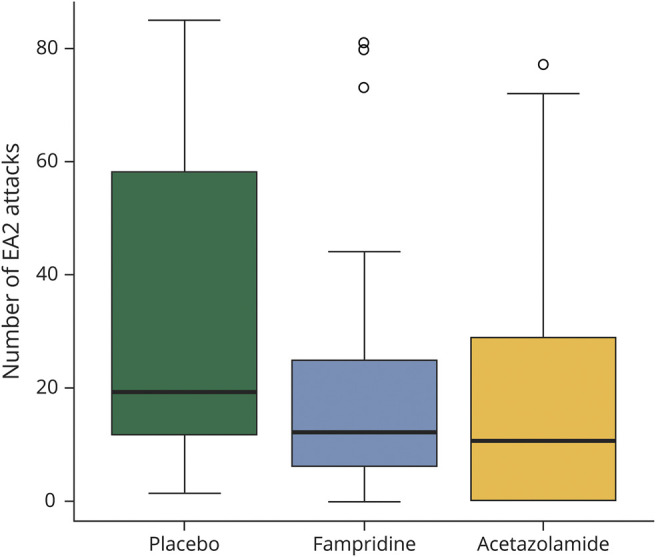
The number of attacks was determined according to the patients' diaries. Boxplots for outcome measures during the placebo, fampridine, and acetazolamide treatment phases (box-whisker plot with 25% and 75% percentiles [borders of the boxes], the median [line], whiskers [extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box], and outliers).
The absolute reduction of attacks (mean and 95% CI) was as follows: Fampridine in comparison to placebo caused – 8.00 (95% CI −17.91 to 1.91), acetazolamide in comparison to placebo −16.11 (95% CI −25.17 to −7.04), and fampridine in comparison to acetazolamide 8.39 (95% CI 3.39–13.39). Fampridine treatment in comparison to placebo led to a median reduction of 3 attacks (IQR −11.0 to 1.5) and acetazolamide to a median reduction of 9 attacks compared with placebo (IQR −20.5 to −4) (figure 3). Figure e-2 (links.lww.com/CPJ/A234) shows the number of EA2 attacks in correlation with the 6 different treatment sequences.
Figure 3. Changes in Number of EA2 Attacks Comparing Fampridine or Acetazolamide Treatment With Placebo.
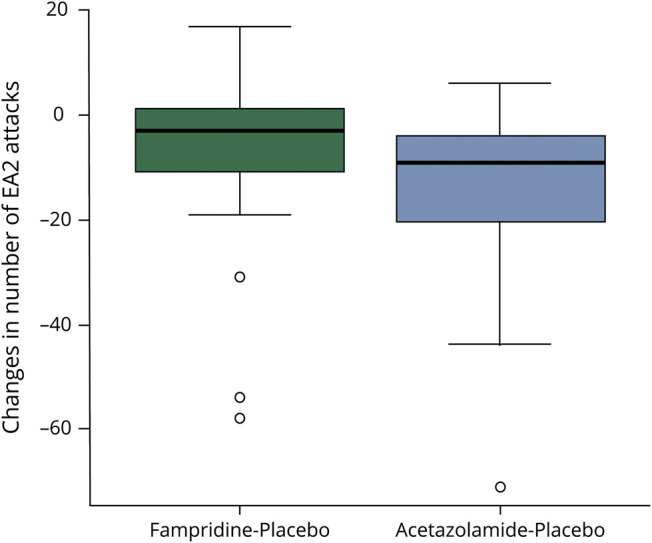
Boxplots of changes in number of EA2 attacks comparing fampridine or acetazolamide treatment with placebo (box-whisker plot with 25% and 75% percentiles [borders of the boxes], the median [line], whiskers [extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box], and outliers).
The Poisson mixed-effects model allowed for quantification of the treatment effect and the evaluation of potential period effects (see Statistical analysis). There is a substantial effect of both treatments compared with placebo, quantified as a factor, which reduces the mean attack rate. Fampridine reduced the number of attacks to 63% (95% CI 54%–74%) and acetazolamide to 52% (95% CI 46%–60%).
The attack duration, the attack severity, the SARA, the coefficient of variability of maximal walking speed, and the quality of life measured by VDADL and EQ-5D-5L were analyzed as secondary outcome measures. In conclusion, there was no evidence for any effect of fampridine or acetazolamide treatment in comparison to placebo with regard to these secondary outcome measures.
Safety and Tolerability
In total, 147 AEs were observed during the trial (table 2). Thirty-nine (26.5%) AEs were observed under fampridine treatment, 66 (44.9%) AEs under acetazolamide, and 42 (28.6%) AEs under placebo. Eight SAEs were observed during the trial (table 2). Five SAEs were observed under treatment with fampridine, 1 SAE under acetazolamide, and 2 SAEs under placebo. One SAE under acetazolamide treatment—kidney stones—led to the patient dropping out. One SAE under fampridine treatment—overdose of study medication—led to the patient dropping out at the instigation of the principal investigator of the study.
Table 2.
AEs and SAEs

Classification of Evidence
The EAT2TREAT study provides Class II evidence that administration of fampridine 20 mg/d and acetazolamide 750 mg/d reduces the number of attacks in patients with EA2. Compared with placebo, fampridine reduced the number of attacks to 63% (95% CI 54%–74%) and acetazolamide to 52% (95% CI 46%–60%).
TAKE-HOME POINTS
→ Both the prolonged-release 4-AP (fampridine, 10 mg twice daily) and acetazolamide (250 mg tid) significantly reduced the number of attacks in patients with EA2 in comparison to placebo.
→ Fampridine had fewer side effects than acetazolamide.
→ Based on these findings, in a 3-arm trial, the effects of 4-AP vs acetazolamide vs the combination of both drugs could be evaluated in the future.
Discussion
The EAT2TREAT trial investigates the efficacy and safety of fampridine and acetazolamide for the treatment of EA2 patients. The results indicate the efficacy of both fampridine and acetazolamide: fampridine reduced the number of EA2 attacks to 63% (95% CI 54%–74%) and acetazolamide to 52% (95% CI 46%–60%).
So far, treatment of patients with EA2 with acetazolamide is based on a serendipitously found effect and clinical experience, however, without proven efficacy based on a randomized controlled trial.6 The results of the EAT2TREAT trial presented here prove the efficacy of acetazolamide for the treatment of patients with EA2 in a randomized controlled setting. The mechanism by which acetazolamide prevents EA2 attacks is likely to be a change in the intracellular pH and subsequently in the transmembrane potential.8
The efficacy of 4-AP in treating patients with EA2 and related familial episodic ataxias was shown in a randomized controlled trial.10 In an observational study, the prolonged-release form of 4-AP (fampridine) was reported to prevent attacks in 2 patients with genetically confirmed EA2.15 However, fampridine, which is approved for the treatment of gait disorders in multiple sclerosis, had not yet been tested in a randomized controlled trial in patients with EA2.12,13 The EAT2TREAT trial also proves the efficacy of fampridine at preventing EA2 attacks. The presumed mode of action of 4-AP in EA2 is most likely by increasing the release of the inhibitory transmitter gamma-aminobutyric acid (GABA) and by increasing the excitability of Purkinje cells by prolonging the duration of action potentials through the blockade of potassium channels, mainly Kv1.5.7,16
Actually, acetazolamide and fampridine show a comparable effect in reducing the number of EA2 attacks (fampridine reduced the number of EA2 attacks to 63% [95% CI 54%–74%] and acetazolamide to 52% [95% CI 46%–60%]).
In terms of safety, most AEs were observed under acetazolamide treatment (66 [44.9%]), whereas the number of AEs observed under fampridine treatment (39 [26.5%]) was similar to placebo (42 [28.6%]). The most common AEs under acetazolamide treatment were tingling paresthesia, taste disturbance, and gastrointestinal complaints. The most common AEs under fampridine treatment were fatigue, gastrointestinal complaints, and tingling paresthesia. The described AEs correspond to the known side effects of acetazolamide and fampridine. Under fampridine, none of the patients discontinued treatment due to an AE. Two patients discontinued treatment due to AEs under acetazolamide.
Eight SAEs were observed during the trial: 5 SAEs occurred under fampridine treatment, 1 SAE under acetazolamide, and 2 SAEs under placebo. The SAE under acetazolamide treatment—kidney stones, known as a side effect of acetazolamide—led to a patient dropping out. One SAE under fampridine treatment—overdose of the study medication by the patient himself—led to the patient dropping out, based on a decision by the principal investigator of the study. Three episodes of kidney stones in 2 patients under fampridine were reported. Kidney stones have not been described as a side effect of fampridine so far. The patient who reported kidney stones twice during the trial had been on long-term treatment with acetazolamide before his trial participation when he received fampridine. In fact, kidney stones had already been a well-known problem in the patient's history for many years, and the occurrence of kidney stones under fampridine treatment is most likely not related to the study medication. The second patient with kidney stones under fampridine was treated with acetazolamide in the preceding treatment period. This may be seen as a carryover effect from the preceding treatment period. One patient had a tremor while being treated with fampridine, which is a known side effect. Therefore, no unknown side effects related to the study medication were observed during the trial.
In conclusion, based on the results of the EAT2TREAT trial, both fampridine and acetazolamide are effective treatments for patients with EA2 in comparison to placebo. During the treatment withacetazolamide, more AEs were observed than during the treatment with fampridine.
The current study has the following limitation, which should be taken into consideration: Of the 30 patients, 18 patients had a genetically confirmed CACNA1A mutation, and 4 patients presented with a family history of episodic ataxia. Eight patients had neither a CACNA1A mutation nor a family history of episodic ataxia. However, they presented with the clinical diagnosis of episodic ataxia and were therefore included in the trial in accordance with the inclusion criteria. Although EA2 is an autosomal dominant hereditary disorder caused by heterozygous mutations of the calcium channel gene CACNA1A on chromosome 19p13,5 in approximately 30%–50% of patients with typical clinical features of EA2, no mutation of the CACNA1A gene can be detected.3,17 Even the lack of family history does not necessarily exclude the diagnosis of EA2, since sporadic cases (spontaneous mutations) have been observed.1 The techniques of whole-exome and whole-genome sequencing have increased the number of genetic variants.2 Furthermore, next-generation sequencing has uncovered mutations in genes not previously associated with episodic ataxia in patients with a typical clinical presentation.2,18,19
Future studies could directly compare the efficacy and long-term effects of fampridine, acetazolamide, and—due to the different mechanism of action—the combination of agents. In addition, because of the typical disease onset between infancy and early childhood, a trial investigating especially the safety profile of the drugs in children should be performed. Furthermore, a combination of a daily medication with fampridine or acetazolamide and a rescue medication with non–prolonged-release 4-AP, especially for situations with known triggers of the EA2 attacks, could be investigated.
This randomized, placebo-controlled, 3-period crossover trial indicates the efficacy of both fampridine 20 mg/d and acetazolamide 750 mg/d in the treatment of patients with EA2 and related familial EA in comparison to placebo. In terms of side effects, patients reported fewer side effects under fampiridine than under acetazolamide in the dosages used in this study.
Acknowledgment
The authors thank Katie Göttlinger for copyediting the manuscript, Ingrid Berger for her assistance in the project management, and Prof. Dr. Hans-Helge Müller for his protocol contributions regarding the design-specific biostatistical concept. They also thank the patients for their participation and the investigators who took part in this trial.
Appendix. Authors
Footnotes
Class of Evidence: NPub.org/coe
Study Funding
This study was an investigator-initiated trial supported by the Federal Ministry of Education and Research (BMBF) together with the foundation of the German Center for Vertigo and Balance Disorders (DSGZ) (grant number 01EO0901). Fampridine (study medication) was provided by Biogen Idec. Biogen Idec had no role in the study design, data collection, data analysis, data interpretation, writing of the report, or the decision to submit the manuscript for publication.
Disclosure
C. Muth, J. Teufel, L. Schöls, M. Synofzik, C. Franke, D. Timmann, and U. Mansmann declare no conflict of interest. M. Strupp is Joint Chief Editor of the Journal of Neurology, Editor in Chief of Frontiers of Neuro-otology, and Section Editor of F1000. He has received speaker's honoraria from Abbott, Actelion, Auris Medical, Biogen, Eisai, Grünenthal, GSK, Henning Pharma, Interacoustics, J&J, MSD, Otometrics, Pierre-Fabre, Teva, and UCB. He is a shareholder of IntraBio. He is the distributor of M glasses and the “positional vertigo app.” He acts as a consultant for Abbott, Actelion, AurisMedical, Heel, IntraBio, and Sensorion. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
References
- 1.Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007;130:2484–2493. [DOI] [PubMed] [Google Scholar]
- 2.Jen JC, Wan J. Episodic ataxias. Handb Clin Neurol 2018;155:205–215. [DOI] [PubMed] [Google Scholar]
- 3.Jen J, Kim GW, Baloh RW. Clinical spectrum of episodic ataxia type 2. Neurology 2004;62:17–22. [DOI] [PubMed] [Google Scholar]
- 4.Denier C, Ducros A, Vahedi K, et al. High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2. Neurology 1999;52:1816. [DOI] [PubMed] [Google Scholar]
- 5.Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca 2 channel gene CACNL1A4. Cell 1996;87:543–552. [DOI] [PubMed] [Google Scholar]
- 6.Griggs RC, Moxley RT, Lafrance RA, McQuillen J. Hereditary paroxysmal ataxia. Neurology 1978;28:1259–1264. [DOI] [PubMed] [Google Scholar]
- 7.Strupp M, Zwergal A, Brandt T. Episodic ataxia type 2. Neurotherapeutics 2007;4:267–273. [DOI] [PubMed] [Google Scholar]
- 8.Zasorin NL, Baloh RW, Myers LB. Acetazolamide‐responsive episodic ataxia syndrome. Neurology 1983;33:1212–1214. [DOI] [PubMed] [Google Scholar]
- 9.Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology 2004;62:1623–1625. [DOI] [PubMed] [Google Scholar]
- 10.Strupp M, Kalla R, Claassen J, et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011;77:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zesiewicz TA, Wilmot G, Han Kuo S, et al. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia. Neurology 2018;90:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodman AD, Brown TR, Krupp LB, et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet 2009;373:732–738. [DOI] [PubMed] [Google Scholar]
- 13.Goodman AD, Brown TR, Edwards KR, et al. A phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann Neurol 2010;68:494–502. [DOI] [PubMed] [Google Scholar]
- 14.Schniepp R, Wuehr M, Neuhaeusser M, et al. Locomotion speed determines gait variability in cerebellar ataxia and vestibular failure. Mov Disord 2012;27:125–131. [DOI] [PubMed] [Google Scholar]
- 15.Claassen J, Teufel J, Kalla R, Spiegel R, Strupp M. Effects of dalfampridine on attacks in patients with episodic ataxia type 2: an observational study. J Neurol 2013;260:668–669. [DOI] [PubMed] [Google Scholar]
- 16.Gao Z, Todorov B, Barrett CF, et al. Cerebellar ataxia by enhanced CaV2.1 currents is alleviated by Ca2+-dependent K+-Channel activators in Cacna1aS218L mutant mice. J Neurosci 2012;32:15533–15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirose H, Arayama T, Takita J, Igarashi T, Hayashi Y, Nagao Y. A family of episodic ataxia type 2: no evidence of genetic linkage to the CACNA1A gene. Int J Mol Med 2003;11:187–189. [PubMed] [Google Scholar]
- 18.Gardiner AR, Bhatia KP, Stamelou M, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology 2012;79:2115–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leach EL, van Karnebeek CDM, Townsend KN, Tarailo-Graovac M, Hukin J, Gibson WT. Episodic ataxia associated with a de novo SCN2A mutation. Eur J Paediatr Neurol 2016;20:772–776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data cannot be shared publicly because participants did not explicitly consent to the sharing of their data as per the European Union's General Data Protection Regulation and the corresponding German privacy laws. Data are available through the Research Ethics Board of the Ludwig Maximilian University, Munich, for researchers who meet the criteria for access to confidential data. Please address your request to ethikkommission@med.uni-muenchen.de.