

Abstract
Background and Objectives
To investigate the total circular RNA (circRNA) profile in patients with relapsing-remitting multiple sclerosis (RRMS) and healthy controls (HCs).
Methods
Hybridization microarray was used to define the circRNA profile in peripheral blood mononuclear cells (PBMCs) from 20 untreated patients with RRMS (10 in relapse and 10 in remission) and 10 HCs. We analyzed close to 14,000 individual circRNAs per sample. The discovery set data were validated using quantitative reverse transcription-PCR with an independent cohort of 47 patients with RRMS (19 in relapse and 28 in remission) and 27 HCs.
Results
Microarray analysis revealed 914 transcripts to be differentially expressed between patients with RRMS in relapse and HCs (p < 0.05). We validated 3 circRNAs from 5 showing highest levels of differential expression in the RRMS relapse vs HC group: hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA_104361. Their expression was significantly increased during relapse in RRMS (p = 0.0002, FC = 2.9; p = 0.01, FC = 1.6; and p = 0.001, FC = 1.5, respectively) and in patients showing gadolinium enhancement on brain MRI (hsa_circRNA_101348, p = 0.0039, FC = 2.4; hsa_circRNA_104361, p = 0.029, FC = 1.7). Bioinformatic analysis revealed 15 microRNAs interacting with these circRNAs in a complementary manner and led to the discovery and validation of 3 protein-coding RNAs upregulated in patients with RRMS during relapse. Two of these, AK2 and IKZF3, have previously been implicated in B-cell function.
Discussion
circRNAs display a distinct profile in PBMCs from patients with RRMS, and our results may implicate circRNA in the known disturbed B-cell activity in RRMS and thus represent a novel biomarker for monitoring relapse activity.
Multiple sclerosis (MS) is a chronic neurologic condition in which the CNS displays inflammation and demyelination.1,2 Although the etiology of MS is still not fully understood, accumulating evidence purports it to be a multifactorial entity with a significant autoimmune component.3
The working model of the pathogenesis of MS implicates interplay between genetic, environmental, and epigenetic factors.4 Results from genome-wide association studies in MS have led to the discovery of more than 200 nonmajor histocompatibility complex genes potentially involved in predisposition to the disease but with a modest odds ratio.5 More recently, attention has turned to epigenetic mechanisms which might contribute to mechanisms in MS. Epigenetic mechanisms identified for the pathogenesis of MS are DNA methylation, histone modification, and microRNA (miRNA) alterations. Because human genome sequencing has progressed, noncoding RNA (ncRNA) has received increasing attention as an important epigenetic component. Several classes of ncRNAs are known, but a particular role in epigenetic modification is attributed to miRNA. These small RNA molecules are considered to be one of the most important posttranscriptional regulators. For example, it was found that several miRNAs control cellular transcription processes and contribute to the development of many disorders.6 Previous studies also implicate miRNA in MS in the induction of autoimmune reactions.7 Because regulation of miRNA expression is highly dynamic and complex, growing evidence supports the existence of yet another higher level of regulation involved in miRNA activity affected by a group of molecules referred to as circular RNA (circRNA).
It has been shown that circRNA molecules represent a novel, unique class of endogenous ncRNA controlling the expression and function of miRNA.8 They are called natural miRNA “sponges” because a single circRNA molecule is able to neutralize several miRNAs simultaneously and thus might determine the availability of miRNA for posttranscriptional regulation. In addition, it was shown that circRNA might influence the posttranscriptional regulation through other miRNA-independent mechanisms, for example, binding and sequestering RNA-binding proteins (RBPs), base pairing with other than miRNA types of transcripts, translation control, and production of proteins.9
Interestingly, circRNAs are characterized by exceptionally high stability, tissue-specific expression, and evolutionary conservation. In recent years, there has occurred steadily growing evidence revealing that the unique features of circRNAs implicate them in a variety of physiologic and pathologic processes.10 The structure of circRNAs in the form of covalently closed continuous loops renders them more resistant to endonucleases than linear RNA forms, and as a result, they might be more suitable as biomarkers than other types of RNAs. Although accumulating evidence reveals circRNA to have an active role in CNS pathology and immune regulation, little is known about their involvement in the pathogenesis of MS.11
In the present exploratory study, we provide novel data on the global expression pattern of circRNA in peripheral blood mononuclear cells (PBMCs) from the blood of patients with MS and correlate the profile with disease stages and activity. We also evaluate the involvement of circRNA in buffering the regulatory activity of miRNAs and consequently their influence on the transcription of annotated protein genes. Our data support an important role for a circRNA-miRNA regulatory network in immune mechanisms in MS at the level of the B cell for which it may serve as a useful biomarker for disease activity.
Methods
Participants and Study Design
A total of 104 participants were enrolled, comprising 67 patients with relapsing-remitting multiple sclerosis (RRMS) and 37 healthy controls (HCs) (Table 1). All patients with MS fulfilled the McDonald criteria, 2017. All patients had not been treated with disease-modifying drugs for at least 6 months. None had a history of anti-CD20 and anti-CD52 treatment. The disability level for neurologic status of MS was assessed by the expanded disability severity scale (EDSS). Relapse was defined as the development of new neurologic symptoms or worsening of existing symptoms leading to an EDSS increase by 1 point over at least 24 hours. Patients in relapse were sampled within 3 days from the beginning of the relapse and before the administration of glucocorticoid treatment. Remission was defined as stable clinical condition at least for 6 months.
Table 1.
Main Characteristics of the Individuals Enrolled in the Study
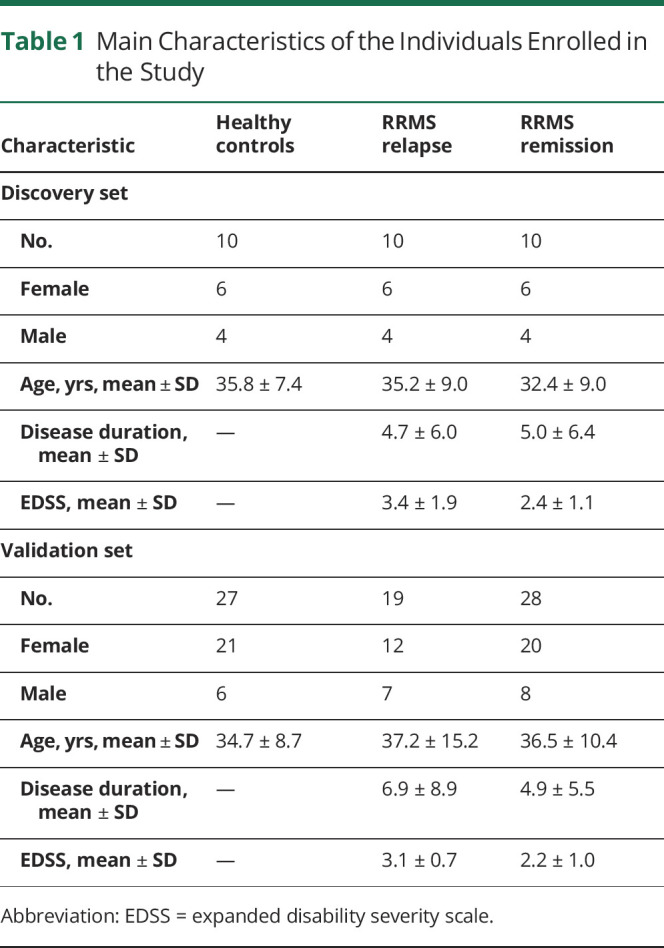
In the exploratory stage of our investigation, we performed microarray hybridization analysis of RNA from the PBMCs of 20 patients with RRMS (10 in relapse and 10 in remission) and 10 HCs. In the second stage, for further validation, RNA samples were isolated from a new cohort of 47 patients with MS and 27 HCs and were subjected to quantitative reverse transcription (qRT)-PCR analysis. Clinical and demographic characteristics of participants are listed in Table 1.
Standard Protocol Approvals, Registrations, and Patient Consents
Study protocols and patient consent forms were approved by the Ethics Committees of the Medical University of Lodz and the University of Warmia and Mazury in Olsztyn, Poland. Written informed consent was obtained from all study participants.
MRI Protocol
Brain MRI data were acquired on a 3.0 T scanner (Siemens, Erlangen, Germany). MRI images used for this study were obtained ±2 weeks from the time of the blood sample collection. To detect focal white matter lesions, MRI used dual-echo (repetition time [TR] = 4,500 milliseconds, echo time [TE] = 22 and 90 milliseconds, 25 slices, slice thickness = 3 mm, 512 × 512 × 44 matrix, and field of view [FOV] = 250 mm) and T1-weighted (TR = 750 milliseconds, TE = 17 milliseconds, 25 slices, slice thickness = 3 mm, 512 × 512 × 44 matrix, and FOV = 250 mm). Gadolinium (Gd)-enhancing T1-weighted lesions were identified from postcontrast T1-weighted spin echo images (TR = 467 milliseconds, TE = 8 milliseconds, 240 × 240 × 132 mm FOV, number of excitations = 1), acquired 5 minutes after administration of a dose of contrast (0.1 mM/kg).
PBMC and RNA Isolation
PBMCs were isolated from whole blood by a density gradient centrifugation method using Ficoll Histopaque (Sigma-Aldrich), as described earlier.23 Total RNA was extracted from PBMCs using the mirVana miRNA Isolation Kit (Life Technologies). The RNA quantity and quality were measured with NanoDrop ND-1000 (Agilent).
Microarray Hybridization
RNA samples were subjected to microarray hybridization with Arraystar Human Circular Array V2 (8 × 15K, Arraystar). Briefly, total RNA was digested with Rnase R (Epicentre, Inc.) to remove linear RNA and to enrich circRNA. Then, the enriched circRNA fraction was amplified and transcribed into fluorescent complementary RNA (cRNA) using a random priming method (Arraystar Super RNA Labeling Kit; Arraystar). The labeled cRNA molecules were purified by the RNeasy Mini Kit (Qiagen). The concentration and specific activity of the labeled cRNA (pmol Cy3/μg cRNA) were measured using a NanoDrop ND-1000 (Thermo Fisher) and a 2100 Bioanalyzer (Agilent). Each labeled cRNA (1 μg) was fragmented by adding 5 μL of 10× blocking agent and 1 μL of 25× fragmentation buffer; then, the mixture was heated to 60°C for 30 minutes, and finally, 25 μL of 2× hybridization buffer was added to dilute the labeled cRNA. Hybridization solution (50 μL) was dispensed into the gasket slide and attached to the circRNA expression microarray slide. The slides were incubated for 17 hours at 65°C in an Agilent Hybridization Oven. The hybridized arrays were washed, fixed, and scanned using an Agilent Scanner G2505C (Agilent Technology).
Agilent Feature Extraction software (version 11.0.1.1) was used to analyze the acquired array images. Quantile normalization and subsequent data processing were performed using the R software limma package.
qRT-PCR Analysis
Quantitative validation of circRNA was performed by qRT-PCR with SYBR Green using specific primers for circRNA identified in the discovery segment of our study. For qRT-PCR, cDNA was synthesized from total RNA using SuperScript III Reverse Transcriptase (Invitrogen) from 1 μg of the total RNA. Subsequently, the qRT-PCR reaction was performed in a total volume of 10 μL, containing 0.5 μL/10 μM of forward/reverse primers, 2 μL of cDNA, 2× SuperArray PCR master mix, and 2 μL of double-distilled water. The primers used in the qRT-PCR were designed such that the amplicon spanned the circular junction site as follows: for hsa_circRNA_010402, 5′ CCCATTACAAGTTGACCTCCAC 3′ and 5′ GCCACCTTCAAAATACCTGTCT 3′; for hsa_circRNA_101348, 5′ GACCTTAGTAAAGCTCAGTTCCAT 3′ and 5′ GATATTGAAAACGTGCTCTCTTCT 3′; for hsa_circRNA_104361, 5′ GCTTTGATGGATGCTTCCAG 3′ and 5′ GCAGTTGACAACAACCCAATT 3′; for hsa_circRNA_102611, 5′ TTTGGATGGTATGCCTTACACT 3′ and 5′ CACACTACAAAGTTGACTTGTGC 3′; and for hsa_circRNA_036391, 5′ TTTCCAAGTGGCAGATTATGG 3′ and 5′ CATGCTCAGCTATTACCAGGGT 3′. The primers used in the qRT-PCR for gene expression measurement of CBX5, DGKH, IKZF3, RNF24, and AK2 were as follows: for CBX5, 5′ GTGGCCGAGGACTTTGATTG 3′ and 5′ CCTGTAACAACGCATCTCATATT 3′; for DGKH, 5′ GTGAGGCAAGTCATTGAGGAA 3′ and 5′ GGTATTGAGCACAGGGAGGTT 3′; for IKZF3, 5′ GGCTCCAACTCAACTCGTCTA 3′ and 5′ TCACAAAGGGAATAAGCAATCT 3′; for RNF24, 5′ GCTCGGATTTCCCACATTA 3′ and 5′ TCTTTGTGTGCTTGATGTCTTAG 3′; and for AK2, 5′ GGCTGATTCACCCCAAGAGT 3′ and 5′ GCAGGCGGATTTTCAAGG 3′.
β-actin was used as an internal control using the following primers: 5′ GTGGCCGAGGACTTTGATTG 3′ and 5′ CCTGTAACAACGCATCTCATATT 3′. Briefly, the cycle parameters for the PCR were as follows: 95°C for 10 minutes, followed by 40 amplification cycles of a denaturing step at the same temperature for 10 seconds and an annealing/extension step of 1 minute at 60°C. A melting curve was performed to confirm that the amplicons were unique and specific. Quantification for each sample was generated using Rotor-Gene Real-Time Analysis software 6.0 (Qiagen). The relative amount of the target gene was determined by calculating the ratio between the target and housekeeping genes using a 2-ddCt formula.
circRNA Bioinformatic Target Prediction
The target miRNAs for differentially expressed circRNAs were predicted using Arraystar miRNA target prediction software based on TargetScan12 and miRanda,13 software tools including composite metrics allowing a search for the presence of circRNA sites that might match the seed region of miRNA. Based on the number and potential strength of the circRNA-miRNA interactions, the top 5 that show the highest expression of the predicted miRNA interacting with a given circRNA are reported.
The classification of protein-coding mRNA putatively controlled by miRNA identified by circRNA analysis was performed with the help of a miRNA target prediction database miRWalk (version 3.0; mirwalk.umm.uni-heidelberg.de/). miRWalk contains information that applies a random forest-based approach to integrate and predict miRNA target sites. The output of the random forest-based model is the predicted probability that a candidate target site is a true target site. Each of the miRNAs predicted to be affected by circRNA is queried into the miRWalk to produce a list of the candidate mRNA with a predicted p < 0.05 value.
Statistical Analysis
In the discovery set, candidate circRNAs were chosen if significant differences (p < 0.05) were observed among the groups using a 1-way analysis of variance (ANOVA) test with the Scheffé post hoc test. Before the ANOVA test, the Levene test for equality of variances was performed. The p values for each analysis of discovery set were also adjusted for multiple comparisons using the approach of Benjamini and Hochberg to control for false discovery rates at 0.05. In the qRT-PCR validation set, the association and the levels of each of the selected circRNA between groups were assessed by ANOVA with the Scheffé post hoc test; the associated p value and the area under the curve (AUC) from receiver operating characteristic (ROC) curve for the full model were calculated. Outlier sensitivity analysis of the circRNA levels was tested using the Tukey method value (Tukey test). R-squared statistic was used to examine the association between circRNA expression and each of the clinical measures.
Data Availability
Anonymized circRNA microarray analysis expression data have been reposited in the GEO database, accession number: GSE171950 (ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171950).
Results
Global Profiling of circRNA in the PBMCs of Patients With MS
To determine the global expression profile of circRNAs from the PBMCs of patients with RRMS, in a discovery set of the study, we used microarrays containing probes for 13,617 human circRNAs. At this stage of the analysis, we tested 30 samples. In total, we have identified on average 11,805 different circRNAs per sample in the RRMS relapse group, 12,077 circRNAs in the RRMS remission groups, and 12,184 in the HC group.
Profile of Differentially Expressed circRNA
To search for unique circRNA in the RRMS profile, we performed global statistical analysis to detect differentially expressed transcripts. We found that expression of individual circRNAs in the RRMS relapse and RRMS remission groups significantly differed from that in controls (Figure 1, A and B). Unsupervised hierarchical clustering based on circRNA expression allowed for clear separation of both RRMS groups, relapse and remission, from controls (Figure 1A and B). However, similar unsupervised hierarchical clustering based on circRNA expression in the RRMS relapse vs remission group did not provide a clear distinction between these 2 stages of the disease (Figure 1C). Thus, microarray data from the discovery set permitted identification of a group of circRNA that was specifically and differentially expressed in patients with RRMS. Individual analysis of circRNA expression revealed 914 transcripts to be differentially expressed between patients with RRMS in relapse and HCs; 595 were upregulated (p < 0.05, fold change [FC] > 2.0), and 319 were downregulated (p < 0.05, FC < 0.5). In the RRMS remission group, there were 622 differentially expressed circRNAs vs HCs; 573 were upregulated and 49 downregulated (p < 0.05, FC > 2.0)—Figure 1, D and E. When the RRMS relapse group was analyzed and compared with the RRMS remission group, we found 15 differentially expressed circRNAs, all of them upregulated in relapse (p < 0.05; FC > 2.0) (Figure 1F). In the subsequent analysis, we focused our research on the upregulated circRNA transcripts.
Figure 1. circRNA Profile in RRMS.

Heatmaps show differentially expressed circRNAs from unsupervised hierarchical clustering present in the PBMCs of patients with RRMS relapse vs controls (A), RRMS remission versus controls (B), and RRMS relapse versus remission (C). Color key represents Z score values; the raw data displayed as a heatmaps are present in Supplementary file, links.lww.com/NXI/A538. Volcano plot graphs for circRNA expression levels in the PBMCs of RRMS relapse vs controls (D), RRMS remission versus controls (E), and RRMS relapse versus remission (F). The vertical green lines correspond to upregulation and downregulation >2-fold. The horizontal green line represents an adjusted p value of 0.05. The red squares indicate differentially expressed circRNAs with statistical significance. (G) Chromosomal distribution of significantly differentially expressed circRNAs in RRMS relapse and RRMS remission versus controls. circRNA= circular RNA; PBMCs = peripheral blood mononuclear cells; RRMS = relapsing-remitting multiple sclerosis.
Differentially expressed circRNAs were widely distributed among chromosomes (Figure 1G). They were most often found on chromosome 1 (8.6% and 8.7%) and chromosome 3 (11.7% and 7.6%) for the RRMS relapse and remission groups vs HCs, respectively.
Validation of Differentially Expressed circRNA
To validate the differentially upregulated circRNA, we tested the 5 circRNAs from the group of the highest differentially expressed circRNA in the RRMS relapse vs control groups in the discovery cohort (Table 2). The validation cohort involved 74 participants: 19 patients with RRMS in relapse, 28 in remission, and 27 HCs. The highest relative amount of circRNA in the validation cohort was found for hsa_circRNA_102611. Interestingly, the results showed that expression levels of hsa_circRNA_101348 (p = 0.0002; FC = 2.9), hsa_circRNA_102611 (p = 0.01; FC = 1.6), and hsa_circRNA_104361 (p = 0.001; FC = 1.5) were higher in patients with RRMS in relapse vs HCs. Importantly, all these 3 circRNAs showed higher expression in RRMS relapse vs remission (hsa_circRNA_101348, p = 0.0006; FC = 2.8; hsa_circRNA_102611, p = 0.01; FC = 1.5; and hsa_circRNA_104361, p = 0.03; FC = 1.8), but not in the remission vs HC group (hsa_circRNA_101348, p = 0.827; FC = 1.0; hsa_circRNA_102611, p = 0.675; FC = 1.1; and hsa_circRNA_104361, p = 0.318; FC = 0.9)—Figure 2A.
Table 2.
List of the 5 Selected circRNAs Differentially Expressed in RRMS Relapse vs Healthy Controls

Figure 2. Validation of the Differential Expression of the Selected circRNA in RRMS.

(A) The expression levels of the indicated circRNA were analyzed using qRT-PCR. Significance was determined by analysis of variance with the Scheffé post hoc test. *p < 0.05, **p < 0.01, and ***p < 0.0001. The bold line represents mean value (±SD). (B) ROCs for the comparison of hsa_circ_101348, hsa_circ_102611, and hsa_circ_104361 expression in (B.a) RRMS relapse vs controls, (B.b) RRMS remission vs controls, and (B.c) RRMS relapse versus remission. circRNA = circular RNA; ROC = receiver operating characteristic; RRMS = relapsing-remitting multiple sclerosis; qRT = quantitative reverse transcription.
To further confirm the significance of the observed differences in expression of hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA_104361 between RRMS groups and controls, we performed ROC curve analysis. ROC confirmed that these circRNAs provided the best AUC values for discriminating patients with RRMS relapse from HCs (for hsa_circRNA_101348, 0.8444; for hsa_circRNA_102611, 0.7194; and for hsa_circRNA_104361, 0.6667) (Figure 2B). ROC curve analysis also confirmed that expression of these 3 circRNAs better predicted RRMS relapse than RRMS remission. Collectively, the above-mentioned data showed that PBMC expression of hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA_104361 provided a clear distinction between RRMS and controls, in addition to RRMS relapse and remission.
circRNA Expression Levels Correlate With MRI and RRMS Clinical Activity
To determine whether expression of the circRNA hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA104361 correlated with RRMS activity, we performed a correlative analysis between the level of circRNA expression and inflammatory activity measured by MRI. We divided MS samples from patients in relapse into 2 categories as follows: with gadolinium-enhancing lesions (Gd+; n = 11) and without (Gd−; n = 13) on MRI. The results showed that patients with Gd+ had higher expression of hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA104361 than patients without enhancing lesions (Figure 3A). More importantly, the differences for hsa_circRNA_101348 and hsa_circRNA_104361 were statistically significant (p = 0.0039, FC = 2.4; p = 0.029, FC = 1.7, respectively). Thus, we confirmed using an independent MRI measure that higher expression of hsa_circRNA_101348 and hsa_circRNA_104361 was associated with inflammatory activity in RRMS.
Figure 3. circRNA Expression in PBMC is Increased in Relation to Magnetic Resonance Imaging Activity in RRMS.

(A) Box and whisker plots of relative expression of indicated circRNA in PBMCs in gadolinium-enhanced (Gd)+ (n = 11) and in (Gd)− (n =13) patients with RRMS. Significance was determined by the Student t test. *p < 0.05. PBMCs (B.a) hsa_circRNA_101348, (B.b) hsa_circRNA_102611, and (B.c) hsa_circRNA_104361 expression levels were plotted against disease duration. Circles represent individual data, and the line represents a correlation trend; p values were calculated by R2 statistic. PBMCs (C.a) hsa_circRNA_101348, (C.b) hsa_circRNA_102611, and (C.c) hsa_circRNA_104361 expression levels were plotted against EDSS. Circles represent individual data, and the line represents a correlation trend; p values were calculated by R2 statistics. circRNA = circular RNA; EDSS = expanded disability severity scale; PBMCs = peripheral blood mononuclear cells; RRMS = relapsing-remitting multiple sclerosis.
We also assessed correlation between hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA104361 expression and disease duration in RRMS (n = 47; Figure 3B), as well as each patient's disability level, as measured by EDSS (n = 47; Figure 3C). circRNA in RRMS showed higher levels in patients with a higher disability score but without statistical significance. The correlative analysis of all 3 circRNAs differentially expressed in patients with MS did not associate with disease duration up to 20 years postdiagnosis.
Identification of the Spectrum of Molecular Targets for hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA_104361
To investigate the potential functions of differentially expressed circRNAs and their networking with miRNA, hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA_104361, we annotated miRNA sequences that are known putative targets of these circRNAs. In Table 3, we list data on the top 5 miRNAs for each of the 3 differentially expressed circRNAs defined by the presence of miRNA response elements.
Table 3.
Validated Differentially Expressed circRNAs in RRMS and the Top 5 miRNAs Predicted to Interact With Them

To further characterize the function of differentially expressed circRNA, we analyzed the potential effect of the circRNA/miRNA interactive complex on protein-coding transcripts. miRNA target database search revealed a number of mRNAs, which might be targeted by miRNA controlled by hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA104361 (Figure 4A). For hsa_circRNA_104361, there were 192 annotated mRNAs simultaneously controlled by all 5 miRNAs predicted to be interacting with this circRNA. For hsa_circRNA_102611, there was only 1 annotated mRNA, and for hsa_circRNA_101348, we found 40 predicted mRNAs. Subsequently, we analyzed shared annotated mRNA for all 3 differentially expressed circRNAs. We found that 5 mRNAs overlapped with 2 circRNAs, hsa_circRNA_101348 and hsa_circRNA104361, and no mRNA overlapped with all 3 circRNAs (Figure 4B).
Figure 4. mRNA Predicted From miRNA Identified as Interacting With CircRNA.

(A) The Venn diagrams display the overlap of the mRNA predicted from the hsa_circRNA_101348–regulated miRNA (hsa-miR-141-5p, hsa-miR-382-3p, hsa-miR-601, hsa-miR-134-3p, and hsa-miR-642a-5p); hsa_circRNA_102611–regulated miRNA (hsa-miR-130b-3p, hsa-miR-130a-3p, hsa-miR-513a-3p, hsa-miR-136-5p, and hsa-miR-301a-3p), and hsa_circRNA_104361–regulated miRNA (hsa-miR-608, hsa-miR-320a, hsa-miR-320b, hsa-miR-320c, and hsa-miR-885-3p). The figures indicate the numbers of the overlapping mRNAs regulated by corresponding miRNAs. (B) The Venn diagram of the overlapping mRNA regulated by all miRNA predicted to interact with hsa_circRNA_101348, hsa_circRNA_102611, or hsa_circRNA_104361. Figures indicate the numbers of the overlapping mRNAs regulated by all miRNAs predicted from a single circRNA. (C) Validation of differential expression of mRNAs in PBMCs in relapsing-remitting multiple sclerosis. Expression levels of the indicated mRNAs were analyzed using qRT-PCR. Significance was determined by analysis of variance with the Scheffé post hoc test. Dashed line represents mean value (±SD). ****p < 10−16. circRNA = circular RNA; miRNA = microRNA; PBMCs = peripheral blood mononuclear cells; RRMS = relapsing-remitting multiple sclerosis; qRT = quantitative reverse transcription.
Thus, we have identified 5 protein-coding transcripts putatively targeted by hsa_circRNA_101348 and hsa_circRNA_104361 through the miRNA circuit in patients with RRMS in relapse (Table 4). Interestingly, within these 5 protein-coding transcripts, we identified 2 mRNAs known to be involved in B-cell function, AK2 and IKZF3. The aforementioned findings provide support for a contribution of circRNA, hsa_circRNA_101348 and hsa_circRNA_104361, highlighted in this study, to the reported B-cell dysfunction in RRMS.
Table 4.
mRNA Predicted to be Induced by hsa_circRNA_101348 and hsa_circRNA_104361 Upregulation

AK2, CBX5, and IKZF3 Expression Is Increased in PBMCs in RRMS During Relapse
To confirm the bioinformatic prediction of the circRNA effect on the mRNA profile in patients with RRMS during relapse, we measured directly the expression levels of AK2, CBX5, DGKH, IKZF3, and RNF24 in PBMCs. This revealed that 3 transcripts showed higher expression in PBMCs in RRMS during relapse compared with HCs (AK2, p = 5.65E-22, FC = 3.1; CBX5, p = 6.86E-22, FC = 2.8; and IKZF3, p = 1.72E-17, FC = 3.0). Furthermore, within these mRNAs, there were 2 B-cell–associated transcripts, AK2 and IKZF3, which were predicted by the above-mentioned bioinformatic analysis. Thus, we have provided the transcription profile of differentially expressed circRNAs in PBMCs from patients with RRMS and controls in addition to information bespeaking a role for these transcripts in B-cell function (Figure 4C).
Discussion
In this report, we have defined the profile of circRNA expression in PBMCs from patients with RRMS and HCs. A distinct pattern was revealed between patients with RRMS and controls. To provide an unbiased profile assessment, we started with a total RNA analysis of PBMCs using a global hybridization assay. From this, we obtained a complete circRNA profile in the PBMCs of patients with RRMS. We identified close to 14,000 circRNA transcripts per sample, and in the discovery set analysis, we found several hundred circRNAs differentially expressed of which the majority were upregulated in RRMS. Our findings were then validated in a separate group of patients with a quantitative PCR method that highlighted and confirmed a significant upregulation of 3 circRNAs, hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA104361 in patients with RRMS in relapse. In addition, these 3 circRNAs showed significant correlation with MRI Gd-enhancing lesions, thus indicating their potential role in inflammatory disease activity.
Apparently, circRNA are a novel, unique class of endogenous RNA, the discovery of which has added yet another layer of amazing complexity to human genetics. circRNAs are characterized by a covalently closed cyclic structure lacking polyadenylated tails. circRNAs have emerged as critical posttranscriptional regulators of gene expression by binding to miRNAs and buffering their repression of mRNA targets.8 Because single circRNA might bind to several miRNAs simultaneously and have several binding sites for an individual miRNA, this buffering repression mechanism has been dubbed a “spongy” type of interaction.10 The spongy type of property linked to circRNA has been implicated in several important biological processes, including brain cell differentiation, macrophage polarization, and several immune processes, including B-cell function.14 The aforementioned activity directly links circRNA to the regulation of immune reactions and immune-mediated disorders. In addition, it was shown that circRNA might influence posttranscriptional regulation in several other miRNA-independent mechanisms. Specifically, circRNA might be responsible for binding and sequestering RBPs,15 which might lead to base pairing with other RNA and regulate translation or even the production of proteins.9
Thus, this study has resulted in the identification of 3 circRNAs and a group of 15 miRNAs, which they might target in patients with RRMS in relapse, a stage known to be heavily involved in mechanisms of autoimmune demyelination. Several miRNAs have been linked to autoimmune processes leading to demyelination in MS.16 We and others have previously identified miRNAs connected to the development and maintenance of encephalitogenic populations of T helper cells.17-19 Other studies have focused on systematic profiling of miRNAs in various immune system compartments in MS. Indeed, reports of miRNA in patients with MS have shown them to be differentially expressed in PBMCs,20 whole blood, T cells, and B cells.17,21 Much effort has been attempted to identify circulating extracellular miRNAs as biomarkers in MS.22-24 It has been found that miRNA expression may be influenced by the clinical type of MS, disease activity, and the therapy being applied.25 In particular, earlier work from our group identified miR-301a as an endogenous regulator of Th17 development in an animal model of MS.21 We have also identified significant changes in serum exosomes expressing miR-301a in MS and have validated it as a biomarker for RRMS during relapse.25 Interestingly, the circRNAs highlighted in this study targeted several miRNAs, which have been previously implicated in MS. miRNA-141 has been found to be modified during T helper–cell activation. miRNA-320a has been localized within MS brain lesions,26 and miRNA-134 has been found to be dysregulated in the blood of patients with MS.22 Of particular interest are data on miRNA-130b and miRNA-320b, which were found to be downregulated in B cells from patients with MS, raising the possibility of their involvement in an increased B-cell transcriptional program. However, it should be recognized that studies of miRNA changes in PBMCs provided heterogenic profiles, rendering them a challenging task to establish a solid and reliable miRNA biomarker for MS.24 Therefore, profiling circRNA, molecules involving a higher level of control over miRNA activity, may yield a more robust correlation, leading to the identification of more specific biomarkers for MS.
The data obtained in this study lend a strong immune-related context to the action of the 3 circRNAs highlighted. Previous analyses have shown that circRNAs are implicated in the induction of innate immunity genes and may confer protection against viral infection.27 Accordingly, circRNAs have already been implicated in autoimmune pathology, including MS28 specifically that circRNA, hsa_circ_0106803, derived from the GSDMB gene, was 2.8-fold upregulated in patients with RRMS. The same group also found a consistent up-regulation in MS of lncRNAMALAT1, known to regulate alternative splicing and generation of circRNA derived from MS-associated genes (IL7R, SP140).29 Iparraguirre et al.30 reported the presence of differentially expressed circRNA in 8 patients with RRMS in remission and 4 HCs. They found that circ_0005402 and circ_0035560, both located inside the ANXA2 gene, are downregulated in patients with RRMS in remission. More recently, the same group reported on RNA-Seq profiling of leukocyte and showed sex-dependent circRNA upregulation in patients with MS.31 Six differentially expressed circRNAs were validated in women with MS and 3 in men with MS. Paraboschi et al. presented a bioinformatic analysis of circRNA enrichments derived from noncoding elements in the MS-associated genome and suggested that they may be possibly involved in susceptibility to the disease.32 In another study, exosomal circRNA within CSF was found to correlate with IgG levels in MS.33
Among the autoimmune aberrations described in RRMS, a significant role for B-cell function is prominent. In recent years, B cells have been proven to play a leading role in antigen presentation within the CNS, the secretion of proinflammatory cytokines,34 and the production of antimyelin antibodies.35 It has also been proposed that ectopic lymphoid tissue aggregates in the leptomeninges are composed of B cells and may determine chronicity of MS disease.36 Finally, recent clinical trials confirmed that B-cell depletion represents an efficient therapeutic approach in RRMS37-39 and primary progressive MS.40 In light of these data on a critical role for B cells in the pathogenesis of MS, it is of particular relevance that within protein-coding genes controlled by the circRNA molecules identified in this study, we have revealed 2 B-cell–related transcripts, AK2 and IKZF3 (Aiolos). These findings were confirmed by direct demonstration of an upregulation of AK2 and IKZF3 in the PBMCs of patients with MS in relapse. The AK2 gene is known to encode phosphotransferase adenylate kinase 2 and meets the cell-specific requirements for the functions of mitochondria, particularly in B-cell activation and antibody production. The role of AK2 has been specifically emphasized in B cells infected with Epstein-Barr virus, long discussed in MS as an MS-associated viral infection.41 IKZF3 encoding for the proteins Aiolos and Ikaros, which are key Ikaros family zinc-finger transcription factors, regulates lymphoid and myeloid cell development and immune homeostasis.42 Aiolos has been shown to play a critical role in the function of mature B cells and is required for the generation of high-affinity Ab-secreting plasma cells.43 Other studies have suggested an essential role of Aiolos at later stages of B-cell differentiation.44 Numerous studies have identified polymorphisms in IKZF3 to be associated with a risk for the development of systemic lupus erythematosus.45 Interestingly, an IKZF3 variant is described as one of the MS risk alleles.46 Thus, modulation of Aiolos expression by circRNA has the potential to contribute to the B-cell–mediated aberrant immune response in MS. Specifically, it might direct B-cell differentiation toward autoantibody secretion. In light of the above-mentioned data, further research involving B-cell populations is warranted. The third transcript, CBX5, predicted from differentially expressed circRNA/miRNA interactions in relapse, has been linked to stem cell self-renewal, lineage commitment, and cancer and development.47
Another aspect of our findings might prompt consideration of the circRNAs identified in this study as potential distinguishing biomarkers for MS. To date, the diagnosis of MS has been largely based on clinical and MRI findings. Of high priority in MS is the identification of specific biomarkers that might improve diagnosis and provide further insight into its pathologic background. All 3 differentially expressed circRNAs shown in this study had an increased presence in PBMCs from patients with RRMS in relapse. Their expression also correlated with inflammatory activity, as assessed by MRI. Significantly, circRNAs have already been found to distinguish several disorders with particular emphasis on cancer and other malignancies.48 Among circRNA properties that make them attractive candidates for disease biomarkers are their unique stability, conservation, and relatively high abundance in blood and body fluids. This possibility stems from the known resistance of circRNA to RNAse activity due to their covalently closed cyclic structure.49 The abundance of circRNA in the CNS suggests that they might have particular relevance to the search for biomarkers in nervous system diseases.50 Thus, it is anticipated that further studies on circRNA might facilitate the delineation of novel diagnostic and prognostic biomarkers in MS.
In summary, we have demonstrated overexpression of the circRNA molecules, hsa_circRNA_101348, hsa_circRNA_102611, and hsa_circRNA104361 in the PBMCs of patients with RRMS in relapse. The expression pattern of circRNAs that interact by a spongy type of activity with miRNA creates a circRNA/miRNA network specific for RRMS. These data hold promise for the development of new biomarkers for RRMS and suggest a previously undescribed role for circRNAs in the regulation of the transcriptional program of B cells in MS.
Acknowledgment
The authors would like to thank Patricia Cobban-Bond for assistance in the preparation of the manuscript. The authors would also like to thank Magdalena Tinsley and Agnieszka Lewandowska for technical assistance.
Glossary
- ANOVA
analysis of variance
- AUC
area under the curve
- circRNA
circular RNA
- cRNA
complementary RNA
- EDSS
expanded disability severity scale
- FOV
field of view
- Gd
gadolinium
- HC
healthy control
- miRNA
microRNA
- MRE
miRNA response element
- MS
multiple sclerosis
- ncRNA
noncoding RNA
- PBMC
peripheral blood mononuclear cell
- qRT
quantitative reverse transcription
- RBP
RNA-binding protein
- ROC
receiver operating characteristic
- RRMS
relapsing-remitting multiple sclerosis
- TE
echo time
- TR
repetition time
Appendix. Authors
Contributor Information
Anna E. Zurawska, Email: anna.elzbieta.zurawska@gmail.com.
Marcin P. Mycko, Email: marcin.mycko@uwm.edu.pl.
Igor Selmaj, Email: igorselmaj@gmail.com.
Cedric S. Raine, Email: cedric.raine@einsteinmed.org.
Study Funding
This study was supported by the National Science Centre Poland (PRELUDIUM 2015/17/N/NZ6/03504 to A.E.Z. and OPUS 2016/23/B/NZ6/02541 to M.P.M.), a CEE Merck grant to K.W.S., and by the University of Warmia and Mazury in Olsztyn internal grants to K.W.S., M.P.M., and A.E.Z. C.S.R. was the Wollowick Professor for MS Research at the Albert Einstein College of Medicine, New York.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/NN for full disclosures.
References
- 1.Frohman EM, Racke MK, Raine CS. Medical progress: multiple sclerosis - the plaque and its pathogenesis. New Engl J Med. 2006;354(9):942-955. [DOI] [PubMed] [Google Scholar]
- 2.Axisa PP, Hafler DA. Multiple sclerosis: genetics, biomarkers, treatments. Curr Opin Neurol. 2016;29(3):345-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 2: CD8+T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016;15(3):317-331. [DOI] [PubMed] [Google Scholar]
- 4.Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. 2017;13(1):25-36. [DOI] [PubMed] [Google Scholar]
- 5.Cotsapas C, Mitrovic M. Genome-wide association studies of multiple sclerosis. Clin Translational Immunol. 2018;7(6):e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwakawa HO, Tomari Y. The functions of MicroRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015;25(11):651-665. [DOI] [PubMed] [Google Scholar]
- 7.Mycko MP, Baranzini SE. microRNA and exosome profiling in multiple sclerosis. Mult Scler. 2020;26(5):599-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384-388. [DOI] [PubMed] [Google Scholar]
- 9.Li HM, Ma XL, Li HG. Intriguing circles: conflicts and controversies in circular RNA research. Wiley Interdiscip Rev RNA. 2019;10(5):e1538. [DOI] [PubMed] [Google Scholar]
- 10.Han B, Chao J, Yao H. Circular RNA and its mechanisms in disease: from the bench to the clinic. Pharmacol Ther. 2018;187:31-44. [DOI] [PubMed] [Google Scholar]
- 11.Zurawska A, Mycko MP, Selmaj KW. Circular RNAs as a novel layer of regulatory mechanism in multiple sclerosis. J Neuroimmunol. 2019;334:576971. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11(8):R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Zhang Y, Li X, Zhang M, Lv K. Microarray analysis of circular RNA expression patterns in polarized macrophages. Int J Mol Med. 2017;39(2):373-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zang J, Lu D, Xu A. The interaction of circRNAs and RNA binding proteins: an important part of circRNA maintenance and function. J Neurosci Res. 2020;98(1):87-97. [DOI] [PubMed] [Google Scholar]
- 16.Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol. 2016;16(5):279-294. [DOI] [PubMed] [Google Scholar]
- 17.Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10(12):1252-1259. [DOI] [PubMed] [Google Scholar]
- 18.Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW. microRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci USA. 2012;109(20):E1248-E1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mycko MP, Cichalewska M, Cwiklinska H, Selmaj KW. miR-155-3p drives the development of autoimmune demyelination by regulation of heat shock protein 40. J Neurosci 2015;35(50):16504-16515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otaegui D, Baranzini SE, Armañanzas R, et al. Differential micro RNA expression in PBMC from multiple sclerosis patients. Plos One 2009;4(7):e6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerau-de-Arellano M, Smith KM, Godlewski J, et al. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain. 2011;134(pt 2):3575-3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandhi R, Healy B, Gholipour T, et al. Circulating MicroRNAs as biomarkers for disease staging in multiple sclerosis. Ann Neurol. 2013;73(6):729-740. [DOI] [PubMed] [Google Scholar]
- 23.Selmaj I, Mycko MP, Raine CS, Selmaj KW. The role of exosomes in CNS inflammation and their involvement in multiple sclerosis. J Neuroimmunol. 2017;306:1-10. [DOI] [PubMed] [Google Scholar]
- 24.Regev K, Healy BC, Paul A, et al. Identification of MS-specific serum miRNAs in an international multicenter study. Neurol Neuroimmunol Neuroinflamm. 2018;5(5):e491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selmaj I, Cichalewska M, Namiecinska M, et al. Global exosome transcriptome profiling reveals biomarkers for multiple sclerosis. Ann Neurol. 2017;81(5):703-717. [DOI] [PubMed] [Google Scholar]
- 26.Junker A, Krumbholz M, Eisele S, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;132(12):3342-3352. [DOI] [PubMed] [Google Scholar]
- 27.Cadena C, Hur S. Antiviral immunity and circular RNA: No end in sight. Mol Cell. 2017;67(2):163-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardamone G, Paraboschi EM, Rimoldi V, Duga S, Soldà G, Asselta R. The characterization of GSDMB splicing and backsplicing profiles identifies novel isoforms and a circular RNA that are dysregulated in multiple sclerosis. Int J Mol Sci. 2017;18(3):576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardamone G, Paraboschi E, Soldà G, et al. Not only cancer: the long non-coding RNA MALAT1 affects the repertoire of alternatively spliced transcripts and circular RNAs in multiple sclerosis. Hum Mol Genet. 2019;28(9):1414-1428. [DOI] [PubMed] [Google Scholar]
- 30.Iparraguirre L, Muñoz-Culla M, Prada-Luengo I, Castillo-Triviño T, Olascoaga J, Otaegui D. Circular RNA profiling reveals that circular RNAs from ANXA2 can be used as new biomarkers for multiple sclerosis. Hum Mol Genet. 2017;26(18):3564-3572. [DOI] [PubMed] [Google Scholar]
- 31.Iparraguirre L, Alberro A, Sepúlveda L, et al. RNA-Seq profiling of leukocytes reveals a sex-dependent global circular RNA upregulation in multiple sclerosis and 6 candidate biomarkers. Hum Mol Genet. 2020;29(20):3361-3372. [DOI] [PubMed] [Google Scholar]
- 32.Paraboschi EM, Cardamone G, Soldà G, Duga S, Asselta R. Interpreting non-coding genetic variation in multiple sclerosis genome-wide associated regions. Front Genet. 2018;9:647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He J, Ren M, Li H, Yang L, Wang X, Yang Q. Exosomal circular RNA as a biomarker platform for the early diagnosis of immune-mediated demyelinating disease. Front Genet. 2019;10:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol. 2018;19(7):696-707. [DOI] [PubMed] [Google Scholar]
- 35.Berger T, Rubner P, Schautzer F, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2003;349(2):139-145. [DOI] [PubMed] [Google Scholar]
- 36.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(4):1089-1104. [DOI] [PubMed] [Google Scholar]
- 37.Cross AH, Klein RS, Piccio L. Rituximab combination therapy in relapsing multiple sclerosis. Ther Adv Neurol Disord. 2012;5(6):311-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376:221-234. [DOI] [PubMed] [Google Scholar]
- 39.Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546-557. [DOI] [PubMed] [Google Scholar]
- 40.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209-220. [DOI] [PubMed] [Google Scholar]
- 41.Chou J, Alazami AM, Jaber F, et al. Hypomorphic variants in AK2 reveal the contribution of mitochondrial function to B-cell activation. J Allergy Clin Immunol. 2019;146(1):192-202. [DOI] [PubMed] [Google Scholar]
- 42.Heizmann B, Kastner P, Chan S. The Ikaros family in lymphocyte development. Curr Opin Immunol. 2018;51:14-23. [DOI] [PubMed] [Google Scholar]
- 43.Cortés M, Georgopoulos K. Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J Exp Med. 2004;199(2):209-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JH, Avitahl N, Cariappa A, et al. Aiolos regulates B cell activation and maturation to effector state. Immunity. 1998;9(4):543-553. [DOI] [PubMed] [Google Scholar]
- 45.Manou-Stathopoulou S, Rivellese F, Mauro D, et al. The transcription factors IKZF1 and IKZF3 control B cell activation and differentiation in systemic lupus erythematosus. Ann Rheum Dis. 2019;78:1548. [Google Scholar]
- 46.Keshari PK, Harbo HF, Myhr KM, Aarseth JH, Bos SD, Berge T. Allelic imbalance of multiple sclerosis susceptibility genes IKZF3 and IQGAP1 in human peripheral blood. BMC Genet. 2016;17(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Wijnen AJ, Bagheri L, Badreldin AA, et al. Biological functions of chromobox (CBX) proteins in stem cell self-renewal, lineage-commitment, cancer and development. Bone. 2020;143:115659. [DOI] [PubMed] [Google Scholar]
- 48.Lei B, Tian Z, Fan W, Ni B. Circular RNA: a novel biomarker and therapeutic target for human cancers. Int J Med Sci. 2019;16(2):292-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32(5):453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu D, Xu AD. Mini review: circular RNAs as potential clinical biomarkers for disorders in the central nervous system. Front Genet. 2016;7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized circRNA microarray analysis expression data have been reposited in the GEO database, accession number: GSE171950 (ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE171950).