

Abstract
Despite the importance of gene regulatory enhancers in human biology and evolution, we lack a comprehensive model of enhancer evolution and function. This substantially limits our understanding of the genetic basis of species divergence and our ability to interpret the effects of noncoding variants on human traits.
To explore enhancer sequence evolution and its relationship to regulatory function, we traced the evolutionary origins of transcribed human enhancer sequences with activity across diverse tissues and cellular contexts from the FANTOM5 consortium. The transcribed enhancers are enriched for sequences of a single evolutionary age (“simple” evolutionary architectures) compared with enhancers that are composites of sequences of multiple evolutionary ages (“complex” evolutionary architectures), likely indicating constraint against genomic rearrangements. Complex enhancers are older, more pleiotropic, and more active across species than simple enhancers. Genetic variants within complex enhancers are also less likely to associate with human traits and biochemical activity. Transposable-element-derived sequences (TEDS) have made diverse contributions to enhancers of both architectures; the majority of TEDS are found in enhancers with simple architectures, while a minority have remodeled older sequences to create complex architectures. Finally, we compare the evolutionary architectures of transcribed enhancers with histone-mark-defined enhancers.
Our results reveal that most human transcribed enhancers are ancient sequences of a single age, and thus the evolution of most human enhancers was not driven by increases in evolutionary complexity over time. Our analyses further suggest that considering enhancer evolutionary histories provides context that can aid interpretation of the effects of variants on enhancer function. Based on these results, we propose a framework for analyzing enhancer evolutionary architecture.
Keywords: human genetics, genome evolution, sequence age, gene regulation, noncoding gene regulatory sequence
Introduction
Enhancers are noncoding DNA sequences bound by transcription factors (TFs) that regulate gene transcription and establish tissue- and cell-specific gene expression patterns (Shlyueva et al. 2014). Rapid turnover of sequences with enhancer activity is a common evolutionary process that contributes to species-specific gene regulation and phenotypic diversity (Wittkopp and Kalay 2012). Despite the importance of gene regulatory enhancers in human biology and evolution, we lack a comprehensive model of their evolutionary and functional dynamics.
Comparative genomic studies have demonstrated that gene regulatory activity turns over rapidly between species. For example, active liver enhancers defined by histone modifications are rarely shared among 20 placental mammals, though most liver enhancer sequences are alignable across diverse species (Villar et al. 2015). Similarly, the majority of liver TF DNA binding events among five vertebrates are private to a single species, and DNA binding site divergence between species is largely explained by lineage-specific mutations that activate and inactivate binding sites (Schmidt et al. 2010).
Although enhancer activity is often species-specific, DNA sequences underlying active enhancers are often alignable across species and originate from a common ancestor. For example, 80% of mouse DNase I hypersensitive site (DHS) sequences originate from the last common ancestor of mice and humans, yet only 36% of DHS sites have shared open-chromatin activity between humans and mice (Vierstra et al. 2014). Similarly, a comparison of human, rhesus, and mouse enhancers involved in embryonic limb development showed that most human-specific gains in enhancer activity occurred in ancient mammalian sequences, most often due to a small number of substitutions (Cotney et al. 2013). These studies indicate that most enhancer sequences do not maintain consistent activity over evolutionary distances and suggest that a common mode of enhancer evolution has relied on the evolution of new functions in DNA sequences with ancient origins (sometimes referred to as exaptation). Thus, it is important to distinguish the evolutionary history of enhancer activity, which is often species-specific, from the history of the underlying DNA sequence, which is often ancient. For brevity, we use the term “enhancer” when discussing sequence with enhancer activity in a context of interest.
Species-specific patterns of enhancer activity can arise from a range of genomic changes. Human-specific adaptive nucleotide substitutions in conserved developmental enhancers have been shown to drive robust in vivo reporter activity in mouse compared with chimpanzee and rhesus orthologs (Prabhakar et al. 2008; Capra, Erwin, et al. 2013). Despite this, most gains of enhancer activity are not under strong positive selection (Pollard et al. 2006; Thurman et al. 2012; Moon et al. 2019). Repetitive sequences derived from transposable elements (TEs) also contribute to species-specific enhancer activity (Chuong et al. 2017). Though important, TE derived sequences (TEDS) are depleted in sequences with enhancer activity compared with the rest of the genome (Emera et al. 2016; Simonti et al. 2017). Together, these results illustrate that enhancer sequence evolution is dynamic and can proceed through different evolutionary trajectories.
Determining evolutionary origins by estimating sequence age—that is, the common ancestor in which a homologous sequence first appeared—has expanded knowledge of enhancer sequence evolution, biological functions, and associations with complex human diseases. Most sequences with human liver enhancer activity are ancient, even though their activity turns over rapidly between species (Villar et al. 2015). Furthermore, regulatory elements of different ages have different gene targets and cross-species analyses have revealed three periods of regulatory sequence innovation during vertebrate evolution (Lowe et al. 2011), suggesting sequences from distinct periods have been co-opted to regulate specific gene pathways. Specific TE insertions provided new TF binding motifs through these evolutionary epochs, expanding gene regulatory regions and, in some cases, driving shifts in nearby gene expression (Marnetto et al. 2018). Enhancer evolutionary origins may also be relevant to their roles in disease, as human enhancers with older sequence ages are more enriched for heritability of complex traits than enhancers in younger sequences, independent of the conservation of enhancer function across species (Hujoel et al. 2019). When interpreting these and our results, we emphasize that estimating the age of sequences with human enhancer activity is not necessarily the age when the sequence first gained enhancer activity.
Further complicating these analyses, regulatory regions can contain sequences of multiple ages, suggesting that the juxtaposition of sequences of different origins may benefit or change enhancer function over time. A pioneering analysis of conserved mammalian neocortical enhancers found that many had composite sequences of multiple ages and origins (Emera et al. 2016). A two-step life cycle model was proposed to explain enhancer sequence evolution. In the first step, short proto-enhancer sequences of a single evolutionary origin gain weak enhancer activity, and most are inactivated over time. In the second step, a fraction of proto-enhancers acquires more stable activity through the integration of younger sequences carrying relevant TF binding sites (TFBSs) that could create or modify TF-complex interactions.
It is unclear whether the juxtaposition of sequences of different origins represents the common mode of enhancer sequence evolution across contexts. Further, how these evolutionary histories influence human enhancer function has not been explored. Previous work has largely overlooked the evolutionary architecture of enhancers—that is, the evolutionary age(s) of sequences with enhancer activity—which more precisely reflects the evolutionary events that produced them. Thus, there is a gap in our understanding of the evolutionary dynamics that result in sequences with enhancer activity and how these histories relate to gene regulatory function.
Here, we build on previous work (Lowe et al. 2011; Emera et al. 2016; Marnetto et al. 2018; Hujoel et al. 2019) to quantify enhancer sequence age architecture—the age of every base pair within a sequence with enhancer activity—across human transcribed enhancers. We then evaluate how sequence age architecture relates to enhancer function, evolutionary stability, and tolerance to human variation. We find that transcribed enhancer sequences have simpler age architectures than expected, with the majority consisting of sequence of a single age and a minority with multi-age evolutionary architectures. Surprisingly, given recent work (Emera et al. 2016), enhancers of both architectures have similar evolutionary conservation after accounting for age differences, suggesting that increasing complexity over time is not required for stable gene regulatory function. Nonetheless, enhancers with different architectures differ in their associated functional features. Pleiotropy and cross-species activity are higher in enhancers with multi-age architectures, while functional differences in enhancer activity due to natural human variation occur slightly more frequently in enhancer sequences of a single age. Based on these observations, we present a model of enhancer sequence evolution and provide a framework for dissecting the evolution and function of human enhancer sequences.
Results
Estimating Enhancer Ages Using Vertebrate Multiple Species Alignments
In this study, our goal is to characterize the evolutionary architecture of human enhancer sequences and associations with regulatory function. In this section, we describe the data sets and strategies we used to define enhancer sequence ages and provide context necessary for interpreting our results. We analyzed 30,438 transcribed human autosomal enhancers identified in 112 cell and tissues based on enhancer RNA (eRNA) data sets from the FANTOM5 consortium (Andersson et al. 2014). We focused on transcribed enhancers, because eRNA are enriched for sequences with functional activity in massively parallel reporter assays (MPRA) and mark sequence boundaries that are sufficient for enhancer function with high specificity (Andersson et al. 2014; Benton et al. 2019; Tippens et al. 2020). We also analyzed the architectures of enhancers identified based on histone modification patterns from the Roadmap Epigenomics Consortium to complement the main eRNA results.
We assigned sequence ages to enhancers based on the evolutionary histories of the overlapping syntenic blocks from the UCSC 46-way alignment of diverse vertebrate species spanning 600 My of evolution (fig. 1; Materials and Methods). For simplicity, we grouped most recent common ancestor (MRCA) nodes into 10 age categories and report sequence age as the oldest ancestral branch on which the sequence first appeared (Materials and Methods). We generated random sets of enhancer-length-matched, chromosome-matched, noncoding genomic sequences throughout to create null distributions for interpreting enhancer attributes (Materials and Methods and supplementary fig. S1, Supplementary Material online).
Fig. 1.

Illustration of the method for mapping enhancer sequence age architecture. We quantify the age of a sequence with human enhancer activity based on the oldest most recent common ancestor (MRCA) in overlapping syntenic blocks from the MultiZ multiple sequence alignments of 46 vertebrates (inset). Enhancer age is assigned as the oldest, overlapping syntenic block age. Millions of years ago (Ma) divergence estimates from TimeTree (Hedges et al. 2015) are annotated in parenthesis in the color key. As expected from previous work, we find that enhancers are older, longer, and more conserved than expected from the genomic background (supplementary fig. S2, Supplementary Material online).
Enhancers Are Older, Longer, and More Conserved Than the Genomic Background
As expected from previous observations (Lowe et al. 2011; Villar et al. 2015; Emera et al. 2016; Marnetto et al. 2018), we find that sequences with human enhancer activity are older, longer, and more conserved than expected from the noncoding genomic background, supporting that they have been maintained due to their regulatory functions. Among human enhancer sequences, 54% originate from the common ancestors of Eutherians, while 35% can be traced to older ancestors, and 11% can be traced to younger ancestors. Human enhancers are significantly older than matched sets of random sequences from across the human genome (supplementary fig. S2A and D, Supplementary Material online). Old enhancer sequences (origins before the Eutherian ancestor) are significantly longer than younger enhancer sequences and longer than expected from age-matched regions from the random genomic background sets (Materials and Methods; supplementary fig. S2B and E, Supplementary Material online). Conversely, younger enhancers are shorter than expected. Similarly, older enhancers are more conserved than younger enhancers and more conserved than expected from the genomic background (supplementary fig. S2C, Supplementary Material online). This highlights that sequence age and conservation provide complementary information; age estimates the origin of the sequence, while conservation estimates constraint on sequence variation.
Enhancers Are Enriched for Simple Evolutionary Sequence Architectures
The majority (65%, N = 19,857) of human transcribed enhancers are found within a single syntenic block (i.e., they are of a single age). The median enhancer length is 292 bp, and the median syntenic block genome-wide is 54 bp (supplementary fig. S3, Supplementary Material online). Thus, it was surprising that only 35% (N = 10,581) of enhancers mapped to more than one syntenic age (fig. 2B). To evaluate whether the sequence age architectures of transcribed enhancers differ from what would be expected given the length distributions of enhancers and syntenic blocks, we compared the number of syntenic bocks with distinct ages in enhancers versus matched non-coding regions from the genomic background (Materials and Methods).
Fig. 2.

Simple and complex enhancers have distinct evolutionary architectures, lengths, and ages. (A) Schematic of simple and complex enhancer architectures based on overlapping syntenic block ages. Simple FANTOM enhancers are composed of sequence of one evolutionary age, while complex FANTOM enhancers contain sequence of multiple ages. Within complex enhancers, the oldest segment is the “core” and younger segments are “derived.” (B) Example simple and complex enhancer architectures from 921 random autosomal FANTOM enhancers. The majority (65%, N = 19,857) have simple architectures. For illustration, the age of each enhancer sequence is summarized across 100 equally spaced bins; color indicates the age of each sequence in each bin. (C) Enhancers have significantly fewer segments of different ages than expected by chance; simple FANTOM enhancers are 1.3-fold enriched versus 100 length-matched random shuffled sets; P = 7.6e−107, Fisher’s Exact Test. (D) Complex enhancers are older at their centers on average. Dividing complex enhancers and length- and architecture-matched genomic background into 100 equally spaced bins, mean complex enhancer sequence age from 10,581 complex enhancers and 17,277 complex genomic background sequences from random shuffle is shown; the light green and light gray regions represent bootstrapped 95% confidence intervals. The middle 50% of complex enhancer bins are significantly older than the outer 50% of bins (0.275 vs. 0.265 substitutions per site; P = 4.9e−166, Mann–Whitney U). This pattern is mainly driven by older enhancers (supplementary figs. S7 and S8, Supplementary Material online). (E) Complex enhancers are longer than simple enhancers (median 347 bp vs. 259 bp; P < 2.2e−308, error bars give 95% bootstrapped confidence intervals). The number of enhancers of each type in each age bin are given below the panel. (F) Complex enhancers are significantly older than simple enhancers (61% complex vs. 19% simple enhancers older than Eutherian).
Human enhancers are enriched for simpler architectures compared with the noncoding genomic background (fig. 2C; 1.3-fold enrichment for a single age; P = 7.6e−107 Fisher’s Exact Test; 0.1–0.5-fold depletion for multiple age segments; P = 7.1e−12). This suggests constraint against insertions and deletions among sequences with gene regulatory potential. These differences were greatest among enhancer architectures with Therian and Eutherian sequence origins (supplementary fig. S5B, Supplementary Material online), and complex architectures are depleted among enhancers of most ages (supplementary fig. S6B, Supplementary Material online). This further supports that enhancer architecture is constrained across ages and does not favor complex architectures.
For simplicity, we refer to enhancer sequences with greater than or equal to the median segments of different ages across enhancers as having complex sequence age architectures (“complex” enhancers). Enhancers with fewer than the median age segments have simple sequence age architectures (“simple” enhancers, fig. 2A). Given that the majority (65%) of transcribed enhancers consist of a single age segment, all enhancer sequences of two or more ages are classified as complex (35%). We assigned complex enhancer ages according to its oldest sequence age, and note that human-specific enhancers can only be classified as simple enhancers because the oldest sequence age maps to the human branch (Materials and Methods).
The Oldest Sequences Occur in the Middle of Complex Enhancers
Among complex enhancer sequences, we define the oldest sequence as the “core” and younger sequences as “derived” segments (fig. 2A). The core is generally at the center of the enhancer, while younger sequences are generally flank core sequences in complex enhancers (fig. 2D; Materials and Methods). This organization is specific to enhancer sequences; we do not observe similar organization in matched regions from the genomic background with complex architectures. Stratifying complex enhancers by core age revealed that this pattern was driven by enhancers with older sequence origins (supplementary fig. S7, Supplementary Material online). Enhancers with three or more age segments also are enriched for the oldest sequence in the middle, further supporting the prevalence of this organization across complex enhancer sequences (supplementary fig. S8, Supplementary Material online). In younger complex enhancers, core sequences are located slightly more towards sequence edges. This may reflect the fact that most young complex enhancers consist of only two ages, one older and one younger (supplementary fig. S5, Supplementary Material online). This suggests that older core sequences and younger flanking sequences are nonrandomly arranged within complex enhancer architectures.
Complex Enhancers Are Longer and Older Than Simple Enhancers
Complex enhancers are significantly longer than simple enhancers (fig. 2E and supplementary fig. S9, Supplementary Material online; median 347 vs. 259 bp; P < 2.2e−308, Mann–Whitney U test). Some length difference is expected based on the definition of complex enhancers, since longer regions are more likely to overlap multiple syntenic blocks by chance. To evaluate whether the length difference between simple and complex enhancers was greater than expected, we shuffled noncoding genomic regions matched on enhancer length and assessed architectures (simple or complex) and ages in the resulting random regions (Materials and Methods; supplementary fig. S1, Supplementary Material online). We observed that complex enhancer sequences are slightly, but significantly, longer than expected (median 347 bp vs. 339 bp; P = 2.5e−06, Mann–Whitney U test) and that complex enhancers have a stronger positive correlation between length and age than expected (supplementary fig. S9B, Supplementary Material online; 10.6 bp/100 My; P = 1.1e−17 vs. 4.3 bp/100 My; P = 3.7e−251, linear regression). In contrast, simple enhancers retain similar lengths over time (–0.7 bp/100 My; P = 0.5 vs. –5.5 bp/100 My, P < 2.2e−308) and are also slightly longer than expected (supplementary fig. S9A, Supplementary Material online, median 259 bp vs. 255 bp; P = 7.3e−05). We note that complex enhancer length plateaus among sequences older than the Mammalian ancestor (supplementary fig. S9A, Supplementary Material online). This pattern also holds when broken down by syntenic block, though complex syntenic blocks are consistently shorter than simple syntenic blocks (supplementary fig. S10, Supplementary Material online).
Next, we compared the sequence age distribution for simple and complex architectures (fig. 2F). Complex enhancers are generally older than simple enhancers. Sixty-eight percent of simple enhancer sequences are derived from the Eutherian ancestor, while 12% are younger and 19% are older. Simple enhancers are enriched for Eutherian sequences and are older than expected overall (supplementary fig. S6A, Supplementary Material online; P < 2.2e−308). Conversely, 30% of complex enhancers are derived from the Eutherian ancestor, 9% are younger than the Eutherian ancestor and 61% of complex enhancers are older. Complex enhancers are enriched for sequences older than Eutherian ancestor and are also older than expected (P < 2.2e−308). Consistent with the overall depletion for complex architectures reported in the previous section, enhancers stratified by age are also depleted of complex architectures and this trend does not appear time-linear (supplementary fig. S6B, Supplementary Material online). The presence of many simple enhancers with old sequence ages suggests that complex evolutionary architecture is not necessary for survival over long periods.
Complex Enhancers Are More Pleiotropic and More Conserved in Activity across Species Than Simple Enhancers
In this section, we evaluate whether simple and complex enhancers have different patterns and breadth of activity across tissues and species. Among tissues and cell types, the enrichment for simple enhancers versus complex varies. Most contexts are enriched for simple enhancers, including many blood cell, brain, and pregnancy-related cell types, while the contexts with complex architecture enrichment include smooth muscle and digestive tissues (supplementary fig. S11, Supplementary Material online).
Enhancers with ancient origins and conserved activity across diverse mammals are known to be more pleiotropic—that is, they have activity across multiple human tissues (Fish et al. 2017). Thus, we hypothesized that complex enhancers would be more pleiotropic than simple enhancers given their older age distribution. To test this, we quantified the overlap of enhancer activity across 112 tissue and cell enhancer data sets and stratified by architecture (Materials and Methods). To control for length differences between simple and complex enhancers in this and subsequent analyses, we trimmed or expanded enhancers around their midpoints to match the data set-wide mean length (310 bp).
Complex enhancers have activity across significantly more biological contexts than simple enhancers (fig. 3A; 7.4 vs. 4.8 contexts; P = 5.9e−199, Mann–Whitney U). Enhancer pleiotropy overall increases with age, and complex enhancers are consistently more pleiotropic than age-matched simple enhancers (fig. 3A). Considering the full length of enhancers, we find that length is similarly correlated with pleiotropy in age-matched simple and complex enhancers (supplementary fig. S12, Supplementary Material online). These results suggest that complex enhancers are more likely to have activity across biological contexts than simple enhancers, and increased length associates with increased pleiotropy in both simple and complex enhancers.
Fig. 3.
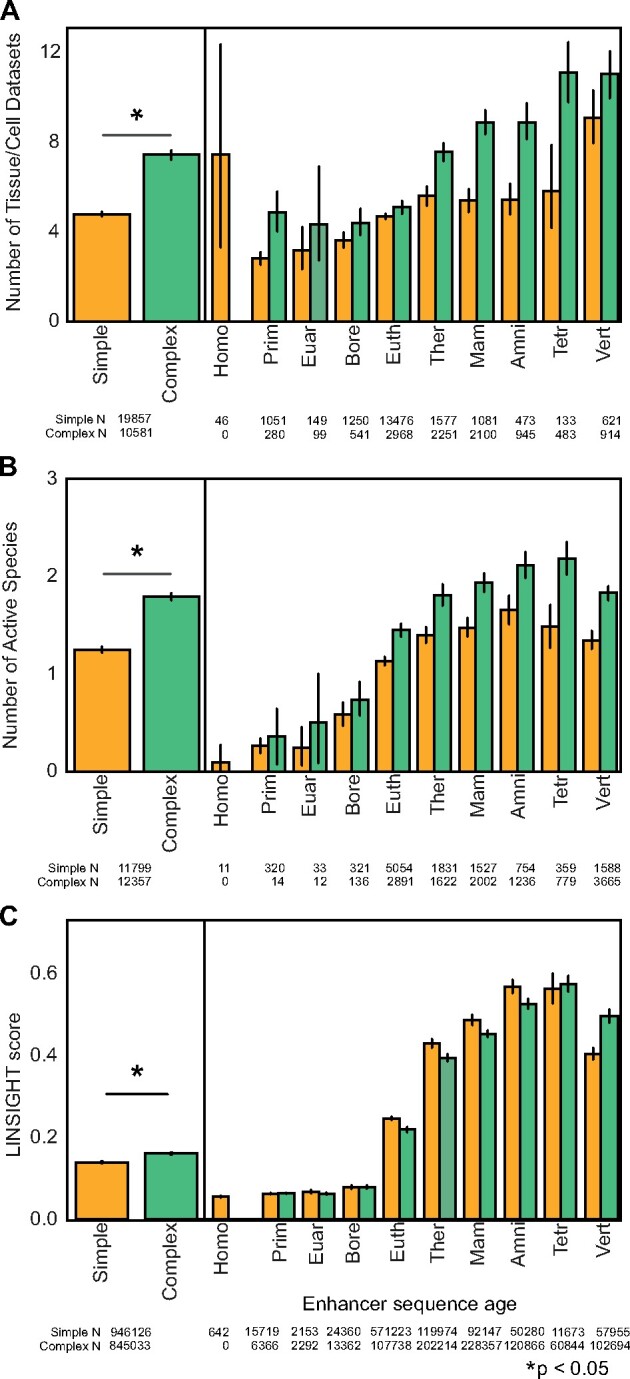
Complex enhancers are more active across tissues and species and under stronger purifying selection than simple enhancers. (A) Complex enhancers are more pleiotropic than simple enhancers. Simple and complex enhancer activity was evaluated across 112 FANTOM enhancer contexts. Overall, simple enhancers are active in 4.8 contexts on average and complex enhancers are active in 7.4 contexts (left, P = 5.9e−199, Mann–Whitney U test). Activity across tissues increases with sequence age, but the effect is stronger for complex enhancers overall and stratified by enhancer age (right). (B) Complex human liver enhancers are active across significantly more species than simple liver enhancers (left, 1.8 vs. 1.2 mean species; P = 5.2e−88). To enable cross-species comparison, this analysis is based on simple enhancers and matched-length complex human liver enhancers defined by H3K27ac+ H3K4me3– ChIP-peaks from Villar 2015 (Materials and Methods, N = 11,799 and 12,357) that were evaluated for enhancer activity across nine placental (Eutherian) mammals. Stratifying by enhancer age reveals that older complex enhancers are active across more species than age-matched simple enhancers (right). (C) Complex enhancers are under slightly stronger purifying selection on the human lineage than simple enhancers (left, 0.16 vs. 0.14 mean LINSIGHT score per bp; P < 2.2e−308). However, estimates stratified by age generally showed similar levels among complex and simple enhancers (right). To account for length differences between architectures, all enhancers were trimmed or expanded to the mean enhancer length of 310 bp. In all panels, error bars represent 95% confidence intervals based on 10,000 bootstraps. Sample size for each age is annotated beneath the x-axis.
We next asked if simple and complex architectures differed in the conservation of enhancer activity across species. This analysis required enhancer maps from the same tissue across species; thus, we assigned age architectures to H3K27ac+ H3K4me3– enhancers identified across liver samples from nine placental mammals (Villar et al. 2015). In this analysis, we used the same median age segment strategy for defining simple and complex enhancers as we used for the FANTOM enhancers (Materials and Methods). To control for differences in length, we matched the length distribution of complex enhancers to simple enhancers (n = 11,799 simple enhancers and n = 12,357 matched-length complex enhancers) and evaluated cross-species activity. As expected from previous studies, human liver enhancers are largely species-specific, but complex liver enhancers are active across significantly more species than simple liver enhancers (fig. 3B left; 1.8 vs. 1.2 mean species; P = 5.2e−88, Mann–Whitney U). In general, older enhancers are more active across species than younger enhancers. Given that younger sequences have fewer opportunities to overlap multiple species than older sequences, we compared cross-species overlap between age-matched sequences (fig. 3B, right). We observe consistent activity differences between age-matched enhancers, indicating that complex enhancer sequence histories are associated with higher cross-species activity compared with simple enhancers from the same age. We also found that human developmental neocortex enhancers with complex architectures (supplementary fig. S13, Supplementary Material online) have more cross-species activity among rhesus macaque and mouse enhancers than simple human neocortex enhancers, though the difference is smaller than for liver enhancers (supplementary fig. S14, Supplementary Material online; 1.29 v. 1.26 species in complex, simple enhancers; P = 7.9e−13), perhaps due to the shallower sampling of these enhancers across species or differences between developmental and adult tissues. These analyses support the conclusion that complex enhancer architecture is associated with more stable activity across species than simple enhancers at each age.
Simple and Complex Enhancers Are Under Similar Levels of Purifying Selection
Given the older ages, greater pleiotropy, and greater cross-species activity observed in complex enhancers, we hypothesized that complex enhancers would be under stronger purifying selection than simple enhancers. To evaluate this, we compared LINSIGHT scores between simple and complex enhancers. Briefly, LINSIGHT estimates the probability of purifying selection on sites in the human genome at a base-pair level using both functional genomics annotations and evolutionary conservation metrics; higher scores indicate stronger purifying selection (Huang et al. 2017). Complex enhancers have slightly higher LINSIGHT scores than simple enhancers overall, suggesting slightly stronger purifying selection in complex enhancers (fig. 3C, left; 0.16 vs. 0.14 mean LINSIGHT score; P < 2.2e−308). Given that simple and complex enhancer sequences have different age distributions, we stratified by age to evaluate whether simple enhancers had lower scores than complex enhancers of the same age (fig. 3C, right). This revealed that per age, simple and complex enhancers do not show a consistent pattern and generally have similar LINSIGHT scores. Similarly, analysis of PhastCons conserved element overlap supports that complex enhancers are overall more conserved than simple enhancers and that the majority of both simple and complex enhancers are highly conserved at older ages (supplementary fig. S15, Supplementary Material online). These results suggest that simple and complex enhancers of similar age experience similar purifying selection pressures.
Genetic Variants in Simple Enhancers Are More Likely to Be Associated with Human Traits and Disease Than Variants in Complex Enhancers
The majority of genetic variants associated with human complex traits and disease are located in functional, non-coding regulatory regions (Maurano et al. 2012; Corradin and Scacheri 2014). Based on the differences in pleiotropy and constraint observed between architectures, we hypothesized that enhancer evolutionary architecture could provide context for interpreting the effects of enhancer variants on traits. To test this, we evaluated enrichment of 55,480 significant (P < 5e−8, linkage disequilibrium expanded at r2 = 1) GWAS Catalog single-nucleotide variants from 2,619 genome-wide association studies (Buniello et al. 2019) in simple and complex enhancer architectures against length- and architecture-matched background regions. We observed GWAS enrichment in both simple enhancers and complex enhancers compared with expected levels (fig. 4A; 1.17-fold-change for simple vs. 1.14-fold-change complex; P = 0.01, two-tailed permutation test). Stratifying by age, we observe GWAS variant enrichment across ages and architectures (supplementary fig. S17, Supplementary Material online). Simple enhancer GWAS enrichment is greater at Primate, Eutherian, and Tetrapod origins, while complex enhancer enrichment is greater in Boreotherian, Mammalian, and Vertebrate origins. This demonstrates that enhancer sequences across different ages and architectures have variant enrichment and association with human traits. More work is needed to evaluate variation in simple and complex enhancer enrichment across tissues, for example by matching the GWAS considered to the different tissue contexts or evaluating variant effect sizes.
Fig. 4.

Simple enhancers are enriched for GWAS hits and variants with significant regulatory activity in massively parallel reporter assays. (A) Simple and complex enhancers are both enriched for GWAS catalog variant overlap compared with random matched regions (1.17-fold enrichment in simple enhancers [N = 690 SNPs], and 1.14-fold enrichment in complex [N = 591 SNPs]). Simple enhancers are more enriched for GWAS variants than complex enhancers (P = 0.01, two-tailed permutation test). Error bars represent 95% confidence intervals based on 10,000 bootstraps. (B) Common genetic variants in simple enhancers are enriched for significant changes in regulatory activity compared with complex enhancers in massively parallel reporter assays (MPRAs). In K562 cells (left), 3.3% of variants in simple enhancers (N = 12,523 variants) and 2.8% of variants in complex enhancers (N = 9,054 variants) exhibit significant changes in MPRA activity compared with 1.7% of all variants tested (simple odds ratio [OR] = 1.9; P = 2.1e−35, and complex OR = 1.6; P = 1.7e−10). This difference in enrichment over background for simple versus complex is significant (OR = 1.2; P = 0.04, Fisher’s Exact Test). In HepG2 cell (right), 4.1% of variants in simple enhancers (N = 568 variants) and 3.8% of variants in complex enhancers (N = 289 variants) produce significant changes in MPRA activity compared with 1.8% of background variants (simple OR = 2.3; P = 1.6e−66 and complex OR =2.1; P = 3.8e−29). The enrichment over background is modestly higher in simple enhancers (OR = 1.1; P = 0.26). The dashed horizontal lines represent the fraction of all variants tested with significant activity per cell line. Error bars represent 95% confidence intervals based on 1,000 bootstraps. Number of overlapping variants are annotated in white.
To explore the patterns of clinically relevant variants in different enhancer architectures, we evaluated ClinVar disease-associated variant enrichment in simple and complex enhancers (Landrum et al. 2018). While GWAS associations reflect variant effects on common, complex diseases, ClinVar pathogenic variants are often the cause of rare Mendelian disorders. Simple enhancer variants overlapped more “pathogenic” annotations while complex enhancers overlapped more “benign” annotations than expected, though these differences were not statistically significant (supplementary fig. S18, Supplementary Material online). Together, these results confirm enrichment for trait and rare disease variants in both complex and simple enhancer architectures compared with regions without enhancer activity; however, known complex trait-associated variation occurs more frequently in simple enhancer architectures.
To complement these findings, we evaluated the enrichment of known expression quantitative trait loci (eQTL). Simple and complex enhancers were similarly enriched for GTEx eQTL across 46 tissues (GTEx Consortium 2017) at ∼1.1× fold-change (supplementary fig. S19, Supplementary Material online; median 1.09× and 1.11× for simple and complex, respectively; P = 0.38, Mann–Whitney U). This indicates that both architecture types are similarly likely to contain variants associated with gene expression variation across individuals.
Genetic Variants in Simple Enhancers Are Enriched for Changes in Biochemical Regulatory Activity Compared with Variants in Complex Enhancers
Given the differences in constraint and complex trait associated variants between simple versus complex enhancers, we hypothesized that there would be architecture-related differences in the effects of variants on gene regulatory biochemical activity. We tested for enrichment of variants that significantly affect biochemical regulatory activity among trimmed simple and complex architectures. We considered >110,000 common human variants shown to affect regulatory activity in recent MPRA performed in K562 and HepG2 cells (van Arensbergen et al. 2019). For both cell lines, variants in annotated enhancers are significantly more likely to have regulatory effects than all background variants tested in the assay (fig. 4B; simple odds ratio [OR] = 1.9; P = 2.1e−35 in K562 and OR = 2.3; P = 1.6e−66 in HepG2; complex OR = 1.6; P = 1.7e−10 in K562 and OR =2.1; P = 3.8e−29 in HepG2, Fisher’s exact test). Simple architectures are more enriched than complex architectures for variants that significantly affect regulatory activity in both K562 (OR = 1.2; P = 0.04) and in HepG2 cells, although the enrichment is smaller (OR = 1.1; P = 0.26). We repeated this analysis using only granulocyte and liver FANTOM enhancers to match the cellular contexts tested and found even stronger enrichment among simple enhancers in these data sets (supplementary fig. S20, Supplementary Material online; liver OR = 1.8; P = 0.08 and granulocyte OR = 1.3; P = 0.13, Fisher’s exact test). These findings indicate that common human variants in simple enhancers are more likely to significantly affect enhancer biochemical regulatory activity than common variants in complex enhancers.
Simple Enhancers Overlap TEDS More Often Than Complex Enhancers
TEDS have enhancer activity across many cellular contexts (Su et al. 2014; Sundaram et al. 2014; Chuong et al. 2017; Simonti et al. 2017; Trizzino et al. 2017; Marnetto et al. 2018;). A previous study identified that TE insertions occur nearby sequence age breaks (Marnetto et al. 2018). We hypothesized that TEDS might have different influences on simple and complex enhancer architectures, and that TEDS integration might contribute to sequence patterns observed in complex architectures. To explore this, we tested TEDS enrichment in simple and complex enhancers against the genomic background. To control for length differences, we evaluated both 310 bp and 1 kb trimmed/expanded enhancers. Both length-control strategies yielded similar results, and we present the 310 bp results below. We intersected the enhancers with genome-wide maps of TEDS (Materials and Methods). We find that 48% of simple enhancers and 42% of complex enhancers contain TEDS. As expected from previous reports (Emera et al. 2016; Simonti et al. 2017), both simple and complex enhancers are depleted of TEDS compared with architecture-matched genomic backgrounds. However, we find that complex enhancers are substantially more depleted (fig. 5A; OR = 0.50 vs. 0.25; P < 2.2e−308, Fisher’s Exact Test). The majority of enhancer sequences younger than the Eutherian ancestor contain TEDs (fig. 5C). Complex enhancers younger than the Therian ancestor and simple enhancers younger than the Eutherian ancestor highly overlap TEDS. This establishes that patterns in both simple and complex enhancers are consistent with previous observations that the majority of young human/primate cis-regulatory elements contain TEDS (Simonti et al. 2017; Trizzino et al. 2017).
Fig. 5.

Simple and complex enhancers are enriched for sequences derived from different transposable element families at different ages. (A) Simple and complex enhancers are significantly depleted of transposable element derived sequences (TEDS) compared with 100 architecture-matched random controls (∼2-fold depleted for simple and ∼4-fold depleted for complex; P < 2.2e−308, Fisher’s Exact Test). The number of TE overlapping elements is annotated in each bar. Within complex enhancers, older core sequences are ∼2-fold-depleted of TEDS compared with the younger, derived sequences (OR = 0.56; P = 9.72e−89). Number of TEDS overlapping elements are annotated per bar. (B) TEDS are enriched in the outer 50% of complex enhancer sequences. TEDS enrichment is quantified as the z-score of TE overlap counts in each normalized enhancer bin across complex enhancers (green, median z-score = 0.17 vs. –0.73, outer v inner 50% bins; P = 6.38e−18, Mann–Whitney U) and simple enhancers (yellow, median z-score = –0.43 vs. –0.43, outer v inner 50% bins; P = 0.47). (C) Percent of simple and complex enhancers overlapping TEDS at each age. Number of TEDS overlapping elements are annotated beneath each bar. (D) Simple and complex enhancers are significantly enriched for sequences derived from different TE families across ages. TEDS enrichment in enhancer architectures was calculated among TEDS-overlapping enhancers of each age using Fisher’s Exact Test. Positive values (green) represent TEDS enrichment in complex enhancers, while negative values represent enrichment in simple enhancers (yellow, * indicates FDR < 0.10). Nonsignificant families are not shown.
TE Sequences Can Both Nucleate and Remodel Enhancers
Sequences with regulatory potential have been hypothesized to nucleate enhancer activity, which can then be expanded and remodeled by the addition of younger sequences (Emera et al. 2016). To explore the role of TEDS in this process, we tested for TEDS enrichment in complex enhancer core sequences versus younger derived sequences. Overall, complex enhancer cores are depleted of TEDS compared with derived sequences (fig. 5A and supplementary fig. S21, Supplementary Material online; OR = 0.56; P = 9.7e−89). We also found strong depletion for TEDS at the centers of complex enhancers and enrichment at their edges (fig. 5B, green; median z-score = –0.73 vs. 0.17, inner vs. outer 50% bins; P = 6.4e−18, Mann–Whitney U). These results are consistent with our finding that younger sequences flank older core sequences in general (fig. 2D), and suggest that TEDS often contribute younger sequences to complex enhancer architectures. However, this general trend is largely driven by old complex enhancers; young complex enhancers (younger than the Therian ancestor) are enriched for TEDS in their cores (supplementary fig. S22, Supplementary Material online). By comparison, TEDS are also enriched at the edges of simple enhancers, though the central regions of simple enhancers do not show strong TEDS depletion (fig. 5B, right panel and supplementary fig. S21, Supplementary Material online). These results support a model where TEDS can both nucleate and remodel enhancer sequences.
Different TE Families Are Enriched in Simple and Complex Enhancers
As discussed above, TE insertions can disrupt functional elements and lead to genome instability. Thus, the probability of TE insertions gaining gene regulatory activity is influenced by their genomic sequence context. We hypothesized that enhancers with different architectures and origins would be enriched for TEDS from specific TE families. Several TE families show biases for simple or complex enhancer architectures at different evolutionary ages (fig. 5D). Complex enhancers are consistently enriched across ages for SINE/Alu, DNA/TcMar-Tigger, and LTR/ERVL-MaLR elements. SINE/Alu elements are abundant in the Primate lineage (Batzer and Deininger 2002), but are also frequently observed in complex enhancers with origins before the Primate ancestor. Integrating young SINE/Alu TEDS with these older sequences may have altered ancient regulatory activity or created new regulatory activity. Simple enhancers are consistently enriched across ages for LINE/CR1, LINE1/L1, and LTR/ERVL elements (fig. 5D). LTR/ERV1 elements are significantly enriched in both older complex and younger simple enhancers, while LINE/L2, DNA/hAT-Charlie, and DNA/hAT-Tip100 are enriched for younger complex enhancers and older simple enhancers. This suggests that these families have contributed sequence to both architectures during different evolutionary phases. Together, different TE families have contributed to enhancer sequences of different origins and evolutionary architectures, and some more often nucleate simple enhancers, while others integrate into complex enhancer architectures.
Age Architectures of Enhancers Identified by Histone Modifications Show Similar Trends
Differences in assays commonly used to identify enhancers influence the sequence resolution, spatiotemporal variability, and many other attributes of the identified enhancers. Both eRNA and histone modification patterns provide imperfect operational definitions for enhancer activity and often disagree with one another (Benton et al. 2019; Gasperini et al. 2020). Given the sequence and temporal specificity of transcribed eRNA enhancers (Tippens et al. 2020), we focused on them throughout the main text. However, we also evaluated our main findings with additional analysis of 2,827,573 autosomal enhancers identified by histone-modification chromatin immunoprecipitation sequencing (ChIP-seq) in 98 cell and tissue contexts from the Roadmap Epigenomics Mapping Consortium (Roadmap Epigenomics Consortium et al. 2015). Histone-mark-identified sequences are more likely to capture an entire regulatory locus, while eRNA-identified sequences capture specific sub-regions with high transcriptional activity (Andersson and Sandelin 2020). Whether the entire length of a putative enhancer sequence is necessary and sufficient for endogenous enhancer function and how this activity is modified by nearby regulatory elements is an area of active research (Gasperini et al. 2020). In this section, we summarize results on Roadmap enhancers and report details in Supplementary Material. Many, but not all, of our findings are consistent between eRNA and Roadmap enhancers identified based on ChIP-seq for histone modifications (supplementary table 1, Supplementary Material online).
Roadmap enhancers are substantially longer than FANTOM enhancers (supplementary fig. S23C, Supplementary Material online; median 2.4 kb vs. 292 bp) and many times the average length of a syntenic block (54 bp). Thus, Roadmap enhancers overlap a median four syntenic blocks (supplementary fig. S24, Supplementary Material online; range 2–8 syntenic blocks per enhancer data set), and enhancers made up of a single syntenic block are rare (2%). To compare Roadmap enhancer architectures to FANTOM enhancers accounting for these differences, we took two complementary approaches. First, we quantified the evolutionary architecture of Roadmap enhancers trimmed to the median FANTOM enhancer length (310 bp centered on the middle of the ChIP-peak). Second, we considered the entire Roadmap enhancer sequence using the same “relative” simple versus complex architecture criterion as we had applied to the FANTOM enhancer; enhancers with fewer syntenic blocks than the median over all enhancers in the context were considered simple (Materials and Methods). As with the FANTOM enhancers, the trimmed Roadmap enhancers exhibit enrichment for simple architectures compared with random regions (supplemental fig. 30A; 58% simple). Under both approaches for analyzing Roadmap enhancers, relative enrichment for simple versus complex enhancer architectures varies across contexts (supplementary figs. S26 and S29, Supplementary Material online). Roadmap enhancers also recapitulate our main findings that complex enhancers exhibit older sequence ages in their centers (supplementary figs. S28–S30, Supplementary Material online), and are more pleiotropic across tissues (supplementary fig. S31A, Supplementary Material online). This relationship between complex enhancers and increased pleiotropy was consistent in both adult and developmental tissues (supplementary fig. S31B, Supplementary Material online). They also support that purifying selection pressures are similar between simple and complex architectures (supplementary fig. S32, Supplementary Material online), while GWAS variant (supplementary fig. S33, Supplementary Material online), ClinVar pathogenic annotations (supplementary fig. S34, Supplementary Material online) and variants affecting biochemical activity (supplementary fig. S20, Supplementary Material online) more often occur in simple enhancers. Thus, evolutionary architecture patterns in histone-mark-defined enhancers largely reflect the findings in transcribed enhancers; however, due to their greater length histone mark-defined enhancers are rarely of a single evolutionary origin.
Discussion
Here, we evaluate the genomic, evolutionary, and functional features associated with human enhancers with different evolutionary age architectures. Human transcribed enhancers have many distinct age architectures—they can consist of sequence of a single origin or complex composites of sequences of many different ages. We demonstrate that simple architectures are favored over complex architectures; however, these patterns vary by cellular context. Functionally, simple and complex architectures show differences in tissue-specific and cross-species activity profiles, but both architectures experience similar selective constraints by age. Simple architectures are slightly more enriched for variants associated with complex traits in GWAS studies, rare pathogenic variants in ClinVar, and variants that significantly alter biochemical activity. Sequences derived from TEs are depleted among all enhancers, but they are more depleted in complex architectures than simple. Nonetheless, these TEDS provided genomic material for many younger enhancers of both architectures and many modified older sequences into complex architectures with enhancer activity. Distinct TE families are enriched in different architectural contexts. Thus, TEDS have made important contributions to the evolution of human enhancers with both simple and complex sequence age architectures. Finally, the consistency of many of these architecture observations across enhancer sequences identified from both eRNA and histone modification patterns (supplementary table 1, Supplementary Material online) supports their generality.
Our work expands current understanding of enhancer sequence evolution in several dimensions. We show that aspects of the two-step proto-enhancer life-cycle model proposed by Emera et al. are present in enhancers across diverse tissues and many of our results hold in their original data set (supplementary figs. S13 and S14, Supplementary Material online). However, the depletion for complex architectures among transcribed enhancer sequences suggests that evolving multi-aged sequence architecture is not necessary for their function and that the juxtaposition of sequences of different origins was not the most common evolutionary history for human transcribed enhancer sequences. Furthermore, several lines of evidence suggest that simple enhancers are not simply a snapshot of proto-enhancers in the first step of the enhancer life cycle: 1) Simple enhancer sequences are often as old as complex enhancers. 2) Simple and complex enhancers of similar ages are under similar levels of purifying selection pressure. 3) Simple enhancers are enriched for tissue-specific functions. 4) Simple enhancers are enriched for GWAS variants, pathogenic ClinVar variants, and variants modifying biochemical activity, implying that simple enhancer variation contributes to human trait variation and changes in molecular function. Together, these results support that enhancers with simple evolutionary architectures play important roles in human gene regulatory biology.
However, simple enhancer sequences may be less evolutionarily stable, as fewer older simple enhancers are observed. In contrast, complex enhancers may be more functionally robust to mutations and evolutionary turnover given their older ages, increased cross-species activity, and trait-associated variant patterns. We speculate that younger derived sequences may protect complex enhancers from inactivating mutations. Future biochemical work could address whether architectural features of complex enhancers may make them more robust to mutations and resistant to evolutionary turnover.
Our analyses consider sequences with human enhancer activity, but enhancer activity often turns over between closely related species (Villar et al. 2015). Thus, we cannot assume that these sequences have maintained enhancer activity since their origin. Highly expressed genes and genes with more evolutionary stable expression patterns are associated with enhancers that have conserved activity across species (Berthelot et al. 2018). When enhancers have evidence of shared activity across species, we show that they are more often complex than simple, even when accounting for age. Many factors likely contribute to this finding. We speculate that older enhancers (whether simple or complex) are more likely to regulate genes with more important and evolutionarily stable expression patterns, and thus experience stronger purifying selection.
Determining how relationships between pleiotropy, cross-species activity, sequence length, and purifying selection pressures shape these enhancer age and architecture observations is challenging. We observed that length is positively correlated with pleiotropy in both simple and complex enhancers (supplementary fig. S12, Supplementary Material online). Thus, we tested whether enhancers with higher pleiotropy are under stronger purifying selection, but found that pleiotropy only weakly correlates with purifying selection in both architectures and fluctuates with age (supplementary fig. S16, Supplementary Material online). This suggests that pleiotropy is not the main driver of enhancer constraint and survival. Dissection of these relationships while controlling for other functional variables must be pursued in future work.
To integrate our findings and provide a framework for future work, we propose a general model for enhancer evolutionary architecture and activity (fig. 6). In our model, inspired by Markov models, sequences occupy either simple or complex architecture states and either active or inactive states. Genomic events (e.g., substitutions and rearrangements) drive transitions between these states over time. Based on our results, we propose that certain paths through the model are common in the enhancer life cycle. Most sequences that ultimately obtain enhancer activity likely begin as inactive or weakly active sequence segments (fig. 6, left). Small-scale genomic events, like point mutations, can strengthen regulatory activity and create simple enhancers (fig. 6, top right). Examples include human accelerated regions, such as HACNS1/HAR2, where human-specific substitutions have created human-specific enhancer activity in limb bud formation (Prabhakar et al. 2008; Cotney et al. 2013). TE insertions also give rise to simple enhancers by integrating sequence with regulatory potential into genomes (Chuong et al. 2017); for example, the mouse-specific RLTR13 endogenous retrovirus sequence is sufficient to drive gene expression in rat placental cells (Chuong et al. 2013).
Fig. 6.
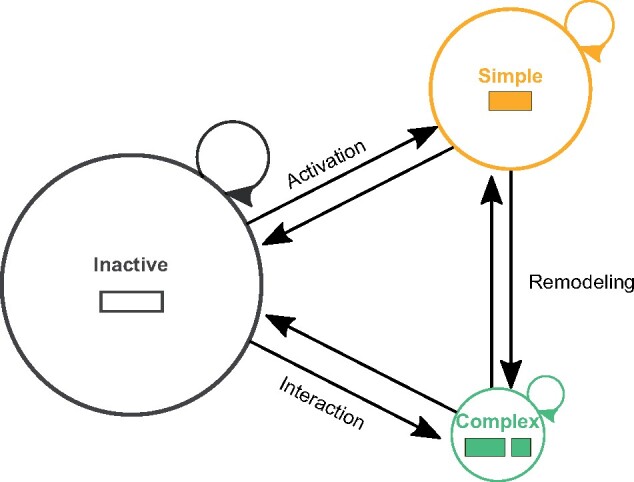
Model of enhancer evolutionary architecture change and activity. Sequences with the potential to be enhancers (rectangles) can occupy active or inactive states and have either simple or complex evolutionary architectures. Sequences transition between these states as a result of large- and small-scale genomic variants. Inactive sequences can become simple enhancers through small-scale genomic changes, such as substitutions that increase activity or nearby chromatin changes that increase accessibility. Complex enhancers sequences of different evolutionary origins are brought together by genomic rearrangements. In some cases, the integration of these sequences and subsequent substitutions produce activity. In others, already active simple enhancers are remodeled into active complex enhancers with presumably different activity patterns. Sequences regularly transition between these states over evolutionary time.
Complex enhancers can emerge from multiple different evolutionary paths. For example, large-scale (greater than a few nucleotides) genomic insertions or rearrangements combined with small-scale substitutions may remodel active simple enhancers into complex enhancers with stronger or different activity patterns (fig. 6, right). Work in Drosophila has demonstrated that small-scale substitutions in complex cross-vein and wing spot enhancers “co-opt” ancestral enhancer activity to develop lineage-specific wing pigmentation patterns (Prud’homme et al. 2006; Koshikawa et al. 2015). Isolated derived segments in these complex enhancers were not sufficient to drive enhancer activity during development, but may function to support lineage-specific enhancer activity in other ways, such as facilitating cooperative or co-activator binding (Long et al. 2016). Complex enhancers can also be created when genomic rearrangements place weakly active sequences of different origins adjacent to each other in such a way that these sequences interact and/or accumulate additional substitutions to create a new active complex enhancer (fig. 6, bottom right). TE insertions can facilitate such interactive effects. For example, the interaction of a LINE/L2 insertion and flanking sequence formed a new enhancer that was both necessary and sufficient for driving increased, lineage-specific GDF6 expression and evolutionary changes in armor-plate size in freshwater stickleback (Indjeian et al. 2016). Older active regulatory sequences may protect TEDS from inactivation by the host genome, creating substrates for complex enhancers to form (Levin and Moran 2011; Varshney et al. 2015; Elbarbary et al. 2016). Finally, deletions can change or inactivate complex and simple sequences with enhancer function. For example, human-specific conserved deletion of a complex enhancer sequence reduces expression of the androgen receptor and is correlated with loss of penile spine and sensory vibrissae anatomy in humans (McLean et al. 2011). Whether complex enhancers undergo deletions to become simple enhancers is not known, and we speculate this rarely occurs. Without experimental dissection, it is currently challenging to trace the history of functional activity, especially for complex enhancer sequences.
We emphasize that most enhancer sequences do not reach a final stable state; sequences continue to change and activity turns over rapidly (Villar et al. 2015). Thus, we constructed our model (fig. 6) to emphasize that sequences regularly transition between these states over evolutionary time. Large comparative regulatory genomics data sets across species and tissues are needed to estimate these transition probabilities. Previous comparisons of both conserved non-coding sequences and TEs suggests that these transition probabilities are not stable over evolutionary time. Instead, there were likely different period of regulatory innovation driven by waves of TE insertions and new cell-signaling modalities (Lowe et al. 2011; Chuong et al. 2013; Lynch et al. 2015). The prevalence of simple architectures indicates many enhancers emerge from a single age, while transitions from simple to complex architecture challenges the idea that enhancers maintain a single function. We hope that future work will enable estimation of rates of simple and complex enhancer emergence, decay, and turnover across other species and over time.
Several limitations must be considered when interpreting our results. First, sequence age estimates are influenced by the accuracy of sequence alignment methods, genome quality, and different rates of sequence divergence across the genome over evolutionary time (Cooper and Brown 2008; Margulies and Birney 2008; Capra, Stolzer, et al. 2013). Assembling and aligning repetitive elements is particularly challenging and may limit TEDS detection (Ewing 2015). Thus, our estimates should be viewed as lower bounds on the actual sequence age. Second, our analyses are limited by the availability and concordance of enhancer data sets. Histone-modification-based ChIP-seq measurements and quantification of eRNA transcription produce enhancer boundary estimates with different resolution and expected functional properties (Andersson et al. 2014; Benton et al. 2019; Tippens et al. 2020) Whether eRNA transcripts represent local enhancer units within larger, multi-cluster chromatin regions, or even sub-regions within “super enhancers” is not resolved (Hay et al. 2016; Moorthy et al. 2017). Further, current enhancer definitions in tissue-level data sets do not capture underlying cellular heterogeneity in epigenetics and expression (Carter and Zhao 2020). Similarly, our cross-species activity analysis is limited by the number of tissues and species assayed, which reduces our power to detect conserved activity. Third, we are limited in our knowledge of human-trait and disease-associated variants. GWAS-variant enrichment reflects tag SNPs and LD-linked loci associated with measurable common human traits; whether the mechanisms underlying their associations to disease pathology or trait variation are mediated by enhancer activity is not clear. The ClinVar variant enrichment analyses are limited by the small number of known pathogenic noncoding variants. As a result, these analyses were underpowered, and the trends for associations between simple architectures and pathogenic variants in both data sets did not reach common thresholds for statistical significance. Finally, we do not explore sequence-level features that distinguish simple and complex architectures. We envision that a thorough analysis of sequence features (e.g., binding site motifs) will reveal distinct sequence patterns between evolutionary periods and evolutionary architectures.
In conclusion, we defined evolutionary architectures of human enhancers and related them to function and genetic variation. Evaluating these architectures revealed different evolutionary origins and evolutionary trajectories among human enhancer sequences. Based on these results, we present a model of enhancer sequence evolution that encompasses the multiple possible evolutionary trajectories. Our work provides a foundation for future studies that dissect the relationships between enhancer evolutionary architecture, sequence patterns, and the consequences on function and noncoding variation in the human genome.
Materials and Methods
Syntenic Block Aging Strategy
The genome-wide hg19 46-way vertebrate multiz multiple species alignment was downloaded from the UCSC genome browser. Each syntenic block was assigned an age based on the MRCA of the species present in the alignment block in the UCSC all species tree model (fig. 1A). For most analyses, we focus on the MRCA-based age, but when a continuous estimate is needed we use evolutionary distances from humans to the MRCA node in the fixed 46-way neutral species phylogenetic tree. Estimates of the divergence times of species pairs in millions of years ago (Ma) were downloaded from TimeTree (Hedges et al. 2015). Sequence age provides a lower-bound on the evolutionary age of the sequence block. Sequence ages could be estimated for 93% of the base pairs (bp) in the human genome.
eRNA Enhancer Identification, Aging, and Architecture Assignment
We considered enhancers called from eRNAs identified across 112 tissue and cell lines by high-resolution cap analysis of gene expression sequencing (CAGE-seq) carried out by the FANTOM5 consortium (Andersson et al. 2014). This yielded a single set of 30,438 autosomal enhancer coordinates. We assigned enhancer ages by intersecting their genomic coordinates with aged syntenic blocks using Bedtools v2.27.1 (Quinlan and Hall 2010). Syntenic blocks that overlapped at least 6 bp of an enhancer sequence (reflecting the minimum size of a TFBS [Lambert et al. 2018]) were considered when assigning the enhancer’s age and architecture. We considered enhancers with one syntenic age as “simple” enhancer architectures and enhancers overlapping more than one syntenic age as “complex” enhancer architectures. Given that some enhancers are composed of multiple sequence ages, we assigned complex enhancer age according to the oldest age. Sequences without an assigned age were excluded from this analysis.
From the human syntenic blocks that could be assigned ages, the plurality (44%) are derived from the placental (Eutherian) ancestor, while 40% are younger than the placental ancestor, and 16% are older (supplementary fig. S3A, Supplementary Material online). This result was consistent with syntenic age estimates using hg38 and 100-way species alignments (Marnetto et al. 2018). Younger syntenic blocks are generally longer than older syntenic blocks (median 128 bp for Primate-specific blocks vs. 42–66 bp for older syntenic blocks) (supplementary fig. S3B, Supplementary Material online).
ChIP-Peak Enhancer Identification, Aging, and Architecture Assignment
We explored the architectures of enhancers identified by the Roadmap Epigenomics Mapping Consortium (Roadmap Epigenomics Consortium et al. 2015) across 98 cellular contexts. Roadmap defined enhancers from histone modification chromatin immunoprecipitation (ChIP-seq) peaks by subtracting H3K4me3+ peaks from H3K27ac+ peaks to exclude active promoters. This resulted in 2,827,573 predicted autosomal enhancers. Enhancers <10 kb in length were considered. Roadmap enhancers were assigned ages as described above for the FANTOM enhancers. Because of increased ChIP-peak lengths, most absolute simple enhancers (i.e., enhancers of a syntenic age) are rare (2%). To account for the differences in the number of possible underlying syntenic blocks, we considered enhancers with less than the median number of syntenic blocks per enhancer (typically one or several syntenic blocks) as “simple” enhancer architectures, while enhancers overlapping equal to or more than the median number of syntenic blocks of different ages have “complex” enhancer architectures. Four age segments per enhancer was the median for multiple Roadmap data sets (supplementary fig. S24, Supplementary Material online), though there was some variation in the median number of age segments per data set.
Trimming and Expansion of ChIP-Peak Enhancer Lengths
For some analyses, we trimmed or expanded Roadmap enhancers to 310 bp to equalize enhancer lengths between ChIP-seq and eRNA sets. However, trimming ChIP peak sequences has limitations. First, it assumes peak centers represent the most stable segment of the enhancer sequence. Second, we exclude flanking sequences that may be important for opening chromatin or recruiting transcriptional machinery. Third, it may bias analysis of complex enhancers toward older sequences, as older sequence ages tend to occur at enhancer centers. Finally, multiple active enhancer subregions might be dispersed throughout a peak or constitute superenhancers.
Human Syntenic Block PhastCons Conservation
PhastCons vertebrate hg19 conserved elements were downloaded from the UCSC genome browser (last accessed April 1, 2017) (Siepel 2005). PhastCons elements were assigned ages using the same MRCA-based strategy described for enhancers. As expected, sequence age is correlated with sequence conservation (R2 = 0.82; P = 0.009), since sequence homology is the basis for estimating both sequence age and sequence conservation. However, these metrics capture complementary information about regions of interest. Sequence conservation summarizes the evidence that purifying selection has acted on the region, and conserved sequences have high similarity across species. Sequence age estimates a lower bound on the evolutionary origin of a sequence and can be assigned both to conserved sequences and neutrally evolving sequences with lower sequence identity among species. For example, only 35% of the oldest syntenic blocks have significant evidence of evolutionarily conservation (Vertebrate PhastCons overlap, supplementary fig. S3C, Supplementary Material online). In other words, not all old sequences have evidence of significant conservation. Thus, even though neutrally evolving sequences become more difficult to accurately age with time (such that age reflects a lower bound estimate of sequence origin), sequence age provides complementary information about sequences shared among vertebrates.
Background Random Genome Regions and Architectures
For FANTOM enhancers, 100 random shuffles of the genomic regions in each data set of interest (e.g., cellular context) were performed using BEDTools. For Roadmap enhancers, each of the 98 tissue data sets was shuffled 10 times, resulting in 980 shuffled data sets total. The shuffled sets were matched on chromosome number and enhancer length, and they excluded both Ensembl exon coordinates (supplementary fig. S28, Supplementary Material online) and ENCODE blacklist regions and genomic gaps as defined by the hg19 UCSC gaps track (Amemiya et al. 2019). Random genomic regions were then assigned ages and architectures with the same strategy used for enhancers described above (supplementary fig. S1, Supplementary Material online). We calculated enrichments by comparing the observed enhancer age and architecture distribution with the expectation from the appropriate sets of shuffled regions.
Enhancer Pleiotropy
To account for the effects of enhancer architecture length differences in quantification of enhancer activity across biological contexts, FANTOM enhancers were trimmed around their midpoints to the mean length of all enhancers in the data set (310 bp). Roadmap enhancers were similarly trimmed to the mean length per data set. Trimmed enhancer data sets were intersected with 112 FANTOM eRNA tissue facets and cell line data sets or with 98 Roadmap ChIP-seq data sets using BEDTools multi-intersect command. We considered an enhancer pleiotropic when at least 50% of the enhancer length overlapped enhancers in other contexts.
Cross-Species Enhancer Activity
Human liver enhancers from a cross-species analysis of vertebrate livers (Villar et al. 2015) were assigned ages and architectures. Briefly, the authors used pairwise lastZ alignments to determine the sequence conservation of H3K27ac+ H3K4me3– peaks from nine placental mammal livers. Sequence conservation was required to map peak accessibility in both species. The authors then evaluated whether sequences were found in active chromatin of either or both species in order to call cross-species activity. In other words, sequence must be sufficiently conserved to identify cross-species activity. Simple architecture was assigned to enhancers with <5 age segments, as five was the median number of age segments in this data set. To account for length differences, complex enhancer lengths were matched to the simple enhancer lengths (N = 11,799 and N = 12,357 matched-length complex and simple enhancers).
Further, we leveraged a H3K27ac ChIP-seq data set assayed in developmental mouse, rhesus macaque, and human neocortex samples from Reilly et al. (2015). The Emera et al data set is derived from the Reilly et al. data set and filtered on human–mouse active enhancer overlap and alignment. Sequence conservation was required to determine if ChIP-peaks were active across species. Enhancer sequences were assigned ages and architectures. Simple architectures were defined as enhancers with <5 age segments per element (data set-wide median number of age segments). Enhancer architectures were matched on length for analysis of cross-species activity (N = 17,670 simple and N = 22,506 complex enhancers).
Enhancer Sequence Constraint
LINSIGHT scores were downloaded from http://compgen.cshl.edu/∼yihuang/LINSIGHT/ (last accessed August 1, 2019). LINSIGHT provides per base pair estimates of negative selection (Huang et al. 2017). Enhancers were intersected with LINSIGHT base pair estimates. 46-way hg19 vertebrate PhastCons elements were downloaded from the UCSC genome browser. Enhancers overlapping any PhastCons element by at least 6 bp were considered conserved.
GWAS Catalog Enrichment
Enrichment for overlap with 55,480 GWAS Catalog variants (P < 5e−8) from 2601 traits (last downloaded September 24, 2019) (Buniello et al. 2019) were linkage disequilibrium expanded (r2 =1.0) using European 1000 Genome phase reference panels (The 1000 Genomes Project Consortium 2015). Enrichment was tested by comparing the observed overlap for a set of regions of interest with overlaps observed across 100 shuffled sets matched on length, sequence age architecture, and chromosome. Median fold-change was calculated based on the GWAS Catalog variants overlapping enhancer architectures compared with these random genomic sets. Confidence intervals (CI = 95%) were generated by bootstrapping the 1,000 random genomic fold-change values 10,000 times. P-values were corrected for multiple hypothesis testing by controlling the false discovery rate (FDR) at 5% using the Benjamini–Hochberg procedure.
ClinVar Variant Enrichment
ClinVar variants in VCF format were downloaded from ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/ (last downloaded February 12, 2019). Trimmed FANTOM and Roadmap enhancers were intersected with ClinVar variants. FANTOM enhancers overlapped 21 annotated variants total (n = 9 simple, n = 12 complex). Among 98 Roadmap tissue enhancer sets, nonexonic enhancers overlapped 24 annotated ClinVar variants (n = 7 simple, n = 17 complex). ClinVar variants were considered pathogenic if annotated with the term “pathogenic” and excluded if annotated with the term “conflicting.” Similar inclusion and exclusion criteria were used for “benign” and “protective.” The fraction of annotated variants per architecture was estimated as the number of “pathogenic,” “benign,” or “protective” annotations versus all ClinVar variants overlapping that architecture.
eQTL Enrichment
Enrichment for GTEx v6 eQTL from 46 tissues (last downloaded July 23, 2019) (GTEx Consortium 2017) in enhancers with simple and complex architectures was tested against a null distribution determined by shuffling observed enhancers using the same strategy as described for GWAS variant enrichment.
MPRA Data
Results from recent MPRAs (van Arensbergen et al. 2019) were downloaded. Significant changes in MPRA activity and p-values were calculated by the authors using a Wilcoxon rank-sum test with a 5% FDR separately identified in K562 and HepG2 cell lines. Trimmed enhancers were intersected with alleles tested in MPRA. Ninety-five percent confidence intervals were estimated with 1,000 bootstraps. Fisher’s Exact Test was used to estimate the odds an allele with significant changes in MPRA activity occurred in a specific architecture compared with the background set of alleles that do not overlap enhancers. Significant allele overlap was also compared between simple and complex enhancer architectures to estimate an OR of enrichment.
TEDS Enrichment
TEDS identified by RepeatMasker were downloaded from the UCSC genome browser and liftedOver to hg19 from hg38 (last downloaded April 14, 2018). Trimmed enhancers (310 bp) were intersected with TEDS coordinates. TEDS overlapping enhancers ≥6 bp were evaluated further for enrichment in FANTOM enhancers of different ages. Enrichment was estimated as the number of TEDS in enhancer architectures compared with random-shuffled regions matched on both length and architecture using Fisher’s Exact Test. We compared enrichment between core and derived segments of complex enhancers by using Fisher’s Exact Test on TEDS overlap counts in core and derived syntenic blocks. To estimate TEDS family enrichment in enhancers with different sequence age architectures, we compared the number of simple/complex enhancers overlapping a TEDS family with the number of simple/complex architectures overlapping any other TEDS family of that age. Enrichment significance was evaluated using Fisher’s Exact Test and FDR controlled at 10%.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank members of the Capra Lab, Emily Hodges, and Tyler Hansen for helpful discussions. This study was supported by the National Institutes of Health grants R35GM127087 to J.A.C. and T32GM080178 to S.L.F.
Data Availability
The syntenic age data underlying this article are available in Zenodo, at https://doi.org/10.5281/zenodo.4734606.
The following data sets were derived from sources in the public domain:
FANTOM5 (Andersson et al. 2014)—http://slidebase.binf.ku.dk/human_enhancers/
ROADMAP (Roadmap Epigenomics Consortium et al. 2015)—https://egg2.wustl.edu/roadmap/web_portal/processed_data.html#ChipSeq_DNaseSeq
Villar (Villar et al. 2015)—https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2633/
Reilly (Reilly et al. 2015)—GSE63649.
Hg19 46-way vertebrate species multiz alignment—https://hgdownload.soe.ucsc.edu/gbdb/hg19/multiz46way/
LINSIGHT (Huang et al. 2017)—http://compgen.cshl.edu/LINSIGHT/LINSIGHT.bw
Phastcons—https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=cons46way
Van Arensbergen (van Arensbergen et al. 2019)—GSE128325.
GWAS (Buniello et al. 2019)—https://www.ebi.ac.uk/gwas/api/search/downloads/full
ClinVar (Landrum et al. 2018)—https://ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh37/
Repeatmasker—http://genome.ucsc.edu/cgi-bin/hgTrackUi?g=rmsk
All data analysis scripts are available at: https://github.com/slifong08/enh_ages/tree/master/age_arch
References
- Amemiya HM, Kundaje A, Boyle AP.. 2019. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep. 9(1):9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2014. An atlas of active enhancers across human cell types and tissues. Nature 507(7493):455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Sandelin A.. 2020. Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet. 21(2):71–87. [DOI] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL.. 2002. Alu repeats and human genomic diversity. Nat Rev Genet. 3(5):370–379. [DOI] [PubMed] [Google Scholar]
- Benton ML, Talipineni SC, Kostka D, Capra JA.. 2019. Genome-wide enhancer annotations differ significantly in genomic distribution, evolution, and function. BMC Genomics 20(1):511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthelot C, Villar D, Horvath JE, Odom DT, Flicek P.. 2018. Complexity and conservation of regulatory landscapes underlie evolutionary resilience of mammalian gene expression. Nat Ecol Evol. 2(1):152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. 2019. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47(D1):D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Erwin GD, McKinsey G, Rubenstein JLR, Pollard KS.. 2013. Many human accelerated regions are developmental enhancers. Philos Trans R Soc B: Biol Sci. 368(1632):20130025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Stolzer M, Durand D, Pollard KS.. 2013. How old is my gene? Trends Genet. 29(11):659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter B, Zhao K.. 2020. The epigenetic basis of cellular heterogeneity. Nat Rev Genet. 22(4):235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C.. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 18(2):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Rumi MAK, Soares MJ, Baker JC.. 2013. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat Genet. 45(3):325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Brown CD.. 2008. Qualifying the relationship between sequence conservation and molecular function. Genome Res. 18(2):201–205. [DOI] [PubMed] [Google Scholar]
- Corradin O, Scacheri PC.. 2014. Enhancer variants: evaluating functions in common disease. Genome Med. 6(10):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Leng J, Yin J, Reilly SK, DeMare LE, Emera D, Ayoub AE, Rakic P, Noonan JP.. 2013. The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154(1):185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary RA, Lucas BA, Maquat LE.. 2016. Retrotransposons as regulators of gene expression. Science (New York, NY). 351(6274):aac7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emera D, Yin J, Reilly SK, Gockley J, Noonan JP.. 2016. Origin and evolution of developmental enhancers in the mammalian neocortex. Proc Natl Acad Sci USA. 113(19):E2617–E2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD.2015. Transposable element detection from whole genome sequence data. Mob DNA. 6(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish A, Chen L, Capra JA.. 2017. Gene regulatory enhancers with evolutionarily conserved activity are more pleiotropic than those with species-specific activity. Genome Biol Evol. 9(10):2615–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini M, Tome JM, Shendure J.. 2020. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat Rev Genet. 21(5):292–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium. 2017. Genetic effects on gene expression across human tissues. Nature 550(7675):204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HayD, , HughesJR, , BabbsC, , DaviesJOJ, , GrahamBJ, , HanssenL, , KassoufMT, , Marieke OudelaarAM, , SharpeJA, , Suciu MC, . et al. 2016. Genetic dissection of the α-globin super-enhancer in vivo. Nat Genet. 48(8):895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Marin J, Suleski M, Paymer M, Kumar S.. 2015. Tree of life reveals clock-like speciation and diversification. Mol Biol Evol. 32(4):835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-F, Gulko B, Siepel A.. 2017. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat Genet. 49(4):618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hujoel MLA, Gazal S, Hormozdiari F, van de Geijn B, Price AL.. 2019. Disease heritability enrichment of regulatory elements is concentrated in elements with ancient sequence age and conserved function across species. Am J Hum Genet. 104(4):611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indjeian VB, Kingman GA, Jones FC, Guenther CA, Grimwood J, Schmutz J, Myers RM, Kingsley DM.. 2016. Evolving new skeletal traits by cis-regulatory changes in bone morphogenetic proteins. Cell 164(1–2):45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshikawa S, Giorgianni MW, Vaccaro K, Kassner VA, Yoder JH, Werner T, Carroll SB.. 2015. Gain of cis-regulatory activities underlies novel domains of wingless gene expression in Drosophila. Proc Natl Acad Sci USA. 112(24):7524–7529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT.. 2018. The human transcription factors. Cell 172(4):650–665. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. 2018. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46(D1):D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, Moran JV.. 2011. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet. 12(9):615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, Wysocka J.. 2016. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167(5):1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe CB, Kellis M, Siepel A, Raney BJ, Clamp M, Salama SR, Kingsley DM, Lindblad-Toh K, Haussler D.. 2011. Three periods of regulatory innovation during vertebrate evolution. Science 333(6045):1019–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch VJ, Nnamani MC, Kapusta A, Brayer K, Plaza SL, Mazur EC, Emera D, Sheikh SZ, Grützner F, Bauersachs S, et al. 2015. Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep. 10(4):551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies EH, Birney E.. 2008. Approaches to comparative sequence analysis: towards a functional view of vertebrate genomes. Nat Rev Genet. 9(4):303–313. [DOI] [PubMed] [Google Scholar]
- Marnetto D, Mantica F, Molineris I, Grassi E, Pesando I, Provero P.. 2018. Evolutionary rewiring of human regulatory networks by waves of genome expansion. Am J Hum Genet. 102(2):207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337(6099):1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, et al. 2011. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471(7337):216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JM, Capra JA, Abbot P, Rokas A.. 2019. Signatures of recent positive selection in enhancers across 41 human tissues. G3 Genes|Genomes|Genetics. 9(8):2761–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MoorthySD, , DavidsonS, , ShchukaVM, , SinghG, , Malek-GilaniN, , LangroudiL, , MartchenkoA, , SoV, , MacphersonNN, , Mitchell JA.. 2017. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 27(2):246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, et al. 2006. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2(10):e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, FitzPatrick DR, Afzal V, et al. 2008. Human-specific gain of function in a developmental enhancer. Science 321(5894):1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud’homme B, Gompel N, Rokas A, Kassner VA, Williams TM, Yeh S-D, True JR, Carroll SB.. 2006. Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature 440(7087):1050–1053. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6):841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SK, Yin J, Ayoub AE, Emera D, Leng J, Cotney J, Sarro R, Rakic P, Noonan JP.. 2015. Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science (New York, NY). 347(6226):1155–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518(7539):317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science (New York, NY). 328(5981):1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A.. 2014. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 15(4):272–286. [DOI] [PubMed] [Google Scholar]
- Siepel A.2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15(8):1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonti CN, Pavličev M, Capra JA.. 2017. Transposable element exaptation into regulatory regions is rare, influenced by evolutionary age, and subject to pleiotropic constraints. Mol Biol Evol. 34(11):2856–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Han D, Boyd-Kirkup J, Yu X, Han J-DJ.. 2014. Evolution of Alu elements toward enhancers. Cell Rep. 7(2):376–385. [DOI] [PubMed] [Google Scholar]
- Sundaram V, Cheng Y, Ma Z, Li D, Xing X, Edge P, Snyder MP, Wang T.. 2014. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 24(12):1963–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489(7414):75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippens ND, Liang J, Leung AK-Y, Wierbowski SD, Ozer A, Booth JG, Lis JT, Yu H.. 2020. Transcription imparts architecture, function and logic to enhancer units. Nat Genet. 52(10):1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trizzino M, Park Y, Holsbach-Beltrame M, Aracena K, Mika K, Caliskan M, Perry GH, Lynch VJ, Brown CD.. 2017. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res. 27(10):1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J, Pagie L, FitzPatrick VD, de Haas M, Baltissen MP, Comoglio F, van der Weide RH, Teunissen H, Võsa U, Franke L, et al. 2019. High-throughput identification of human SNPs affecting regulatory element activity. Nat Genet. 51(7):1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney D, Vavrova-Anderson J, Oler AJ, Cowling VH, Cairns BR, White RJ.. 2015. SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation. Nat Commun. 6(1):6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J, Rynes E, Sandstrom R, Zhang M, Canfield T, Hansen RS, Stehling-Sun S, Sabo PJ, Byron R, Humbert R, et al. 2014. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science (New York, NY). 346(6212):1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, et al. 2015. Enhancer evolution across 20 mammalian species. Cell 160(3):554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Kalay G.. 2012. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet. 13(1):59–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The syntenic age data underlying this article are available in Zenodo, at https://doi.org/10.5281/zenodo.4734606.
The following data sets were derived from sources in the public domain:
FANTOM5 (Andersson et al. 2014)—http://slidebase.binf.ku.dk/human_enhancers/
ROADMAP (Roadmap Epigenomics Consortium et al. 2015)—https://egg2.wustl.edu/roadmap/web_portal/processed_data.html#ChipSeq_DNaseSeq
Villar (Villar et al. 2015)—https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2633/
Reilly (Reilly et al. 2015)—GSE63649.
Hg19 46-way vertebrate species multiz alignment—https://hgdownload.soe.ucsc.edu/gbdb/hg19/multiz46way/
LINSIGHT (Huang et al. 2017)—http://compgen.cshl.edu/LINSIGHT/LINSIGHT.bw
Phastcons—https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=cons46way
Van Arensbergen (van Arensbergen et al. 2019)—GSE128325.
GWAS (Buniello et al. 2019)—https://www.ebi.ac.uk/gwas/api/search/downloads/full
ClinVar (Landrum et al. 2018)—https://ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh37/
Repeatmasker—http://genome.ucsc.edu/cgi-bin/hgTrackUi?g=rmsk
All data analysis scripts are available at: https://github.com/slifong08/enh_ages/tree/master/age_arch