

Abstract
Whole genome duplication (WGD) can promote adaptation but is disruptive to conserved processes, especially meiosis. Studies in Arabidopsis arenosa revealed a coordinated evolutionary response to WGD involving interacting proteins controlling meiotic crossovers, which are minimized in an autotetraploid (within-species polyploid) to avoid missegregation. Here, we test whether this surprising flexibility of a conserved essential process, meiosis, is recapitulated in an independent WGD system, Cardamine amara, 17 My diverged from A. arenosa. We assess meiotic stability and perform population-based scans for positive selection, contrasting the genomic response to WGD in C. amara with that of A. arenosa. We found in C. amara the strongest selection signals at genes with predicted functions thought important to adaptation to WGD: meiosis, chromosome remodeling, cell cycle, and ion transport. However, genomic responses to WGD in the two species differ: minimal ortholog-level convergence emerged, with none of the meiosis genes found in A. arenosa exhibiting strong signal in C. amara. This is consistent with our observations of lower meiotic stability and occasional clonal spreading in diploid C. amara, suggesting that nascent C. amara autotetraploid lineages were preadapted by their diploid lifestyle to survive while enduring reduced meiotic fidelity. However, in contrast to a lack of ortholog convergence, we see process-level and network convergence in DNA management, chromosome organization, stress signaling, and ion homeostasis processes. This gives the first insight into the salient adaptations required to meet the challenges of a WGD state and shows that autopolyploids can utilize multiple evolutionary trajectories to adapt to WGD.
Keywords: polyploidy, convergence, genome duplication, adaptation
Introduction
Whole genome duplication (WGD) is both a massive mutation and a powerful force in evolution. The opportunities and challenges presented by WGD emerge immediately, realized in a single generation. As such, WGD comes as a shock to the system. Autopolyploids, formed by within-species WGD (without hybridization), result from the chance encounter of unreduced gametes (with diverse underlying factors, see Mason and Pires 2015). Thus, they typically harbor four full haploid genomes that are similar in all pairwise combinations, resulting in a lack of pairing partner preferences at meiosis. This, combined with multiple crossover events per chromosome pair, can result in multivalents among three or more homologs at anaphase, increasing the likelihood of missegregation or chromosome breakage, leading to aneuploidy (Bomblies and Madlung 2014; Bomblies et al. 2016). Beyond this, WGD presents a suddenly transformed intracellular landscape to the conserved workings of the cell, such as altered ion homeostasis and a host of nucleotypic factors related to cell size, volume, and cell cycle progression (Chao et al. 2013; Yant and Bomblies 2015; Doyle and Coate 2019; Bomblies 2020).
Despite this, some lineages survive this early trauma and successfully speciate, with direct empirical evidence of the increased adaptability of autopolyploid lineages from in vitro evolutionary competition experiments in yeast (Selmecki et al. 2015). With increased ploidy, genetic variability can be maintained in a masked state, with evidence of young WGD lineages further recruiting diverse alleles by gene flow across ploidies, and indeed, species (Arnold et al. 2016; Marburger et al. 2019; Monnahan et al. 2019). At the genomic level, recent detailed understanding of gene flow following WGD supports the idea that WGD can cause the breakdown of species barriers present in diploids. Evidence for this has come from both plants (Arabidopsis arenosa/Arabidopsis lyrata [Schmickl and Koch 2011]) and animals (the frog genus Neobatrachus [Novikova et al. 2020], reviewed in Schmickl and Yant 2021). In both examples WGD led to niche expansion (Molina-Henao and Hopkins 2019; Novikova et al. 2020) and the invasion of particularly challenging environments relative to the diploid: in the case of polyploid frogs, the desert (Novikova et al. 2020) and polyploid A. arenosa, metal-contaminated mines and serpentine barrens (Arnold et al. 2016; Preite et al. 2019; Konečná et al. 2021). Thus, although clear challenges must be overcome to function as a polyploid (Bomblies et al. 2015; Yant and Bomblies 2015; Baduel, Hunter, et al. 2018), novel population genomic and ecological opportunities await a lineage that successfully adapts to a WGD state (Yant and Bomblies 2015; Baduel, Hunter, et al. 2018).
The functional and genomic basis for adaptation to WGD has been closely investigated in A. arenosa, which exists as both diploid and young autotetraploid lineages (∼20,000 generations old; Arnold et al. 2015; Kolář et al. 2016). Population genomic scans for selection using a diversity of metrics have shown the strongest signals of positive selection following WGD in A. arenosa as sharp, single-gene peaks over 10 genes that physically and functionally interact to control meiotic chromosome crossovers (Hollister et al. 2012; Yant et al. 2013; Bohutínská et al. 2021a). During early meiotic chromosome crossover formation in an autotetraploid, the four copies of each chromosome are impossible to distinguish. Thus, crossovers can occur haphazardly in any pairwise manner. If more than one crossover per chromosome pair is allowed to occur, multivalent associations can result, leading to aneuploidy at anaphase. Thus a reduction in the number of meiotic crossovers to one per chromosome pair stands as the leading candidate process mediating adaptation to WGD (Bomblies et al. 2016). In the young A. arenosa autotetraploids harboring these derived alleles, we observed a decrease in meiotic crossover number as well as fewer multivalents relative to synthetic autopolyploids with ancestral-like diploid alleles (Yant et al. 2013). Recent work found that the closely related sister species A. lyrata, which contains a younger autotetraploid lineage, also harbors many of the same selected alleles discovered in A. arenosa (Marburger et al. 2019). Moreover, from a joint population genomic analysis of both species across an established natural hybrid zone between A. arenosa and A. lyrata, clear gene sized signals of directional adaptive gene flow and positive selection emerge precisely at these alleles specifically between the two tetraploids (Marburger et al. 2019; Seear et al. 2020), indicating that A. lyrata and A. arenosa WGD stabilization events are not fully independent. Among these candidate adaptive alleles at least one has been functionally shown to modulate adaptive decreases in crossover numbers (Morgan et al. 2020; Seear et al. 2020),
Here, we use an independent system, approximately 17 My diverged from both A. arenosa and A. lyrata (Huang et al. 2020), to test the hypothesis that this solution of meiosis gene evolution is repeated, and if not, whether changes in other genes from analogous processes are associated with adaptation to WGD. Given the clear results in A. arenosa and A. lyrata, we hypothesized that the adaptive trajectories which are available to mediate adaptation to a WGD state are constrained, leading to repeated selection of the same suite of meiosis genes. Such a result would offer a striking case of convergent evolution in core cellular processes. To test this hypothesis, we take advantage of a well-characterized model, Cardamine amara (Brassicaceae, tribe Cardamineae). A large-scale cytotyping survey of over approximately 3,300 individuals in 302 populations and genetic analysis detail the demographic relationships of this diploid/tetraploid complex in the Eastern and Central Alps (Zozomová-Lihová et al. 2015). Comparison of genotyping results of this study with simulations indicates a single autotetraploid origin. Importantly, C. amara is a perennial herb harboring a high level of genetic diversity and shares with A. arenosa a similar distribution range and evolutionary history, with a likely single geographic origin, followed by autotetraploid expansion associated with glacial oscillations (Marhold et al. 2002; Zozomová-Lihová et al. 2015).
To test our hypothesis that gene-level evolutionary convergence is likely following WGD, we performed genome scans for positive selection in both C. amara and A. arenosa, contrasting natural autotetraploid and diploid populations in both species. Because there was no reference genome available for C. amara, we first generated a novel quality reference. We then tested for convergence in the evolutionary response to WGD at the level of the ortholog, process, and network in a sampling of 100 C. amara and 120 A. arenosa individuals from well-assessed ranges (Arnold et al. 2015; Zozomová-Lihová et al. 2015; Kolář et al. 2016; Monnahan et al. 2019). Overall, we found that the evolutionary response to WGD in C. amara is very different to that of A. arenosa, with none of the orthologous genes that control meiotic chromosome crossovers in A. arenosa under strong selection in C. amara. In contrast, we find a clear signal of process-level convergence in core pathways controlling DNA management and chromosome organization.
Results and Discussion
Reference Genome, Population Selection, Sampling, and Genetic Structure
Because C. amara is approximately 17 My diverged from A. arenosa (Huang et al. 2020), using the same reference genome for mapping reads of both species would result in unacceptably low mapping efficiencies and missing data. We therefore first generated a novel reference genome for C. amara (N50 = 1.82 Mb, 95% complete BUSCOs; see Materials and Methods). We then resequenced in triplicate four populations of contrasting ploidy, sampling 100 individuals: two diploid (LUZ, VRK) and two autotetraploid (CEZ, PIC; fig. 1a;supplementary table 1, Supplementary Material online). We chose these populations based on a comprehensive cytological and demographic survey of approximately 3,300 C. amara samples throughout the Czech Republic (Zozomová-Lihová et al. 2015). Sampled plants were spaced at least 3 m apart, as this distance was sufficient to avoid resampling of identical clones in that study. We chose populations to represent core areas of each cytotype, away from potential hybrid zones and distant from any triploid-containing populations based on (Zozomová-Lihová et al. 2015). Further, we performed flow cytometry on every sample sequenced to verify expected ploidy.
Fig. 1.
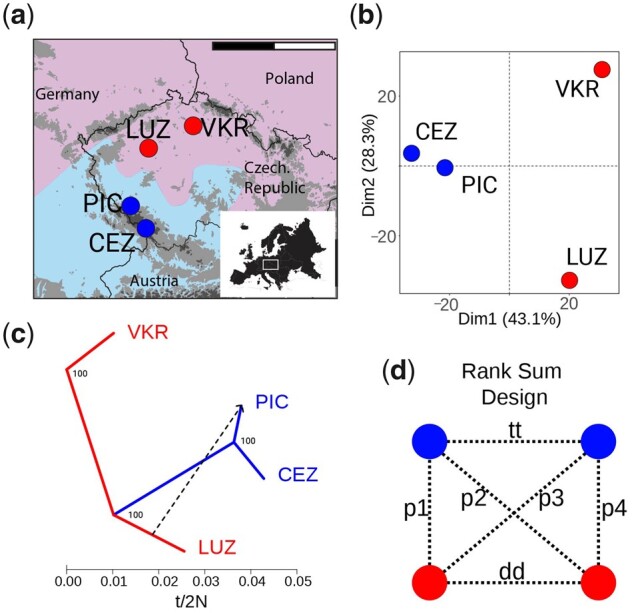
Sampling and population structure of Cardamine amara. (a) Locations of diploid (red) and autotetraploid (blue) C. amara populations sampled. Scale bar corresponds to 200 km; shaded area represents each cytotype range in Zozomová-Lihová et al. (2015). (b) Population differentiation represented by Principal Component Analysis of approximately 124,000 4-fold degenerate SNPs. (c) Phylogenetic relationships and migration events between populations inferred by TreeMix analysis. X-axis shows the drift estimation, corresponding to the number of generations separating the two populations (t), and effective population size (N) (Pickrell and Pritchard 2012). Node labels show bootstrap support, and the arrow indicates the most likely migration event (migration weight, which can be interpreted as a moderate degree of admixture = 0.18, similar to Arabidopsis arenosa, shown in supplementary fig. 1, Supplementary Material online). Additional migration events did not improve the model likelihood. (d) Rank Sum design used in divergence scans to minimize potential bias of population-specific divergence. p1–p4 represent the between-ploidy contrasts used for the rank sum calculations. dd and tt represent within-ploidy contrasts used to subtract signal of local population history within each cytotype.
To obtain robust population allele frequency (AF) estimates across genomes, we performed a replicated pooled sequencing approach. From every population we pooled DNA from 25 individuals and generated on average 31 million reads per sample (for all samples we generated triplicate DNA preps, pooling, and sequencing to control for potential sampling error: details in Materials and Methods) and mapped reads to our new C. amara assembly (mean coverage per population = 86, supplementary table 2, Supplementary Material online). After mapping, variant calling and quality filtration, we obtained a final data set of 2,477,517 SNPs.
The first PCA axis dominantly explained 43% of variation (fig. 1b) and was consistent with differentiation primarily by geographic distribution or ploidy (which coincide), followed by differentiation between the two diploid populations from each other (second axis explaining 28% of variation). The two autotetraploid populations clustered together in the TreeMix graph (fig. 1c) and had the lowest genetic differentiation of all contrasts (Fst = 0.04, mean AF difference = 0.06, table 1) and lacked any fixed SNP difference whatsoever (table 1). This high genetic similarity and spatial arrangement (the populations represent part of a continuous range of the autotetraploid cytotype), suggest that both autotetraploid populations represent the outcome of a single polyploidization event, in line with previous assessments (Marhold et al. 2002; Zozomová-Lihová et al. 2015), although multiple tetraploid origins cannot be ruled out. The absence of individual-level genotype information did not allow for exact dating, but nearly identical levels of interploidy divergence in both C. amara and A. arenosa (average Fst between diploids and autotetraploids = 0.10 and 0.11, respectively) and comparable drift estimates in TreeMix (supplementary fig. 1, Supplementary Material online), suggested that the polyploidization may be roughly the same age (table 1). Supporting this, both WGD events were estimated to correspond with the end of the last European glaciation (Marhold et al. 2002; Arnold et al. 2015; Zozomová-Lihová et al. 2015).
Table 1.
Measures of Genome-wide Differentiation between Cardamine amara and Arabidopsis arenosa Populations.
| Populations | Ploidies | Mean AFD | Fixed Diffs | Mean Fst | No. of SNPs |
|---|---|---|---|---|---|
| PIC–VKR | 4× – 2× | 0.09 | 30 | 0.09 | 2,326,315 |
| PIC–LUZ | 4× – 2× | 0.09 | 2 | 0.08 | 2,314,229 |
| CEZ–VKR | 4× – 2× | 0.11 | 120 | 0.12 | 2,333,538 |
| CEZ–LUZ | 4× – 2× | 0.11 | 86 | 0.11 | 2,335,004 |
| CEZ–PIC | 4× – 4× | 0.06 | 0 | 0.04 | 2,297,229 |
| LUZ–VKR | 2× – 2× | 0.10 | 6 | 0.09 | 2,018,892 |
| A. arenosa tetraploids–A. arenosa diploids | 4× – 2× | 0.05 | 21 | 0.11 | 7,106,848 |
Note.—Differentiation metrics shown are genome-wide mean allele frequency difference between populations (Mean AFD), the number of fixed differences (Fixed diffs) and mean Fst (Nei 1972). In the case of A. arenosa, Fst in diploids is calculated as a mean over all pairwise Fst measurements between the five previously characterized diploid lineages (Monnahan et al. 2019).
Selection Specifically Associated with WGD in C. amara
To minimize false positives due to local population history we leveraged a quartet-based design (Vijay et al. 2016), consisting of two diploid and two autotetraploid populations (details in Materials and Methods). The mean number of SNPs per population contrast was 2,270,868 (table 1). We calculated Fst for 1-kb windows with a minimum 20 SNPs for all six possible population contrasts (fig. 1d), and ranked windows based on Fst values. To focus on WGD-associated adaptation, we first assigned ranks to each window based on the Fst values in each of four possible pairwise diploid–autotetraploid contrasts and identified windows in the top 1% outliers of the resultant combined rank sum (fig. 1d, contrasts p1–p4). We then excluded any window which was also present in the top 1% Fst outliers in diploid–diploid or autotetraploid–autotetraploid population contrasts to avoid misattribution caused by local population history (fig. 1c, contrasts tt and dd). By this approach, we identified 440 windows that intersected 229 gene coding loci (supplementary data set 1, Supplementary Material online; termed WGD adaptation candidates below). To control for possible biases due to suboptimal window size selection, we recalculated Fst on a SNP-by-SNP basis, considering genes with 5 or more SNPs. This approach resulted in the comparable candidate list to the window-based analysis (see Materials and Methods). Larger windows (50 kb) failed to detect peaks of divergence.
Among these 229 gene coding loci, a Gene Ontology (GO) term analysis yielded 22 significantly enriched biological processes (Fisher’s exact test with conservative “elim” method, P < 0.05, supplementary table 3, Supplementary Material online). To further control for false positives and refine this candidate list to putatively functional candidates, we complemented these differentiation measures with a quantitative estimate that incorporates potential functional impact of encoded derived amino acid changes, following the FineMAV method (Szpak et al. 2018) (see Materials and Methods for a full description). In short, as an orthogonal complement to Fst scans above, FineMAV assigns SNPs a score based on the predicted functional consequences of resultant amino acid substitutions using Grantham scores, and amplifies these by the per-cytotype AF difference between the two amino acids (Szpak et al. 2018, Bohutínská et al. 2021a). This allowed us to focus on radical amino acid changes driven to high frequency specifically in the autotetraploids. From our 229 Fst window-based WGD adaptation candidates, 120 contained at least one 1% FineMAV outlier amino acid substitution (supplementary data sets 1 and 2, Supplementary Material online).
DNA Maintenance (Repair, Chromosome Organization) and Meiosis under Selection in C. amara
Of the 22 significantly enriched GO processes, the most enriched by far was DNA metabolic process (P-value = 6.50E-08, vs. 0.00021 for the next most confident enrichment), although there was also enrichment for chromosome organization and meiotic cell cycle. The 40 genes contributing to these categories showed highly localized peaks of differentiation (fig. 2), as well as 1% FineMAV outlier SNPs in coding regions (fig. 2, supplementary data sets 1 and 2, Supplementary Material online). These genes also clustered in STRING interaction networks, suggesting coevolutionary dynamics driving the observed selection signals (supplementary fig. 2, Supplementary Material online; see Materials and Methods). The largest cluster comprised of MSH6, PDS5e, SMC2, MS5, PKL, HDA18, CRC, and homologs of two uncharacterized, but putative DNA repair related loci AT1G52950 and AT3G02820 (containing SWI3 domain). MutS Homolog 6 (MSH6) is a component of the postreplicative DNA mismatch repair system. It forms a heterodimer with MSH2 which binds to DNA mismatches (Culligan and Hays 2000; Wu et al. 2003), enhancing mismatch recognition. MutS homologs have also been shown to control crossover number in Arabidopsis thaliana (Lu et al. 2008). The C. amara ortholog of AT1G15940 is a close homolog of PDS5, a protein required in fungi and animals for formation of the synaptonemal complex and sister chromatid cohesion (Panizza et al. 2000). Structural Maintenance Of Chromosomes 2 (SMC2/TTN3) is a central component of the condensin complex, which is required for segregation of homologous chromosomes at meiosis (Siddiqui et al. 2003) and stable mitosis (Liu and Meinke 1998). PICKLE (PKL) is a SWI/SWF nuclear-localized chromatin remodeling factor (Ogas et al. 1999; Shaked et al. 2006) that also has highly pleiotropic roles in osmotic stress response (Perruc et al. 2007), stomatal aperture (Kang et al. 2018), root meristem activity (Aichinger et al. 2011), and flowering time (Jing et al. 2019). Beyond this cluster, other related DNA metabolism genes among our top outliers include DAYSLEEPER (fig. 2), a domesticated transposase that is essential for development, first isolated as binding the Kubox1 motif upstream of the DNA repair gene Ku70 (Bundock and Hooykaas 2005). The complex Ku70/Ku80 regulate nonhomologous end joining (NHEJ) double-strand break repair (Tamura et al. 2002). Consistent with this, DAYSLEEPER mutants accumulate DNA damage (Knip 2012), but the exact role of DAYSLEEPER in normal DNA maintenance is not yet understood. Interesting also is the identification of MALE-STERILE 5 (MS5/TDM1), which is required for cell cycle exit after meiosis II. As the name implies, MS5 mutants are male sterile, with pollen tetrads undergoing an extra round of division after meiosis II without chromosome replication (Glover et al. 1998). MS5/TDM1 may be an APC/C component whose function is to ensure meiosis termination at the end of meiosis II (Cifuentes et al. 2016). Together, this set of DNA management loci exhibiting the strongest signals of selection points to widespread modulation of DNA repair and chromosome management following WGD in C. amara.
Fig. 2.

Selective sweep signatures at DNA management and ion homeostasis loci. Examples of selective sweep signatures among four candidate loci (red arrows). X-axis gives scaffold position in base pairs. Y-axis gives Fst values at single nucleotide polymorphisms (dots) between diploid and autotetraploid Cardamine amara. Red dots indicate FineMAV outlier SNPs. Red arrows indicate gene models overlapping top 1% Fst windows and gray lines indicate neighboring gene coding loci.
Evolution of Stress Signaling and Ion Homeostasis Genes
The remainder of the enriched GO categories in C. amara revolved around a diversity of cellular processes, including stress response, protein phosphorylation, root development, ABA signaling, and ion homeostasis. The intersection of these processes was often represented by several genes. For example, two of the top 20 highest-scoring SNPs in the genome-wide FineMAV analysis reside in SNF1-related protein kinase SnRK2.9 (supplementary data set 2, Supplementary Material online). SnRKs have been implicated in osmotic stress and root development (Fujii et al. 2011; Kawa et al. 2020), and their activity also mediates the prominent roles of Clade A protein phosphatase 2C proteins in ABA and stress signaling (Cutler et al. 2010). Interesting in this respect is a strong signature of selection in HIGHLY ABA-INDUCED PP2C GENE 1, a clade A PP2C protein (supplementary data set 1, Supplementary Material online). Stress-related phosphoinositide phosphatases are represented by SAC9, mutants of which exhibit constitutive stress responses (Williams et al. 2005). Diverse other genes related to these categories exhibit the strongest signatures of selection, such as PP2-A8 (Meyers et al. 2002) and AT4G19090, a transmembrane protein strongly expressed in young buds (Klepikova et al. 2016) (fig. 2).
Given the observed increase in potassium and dehydration stress tolerance in first generation autotetraploid A. thaliana (Chao et al. 2013), it is very interesting that our window-based outliers included an especially dramatic selective sweep at K+ Efflux Antiporter 2 (KEA2, fig. 2), a K+ antiporter that modulates osmoregulation, ion, and pH homeostasis (Kunz et al. 2014). Recent evidence indicates that KEA2 is important for eliciting a rapid hyperosmotic-induced Ca2+ response to water limitation imposed by osmotic stress (Stephan et al. 2016). The KEA2 locus in autotetraploid C. amara features an exceptional ten FineMAV-outlier SNPs (fig. 2, supplementary data sets 1 and 2, Supplementary Material online), indicating that the sweep contains a run of radical amino acid changes at high AF difference between the ploidies, pointing to a potential functional change. We also detected cation-chloride co-transporter 1 (HAP 5) a Na+, K+, Cl− cotransporter, involved in diverse developmental processes and Cl− homeostasis (Colmenero-Flores et al. 2007). These cellular processes map well onto increasingly recognized changes that occur in polyploids, most comprehensively reviewed by (Bomblies 2020).
Limited Gene Ortholog-Level Convergence between C. amara and A. arenosa
We hypothesized that WGD imposed strong, specific selection pressures leading to convergent directional selection on the same genes or at least on different genes playing a role in the same process (ortholog- or function-level convergence, respectively) between C. amara and A. arenosa. To test for this, we complemented our C. amara genome scan with an analysis of A. arenosa divergence outliers based on an expanded sampling relative to the original A. arenosa genome scan studies. We selected the 80 diploid and 40 autotetraploid individuals sequenced most deeply in a recent range-wide survey (Monnahan et al. 2019, subsampling following Bohutínská et al. 2021a) of genomic variation in A. arenosa (mean coverage depth per individual = 18; 160 haploid genomes sampled of each ploidy), and scanned for Fst outliers in 1-kb windows, as we did for C. amara. We identified 696 windows among 1% Fst outliers, overlapping 452 gene-coding loci (supplementary data set 3, Supplementary Material online), recovering results similar to (Yant et al. 2013, Bohutínská et al. 2021a), including the interacting set of loci that govern meiotic chromosome crossovers, despite radically different sampling in each of the A. arenosa studies. From this entire list of 452 A. arenosa WGD adaptation candidates, only six orthologous loci were shared with our 229 C. amara WGD adaptation candidates (table 2). Although it is possible that these six genes may be convergently evolving in each species, this degree of overlap was not significant (P = 0.42, Fisher’s exact test), indicating no excess convergence at the level of orthologous genes beyond the quantity expected by chance. Re-analysis with candidate genes detected using the SNP-by-SNP divergence scan did not identify any additional convergent gene. Similarly, there was no excess overlap among genes which harbor at least one candidate FineMAV substitution (3 overlapping candidate genes out of 120 in C. amara and 303 in A. arenosa; P = 0.27, Fisher’s exact test). This lack of excess convergence at the ortholog level may come as a surprise given the expected shared physiological challenges attendant to WGD (Yant and Bomblies 2015; Baduel, Bray, et al. 2018; Bomblies 2020).
Table 2.
WGD Adaptation Candidates in Both Arabidopsis arenosa and Cardamine amara.
| C. amara ID | Arabidopsis thaliana ID | A. arenosa ID | Name | Function (TAIR) |
|---|---|---|---|---|
| CAg1480 | AT1G16460 | AL1G28600 | MST2/RDH2 | Embryo/seed development |
| CAg20214 | AT2G45120 | AL4G44210 | C2H2-like zinc finger | Stress response |
| CAg11103 | AT3G42170 | AL3G27110 | DAYSLEEPER | DNA repair |
| CAg16465 | AT3G62850 | AL1G11960 | Zinc finger-like | Unknown |
| CAg4024 | AT5G05480 | AL6G15370 | Asparagine amidase A | Growth and development |
| CAg5641 | AT5G23570 | AL6G34840 | SGS3 | Posttranscriptional gene silencing |
Note.—The number of genes does not exceed random expectations for the overlap of candidate gene lists from each species, indicating a lack of gene-level convergence. Underlined genes also harbor at least one candidate FineMAV SNP in both species.
To determine whether we may have failed to detect convergent loci due to missing data or if top outliers in A. arenosa had few, but potentially functionally implicated, differentiated SNPs in C. amara, we performed a targeted search in C. amara for the interacting set of meiosis proteins found to exhibit the most robust signatures of selection in A. arenosa (Yant et al. 2013; Bohutínská et al. 2021a) (supplementary table 4, Supplementary Material online). All meiosis-related orthologs in C. amara that exhibit selection signatures in A. arenosa (13 in total) passed our data quality criteria and were included in our analyses. Only three showed any signal by FineMAV analysis: PDS5b harbors an unusually high three fineMAV outlier SNPs, although it is not a Fst outlier. ASY3, which controls crossover distribution at meiosis, has only one FineMAV outlier SNP. Finally, a regulator of endoreduplication, CYCA2; 3, also harbors a single FineMAV 1% outlier in C. amara, although it was not included in the Fst window analysis (the window overlapping it contained only 7 SNPs, below the 20 SNP minimum cut-off for inclusion in the Fst window analysis). However, these 7 SNPs exhibited high mean Fst (0.55). Thus, although we detect varying signal in these three meiosis-related genes following WGD (supplementary table 4, Supplementary Material online), we do not see widespread signals of selection in the set of interacting crossover-controlling genes that were so conspicuous in A. arenosa (Yant et al. 2013).
Meiotic Stability in C. amara
Despite our broad overall analysis of selection in C. amara, as well as a targeted assessment of particular meiosis genes, we did not detect strong signal of selection in meiosis genes in C. amara (supplementary table 4, Supplementary Material online). The C. amara autotetraploid is a fertile, outcrossing, well-established lineage, but we still wondered if some contrast in meiotic behavior underlies this difference in specific loci under selection. We therefore cytologically assessed the degree of male meiotic stability in C. amara (fig. 3a). A reduction in crossover number to one per bivalent is indicated as a leading mechanism for meiotic diploidization in autopolyploids because this limits multivalent associations (which increase the propensity toward breakage and aneuploidy vs. bivalents [Cifuentes et al. 2010; Le Comber et al. 2010; Bomblies et al. 2016]), so we use proportion of bivalents to multivalents as our estimator (Materials and Methods). This revealed a highly variable degree of stability in both C. amara cytotypes (mean proportion stable metaphase I cells in diploid maternal seed lines = 0.38–0.69, n = 133 scored cells; in tetraploids = 0.03–0.38; n = 348 scored cells; supplementary table 5, Supplementary Material online). Indeed, while still highly variable, the overall degree of stability was lower in autotetraploids versus diploids (differing proportion of stable to unstable meiotic cells for each ploidy; D = 62.7, df = 1, P < 0.0001, GLM with binomial errors; fig. 3b, supplementary table 5, Supplementary Material online), corresponding with the lack of selection signal in crossover-controlling meiosis genes. Interestingly, the broad variation in stability estimates within both cytotypes suggests widespread standing variation controlling this trait. In contrast, higher frequencies of stable metaphase I cells (>80%) have been commonly observed for diploid and autotetraploid A. arenosa (Marburger et al. 2019), although wider estimates of meiotic variation have also been observed in populations hybridizing with A. lyrata (Seear et al. 2020). Taken together with the observation of occasional clonal spreading of C. amara (Hejný et al. 1992; Tedder et al. 2015; Zozomová-Lihová et al. 2015), this indicates an ability to maintain stable populations, thus perhaps decreasing the immediate necessity to fully stabilize meiosis in either cytotype. Vegetative reproduction is often seen in polyploids (Herben et al. 2017; Van Drunen and Husband 2019) and in turn may have facilitated the establishment of the autotetraploid cytotypes. We note finally that the tetraploid populations are still highly fertile, consistent with observations across the range (Koch et al. 2003).
Fig. 3.
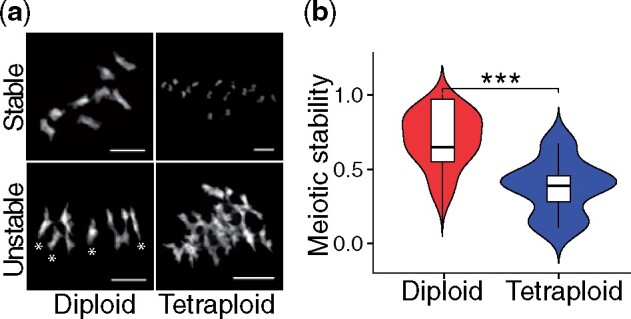
Variable meiotic stability in Cardamine amara. (a) An example of stable and unstable diploid and autotetraploid DAPI-stained meiotic chromosomes (diakinesis and metaphase I). Unstable meiosis is characterized by multivalent formation and interchromosomal connections, so we use the proportion of bivalents to multivalents as a proxy to estimate stability. In this example, the stable and unstable diploids (left panels) pictured contain 8 and 4 bivalents, respectively, whereas the stable and unstable tetraploids (right panels) show 16 and 0 bivalents, respectively. Thus all chromosomes pictured in these “Stable” examples are present as bivalents, whereas in the “Unstable” examples, only the four with asterisks (*) are bivalents, whereas the rest are mulivalents. Scale bar corresponds to 10 µm. For a complete overview of all scored chromosome spreads see supplementary figure 5, Supplementary Material online. (b) Distribution of meiotic stability (calculated as proportion of stable and partly stable to all scored meiotic spreads) in diploid and autotetraploid individuals of C. amara. *** P < 0.001, GLM with binomial errors.
Evidence for Process-level Convergence
Although we found no excess convergence at the level of orthologous genes under selection, we speculated that convergence may occur nevertheless at the level of functional processes. To test this, we used two complementary approaches: overlap of GO term enrichment and evidence of shared protein function from interaction networks. First, of 73 significantly (P < 0.05) enriched GO terms in A. arenosa (supplementary table 6, Supplementary Material online), we found that five were identical to those significantly enriched in C. amara, which is more than expected by chance (P < 0.001, Fisher’s exact test; table 3). In addition, some processes were found in both species, but were represented by slightly different terms, especially in the case of meiosis (“meiotic cell cycle” in C. amara, “meiotic cell cycle process” in A. arenosa: supplementary tables 3 and 6, Supplementary Material online). Remarkably, the relative ranking of enrichments of all five convergent terms was identical in both C. amara and A. arenosa (table 3). This stands in strong contrast to the fact that A. arenosa presented an obvious set of physically and functionally interacting genes in the top two categories (DNA metabolic process and chromosome organization), whereas the genes in these categories in C. amara are implicated in more diverse DNA management roles.
Table 3.
Convergent Processes under Selection in Both Cardamine amara and Arabidopsis arenosa Following WGD.
| GO ID | Term | P-value(C. amara) | P-value(A. arenosa) | Enrichment(C. amara) | Enrichment(A. arenosa) |
|---|---|---|---|---|---|
| GO:0006259 | DNA metabolic process | 6.50E-08 | 8.20E-04 | 3.72 | 2.46 |
| GO:0051276 | Chromosome organization | 0.019 | 2.10E-04 | 1.98 | 2.01 |
| GO:0009738 | Abscisic acid-activated signaling pathway | 0.032 | 0.022 | 2.54 | 2.10 |
| GO:0071215 | Cellular response to abscisic acid stimulation | 0.048 | 0.04 | 2.30 | 1.90 |
| GO:0097306 | Cellular response to alcohol | 0.048 | 0.04 | 2.30 | 1.90 |
Note.—P-values given are Fisher’s exact test, which tests for enrichment of terms from the GO hierarchy. Enrichment refers to fold enrichment.
Second, we sought for evidence that genes under selection in C. amara might interact with those found under selection in A. arenosa, which would further support process-level convergence between the species. Thus, we took advantage of protein interaction information from the STRING database, which provides an estimate of proteins’ joint contributions to a shared function (Szklarczyk et al. 2015). For each C. amara WGD adaptation candidate we searched for the presence of STRING interactors among the A. arenosa WGD adaptation candidates, reasoning that finding such an association between candidates in two species may suggest that directional selection has targeted the same processes in both species through different genes. Following this approach, we found that out of the 229 C. amara WGD adaptation candidates, 90 were predicted to interact with at least one of the 452 WGD adaptation candidates in A. arenosa. In fact, 57 likely interacted with more than one A. arenosa candidate protein (fig. 4 and supplementary table 7, Supplementary Material online). This level of overlap was greater than expected by chance (P = 0.001 for both “any interaction” and “more-than-one interaction,” as determined by permutation tests with the same database and 1,000 randomly generated candidate lists).
Fig. 4.

Evidence for functional convergence between Cardamine amara and Arabidopsis arenosa following independent WGDs. Plots show C. amara candidate genes in blue and STRING-associated A. arenosa candidate genes in gray. We used only medium confidence associations and higher (increasing thickness of lines connecting genes indicates greater confidence). (a) Meiosis- and chromatin remodeling-related genes. (b) Ion transport-related genes.
Several large STRING clusters were evident among WGD adaptation candidates in C. amara and A. arenosa (fig. 4). The largest of these clusters center on genome maintenance, specifically meiosis and chromatin remodeling (fig. 4a), and ion homeostasis (especially K+ and Ca2+), along with stress (ABA) signaling (fig. 4b), consistent with the results of GO analysis. Taken together, both STRING and GO analyses support our hypothesis of functional convergence of these processes following WGD in C. amara and A. arenosa.
Conclusions
Given the expected shared challenges attendant to WGD in C. amara and A. arenosa, we hypothesized at least partially convergent evolutionary responses to WGD. Although we found obvious convergent recruitment at the level of functional processes, we did not detect excess convergence at the gene level. This was consistent with the probable absence of shared standing variation between these species (Hudson and Coyne 2002), which are 17 My diverged. Nevertheless, we note that if any shared variation has persisted, it was not selected upon convergently in both young autotetraploids, thus strengthening the conclusion that the genes selected in response to WGD are not highly constrained.
The most prominent difference we observed here is the lack of an obvious coordinated evolutionary response in genes stabilizing early meiotic chromosome segregation in C. amara, relative to the striking coevolution of physically and functionally interacting proteins governing crossover formation in A. arenosa. This might be explained to some extent by our observation that in C. amara both diploids and autotetraploids are somewhat less meiotically stable than either cytotype in A. arenosa, and this instability may preadapt the autotetraploids to enjoy a less strict reliance on the generation of a high percentage of euploid gametes, by forcing occasional reliance on vegetative reproduction, as has been observed (Herben et al. 2017). This then may allow a decoupling of crossover number reduction from broader changes across meiosis and other processes we observe. This is not to say that we see no signal of WGD adaptation in C. amara: factors governing timing during later meiosis, especially the exit from meiotic divisions as evidenced by the interacting trio of SMG7, SDS, and MS5, along with other chromatin remodeling factors and DNA repair-related proteins, such as MSH6 and DAYSLEEPER give very strong signals. The convergent functions we did detect (other meiotic processes, chromosome organization/chromatin remodeling, ABA signaling and ion transport) provide first insights into the salient challenges associated with WGD. We note also that tetraploid populations of both C. amara and A. arenosa are found in slightly colder environments than conspecific diploids (Zozomová-Lihová et al. 2015; Molina-Henao and Hopkins 2019), so some of these processes (e.g., ABA signaling and ion transport) might be linked to ecological adaptation following WGD.
Overall, our results provide contrast to widespread reports of gene-level convergence (reviewed, e.g., in Elmer and Meyer 2011; Martin and Orgogozo 2013; Blount et al. 2018) and support the idea that pathway-level convergence becomes dominant when the divergence between species is high (Takuno et al. 2015; Birkeland et al. 2020; Bohutínská et al. 2021b). This could be due to the absence of shared low-frequency alleles (acquired via gene flow or from standing variation) in species diverging millions of years ago, as was shown in alpine adaptation of different Brassicaceae species (Bohutínská et al. 2020). Alternatively, WGD provides complex multi-factorial challenge (Bomblies et al. 2015; Baduel, Bray, et al. 2018; Bomblies 2020) and the possible solutions to overcome such challenge may in fact be diverse. The result would be multiple alternative genetic paths to adaptation, with limited gene-level convergence due to the low diversity constraints (Yeaman et al. 2018). Finally, we note that the lack of gene-level convergence in meiosis genes suggests that the genomic changes associated with meiosis stabilization after WGD might not be as constrained as would be expected based on its functional conservation across eukaryotes (Grishaeva and Bogdanov 2014; Rosenberg and Corbett 2015; Baker et al. 2017).
We conclude that evolutionary solutions to WGD-associated challenges vary strongly from case to case, suggesting less functional constraint than one may expect based on the fact that these processes are conserved and essential. This may help explain how it is that many species manage to thrive following WGD and, once established as polyploids, experience evolutionary success. In fact, we envision that the meiotic instability experienced by some WGD lineages, such as C. amara, could serve as a diversity-generating engine promoting large effect genomic structural variation, as has been observed in aggressive polyploid gliomas (Yant and Bomblies 2015).
Materials and Methods
Reference Genome Assembly and Alignment
We generated a de novo assembly using the 10× Genomics Chromium approach. In brief, a single diploid individual from pop LUZ (supplementary table 8, Supplementary Material online) was used to generate one Chromium library, sequenced using 250PE mode on an Illumina sequencer, and assembled with Supernova version 2.0.0. This assembly had an overall scaffold N50 of 1.82 Mb. An assessment of genome completeness using BUSCO (version 3.0.2) (Seppey et al. 2019) for the 2,251 contigs ≥ 10 kb was estimated at 94.8% (1,365/1,440 BUSCO groups; supplementary table 9, Supplementary Material online).
BioNano Plant Extraction Protocol
Fresh young leaves of the C. amara accession LUZ were collected after 48-h dark treatment. DNA was extracted by the Earlham Institute’s Platforms and Pipelines group following an IrysPrep “FixnBlend” Plant DNA extraction protocol supplied by BioNano Genomics. First 2.5 g of fresh young leaves were fixed with 2% formaldehyde. After washing, leaves were disrupted and homogenized in the presence of an isolation buffer containing PVP10 and BME to prevent polyphenol oxidation. Triton X-100 was added to facilitate nuclei release. Nuclei were then purified on a Percoll cushion. The nuclear phase was taken and washed in isolation buffer before embedding into low melting point agarose. Two plugs of 90 μl were cast using the CHEF Mammalian Genomic DNA Plug Kit (Bio-Rad 170-3591). Once set at 4 °C the plugs were added to a lysis solution containing 200 μl proteinase K (QIAGEN 158920) and 2.5 ml of BioNano lysis buffer in a 50 ml conical tube. These were put at 50 °C for 2 h on a thermomixer, making a fresh proteinase K solution to incubate overnight. Samples were then removed from the thermomixer for 5 min before 50 μl RNAse A (Qiagen158924) was added and the tubes incubated for a further hour at 37 °C. Plugs were then washed 7 times in the Wash Buffer supplied in the Chef kit and 7 times in 1×TE. One plug was removed and melted for 2 min at 70 °C followed by 5 min at 43 °C before adding 10 μl of 0.2 U/μl of GELase (Cambio Ltd G31200). After 45 min at 43 °C the melted plug was dialyzed on a 0.1 μM membrane (Millipore VCWP04700) sitting on 15 ml of 1×TE in a small petri dish. After 2 h the sample was removed with a wide bore tip and mixed gently and left overnight at 4 °C.
10× Library Construction
DNA material was diluted to 0.5 ng/µl with EB (Qiagen) and checked with a QuBit Flourometer 2.0 (Invitrogen) using the QuBit dsDNA HS Assay kit (supplementary table 8, Supplementary Material online). The Chromium User Guide was followed as per the manufacturer’s instructions (10× Genomics, CG00043, Rev A). The final library was quantified using qPCR (KAPA Library Quant kit [Illumina] and ABI Prism qPCR Mix, Kapa Biosystems). Sizing of the library fragments was checked using a Bioanalyzer (High Sensitivity DNA Reagents, Agilent). Samples were pooled based on the molarities calculated using the two QC measurements. The library was clustered at 8 pM with a 1% spike in of PhiX library (Illumina). The pool was run on a HiSeq2500 250 bp Rapid Run V2 mode (Illumina).
Sequencing and Assembly
Reads were subsampled to 90 M reads and assembled with Supernova 2.0.0 (10× Genomics), giving a raw coverage of 60.30× and an effective coverage of 47.43×. The estimated molecule length was 44.15 kb. The assembly size, considering only scaffolds longer than 10 kb was 159.53 Mb and the Scaffold N50 was 1.82 Mb. Genome size estimate by kmer analysis was 225.39 Mb, hence we estimate we are missing 16.61% from the assembly. Because the diploid individual used for reference genome sequencing was not homozygous, we sought to confirm whether the assembly harbored evidence of uncollapsed haplotypes by using a reciprocal BLAST (Camacho et al. 2009) best hits approach. A small proportion (1.7%) of scaffolds exhibited substantial homology (90% or greater identity to another scaffold over 90% of their length), indicating that very few alternate alleles at heterozygous loci were misinferred as separate genomic loci in the diploid assembly. Manual investigation of a suite of meiosis-related loci indicated no cases of false negatives in the data set caused by alternate alleles aligning to separate scaffolds. We further scaffolded the assembly using the published Cardamine hirsuta genome using graphAlign (Spalding and Lammers 2004) and Nucmer (Marçais et al. 2018).
Gene Calling and Annotation
The plants set database embryophyta_odb9.tar.gz was downloaded from http://busco.ezlab.org/ and used to assess orthologue presence/absence in our C. amara genome annotation. Running BUSCO gave Augustus (Stanke and Waack 2003) results via BUSCO HMMs to infer where genes lie in the assembly and to infer protein sequences. Augustus was used to generate a gff annotation file using “arabidopsis” as the training option. A BLAST (v. 2.2.4) database was built for Brassicales (taxid: 3699) by downloading approximately 1.26 M protein sequences from https://www.ncbi.nlm.nih.gov/taxonomy/ and the Augustus-predicted proteins were annotated via Interproscan (Quevillon et al. 2005) and blast2go (Conesa and Götz 2008).
Functional Annotation of C. amara Genes
To functionally annotate C. amara genes we performed an orthogrouping analysis using Orthofinder version 2.3.3 (Emms and Kelly 2018), inferring orthologous groups (OGs) from four species (C. amara, A. lyrata, A. thaliana, Cochlearia pyrenaica). A total of 21,618 OGs were found. Best reciprocal BLAST hits (RBHs) for C. amara and A. thaliana genes were found using BLAST version 2.9.0.
Cardamine amara genes were then assigned an A. thaliana gene ID for GO enrichment analysis via the following protocol: 1) if the C. amara gene was in an OG with only one A. thaliana gene, that A. thaliana ID was used; 2) if the C. amara gene was in an OG with more than one A. thaliana gene, then the RBH, provided it was in the same OG with the C. amara gene, was used; 3) if the C. amara gene was in an OG that contained more than one A. thaliana gene, none of which was the RBH, then the A. thaliana gene from that OG with the lowest BLAST E-value was taken; 4) if the C. amara gene was in an OG group that lacked A. thaliana genes, then the RBH was taken instead; and 5) finally, if the C. amara gene was in an OG group without any A. thaliana genes and there was no RBH, then the gene with the lowest E-value in a BLASTs versus the TAIR10 database was used. BLASTs versus the TAIR10 database were performed during December 2019.
Sampling, Sequencing, and Genetic Structure Analysis
Sampling
A total of 100 plants were sampled from four populations (fig. 1d): 25 individuals for each of CEZ (4×), PIC (4×), VKR (2×), and LUZ (2×). Sampled plants were spaced at least 3 m apart, as such distance was enough to avoid resampling of identical clones according to analysis in a study sampling approximately 3,300 individuals across the C. amara range, including these populations (Zozomová-Lihová et al. 2015).
Flow Cytometry
All plants used for DNA extraction were verified for expected ploidy by flow cytometry. Approximately 1 square cm of leaf material was diced alongside an internal reference using a razor blade in 1 ml ice cold extraction buffer (45 mM MgCl2, 30 mM sodium citrate, 20 mM MOPS, 1% Triton-100, pH 7 with NaOH). The resultant slurry was then filtered through a 40-μm nylon mesh before the nuclei were stained with the addition of 1 ml staining buffer (either CyStain UV precise P [Sysmex, Fluorescence emission: 435–500 nm] for relative ploidy, or Otto 2 buffer [0.4 M Na2HPO4·12H2O, Propidium iodide 50 μg/ml, RNase 50 μg/ml], for absolute DNA content). After 1 min of incubation at room temperature the sample was run for 5,000 particles on either a Partec PA II flow cytometer or a BD FACS Melody. Histograms were evaluated using FlowJo software version 10.6.1.
DNA Isolation, Library Preparation, and Sequencing
A replicated approach was used for the DNA isolation, pooling, and sequencing to reduce variation that may be associated with Pool-Seq data. DNA isolations were performed in triplicate for every plant and then each replicate was pooled with samples from the other 24 replicates in each population, generating three independently extracted and pooled replicates for every population. DNA was extracted with the RNeasy Plant Mini Kit (Qiagen). Each of the 12 resultant pools for the four populations was used as input for library construction with the Illumina Truseq kit (Illumina, Inc.), and then sequenced on an Illumina NextSeq (150 bp paired end specification).
Data Preparation, Alignment, and Genotyping
Fastq files from two runs on the Illumina NextSeq concatenated to give an average of 30.5 million reads per sample. Adapter sequences were removed using cutadapt (version 1.9.1) (Martin 2011) and quality trimmed via Sickle (version 33) (Joshi and Fass 2011) to generate only high-quality reads (Phred score ≥30) of 30 bp or more, resulting in an average of 27.9 million reads per sample. Reads were then aligned with (Li et al. 2009) BWA (version 0.7.12) (Li and Durbin 2009) and processed with Samtools (version 1.7) (Li et al. 2009). Using Picard (version 1.134) (Broad Institute 2009), duplicate reads were removed via MarkDuplicates followed by the addition of read group IDs to the bam files via AddOrReplaceReadGroups. Finally, to handle the presence of indels, GATK (version 3.6.0) (McKenna et al. 2010) was used to realign reads using IndelRealigner.
Variant Calling
Variants were called for the 12 bam files (three replicates per population) using Freebayes (version 1.1.0.46) (Garrison and Marth 2012) to generate a single VCF output file. Freebayes was run with default parameters, except we specified “--pooled-discrete” to indicate samples were pooled, “--use-best-n-alleles 2” to restrict to biallelic sites, and “--no-indels” to exclude indels. The resultant VCF was then filtered with BCFtools (version 1.8) (Narasimhan et al. 2016) to remove sites where the read depth was <10, or >1.6× the second mode (determined as 1.6 × 31 = 50, supplementary fig. 3, Supplementary Material online) in order to remove from the analysis regions exhibiting heterozygous deletions or where multiple genomic regions may have mapped to the reference due to, for example, paralogous duplications n the sequenced individuals.
Population Genetic Structure
We first calculated genome-wide between-population metrics (Nei’s Fst [Nei 1972] and AF difference). The AF in individual replicate pools was calculated as the fraction of the total number of reads supporting the alternative allele (Anand et al. 2016). For each population the average AF was then calculated from the three replicates and used for all further calculations. We used the python3 PoolSeqBPM pipeline, designed to input pooled data (https://github.com/mbohutinska/PoolSeqBPM). Then we inferred relationships between populations over putatively neutral 4-fold degenerate SNPs using PCA as implemented in adegenet (Jombart and Ahmed 2011). Finally, we inferred relationships between populations using AF covariance graphs implemented in TreeMix (Pickrell and Pritchard 2012). We ran TreeMix allowing a range of migration events; and presented one additional migration edge, as it represented points of log-likelihood saturation. To obtain confidence in the reconstructed topology, we bootstrapped the scenario with zero events (the tree topology had not changed when considering the migration events) choosing a bootstrap block size of 1,000 bp, equivalent to the window size in our selection scan, and 100 replicates.
Genome Scans for Selection
To detect signals of selection, we used a combination of two different selection scan approaches. First, we calculated pairwise window-based Fst between diploid and populations and used minimum sum of ranks between informative contrasts in a quartet design (below). To further control for false positives and refine the gene list to putatively functional candidates we complemented these differentiation measures with a functional score estimate following the FineMAV method (below). Both approaches are based on population AFs and allow analysis of diploid and autopolyploid populations.
Window-Based Selection Scan Using a Quartet Design
We performed a window-based Fst (Nei 1972) scan for directional selection in C. amara, taking advantage of quartet sampling of two diploid and two autotetraploid populations (fig. 1d). Using this design, we identified top candidate windows for selective sweeps associated with ploidy differentiation, while excluding differentiation patterns private to a single population or ploidy-uninformative selective sweeps. Thus comparisons between populations of the same ploidy constitute a null model for shared heterogeneity in genetic differentiation arising through processes unrelated to WGD (following an approach successfully applied in Vijay et al. 2016). To do this, we calculated Fst for 1-kb windows with minimum 20 SNPs for all six population pairs in the quartet (fig. 1d) and ranked windows based on their Fst value. We excluded windows which were top 1% outliers in diploid–diploid (dd in fig. 1d) or autotetraploid–autotetraploid (tt) population contrasts, as they represent variation inconsistent with diploid–autotetraploid divergence but rather signal local differentiation within a cytotype. Next, we assigned ranks to each window based on the Fst values in four diploid–autotetraploid contrasts and identified windows being top 1% outliers of minimum rank sum.
Because candidate detection could be biased by arbitrary window size choice, we re-analyzed our differentiation scans changing two parameters: 1) using a SNP-by-SNP basis (requiring at least five SNPs per gene for inclusion); and 2) using larger, 50-kb windows. Doing this, we found that SNP-level and 1-kb-window scans resulted in comparable candidate gene lists, whereas 50-kb windows were too wide to identify local peaks of differentiation. Thus, we decided to use scans with a window size of 1 kb, which best corresponded to the average length of selective sweep signatures in differentiation plots (e.g., fig. 2), and allowed to locate the candidate selected region while still providing enough polymorphisms to robustly estimate differentiation between ploidies.
To account for possible confounding effect of comparing windows from genic and nongenic regions, we calculated the number of base pairs overlapping with any gene within each window. There was no relationship between the proportion of genic space within a window and Fst (Pearsons r = −0.057, supplementary fig. 4, Supplementary Material online), indicating that our analyses were unaffected by unequal proportion of genic space in a window.
In A. arenosa, we performed window-based Fst scan for directional selection using the same criteria as for C. amara (1-kb windows, min 20 SNPs per window). We did not use the quartet design as the range-wide data set of 80 diploid and 40 autotetraploid individuals drawn from the entire A. arenosa range (15 diploid and 24 autotetraploid populations) assured power to detect genomic regions with WGD-associated differentiation. This A. arenosa analysis gave very similar results to (Yant et al. 2013), which used only two diploid and four autotetraploid populations, indicating minimal dependence on sampling to detect these strongest signatures of selection in the A. arenosa system.
FineMAV
We adopted the approach, Fine-Mapping of Adaptive Variation, FineMAV (Szpak et al. 2018), using our C. amara annotation (following approach successfully applied to nonhuman genome in Bohutínská et al. 2021a). To functionally annotate each amino acid change, we used the Grantham score (Grantham 1974), a theoretical amino acid substitution value, encoded in the Grantham matrix, where each element shows the differences of physicochemical properties between two amino acids. We used SnpEff (version 4.3) (Cingolani et al. 2012) to annotate our SNP data set by applying our Augustus-generated C. amara annotation (“Gene Calling and Annotation,” above). We estimated the population genetic component of FineMAV (see [Szpak et al. 2018] for details on calculations) using AF information at each site (considering minor frequency alleles as derived) and derived allele purity (DAP) parameter of 3.5, a measure of population differentiation, which describes how unequally the derived allele is distributed among populations. The advantage is that DAP can summarize differentiation across many populations in a single measure for each variant. Finally, for each amino acid substitution, we assigned Grantham scores, together with population genetic component of FineMAV, using custom scripts in Python 2.7.10 and Biopython version 1.69. We identified the top 1% outliers as FineMAV candidates. All calculations were performed using code available at (github.com/paajanen/meiosis_protein_evolution).
Arabidopsis arenosa Population Genomic Data Set
Our selection analysis in A. arenosa was based on an expanded sampling (Monnahan et al. 2019) relative to Yant et al. (2013), who sampled 24 individuals (from two diploid and four tetraploid populations, sourced from a fraction of now known lineages). This expanded sampling covered all known lineages, across the entire range of the species, including 39 populations: 15 diploid populations (105 individually resequenced plants) and 24 tetraploid populations (182 individually resequenced plants) (Monnahan et al. 2019). We aligned PE Illumina data to the A. lyrata reference (Hu et al. 2011), called variants and filtered as previously (Monnahan et al. 2019) using GATK 3.5 (McKenna et al. 2010). We used a subset of the data consisting of 80 diploid individuals and 40 tetraploid individuals from populations unaffected by secondary introgression from diploid lineages (following Bohutínská et al. 2021a; samples selected based on the highest mean depth of coverage). Such subsampling gave us a balanced number of 160 high-quality haploid genomes of each ploidy suitable for selection scans. Finally, we filtered each subsampled data set for genotype read depth >8 and maximum fraction of missing genotypes < 0.5 in each lineage. We calculated Fst using python3 ScanTools pipeline (github.com/mbohutinska/ScanTools_ProtEvol). All subsequent analyses were performed following the same procedure as with C. amara data.
GO Enrichment Analysis
To infer functions significantly associated with directional selection following WGD, we performed a GO enrichment on the gene list using the R package topGO (Tilford and Siemers 2009), using A. thaliana orthologs of C. amara/A. lyrata genes, obtained using biomaRt (Smedley et al. 2009). We used Fisher’s exact test with conservative “elim” method, which tests for enrichment of terms from the bottom of the GO hierarchy to the top and discards any genes that are significantly enriched in a descendant GO terms (Grossmann et al. 2007). Re-analysis with the “classic” method did not identify any additional convergently enriched GO terms. We used biological process ontology with minimum node size of 150 genes.
Protein Associations from STRING Database
We searched for potential functional associations among C. amara and A. arenosa candidate genes using STRING (Szklarczyk et al. 2015). Genes were assigned an A. thaliana gene ID as described above. We used the “multiple proteins” search in A. thaliana, with text mining, experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence as information sources. We used minimum confidence 0.4 and retained only 1st shell associations (proteins that are directly associated with the candidate protein: i.e., immediately neighboring network circles).
Quantifying Convergence
We considered convergent any candidates or enriched GO categories that overlapped across both species. Convergent candidate genes had to be members of the same orthogroups (Emms and Kelly 2018). To test for higher than random number of overlapping items we used Fisher’s Exact Test for Count Data in R (R Development Core Team 2011).
Cytological Assessment of Meiotic Stability
We cytologically estimated the degree of male meiotic stability in C. amara by counting the number of bivalent chromosome associations in each metaphase event. A lower number of bivalents and a higher number of multivalents are taken as a proxy for reduced meiotic stability. The reasoning behind this is that a reduction in crossover number to one per bivalent is strongly indicated as a leading mechanism for meiotic diploidization in autopolyploids as this limits multivalent associations (which increase the propensity toward breakage and aneuploidy vs. bivalents [Cifuentes et al. 2010; Le Comber et al. 2010; Bomblies et al. 2016]).
Chromosome Preparation
Whole young inflorescences were fixed in freshly prepared ethanol: acetic acid (3:1) overnight, transferred into 70% ethanol and stored at –20 °C until use. Meiotic chromosome spreads were prepared from anthers according to Mandáková et al. (2014). Briefly, after washing in citrate buffer (10 mM sodium citrate, pH 4.8), selected flower buds were digested using a 0.3% mix of pectolytic enzymes (cellulase, cytohelisase, pectolyase; Sigma–Aldrich Corp., St. Louis, MO) in citrate buffer for 3 h. Individual anthers were dissected and spread in 20 µl of 60% acetic acid on a microscope slide placed on a metal hot plate (50 °C), fixed by ethanol: acetic acid (3:1) and the preparation was dried using a hair dryer. Slides were postfixed in freshly prepared 4% formaldehyde in distilled water for 10 min and air-dried. The preparations were stained with 4′,6-diamidino-2-phenylindole (DAPI; 2 µg/ml) in Vectashield (Vector Laboratories, Peterborough, UK). Fluorescence signals were analyzed using an Axioimager Z2 epifluorescence microscope (Zeiss, Oberkochen, Germany) and CoolCube CCD camera (MetaSystems, Newton, MA).
Meiotic Stability Assessments
In diploids, chromosome spreads with 8 bivalents were scored as “stable meiosis,” 7-6 as “partly stable,” 5-4 as “partly unstable,” and <4 as “unstable.” In autotetraploids, chromosome spreads with 16 bivalents were scored as “stable meiosis,” 14-12 as “partly stable,” 10-8 as “partly unstable,” and <8 as “unstable.” We report a mean value of meiotic stability for each ploidy calculated over “stable meiosis” and over sum of “stable meiosis” and “partly stable” categories. Differences in meiotic stability between diploids and autotetraploids (fig. 3b) are reported for the sum of “stable” and “partly stable” categories. However, considering only the “stable meiosis” category does not qualitatively affect the results (i.e., the degree of meiotic stability is significantly lower in tetraploids, D = 125.7, df = 1, P < 0.0001, GLM with binomial errors). Photos of all spreads scored are supplied in supplementary figure 4, Supplementary Material online.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors thank Veronika Konečná for assistance with the map figure, Doubravka Požárová and Paolo Bartolić for help with field collections and Karol Marhold for useful discussions. We also thank the anonymous reviewers for their constructive comments. This study was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme [grant number ERC-StG 679056 HOTSPOT], via a grant to L.Y and UK Biological and Biotechnology Research Council (BBSRC) Grant BB/P013511/1. Additional support was provided by Czech Science Foundation (project 20-22783S to F.K., 19-03442S to T.M., and 19-06632S), by Charles University (project Primus/SCI/35 to F.K.), by the long-term research development project No. RVO 67985939 of the Czech Academy of Sciences and by the CEITEC 2020 project (grant LQ1601). Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
L.Y. conceived of and directed the study. M.B., S.B., M.A., P.P., T.M., and P.M. performed analyses. P.M. and T.M. performed laboratory experiments. P.M., F.K., S.B., and M.B. performed field collections. L.Y. and M.B. wrote the manuscript with input from all authors. All authors approved of the final manuscript.
Data Availability
Sequence data are available at the European Nucleotide Archive at http://www.ebi.ac.uk/ena/data/view/PRJEB39872 (Cardamine amara) and the Sequence Read Archive at https://www.ncbi.nlm.nih.gov/sra/, study code SRP156117 (Arabidopsis arenosa data). All scripts are available at https://github.com/mbohutinska/PoolSeqBPM (Fst-based selection scans and all following analyses) and https://github.com/paajanen/meiosis_protein_evolution (FineMAV scan).
References
- Aichinger E, Villar CBR, di Mambro R, Sabatini S, Köhler C.. 2011. The CHD3 chromatin remodeler PICKLE and polycomb group proteins antagonistically regulate meristem activity in the Arabidopsis root. Plant Cell. 23(3):1047–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand S, Mangano E, Barizzone N, Bordoni R, Sorosina M, Clarelli F, Corrado L, Boneschi FM, D’Alfonso S, De BG.. 2016. Next generation sequencing of pooled samples: guideline for variants’ filtering. Sci Rep. 6:33735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold B, Kim ST, Bomblies K.. 2015. Single geographic origin of a widespread autotetraploid Arabidopsis arenosa lineage followed by interploidy admixture. Mol Biol Evol. 32(6):1382–1395. [DOI] [PubMed] [Google Scholar]
- Arnold BJ, Lahner B, DaCosta JM, Weisman CM, Hollister JD, Salt DE, Bomblies K, Yant L.. 2016. Borrowed alleles and convergence in serpentine adaptation. Proc Natl Acad Sci USA. 113(29):8320–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baduel P, Bray S, Vallejo-Marin M, Kolář F, Yant L.. 2018. The “Polyploid Hop”: shifting challenges and opportunities over the evolutionary lifespan of genome duplications. Front Ecol Evol. 6:117. [Google Scholar]
- Baduel P, Hunter B, Yeola S, Bomblies K.. 2018. Genetic basis and evolution of rapid cycling in railway populations of tetraploid Arabidopsis arenosa. PLoS Genet. 14(7):e1007510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker Z, Schumer M, Haba Y, Bashkirova L, Holland C, Rosenthal GG, Przeworski M.. 2017. Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. Elife 6:e24133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkeland S, Gustafsson ALS, Brysting AK, Brochmann C, Nowak MD, Purugganan M.. 2020. Multiple genetic trajectories to extreme abiotic stress adaptation in arctic Brassicaceae. Mol Biol Evol. 37(7):2052–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL.. 2009. BLAST+: architecture and applications. BMC Bioinformatics. 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount ZD, Lenski RE, Losos JB.. 2018. Contingency and determinism in evolution: replaying life’s tape. Science 362(6415):eaam5979. [DOI] [PubMed] [Google Scholar]
- Bohutínská M, Handrick V, Yant L, Schmickl R, Kolář F, Bomblies K, Paajanen P.. 2021a. De-novo mutation and rapid protein (co-)evolution during meiotic adaptation in Arabidopsis arenosa. Mol Biol Evol. 38(5):1980–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohutínská M, Vlček J, Yair S, Laenen B, Konečná V, Fracassetti M, Slotte T, Kolář F.. 2021b. Genomic basis of parallel adaptation varies with divergence in Arabidopsis and its relatives. Proc Natl Acad Sci U S A. 118(21)e2022713118. [DOI] [PMC free article] [PubMed]
- Bomblies K.2020. When everything changes at once: finding a new normal after genome duplication: evolutionary response to polyploidy. Proc R Soc B Biol Sci. 287: 20202154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K, Higgins JD, Yant L.. 2015. Meiosis evolves: adaptation to external and internal environments. New Phytol. 208(2):306–323. [DOI] [PubMed] [Google Scholar]
- Bomblies K, Jones G, Franklin C, Zickler D, Kleckner N.. 2016. The challenge of evolving stable polyploidy: could an increase in “crossover interference distance” play a central role? Chromosoma 125(2):287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K, Madlung A.. 2014. Polyploidy in the Arabidopsis genus. Chromosome Res. 22(2):117–134. [DOI] [PubMed] [Google Scholar]
- Broad Institute 2009. “Picard Tools.” Broad Institute, GitHub repository. Available from: http://broadinstitute.github.io/picard/. [Google Scholar]
- Bundock P, Hooykaas P.. 2005. An Arabidopsis hAT-like transposase is essential for plant development. Nature 436(7048):282–284. [DOI] [PubMed] [Google Scholar]
- Chao DY, Dilkes B, Luo H, Douglas A, Yakubova E, Lahner B, Salt DE.. 2013. Polyploids exhibit higher potassium uptake and salinity tolerance in Arabidopsis. Science 341(6146):658–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes M, Grandont L, Moore G, Chèvre AM, Jenczewski E.. 2010. Genetic regulation of meiosis in polyploid species: new insights into an old question. New Phytol. 186(1):29–36. [DOI] [PubMed] [Google Scholar]
- Cifuentes M, Jolivet S, Cromer L, Harashima H, Bulankova P, Renne C, Crismani W, Nomura Y, Nakagami H, Sugimoto K, et al. 2016. TDM1 regulation determines the number of meiotic divisions. PLoS Genet. 12(2):e1005856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM.. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmenero-Flores JM, Martínez G, Gamba G, Vázquez N, Iglesias DJ, Brumós J, Talón M.. 2007. Identification and functional characterization of cation-chloride cotransporters in plants. Plant J. 50(2):278–292. [DOI] [PubMed] [Google Scholar]
- Conesa A, Götz S.. 2008. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008:619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culligan KM, Hays JB.. 2000. Arabidopsis MutS homologs – AtMSH2, AtMSH3, AtMSH6, and a novel AtMSH7 – form three distinct protein heterodimers with different specificities for mismatched DNA. Plant Cell. 12:991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler SR, Rodriguez PL, Finkelstein RR, Abrams SR.. 2010. Abscisic acid: emergence of a core signaling network. Annu Rev Plant Biol. 61:651–679. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Coate JE.. 2019. Polyploidy, the nucleotype, and novelty: the impact of genome doubling on the biology of the cell. Int J Plant Sci. 180(1):1–52. [Google Scholar]
- Elmer KR, Meyer A.. 2011. Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends Ecol Evol. 26(6):298–306. [DOI] [PubMed] [Google Scholar]
- Emms DM, Kelly S.. 2018. OrthoFinder: Phylogenetic orthology inference for comparative genomics. bioRxiv 466201. Available from: https://www.biorxiv.org/content/10.1101/466201v1. [DOI] [PMC free article] [PubMed]
- Fujii H, Verslues PE, Zhu JK.. 2011. Arabidopsis decuple mutant reveals the importance of SnRK2 kinases in osmotic stress responses in vivo. Proc Natl Acad Sci USA. 108(4):1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G.. 2012. Haplotype-based variant detection from short-read sequencing. Cornell University. Available from: http://arxiv.org/abs/1207.3907.
- Glover J, Grelon M, Craig S, Chaudhury A, Dennis E.. 1998. Cloning and characterization of MS5 from Arabidopsis: a gene critical in male meiosis. Plant J. 15(3):345–356. [DOI] [PubMed] [Google Scholar]
- Grantham R.1974. Amino acid difference formula to help explain protein evolution. Science 185(4154):862–864. [DOI] [PubMed] [Google Scholar]
- Grishaeva TM, Bogdanov YF.. 2014. Conservation and variability of synaptonemal complex proteins in phylogenesis of Eukaryotes. Int J Evol Biol. 2014:856230–856216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann S, Bauer S, Robinson PN, Vingron M.. 2007. Improved detection of overrepresentation of Gene-Ontology annotations with parent-child analysis. Bioinformatics 23(22):3024–3031. [DOI] [PubMed] [Google Scholar]
- Hejný S, Slavík B, Kirschner J, Křísa B.. 1992. Květena České republiky 3. Prague, Czech Republic: Academia.
- Herben T, Suda J, Klimešová J.. 2017. Polyploid species rely on vegetative reproduction more than diploids: a re-examination of the old hypothesis. Ann Bot. 120(2):341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, Arnold BJ, Svedin E, Xue KS, Dilkes BP, Bomblies K.. 2012. Genetic adaptation associated with genome-doubling in autotetraploid Arabidopsis arenosa. PLoS Genet. 8(12):e1003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu TT, Pattyn P, Bakker EG, Cao J, Cheng JF, Clark RM, Fahlgren N, Fawcett JA, Grimwood J, Gundlach H, et al. 2011. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 43(5):476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XC, German DA, Koch MA.. 2020. Temporal patterns of diversification in Brassicaceae demonstrate decoupling of rate shifts and mesopolyploidization events. Ann Bot. 125(1):29–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Coyne JA.. 2002. Mathematical consequences of the genealogical species concept. Evolution (NY) 56:1557–1565. [DOI] [PubMed] [Google Scholar]
- Jing Y, Guo Q, Lin R.. 2019. The chromatin-remodeling factor pickle antagonizes polycomb repression of FT to promote flowering. Plant Physiol. 181(2):656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T, Ahmed I.. 2011. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27(21):3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Fass J.. 2011. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. Available from: https://github.com/najoshi/sickle.:2011.
- Kang X, Xu G, Lee B, Chen C, Zhang H, Kuang R, Ni M.. 2018. HRB2 and BBX21 interaction modulates Arabidopsis ABI5 locus and stomatal aperture. Plant Cell Environ. 41(8):1912–1925. [DOI] [PubMed] [Google Scholar]
- Kawa D, Meyer AJ, Dekker HL, Abd-El-Haliem AM, Gevaert K, Van De Slijke E, Maszkowska J, Bucholc M, Dobrowolska G, De Jaeger G, et al. 2020. SnRK2 protein kinases and mRNA decapping machinery control root development and response to salt. Plant Physiol. 182(1):361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepikova AV, Kasianov AS, Gerasimov ES, Logacheva MD, Penin AA.. 2016. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 88(6):1058–1070. [DOI] [PubMed] [Google Scholar]
- Knip M. (Leiden U). 2012. Daysleeper: from genomic parasite to indispensable gene. Leiden: LEI Universiteit. Available from: https://hdl.handle.net/1887/20170
- Koch M, Huthmann M, Bernhardt KG.. 2003. Cardamine amara L. (Brassicaceae) in dynamic habitats: genetic composition and diversity of seed bank and established populations. Basic Appl Ecol. 4(4):339–348. [Google Scholar]
- Kolář F, Fuxová G, Záveská E, Nagano AJ, Hyklová L, Lučanová M, Kudoh H, Marhold K.. 2016. Northern glacial refugia and altitudinal niche divergence shape genome-wide differentiation in the emerging plant model Arabidopsis arenosa. Mol Ecol. 25(16):3929–3949. [DOI] [PubMed] [Google Scholar]
- Konečná V, Bray S, Vlček J, Bohutínská M, Požárová D, Choudhury RR, Bollmann-Giolai A, Flis P, Salt DE Parisod C, et al. 2021. Parallel adaptation in autopolyploid Arabidopsis arenosa is dominated by repeated recruitment of shared alleles. bioRxiv [Internet]:2021.01.15.426785. Available from: http://biorxiv.org/content/early/2021/01/17/2021.01.15.426785.abstract. [DOI] [PMC free article] [PubMed]
- Kunz HH, Gierth M, Herdean A, Satoh-Cruz M, Kramer DM, Spetea C, Schroeder JI.. 2014. Plastidial transporters KEA1, -2, and -3 are essential for chloroplast osmoregulation, integrity, and pH regulation in Arabidopsis. Proc Natl Acad Sci USA. 111(20):7480–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Comber SC, Ainouche ML, Kovarik A, Leitch AR.. 2010. Making a functional diploid: from polysomic to disomic inheritance. New Phytol. 186(1):113–122. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CM, Meinke DW.. 1998. The titan mutants of Arabidopsis are disrupted in mitosis and cell cycle control during seed development. Plant J. 16(1):21–31. [DOI] [PubMed] [Google Scholar]
- Lu X, Liu X, An L, Zhang W, Sun J, Pei H, Meng H, Fan Y, Zhang C.. 2008. The Arabidopsis MutS homolog AtMSH5 is required for normal meiosis. Cell Res. 18(5):589–599. [DOI] [PubMed] [Google Scholar]
- Mandáková T, Marhold K, Lysak MA.. 2014. The widespread crucifer species Cardamine flexuosa is an allotetraploid with a conserved subgenomic structure. New Phytol. 201(3):982–992. [DOI] [PubMed] [Google Scholar]
- Marburger S, Monnahan P, Seear PJ, Martin SH, Koch J, Paajanen P, Bohutínská M, Higgins JD, Schmickl R, Yant L.. 2019. Interspecific introgression mediates adaptation to whole genome duplication. Nat Commun. 10: 5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A.. 2018. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 14(1):e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marhold K, Huthmann M, Hurka H.. 2002. Evolutionary history of the polyploid complex of Cardamine amara (Brassicaceae): Isozyme evidence. Plant Syst Evol. 233(1–2):15–28. [Google Scholar]
- Martin A, Orgogozo V.. 2013. The loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution 67(5):1235–1250. [DOI] [PubMed] [Google Scholar]
- Martin M.2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 17(1):10. [Google Scholar]
- Mason AS, Pires JC.. 2015. Unreduced gametes: meiotic mishap or evolutionary mechanism? Trends Genet. 31(1):5–10. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers BC, Morgante M, Michelmore RW.. 2002. TIR-X and TIR-NBS proteins: two new families related to disease resistance TIR-NBS-LRR proteins encoded in Arabidopsis and other plant genomes. Plant J. 32(1):77–92. [DOI] [PubMed] [Google Scholar]
- Molina-Henao YF, Hopkins R.. 2019. Autopolyploid lineage shows climatic niche expansion but not divergence in Arabidopsis arenosa. Am J Bot. 106(1):61–70. [DOI] [PubMed] [Google Scholar]
- Monnahan P, Kolář F, Baduel P, Sailer C, Koch J, Horvath R, Laenen B, Schmickl R, Paajanen P, Šrámková G, et al. 2019. Pervasive population genomic consequences of genome duplication in Arabidopsis arenosa. Nat Ecol Evol. 3(3):457–468. [DOI] [PubMed] [Google Scholar]
- Morgan C, Zhang H, Henry CE, Franklin CFH, Bomblies K.. 2020. Derived alleles of two axis proteins affect meiotic traits in autotetraploid Arabidopsis arenosa. Proc Natl Acad Sci USA. 117(16):8980–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R.. 2016. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32(11):1749–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M.1972. Genetic Distance between Populations. Am Nat. 106(949):283–292. [Google Scholar]
- Novikova PY, Brennan IG, Booker W, Mahony M, Doughty P, Lemmon AR, Lemmon EM, Dale Roberts J, Yant L, Van de Peer Y, et al. 2020. Polyploidy breaks speciation barriers in Australian burrowing frogs Neobatrachus. PLoS Genet. 16(5):e1008769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogas J, Kaufmann S, Henderson J, Somerville C.. 1999. PICKLE is a CHD3 chromatin-remodeling factor that regulates the transition from embryonic to vegetative development in Arabidopsis. Proc Natl Acad Sci USA. 96(24):13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizza S, Tanaka T, Hochwagen A, Eisenhaber F, Nasmyth K.. 2000. Pds5 cooperates with cohesion in maintaining sister chromatid cohesion. Curr Biol. 10(24):1557–1564. [DOI] [PubMed] [Google Scholar]
- Perruc E, Kinoshita N, Lopez-Molina L.. 2007. The role of chromatin-remodeling factor PKL in balancing osmotic stress responses during Arabidopsis seed germination. Plant J. 52(5):927–936. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Pritchard JK.. 2012. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8(11):e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preite V, Sailer C, Syllwasschy L, Bray S, Ahmadi H, Krämer U, Yant L.. 2019. Convergent evolution in Arabidopsis halleri and Arabidopsis arenosa on calamine metalliferous soils. Philos Trans R Soc B Biol Sci. 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R.. 2005. InterProScan: protein domains identifier. Nucleic Acids Res. 33(Web Server issue):W116-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team R 2011. R: A Language and Environment for Statistical Computing. Available from: http://www.r-project.org.
- Rosenberg SC, Corbett KD.. 2015. The multifaceted roles of the HOR MA domain in cellular signaling. J Cell Biol. 211(4):745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmickl R, Koch MA.. 2011. Arabidopsis hybrid speciation processes. Proc Natl Acad Sci USA. 108(34):14192–14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmickl R, Yant L.. 2021. Adaptive introgression: how polyploidy reshapes gene flow landscapes. New Phytol. 230(2):457–461. [DOI] [PubMed] [Google Scholar]
- Seear P, France M, Gregory C, Heavens D, Schmickl R, Yant L, Higgins J.. 2020. A novel allele of ASY3 promotes meiotic stability in autotetraploid Arabidopsis lyrata. PLoS Genet. [Internet] 16(7):e1008900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, et al. 2015. Polyploidy can drive rapid adaptation in yeast. Nature 519(7543):349–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppey M, Manni M, Zdobnov EM.. 2019. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol. 1962:227–245. [DOI] [PubMed] [Google Scholar]
- Shaked H, Avivi-Ragolsky N, Levy AA.. 2006. Involvement of the arabidopsis SWI2/SNF2 chromatin remodeling gene family in DNA damage response and recombination. Genetics 173(2):985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui NU, Stronghill PE, Dengler RE, Hasenkampf CA, Riggs CD.. 2003. Mutations in Arabidopsis condensin genes disrupt embryogenesis, meristem organization and segregation of homologous chromosomes during meiosis. Development 130(14):3283–3295. [DOI] [PubMed] [Google Scholar]
- Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A.. 2009. BioMart – biological queries made easy. BMC Genomics 10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding JB, Lammers PJ.. 2004. BLAST Filter and GraphicAlign: rule-based formation and analysis of sets of related DNA and protein sequences. Nucleic Acids Res. 32(1):W22–W36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S.. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2):ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Stephan AB, Kunz HH, Yang E, Schroeder JI.. 2016. Rapid hyperosmotic-induced Ca2+ responses in Arabidopsis thaliana exhibit sensory potentiation and involvement of plastidial KEA transporters. Proc Natl Acad Sci USA. 113(35):E5242–E5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43(Database issue):D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpak M, Mezzavilla M, Ayub Q, Chen Y, Xue Y, Tyler-Smith C.. 2018. FineMAV: prioritizing candidate genetic variants driving local adaptations in human populations. Genome Biol. 19(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno S, Ralph P, Swart K, Elshire RJ, Glaubitz JC, Buckler ES, Hufford MB, Ross-Ibarra J.. 2015. Independent molecular basis of convergent highland adaptation in maize. Genetics 200(4):1297–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Adachi Y, Chiba K, Oguchi K, Takahashi H.. 2002. Identification of Ku70 and Ku80 homologues in Arabidopsis thaliana: evidence for a role in the repair of DNA double-strand breaks. Plant J. 29(6):771–781. [DOI] [PubMed] [Google Scholar]
- Tedder A, Helling M, Pannell JR, Shimizu-Inatsugi R, Kawagoe T, Van Campen J, Sese J, Shimizu KK.. 2015. Female sterility associated with increased clonal propagation suggests a unique combination of androdioecy and asexual reproduction in populations of Cardamine amara (Brassicaceae). Ann Bot. 115(5):763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilford CA, Siemers NO.. 2009. Gene set enrichment analysis. Methods Mol Biol. 563:99–121. [DOI] [PubMed] [Google Scholar]
- Van Drunen WE, Husband BC.. 2019. Evolutionary associations between polyploidy, clonal reproduction, and perenniality in the angiosperms. New Phytol. 224(3):1266–1277. [DOI] [PubMed] [Google Scholar]
- Vijay N, Bossu CM, Poelstra JW, Weissensteiner MH, Suh A, Kryukov AP, Wolf JBW.. 2016. Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat Commun. 7:13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Torabinejad J, Cohick E, Parker K, Drake EJ, Thompson JE, Hortter M, DeWald DB.. 2005. Mutations in the Arabidopsis phosphoinositide phosphatase gene SAC9 lead to overaccumulation of PtdIns(4,5)P2 and constitutive expression of the stress-response pathway. Plant Physiol. 138(2):686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Culligan K, Lamers M, Hays J.. 2003. Dissimilar mispair-recognition spectra of Arabidopsis DNA-mismatch-repair proteins MSH2·MSH6 (MutSα) and MSH2·MSH7 (MutSγ). Nucleic Acids Res. 31(20):6027–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant L, Bomblies K.. 2015. Genome management and mismanagement—cell-level opportunities and challenges of whole-genome duplication. Genes Dev. 29(23):2405–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant L, Hollister JD, Wright KM, Arnold BJ, Higgins JD, Franklin FCH, Bomblies K.. 2013. Meiotic adaptation to genome duplication in Arabidopsis arenosa. Curr Biol. 23(21):2151–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman S, Gerstein AC, Hodgins KA, Whitlock MC.. 2018. Quantifying how constraints limit the diversity of viable routes to adaptation. PLoS Genet. 14(10):e1007717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zozomová-Lihová J, Malánová-Krásná I, Vít P, Urfus T, Senko D, Svitok M, Kempa M, Marhold K.. 2015. Cytotype distribution patterns, ecological differentiation, and genetic structure in a diploid–tetraploid contact zone of Cardamine amara. Am J Bot. 102(8):1380–1395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data are available at the European Nucleotide Archive at http://www.ebi.ac.uk/ena/data/view/PRJEB39872 (Cardamine amara) and the Sequence Read Archive at https://www.ncbi.nlm.nih.gov/sra/, study code SRP156117 (Arabidopsis arenosa data). All scripts are available at https://github.com/mbohutinska/PoolSeqBPM (Fst-based selection scans and all following analyses) and https://github.com/paajanen/meiosis_protein_evolution (FineMAV scan).