

Abstract
An N-terminal-domain (NTD) and adjacent catalytic body (CB) make up subunit-α of ribonucleotide reductase (RNR), the rate-limiting enzyme for de novo dNTP biosynthesis. A strong linkage exists between ligand binding at the NTD and oligomerization-coupled RNR inhibition, inducible by both dATP and nucleotide chemotherapeutics. These observations have distinguished the NTD as an oligomeric regulation domain dictating the assembly of inactive RNR oligomers. Inactive states of RNR differ between eukaryotes and prokaryotes (α6 in human versus α4β4 in Escherichia coli, wherein β is RNR’s other subunit); however, the NTD structurally interconnects individual α2 or α2 and β2 dimeric motifs within the respective α6 or α4β4 complexes. To elucidate the influence of NTD ligand binding on RNR allosteric and oligomeric regulation, we engineered a human–E. coli hybrid enzyme (HE) where human-NTD is fused to E. coli-CB. Both the NTD and the CB of the HE bind dATP. The HE specifically partners with E. coli-β to form an active holocomplex. However, although the NTD is the sole physical tether to support α2 and/or β2 associations in the dATP-bound α6 or α4β4 fully inhibited RNR complexes, the binding of dATP to the HE NTD only partially suppresses HE activity and fully precludes formation of higher-order HE oligomers. We postulate that oligomeric regulation is the ultimate mechanism for potent RNR inhibition, requiring species-specific NTD–CB interactions. Such interdomain cooperativity in RNR oligomerization is unexpected from structural studies alone or biochemical studies of point mutants.
Graphical Abstract
The catalytic chemistry of enzymes is predominantly understood using a mechanistic arrow-pushing analysis.1,2 In this way, the chemical steps of catalysis by proteases and kinases appear widely in the literature and textbooks.1–3 However, unlike simple small-molecule catalysts, enzymes are complex structures in which only a small fraction of the entire catalyst scaffold usually interacts with the substrate.4,5 This property allows enzymes to exhibit complexities such as allosteric regulation and changes in quaternary associations, which can modulate the selection or ordering of substrates, and the rate or timing of catalysis.1,2,6 Although an appreciation of enzyme allostery is key to understanding the various aspects of feedback, crosstalk, and regulation that form cornerstones of complex living systems,7 little is known about modular allostery in regulatory proteins with multidomain architecture. In this study, we used domain swapping between a prokaryotic and a eukaryotic enzyme that differ considerably in their regulation to challenge various literature models of quaternary regulation coupled with enzyme allostery. This technique may prove useful to examine other multidomain proteins involved in signaling8 or metabolic9 pathways in which significant differences exist between the regulatory elements within two related enzymes.
Ribonucleotide reductase (RNR) is a paradigm for allosteric regulation.10–12 RNR is an essential enzyme that is uniquely responsible for the reduction of nucleotides to 2′-deoxynucleo-tides in all organisms.12 RNR is also a key player in maintaining dNTP pool homeostasis, the imbalance of which is a major cause of genomic instability, with linkage to multiple human disorders.13,14 Not surprisingly, RNR activity is positively correlated with cancer cell proliferation15 and the enzyme is a validated target of several chemotherapeutics in clinical use.16–18
The role of RNR in the maintenance of dNTP pools stems from its capability to sense both absolute levels and relative ratios of monomeric DNA/RNA precursor nucleotides that allosterically fine-tune RNR activity in response to cellular demand.10,14 Fortuitously, in Escherichia coli, Saccharomyces cerevisiae, mouse, and human RNRs, this nucleotide-driven allosteric regulation occurs exclusively on one of the two subunits (α) of the dual-subunit enzyme RNR,10 allowing the intricate interplay of small-molecule-induced allosteric modulations to be studied in a controlled manner.
RNR-α can be sectioned into two separate domains: a smaller N-terminal-domain (NTD) and an adjacent catalytic body (CB). The NTD houses the first allosteric activity (A) site, and the CB houses the second allosteric specificity (S) site and the catalytic (C) site. ATP binding to the A site in NTD augments RNR activity, whereas dATP results in activity suppression. The S site binds ATP, dATP, dGTP, and dTTP that respectively enable selective recruitment of CDP, UDP, ADP, and GDP substrates to the C site (active site) on α. Active, phosphorylated forms of clinically applied nucleoside analog prodrugs such as clofarabine (ClF) and gemcitabine (F2C) inhibit RNR activity through direct interaction with the A and C sites.19–21
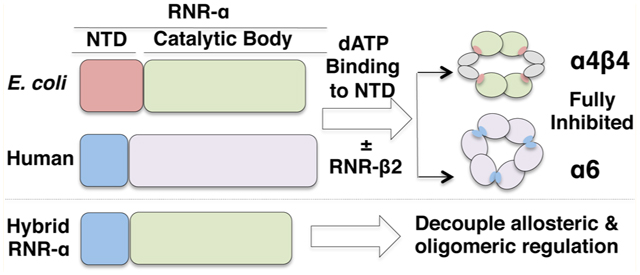
Mounting evidence supports a connection between RNR allostery and enzyme quaternary states. Although the quaternary state of the active RNR holoenzyme in all organisms remains debatable, the latest lines of investigations strongly suggest α2β2 as a catalytically relevant state of E. coli RNR.22,23 The oligomeric state representation of the active E. coli RNR holocomplex is generally referred to as a “docking model”24 (Figure 1A, inset). In eukaryotes, the active form is unknown. Conversely, dATP repression through dATP binding at the A site gives an inert α4β4 holocomplex in prokaryotes22,25,26 (Figure 1A). Importantly, in E. coli RNR, the presence of β is necessary to achieve higher-order complexes. In human, yeast, and mouse RNRs, dATP inhibition is clearly coupled to α-hexamerization. Interestingly, dATP can drive α-hexamerization independently of β19,27–29 (Figure 1A), which may constitute an evolutionary advantage that enables the two subunits to be regulated differentially in eukaryotes.
Figure 1.

(A) Quaternary state of active RNR is unknown, whereas dATP-induced inhibition leads to species-specific changes in oligomeric states. Ribbon representations of previously determined 3.95 and 6.61 Å X-ray structures, respectively, of inhibited RNR complexes, α4β4 and α6 from E. coli (4ERM26) and S. Cerevisiae (3PAW28): red, NTD of α; green, CB of α; green + red = entire α monomer; blue, β monomer. Cartoon representations were included for clarity. The inset shows proposed docking model for the active RNR α2β2 holocomplex, based on the structures of individual α and β subunits from E. coli.24 S, A, and C, respectively, designate allosteric specificity (S), activity (A) sites, and catalytic (C) sites. (B) Structure-based sequence alignment of α-subunits from human (H1), mouse (M1), yeast (Y1 and Y3), and E. coli (E1) RNR featuring the first 56(8) residues. In E1 (E. coli α), residues in vertical red boxes are specific residues at the α–β interface necessary for adopting α4β4 inhibited holocomplex. These residues are absent in eukaryotes. (C) Domain arrangement including three nucleotide-binding sites in the engineered 759-amino acid-long functional HE RNR-α. NTD (red) houses the A site. CB, composed of catalytic barrel (blue) and C-terminal tail (green), houses the S and C sites.
The globular NTD consisting of four α-helices and three β-strands24,30 is a common feature for all Class Ia RNRs to which mouse, human, yeast, and E. coli RNRs belong. In line with the general acceptance that evolutionary pressure is less severe at allosteric sites, giving rise to their flexibility, an iterative search, by using position-specific iterative basic local alignment search tool (PSI-BLAST), on the origin of RNR-α NTD has suggested evolutionarily independent acquisition of NTD by various classes of RNR, highlighting domain plasticity.30 However, the actual mobility or modularity of the NTD has not been studied experimentally. A thoughtful analysis has been made on Class Ia RNR from the eubacterial species Pseudomonas aeruginosa in which RNR-α has two NTDs derived via gene duplication.31 In this case, the distal NTD (farthest from the CB) is dispensable for enzymatic activity, whereas the loss of both NTDs results in a catalytically inactive enzyme. On the basis of these results, RNR has been inferred to be a modular protein, and NTD modularity is considered an important diversification platform within the RNR family.31 However, the indispensable nature of the proximal NTD (contiguous with the CB) observed in this study begs the question about the genuine plasticity in the RNR-α NTD, particularly because only a single NTD is manifested in the RNR-α from most organisms.10 Importantly, the functional significance of how nucleotide binding at the NTD of RNR from one species might choreograph RNR quaternary equilibria and/or allosteric regulatory capability in a distantly related species has never been investigated.
Although dATP-downregulated states between the two classes are clearly distinct with either inclusion or exclusion of β (Figure 1A), nucleotide binding at the NTD is believed to be obligatory in the formation of inert, higher-order homo- and hetero-oligomers.10 The available X-ray and electron microscopy (EM) structural data of dATP-inhibited α4β4 and α6 oligomers of E. coli22,26 and yeast or human RNR,28,29 respectively, have suggested that the NTD plays a critical role in physically interconnecting the individual α2 and/or β2 dimeric motifs (Figure 1A). By contrast, if the α2β2 docking model (Figure 1A, inset) serve as the basis for a catalytically competent state in which physical interaction between the two subunits occurs exclusively between the CB and β, species specificity in the NTD should be dispensable. We thus envisioned that various aspects of RNR regulation, contributions of the two allosteric sites, and interdomain crosstalk could be investigated by creating a hybrid RNR-α, HE, consisting of human NTD fused directly to the CB of E. coli RNR-α.
In this study, successful construction of and subsequent biochemical investigations into HE (Figure 1C and S1, Supporting Information) revealed the influences each domain has on the capability of RNR to adopt catalytically viable versus nonviable quaternary states. The catalytically functional hybrid is folded correctly, with both the NTD and the CB showing expected affinity for natural ligands. The data are most consistent with the proposed α2β2 docking model (Figure 1A, inset) being the active state of the RNR holocomplex. The evolutionarily mobile NTD30 is found to be transposable between the two species that have only 27% sequence identity. This feature applies to holocomplex formation as well as causing enzyme inactivation driven by the mechanism-based inactivator gemcitabine diphosphate (F2CDP). We additionally engineered E. coli-α lacking an NTD and bearing the CB alone (hereafter, E-CB). Although E-CB remains functional in terms of ligand binding and binding to E. coli-β, E-CB alone is catalytically inactive, suggesting that human NTD can rescue the activity in the HE. By contrast, the NTD alone in the HE is insufficient to promote assembly of inert humanlike HE RNR α6 complexes induced by dATP, despite the fact that the NTD is the sole structural tether between individual dimeric units within the inactive trimer-of-dimer [(α2)3] states (Figure 1A).28,29 Interestingly, allosteric regulation from the A site in NTD is weakened in the hybrid protein. The binding of dATP in saturating amounts at the NTD only partially inhibits the HE, compared with the near-complete inhibition previously observed with canonical RNR-αs.19,22 Although crosstalk between the human NTD and E. coli CB is possible, we observed no formation of higher-order oligomers of the HE under any conditions. We thus propose that the formation of species-specific higher-order oligomers is an ultimate regulatory route that engenders potent RNR inhibition, whereas inhibitor binding at the NTD exerts only a partial effect.
MATERIALS AND METHODS
Materials.
BL21-CodonPlus(DE3)-RIL competent cells were from Stratagene. Complete EDTA-free protease inhibitor tablets and calf intestine alkaline phosphatase were from Roche. NH4+ salt of [5-3H]-CDP was from Vitrax. F2C was from Selleckchem. F2CDP was synthesized as previously reported.20 Texas Red-5-dATP and liquid scintillation counting cocktail (Emulsifier-Safe) were from Perkin-Elmer. All primers were from IDT. Fusion HotStartII polymerase was from Thermo. DNase I and all the restriction enzymes were from NEB. TALON metal affinity resin was from Clontech. Sephadex G-25 resin was from GE Healthcare. Ultrafiltration membrane Amicon ultracentrifugal devices were from Millipore. dATP was from Invitrogen and ATP was from Acros Organics. NADPH was from MP Biomedicals. Streptomycin sulfate and isopropyl β-d-1-thiogalactopyranoside (IPTG) were from Gold Biotechnology. All other chemicals were from Sigma. E. coli thioredoxin (TRx), E. coli RNR subunits α and β (E-α and E-β), human RNR subunits α (H-α), and human D57N-α (HD57N-α) were isolated as previously described.19,20,32 A pET28a vector encoding E. coli thioredoxin reductase (TrxR) gene was generated from the plasmid pTrR301 bearing wild-type E. coli TrxR gene (a kind gift of Dr. Scott Mulrooney, Michigan State University). His6–TrxR was subsequently expressed in E. coli and purified to homogeneity by TALON-affinity chromatography. Specific activity of the isolated His6–TrxR was 13 700 ± 1400 U/mg (7.77 ± 0.79 s−1), determined by the coupled assay using Ellman’s reagent.33 Protein concentrations were determined using ε280nm/M−1 cm−1: 119 160 and 45 900 for H-α and H-β; 94 500 and 65 500 for E-α and E-β; 81 600 for HE hybrid; 77 755 for E-CB alone (calculated from the amino acid sequence using the ProtParam tool (http://web.expasy.org/protparam/). All concentrations reported are for monomers.
Hybrid Gene Constructions and Generation of Recombinant Expression Plasmids.
NTD and CB (Figure 1C) were separately amplified from the plasmids encoding either wild-type E-α or H-α genes (NrdA or RRM1), using the primers shown in Table S1A,B of the Supporting Information. The hybrid gene, HE, was subsequently constructed by PCR ligation of the two respective domains. The resultant gene was finally cloned into an empty pET28a vector. pET28a H- and E-NTD-alone constructs were also generated during these cloning efforts. In addition, construction of plasmid encoding E-CB alone was achieved via analogous procedures using the primers shown in Table S1C of the Supporting Information to generate pET28a E-CB. The genes were confirmed by sequencing of the entire HE, E-CB, H-NTD, and E-NTD genes at the Cornell University Life Science Core Laboratories Center.
Enzyme Activity and Inhibition Assays.
Subsequent to metal-affinity chromatographic purification of the proteins (see Supporting Information), all assays were carried out at 37 °C. α-Subunit-specific radioactive assays for E- and H-α and time-dependent inhibition analyses were performed as previously described.19,20,32,34 Corresponding assay mixture for hybrids or E-CB contained 1.0 μM (or 2.0 μM) α, 4.0 μM (or 8.0 μM) β, 3 mM ATP, 15 mM MgCl2, 0.5 mM [5-3H]-CDP (SA: 25 000 cpm/nmol), 100 μM E. coli Trx, 1.0 μM E. coli His6–TrxR, and 2 mM NADPH in 50 mM Hepes buffer (pH 7.6). Time-dependent inhibition analysis of HE by F2CDP was carried out as previously reported.20
CD Measurements.
Circular dichroism (CD) spectra were recorded as the average of three scans on a AVIV CD spectrometer (Model 62ADS) using a 1.0 mm path-length quartz cuvette. Bandwidth was 1 nm, and signal averaging time was 10 s. The sample temperature was set at 25 °C with a digital circulating water bath. Protein samples were prepared just before use and filtered through 0.22 μm syringe filters (Millex low protein binding PVDF membrane, Millipore). Each 400 μL sample contained in final concentrations 10 mM NaH2PO4 (pH 7.6), 0.2 mM DTT, 150 mM NaCl, and 1.5 μM of HE, E-CB, E- or H-α.
Fluorescence Anisotropy Assays.
Anisotropy measurements were performed on a Varian Cary Eclipse spectro-fluorometer equipped with a manual polarizer accessory. The total fluorescence and anisotropy were measured using an excitation wavelength of 585 nm and an emission wavelength of 612 nm. The excitation and emission slit widths were 10 and 20 nm, respectively. All measurements were carried out in 50 mM Tris (pH = 7.6) in the presence of 15 mM MgCl2, 100 mM KCl, 5 mM BME, and 5% glycerol at 25 °C. Each sample contained in final concentrations 0.5 μM Texas Red-5-dATP and either 1–500 μM H-NTD or 0.1–160 μM E-CB. The samples were incubated for 5 min at 25 °C prior to anisotropy measurements.
Gel Filtration Analyses.
Gel filtration was performed on a Superdex 200 10/300 column (GE Healthcare) using a Shimadzu Prominence SIL-20ACHT HPLC. The incubation mixture contained 15 mM MgCl2, 3 μM α, ±3 μM β, 5 mM DTT, ±1 mM dATP and ±1 mM CDP in 50 mM Hepes (pH 7.6). Prior to injection, the reaction mixture was incubated at 37 °C for 2 min and filtered through 0.22 μm syringe filters (Millex low protein binding PVDF membrane, Millipore). A 100 mL amount of reaction mixture was injected onto the column that had been pre-equilibrated with the running buffer [15 mM MgCl2, 150 mM NaCl, ±20 or 300 μM dATP and ±1 mM CDP in 50 mM Hepes (pH 7.6)]. The flow rate was 0.5 mL/min. Molecular mass standards (GE Healthcare): ovalbumin, 44 kDa; conalbumin, 75 kDa; aldolase, 158 kDa; ferritin, 440 kDa; thyroglobulin, 669 kDa were run under identical conditions at the end of each run.
Electron Microscopy (EM).
Samples containing 3 μM E-α or HE, 3 μM E-β, 15 mM MgCl2, 5 mM DTT, 1 mM CDP, and 300 μM dATP in 50 mM Hepes (pH 7.6) were freshly prepared and incubated for 5–10 min at room temperature. The samples were diluted 20-fold into “Buffer A” [15 mM MgCl2, 1 mM CDP, 300 μM dATP in 50 mM Hepes (pH 7.6)], and the diluted samples (3 μL from each) were applied to continuous carbon-coated grids that had been glow-discharged for 2 min at −10 mA just before use. The samples were subsequently allowed to equilibrate for 30–60 s at room temperature. The grids were quickly washed and blotted once with Buffer A, followed by staining with a drop of 0.75% uranyl formate. Stain was applied a second time, and the grids were blotted and air-dried. The specimens were imaged at 200 kV on an FEI Tecnai F20 electron microscope using a Gatan US4000 CCD at a nominal defocus of −2.4 μm. The magnification was 50,000×, and the pixel size was 1.88 Å.
RESULTS
Hybrid Enzyme Design and Construction of Expression Plasmids.
The HE fusion gene was designed by swapping the NTD of human α directly with the CB of E. coli α (Figures 1C and S1A of the Supporting Information). No linker was placed between the two domains, such that the end residue of the human (H) NTD, K91, is contiguous with G94, the first residue of the E. coli (E) CB. The previously constructed plasmids that respectively encode H- and E-α20 are the sources for the requisite hybrid gene. The hybridized gene was then inserted into the empty pET28a vector such that the N-terminal His6-tagged hybrid gene product, HE, could be recombinantly expressed and isolated from E. coli. During this cloning process, constructs that encoded either the NTD alone of H- and E-α or the CB alone of E-α were also generated. We achieved expression and isolation of the HE, the E. coli CB alone (E-CB), and the human NTD alone (H-NTD) (Figures 2A and S1B of the Supporting Information), although E. coli NTD alone failed to express. These data suggest that the H-NTD is stable and correctly folds independently of its CB.
Figure 2.
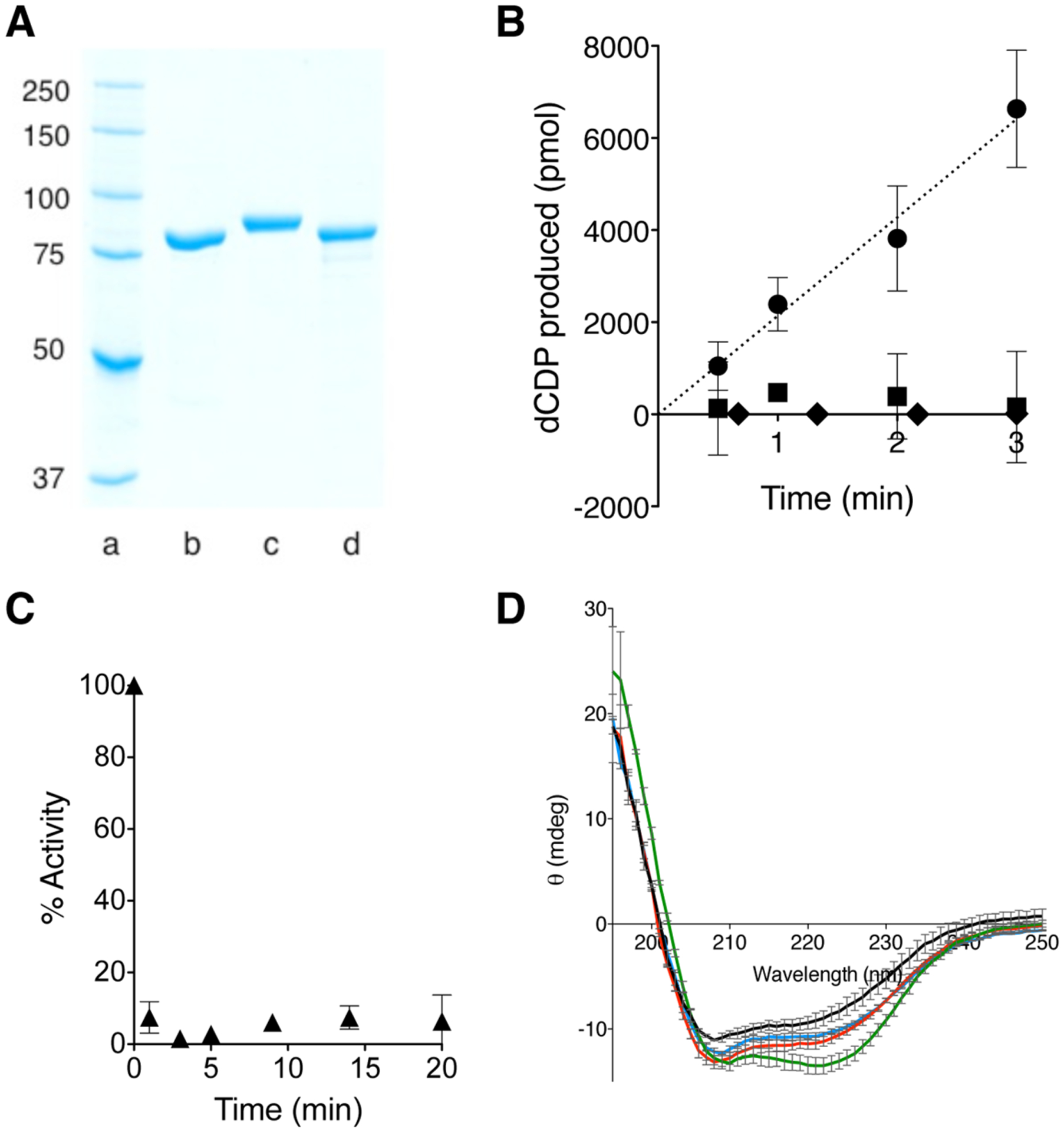
Biochemical characterizations of functional HE. (A) SDS–PAGE analysis of isolated HE in comparison with E- and H-α. Lanes a → d: ladder, E-α (86 kDa), His6–H-α (92 kDa), His6–HE (87 kDa). (B) Time dependent [5-3H]-dCDP production catalyzed by 1 nmol of HE and either E- or H-β (● or ■, respectively). Results under identical conditions except E-CB replaces HE (⧫). Error bars represent standard deviation (N = 3). (C) Time-dependent inhibition analysis of HE in the presence of stoichiometric amount of F2CDP. Data has been adjusted for inherent enzyme decay under assay conditions. Error range was derived from N = 2. (D) CD spectrum of 1.5 μM HE (red), E-α (blue), H-α (green) or E-CB (black) in 10 mM NaH2PO4 (pH 7.6), 0.2 mM DTT and 150 mM NaCl at 25 °C (1.0 mm path length, 1 nm bandwidth, 10 s signal averaging time). The vertical scale is elipticity in millidegrees.
The Catalytic Body Alone of E. coli α Is Catalytically Inert.
Prior to evaluating the contributions of the human NTD in moderating RNR allosteric and quaternary regulation, the biochemical study of the E-CB alone was first attempted. The E-CB in the presence of saturating amounts of E-β, CDP, Mg· ATP, and turnover-generating enzymes showed no reductase capacity (Figure 2B, ◆). Assay conditions were such that activity as low as ~0.1 U/mg (0.0001 s−1) would be detectable. It was further confirmed (with circular dichroism spectroscopy; see below and Figure 2D) that the observed inertness of the E-CB is, first, unrelated to misfolding. Second, the E-CB alone is not defective in ligand binding, as demonstrated with fluorescence anisotropy assays using Texas Red-5-dATP (Figure S2A of the Supporting Information); the Kd was calculated to be 5.3 ± 0.3 μM, a value within the range previously reported for the affinity of unlabeled dATP to the S site of canonical E-α (0.5 μM).38 Third, the E-CB can competitively bind to E-β, as shown by competition assays with a calculated IC50 of 7.9 ± 1.4 μM (Figure S2B of the Supporting Information).
Expressed HE Protein Is Active in the Presence of E-β but Not H-β.
In contrast to the E-CB, the isolated HE enzyme had an α-subunit-specific CDP reductase activity of 21.2 ± 2.4 U/mg (0.0307 ± 0.0035 s−1), measured under identical conditions (Figure 2B, ●), suggesting that the H-NTD can rescue the catalytic activity in the CB.
Although HE-specific activity is less than the activities measured for the canonical H- or E-α, it lies within the range of reported RNR-α and RNR-β subunit-specific activities from various species, such as P. aeruginosa RNR-α [88 U/mg (0.16 s−1)],31 Bacillus subtilis RNR-β [22 U/mg (0.016 s−1)],35 and catalytically competent radical transfer pathway mutants of E. coli RNR-α (3–5% of wild-type RNR-α).36 Subsequent analysis of effects of dATP on HE (discussed below) also ruled out the possibility of measured HE activity being due to contaminating E-α.
When the activity assays were replicated with H-β [an in vitro reconstituted protein bearing 1.2 tyrosyl radicals (Y·) per β2 dimer],19,20 we observed no reductase activity for the HE (Figure 2B, ■). These data collectively suggest that to achieve a basal level of proton-coupled electron transfer (PCET)-mediated RNR catalysis,12 the N-termini need not be “matched” for a particular organism, whereas the CB has a high species specificity in interacting with partner subunit β. As a corollary, we assayed the activity of E-α in the presence of H-β, and in parallel, the activity of H-α in the presence of E-β and observed no measurable activity in either case.
HE Is Inactivated by the Mechanism-Based Inactivator Gemcitabine Diphosphate (F2CDP).
In canonical E-α–E-β or H-α–H-β pairs, potent F2CDP inactivation stems from the capability of the drug to hijack the thiyl radical within the C site of the CB. Formation of the transient thiyl radical during the catalytic cycle is mediated by forward PCET from the active site of β.12 Addition of stoichiometric amounts of F2CDP to the HE led to inactivation of the HE–E-β pair (Figure 2C). The data imply that despite HE having the human NTD, thiyl radical formation within the C site of the HE CB is achievable via forward PCET initiated from E-β. This implication is consistent with the preceding HE activity data suggesting that species-specific NTD is not required for PCET-mediated RNR catalysis within the holocomplex.
HE Is Not Misfolded.
To verify that low intrinsic activity of HE is not due to the majority of the sample being improperly folded, a far-UV CD spectrum was obtained for HE. Corresponding spectra for E-α and H-α collected under identical conditions showed HE adopts a similar secondary structure (Figure 2D): E-α and H-α differ considerably in the CD spectra, and the HE chimera showed a spectrum with much closer resemblance to that of E-α, as would be expected because HE bears a greater sequence similarity to E-α than to H-α (Figures 1C and S1 of the Supporting Information). E-CB was also folded but distinct from HE, E- and H-α.
HE is Partially Responsive to Allosteric Activity Suppressor dATP at the A Site.
In Class Ia RNRs, to which that for human, mouse, yeast, and E. coli RNRs belong, dATP provides a universal feedback loop through which the cell prevents overproduction of dNTPs.10 dATP exerts this downregulatory effect on RNR through binding at the A site within the NTD (Figure 1A).10,11,19,22,23,25–29 Compared with the basal (no dATP) level, >70–90% inhibition of RNR activity is observed at 100–600 μM dATP in yeast28 mouse,27,37 and human19 α’s (Figure S3 of the Supporting Information) and at 200–1000 μM dATP in E. coli α.22 Binding affinities of dATP to the A site are reportedly 5 and 54 μM in E. coli38 and mouse α’s,39 respectively. From Figure S3 of the Supporting Information, the EC50 of dATP-induced inhibition of H-α was calculated to be 33 ± 7 μM. Because dATP is the major determinant in RNR activity regulation, its effect on the HE was explored. Titration of the HE with dATP showed a stimulatory effect at low dATP concentrations (Figure 3A). The upregulation of enzyme activity at low dATP concentrations through S-site binding has previously been observed for E-α [S-site Kd for dATP = 0.5 μM38],22,25 and H-α (Figure S3 of the Supporting Information). The dATP-induced activity enhancement in HE shows that allosteric communication between the S and C sites within the CB is operational in the HE.
Figure 3.
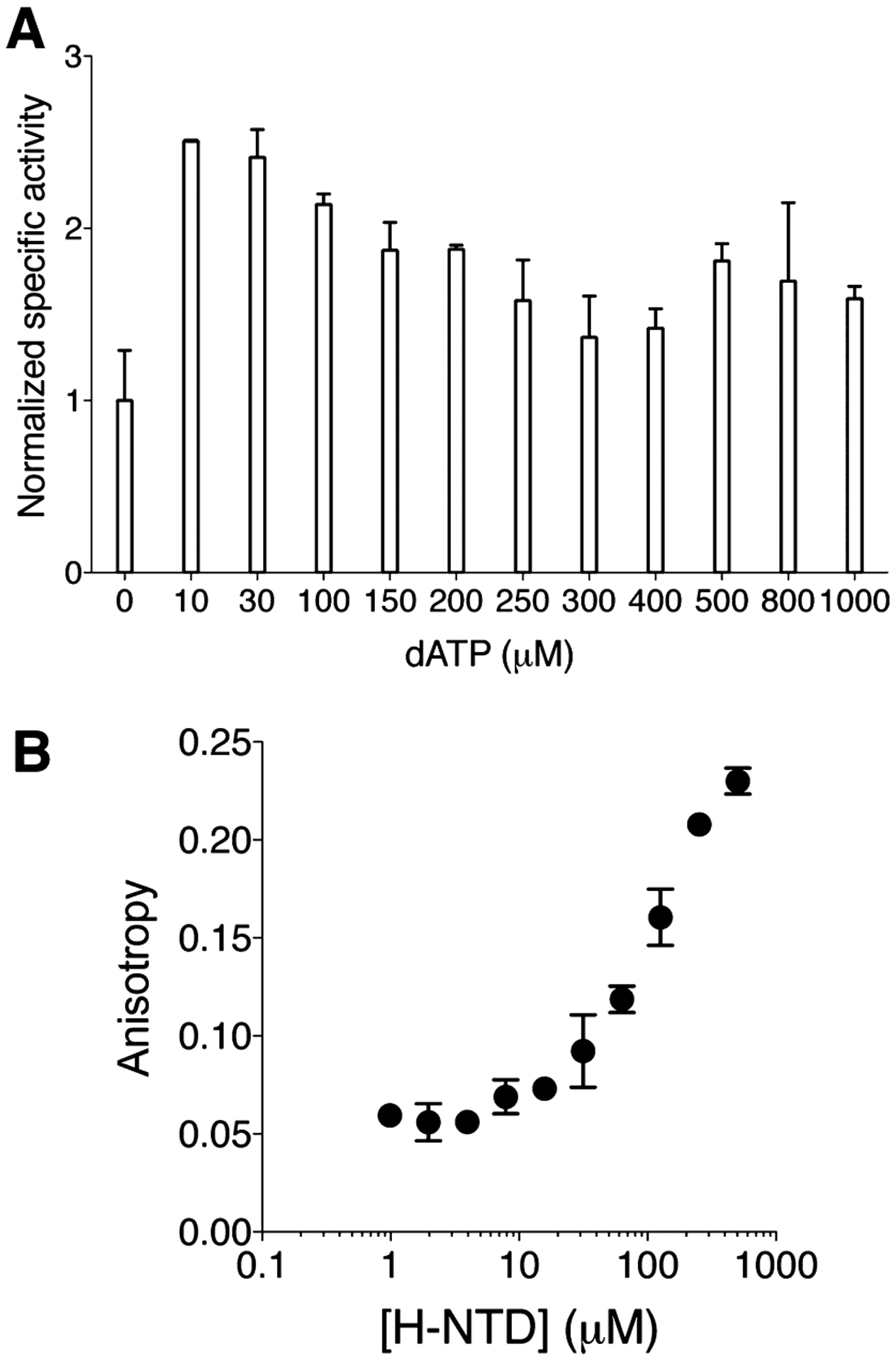
(A) Titration of dATP to HE. Allosteric activity promotion was observed at low [dATP], consistent with Kd = 0.5 μM for the S site of E-α.38 dATP-promoted partial allosteric inhibition of HE, and saturation of dATP binding to the A site of HE, were seen with high [dATP]. Kd of 54 μM previously reported as the affinity of dATP to the A site of mouse α39 is comparable to that of the HE estimated from these data (~90 μM). Normalized activity of 1.0 corresponds to 21 nmol min−1 of [5-3H]-dCDP produced per mg of HE. (Note that assays contained no allosteric promoter ATP). The Error range was derived from at least N = 2. (B) Fluorescence anisotropy resulting from titration of 1–500 μM H-NTD and 0.5 μM Texas Red-5-dATP. Error range was derived from N = 2. Titration to higher concentrations was precluded by poor solubility of H-NTD. Estimated binding affinity of Texas Red-5-dATP to H-NTD = 153 ± 19 μM.
Increasing the dATP concentration decreased (reduction in stimulation of) HE activity, indicating that dATP can bind to the HE NTD, downregulating the activity. The observed inhibitory effect saturated at ~300 μM dATP (EC50 ~ 90 μM) (Figure 3A). At saturation, the activity returned only to a level close to the basal (no dATP) level, indicating that dATP is unable to inhibit the enzyme fully, unlike in the canonical E- or H-α (Figure S3 of the Supporting Information)19,27,28,37. This interesting result is also consistent with the attribution of the activity in the HE sample to the inherent property of the HE and not to contaminating E. coli α.
To validate further the estimated dATP affinity to the A site and confirm that the NTD of the HE is functional for ligand binding, we measured the affinity of the isolated H-NTD alone to Texas Red-5-dATP (Figure 3B). The Kd was calculated to be 153 ± 19 μM. Thus, the CB of E. coli has no significant impact on the human NTD binding of dATP, even though partial allosteric modulatory effects through the NTD is possible. Furthermore, the known affinity of mammalian RNR-α NTD for dATP is also within this concentration range (reportedly 54 μM using unlabeled dATP).39 [A similar range of affinity can be seen from our dATP titration data to H-α (Figure S3 of the Supporting Information)]. These data establish clearly that the human NTD binds nucleotides with similar efficiency irrespective of the presence of a (mis)matched catalytic body. From the observed dATP stimulation, the affinity of the S site for the CB within the HE must also be within an order of magnitude of the canonical E. coli α. Because both the NTD and the CB are similarly competent to bind dATP by this metric, and the HE features partial allosteric regulation through dATP, we sought another explanation for the impaired dATP downregulation in HE.
HE Underscores the Importance of Interdomain Allosteric Communications in Modulating RNR Oligomeric Equilibria: Oligomerization Studies on HE Alone.
dATP-Bound inert RNR complexes adopt hexameric [α6-(±β2)] states in yeast,28 human,19,29 and mouse RNRs27,37 and octameric (α4β4) states in E. coli RNRs.22,25,26 A unified stipulation exists that NTD is essential for dATP-driven RNR oligomerization and inhibition (Figure 1A). However, the molecular basis of nucleotide-induced RNR oligomerization, such as the role of the CB or interdomain allosteric crosstalk remains unknown. We thus evaluated the effect of dATP on the quaternary state of HE, in the presence or absence of E-β and compared the outcomes with canonical H-and E-α. Comparison was also made with the known H-α point mutant, H-D57N-α, which does not undergo dATP-induced inhibition and α-hexamerization.19,28 When α alone was analyzed by gel filtration in the absence of β, we observed that H-α and E-α, respectively, hexamerized and dimerized in the presence of 20 μM dATP in running buffer (Table 1), consistent with previous reports.19,25,28,37 Hexamerization is also a commonly accepted mechanism of eukaryotic RNR α inhibition that is independent of β.19,27–29 On the other hand, the HE failed to hexamerize but reproducibly resolved to a single peak corresponding to a dimer, a behavior observed for H-D57N-α. Dimer formation has also been observed with two additional H-α point mutants, H-D16R-α and H-H2E-α, which also completely resist dATP inhibition.28 The dimeric quaternary state of the HE was maintained up to 300 μM dATP. This value lies in the concentration range in which dATP binding to the A site of HE is saturated (Figure 3A). Importantly, this observation rules out dissolution of the dimeric state as a reason for the observed partial inhibition at high dATP concentrations. The dimerization of HE induced by dATP at concentrations as low as 20 μM dATP is in line with the stimulatory effect on HE activity (Figure 3A) that we propose is caused by S-site binding of the nucleotide. A similar stimulatory effect has been noted for the canonical E. coli22,25 and human RNR-α (Figure S3 of the Supporting Information).19 These data on HE provide the first evidence that although the N-terminus appears to be the only structural element physically linking individual dimeric α2 motifs within the hexameric framework (Figure 1A), a functional NTD–CB interface is obligatory in the molecular mechanism of α-hexamerization. This observation was unexpected based on structural data alone.28,29
Table 1.
Gel Filtration Analysis
| α | dATP (20 μM) | retention time (min) | extracted MW (kDa) | theoretical MW (kDa) |
|---|---|---|---|---|
| HE | − | 27.9 | 82 | 87 (α1) |
| + | 24.6 | 171 | 174 (α2) | |
| E-α | − | 28.2 | 78 | 86 (α1) |
| + | 24.9 | 160 | 172 (α2) | |
| H-α | − | 27.5 | 89 | 92 (α1) |
| + | 19.6 | 561 | 552 (α6) | |
| H-D57N-α | − | 27.9 | 82 | 92 (α1) |
| + | 24.1 | 191 | 184 (α2) |
Oligomerization Studies in the Presence of E-β.
In prokaryotic Class Ia RNRs such as E. coli, β must be present to form the dATP-bound inert complex.22,25,26 Studies using gas-phase electrophoretic mobility analysis have identified α4β4 assembly in E. coli RNR in the presence of dATP at the A site.25 The presence of CDP at the C site was shown to have a stimulatory effect on octamerization.25 The latter finding is also consistent with crosstalk between the two domains. An independent study using multiple biophysical and structural methods has additionally demonstrated that the α4β4 holocomplex is an inactive state of E. coli RNR in the presence of 175 μM dATP and 1 mM CDP22,26 (Figure 1A). The X-ray structure of this holocomplex at 3.95 Å resolution revealed key α–β interface residues proximal to the A site within the individual α2 motif that is connected to two distinct β2 molecules (Figure 1A).26 Because these residues are absent in the NTD of eukaryotic RNRs (Figure 1B), we propose that octamerization is infeasible with the HE and E-β. Consequently, dATP-induced partial inhibition of the HE (Figure 3A) may be linked to the inability to form HE–E-β octamers.
The impact of the NTD on the dATP-induced RNR oligomerization in the presence of β subunit was thus addressed with the HE. By gel filtration chromatography, a 1:1 ratio of E-α:E-β was eluted in the presence of a saturating amount dATP (300 μM) and 1 mM CDP in the running buffer. Fractions were collected and subsequent SDS–PAGE analysis of the fractions showed coelution of the two subunits beginning as early as 19 min (Figure 4A). This retention time translates to 544 kDa (theoretical α4β4 = 517 kDa). The coelution of the two subunits is consistent with the 30–50 times tighter α–β intersubunit affinity reported for the inactive α4β4 complex compared with that of the active holocomplex.25 When the same experiment was performed with HE in place of E-α, we observed no coelution (Figure 4A). In addition, HE eluted much later around 24 min, corresponding to a dimer, whereas E-β separately migrated at the 27 min expected for β2 dimer. We note that the HE protein we isolated consistently had a lower molecular weight impurity band (<5% by densitometric quantitation, Figure 2A). This impurity was, however, concentrated as a large aggregate eluting around 19 min on gel filtration (Figure 4A).
Figure 4.

(A) SDS–PAGE analysis of eluted fractions from gel filtration analysis of 3 μM either E-α or HE in the presence of 3 μM E-β and 300 μM dATP. Coelution of the two subunits α and β is observed only in the case of 1:1 E-α:E-β (top) but not in the case of 1:1 HE:E-β (bottom). Failure to coelute HE and E-β is consistent with the inability of HE to form α4β4 holocomplex in the presence of saturating amount of dATP. The requirement for high concentrations of nucleotides in the running buffer limits direct determination of elution time by the analysis of protein absorbance peaks in the elution profile. An asterisk indicates molecular weight ladders (100, 75, and 50 kDa from top to bottom in each case). E-α = 86, HE = 87, and E-β = 44 kDa. The identity of a ~70 kDa impurity band that elutes at 19 min in the case with hybrid (bottom) has not been determined. (B) EM image of α4β4 rings formed by 1:1 E-α:E-β in the presence of 300 μM dATP and 1 mM CDP (left). Scale bar shown corresponds to 100 nm. Under identical conditions, α4β4 rings were absent when E-α was replaced with HE (right).
In parallel, EM imaging studies were performed to validate independently the gel filtration data. Whereas a 0.15 μM solution of 1:1 E-α:E-β in the presence of saturating dATP (300 μM) resulted in intact α4β4 ring structures, consistent with previous reports,22,26 replacement of E-α with the HE under identical conditions led to collapse of these ring complexes (Figure 4B). These data together provide strong evidence that failure by dATP to inhibit HE fully is linked to restricted access to the α4β4 inactive oligomeric assembly. The data imply that oligomerization is an effective mechanism for potent RNR inhibition beyond allosteric activity suppression.
DISCUSSION
It is generally accepted that nature has employed “domain swapping” to optimally evolve new functional dynamics in proteins.40,41 The past decade has witnessed a resurgence of intense interest in protein remodeling through rational or combinatorial approaches and exploration of the interdependence of protein oligomeric equilibria and functional properties.42 Research has shown that plasticity in the domain architecture typically found with most proteins has enabled facile transposition of allosteric domains in engineering artificial proteins with acquired allosteric functionality. Stemming from these discoveries, applications including the creation of molecular biosensors,43 conditional allosteric switches, and “novel enzymes” with unrelated catalytic activity have been demonstrated with modular allosteric properties in multidomain proteins.44–47
Importantly, mechanistic knowledge of allostery has also remained a key element in pharmacological drug design.41,48,49 RNR-α is a validated target of clinically approved chemotherapeutics, such as clofarabine (ClF), cladribine, fludarabine, and gemcitabine (F2C).17,18,50 Although these drugs belong to nucleoside antimetabolites, the identity of the actual phosphorylated form(s) that inhibit RNR and the mechanism of inhibition can be highly variable and unpredictable.19–21,29 Even in the case of natural substrates and effectors, mechanisms of site-to-site communications or “cooperativity” remain largely elusive for RNR, especially insight into how ligand-induced allostery orchestrates oligomeric heterogeneity. Ultimately, distinct oligomeric states of RNR can result in varying catalytic competency. Thus, biochemical knowledge about RNR domain modularity, and the influence of allosteric or orthosteric ligand binding not only on interdomain crosstalk but also on the perturbation of RNR quaternary state should prove useful.
Based on BLAST, a 27% identity exists between H-α and E-α. Comparing the two individual domains, we observed that NTDs have slightly higher identity with respect to catalytic body components: 33% and 28%, respectively [identity between the C-termini (Figure 1C) is below the level of significance]. On the basis of macromolecular size proportions, HE was expected to feature greater similarity to E-α over H-α. This expectation was consistent with the CD data (Figure 2D).
Implications for the Active State of RNR Holocomplex.
In bacterial RNRs, although the active state is believed to be α2β2 heterotetramer, the α–β interface-locking mechanisms have long been debated. An X-ray structure of an active RNR holocomplex in any species remains to be achieved. The “docking model” (Figure 1A, inset) reconstructed based on shape complementarity between individual E-α and E-β X-ray structures has been the preferred model.24 In-depth data accumulated from biophysical, chemical, and most recently structural studies using PCET pathway mutants of E. coli RNR subunits23,51,52 have provided strong evidence of the validity of the docking model in which a lack of physical interaction between the α-NTD and β appears obvious (Figure 1A, inset). To date, the only available crystal structure of the α2β2 holocomplex obtained is from Salmonella typhimurium RNR that belongs to Class Ib enzymes that do not house an NTD.53 Nevertheless, the α–β interaction formed in this crystal showed only one β molecule within the β2 dimer associating primarily with the C-site hydrophobic cleft of one α molecule within the α2 dimer, the stage of which authors describe as an intermediate along the catalytic cycle.
The exclusive reductase activity of the HE with E-β and potent inactivation of the HE–E-β holocomplex by the mechanism-based inactivator F2CDP (Figure 2C) imply that within the active RNR holocomplex, adequate association with β that drives PCET catalysis can be achieved without the requirement of an α-NTD from the same species (hereafter, “matched” NTD). Our data are thus more consistent with the docking model in which key interactions in the active holocomplex can be most simply viewed between E-CB and E-β (Figure 1A, inset). The fact that the E-CB alone is inactive (Figure 2B) despite having the ability to bind E-β (Figure S2B of the Supporting Information) and ligands, as exemplified by dATP (Figure S2A of the Supporting Information), further implies that the presence of the NTD is necessary to gain catalytic activity. We thus propose a refined model for the catalytically active state of Class Ia RNRs to which human, mouse, yeast, and E. coli RNRs belong: (1) the NTD, irrespective of species origin (i.e., E. coli versus mammals), is required to induce conformational transition of the E-CB that leads to productive E-β-binding during turnover; (2) NTD species-specificity is dispensable in achieving functional catalysis. The latter proposition is consistent with the existing docking model of an α2β2 active state wherein physical interaction between the NTD and E-β is absent (Figure 1A, inset). These data present a starting point to understanding the allosteric evolution of RNR regulation.
Implications for the Inactive State.
HE data collectively reinforce the current model of the inactive state of E. coli holocomplexes. X-ray structural analysis of the dATP-bound inactivated E. coli holocomplex has previously revealed a set of strategically located amino acid residues on the NTD of E-α that are responsible for the noncovalent complexation between α2–β2 dimeric units within the inert α4β4 complex (Figure 1A).26 These hot spot residues may serve as messengers in mediating intersubunit allosteric communication during dATP-induced octamerization of E. coli RNR. Amino acid sequence alignment (Figure 1B) reveals a different set of conserved residues exhibited by the eukaryotic RNR-α (and HE). Inability of the HE to octamerize with E-β (Figure 4A,B) and its capability to resist potent inhibition by dATP (Figure 3A) revealed the requirements of NTD-mediated species-specific mechanisms in RNR oligomerization-coupled inactivation. Our data suggest a model in which the potency of dATP-induced inhibition of prokaryotic Class Ia RNR represented by the E. coli enzyme is exclusively brought about by specific interaction between the E-α NTD and E-β required for α4β4 octamerization. In this model, allosteric ligand binding alone is insufficient to drive the oligomerization-coupled enzyme inhibition, suggesting that additional intraprotein or interprotein interactions may be required.
Conversely, failure of HE to hexamerize by the known H-α hexamerization inducer, dATP that interacts with the NTD (Table 1) also suggests that hexamerization, a commonly accepted mechanism for eukaryotic RNR-α inhibition, is not simply dominated by allosteric activity suppression through ligand binding at NTD. It is conceivable that energetically favored pathways for efficient propagation of oligomeric regulatory responses from dATP binding of NTD, to the CB, are missing in the HE because of the mismatched recipient domain, precluding access to a hexameric state. These studies highlight the importance of species-specific interdomain interactions in nucleotide-induced RNR conformational and quaternary structural changes. We thus postulate that a network of amino acids exists within the interdomain interface that can effectively support site-to-site communication to promote eukaryotic RNR inhibition through protein hexamerization.
In summary, successful creation and biochemical investigations of a hybrid HE RNR-α highlighted the species-specific interdependence of the two distinct domains in modulating allosteric and oligomeric control of RNR enzyme activity. These insights were previously not revealed by studies of the inactive states of canonical RNRs19,22,25–29 or hexamerization-defective NTD point mutants alone.19,28 Study of HE enabled an appreciation of species specificity and dual-domain choreography in regulating RNR oligomerization.
Supplementary Material
ACKNOWLEDGMENTS
Professor Niko Grigorieff, Brandeis University and HHMI, is gratefully acknowledged for providing resources for EM sample preparation and data collection. We thank Professor JoAnne Stubbe and the Stubbe Laboratory, MIT, and Dr. Scott Mulrooney, Michigan State University, for the plasmids encoding wild-type human and E. coli RNR-α genes, and the pTrR300 plasmid, respectively. We thank the laboratories of Professors Peng Chen, Brian Crane, and Steve Ealick, Cornell University, for the use of fluorometer, CD spectrometer, and inert atmosphere chamber, respectively. ESR data were collected at the National Biomedical Center for ADVANCED ESR Technology (ACERT): supported by NIH/NIGMS Grant P41GM103721 [at Cornell University, Professor Jack Freed (PI); we thank Dr. Boris Dzikovski for his aid]. This research was supported by an HHMI international student predoctoral fellowship (to M.J.C.L.), by a Hill undergraduate research fellowship (to W.A.B.), and by the Cornell University junior faculty startup funds, the Milstein new faculty fellowship, the Affinito-Stewart grant from the President’s Council of Cornell Women, and the faculty development grant from the ACCEL: Advancing Cornell’s Commitment to Excellence and Leadership program supported by NSF (SBE-0547373, PI: Kent Fuchs) (to Y.A.).
ABBREVIATIONS
- CB
catalytic body
- dNTP
deoxynucleoside triphosphate
- E
E. coli
- EM
electron microscopy
- F2CDP
gemcitabine diphosphate
- H
human
- NTD
N-terminal-domain
- RNR
ribonucleotide reductase
Footnotes
Supporting Information
Supporting methods information including recombinant expression of HE and single domains of RNR-α, isolation of HE and E-α-CB, pull-down of E-NTD, H-NTD, and HE, competition assays. Table S1 showing primers used for the construction of pET-28a −HE and E-CB (E = E. coli, CB = catalytic body). Figures S1–4 showing amino acid sequence of Hybrid His6–HE and pull-down analysis validating expression and solubility of His6–H–NTD, E. Coli binding, titration of dATP to canonical H-α, and gel filtration standards. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
NOTE ADDED AFTER ASAP PUBLICATION
This paper was published ASAP September 23, 2013, with an error in the second to last sentence of the first paragraph of the Materials section and errors in the head row and the bottom row of the second column in Table 1. The corrected version was reposted on September 25, 2013.
REFERENCES
- (1).Fersht A (1999) Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding, W. H. Freeman, New York. [Google Scholar]
- (2).Walsh C (1979) Enzymatic reaction mechanisms, W. H. Freeman, San Francisco. [Google Scholar]
- (3).Hedstrom L (2002) Serine protease mechanism and specificity. Chem. Rev. (Washington, DC, U. S.) 102, 4501–4524. [DOI] [PubMed] [Google Scholar]
- (4).Britt BM (1997) For enzymes, bigger is better. Biophys. Chem 69, 63–70. [DOI] [PubMed] [Google Scholar]
- (5).Srere PA (1984) Why are enzymes so big? Trends Biochem. Sci 9, 387–390. [Google Scholar]
- (6).Kirsch JF (2008) Enzyme kinetics and mechanism, by Paul F. Cook and W. W. Cleland. Protein Sci. 17, 380–381. [Google Scholar]
- (7).Changeux JP (2012) Allostery and the Monod-Wyman-Changeux model after 50 years. Annu. Rev. Biophys 41, 103–133. [DOI] [PubMed] [Google Scholar]
- (8).Tsai CJ, Del Sol A, and Nussinov R (2009) Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Mol. BioSyst 5, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brzovic PS, Ngo K, and Dunn MF (1992) Allosteric interactions coordinate catalytic activity between successive metabolic enzymes in the tryptophan synthase bienzyme complex. Biochemistry 31, 3831–3839. [DOI] [PubMed] [Google Scholar]
- (10).Hofer A, Crona M, Logan DT, and Sjoberg BM (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol 47, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nordlund P, and Reichard P (2006) Ribonucleotide reductases. Annu. Rev. Biochem 75, 681–706. [DOI] [PubMed] [Google Scholar]
- (12).Stubbe J, and van Der Donk WA (1998) Protein Radicals in Enzyme Catalysis. Chem. Rev. (Washington, DC, U. S.) 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (13).Thelander L (2007) Ribonucleotide reductase and mitochondrial DNA synthesis. Nat. Genet 39, 703–704. [DOI] [PubMed] [Google Scholar]
- (14).Zhou BB, and Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439. [DOI] [PubMed] [Google Scholar]
- (15).Elford HL, Freese M, Passamani E, and Morris HP (1970) Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J. Biol. Chem 245, 5228–5233. [PubMed] [Google Scholar]
- (16).Shao J, Zhou B, Chu B, and Yen Y (2006) Ribonucleotide reductase inhibitors and future drug design. Curr. Cancer Drug Targets 6, 409–431. [DOI] [PubMed] [Google Scholar]
- (17).Bonate PL, Arthaud L, Cantrell WR Jr., Stephenson K, Secrist JA 3rd, and Weitman S (2006) Discovery and development of clofarabine: a nucleoside analogue for treating cancer. Nature reviews. Drug Discovery Dev. 5, 855–863. [DOI] [PubMed] [Google Scholar]
- (18).Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC, and Grindey GB (1990) Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res. 50, 4417–4422. [PubMed] [Google Scholar]
- (19).Aye Y, and Stubbe J (2011) Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. U. S. A 108, 9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang J, Lohman GJ, and Stubbe J (2007) Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. U. S. A 104, 14324–14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Licht S, and Stubbe J (1999) Mechanistic investigation of ribonucleotide reductase. in Comprehensive Natural Product Chemistry (Poulter CD, ), pp 163–204, Elsevier, Amsterdam. [Google Scholar]
- (22).Ando N, Brignole EJ, Zimanyi CM, Funk MA, Yokoyama K, Asturias FJ, Stubbe J, and Drennan CL (2011) Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U. S. A 108, 21046–21051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Minnihan EC, Ando N, Brignole EJ, Olshansky L, Chittuluru J, Asturias FJ, Drennan CL, Nocera DG, and Stubbe J (2013) Generation of a stable, aminotyrosyl radical-induced alpha2beta2 complex of Escherichia coli class Ia ribonucleotide reductase. Proc. Natl. Acad. Sci. U. S. A 110, 3835–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Uhlin U, and Eklund H (1994) Structure of ribonucleotide reductase protein R1. Nature 370, 533–539. [DOI] [PubMed] [Google Scholar]
- (25).Rofougaran R, Crona M, Vodnala M, Sjoberg BM, and Hofer A (2008) Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem 283, 35310–35318. [DOI] [PubMed] [Google Scholar]
- (26).Zimanyi CM, Ando N, Brignole EJ, Asturias FJ, Stubbe J, and Drennan CL (2012) Tangled up in knots: structures of inactivated forms of E. coli class Ia ribonucleotide reductase. Structure 20, 1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rofougaran R, Vodnala M, and Hofer A (2006) Enzymatically active mammalian ribonucleotide reductase exists primarily as an alpha6beta2 octamer. J. Biol. Chem 281, 27705–27711. [DOI] [PubMed] [Google Scholar]
- (28).Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, Nordlund P, Li Z, Walz T, and Dealwis CG (2011) Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol 18, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Aye Y, Brignole Edward, J., Long Marcus, J. C., Chittuluru J, Drennan Catherine, L., Asturias Francisco, J., and Stubbe J (2012) Clofarabine Targets the Large Subunit (±) of Human Ribonucleotide Reductase in Live Cells by Assembly into Persistent Hexamers. Chem. Biol. (Oxford, U. K.) 19, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Aravind L, Wolf YI, and Koonin EV (2000) The ATP-cone: an evolutionarily mobile, ATP-binding regulatory domain. J. Mol. Microbiol. Biotechnol 2, 191–194. [PubMed] [Google Scholar]
- (31).Torrents E, Westman M, Sahlin M, and Sjoberg BM (2006) Ribonucleotide reductase modularity: Atypical duplication of the ATP-cone domain in Pseudomonas aeruginosa. J. Biol. Chem 281, 25287–25296. [DOI] [PubMed] [Google Scholar]
- (32).Ge J, Yu G, Ator MA, and Stubbe J (2003) Pre-steady-state and steady-state kinetic analysis of E. coli class I ribonucleotide reductase. Biochemistry 42, 10071–10083. [DOI] [PubMed] [Google Scholar]
- (33).Ellman GL (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys 82, 70–77. [DOI] [PubMed] [Google Scholar]
- (34).Steeper JR, and Steuart CD (1970) A rapid assay for CDP reductase activity in mammalian cell extracts. Anal. Biochem 34, 123–130. [DOI] [PubMed] [Google Scholar]
- (35).Zhang Y, and Stubbe J (2011) Bacillus subtilis class Ib ribonucleotide reductase is a dimanganese(III)-tyrosyl radical enzyme. Biochemistry 50, 5615–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Seyedsayamdost MR, and Stubbe J (2007) Forward and Reverse Electron Transfer with the Y356DOPA-β2 Heterodimer of E. coli Ribonucleotide Reductase. J. Am. Chem. Soc 129, 2226–2227. [DOI] [PubMed] [Google Scholar]
- (37).Kashlan OB, and Cooperman BS (2003) Comprehensive model for allosteric regulation of mammalian ribonucleotide reductase: refinements and consequences. Biochemistry 42, 1696–1706. [DOI] [PubMed] [Google Scholar]
- (38).Brown NC, and Reichard P (1969) Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J. Mol. Biol 46, 39–55. [DOI] [PubMed] [Google Scholar]
- (39).Chimploy K, and Mathews CK (2001) Mouse ribonucleotide reductase control: influence of substrate binding upon interactions with allosteric effectors. J. Biol. Chem 276, 7093–7100. [DOI] [PubMed] [Google Scholar]
- (40).Ostermeier M, and Benkovic SJ (2000) Evolution of protein function by domain swapping. Adv. Protein Chem 55, 29–77. [DOI] [PubMed] [Google Scholar]
- (41).Vogel C, Bashton M, Kerrison ND, Chothia C, and Teichmann SA (2004) Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol 14, 208–216. [DOI] [PubMed] [Google Scholar]
- (42).Selwood T, and Jaffe EK (2012) Dynamic dissociating homo-oligomers and the control of protein function. Arch. Biochem. Biophys 519, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Doi N, and Yanagawa H (1999) Design of generic biosensors based on green fluorescent proteins with allosteric sites by directed evolution. FEBS Lett. 453, 305–307. [DOI] [PubMed] [Google Scholar]
- (44).Dueber JE, Yeh BJ, Chak K, and Lim WA (2003) Reprogramming control of an allosteric signaling switch through modular recombination. Science 301, 1904–1908. [DOI] [PubMed] [Google Scholar]
- (45).Guntas G, Mitchell SF, and Ostermeier M (2004) A molecular switch created by in vitro recombination of nonhomologous genes. Chem. Biol 11, 1483–1487. [DOI] [PubMed] [Google Scholar]
- (46).Ostermeier M (2005) Engineering allosteric protein switches by domain insertion. Protein Eng., Des. Sel 18, 359–364. [DOI] [PubMed] [Google Scholar]
- (47).Dueber JE, Mirsky EA, and Lim WA (2007) Engineering synthetic signaling proteins with ultrasensitive input/output control. Nat. Biotechnol 25, 660–662. [DOI] [PubMed] [Google Scholar]
- (48).Peracchi A, and Mozzarelli A (2011) Exploring and exploiting allostery: Models, evolution, and drug targeting. Biochim. Biophys. Acta 1814, 922–933. [DOI] [PubMed] [Google Scholar]
- (49).Arkin MR, and Whitty A (2009) The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr. Opin. Chem. Biol 13, 284–290. [DOI] [PubMed] [Google Scholar]
- (50).Gandhi V, P. W (2006) Clofarabine: Mechanism of Action, pharmacology and clinical investigations. in Cancer Drug Discovery and Development: Deoxynucleosides Analogues in Cancer Therapy (Peters GJ, ), pp 153–171, Humana Press, Totowa, NJ. [Google Scholar]
- (51).Stubbe J, Nocera DG, Yee CS, and Chang MC (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. (Washington, DC, U. S.) 103, 2167–2201. [DOI] [PubMed] [Google Scholar]
- (52).Minnihan EC, Nocera DG, and Stubbe J (2013) Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res, DOI: 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Uppsten M, Farnegårdh M, Domkin V, and Uhlin U (2006) The First Holocomplex Structure of Ribonucleotide Reductase Gives New Insight into its Mechanism of Action. J. Mol. Biol 359, 365–377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.