

Summary
Ritter reaction has been recognized as an elegant strategy to construct the C−N bond. Its key feature is forming the carbocation for nucleophilic attack by nitriles. Herein, we report a complementary visible-light-induced three-component Ritter reaction of alkenes, nitriles, and α-bromo nitriles/esters, thereby providing mild and rapid access to various γ-amino nitriles/acids. Mechanistic studies indicated that traceless fluoride relay, transforming KF into imidoyl fluoride intermediate, is critical for the efficient reaction switch from atom transfer radical addition (ATRA) to the Ritter reaction. This approach to amino-alkylation of alkenes is chemoselective and operationally simple.
Subject areas: Chemistry, Organic chemistry, Organic synthesis
Graphical abstract

Highlights
-
•
Using light irradiation to promote amino-alkylation of alkenes
-
•
Using KF to facilitate three-component Ritter reaction
-
•
Access functionalized amides under mild conditions
Chemistry; Organic chemistry; Organic synthesis
Introduction
Since its discovery in 1948 (Ritter and Kalish, 1948; Ritter and Minieri, 1948), Ritter reaction has been recognized as one of the most powerful methods for amide synthesis through the formation of the C−N bond (Scheme 1A) (Bolsakova and Jirgensons, 2017; Crouch, 1994; Guérinot et al., 2012; Jiang et al., 2014; Kürti and Czakó, 2005; Li-Zhulanov et al., 2020; Mohammadi Ziarani et al., 2020; Pronin et al., 2013; Qu et al., 2012; Zheng et al., 2015). This two-component protocol usually involves the generation of carbocation intermediates from tertiary, secondary, and benzylic alcohols under acidic conditions (Kürti and Czakó, 2005). As basic feedstock chemicals, simple alkenes have also been widely used as carbocation precursors in Ritter reaction (Eren and Kusefoglu, 2005; Huang et al., 2012; Jiang and Studer, 2020; Nandy et al., 2020; Park et al., 2018; Shi et al., 2015; Subba Reddy et al., 2010; Welniak, 1996; Williams et al., 2017; Xu et al., 2017; Yang et al., 2018; Yasuda and Obora, 2015; Zhang et al., 2020). Of particular interest is the three-component Ritter reaction, which can efficiently incorporate two distinct functional groups onto the carbon-carbon double bonds in one-step (Abe et al., 2010, 2017; Ahmed et al., 2020; Ai et al., 2015; Bao et al., 2019; Chen et al., 2016; Feng et al., 2018; Liu and Klussmann, 2020; Qian et al., 2017; Zhu et al., 2017). Nowadays, photoredox catalysis (Hopkinson et al., 2016; Marzo et al., 2018; Narayanam and Stephenson, 2011; Prier et al., 2013; Romero and Nicewicz, 2016; Shaw et al., 2016; Skubi et al., 2016; Tellis et al., 2016; Twilton et al., 2017; Xuan and Xiao, 2012; Yu et al., 2020, 2021) for simultaneously constructing C−C and C−X bonds has become a new paradigm of alkene difunctionalizations (Badir and Molander, 2020; Chen et al., 2018; Koike and Akita, 2016; Lipp et al., 2021; Protti et al., 2016; Yin et al., 2020; Zhu et al., 2020). With the help of cationic precursors (Umemoto's reagent, iodonium salt, or diazonium salt), Akita, Greaney, and König developed elegant three-component Ritter reactions of alkenes under visible light irradiation (Scheme 1B) (Fumagalli et al., 2013; Prasad Hari et al., 2014; Yasu et al., 2013; Zong et al., 2019). Notably, the introduction of a corresponding counterion (BF4−) with weak nucleophilicity could spare the active carbocation intermediates to be exclusively attacked by nitrile partners. Therefore, the development of photo-induced Ritter reaction from neutral precursors with competitive nucleophiles is challenging and appealing.
Scheme 1.

Design of photo-induced three-component Ritter reaction via fluoride
(A) Classic Ritter reaction for the synthesis of amides.
(B) Previous work: photo-induced Ritter reaction with cationic precursors.
(C) This work: inorganic fluoride-induced chemoselective Ritter reaction.
As is well known, photo-induced atom transfer radical addition (ATRA) with neutral precursors is a well-established protocol for alkene difunctionalizations (Courant and Masson, 2016; Magagnano et al., 2017; Mao and Cong, 2017; Ouyang et al., 2018; Pu et al., 2019; Rawner et al., 2018). The high chemoselectivity of ATRA is mostly attributed to the existence of a single nucleophile which attacked carbocation. Regarding the use of the widely available alkylbromides as precursors for carbocation intermediates in the Ritter reaction, which nucleophile would display stronger affinity toward carbocation, bromides or nitriles? Intrigued by the aforementioned issue and our long-standing interests in functionalization of alkenes (Ji et al., 2019; Jiang et al., 2021; Kuai et al., 2020; Min et al., 2021; Yang et al., 2019), we sought to develop photo-induced three-component Ritter reaction of alkenes with alkylbromides and nitriles. Herein, we demonstrated an unprecedented role of fluoride salts for the enhancement of Ritter reaction and inhibition of ATRA (Scheme 1C).
Results
Optimization reaction conditions
Initially, with Ir(ppy)3 (1 mol%) as a photocatalyst, styrene (1a) and 2-bromoacetonitrile (2a) were chosen as the model substrates to test our hypothesis (see Tables 1, S1). Without any additive, ATRA of 1a proceeded smoothly as expected to give 4-bromo-4-phenylbutanenitrile (4a) at 71% yield (entry 1). Despite the favor for 4a, the use of KBF4 as the additive accidentally gave 3a as a minor product (entry 2). It inspired us to use other additives containing fluorine atoms (entries 3–8). Trifluoroacetic acid (TFA) showed no enhancement on the selectivity of 3a (entry 3). Fortunately, NEt3·3HF and NaF could facilitate the formation of γ-amino nitrile (3a) with moderate selectivity (entries 4 and 5). Particularly, KF and CsF proved to be suitable additives and the target product 3a was delivered in satisfactory yields and good selectivity (entries 6–7). In the case of quaternary ammonium salt NBu4F, the reaction did not occur (entry 8). With respect to the anion effect of additives, potassium salts with other halide ions (KCl, KBr, and KI) contrarily gave 4a as the main product (entries 9–11). The aforementioned results showed the significance of fluoride anion for high selectivity of 3a. Other common metallaphotoredox catalysts, such as [Ir(ppy)2dtbbpy]PF6 and [Ru(bpy)3]Cl2, yielded no desired product 3a (entries 12 and 13). When switching to organic photocatalyst Eosin Y, the reaction could not take place (entry 14). In addition, the control experiments confirmed the essential roles of the iridium catalyst and the visible light irradiation for this protocol (entries 15 and 16). If H2O was present from the beginning in the similar condition under air atmosphere, 3a was obtained in 57% yield, albeit with a small amount of 4-hydroxy-4-phenylbutyronitrile (entry 17). No 3a was observed in the absence of KF (entry 18). The aforementioned results indicate that KF plays an indispensable role to create target product 3a.
Table 1.
Selected optimization studies
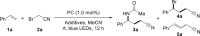 | |||||
|---|---|---|---|---|---|
| Entry | Photocatalyst | Additive | Yield (%)a |
||
| 3a | 4a | 5a | |||
| 1 | Ir(ppy)3 | – | 1 | 71 | 6 |
| 2 | Ir(ppy)3 | KBF4 | 8 | 59 | 7 |
| 3b | Ir(ppy)3 | TFA | 5 | 73 | 1 |
| 4c | Ir(ppy)3 | NEt3·3HF | 51 | 22 | 6 |
| 5 | Ir(ppy)3 | NaF | 63 | 12 | 9 |
| 6 | Ir(ppy)3 | KF | 95 | 0 | 0 |
| 7 | Ir(ppy)3 | CsF | 85 | 0 | 1 |
| 8 | Ir(ppy)3 | NBu4F | – | – | – |
| 9 | Ir(ppy)3 | KCl | 16 | 37 | 13 |
| 10 | Ir(ppy)3 | KBr | 0 | 63 | 19 |
| 11 | Ir(ppy)3 | KI | 1 | 12 | 5 |
| 12 | [Ir(ppy)2(dtbbpy)]PF6 | KF | 0 | 7 | 1 |
| 13 | [Ru(bpy)3]Cl2 | KF | 0 | 8 | 0 |
| 14 | Eosin Y | KF | – | – | – |
| 15 | – | KF | – | – | – |
| 16 | Ir(ppy)3 (In dark) | KF | – | – | – |
| 17d | Ir(ppy)3 (air, H2O) | KF | 57 | 0 | 1 |
| 18d | Ir(ppy)3 (air, H2O) | – | 0 | 55 | 2 |
Reaction conditions: 1a (0.21 mmol), 2a (0.20 mmol), photocatalyst (1.0 mol%), additives (0.40 mmol), MeCN (0.8 mL), rt, under N2, 10 W blue LEDs, 12 h. Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. See also Tables S1.
TFA = trifluoroacetic acid, 0.3 equiv.
1.0 equiv.
H2O (3.0 equiv.) was added.
Substrate scope study
With the optimized conditions in hand, the generality of alkene substrates was subsequently investigated. As shown in Figure 1, various substituted styrenes were suitable for this photo-induced Ritter reaction, affording γ-cyano acetamides with moderate to excellent yields. Aryl alkenes 1 bearing substituents at para position, such as Me, tBu, and Ph groups, all afforded the desired products in good yields (83–99%, 3b-3d, 3i). Substrates with halides, including F, Cl, or Br on the phenyl ring, were well tolerated under the current protocol regardless of the substitution position (3e-3g, 3j-3l). It should be noted that benzyl chloride, which was easily attacked by nucleophiles, also remained intact, giving the target product an 84% yield (3h). When a naphthyl alkene was subjected to the standard condition, the corresponding product 3m was obtained in moderate yield. Notably, 1,2-disubstituted alkenes such as cyclic (3n) and acyclic alkenes (3o) were well compatible to give desired products in good yields with acceptable diastereoisomeric ratios (dr).
Figure 1.

Substrate scope with respect to alkenes
Reaction conditions: 1 (0.21 mmol), 2a (0.20 mmol), Ir(ppy)3 (1.0 mol%), KF (0.40 mmol), MeCN (0.8 mL), rt, under N2, 10 W blue LEDs, 12 h. Isolated yield was given. Diastereoisomeric ratio (dr) was determined by 1H NMR analysis.
In order to enrich the category of products, we next studied substrate scope with respect to radical precursors, α-bromoesters (Figure 2). Substituted vinylarenes with the Br or Cl group all reacted successfully with ethyl bromoacetate, furnishing the target products with moderate to good yields (6a-6e). The molecular structure of 6b was confirmed by X-ray crystallographic analysis. Interestingly, α-substituted C-radicals bearing electron-withdrawing groups such as mono-fluoro, gem-difluoro groups worked well to provide the corresponding products 6f-6i in 37–68% yields. In the case of methyl 2-bromopropanoate, the product 6j was isolated at 45% yield, together with 3:1 dr. Furthermore, the reaction with phenyl 2-bromoacetate also performed smoothly to give the desired product with 63% yield (6k).
Figure 2.

Substrate scope with respect to radical precursors
Reaction conditions: 1 (0.21 mmol), 2a (0.20 mmol), Ir(ppy)3 (1.0 mol%), KF (0.30 mmol), MeCN (0.8 mL), rt, under N2, 10 W blue LEDs, 12 h. Isolated yield was given. Diastereoisomeric ratio (dr) was determined by 1H NMR analysis.
In addition, various nitriles were evaluated as well (Figure 3). To our delight, benzonitrile was a suitable partner, giving rise to the product 8a with a 46% yield. In terms of reactions with o- and m-tolunitriles, CsF was found to be a better additive (8b, 8c). Under the similar conditions, isobutyronitrile and valeronitrile could also be readily transformed into the corresponding products (8d, 8e) in moderate yields. Isovaleronitrile was also applicable to the process, forming the product 8f with 46% yield. Especially, bulkier pivalonitrile also worked to afford the product 8g with 32% yield. These lower yields than that of acetonitrile might be attributed to steric hindrance and weaker nucleophilic ability.
Figure 3.

Substrate scope with respect to nitriles
Reaction conditions: 1a (0.21 mmol), 2a (0.20 mmol), Ir(ppy)3 (1.0 mol%), KF (0.40 mmol), nitriles 7 (0.8 mL), rt, under N2, 10 W blue LEDs, 12 h. Isolated yield was given.
Scale-up synthesis and transformations
To demonstrate the potential utility of this methodology, a gram scale reaction of 1a and 2a in acetonitrile was performed. We were glad to find that product 3a was obtained with high yield even when the catalyst loading of Ir(ppy)3 was decreased to 0.2 mol% (Scheme 2A). In addition, further synthetic transformations of the products toward cyclic amine and amide were studied (Scheme 2B). By using commercially available acetaldoxime and nickel salts in water, 3a was hydrated into the corresponding amide 9 with 64% yield (Ma et al., 2012). A treatment of 3a with sodium in butanol delivered 2-phenylpyrrolidine 10 with 76% yield (Zhu et al., 2017). In the presence of Cs2CO3, γ-amino ester 6a could be readily transformed into 5-phenylpyrrolidin-2-one 11 with 81% yield.
Scheme 2.

Gram-scale reaction and synthetic transformations
(A) Gram-scale reaction with low catalyst loading.
(B) Synthetic transformations.
Discussion
Mechanism of the study
To probe the importance of photonic input, the light dependence of the reaction was examined (see Table S3 and Figure S81 for light on/off experiments). It is shown that continuous irradiation of visible light is required for effective formation of product 3a which rapidly ceases in the absence of light. Furthermore, we calculated a quantum yield value (Cismesia and Yoon, 2015) of Ф = 0.35 (see Figures S82 and S83). This observation indicates that this protocol probably does not involve a light-initiated radical chain pathway.
Next, several control experiments have been conducted to probe into the generation of carbocation intermediates under this protocol. When CD3CN was used as the solvent, the target deuterated product 3a-d3 was obtained in 97% yield with >99% deuterium at the methyl group (Scheme 3A). This deuterium labeling result indicates that 2-bromo-acetonitrile 2a serves as a radical precursor and the carbocation intermediate is exclusively trapped by acetonitrile. Instead of 3a, 4-methoxy-4-phenylbutane-nitrile 12 was formed at 90% yield in the presence of methanol. The stronger nucleophilicity of methanol brought about this product variation supporting the existence of conceivable carbocation intermediate (Scheme 3B). In the presence of radical scavenger [(2,2,6,6-tetramethylpipe-ridin-1-yl)oxyl] (TEMPO), the reaction was totally suppressed. This radical trapping experiment suggests that a radical pathway is probably involved for generation of carbocation intermediates (Scheme 3C).
Scheme 3.

Mechanistic studies regarding the carbocation intermediate
(A) Deuterium labeling experiment.
(B) Carbocation attacked by heteroatom nucleophiles.
(C) Radical trapping experiment. See also Scheme 1, Scheme 2, Scheme 3.
Encouraged by these significant results on the nature of the photo-induced Ritter reaction, we were curious about the effect of KF. The usage amount of KF on the control of product selectivity was further examined (Figure 4A, Table S2). ATRA product 4a was favored as a major product in the absence of KF. With the increasing loading of KF (0.5–2.0 equiv.), product 3a gradually dominated in the product distributions (23–95% yield). Taken together, we wondered whether 4a could be transformed into 3a with appropriate use of KF. Thus, the transformation of 4a was carried out under the standard condition (Figure 4B, entry 1, Table S4). As expected, product 3a was isolated at 77% yield after 12 h. In the absence of the iridium catalyst, visible light irradiation, or KF, no 3a was generated (entries 2–4). These results suggest that KF plays an important role in the orientation of intermediate trapping to control product selectivity. Furthermore, the inseparable mixture of 4-fluoro-4-phenylbutyronitrile 13 and 4a as reactants were carried out with the standard condition. As a result, 13 showed an inert substrate and was fully recovered (Figure 4C and Scheme S5). This observation suggests 13 is not the resting intermediate for the formation of target product 3a.
Figure 4.

The effect of fluoride on the product selectivity
Given the dramatic effect of KF on the product selectivity (Figure 4), the fate of fluoride during the reaction was further investigated (Figure 5). Fortunately, fluorine-19 nuclear magnetic resonance (19F NMR) provides an effective means for analyzing fluoride species. The comparison of 19F NMR spectra between 0 and 10 min showed a clear change from −122.10 ppm to −20.45 ppm (Figures 5A, I, II, S93, and S96). This downfield shift suggests a strong decrease of electron density on fluorine atoms. Both F signals (−19.12 and −20.45 ppm) were split into a quartet with similar coupling constants (11.2 and 11.4 Hz), indicating the existence of three neighboring hydrogen atoms (Figure 5B). These characteristics suggest the mysterious intermediate is an organic fluoride.
Figure 5.

The characterization of the fluoride intermediate
(A) 19F NMR spectra of reaction mixture at different reaction stages.
(B) Summarized chemical shifts and spin coupling constants for the fluoride intermediate.
(C) HMQC spectrum at 10 min.
(D) HMBC spectrum at 10 min.
(E) Further transformation of imidoyl fluoride.
Subsequently, 1H, 13C, 19F NMR, 1H-13C heteronuclear multiple quantum coherence (HMQC), and 1H-13C heteronuclear multiple bond correlation (HMBC) spectra were collected for comprehensive analysis of the structure of fluoride intermediate (see Figures S94–S98). To cut a long story short, representative chemical shifts and coupling constants are summarized in Figure 5B. Notably, 13C NMR spectra displayed two sets of doublets at 152.3 and 159.9 ppm characterized by very large 1JC-F coupling constants (340.0 and 254.0 Hz). Obvious remote heteronuclear J-couplings (2JC-F and 3JC-F) were also observed at the aliphatic carbon region (13.9, 18.5, 58.5, and 61.8 ppm). The analysis of 1H-13C HMQC and 1H-13C HMBC spectra (Figures 5C and 5D) was carried out to determine the space connectivity between diagnostic carbons (58.5, 61.8 152.3, and 159.9 ppm) and hydrogens (4.90 and 4.48 ppm). With these self-consistent correlations, we inferred a formation of imidoyl fluoride intermediate Int-F. The significant difference in the 1JC-F coupling constants (340.0 and 254.0 Hz) probably results from the geometrical effect (Z/E isomers) of Int-F on the heteronuclear Cα−F interaction (Norell, 1970; Rowe et al., 1999). Moreover, 1H NMR, 13C NMR, 1H-13C HMQC, and 1H-13C HMBC spectra were collected and analyzed to further support the proposed imidoyl fluoride Int-F using CD3CN as the deuterium-labeling reactant (see Figures S99–S103). Eventually, the desired product 3a was generated from the quench of intermediate Int-F by H2O in the NMR tube (see Figures S104–S106). Meanwhile, a dramatic upfield shift in 19F NMR from −20.45 ppm to −150.70 ppm also suggests the cleavage of the Cα−F bond on intermediate Int-F (Figures 5A, III, and S106). In addition, intermediate Int-F could be captured by N-methylbenzylamine to afford the corresponding amidine 14 (Gurjar and Fokin, 2020) in 60% NMR yield (Figures 5E, S107, and S108). This result further supports the formation of intermediate Int-F.
Based on the aforementioned results and literature reports on photo-induced reactions (Courant and Masson, 2016; Fumagalli et al., 2013; Prasad Hari et al., 2014; Yasu et al., 2013; Zong et al., 2019), a plausible mechanism is shown in Scheme 4. Metallaphotoredox catalyst Ir(ppy)3 (E1/2M+/M∗ = −1.73 V vs. SCE) (Shih et al., 2010) is excited by visible light irradiation to generate the excited species ∗IrIII. A subsequent single-electron transfer (SET) process (Yi et al., 2014) yields acetonitrile radical A (bromoacetonitrile: E1/2red = −0.69 V vs. SCE) (Isse and Gennaro, 2004), a bromide anion and IrIV species. Then the radical addition of A onto alkene 1 affords the C−C coupling adduct B, which is oxidized by the IrIV to form the carbocation intermediate C through another SET process. Intermediate acetimidoyl fluoride D is generated from the nucleophilic attack of acetonitrile onto carbocation C. The final hydrolysis workup delivers the expected product 3. Alternatively, atom transfer radical addition between bromo nitrile 2a and alkene 1 yields the adduct 4 which can return the cycle through a photoredox pathway. Side product 5 could be generated from the β-proton elimination of carbocation C.
Scheme 4.

Proposed mechanism
Conclusion
In conclusion, we have developed a three-component Ritter reaction of alkenes, nitriles, and alkylbromides through photoredox catalysis. A variety of synthetically useful γ-amino nitriles/acids were easily prepared. Through the selective capture of carbocation by nitrile and KF, the formation of imidoyl fluoride intermediate diverts the reaction from undesired atom transfer radical addition to the expected Ritter reaction. The salient features of this protocol include mild reaction conditions, good synthetic utility, and easy scalability. This photoredox catalysis serves as a complementary protocol for conventional thermal or acid-promoted Ritter reaction. Further investigations on the utilization of this mild approach are in progress in our laboratory.
Limitations of the study
The 1,1-disubstituted styrenes, alkyl-substituted alkenes, and other alkyl halides were not suitable in this methodology (See Figure S85 for details).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals | ||
| Ir(ppy)3 | Energy Chemical | Cat#94928-86-6 |
| Eosin Y | TCI | Cat#17372-87-1 |
| Styrene | Sinopharm Chemical Reagent Co. LTD | Cat#100-42-5 |
| 4-Bromostyrene | Accela ChemBio Co., Ltd. | Cat#2039-82-9 |
| 4-Chlorostyrene | Chembee | Cat#1073-67-2 |
| 3-Chlorostyrene | Alfa Aesar | Cat#2039-85-2 |
| 4-tert-Butylstyrene | Macklin | Cat#1746-23-2 |
| 4-Methylstyrene | TCI | Cat#622-97-9 |
| 4-Fluorostyrene | J&K Scientific | Cat#405-99-2 |
| 1-(Chloromethyl)-4-vinylbenzene | Energy Chemical | Cat#1592-20-7 |
| 4-Vinyl-1,1'-biphenyl | 9Dingchem | Cat#2350-89-2 |
| 3-Methylstyrene | Energy Chemical | Cat#100-80-1 |
| 2-Bromostyrene | Innochem | Cat#2039-88-5 |
| 2-Chlorostyrene | Alfa Aesar | Cat#2039-87-4 |
| Bromoacetonitrile | Accela ChemBio Co., Ltd. | Cat#590-17-0 |
| Ethyl bromoacetate | Sinopharm Chemical Reagent Co. LTD | Cat#105-36-2 |
| Ethyl 2-bromo-2-fluoro-acetate | Energy Chemical | Cat#401-55-8 |
| Ethyl 2-bromo-2,2-difluoroacetate | Energy Chemical | Cat#667-27-6 |
| Methyl 2-bromopropano-ate | Alfa Aesar | Cat#5445-17-0 |
| Phenyl 2-bromoacetate | TCI | Cat#620-72-4 |
| SuperDry acetonitrile | J&K Scientific | Cat#75-05-8 |
| Benzonitrile | Sinopharm Chemical Reagent Co. LTD | Cat#100-47-0 |
| o-Tolunitrile | Energy Chemical | Cat#529-19-1 |
| m-Tolunitrile | Aladdin | Cat#620-22-4 |
| 3-Methylbutanenitrile | Acros | Cat#625-28-5 |
| Valeronitrile | Sigma-Aldrich | Cat#110-59-8 |
| Isobutyronitrile | TCI | Cat#78-82-0 |
| Pivalonitrile | Energy Chemical | Cat#630-18-2 |
| NEt3·3HF | Energy Chemical | Cat#73602-61-6 |
| Potassium fluoride | Aladdin | Cat#7789-23-3 |
| Cesium fluoride | Energy Chemical | Cat#13400-13-0 |
| Deposited data | ||
| CIF of 6b | CCDC 2039204 | https://www.ccdc.cam.ac.uk/structures/ |
| Other | ||
| Blue LED lamps (40W, peak wavelength of 456 nm) | GeAo Chemical | http://www.geaochem.com/ |
| Silica gel (200-300 mesh) | Xinchengsilicagel | http://www.ytsilica-gel.com |
| thin layer chromatography using TLC silica gel plates with phosphomolybdic acid chromogenic agent | Xinchengsilicagel | http://www.ytsilica-gel.com |
| AVANCE III 400 MHz | Bruker | https://bruker.com |
| AVAVCE III HD 700 MHz | Bruker | https://bruker.com |
| X-ray diffraction | Agilent GeminiUltra | https://www.agilent.com.cn/ |
| HRMS data of new compounds | Agilent Q-TOF 6540 & Agilent 8890-7250 GC/Q-TOF | https://www.agilent.com.cn/ |
| UV/Vis absorption spectra | Lambda 950 | https://www.perkinelmer.com.cn/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Qing-An Chen (qachen@dicp.ac.cn).
Materials availability
All other data supporting the findings of this study are available within the article and the supplemental information or from the lead contact upon reasonable request.
Method details
Initial trials and reaction optimization (see Table S1)
In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of additive (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in acetonitrile (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue light-emitting diodes (LEDs) at room temperature for 12 h. After the reaction completed and was quenched by H2O, yield was determined by 1H NMR analysis of crude mixture using 1,3,5-trimethoxybenzene (8.4 mg, 0.05 mmol) as an internal standard.
General procedure A

In the glove box, alkenes 1 (0.21 mmol) and radical precursors 2 (0.20 mmol) were added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in nitriles (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. The reaction was quenched by exposure to air, with reaction mixture added into 0.5 mL water and extracted with EtOAc (3 x 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate: 8/1-1/1 v/v) to afford products 3 (Zhu et al., 2017), 6, 8.
Light on/off experiments
According to the photo-induced condition (Table S1, entry 6), a reaction containing naphthalene as an internal standard was set up and placed in a blue LED reactor. The reaction was sequentially stirred under visible light irradiation and in the absence of light. Every 30 s, an aliquot of 10 μL was collected via syringe and analyzed by GC-FID. After 240 s, the determined yields were plotted against the reaction time (see Table S2, Figure S81).
Quantum yield measurements
| Equation 1 |
| Equation 2 |
| Equation 3 |
The photon flux of the spectrophotometer was determined by standard ferrioxalate actinometry (Cismesia and Yoon, 2015; Hatchard and Parker, 1956; Kuhn et al., 2004). A 0.15 M solution of ferrioxalate was prepared by dissolving 2.21 g of potassium ferrioxalate hydrate in 30 mL of 0.05 M H2SO4. A buffered solution of phenanthroline was prepared by dissolving 50 mg of phenanthroline and 11.25 g of sodium acetate in 50 mL of 0.5 M H2SO4. Both solutions were stored in dark. To determine the photon flux of the spectrophotometer, 2.0 mL of the ferrioxalate solution was placed in a cuvette and irradiated for 90.0 s at λ = 436 nm with an emission slit width at 2.0 nm. After irradiation, 0.35 mL of the phenanthroline solution was added to the cuvette. The solution was then allowed to rest for 1 h to allow the ferrous ions to completely coordinate to the phenanthroline. The absorbance of the solution was measured at 510 nm. A nonirradiated sample was also prepared, and the absorbance at 510 nm measured. Conversion was calculated using Equation 1, where V is the total volume (0.00235 L) of the solution after addition of phenanthroline, ΔA is the difference in absorbance at 510 nm between the irradiated and nonirradiated solutions, l is the path length (1.000 cm), and ε is the molar absorptivity at 510 nm (11,100 L mol−1 cm−1) (Hatchard and Parker, 1956). The photon flux can be calculated using Equation 3, where Φ is the quantum yield for the ferrioxalate actinometer (1.01 for a 0.15 M solution at λ = 436 nm) (Hatchard and Parker, 1956), t is the time (90.0 s), and f is the fraction of light absorbed at λ = 436 nm (0.9997, Equation 2) (see Figure S82).
| Equation 4 |
A cuvette was charged with styrene 1a (0.21 mmol), and bromoacetonitrile 2a (0.20 mmol) was added to a solution of KF (0.30 mmol) and Ir(ppy)3 (0.001 mmol, 0.5 mol%) in acetonitrile (3.0 mL). The cuvette was then capped with a polytetrafluoroethylene stopper. The sample was stirred and irradiated (λ = 436 nm, slit width = 2.0 nm) for 10,800 s (3 h). After irradiation, internal standard (naphthalene, 9.4 mg) was added. The yield of product formed was determined by GC-FID. The quantum yield was determined using Equation 4 (see Figure S83).
Characterization of products 3a-3o
N-(3-cyano-1-phenylpropyl)acetamide (3a)

According to the general procedure A. Known compound, white solid, mp 95–96°C, 36.9 mg, 91% yield, Rf= 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.37–7.26 (m, 5H), 6.53 (d, J = 8.0 Hz, 1H), 5.04–4.99 (m, 1H), 2.34–2.31 (m, 2H), 2.24–2.15 (m, 1H), 2.15–2.07 (m, 1H), 1.97 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.0, 140.1, 129.0, 128.1, 126.4, 119.3, 52.7, 31.6, 23.1, 14.4.
N-(3-cyano-1-(p-tolyl)propyl)acetamide (3b)

According to the general procedure A. Known compound, white solid, mp 103–104°C, 36.0 mg, 83% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.19–7.15 (m, 4H), 6.11 (d, J = 7.8 Hz, 1H), 5.02–4.96 (m, 1H), 2.35–2.30 (m, 5H), 2.27–2.20 (m, 1H), 2.12–2.07 (m, 1H), 1.98 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.8, 138.0, 136.9, 129.7, 126.4, 119.3, 52.5, 31.6, 23.3, 21.0, 14.4.
N-(1-(4-(tert-butyl)phenyl)-3-cyanopropyl)acetamide (3c)

According to the general procedure A. Known compound, white solid, mp 125–126°C, 43.5 mg, 84% yield, Rf = 0.4 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.3 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 5.89 (d, J = 7.2 Hz, 1H), 5.05–4.99 (m, 1H), 2.36–2.29 (m, 2H), 2.29–2.21 (m, 1H), 2.17–2.10 (m, 1H), 1.99 (s, 3H), 1.31 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 169.7, 151.3, 136.8, 126.2, 126.0, 119.3, 52.4, 34.5, 31.6, 31.2, 23.3, 14.5.
N-(1-([1,1′-biphenyl]-4-yl)-3-cyanopropyl)acetamide (3d)

According to the general procedure A. Known compound, white solid, mp 197–198°C, 46.2 mg, 83% yield, Rf = 0.1 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, DMSO-d6) δ 8.40 (d, J = 8.4 Hz, 1H), 7.66–7.63 (m, 4H),7.46 (t, J = 7.6 Hz, 2H), 7.41–7.34 (m, 3H), 4.94–4.88 (m, 1H), 2.54–2.48 (m, 2H), 2.03–1.97 (m, 2H), 1.89 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.4, 142.1, 140.4, 139.5, 129.4, 127.8, 127.5, 127.2, 127.1, 120.7, 51.7, 31.9, 23.2, 14.5.
N-(3-cyano-1-(4-fluorophenyl)propyl)acetamide (3e)

According to the general procedure A. Known compound, white solid, mp 122–123°C, 36.6 mg, 83% yield, Rf = 0.4 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.28–7.25 (m, 2H), 7.08–7.03 (m, 2H), 6.11 (d, J = 8.0 Hz, 1H), 5.06–5.01 (m, 1H), 2.36 (t, J = 7.3 Hz, 2H), 2.27–2.17 (m, 1H), 2.15–2.06 (m, 1H), 2.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.8, 162.3 (d, J = 247.3 Hz), 135.9 (d, J = 3.3 Hz),128.2 (d, J = 8.1 Hz), 119.1, 116.0 (d, J = 21.5 Hz), 52.1, 31.6, 23.3, 14.5. 19F NMR (376 MHz, CDCl3) δ −113.53.
N-(1-(4-chlorophenyl)-3-cyanopropyl)acetamide (3f)

According to the general procedure A. Known compound, white solid, mp 148–149°C, 45.0 mg, 95% yield, Rf = 0.4 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.35–7.33 (m, 2H), 7.24–7.21 (m, 2H), 6.23 (d, J = 7.6 Hz, 1H), 5.05–4.99 (m, 1H), 2.36 (t, J = 7.3 Hz, 2H), 2.24–2.16 (m, 1H), 2.14–2.07 (m, 1H), 2.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.9, 138.6, 134.0, 129.2, 127.9, 119.1, 52.1, 31.4, 23.2, 14.5.
N-(1-(4-bromophenyl)-3-cyanopropyl)acetamide (3g)

According to the general procedure A. Known compound, off-white solid, mp 144–145°C, 55.3 mg, 98% yield, Rf = 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 6.30 (d, J = 8.2 Hz, 1H), 5.03–4.98 (m, 1H), 2.36 (t, J = 7.4 Hz, 2H), 2.22–2.04 (m, 2H), 1.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.0, 139.3, 132.2, 128.3, 122.1, 119.2, 52.2, 31.4, 23.3, 14.5.
N-(1-(4-(chloromethyl)phenyl)-3-cyanopropyl)acetamide (3h)

According to the general procedure A. Known compound, white solid, mp 131–132°C, 41.9 mg, 84% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 6.2 Hz, 2H), 6.33 (d, J = 7.1 Hz, 1H), 5.07–5.01 (m, 1H), 4.57 (s, 2H), 2.34 (t, J = 7.4 Hz, 2H), 2.22–2.08 (m, 2H), 1.98 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.9, 140.4, 137.4, 129.3, 126.9, 119.2, 52.4, 45.6, 31.5, 23.2, 14.5.
N-(3-cyano-1-(m-tolyl)propyl)acetamide (3i)

According to the general procedure A. Known compound, white solid, mp 96–98°C, 36.6 mg, 85% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.27–7.22 (m, 1H), 7.12–7.06 (m, 3H), 6.38 (d, J = 8.1 Hz, 1H), 5.00–4.95 (m, 1H), 2.34 (s, 3H), 2.33–2.30 (m, 2H), 2.23–2.18 (m, 1H), 2.13–2.06 (m, 1H), 1.98 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.8, 139.9, 138.8, 128.9, 128.8, 127.3, 123.3, 119.3, 52.7, 31.6, 23.2, 21.3, 14.4.
N-(1-(3-chlorophenyl)-3-cyanopropyl)acetamide (3j)

According to the general procedure A. Known compound, white solid, mp 117–118°C, 46.8 mg, 99% yield, Rf = 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.31–7.26 (m, 3H), 7.18–7.16 (m, 1H), 6.74 (d, J = 8.2 Hz, 1H), 5.04–4.98 (m, 1H), 2.36 (t, J = 7.2 Hz, 2H), 2.20–2.05 (m, 2H), 1.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.1, 142.4, 134.8, 130.3, 128.2, 126.5, 124.8, 119.1, 52.2, 31.5, 23.1, 14.5.
N-(1-(2-chlorophenyl)-3-cyanopropyl)acetamide (3k)

According to the general procedure A. Known compound, white solid, mp 128–129°C, 42.4 mg, 90% yield, Rf= 0.3 (petroleum ether/EtOAc1/1). 1H NMR (400 MHz, CDCl3) δ 7.38–7.36 (m, 1H), 7.33–7.31 (m, 1H), 7.28–7.21 (m, 2H), 6.85 (d, J = 8.3 Hz, 1H), 5.42–5.36 (m, 1H), 2.45–2.36 (m, 2H), 2.20–2.10 (m, 2H), 2.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.0, 137.8, 132.7, 130.2, 129.1, 127.8, 127.4, 119.2, 50.6, 30.6, 23.1, 14.5.
N-(1-(2-bromophenyl)-3-cyanopropyl)acetamide (3l)

According to the general procedure A. Unknown compound, white solid, mp 134–135°C, 52.9 mg, 94% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, DMSO-d6) δ 8.50 (d, J = 8.1 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.44–7.37 (m, 2H), 7.23–7.19 (m, 1H), 5.20–5.15 (m, 1H), 2.59 (t, J = 7.0 Hz, 2H), 1.98–1.92 (m, 1H), 1.90 (s, 3H), 1.85–1.76 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.6, 142.2, 133.1, 129.6, 128.6, 127.8, 122.6, 120.3, 51.6, 31.0, 23.1, 14.5. HRMS calculated for C12H14BrN2O+ [M + H]+ 281.0284, found: 281.0281.
N-(3-cyano-1-(naphthalen-2-yl)propyl)acetamide (3m)

According to the general procedure A. Known compound, white solid, mp 137–139°C, 23.8 mg, 47% yield, Rf = 0.1 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.84–7.78 (m, 3H), 7.72 (s, 1H), 7.50–7.48 (m, 2H), 7.36 (dd, J = 8.5, 1.6 Hz, 1H), 6.31 (d, J = 8.1 Hz, 1H), 5.21–5.16 (m, 1H), 2.35–2.30 (m, 2H), 2.28–2.16 (m, 2H), 1.99 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.9, 137.2, 133.2, 132.9, 129.1, 127.8, 127.6, 126.6, 126.4, 125.5, 124.1, 119.3, 52.8, 31.4, 23.3, 14.5.
N-(2-(cyanomethyl)-2,3-dihydro-1H-inden-1-yl)acetamide (3n)

According to the general procedure A. Known compound, white solid, mp 107–109°C, 35.6 mg, 83% yield, 8:1 dr, Rf = 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.31–7.21 (m, 4.5H), 6.15 (d, J = 8.5 Hz, 0.13H), 6.01 (d, J = 8.0 Hz, 1H), 5.47–5.44 (m, 1H), 5.20–5.16 (m, 0.13H), 3.27–3.21 (m, 0.13H), 3.19–3.12 (m, 1H), 2.98–2.89 (m, 2H), 2.82–2.73 (m, 0.26H), 2.61–2.53 (m, 1.13H), 2.51–2.46 (m, 0.13H), 2.44–2.38 (m, 1H), 2.04 (s, 0.38H), 2.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.61, 170.55, 141.4, 141.2, 140.7, 140.6, 128.9, 128.5, 127.4, 127.3, 125.1, 124.8, 124.5, 123.5, 119.0, 118.5, 59.0, 56.2, 45.5, 39.3, 35.9, 35.7, 23.1, 22.9, 20.8, 17.9. HRMS calculated for C13H15N2O+ [M + H]+ 215.1179, found 215.1184.



N-(3-cyano-2-methyl-1-phenylpropyl)acetamide (3o)

Prepared according to the general procedure A, KF (1.5 equiv., 17.4 mg); known compound, white solid, mp 95–97°C, 38.6 mg, 89% yield, 4:1 dr, Rf = 0.2 (petroleum ether/EtOAc 1/1.5). 1H NMR (400 MHz, CDCl3) δ 7.36–7.23 (m, 6.15H), 6.72 (d, J = 9.0 Hz, 1.22H), 4.99–4.95 (m, 0.23H), 4.84–4.79 (m, 1H), 2.60–2.53 (m, 1H), 2.34–2.26 (m, 2.23H), 2.13–2.06 (m, 0.46H), 2.00 (s, 0.69H), 1.97 (s, 3H), 1.15 (d, J = 6.7 Hz, 0.69H), 0.96 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.02, 169.97, 139.7, 139.5, 128.9, 128.9, 127.9, 126.9, 126.7, 119.2, 118.5, 57.8, 56.9, 36.1, 35.8, 23.2, 22.1, 21.7, 17.1, 15.6. HRMS calculated for C13H17N2O+ [M + H]+ 217.1335, found 217.1337.
Characterization of products 6a–6k
Ethyl 4-acetamido-4-phenylbutanoate (6a)

Prepared according to the general procedure A, KF (1.5 equiv., 17.4 mg); known compound (Giedyk et al., 2016), off-white solid, mp 116–117°C, 35.5 mg, 71% yield, Rf = 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.35–7.23 (m, 5H), 6.31 (d, J = 8.0 Hz, 1H), 5.00–4.95 (m, 1H), 4.11 (q, J = 7.1 Hz, 2H), 2.40–2.26 (m, 2H), 2.19–2.05 (m, 2H), 1.95 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.5, 169.4, 141.5, 128.7, 127.5, 126.4, 60.6, 53.1, 31.2, 30.8, 23.2, 14.1. HRMS calculated for C14H20NO3+ [M + H]+ 250.1438, found 250.1445.
Ethyl 4-acetamido-4-(4-bromophenyl)butanoate (6b)

Prepared according to the general procedure A, KF (1.5 equiv., 17.4 mg); unknown compound, light yellow solid, mp 131–133°C, 37.2 mg, 57% yield, Rf = 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 6.40 (d, J = 7.8 Hz, 1H), 4.95–4.89 (m, 1H), 4.12 (q, J = 7.1 Hz, 2H), 2.38–2.29 (m, 2H), 2.13–2.01 (m, 2H), 1.96 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.5, 169.5, 140.8, 131.7, 128.2, 121.2, 60.7, 52.7, 31.1, 30.6, 23.2, 14.1. HRMS calculated for C14H19BrNO3+ [M + H]+ 328.0543, found 328.0553.
Ethyl 4-acetamido-4-(4-chlorophenyl)butanoate (6c)

Prepared according to the general procedure A, KF (1.5 equiv., 17.4 mg); known compound, white solid, mp 112–113°C, 28.8 mg, 51% yield, Rf = 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.31–7.27 (m, 2H), 7.22–7.20 (m, 2H), 6.37 (d, J = 7.8 Hz, 1H), 4.96–4.91 (m, 1H), 4.12 (q, J = 7.1 Hz, 2H), 2.38–2.29 (m, 2H), 2.15–2.01 (m, 2H), 1.96 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.5, 169.5, 140.2, 133.2, 128.8, 127.8, 60.7, 52.6, 31.1, 30.6, 23.2, 14.1. HRMS calculated for C14H19ClNO3+ [M + H]+ 284.1048, found 284.1055.
Ethyl 4-acetamido-4-(3-chlorophenyl)butanoate (6d)

Prepared according to the general procedure A, KF (1.5 equiv., 17.4 mg); unknown compound, white solid, mp 77–79°C, 41.8 mg, 74% yield, Rf = 0.3 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.27–7.21 (m, 3H), 7.17–7.15 (m, 1H), 6.46 (d, J = 7.9 Hz, 1H), 4.97–4.91 (m, 1H), 4.12 (q, J = 7.1 Hz, 2H), 2.39–2.30 (m, 2H), 2.13–2.02 (m, 2H), 1.97 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.5, 169.6, 143.9, 134.5, 129.9, 127.6, 126.5, 124.8, 60.7, 52.8, 31.1, 30.7, 23.2, 14.1. HRMS calculated for C14H19ClNO3+ [M + H]+ 284.1048, found 284.1055.
Ethyl 4-acetamido-4-(2-bromophenyl)butanoate (6e)

Prepared according to the general procedure A, KF (1.5 equiv., 17.4 mg); unknown compound, off-white solid, mp 75–77°C, 47.7 mg, 73% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 8.0 Hz, 1H), 7.28–7.27 (m, 2H), 7.13–7.09 (m, 1H), 6.72 (d, J = 7.5 Hz, 1H), 5.29–5.24 (m, 1H), 4.13 (q, J = 7.1 Hz, 2H), 2.47–2.32 (m, 2H), 2.13–2.08 (m, 2H), 1.97 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.8, 169.5, 140.7, 133.3, 128.8, 127.7, 127.6, 122.9, 60.7, 53.3, 31.2, 29.6, 23.1, 14.1. HRMS calculated for C14H19BrNO3+ [M + H]+ 328.0543, found 328.0535.
Ethyl 4-acetamido-2-fluoro-4-phenylbutanoate (6f)

Prepared according to the general procedure A, KF (1.5 equiv., 17.4 mg); unknown compound, off-white solid, mp 82–83°C, 36.5 mg, 68% yield, 3:1 dr, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.37–7.25 (m, 5H), 6.49 (d, J = 8.3 Hz, 0.72H), 6.37 (d, J = 7.9 Hz, 0.24H), 5.35–5.30 (m, 0.73H), 5.27–5.21 (m, 0.25H), 4.92 (ddd, J = 49.0, 8.7, 4.1 Hz, 0.74H), 4.76 (ddd, J = 48.8, 8.8, 3.6 Hz, 0.25H), 4.23–4.17 (m, 2H), 2.42–2.30 (m, 2H), 1.99 (s, 2.20H), 1.94 (s, 0.74H), 1.30–1.26 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 169.6, 169.5, 169.4, 169.3, 140.6, 140.3, 128.9, 128.7, 127.9, 127.6, 126.6, 126.3, 86.9 (d, J = 184.0 Hz), 86.6 (d, J = 184.0 Hz), 61.75, 61.70, 50.5 (d, J = 2.5 Hz), 49.6 (d, J = 1.7 Hz), 38.5 (d, J = 20.4 Hz), 38.3 (d, J = 20.4 Hz), 23.22, 23.18, 14.0. 19F NMR (376 MHz, CDCl3) δ −190.65, −191.54. HRMS calculated for C14H19FNO3+ [M + H]+ 268.1343, found 268.1328.
Ethyl 4-acetamido-2,2-difluoro-4-phenylbutanoate (6g)

Prepared according to the general procedure A, KF (1.1 equiv., 12.8 mg); known compound (Yang et al., 2020), white solid, mp 59–61°C, 30.1 mg, 53% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.36–7.27 (m, 5H), 6.00 (d, J = 7.7 Hz, 1H), 5.34–5.28 (m, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.78–2.53 (m, 2H), 1.96 (s, 3H), 1.31 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.1, 163.7 (t, J = 32.3 Hz), 140.4, 128.8, 128.0, 126.4, 114.9 (t, J = 251.8 Hz), 63.1, 48.2 (t, J = 4.8 Hz), 40.2 (t, J = 22.8 Hz), 23.2, 13.8. 19F NMR (376 MHz, CDCl3) δ −102.97, −103.05. HRMS calculated for C14H18F2NO3+ [M + H]+ 286.1249, found 286.1255.
Ethyl 4-acetamido-4-(4-bromophenyl)-2,2-difluorobutanoate (6h)

Prepared according to the general procedure A, KF (1.1 equiv., 12.8 mg); unknown compound, off-white solid, mp 97–98°C, 26.6 mg, 37% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 6.20 (d, J = 7.8 Hz, 1H), 5.28–5.23 (m, 1H), 4.22 (q, J = 7.1 Hz, 2H), 2.73–2.48 (m, 2H), 1.96 (s, 3H), 1.32 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.3, 163.6 (t, J = 32.0 Hz), 139.6, 131.9, 128.2, 121.8, 114.8 (t, J = 252.2 Hz), 63.3, 47.7 (t, J = 4.7 Hz), 39.9 (t, J = 22.8 Hz), 23.1, 13.8. 19F NMR (376 MHz, CDCl3) δ −103.14. HRMS calculated for C14H17BrF2NO3+ [M + H]+ 364.0354, found 364.0352.
Ethyl 4-acetamido-4-(2-bromophenyl)-2,2-difluorobutanoate (6i)

Prepared according to the general procedure A, KF (1.1 equiv., 12.8 mg); unknown compound, light yellow solid, mp 116–117°C, 42.1 mg, 58% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.54 (dd, J = 8.0, 0.9 Hz, 1H), 7.35–7.28 (m, 2H), 7.15–7.11 (m, 1H), 6.53 (d, J = 7.7 Hz, 1H), 5.61–5.55 (m, 1H), 4.23 (q, J = 7.1 Hz, 2H), 2.72–2.61 (m, 2H), 1.98 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.2, 163.5 (t, J = 32.3 Hz), 139.3, 133.5, 129.3, 128.4, 127.8, 122.4, 114.9 (t, J = 252.0 Hz), 63.2, 48.5 (t, J = 4.7 Hz), 38.6 (t, J = 22.9 Hz), 23.1, 13.8. 19F NMR (376 MHz, CDCl3) δ −102.99, −103.04. HRMS calculated for C18H19ClNO3+ [M + H]+ 364.0354, found 364.0354.
Methyl 4-acetamido-2-methyl-4-phenylbutanoate (6j)

Prepared according to the general procedure A, KF (1.1 equiv., 12.8 mg); unknown compound, sticky oil, 22.2 mg, 45% yield, 3:1 dr, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.34–7.23 (m, 5H), 6.09 (d, J = 7.7 Hz, 0.27H), 5.98 (d, J = 8.4 Hz, 0.75H), 5.09–5.02 (m, 1H), 3.68 (s, 2.24H), 3.60 (s, 0.79H), 2.57–2.42 (m, 1H), 2.29–2.17 (m, 1H), 1.95–1.88 (m, 3.55H), 1.82–1.75 (m, 0.86H), 1.23–1.19 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 177.2, 176.7, 169.3, 169.1, 142.2, 141.3, 128.7, 128.7, 127.5, 127.4, 126.6, 126.3, 51.8, 39.8, 39.2, 37.2, 36.4, 23.31, 23.26, 17.7, 17.2. HRMS calculated for C18H19ClNO3+ [M + H]+ 250.1438, found 250.1441.
Phenyl 4-acetamido-4-(3-chlorophenyl)butanoate (6k)

Prepared according to the general procedure A, KF (1.1 equiv., 12.8 mg); unknown compound, sticky oil, 41.5 mg, 63% yield, Rf = 0.2 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.39–7.35 (m, 2H), 7.29–7.17 (m, 5H), 7.07–7.04 (m, 2H), 6.38 (d, J = 8.1 Hz, 1H), 5.07–5.01 (m, 1H), 2.63–2.56 (m, 2H), 2.21–2.14 (m, 2H), 1.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.0, 169.7, 150.5, 143.6, 134.6, 130.1, 129.4, 127.8, 126.5, 125.9, 124.8, 121.4, 52.6, 31.2, 30.6, 23.2. HRMS calculated for C18H19ClNO3+ [M + H]+ 332.1048, found 332.1041.
Characterization of products 8a–8g
N-(3-cyano-1-phenylpropyl)benzamide (8a)

Prepared according to the general procedure A, 24 hr; unknown compound, white solid, mp 136–137°C, 24.3 mg, 46% yield, Rf = 0.5 (petroleum ether/EtOAc 2/1). 1H NMR (400 MHz, DMSO-d6) δ 8.87 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 7.6 Hz, 2H), 7.55 (t, J = 7.1 Hz, 1H), 7.48 (t, J = 7.5 Hz, 2H), 7.42–7.40 (m, 2H), 7.35 (t, J = 7.5 Hz, 2H), 7.26 (t, J = 7.0 Hz, 1H), 5.14–5.08 (m, 1H), 2.65–2.54 (m, 2H), 2.25–2.15 (m, 1H), 2.12–2.04 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.6, 143.3, 134.8, 131.8, 128.9, 128.7, 127.9, 127.6, 126.9, 120.7, 52.8, 31.7, 14.7. HRMS calculated for C17H17N2O+ [M + H]+ 265.1335, found 265.1335.
N-(3-cyano-1-phenylpropyl)-2-methylbenzamide (8b)

Prepared according to the general procedure A, CsF (2.0 equiv., 60.8 mg), 27 h; unknown compound, white solid, mp 117–118°C, 26.1 mg, 47% yield, Rf = 0.5 (petroleum ether/EtOAc 2/1). 1H NMR (400 MHz, CDCl3) δ 7.42–7.29 (m, 7H), 7.21–7.16 (m, 2H), 6.16 (d, J = 7.6 Hz, 1H), 5.24–5.18 (m, 1H), 2.44–2.32 (m, 6H), 2.27–2.18 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 169.6, 139.7, 136.2, 135.7, 131.1, 130.2, 129.3, 128.4, 126.6, 126.5, 125.8, 119.1, 53.1, 31.7, 19.8, 14.5. HRMS calculated for C18H19N2O+ [M + H]+ 279.1492, found 279.1489.
N-(3-cyano-1-phenylpropyl)-3-methylbenzamide (8c)

Prepared according to the general procedure A, CsF (2.0 equiv., 60.8 mg), 27 h; unknown compound, white solid, mp 97–99°C, 24.0 mg, 43% yield, Rf = 0.7 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.55–7.53 (m, 1H), 7.41–7.29 (m, 7H), 6.59 (d, J = 7.9 Hz, 1H), 5.27–5.21 (m, 1H), 2.40–2.33 (m, 6H), 2.29–2.20 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 167.3, 139.9, 138.5, 133.9, 132.5, 129.2, 128.5, 128.3, 127.7, 126.5, 123.9, 119.3, 53.1, 31.7, 21.3, 14.5. HRMS calculated for C18H19N2O+ [M + H]+ 279.1492, found 279.1501.
N-(3-cyano-1-phenylpropyl)isobutyramide (8d)

Prepared according to the general procedure A, CsF (2.0 equiv., 60.8 mg), 27 h; unknown compound, white solid, mp 113–115°C, 22.6 mg, 49% yield, Rf = 0.4 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.39–7.27 (m, 5H), 5.99 (d, J = 7.8 Hz, 1H), 5.07–5.01 (m, 1H), 2.41–2.31 (m, 3H), 2.28–2.21 (m, 1H), 2.19–2.11 (m, 1H), 1.17 (d, J = 6.9 Hz, 3H), 1.12 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 176.7, 140.1, 129.1, 128.1, 126.4, 119.3, 52.4, 35.6, 31.7, 19.5, 19.4, 14.4. HRMS calculated for C14H19N2O+ [M + H]+231.1492, found 231.1486.
N-(3-cyano-1-phenylpropyl)pentanamide (8e)

Prepared according to the general procedure A, CsF (2.0 equiv., 60.8 mg); unknown compound, white solid, mp 59–60°C, 22.4 mg, 46% yield, Rf = 0.6 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.40–7.27 (m, 5H), 5.83 (d, J = 7.6 Hz, 1H), 5.08–5.03 (m, 1H), 2.36–2.10 (m, 6H), 1.65–1.57 (m, 2H), 1.37–1.28 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 172.8, 140.0, 129.2, 128.2, 126.5, 119.3, 52.6, 36.4, 31.7, 27.6, 22.3, 14.5, 13.7. HRMS calculated for C15H21N2O+ [M + H]+ 245.1648, found 245.1647.
N-(3-cyano-1-phenylpropyl)-3-methylbutanamide (8f)

Prepared according to the general procedure A, 27 h; unknown compound, white solid, mp 80–81°C, 22.3 mg, 46% yield, Rf = 0.6 (petroleum ether/EtOAc 1/1). 1H NMR (400 MHz, CDCl3) δ 7.39–7.37 (m, 5H), 5.99 (d, J = 7.8 Hz, 1H), 5.08–5.02 (m, 1H), 2.35–2.28 (m, 2H), 2.28–2.03 (m, 5H), 0.94 (d, J = 6.2 Hz, 3H), 0.91 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 172.2, 140.0, 129.1, 128.2, 126.5, 119.2, 52.6, 45.9, 31.6, 26.1, 22.40, 22.36, 14.5. HRMS calculated for C15H21N2O+ [M + H]+ 245.1648, found 245.1654.
N-(3-cyano-1-phenylpropyl)pivalamide (8g)

Prepared according to the general procedure A, CsF (2.0 equiv., 60.8 mg), 27 h; unknown compound, white solid, 15.4 mg, 32% yield, Rf = 0.3 (petroleum ether/EtOAc 2/1). 1H NMR (400 MHz, CDCl3) δ 7.40–7.37 (m, 2H), 7.34–7.32 (m, 1H), 7.30–7.26 (m, 2H), 5.94 (d, J = 7.4 Hz, 1H), 5.07–5.02 (m, 1H), 2.35–2.29 (m, 2H), 2.27–2.13 (m, 2H), 1.21 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 178.1, 140.1, 129.2, 128.1, 126.3, 119.4, 52.6, 38.8, 31.7, 27.4, 14.4. HRMS calculated for C15H21N2O+ [M + H]+ 245.1648, found 245.1649.
Gram-scale reaction

In the glove-box, styrene 1a (6.1 mmol) and bromoacetonitrile 2a (6.0 mmol) was added to a solution of KF (9.0 mmol) and Ir(ppy)3 (0.012 mmol, 0.2 mol%) in acetonitrile (8.0 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10 W blue LEDs at room temperature for 18 h. After the reaction completed, the crude product was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate: 8/1-1/1 v/v) to afford the corresponding product 3a (0.978 g, 81% yield).
Synthetic transformations

To a 25 mL round-bottom flask equipped with magnetic stirrer were added 3a (0.5 mmol, 1 equiv.), acetaldoxime (1 mmol, 2 equiv.), nickel(II) chloride hexahydrate (0.05 mmol) and H2O (5 mL). The mixture was heated to reflux for 24 h. After cooling to room temperature, the solution was directly evaporated to dryness and the residue was purified by column chromatography on silica gel (ethyl acetate/n-hexane = 1:1) to give 4-acetamido-4-phenylbutanamide 9 (66.2 mg, 64%) as white solid (Ma et al., 2012). Unknown compound, 1H NMR (400 MHz, CD3OD) δ 7.36–7.32 (m, 4H), 7.29–7.23 (m, 1H), 4.91–4.88 (m, 1H), 2.31–2.18 (m, 2H), 2.09–2.03 (m, 2H), 1.99 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 176.7, 171.3, 142.3, 128.2, 126.9, 126.2, 53.1, 32.0, 31.8, 21.3. HRMS calculated for C12H17N2O2+ [M + H]+ 221.1285, found 221.1296.

Na (8.0 mmol, 40 equiv.), nBuOH (1.0 mL) and 3a (0.20 mmol, 1 equiv.) were added into a flame-dried Schlenk tube with a stirring bar under nitrogen. The reaction mixture was heated to 120°C for 1 h. Then, nBuOH (1 mL) and Na (4.0 mmol, 20 equiv.) were added into the reaction and continued the reaction at 120°C for another 2 h. Then, the reaction mixture was cooled to ambient temperature. The reaction solution was washed by water and extraction with (30 mL × 3) CH2Cl2. The organic layer was dried over magnesium sulfate, filtered and concentrated by rotary evaporator under reduced pressure and the residue was purified by column chromatography on silica gel (PE/EA = 50/50-0/100) to yield 2-phenylpyrrolidine 10 (22.4 mg, 76%) as colorless oil. Known compound (Zhu et al., 2017), 1H NMR (400 MHz, CDCl3) δ 7.37–7.29 (m, 4H), 7.25–7.21 (m, 1H), 4.11 (t, J = 7.7 Hz, 1H), 3.24–3.18 (m, 1H), 3.04–2.98 (m, 1H), 2.23–2.15 (m, 1H), 2.05–2.02 (m, 1H), 1.97–1.80 (m, 2H), 1.72–1.63 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 144.8, 128.3, 126.7, 126.5, 62.6, 47.0, 34.3, 25.6.

To a stirred solution of 6a (0.1 mmol) in CH3OH (1.0 mL) was added Cs2CO3 (0.15 mmol, 1.5 equiv) at room temperature and the mixture was stirred at 50°C for 23 hours. After completion of reaction as checked by TLC, the solvent was evaporated and the residue was purified directly by flash column chromatograph (petroleum ether/ethyl acetate, 1:1 v/v) to give 5-phenylpyrrolidin-2-one 11 (13.1 mg, 81%) as white solid (Zheng and Studer, 2019). Known compound (Shi et al., 2020), 1H NMR (400 MHz, CDCl3) δ 7.39–7.35 (m, 2H), 7.31–7.29 (m, 3H), 6.37 (s, 1H), 4.75 (t, J = 7.1 Hz, 1H), 2.62–2.53 (m, 1H), 2.51–2.36 (m, 2H), 2.02–1.95 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 178.5, 142.5, 128.9, 127.9, 125.6, 58.0, 31.3, 30.3.
Mechanistic study
Radical trapping experiment

In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of KF (0.40 mmol), Ir(ppy)3 (0.002 mmol, 1.0 mol%), and TEMPO (0.40 mmol, 2.0 equiv.) in acetonitrile (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. After the reaction completed, yield was determined by 1H NMR analysis of crude mixture using 1,3,5-trimethoxybenzene (8.4 mg, 0.05 mmol) as an internal standard (see Figures S86 and S87).
Deuterium labeling experiment

In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in CD3CN (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. After the reaction completed, the crude product was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate, 8/1-1/1 v/v) to afford the corresponding product 3a-d3 (39.9 mg, 97% yield). 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 6.49 (d, J = 8.0 Hz, 1H), 5.05–4.99 (m, 1H), 2.34–2.30 (m, 2H), 2.25–2.15 (m, 1H), 2.15–2.07 (m, 1H). 2D NMR (700 MHz, CDCl3) δ 1.97. 13C NMR (100 MHz, CDCl3) δ 170.0, 140.1, 129.0, 128.1, 126.4, 119.3, 52.6, 31.6, 22.8–22.0 (m), 14.4. HRMS calculated for C12H12D3N2O+ [M + H]+ 206.1367, found 206.1367 (see Figure S88).
Heteroatom nucleophiles

In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in CH3CN/MeOH (0.4 mL/0.4 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. After the reaction completed, yield was determined by 1H NMR analysis of crude mixture using 1,3,5-trimethoxybenzene (8.4 mg, 0.05 mmol) as an internal standard. Compared with reported literature (Yi et al., 2014), 4-methoxy-4-phenylbutanenitrile was obtained with 90% yield (see Figure S89).
Control experiments
General procedure B

In the glove box, 4-bromo-4-phenylbutanenitrile 4a (0.20 mmol) was added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in CH3CN (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. After the reaction completed, the crude product was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate, 8/1-1/1 v/v) to afford the corresponding product (see Table S4).

In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of NEt3·3HF (0.60 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in DCE (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. After the reaction completed, the crude product was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate, 12/1-10/1 v/v) to afford the corresponding product (75% yield, 13:4a 2.5:1) (Dauncey et al., 2018) (see Figures S90 and S91).

In the glove box, 4-fluoro-4-phenylbutanenitrile 13 (0.10 mmol) and 4a (0.04 mmol) were added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.0014 mmol, 1.0 mol%) in CH3CN (0.6 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. After the reaction completed, yield was determined by 1H NMR analysis of crude mixture using 1,3,5-trimethoxybenzene (8.4 mg, 0.05 mmol) as an internal standard (see Figure S92).
19F NMR of reaction profiles
In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in CH3CN (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature. After the reaction completed, 19F NMR was tested after quenching by H2O (see Figure S93).
In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in CH3CN (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 10 min. Then, with additional D2O, 1H NMR, 13C NMR, 19F NMR, HMQC and HMBC was tested (see Figures S94–S98). HRMS calculated for C12H13FN2+ [M]+ 204.1057, found 204.1060. In addition, using CD3CN instead of CH3CN, the same procedure was conducted to further confirm the structure of the intermediate (see Figures S99–S103). Furthermore, spectra of 10 min reaction time with the standard condition quenched by H2O (20 μL) were also tested (see Figures S104–S106).
Further transformation of imidoyl fluoride
In the glove box, styrene 1a (0.21 mmol) and bromoacetonitrile 2a (0.20 mmol) were added to a solution of KF (0.40 mmol) and Ir(ppy)3 (0.002 mmol, 1.0 mol%) in CH3CN (0.8 mL). Subsequently, the reaction mixture was stirred under the irradiation of 10-W blue LEDs at room temperature for 12 h. Then, N-methylbenzylamine (0.22 mmol) was added to the mixture in the glove box, stirring at rt for 26 h. Next, yield of N-benzyl-N'-(3-cyano-1-phenylpropyl)-N- methylacetimidamide 14 (see Figures S107 and S108) was determined by 1H NMR spectroscopy using 1,3,5-trimethoxy-benzene (8.4 mg, 0.05 mmol) as the internal standard. HRMS calculated for C20H24N3+ [M + H]+ 306.1965, found 306.1960.
Acknowledgments
We are grateful for the Dalian Institute of Chemical Physics (DICPI201902), Dalian Outstanding Young Scientific Talent, and China Postdoctoral Science Foundation (2019M661137) for financial support. We thank Xiujuan Chi (DICP) for expert advice with the NMR spectroscopic measurements.
Author contributions
Q.-A.C conceived and supervised the project. Y.-Q.G. discovered the reported process and designed and carried out almost all the experiments. X.-T.M. participated in synthesizing partial Ritter products and synthetic transformations. G.-C.H. synthesized partial Ritter products. D.-W.J. and S.-Y.G. helped in analyzing the data. Y.-Q.G., Y.-C.H., and Q.-A.C wrote the manuscript. Y.-Q.G. wrote supporting information. All the authors discussed the results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102969.
Supplemental information
Data and code availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC 2039204 (6b). Copies of the data can be obtained free of charge from https://www.ccdc.cam.ac.uk/structures/. All other data are available from the Lead Contact upon reasonable request.
References
- Abe T., Kida K., Yamada K. A copper-catalyzed ritter-type cascade via iminoketene for the synthesis of quinazolin-4(3H)-ones and diazocines. Chem. Commun. 2017;53:4362–4365. doi: 10.1039/c7cc01406f. [DOI] [PubMed] [Google Scholar]
- Abe T., Takeda H., Miwa Y., Yamada K., Yanada R., Ishikura M. Copper-catalyzed Ritter-type reaction of unactivated alkenes with dichloramine-T. Helv. Chim. Acta. 2010;93:233–241. [Google Scholar]
- Ahmed W., Zhang S., Feng X., Yu X., Yamamoto Y., Bao M. Cooperative catalysis of copper, silver, and Brønsted acid for three-component carboamination of arylalkenes with allylic alcohols and nitriles. ChemCatChem. 2020;12:5200–5208. [Google Scholar]
- Ai W., Shi R., Zhu L., Jiang D., Ma X., Yuan J., Wang Z. One-pot protocol to synthesize N-(β-nitro)amides by tandem Henry/Ritter reaction. RSC Adv. 2015;5:24044–24048. [Google Scholar]
- Badir S.O., Molander G.A. Developments in photoredox/nickel dual-catalyzed 1,2-difunctionalizations. Chem. 2020;6:1327–1339. doi: 10.1016/j.chempr.2020.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H., Zhou B., Jin H., Liu Y. Copper-catalyzed three-component reaction of N-heteroaryl aldehydes, nitriles, and water. Org. Biomol. Chem. 2019;17:5021–5028. doi: 10.1039/c9ob00599d. [DOI] [PubMed] [Google Scholar]
- Bolsakova J., Jirgensons A. The Ritter reaction for the synthesis of heterocycles. Chem. Heterocycl. Compd. 2017;53:1167–1177. [Google Scholar]
- Chen G.-G., Wei J.-Q., Yang X., Yao Z.-J. Convenient one-step synthesis of benzo[c]phenanthridines by three-component reactions of isochromenylium tetrafluoroborates and stilbenes in acetonitrile. Org. Lett. 2016;18:1502–1505. doi: 10.1021/acs.orglett.6b00010. [DOI] [PubMed] [Google Scholar]
- Chen Y., Lu L.-Q., Yu D.-G., Zhu C.-J., Xiao W.-J. Visible light-driven organic photochemical synthesis in China. Sci. China Chem. 2018;62:24–57. [Google Scholar]
- Cismesia M.A., Yoon T.P. Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015;6:5426–5434. doi: 10.1039/c5sc02185e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courant T., Masson G. Recent progress in visible-light photoredox-catalyzed intermolecular 1,2-difunctionalization of double bonds via an ATRA-type mechanism. J. Org. Chem. 2016;81:6945–6952. doi: 10.1021/acs.joc.6b01058. [DOI] [PubMed] [Google Scholar]
- Crouch R.D. The Ritter reaction: trapping a carbocation with a nitrile. J. Chem. Educ. 1994;71:A200–A202. [Google Scholar]
- Dauncey E.M., Morcillo S.P., Douglas J.J., Sheikh N.S., Leonori D. Photoinduced remote functionalisations by iminyl radical promoted C-C and C-H bond cleavage cascades. Angew. Chem. Int. Ed. 2018;57:744–748. doi: 10.1002/anie.201710790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren T., Kusefoglu S.H. Synthesis and polymerization of the acrylamide derivatives of fatty compounds. J. Appl. Polym. Sci. 2005;97:2264–2272. [Google Scholar]
- Feng C., Li Y., Sheng X., Pan L., Liu Q. A Ritter-type route to N-benzylamides by multicomponent reaction based on p-(trifluoromethyl)-p-quinols. Org. Lett. 2018;20:6449–6452. doi: 10.1021/acs.orglett.8b02762. [DOI] [PubMed] [Google Scholar]
- Fumagalli G., Boyd S., Greaney M.F. Oxyarylation and aminoarylation of styrenes using photoredox catalysis. Org. Lett. 2013;15:4398–4401. doi: 10.1021/ol401940c. [DOI] [PubMed] [Google Scholar]
- Giedyk M., Goliszewska K., ó Proinsias K., Gryko D. Cobalt(I)-catalysed CH-alkylation of terminal olefins, and beyond. Chem. Commun. 2016;52:1389–1392. doi: 10.1039/c5cc07363d. [DOI] [PubMed] [Google Scholar]
- Guérinot A., Reymond S., Cossy J. Ritter reaction: Recent catalytic developments. Eur. J. Org. Chem. 2012:19–28. [Google Scholar]
- Gurjar J., Fokin V.V. Sulfuryl fluoride mediated synthesis of amides and amidines from ketoximes via Beckmann rearrangement. Chem. Eur. J. 2020;26:10402–10405. doi: 10.1002/chem.201905358. [DOI] [PubMed] [Google Scholar]
- Hatchard C.G., Parker C.A. A new sensitive chemical actinometer. II. Potassium ferrioxalate as a standard chemical actinometer. Proc. R. Soc. Lond. Ser. A. 1956;235:518–536. [Google Scholar]
- Hopkinson M.N., Tlahuext-Aca A., Glorius F. Merging visible light photoredox and gold catalysis. Acc. Chem. Res. 2016;49:2261–2272. doi: 10.1021/acs.accounts.6b00351. [DOI] [PubMed] [Google Scholar]
- Huang J.-M., Ye Z.-J., Chen D.-S., Zhu H. Iodine mediated/Brønsted acid-catalyzed dimerization of vinylarenes: a tandem reaction through Ritter trapping to produce N-(4-iodo-1,3-diarylbutyl) acetamides. Org. Biomol. Chem. 2012;10:3610–3612. doi: 10.1039/c2ob25142f. [DOI] [PubMed] [Google Scholar]
- Isse A.A., Gennaro A. Homogeneous reduction of haloacetonitriles by electrogenerated aromatic radical anions: determination of the reduction potential of ·CH2CN. J. Phys. Chem. A. 2004;108:4180–4186. [Google Scholar]
- Ji D.-W., Hu Y.-C., Zheng H., Zhao C.-Y., Chen Q.-A., Dong V.-M. A regioselectivity switch in Pd-catalyzed hydroallylation of alkynes. Chem. Sci. 2019;10:6311–6315. doi: 10.1039/c9sc01527b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D., He T., Ma L., Wang Z. Recent developments in Ritter reaction. RSC Adv. 2014;4:64936–64946. [Google Scholar]
- Jiang H., Studer A. Intermolecular radical carboamination of alkenes. Chem. Soc. Rev. 2020;49:1790–1811. doi: 10.1039/c9cs00692c. [DOI] [PubMed] [Google Scholar]
- Jiang W.-S., Jiang X.-L., Ji D.-W., Zhang W.-S., Zhang G., Min X.-T., Hu Y.-C., Chen Q.-A. Orthogonal regulation of nucleophilic and electrophilic sites in Pd-catalyzed regiodivergent couplings between indazoles and isoprene. Angew. Chem. Int. Ed. 2021;60:8321–8328. doi: 10.1002/anie.202100137. [DOI] [PubMed] [Google Scholar]
- Koike T., Akita M. A versatile strategy for difunctionalization of carbon-carbon multiple bonds by photoredox catalysis. Org. Chem. Front. 2016;3:1345–1349. [Google Scholar]
- Kuai C.S., Ji D.-W., Zhao C.-Y., Liu H., Hu Y.-C., Chen Q.-A. Ligand-regulated regiodivergent hydrosilylation of isoprene under iron catalysis. Angew. Chem. Int. Ed. 2020;59:19115–19120. doi: 10.1002/anie.202007930. [DOI] [PubMed] [Google Scholar]
- Kuhn H.J., Braslavsky S.E., Schmidt R. Chemical actinometry (IUPAC technical report) Pure Appl. Chem. 2004;76:2105–2146. [Google Scholar]
- Kürti L., Czakó B. Elsevier Inc; 2005. Strategic Applications of Named Reactions in Organic Synthesis. [Google Scholar]
- Li-Zhulanov N.S., Pavlova A.V., Korchagina D.V., Gatilov Y.V., Volcho K.P., Tolstikova T.G., Salakhutdinov N.F. Synthesis of 1,3-oxazine derivatives based on (–)-isopulegol using the Ritter reaction and study of their analgesic activity. Chem. Heterocycl. Compd. 2020;56:936–941. [Google Scholar]
- Lipp A., Badir S.O., Molander G.A. Stereoinduction in metallaphotoredox catalysis. Angew. Chem. Int. Ed. 2021;60:1714–1726. doi: 10.1002/anie.202007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Klussmann M. Acid promoted radical-chain difunctionalization of styrenes with stabilized radicals and (N,O)-nucleophiles. Chem. Commun. 2020;56:1557–1560. doi: 10.1039/c9cc09369a. [DOI] [PubMed] [Google Scholar]
- Ma X., He Y., Wang P., Lu M. The hydration of nitriles catalyzed by simple transition metal salt of the fourth period with the aid of acetaldoxime. Appl. Organometal. Chem. 2012;26:377–382. [Google Scholar]
- Magagnano G., Gualandi A., Marchini M., Mengozzi L., Ceroni P., Cozzi P.G. Photocatalytic ATRA reaction promoted by iodo-bodipy and sodium ascorbate. Chem. Commun. 2017;53:1591–1594. doi: 10.1039/c6cc09387f. [DOI] [PubMed] [Google Scholar]
- Mao L.-L., Cong H. Atom-transfer radical addition to unactivated alkenes by using heterogeneous visible-light photocatalysis. ChemSusChem. 2017;10:4461–4464. doi: 10.1002/cssc.201701382. [DOI] [PubMed] [Google Scholar]
- Marzo L., Pagire S.K., Reiser O., König B. Visible-light photocatalysis: does it make a difference in organic synthesis? Angew. Chem. Int. Ed. 2018;57:10034–10072. doi: 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- Min X.-T., Ji D.-W., Guan Y.-Q., Guo S.-Y., Hu Y.-C., Wan B., Chen Q.-A. Visible light induced bifunctional rhodium catalysis for decarbonylative coupling of imides with alkynes. Angew. Chem. Int. Ed. 2021;60:1583–1587. doi: 10.1002/anie.202010782. [DOI] [PubMed] [Google Scholar]
- Mohammadi Ziarani G., Soltani Hasankiadeh F., Mohajer F. Recent applications of Ritter reactions in organic syntheses. ChemistrySelect. 2020;5:14349–14379. [Google Scholar]
- Nandy S., Das A.K., Bhar S. Chemoselective formation of C–N bond in wet acetonitrile using Amberlyst®-15(H) as a Recyclable catalyst. Synth. Commun. 2020;50:3326–3336. [Google Scholar]
- Narayanam J.M.R., Stephenson C.R.J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- Norell J.R. Organic reactions in liquid hydrogen fluoride. II. Synthesis of imidoyl fluorides and N,N'-dialkyl-2-alkylaminomalon- amides. J. Org. Chem. 1970;35:1619–1925. [Google Scholar]
- Ouyang X.-H., Song R.-J., Li J.-H. Developments in the chemistry of α-carbonyl alkyl bromides. Chem. -Asian J. 2018;13:2316–2332. doi: 10.1002/asia.201800630. [DOI] [PubMed] [Google Scholar]
- Park S.W., Kim S.-H., Song J., Park G.Y., Kim D., Nam T.-G., Hong K.B. Hypervalent iodine-mediated Ritter-type amidation of terminal alkenes: the synthesis of isoxazoline and pyrazoline cores. Beilstein J. Org. Chem. 2018;14:1028–1033. doi: 10.3762/bjoc.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad Hari D., Hering T., König B. The photoredox-catalyzed Meerwein addition reaction: intermolecular amino-arylation of alkenes. Angew. Chem. Int. Ed. 2014;53:725–728. doi: 10.1002/anie.201307051. [DOI] [PubMed] [Google Scholar]
- Prier C.K., Rankic D.A., MacMillan D.W.C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronin S.V., Reiher C.A., Shenvi R.A. Stereoinversion of tertiary alcohols to tertiary-alkyl isonitriles and amines. Nature. 2013;501:195–199. doi: 10.1038/nature12472. [DOI] [PubMed] [Google Scholar]
- Protti S., Garbarino S., Ravelli D., Basso A. Photoinduced multicomponent reactions. Angew. Chem. Int. Ed. 2016;55:15476–15484. doi: 10.1002/anie.201605288. [DOI] [PubMed] [Google Scholar]
- Pu W., Sun D., Fan W., Pan W., Chai Q., Wang X., Lv Y. Cu-Catalyzed atom transfer radical addition reactions of alkenes with alpha-bromoacetonitrile. Chem. Commun. 2019;55:4821–4824. doi: 10.1039/c9cc01988j. [DOI] [PubMed] [Google Scholar]
- Qian B., Chen S., Wang T., Zhang X., Bao H. Iron-catalyzed carboamination of olefins: synthesis of amines and disubstituted beta-amino acids. J. Am. Chem. Soc. 2017;139:13076–13082. doi: 10.1021/jacs.7b06590. [DOI] [PubMed] [Google Scholar]
- Qu G.-R., Song Y.-W., Niu H.-Y., Guo H.-M., Fossey J.S. Cu(OTf)2-catalysed Ritter reaction: efficient synthesis of amides from nitriles and halohydrocarbons in water. RSC Adv. 2012;2:6161–6163. [Google Scholar]
- Rawner T., Lutsker E., Kaiser C.A., Reiser O. The different faces of photoredox catalysts: visible-light-mediated atom transfer radical addition (ATRA) reactions of perfluoroalkyl iodides with styrenes and phenylacetylenes. ACS Catal. 2018;8:3950–3956. [Google Scholar]
- Ritter J.J., Kalish J. New reaction of nitriles. II. Synthesis of t-Carbinamines. J. Am. Chem. Soc. 1948;70:4048–4050. doi: 10.1021/ja01192a023. [DOI] [PubMed] [Google Scholar]
- Ritter J.J., Minieri P.P. New reaction of nitriles. I. Amides from alkenes and mononitriles. J. Am. Chem. Soc. 1948;70:4045–4048. doi: 10.1021/ja01192a022. [DOI] [PubMed] [Google Scholar]
- Romero N.A., Nicewicz D.A. Organic photoredox catalysis. Chem. Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Rowe J.E., Lee K., Dolliver D.D., Johnson J.E. Preparation of imidoyl fluorides. Aust. J. Chem. 1999;52:807–811. [Google Scholar]
- Shaw M.H., Twilton J., MacMillan D.W.C. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Ai W., He T., Ma X., Qian S., Wang Z. An efficient way to synthesize N-(β-nitroalkyl) amides through Ritter reaction. Lett. Org. Chem. 2015;12:720–726. [Google Scholar]
- Shi Y., Tan X., Gao S., Zhang Y., Wang J., Zhang X., Yin Q. Direct synthesis of chiral NH lactams via Ru-catalyzed asymmetric reductive amination/cyclization cascade of keto acids/esters. Org. Lett. 2020;22:2707–2713. doi: 10.1021/acs.orglett.0c00669. [DOI] [PubMed] [Google Scholar]
- Shih H.-W., Vander Wal M.N., Grange R.L., MacMillan D.W.C. Enantioselective α-benzylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc. 2010;132:13600–13603. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skubi K.L., Blum T.R., Yoon T.P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subba Reddy B.V., Sivasankar Reddy N., Madan C., Yadav J.S. HBF4·OEt2 as a mild and versatile reagent for the Ritter amidation of olefins: a facile synthesis of secondary amides. Tetrahedron Lett. 2010;51:4827–4829. [Google Scholar]
- Tellis J.C., Kelly C.B., Primer D.N., Jouffroy M., Patel N.R., Molander G.A. Single-electron transmetalation via photoredox/nickel dual catalysis: unlocking a new paradigm for sp3-sp2 Cross-Coupling. Acc. Chem. Res. 2016;49:1429–1439. doi: 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twilton J., Le C., Zhang P., Shaw M.H., Evans R.W., MacMillan D.W.C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017;1:0052. [Google Scholar]
- Welniak M. The Ritter reaction of terpenes. Part 1. The isotricyclene system. Pol. J. Chem. 1996;70:752–758. [Google Scholar]
- Williams S.G., Bhadbhade M., Bishop R., Ung A.T. An alkaloid-like 3-azabicyclo[3.3.1]non-3-ene library obtained from the bridged Ritter reaction. Tetrahedron. 2017;73:116–128. [Google Scholar]
- Xu S.-C., Zhu S.-J., Chen Y.-X., Wang J., Bi L.-W., Lu Y.-J., Gu Y., Zhao Z.-D. Skeletal rearrangement in the Ritter reaction of turpentine: a novel synthesis of p-menthane diamides. J. Chem. Res. 2017;41:124–127. [Google Scholar]
- Xuan J., Xiao W.-J. Visible-light photoredox catalysis. Angew. Chem. Int. Ed. 2012;51:6828–6838. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]
- Yang J., Ji D.-W., Hu Y.-C., Min X.-T., Zhou X., Chen Q.-A. Cobalt-catalyzed hydroxymethylarylation of terpenes with formaldehyde and arenes. Chem. Sci. 2019;10:9560–9564. doi: 10.1039/c9sc03747k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Fan W.X., Lin E., Tan D.H., Li Q., Wang H. Synthesis of α-CF3 and α-CF2H amines via the aminofluorination of fluorinated alkenes. Chem. Commun. 2018;54:5907–5910. doi: 10.1039/c8cc03364a. [DOI] [PubMed] [Google Scholar]
- Yang Z.F., Xu C., Zheng X., Zhang X. Nickel-catalyzed carbodifunctionalization of N-vinylamides enables access to gamma-amino acids. Chem. Commun. 2020;56:2642–2645. doi: 10.1039/c9cc09866f. [DOI] [PubMed] [Google Scholar]
- Yasu Y., Koike T., Akita M. Intermolecular aminotrifluoromethylation of alkenes by visible-light-driven photoredox catalysis. Org. Lett. 2013;15:2136–2139. doi: 10.1021/ol4006272. [DOI] [PubMed] [Google Scholar]
- Yasuda K., Obora Y. NbCl5-mediated amidation of olefins with nitriles to secondary amides. J. Organomet. Chem. 2015;775:33–38. [Google Scholar]
- Yi H., Zhang X., Qin C., Liao Z., Liu J., Lei A. Visible light-induced γ-alkoxynitrile synthesis via three-component alkoxycyanomethylation of alkenes. Adv. Synth. Catal. 2014;356:2873–2877. [Google Scholar]
- Yin Y., Zhao X., Jiang Z. Advances in the synthesis of imine-containing azaarene derivatives via photoredox catalysis. ChemCatChem. 2020;12:4471–4489. [Google Scholar]
- Yu X.Y., Chen J.R., Xiao W.J. Visible light-driven radical-mediated C-C bond cleavage/functionalization in organic synthesis. Chem. Rev. 2021;121:506–561. doi: 10.1021/acs.chemrev.0c00030. [DOI] [PubMed] [Google Scholar]
- Yu X.-Y., Zhao Q.-Q., Chen J., Xiao W.-J., Chen J.-R. When light meets nitrogen-centered radicals: from reagents to catalysts. Acc. Chem. Res. 2020;53:1066–1083. doi: 10.1021/acs.accounts.0c00090. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Chen S., Liu Y., Wang Q. Route evaluation and Ritter reaction based synthesis of oxazoline acaricide candidates FET-II-L and NK-12. Org. Process. Res. Dev. 2020;24:216–227. [Google Scholar]
- Zheng D., Studer A. Asymmetric synthesis of heterocyclic gamma-amino-acid and diamine derivatives by three-component radical cascade reactions. Angew. Chem. Int. Ed. 2019;58:15803–15807. doi: 10.1002/anie.201908987. [DOI] [PubMed] [Google Scholar]
- Zheng Y., He Y., Rong G., Zhang X., Weng Y., Dong K., Xu X., Mao J. NaI-mediated acetamidosulphenylation of alkenes with nitriles as the nucleophiles: a direct access to acetamidosulfides. Org. Lett. 2015;17:5444–5447. doi: 10.1021/acs.orglett.5b02752. [DOI] [PubMed] [Google Scholar]
- Zhu C., Yue H., Chu L., Rueping M. Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity. Chem. Sci. 2020;11:4051–4064. doi: 10.1039/d0sc00712a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Wang T., Ge L., Li Y., Zhang X., Bao H. γ-Amino butyric acid (GABA) synthesis enabled by copper-catalyzed carboamination of alkenes. Org. Lett. 2017;19:4718–4721. doi: 10.1021/acs.orglett.7b01969. [DOI] [PubMed] [Google Scholar]
- Zong Y., Lang Y., Yang M., Li X., Fan X., Wu J. Synthesis of β-sulfonyl amides through a multicomponent reaction with the insertion of sulfur dioxide under visible light irradiation. Org. Lett. 2019;21:1935–1938. doi: 10.1021/acs.orglett.9b00620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC 2039204 (6b). Copies of the data can be obtained free of charge from https://www.ccdc.cam.ac.uk/structures/. All other data are available from the Lead Contact upon reasonable request.