

Summary
Cryo-electron tomography has stepped fully into the spotlight. Enthusiasm is high. Fortunately for us, this is an exciting time to be a cryotomographer, but there is still a way to go before declaring victory. Despite its potential, cryo-electron tomography possesses many inherent challenges. How do we image through thick cell samples, and possibly even tissue? How do we identify a protein of interest amidst the noisy, crowded environment of the cytoplasm? How do we target specific moments of a dynamic cellular process for tomographic imaging? In this review, we cover the history of cryo-electron tomography and how it came to be, roughly speaking, as well as the many approaches that have been developed to overcome its intrinsic limitations.
Subject areas: Biochemistry, Cell biology, Structural biology
Graphical abstract
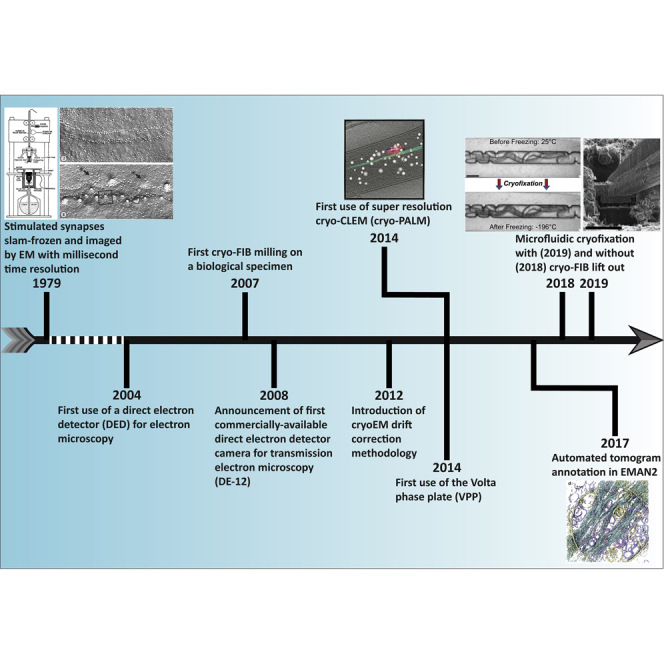
Timeline of major cryo-EM milestones discussed in the text. Citations for each event are as follows: (1) (1979): Heuser et al., 1979; (2) (2004): Xuong et al., 2004; (3) (2007): Marko et al., 2007; (4) (2008): NA; (5) (2012): Brilot et al., 2012 and Campbell et al., 2012; (6) (2014/VPP): Danev et al., 2014; (7) (2014/cryo-PALM): Chang et al., 2014; (8) (2018): Fuest et al., 2018; (9) (2019): Fuest et al., 2019; (10) (2017): Chen et al., 2017.
Biochemistry; Cell biology; Structural biology
Introduction
In the past several decades, cryogenic electron microscopy (cryo-EM) has developed from a niche practice to a fully matured approach for studying biological machinery. In recent memory, it was often referred to as “blobology,” owing to the low-resolution, blob-like density maps that made their way into the literature. As cryo-microscopists, we were excited by these blobs and the things we were learning, as well as the potential the method held to reveal more biological complexity. Over time, cryo-EM density maps took on more features as both microscopic technique and data processing algorithms developed, bringing more and more to bear on the field of structural biology. In more recent years, hardware developments have pushed data collection squarely into a regime capable of reaching near-atomic resolution on a routine basis (Vinothkumar and Henderson, 2016), and just recently to atomic resolution (Nakane et al., 2020). The battle to have the highest resolution structure by cryo-EM has slowed from making progress on the nanometer scale to fractions of an angstrom, and the number of laboratories with access to high-end cryo-EM facilities has grown exponentially. It is no wonder why cryo-EM has taken the field of biology by storm, and there is not a clear end in sight.
The fight for resolution has moved largely from the test tube into the cell and is slowly making its way into tissue via the use of cryo-electron tomography (cryo-ET) (for reviews, refer to the studies by Wan and Briggs, 2016 and Zhang, 2019). Tomography is a three-dimensional (3D) imaging method that functions by collecting a series of projection images through an object from different angles (Gan and Jensen, 2012). Using this tilt series of projections, a 3D image can then be computationally reconstructed into a volume known as a tomogram. The power of tomography is that it can achieve a relatively high resolution (~3 nm), with no averaging, and is especially good for studying unique biological specimens such as whole cells or organelles. Cryo-ET allows for views of cellular structures at unprecedented resolution and clarity. The lack of chemical fixatives and stains allow for direct imaging of the biological molecules in their hydrated state and often provides fresh new details about well-studied cellular ultrastructure (Figure 1). High-resolution molecular structures can also be elucidated, particularly with the use of subtomogram averaging. Here subvolumes within a tomogram containing an object of interest are aligned and averaged in an iterative fashion to produce density maps, often with resolutions below 1 nm (Chen et al., 2019; Sanchez et al., 2020; Schur et al., 2016; Turoňová et al., 2020; Turoňová et al., 2017). Further information on subtomogram averaging can be found in recent reviews written on the topic (Castaño-Díez and Zanetti, 2019; Leigh et al., 2019; Wan and Briggs, 2016).
Figure 1.

Comparison of traditional and cryo-electron microscopy
(A and B) (A) TEM image of the Golgi apparatus from traditionally prepared cellular material and (B) a digital slice rendered from a cryo-ET reconstruction. While the overall morphology is well preserved in both cases, close inspection of the cryo-preserved sample reveals connecting densities between membranes within Golgi stacks (yellow square), a feature never observed before (Engel et al., 2015a).
(C and D) (C) TEM image of a neurite from primary cultured neurons prepared by traditional EM methods (Bartlett and Banker, 1984) and (D) a digital slice rendered from a cryo-ET reconstruction. As a comparison, the microtubules in (D) reveal protofilaments running along their length (small arrows) as well as intralumenal particles within the microtubule (large arrows). Scale bars represent 100 nm.
It is easy to speak highly of cryo-ET and to find countless beautiful examples of its power, but there are some major limitations worth noting. (1) Sample thickness is limited to ~500 nm, making many bacterial, most archeal, and nearly all eukaryotic samples unamenable to imaging. (2) Unless one has prior knowledge about the structure of interest, it is difficult to identify specific molecules within the greyscale volumes produced by cryo-ET. Finally, (3) the static nature of cryo-ET data makes it difficult to determine which dynamic state a cell is in at the moment of freezing. In this review, we will discuss progress in the field toward overcoming all of these challenges. As we will see, some of them have been largely solved while others need continued development.
Going beyond bacteria
In the early days of cryo-ET, purified protein complexes, cellular organelles, and bacterial cells were straightforward targets after directly plunge-freezing in liquid ethane, owing to their small size (Bohm et al., 2001; Grünewald et al., 2003; Nicastro et al., 2000; Nickell et al., 2003; Sandin et al., 2004; Stoffler et al., 2003; Q. Zhao, Ofverstedt, Skoglund and Isaksson, 2004). These studies laid the groundwork and proved that cryo-ET was worthy of everyone's attention. From a cellular perspective, bacteria were king. They are naturally thin and full of discoveries waiting to happen. For instance, the bacterial cytoskeleton, or lack thereof, was an often-cited distinguishing factor between the bacterial and eukaryotic world, and the cytoplasmic preservation of plunge-freezing was key to proving that a variety of cytoplasmic filaments were present in bacteria (Briegel et al., 2006; Kürner et al., 2004; Li et al., 2007; Pilhofer and Jensen, 2013; Pilhofer et al., 2011). The bacterial cytoskeleton is now a thriving field of research and relies heavily on cryo-ET (Pilhofer and Jensen, 2013).
While some work was able to be done at the thin edges of adherent eukaryotic cells in culture (Schwartz et al., 2007), cryo-sectioning of vitreous high-pressure frozen (HPF) material was the first methodological approach to thinning larger samples. In this technique, tissue or cell pellets are put into a small planchet and frozen at ~2000 bars of pressure, allowing the larger mass to vitrify. The planchet is opened, a block-face is trimmed, and a diamond knife is used to cut thin sections (50- to 150-nm thick), all at cryogenic temperatures to preserve the vitreous state of the sample. If you are a seasoned ultramicrotomist, it may be the most straightforward method for obtaining and imaging large vitreous cells and tissue. However, despite the fact that multiple advances have been made to make it more accessible, it requires a lot of practice and dedication (Ladinsky, 2010; Studer et al., 2014). Perhaps more important for the study of cellular structures, however, is that ribbons cut from vitreous samples suffer cutting artifacts such as compression and crevassing that damage the underlying structures (Figure 2A). The artifacts can be substantial, but the high skill ceiling is the more likely culprit for why cryo-sectioning and tomography are not combined more often.
Figure 2.

Comparison of cryosectioning with FIB milling
(A and B) (A) Tomographic slice through dividing S. pombe nucleus generated from a cryosectioned ribbon and (B) a cryo-FIB milled lamella, each ∼150-nm thick. In both slices, the nuclear membrane is visible near the center and is surrounding the microtubules (MT) of the mitotic spindle. Cytoplasmic vesicles (∗) and nuclear pore complexes (arrows) are also visible in both. While the general morphology is captured in both tomograms, the cryosection shows damage from compression (oval vesicles) and cracked membranes and MTs (arrowheads). Red arrows point to crevassing artifacts in the tomograms. Scale bars represent 100 nm.
In 2007, the first article implementing cryo-focused ion beam (cryo-FIB) milling on biological specimens for tomography was published (Marko et al., 2007). FIB milling is a technique commonly used by materials scientists for preparing thin lamellae for inspection by transmission electron microscopy (TEM), and this study demonstrated its potential as an alternative approach to cryo-sectioning. Instead of a diamond blade, cryo-FIB milling uses a beam of ions (typically gallium) to gently ablate regions of a vitreous sample, leaving behind a well-preserved lamella of vitreous cytoplasm (Figure 2B). In 2012, two seminal articles that described a low-angle approach to milling directly on EM grids were published, which made the technique widely available to the rest of the field (Rigort et al., 2012; Wang, Strunk, Zhao, Gray and Zhang, 2012). Since then, the technique has become the standard for gaining access to large eukaryotic cells (Engel et al., 2015a; Engel et al., 2015b; Mahamid et al., 2016; Mahamid et al., 2015; Rigort and Plitzko, 2015; Swulius et al., 2018; Watanabe et al., 2020) and has been both refined (Schaffer et al., 2017; Wolff et al., 2019) and automated (Buckley et al., 2020), allowing routine high-throughput milling (Figure 3A). Additionally, micropatterning techniques have been developed to allow specific positioning of cells on the surface of EM grids to make milling even more straightforward (Engel et al., 2019; Toro-Nahuelpan et al., 2020). In the beginning, it was a victory to get a few nice lamellae milled with a hard day's work, but current instruments and strategies allow for tens of lamellae to be milled every day. In fact, a recent publication demonstrated a workflow for automated cryo-FIB milling that enabled a reduction in machine time needed to mill a single target from 30-45 min to 25.75 min (Zachs et al., 2020). Moreover, this workflow reduced a human operator's time invested to mill 16 targets from ~10 hr to just 2.4 hr.
Figure 3.

Automated FIB and cryo lift outs
(A) Cryo-SEM overview of an EM grid covered in clusters of budding yeast. Red asterisks (∗) mark the location of automatically milled lamellae (the inset to the right shows a blown-up view of the region marked by the red dashed box; the insets to the left show the FIB beam's view of a yeast cluster before and after milling and the SEM view of the lamella from the top) (Zachs et al., 2020).
(B) FIB pattern for milling vertical lamella into the top of a vitrified C. elegans worm.
(C) Vertical lamella after milling with cryo-probe attached at the top left corner prior to lift out.
(D) Final lamella after being thinned to ∼300 nm (Mahamid et al., 2015).
Scale bars in the dashed box in (A) and the left inset represent 200 μm and 5 μm, respectively. Scale bars in (B–D) represent 50 μm.
The holy grail of in situ structural biology is tissue, and cryo-FIB milling is not currently suited for ablating large amounts of material, which is why most studies are done on individual cells in culture. Even if it could, there are structural limitations to consider, such as how do you collect a tilt series on a lamella deep within tissue. To get around this, biologists are borrowing another trick from the materials scientists known as a lift out (Figures 3B–3D). In this approach, a vertical lamella is milled from the tissue block and lifted out via a small probe that is “welded” to the lamella with platinum or water (Mahamid et al., 2015; Zachman et al., 2016). The lamella is then rotated and attached to an EM grid horizontally, so that it is amenable to cryo-ET. While this approach was initially quite cumbersome, recent workflows showing great promise to increase throughput and reliability have been published (Schaffer et al., 2019).
Identifying structures and targeted milling
As mentioned earlier, one of the great challenges of cryo-ET is identifying specific structures within the greyscale tomogram. Without prior knowledge of what specific molecular components look like or how they are arranged, you may not be able to say with confidence that you know what you are looking at, or which subtomogram densities should be averaged together to get to higher resolution. This is the exact opposite challenge associated with fluorescence microscopy, which produces spatial information about where specific molecules are, but often without the context of the surrounding molecular environment. Initially, there was much enthusiasm for the development of a “GFP-like” molecule for use in EM. There were serious attempts to use metal-chelating proteins as genetically encoded electron-dense tags (Bouchet-Marquis et al., 2012; Wang et al., 2011), but success has been limited to those particular proofs of principle. This is likely because the addition of large densities to the object of interest often interferes with the assembly or visualization of the structure and begins to negate the reason for using cryo-ET in the first place.
So far, the most broadly successful approach to identifying molecular components within ET data has been correlated fluorescence (Figure 4). It combines the best of both techniques to overcome the challenges in each, while creating some of its own. This approach is reviewed elsewhere (Begemann and Galic, 2016; de Boer et al., 2015; Kobayashi et al., 2016; Sjollema et al., 2012), so we will only describe its development briefly. In this approach, the protein of interest is tagged with a fluorescent probe and the cells expressing the fluorescent protein are plunge-frozen on the surface of an EM grid. The vitrified sample is loaded onto a specialized fluorescent microscope outfitted with a cryo-stage capable of maintaining liquid nitrogen temperatures. Once the fluorescence signal is located and imaged, the sample is withdrawn and transferred under liquid nitrogen to the cryo-TEM, where the fluorescence data guide target selection for tomographic data collection. If high-precision correlation is needed, fiducial markers that are visible across both imaging modalities and multiple magnifications must be added to the sample prior to plunge-freezing (Schorb and Briggs, 2014), and postprocessing must be done to provide high precision correlation.
Figure 4.

Evolution of cryo-fluorescence microscopy
(A and B) (A) Cryo-fluorescence microscopy accomplished with a cryo-stage attached to a wide-field microscope on a finder grid (Swulius et al., 2011). In this image V. cholerae cells are carrying a GFP-tagged cytoskeletal protein (MreB) and their membranes are dyed with FM4-64 (red). Individual cells were targeted (B, inset) for cryo-ET (B). The MreB bundle is seen (arrows) within the relatively large outline of the GFP signal, which also encompasses other cellular densities.
(C) Cryo-PALM is capable of breaking the resolution limit (∼120-nm resolution) and identifying specific protein complexes within a crowded cellular environment (Chang et al., 2014).
(D) 3D cryo-PALM with drift correction can identify the precise location (∼20-nm resolution) of mitochondrial membrane proteins in the X, Y, and Z dimension (Liu et al., 2015). (Left) Cryo-fluorescence field view of mitochondria within cryosection. (Right-top) Fluorescence map of individual mitochondrion. (Right-bottom) Merging of fluorescence data (green model points) with model reconstructed from cryo-ET of cryosection. Scale bars in (B) and its inset are 200 nm and 1μm, respectively. Scale bars in (D) and the bottom-right inset are 1 μm and 200 nm, respectively.
One challenge of cryo-fluorescence microscopy is that the warm objective cannot touch the vitreous sample or it will thaw and ruin. This has led to the use of long-working-distance objectives with low numerical apertures and relatively poor resolution. This is good enough for detecting which cell is fluorescing or, under the right circumstances, the shape/location of a subcellular object (Figures 4A and 4B). In order to regain the lost resolution, cryo-photoactivated localization microscopy (PALM) was demonstrated (Chang et al., 2014). One advantage of cryo-fluorescence is that the rate of photobleaching is greatly diminished at such low temperatures. In theory, the increased photon count this permits should lead to a higher localization precision than is typical at room temperature. Despite the extra photons, however, thermal vibrations in the cryo-stage from liquid nitrogen bubbling limited the resolution to ~150 nm, an order of magnitude below room-temperature PALM (Figure 4C). Regardless, the study greatly improved the resolution beyond previous correlated fluorescence work and could clearly distinguish between objects within the same bacterium. This resolution barrier was improved by the addition of Piezo-driven thermal drift correction. With this technology, Liu et al. were able to achieve ~20-nm resolution in X, Y, and Z (Liu et al., 2015). The high-resolution Z-dimension information was achieved through the addition of a half-cylindrical lens that altered the shape of the point spread function as a function of its Z-position. With this setup, individual membrane proteins on the surface of mitochondria could be localized with help from the EM map (Figure 4D).
One of the most promising uses of correlated light and electron microscopy (CLEM) comes from combining it with cryo-FIB milling. In this approach, the location of specific objects or cellular compartments is targeted for cryo-FIB milling using cryo-fluorescence data. This was first achieved using a cryo-confocal microscope to obtain 3D fluorescence information that was overlaid on the cryo-scanning electron microscopy (cryo-SEM) image while setting up the milling process (Figures 5A and 5B). The authors were able to target fluorescent lipid droplets in cells grown and plunge-frozen on EM grids (Figure 5C) (Arnold et al., 2016). Combining all of these methods is not a simple process, but more streamlined workflows are being developed (Fuest et al., 2019; Wu et al., 2020), and it is only a matter of time before it is commonplace. For instance, recently a correlative approach was described that starts with cryo-confocal microscopy, and is followed by using a cryo-mill-and-view method for volume imaging, and finally a lamella is prepped for cryo-ET at a chosen point within the volume (Wu et al., 2020) (Figures 5D–5F).
Figure 5.

Guided FIB milling
(A) Cryo-confocal image of lipid droplets inside of cells grown on an EM grid and plunge-frozen. Red circles mark large fiducial beads that are on the grid bars, which can be seen by both fluorescence and SEM (B).
(B) Overlay of fluorescence onto cryo-SEM image of grid square.
(C) Fluorescence overlay on thin lamella postmilling marks where lipid droplets can be seen by EM. (A–C) are adapted from Arnold et al. (2016).
(D) Cryo-SEM image of lamella containing yeast with Zeiss Cross-beam.
(E) Cryo-fluorescence image of lamella from (D) which contains GFP-Hsp104 inclusion bodies (red square).
(F) Slice through tomogram of region in (E) showing densities of inclusion body. Insets show high-contrast cryo-SEM of yeast cell during milling and how the information obtained during the milling process was used in conjunction with tomography data to model the cell interior. (D–F) are adapted from Wu et al. (2020). Scale bars in (D) and (E) are 5 μm. Scale bar in (F) is 100 nm.
An even more integrated workflow was recently demonstrated using a “Photon Ion Electron” microscope (PIE-scope) (Gorelick et al., 2019). In this case, one microscope houses the hardware necessary for both fluorescence microscopy and FIB/SEM. This setup reduces sample contamination by decreasing the number of cryo-transfers between microscopes, and it allows for easy verification that the target has not been lost during milling by quickly translating the grid back under the light objective.
In a third approach, Klein et al. (Klein et al., 2021) collected cryo-fluorescence data both before milling, to target cells for lamella preparation, and again after cryo-ET data collection. By this method, the fluorescence data collected from the milled lamella could be superimposed back on the tomographic reconstruction. While the two microscopes are not integrated in this method, it has the advantage of being usable on any commercially available CLEM/FIB system.
Dynamics and EM
Living cells are not in equilibrium, and perhaps the most difficult task is to determine what state the cell was in when it was statically captured for imaging by EM. There is a wide variety of approaches one can take, as you will see below. In every case, however, it is important to know when to fix/freeze the sample and where to look.
Swulius et al. used a very straightforward dynamic CLEM approach to investigate the structure of the actomyosin ring during cell division in fission yeast (Swulius et al., 2018). Here the light chain of myosin II was tagged with GFP and cells were visualized by epifluorescence. When ~90% of the cells were undergoing mitosis, they were plunge frozen prior to cryosectioning or FIB milling and cryo-ET (Figure 6A). Cytokinesis in these yeast takes ~10 min, so it provided plenty of time to go from imaging to the plunge-freezer.
Figure 6.

Dynamic CLEM
(A) Top panel: Widefield image of GFP-labeled actomyosin rings during synchronized constriction. Dashed outlines show the rod-shaped body of S. pombe. Pairs of fluorescent puncta mark the optical cross section through the actomyosin ring (Swulius et al., 2018). Bottom panel: Side (left) and top (right) views of a 3D model of the membrane (blue) and actin filaments (orange). Arrowheads point to filament ends.
(B) 3D model of ARP2/3-initiated actin wave extracted from a cryotomogram. The color of the filament represents the divergence of its angle from a flat position (Jasnin et al., 2019). Scale bar in the bottom panel of (A) is 100 nm.
A more sophisticated approach was used by Stepanek and Pigino (Stepanek and Pigino, 2017), where they attached a syringe with fixative to a tube that enabled rapid addition of fixative after a cellular event was seen by light microscopy. This was followed by resin embedding and thin sectioning before imaging in a transmission electron microscope. Using this technique, they were able to characterize the directional movement of intraflagellar transport trains in Chlamydomonas reinhardtii flagella.
Recently, ARP2/3-mediated actin waves were imaged by cryo-ET (Figure 6B) (Jasnin et al., 2019). In this case, a fluorescent-protein-tagged LimEΔ protein that labeled F-actin was used to determine which cells were “profusely” generating waves just prior to vitrification. These cells were then targeted for cryo-FIB milling, and cryo-fluorescence was used after milling to identify regions in and around the actin wave (Figure 6B). Filaments at the wave front were seen to be a mixture of horizontal filaments acting as seeds for vertical filaments nucleated by ARP2/3 complexes, which were verified by subtomogram averaging.
In another study of the actin cytoskeleton, filopodia expressing EGFP-cofilin and mCherry-fascin were imaged via live-cell fluorescence before chemical fixation and negative staining (Breitsprecher et al., 2011). The same filopodia from the movies were then targeted for tomography, and it was shown that cofilin and fascin work synergistically to sever filopodial actin filaments.
Using another approach, Kukulski et al. were able to build a spatiotemporal model of endocytosis in yeast. To do this, a set of three proteins involved in different stages of endocytosis were selectively labeled by fluorescence (Kukulski et al., 2012). By changing which proteins were targeted, it was possible to slide the time window for the endocytic event from start to finish. Instead of imaging vitreous cells, yeast was high-pressure frozen, freeze-substituted, and embedded in resin and sectioned prior to fluorescence imaging. CLEM was used to interpret ET data.
Temporally targeted freezing
In 1979, the first machine built for capturing millisecond-timescale events for EM analysis was described (Heuser et al., 1979) (Figure 7A). The device was comprised of a helium-cooled copper block mounted beneath a gravity-powered plunger. The plunger held the freezing head, on which the tissue was mounted, where stimulating wires were attached. Along the length of the plunger, a small protrusion flipped the stimulation trigger as it fell at the precise timing needed before the sample slammed against the cold copper block. Using freeze-fracturing methods on the slam-frozen sample, they were able to image synaptic vesicles fusing with the membrane in frog neuromuscular junctions in direct response to electrical stimulation (Figures 7B and 7C). This device allowed them to resolve ultrastructural changes in the membrane that occurred within a 2-ms window. The correlation with electrophysiological recordings of neurotransmitter release is quite striking even to this day.
Figure 7.

Temporally controlled freezing
(A) Diagram of slam freezer used to capture synaptic vesicles endocytosing at the frog neuromuscular junction.
(B and C) Images of freeze-fractured synaptic contacts frozen 3 ms (B) and 5 ms (C) after stimulation (Heuser et al., 1979).
A more modern approach that makes use of advances in optogenetics called “flash-and-freeze” was first demonstrated by Watanabe et al. (Watanabe et al., 2013a; Watanabe et al., 2013b). In this case, an exogenously expressed channelrhodopsin was controlled by a pulse of blue light to stimulate action potentials in mouse or Caenorhabditis elegans neurons.
To correlate ultrastructural changes with stimulation of the channelrhodopsin, they programmed a high-pressure freezer to vitrify their sample at a precise moment following channel activation, anywhere from 10 ms to 20 s after stimulus. Afterward, the cells underwent freeze-substitution, plastic embedding, sectioning, and finally imaging on a transmission electron microscope. Because of the rapid and precise freezing after stimulation, a new form of “ultrafast endocytosis” was identified using this method.
A similar approach was used by Chakrabarti et al. (2018) on inner hair cells, which makes specialized ribbon synapses with their target neurons. Instead of using optogenetics, however, hair cells were placed in a stimulatory buffer and subsequently underwent high-pressure freezing and freeze-substitution (Chakrabarti et al., 2018). It was shown that stimulation led to discernable subpools of synaptic vesicles associated with each other, with the ribbon through multiple tethers, or with the presynaptic membrane through individual tethers. This separation into subpools was dependent on the expression of the protein otoferlin, a protein for which KO leads to deafness.
Integrated approaches
In order to achieve better spatial and temporal resolution with correlated live-cell fluorescence and cryo-ET, it is likely that an approach that integrates dynamic CLEM and targeted freezing will be necessary. One approach is to mount the fluorescent microscope onto the freezing apparatus, the tradeoff being that the optical path is not optimized for high-resolution dynamic imaging. Another approach is to build a freezing apparatus onto the stage of a high-quality fluorescence microscope, the tradeoff being that the freezing method needs to be reinvented. The former approach was taken by the inventors of MAVIS (Microscopy and Vitrification Integrated System) (Koning et al., 2014), which was a commercially available plunge-freezing robot (Vitrobot, Thermo Fisher) with an objective lens system built into the freezing chamber (Figures 8A and 8B). Using this approach, they were able to achieve cryofixation after live-cell imaging within 4 s. Resolution was limited to ~480 nm despite a theoretical limit of 256 nm, but it is not clear what is limiting.
Figure 8.

Integrated approaches
(A) Live-cell fluorescence image of EM grid with cells growing on it (red) at the end of imaging session in MAVIS (Koning et al., 2014).
(B) Cryo-fluorescence image of the same cells after blotting and plunge-freezing.
(C) DIC image of C. elegans within the microfluidic chamber before and after freezing.
(D) Lamella from vitrified C. elegans during cryo lift out (Fuest et al., 2019). Scale bars in (C) and (D) are 30 μm and 20 μm, respectively.
In order to push time resolution into the millisecond range, a microfluidics flash-freezing approach was developed that works directly on the stage of a commercial live-cell microscope (Fuest et al., 2018). It functions by placing a semiconductor heating layer between the microfluidic chamber and a pool of liquid nitrogen below. With electrical current running through the semiconductor layer, the chamber above could be kept at 37°C until the current was removed. The rate of freezing was demonstrated by watching latex spheres come to a halt within milliseconds following the switch. Since then, an adaptation of this device has been used to freeze C. elegans prior to fluorescence guided cryo-FIB lift out followed by cryo-ET (Fuest et al., 2019) (Figures 8C and 8D).
There is much promise in these approaches. If perfected, they would allow for all of the dynamic data collected up to the moment of freezing to be included in the interpretation of the static structural data from cryo-ET. Integrated with optogenetics or other approaches for controlled stimulation of cells, such an approach could be very powerful.
Hardware developments
No account of cryo-ET's rise to fame would be complete without mentioning certain advances in TEM hardware that have revolutionized cryo-EM in general. For instance, the development of direct electron detectors (DEDs) is largely responsible for the recent “resolution revolution” in both single-particle analysis and subtomogram averaging.
In the not-too-distant past, TEM cameras were typically charge-coupled devices (CCDs) using a scintillator that converted incoming electrons to photons before they hit the camera. This was largely due to the fact that CCD cameras are prone to damage from the high-energy electron beam, but this conversion also generates noise in the image as each individual electron creates a plume of photons. In the last decade, CCDs have been largely replaced with DEDs, which are more resistant to the radiation damage (McMullan et al., 2016). The first prototype direct detector for EM was used in 2004 (Xuong et al., 2004), with the first commercially available one being released a few years later (DE-12, Direct Electron). Direct detection of incoming electrons enables single-electron sensitivity and, therefore, a much higher signal-to-noise ratio (SNR) than CCDs. This is especially the case when using electron counting mode, which facilitates the localization of electrons to a single pixel on the camera and is helpful during low-dose applications such as cryo-ET.
Another benefit of DEDs is their fast readout time which allows researchers to partition the total electron dose over many frames, resulting in movies for each tilt image (Brilot et al., 2012; Campbell et al., 2012). These movies can later be aligned to create one “drift-corrected” image. This is crucial as the electron beam is known to induce motion of particles in the vitrified ice on holey carbon grids (Brilot et al., 2012). This drift-correction process sharpens each image and provides higher resolution (Figures 9A and 9B).
Figure 9.

Hardware developments for cryo-ET
(A and B) Movie frames of viral particles were collected on a direct electron detector. Translational alignment of movie frames, as in B, preserves high-resolution information, unlike in the unaligned image in A (Brilot et al., 2012).
(C and D) Single-tilt images of a neurite imaged in a TEM near focus (500-nm under focus) with (D) or without (C) a Volta phase plate. Contrast is much higher with the use of the phase plate, as microtubules and membrane can be seen in (D), but not in (C). Scale bars in (C) and (D) are 100 nm.
More recently, the Volta phase plate (VPP) was developed to increase image contrast (Danev et al., 2014). In cryo-EM, most information in the image is produced by phase contrast, which is caused by the interference between scattered and unscattered electrons (Wagner et al., 2017). Phase plates enhance contrast by making this interference larger. Importantly, they enable high-contrast imaging at or near focus (Figures 9C and 9D), unlike normal EM imaging where much of the contrast comes from heavy defocusing. Previous iterations of phase plates have been used in cryo-EM (Danev and Nagayama, 2010; Guerrero-Ferreira and Wright, 2014), but the VPP has so far proven to be the most robust to date and has been used successfully in both single particle analysis (SPA) and cryo-ET (Danev et al., 2017; Imhof et al., 2019; Liang et al., 2017; Winter and Chlanda, 2021). These and other technical advancements in cryo-EM have been extensively reviewed elsewhere (Kühlbrandt, 2014; McMullan et al., 2016; Nogales and Scheres, 2015; Wagner et al., 2017).
Software developments
Ascension of cryo-ET was also assisted by many software advances that improve the interpretability of tomograms. While the full breadth of these advancements is much too vast for this review to cover, we want to highlight a few developments especially relevant to cryo-ET. For example, subtomogram averaging now frequently achieves subnanometer resolution (Bharat et al., 2015; M. Chen et al., 2019; Kunz and Frangakis, 2017; Sanchez et al., 2020; Turoňová et al., 2017, 2020), and one reason for this is the development of 3D contrast transfer function (CTF) correction. Traditionally, CTF correction is performed on each image of a tilt series, in two dimensions, which means the same defocus is applied across the entire image. In reality, however, there is a defocus gradient across the width of the image perpendicular to the tilt axis, and this gradient gets sharper at more extreme tilt angles. To solve for this problem, a number of groups have implemented 3D CTF correction (Bharat et al., 2015; Kunz and Frangakis, 2017), and a user-friendly software tool, NovaCTF, was recently introduced that appreciably improved the resolution of subtomogram averages (Turoňová et al., 2017).
Machine learning is one of the most exciting developments in recent years, and it is already being applied to challenges in cryo-EM. For instance, a deep learning denoising model was recently added to the particle picking software Topaz (Bepler et al., 2020). The authors showed improved SNR in cryo-tomograms and a reduced exposure time needed for TEM imaging (Figures 10A and 10B). Another emerging use of machine learning is in tomogram annotation and segmentation, which enables easier visualization and analysis of intracellular environments. This has historically been done by hand and is extremely time-consuming. With the increasingly high throughput of TEMs, however, postprocessing and analysis need to keep up the pace. Fortunately, a machine learning segmentation tool was recently added to the EMAN2 software package (Chen et al., 2017). Here a human user defines a ground truth for any number of cellular features and a convolutional neural network is trained to identify these features in cellular tomograms (Figures 10C–10E). The resulting network can then be applied to other tomograms imaged with the same magnification settings for a high-throughput segmentation workflow following a relatively small amount of human effort. This segmentation workflow has been further demonstrated in cultured neurons (Hylton et al., 2021) and has been added to an integrated workflow within EMAN2 where neural-network-based annotations are converted into subtomogram coordinates for averaging (M. Chen et al., 2019).
Figure 10.

Software developments for cryo-ET
(A and B) Zoomed-in views from a micrograph of T20S proteasomes. In A, the micrograph was low-pass filtered with a binning factor of 16. In B, the micrograph was instead denoised using the U-net denoising model from Topaz-Denoise. The background noise is smoothed substantially more when the Topaz denoising model is used (Bepler et al., 2020).
(C–E) Convolutional-neural-network-assisted automatic segmentation of cellular tomograms in EMAN2. After positive and negative training examples are defined by a human user, a neural network is trained to identify a single cellular feature. (C) shows a section of a tomogram that a neural network was trained on, and (D) shows microtubules segmented following training. (E) is a merged annotation from four networks trained to segment different cellular components (mitochondria, yellow; vesicles, blue; microtubules, cyan; and ribosomes, pink) (Chen et al., 2017).
Conclusions and the future
In the past few years, it has been demonstrated more than once that rapid tilt series could be collected using high-frame-rate cameras and high-precision goniometers (Chreifi et al., 2019; Eisenstein et al., 2019). Now, with a state-of-the art cryo-transmission electron microscope, it is possible to collect a single −60° to +60° tilt series in under one minute. While there are still hurdles to be overcome to make this the norm, it is clear that the levy is about to break, and we will soon need new ways to prepare samples and target areas for high-throughput cryo-ET. Additionally, data processing pipelines will need to be able to handle the onslaught of information. We have still only extracted a fraction of the information from the tomograms that already exist!
Artificial intelligence (AI) will likely play a role here, both at the level of data collection and processing/analyzing. Convolutional neural networks have already shown promise for identifying objects in noisy cryo-EM data sets and segmenting them (Chen et al., 2017; Zhou et al., 2020) and could be used to screen grids for targets while we focus on other things (Du et al., 2021). Similar to all neural networks, defining the ground truth is the most important thing to get right. If done correctly, however, the possibilities carry broad impact. Imagine being able to extract all of the information possible from each tomogram while your microscope generates hundreds of data sets per day.
Taken together, this brief account of the history of cryo-ET and its accompanying technologies provides plenty of reasons to be optimistic. We are currently in a very exciting stage of its development, and we are poised to fill many of the knowledge gaps in biology that are emergent properties of the cell's underlying structural complexity.
Acknowledgments
We would like to acknowledge Han Chen at the Penn State College of Medicine TEM Core for providing the image for Figure 1A, Grant Jensen at Caltech for providing images for Figure 2, and the Penn State College of Medicine Cryo-EM Core for providing images for Figure 1D, as well as figures 9C and 9D.
Author contributions
R.H. wrote and revised the manuscript and prepared figures. M.S. conceptualized, wrote, and revised the manuscript.
Declaration of interests
There are no conflicts of interest.
References
- Arnold J., Mahamid J., Lucić V., de Marco A., Fernández J.J., Laugks T., Mayer T., Hyman A.A., Baumeister W., Plitzko J.M. Site-specific cryo-focused ion beam sample preparation guided by 3D correlative microscopy. Biophys. J. 2016;110:860–869. doi: 10.1016/j.bpj.2015.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett W.P., Banker G.A. An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. I. Cells which develop without intercellular contacts. J. Neurosci. 1984;4:1944–1953. doi: 10.1523/JNEUROSCI.04-08-01944.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann I., Galic M. Correlative light electron microscopy: connecting synaptic structure and function. Front. Synaptic Neurosci. 2016;8:28. doi: 10.3389/fnsyn.2016.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T., Kelley K., Noble A.J., Berger B. Topaz-Denoise: general deep denoising models for cryoEM and cryoET. Nat. Commun. 2020;11:5208. doi: 10.1038/s41467-020-18952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat T.A.M., Russo C.J., Löwe J., Passmore L.A., Scheres S.H.W. Advances in single-particle electron cryomicroscopy structure determination applied to sub-tomogram averaging. Structure. 2015;23:1743–1753. doi: 10.1016/j.str.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm J., Lambert O., Frangakis A.S., Letellier L., Baumeister W., Rigaud J.L. FhuA-mediated phage genome transfer into liposomes: a cryo-electron tomography study. Curr. Biol. 2001;11:1168–1175. doi: 10.1016/s0960-9822(01)00349-9. [DOI] [PubMed] [Google Scholar]
- Bouchet-Marquis C., Pagratis M., Kirmse R., Hoenger A. Metallothionein as a clonable high-density marker for cryo-electron microscopy. J. Struct. Biol. 2012;177:119–127. doi: 10.1016/j.jsb.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D., Koestler S.A., Chizhov I., Nemethova M., Mueller J., Goode B.L., Small J.V., Rottner K., Faix J. Cofilin cooperates with fascin to disassemble filopodial actin filaments. J. Cell Sci. 2011;124:3305–3318. doi: 10.1242/jcs.086934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briegel A., Dias D.P., Li Z., Jensen R.B., Frangakis A.S., Jensen G.J. Multiple large filament bundles observed in Caulobacter crescentus by electron cryotomography. Mol. Microbiol. 2006;62:5–14. doi: 10.1111/j.1365-2958.2006.05355.x. [DOI] [PubMed] [Google Scholar]
- Brilot A.F., Chen J.Z., Cheng A., Pan J., Harrison S.C., Potter C.S., Carragher B., Henderson R., Grigorieff N. Beam-induced motion of vitrified specimen on holey carbon film. J. Struct. Biol. 2012;177:630–637. doi: 10.1016/j.jsb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley G., Gervinskas G., Taveneau C., Venugopal H., Whisstock J.C., de Marco A. Automated cryo-lamella preparation for high-throughput in-situ structural biology. J. Struct. Biol. 2020;210:107488. doi: 10.1016/j.jsb.2020.107488. [DOI] [PubMed] [Google Scholar]
- Campbell M.G., Cheng A., Brilot A.F., Moeller A., Lyumkis D., Veesler D., Pan J., Harrison S.C., Potter C.S., Carragher B., Grigorieff N. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 2012;20:1823–1828. doi: 10.1016/j.str.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño-Díez D., Zanetti G. In situ structure determination by subtomogram averaging. Curr. Opin. Struct. Biol. 2019;58:68–75. doi: 10.1016/j.sbi.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R., Michanski S., Wichmann C. Vesicle sub-pool organization at inner hair cell ribbon synapses. EMBO Rep. 2018:e44937. doi: 10.15252/embr.201744937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.-W., Chen S., Tocheva E.I., Treuner-Lange A., Löbach S., Søgaard-Andersen L., Jensen G.J. Correlated cryogenic photoactivated localization microscopy and cryo-electron tomography. Nat. Methods. 2014;11:737–739. doi: 10.1038/nmeth.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Bell J.M., Shi X., Sun S.Y., Wang Z., Ludtke S.J. A complete data processing workflow for cryo-ET and subtomogram averaging. Nat. Methods. 2019;16:1161–1168. doi: 10.1038/s41592-019-0591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Dai W., Sun S.Y., Jonasch D., He C.Y., Schmid M.F., Chiu W., Ludtke S.J. Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat. Methods. 2017;14:983–985. doi: 10.1038/nmeth.4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chreifi G., Chen S., Metskas L.A., Kaplan M., Jensen G.J. Rapid tilt-series acquisition for electron cryotomography. J. Struct. Biol. 2019;205(2):163–169. doi: 10.1016/j.jsb.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R., Nagayama K. Phase plates for transmission electron microscopy. Methods Enzymol. 2010;481:343–369. doi: 10.1016/S0076-6879(10)81014-6. [DOI] [PubMed] [Google Scholar]
- Danev R., Buijsse B., Khoshouei M., Plitzko J.M., Baumeister W. Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc. Natl. Acad. Sci. U S A. 2014;111:15635–15640. doi: 10.1073/pnas.1418377111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R., Tegunov D., Baumeister W. Using the Volta phase plate with defocus for cryo-EM single particle analysis. eLife. 2017;6 doi: 10.7554/eLife.23006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer P., Hoogenboom J.P., Giepmans B.N.G. Correlated light and electron microscopy: ultrastructure lights up! Nat. Methods. 2015;12:503–513. doi: 10.1038/nmeth.3400. [DOI] [PubMed] [Google Scholar]
- Du X., Wang H., Zhu Z., Zeng X., Chang Y.-W., Zhang J., Xing E., Xu M. Active learning to classify macromolecular structures in situ for less supervision in cryo-electron tomography. Bioinformatics. 2021 doi: 10.1093/bioinformatics/btab123. [DOI] [PubMed] [Google Scholar]
- Eisenstein F., Danev R., Pilhofer M. Improved applicability and robustness of fast cryo-electron tomography data acquisition. J. Struct. Biol. 2019;208:107–114. doi: 10.1016/j.jsb.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel L., Gaietta G., Dow L.P., Swift M.F., Pardon G., Volkmann N., Weis W.I., Hanein D., Pruitt B.L. Extracellular matrix micropatterning technology for whole cell cryogenic electron microscopy studies. J. Micromech. Microeng. 2019;29:115018. doi: 10.1088/1361-6439/ab419a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel B.D., Schaffer M., Albert S., Asano S., Plitzko J.M., Baumeister W. In situ structural analysis of Golgi intracisternal protein arrays. Proc. Natl. Acad. Sci. U S A. 2015;112:11264–11269. doi: 10.1073/pnas.1515337112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel B.D., Schaffer M., Kuhn Cuellar L., Villa E., Plitzko J.M., Baumeister W. Native architecture of the Chlamydomonas chloroplast revealed by in situ cryo-electron tomography. eLife. 2015;4:3583. doi: 10.7554/eLife.04889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuest M., Nocera G.M., Modena M.M., Riedel D., Mejia Y.X., Burg T.P. Cryofixation during live-imaging enables millisecond time-correlated light and electron microscopy. J. Microsc. 2018;272:87–95. doi: 10.1111/jmi.12747. [DOI] [PubMed] [Google Scholar]
- Fuest M., Schaffer M., Nocera G.M., Galilea-Kleinsteuber R.I., Messling J.-E., Heymann M., Plitzko J.M., Burg T.P. In situ microfluidic cryofixation for cryo focused ion beam milling and cryo electron tomography. Sci. Rep. 2019;9:19133–19210. doi: 10.1038/s41598-019-55413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L., Jensen G.J. Electron tomography of cells. Q. Rev. Biophys. 2012;45:27–56. doi: 10.1017/S0033583511000102. [DOI] [PubMed] [Google Scholar]
- Gorelick S., Buckley G., Gervinskas G., Johnson T.K., Handley A., Caggiano M.P., Whisstock J.C., Pocock R., de Marco A. PIE-scope, integrated cryo-correlative light and FIB/SEM microscopy. eLife. 2019;8 doi: 10.7554/eLife.45919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald K., Desai P., Winkler D.C., Heymann J.B., Belnap D.M., Baumeister W., Steven A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science. 2003;302:1396–1398. doi: 10.1126/science.1090284. [DOI] [PubMed] [Google Scholar]
- Guerrero-Ferreira R.C., Wright E.R. Zernike phase contrast cryo-electron tomography of whole bacterial cells. J. Struct. Biol. 2014;185:129–133. doi: 10.1016/j.jsb.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J.E., Reese T.S., Dennis M.J., Jan Y., Jan L., Evans L. Synaptic vesicle exocytosis captured by quick freezing and correlated with quantal transmitter release. J. Cell Biol. 1979;81:275–300. doi: 10.1083/jcb.81.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylton R.K., Seader V.H., Swulius M.T. Cryo-electron tomography and automatic segmentation of cultured hippocampal neurons. Methods Mol. Biol. 2021;2215:25–48. doi: 10.1007/978-1-0716-0966-8_2. [DOI] [PubMed] [Google Scholar]
- Imhof S., Zhang J., Wang H., Bui K.H., Nguyen H., Atanasov I., Hui W.H., Yang S.K., Zhou Z.H. Cryo electron tomography with volta phase plate reveals novel structural foundations of the 96-nm axonemal repeat in the pathogen Trypanosoma brucei. eLife. 2019;8 doi: 10.7554/eLife.52058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasnin M., Beck F., Ecke M., Fukuda Y., Martinez-Sanchez A., Baumeister W., Gerisch G. The architecture of traveling actin waves revealed by cryo-electron tomography. Structure. 2019;27:1211–1223.e5. doi: 10.1016/j.str.2019.05.009. [DOI] [PubMed] [Google Scholar]
- Klein S., Wimmer B.H., Winter S.L., Kolovou A., Laketa V., Chlanda P. Post-correlation on-lamella cryo-CLEM reveals the membrane architecture of lamellar bodies. Commun. Biol. 2021;4:137–212. doi: 10.1038/s42003-020-01567-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S., Iwamoto M., Haraguchi T. Live correlative light-electron microscopy to observe molecular dynamics in high resolution. Microscopy. 2016;65:296–308. doi: 10.1093/jmicro/dfw024. [DOI] [PubMed] [Google Scholar]
- Kukulski W., Schorb M., Kaksonen M., Briggs J.A.G. Plasma membrane reshaping during endocytosis is revealed by time-resolved electron tomography. Cell. 2012;150:508–520. doi: 10.1016/j.cell.2012.05.046. [DOI] [PubMed] [Google Scholar]
- Kunz M., Frangakis A.S. Three-dimensional CTF correction improves the resolution of electron tomograms. J. Struct. Biol. 2017;197:114–122. doi: 10.1016/j.jsb.2016.06.016. [DOI] [PubMed] [Google Scholar]
- Koning R.I., Faas F.G., Boonekamp M., de Visser B., Janse J., Wiegant J.C., de Breij A., Willemse J., Nibbering P.H., Tanke H.J., Koster A.J. MAVIS: an integrated system for live microscopy and vitrification. Ultramicroscopy. 2014;143:67–76. doi: 10.1016/j.ultramic.2013.10.007. [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W. Biochemistry. The resolution revolution. Science. 2014;343:1443–1444. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- Kürner J., Medalia O., Linaroudis A.A., Baumeister W. New insights into the structural organization of eukaryotic and prokaryotic cytoskeletons using cryo-electron tomography. Exp. Cell Res. 2004;301:38–42. doi: 10.1016/j.yexcr.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Ladinsky M.S. Micromanipulator-assisted vitreous cryosectioning and sample preparation by high-pressure freezing. Methods Enzymol. 2010;481:165–194. doi: 10.1016/S0076-6879(10)81008-0. [DOI] [PubMed] [Google Scholar]
- Leigh K.E., Navarro P.P., Scaramuzza S., Chen W., Zhang Y., Castaño-Díez D., Kudryashev M. Subtomogram averaging from cryo-electron tomograms. Methods Cell Biol. 2019;152:217–259. doi: 10.1016/bs.mcb.2019.04.003. [DOI] [PubMed] [Google Scholar]
- Li Z., Trimble M.J., Brun Y.V., Jensen G.J. The structure of FtsZ filaments in vivo suggests a force-generating role in cell division. EMBO J. 2007;26:4694–4708. doi: 10.1038/sj.emboj.7601895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.-L., Khoshouei M., Radjainia M., Zhang Y., Glukhova A., Tarrasch J., Thal D.M., Furness S.G.B., Christopoulos G., Coudrat T. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017;546:118–123. doi: 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Xue Y., Zhao W., Chen Y., Fan C., Gu L., Zhang Y., Zhang X., Sun L., Huang X. Three-dimensional super-resolution protein localization correlated with vitrified cellular context. Sci. Rep. 2015;5:13017. doi: 10.1038/srep13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamid J., Pfeffer S., Schaffer M., Villa E., Danev R., Cuellar L.K., Förster F., Hyman A.A., Plitzko J.M., Baumeister W. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 2016;351:969–972. doi: 10.1126/science.aad8857. [DOI] [PubMed] [Google Scholar]
- Mahamid J., Schampers R., Persoon H., Hyman A.A., Baumeister W., Plitzko J.M. A focused ion beam milling and lift-out approach for site-specific preparation of frozen-hydrated lamellas from multicellular organisms. J. Struct. Biol. 2015;192:262–269. doi: 10.1016/j.jsb.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Marko M., Hsieh C., Schalek R., Frank J., Mannella C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat. Methods. 2007;4:215–217. doi: 10.1038/nmeth1014. [DOI] [PubMed] [Google Scholar]
- McMullan G., Faruqi A.R., Henderson R. Direct electron detectors. Methods Enzymol. 2016;579:1–17. doi: 10.1016/bs.mie.2016.05.056. [DOI] [PubMed] [Google Scholar]
- Nakane T., Kotecha A., Sente A., McMullan G., Masiulis S., Brown P.M.G.E., Grigoras I.T., Malinauskaite L., Malinauskas T., Miehling J. Single-particle cryo-EM at atomic resolution. Nature. 2020;587:152–156. doi: 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D., Frangakis A.S., Typke D., Baumeister W. Cryo-electron tomography of neurospora mitochondria. J. Struct. Biol. 2000;129:48–56. doi: 10.1006/jsbi.1999.4204. [DOI] [PubMed] [Google Scholar]
- Nickell S., Hegerl R., Baumeister W., Rachel R. Pyrodictium cannulae enter the periplasmic space but do not enter the cytoplasm, as revealed by cryo-electron tomography. J. Struct. Biol. 2003;141:34–42. doi: 10.1016/s1047-8477(02)00581-6. [DOI] [PubMed] [Google Scholar]
- Nogales E., Scheres S.H.W. Cryo-EM: a unique tool for the visualization of macromolecular complexity. Mol. Cell. 2015;58:677–689. doi: 10.1016/j.molcel.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilhofer M., Jensen G.J. The bacterial cytoskeleton: more than twisted filaments. Curr. Opin. Cell Biol. 2013;25:125–133. doi: 10.1016/j.ceb.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilhofer M., Ladinsky M.S., McDowall A.W., Petroni G., Jensen G.J. Microtubules in bacteria: ancient tubulins build a five-protofilament homolog of the eukaryotic cytoskeleton. PLoS Biol. 2011;9:e1001213. doi: 10.1371/journal.pbio.1001213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigort A., Plitzko J.M. Cryo-focused-ion-beam applications in structural biology. Arch. Biochem. Biophys. 2015;581:122–130. doi: 10.1016/j.abb.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Rigort A., Bauerlein F.J.B., Villa E., Eibauer M., Laugks T., Baumeister W., Plitzko J.M. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proc. Natl. Acad. Sci. U S A. 2012;109:4449–4454. doi: 10.1073/pnas.1201333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R.M., Zhang Y., Chen W., Dietrich L., Kudryashev M. Subnanometer-resolution structure determination in situ by hybrid subtomogram averaging - single particle cryo-EM. Nat. Commun. 2020;11:3709. doi: 10.1038/s41467-020-17466-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S., Ofverstedt L.-G., Wikström A.-C., Wrange O., Skoglund U. Structure and flexibility of individual immunoglobulin G molecules in solution. Structure. 2004;12:409–415. doi: 10.1016/j.str.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Schaffer M., Mahamid J., Engel B.D., Laugks T., Baumeister W., Plitzko J.M. Optimized cryo-focused ion beam sample preparation aimed at in situ structural studies of membrane proteins. J. Struct. Biol. 2017;197:73–82. doi: 10.1016/j.jsb.2016.07.010. [DOI] [PubMed] [Google Scholar]
- Schaffer M., Pfeffer S., Mahamid J., Kleindiek S., Laugks T., Albert S., Engel B.D., Rummel A., Smith A.J., Baumeister W., Plitzko J.M. A cryo-FIB lift-out technique enables molecular-resolution cryo-ET within native Caenorhabditis elegans tissue. Nat. Methods. 2019;16:757–762. doi: 10.1038/s41592-019-0497-5. [DOI] [PubMed] [Google Scholar]
- Schorb M., Briggs J.A.G. Correlated cryo-fluorescence and cryo-electron microscopy with high spatial precision and improved sensitivity. Ultramicroscopy. 2014;143:24–32. doi: 10.1016/j.ultramic.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schur F.K.M., Obr M., Hagen W.J.H., Wan W., Jakobi A.J., Kirkpatrick J.M., Sachse C., Kräusslich H.-G., Briggs J.A.G. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science. 2016;353:506–508. doi: 10.1126/science.aaf9620. [DOI] [PubMed] [Google Scholar]
- Schwartz C.L., Sarbash V.I., Ataullakhanov F.I., McIntosh J.R., Nicastro D. Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. J. Microsc. 2007;227:98–109. doi: 10.1111/j.1365-2818.2007.01794.x. [DOI] [PubMed] [Google Scholar]
- Sjollema K.A., Schnell U., Kuipers J., Kalicharan R., Giepmans B.N.G. Correlated light microscopy and electron microscopy. Methods Cell Biol. 2012;111:157–173. doi: 10.1016/B978-0-12-416026-2.00009-1. [DOI] [PubMed] [Google Scholar]
- Stepanek L., Pigino G. Millisecond time resolution correlative light and electron microscopy for dynamic cellular processes. Methods Cell Biol. 2017;140:1–20. doi: 10.1016/bs.mcb.2017.03.003. [DOI] [PubMed] [Google Scholar]
- Stoffler D., Feja B., Fahrenkrog B., Walz J., Typke D., Aebi U. Cryo-electron tomography provides novel insights into nuclear pore architecture: implications for nucleocytoplasmic transport. J. Mol. Biol. 2003;328:119–130. doi: 10.1016/s0022-2836(03)00266-3. [DOI] [PubMed] [Google Scholar]
- Studer D., Klein A., Iacovache I., Gnaegi H., Zuber B. A new tool based on two micromanipulators facilitates the handling of ultrathin cryosection ribbons. J. Struct. Biol. 2014;185:125–128. doi: 10.1016/j.jsb.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Swulius M.T., Chen S., Jane Ding H., Li Z., Briegel A., Pilhofer M., Tocheva E.I., Lybarger S.R., Johnson T.L., Sandkvist M., Jensen G.J. Long helical filaments are not seen encircling cells in electron cryotomograms of rod-shaped bacteria. Biochem. Biophys. Res. Commun. 2011;407:650–655. doi: 10.1016/j.bbrc.2011.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swulius M.T., Nguyen L.T., Ladinsky M.S., Ortega D.R., Aich S., Mishra M., Jensen G.J. Structure of the fission yeast actomyosin ring during constriction. Proc. Natl. Acad. Sci. U S A. 2018;94:201711218–E1464. doi: 10.1073/pnas.1711218115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro-Nahuelpan M., Zagoriy I., Senger F., Blanchoin L., Théry M., Mahamid J. Tailoring cryo-electron microscopy grids by photo-micropatterning for in-cell structural studies. Nat. Methods. 2020;17:50–54. doi: 10.1038/s41592-019-0630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B., Hagen W.J.H., Obr M., Mosalaganti S., Beugelink J.W., Zimmerli C.E., Kräusslich H.-G., Beck M. Benchmarking tomographic acquisition schemes for high-resolution structural biology. Nat. Commun. 2020;11:876–879. doi: 10.1038/s41467-020-14535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoňová B., Schur F.K.M., Wan W., Briggs J.A.G. Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4Å. J. Struct. Biol. 2017;199:187–195. doi: 10.1016/j.jsb.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar K.R., Henderson R. Single particle electron cryomicroscopy: trends, issues and future perspective. Q. Rev. Biophys. 2016;49:e13. doi: 10.1017/S0033583516000068. [DOI] [PubMed] [Google Scholar]
- Wagner J., Schaffer M., Fernández-Busnadiego R. Cryo-electron tomography-the cell biology that came in from the cold. FEBS Lett. 2017;591:2520–2533. doi: 10.1002/1873-3468.12757. [DOI] [PubMed] [Google Scholar]
- Wan W., Briggs J.A.G. In: Anthony Crowther R., editor. Vol. 579. Elsevier; 2016. Cryo-electron tomography and subtomogram averaging; pp. 329–367. (The Resolution Revolution: Recent Advances in cryoEM). [DOI] [PubMed] [Google Scholar]
- Wang K., Strunk K., Zhao G., Gray J.L., Zhang P. 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. J. Struct. Biol. 2012;180:318–326. doi: 10.1016/j.jsb.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Mercogliano C.P., Löwe J. A ferritin-based label for cellular electron cryotomography. Structure. 2011;19:147–154. doi: 10.1016/j.str.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Watanabe R., Buschauer R., Böhning J., Audagnotto M., Lasker K., Lu T.-W., Boassa D., Taylor S., Villa E. The in situ structure of Parkinson’s disease-linked LRRK2. Cell. 2020;182:1508–1518.e16. doi: 10.1016/j.cell.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Liu Q., Davis M.W., Hollopeter G., Thomas N., Jorgensen N.B., Jorgensen E.M. Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. eLife. 2013;2:e00723. doi: 10.7554/eLife.00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S., Rost B.R., Camacho-Pérez M., Davis M.W., Söhl-Kielczynski B., Rosenmund C., Jorgensen E.M. Ultrafast endocytosis at mouse hippocampal synapses. Nature. 2013;504:242–247. doi: 10.1038/nature12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S.L., Chlanda P. Dual-axis Volta phase plate cryo-electron tomography of Ebola virus-like particles reveals actin-VP40 interactions. J. Struct. Biol. 2021;213:107742. doi: 10.1016/j.jsb.2021.107742. [DOI] [PubMed] [Google Scholar]
- Wolff G., Limpens R.W.A.L., Zheng S., Snijder E.J., Agard D.A., Koster A.J., Bárcena M. Mind the gap: micro-expansion joints drastically decrease the bending of FIB-milled cryo-lamellae. J. Struct. Biol. 2019;208:107389. doi: 10.1016/j.jsb.2019.09.006. [DOI] [PubMed] [Google Scholar]
- Wu G.-H., Mitchell P.G., Galaz-Montoya J.G., Hecksel C.W., Sontag E.M., Gangadharan V., Marshman J., Mankus D., Bisher M.E., Lytton-Jean A.K.R. Multi-scale 3D cryo-correlative microscopy for vitrified cells. Structure. 2020 doi: 10.1016/j.str.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuong N.-H., Milazzo A.-C., LeBlanc P., Duttweiler F., Bouwer J., Peltier S., Ellisman M., Denes P., Bieser F., Matis H.S. Vol. 5301. Proc.SPIE; 2004. (First Use of a High-Sensitivity Active Pixel Sensor Array as a Detector for Electron Microscopy). [DOI] [PubMed] [Google Scholar]
- Zachman M.J., Asenath-Smith E., Estroff L.A., Kourkoutis L.F. Site-specific preparation of intact solid-liquid interfaces by label-free in situ localization and cryo-focused ion beam lift-out. Microsc. Microanal. 2016;22:1338–1349. doi: 10.1017/S1431927616011892. [DOI] [PubMed] [Google Scholar]
- Zachs T., Schertel A., Medeiros J., Weiss G.L., Hugener J., Matos J., Pilhofer M. Fully automated, sequential focused ion beam milling for cryo-electron tomography. eLife. 2020;9:13726. doi: 10.7554/eLife.52286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P. Advances in cryo-electron tomography and subtomogram averaging and classification. Curr. Opin. Struct. Biol. 2019;58:249–258. doi: 10.1016/j.sbi.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Ofverstedt L.-G., Skoglund U., Isaksson L.A. Morphological variation of individual Escherichia coli 30S ribosomal subunits in vitro and in situ, as revealed by cryo-electron tomography. Exp. Cell Res. 2004;297:495–507. doi: 10.1016/j.yexcr.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Zhou B., Yu H., Zeng X., Yang X., Zhang J., Xu M. One-shot learning with attention-guided segmentation in cryo-electron tomography. Front. Mol. Biosci. 2020;7:613347. doi: 10.3389/fmolb.2020.613347. [DOI] [PMC free article] [PubMed] [Google Scholar]