

Abstract
Cystic fibrosis (CF) is a common, lethal genetic disease, which is due to mutations in the CFTR gene. The CF lung expresses a profoundly proinflammatory phenotype, due to constitutive hypersecretion of IL-8 from epithelial cells lining the airways. In a systematic search for candidate drugs that might be used therapeutically to suppress IL-8 secretion from these cells, we have identified a potent and efficacious series of amphiphilic pyridinium salts. The most potent of these salts is MRS2481, an (R)-1-phenylpropionic acid ester, with an IC50 of ca. 1 μM. We have synthesized 21 analogues of MRS2481, which have proven sufficient to develop a preliminary structure–activity relationship (SAR). For optimal activity, we have found that the ester must be connected to the pyridinium derivative by an eight-carbon chain. An optical isomer of the lead compound, containing an (S)-1-phenylpropionic acid ester, has been found to be a much less active. The mechanism of action of MRS2481 appears to involve inhibition of signaling of the NFκB and AP-1 transcription factors to the IL-8 promoter. MRS2481 is a potent inhibitor of TNFα-induced phosphorylation and proteosomal destruction of IκBα. Inasmuch as IκBα is the principal inhibitor of the NFκB signaling pathway, preservation of intact IκBα would serve to keep the IL-8 promoter silent. We also find that MRS2481 blocks TNFα-activated phosphorylation of JNK, the c-JUN kinase. The IL-8 promoter is also activated by an AP-1 site, which requires a phospho-c-JUN/c-FOS dimer for activity. We therefore interpret these data to suggest that the mechanism of MRS2481 action is to inhibit both NFκB and AP-1 signaling on the IL-8 promoter. Given the medicinally promising properties of water-solubility, potency in the low μM concentration range, and high efficacy, we anticipate that MRS2481, or a further optimized derivative, may find an important place in the armamentarium of pharmaceutical strategies yet to be arrayed against the inflammatory phenotype of the CF lung.
Keywords: Cystic fibrosis, Inflammation, Lung, Interleukin 8, Epithelium, NFκB, AP-1
1. Introduction
Cystic fibrosis (CF) is the most common, lethal auto-somal recessive disease in the United States and Europe [1,2]. The disease is due to mutations in the CFTR gene, which causes a profoundly proinflammatory phenotype in the CF lung. The proinflammatory mechanism is based on constitutive hypersecretion of interleukin-8 (IL-8) from the epithelial cells lining the CF airways [3–10]. IL-8 is the most potent known chemoattractor for leucocytes into inflammatory foci [11]. The IL-8 gene is principally driven by the NFκB signaling pathway, and to a lesser extent by the AP-1 (c-FOS/c-JUN) pathway, and certain other transcription factors [12,13]. The most common CF mutation, [ΔF508]CFTR, generates a mutant CFTR protein that fails to traffick properly to apical plasma membranes in epithelial cells of the CF airway [14]. The cAMP-activated chloride channel activity of the CFTR protein is thereby lost. However, while this mutant trafficking defect appears to be correlated with the pro-inflammatory disease phenotype, it is possible to reduce IL-8 hypersecretion pharmaceutically from CF lung cells with little or no correction of the trafficking defect.
The candidate CF drug CPX illustrates how a close relationship can exist between correction of the trafficking defect, activation of the cAMP-dependent chloride channel activity, and suppression of IL-8 hypersecretion [15–22]. CPX is a xanthine with a ka for trafficking rescue of ca. 1–5 μM, which also suppresses IL-8 hyperexpression. The mechanism is by inhibition of the TNFα/NFκB signaling pathway [22]. The same mechanism is responsible for inhibition of IL-8 secretion from CF lung epithelial cells by gene therapy with [wildtype]CFTR [22].
The candidate CF drug digitoxin is an example of how IL-8 hypersecretion can be suppressed in CF cells with only modest rescue of mutant CFTR [23]. Digitoxin potently suppresses hyperexpression of IL-8 (ka = ca. 1 nM; Ref. [23]). Nonetheless, digitoxin also returns to control levels 62% of the genes that are affected in expression by the mutant CFTR [23]. The mechanism of digitoxin action on the expression of IL-8 is also to block the TNFα/NFκB signaling pathway. Other compounds have been studied with respect to their ability to correct trafficking and/or channel activity of mutant CFTR, but have not yet been characterized in terms of their capacity to suppress IL-8 hypersecretion from CF cells [24–29].
The fundamental importance of the inflammatory arm of CF for lung disease is particularly demonstrated by clinical experience with ibuprofen. For those CF patients able to tolerate the required massively high doses, ibuprofen stops the progressive loss of lung function typical of the disease [30]. The mechanism of action of ibuprofen is to reduce the number of inflammatory leucocytes in the circulation that are available to be recruited into sites of IL-8 expression in the CF airway. We have therefore interpreted these data to suggest that successful CF drug discovery can be achieved by specifically targeting constitutive hypersecretion of IL-8 by CF lung epithelial cells. Accurso et al. [31] have emphasized that the most important property of a clinically useful CF drug is the ability to specifically suppress the pro-inflammatory CF phenotype.
In this paper we describe the discovery of MRS2481, a new amphiphilic pyridinium salt that potently and efficaciously suppresses IL-8 secretion. Although somewhat less potent than digitoxin, MRS2481 is analogous to digitoxin in that its mechanism involves the blockade of the TNFα/NFκB signaling pathway. Furthermore, like digitoxin, MRS2481 has only modest correction activity on the mutant CFTR trafficking defect. The structural requirements responsible for MRS2481 potency and efficacy have been investigated in order to establish a structure–activity relationship (SAR). To this end we synthesized 22 different MRS2481 homologues by the methods of classical organic chemical synthesis. We observed substantial and informative variation in activities. We anticipate that MRS2481 may not only be useful as a tool to investigate the mechanism of the CF pro-inflammatory phenotype, but may also prove to be a prototype for a novel CF pharmaceutical.
2. Materials and methods
2.1. Chemical synthesis
2.1.1. Chemical reagents and instrumentation
Reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). 1H NMR spectra were obtained with a Varian Gemini-300 spectrometer (300 MHz) with D2O, CDCl3, CD3OD, and DMSO-d6 as a solvent. The chemical shifts are expressed as ppm downfield from tetramethylsilane. Purity of compounds was checked with a Hewlett-Packard 1090 HPLC equipped with a Luna 5 μ RP-C18(2) analytical column (250 mm × 4.6 mm; Phenomenex, Torrance, CA). System A: linear gradient solvent system: H2O/CH3CN from 95/5 to 20/80 in 20 min; the flow rate was 1 ml/min. System B: linear gradient solvent system: 5 mM tetrabutylammonium phosphate/CH3CN from 80/20 to 20/80 in 20 min, then isocratic for 2 min; the flow rate was 1 ml/min. Peaks were detected by UV absorption with a diode array detector. All derivatives tested for biological activity showed >96% purity in the HPLC systems. TLC analysis was carried out on aluminum sheets precoated with silica gel F254 (0.2 mm) from Aldrich. Low-resolution FAB mass spectrometry was performed with a JEOL SX102 spectrometer with 6 kV Xe atoms following desorption from a glycerol matrix.
2.1.1.1. Method A.
General procedure for the synthesis of the compounds 1–14 and 16–18.
R-8-Bromo-n-octyl-α-methyl-2-phenylacetate (25).
R-(−)-2-Phenylpropionic acid (210 mg, 1.4 mmol) and 1,8-dibromo-octane (0.26 ml, 1.4 mmol) were put in a 5 ml round bottom flask with a methanolic solution of benzyltrimethylammonium hydroxide (0.65 ml, 40%). Tetrabutylammonium iodide (14 mg, 0.038 mmol) was added and the mixture was stirred for 3 days. The mixture was poured in water (30 ml), and the aqueous solution was extracted with ethyl acetate (10 ml 3×). The organic phase was dried over Na2SO4, filtered and concentrated. The residue was purified using preparative thin layer chromatography (silica gel, eluting with petroleum ether:ethyl acetate, 20:1) obtaining 181 mg of pure 25 (yield 38%).
1H NMR (CDCl3): δ 7.34–7.24 (m, 5H), 4.05 (t, J = 6.3 Hz, 2H), 3.70 (q, J = 6.9 Hz), 3.41 (t, J = 6.3 Hz, 2H), 1.90–1.80 (m, 1H), 1.56–1.37 (m, 7H), 1.35–1.20 (m, 6H). MS (m/e) (positive FAB) 341.1 (M + H)+.
R-8-Iodo-n-octyl-α-methyl-2-phenylacetate (26).
25 (200 mg, 0.58 mmol) was dissolved in acetone (5 ml) and NaI (mg 90, 10.6 mmol) was added. The mixture was stirred at r.t. overnight. The solvent was concentrated and water (20 ml) was added, and the aqueous phase was extracted with ethyl acetate (10 ml × 3). The organic phase was dried over Na2SO4, filtered and concentrated. The product was sufficiently pure to be used without further purification.
1H NMR (CDCl3): δ 7.32–7.24 (m, 5H), 4.05 (t, J = 6.3 Hz, 2H), 3.70 (q, J = 6.9 Hz), 3.18 (t, J = 6.3 Hz, 2H), 1.84–1.79 (m, 2H), 1.56–1.37 (m, 7H), 1.35–1.23 (m, 6H). MS (m/e) (positive FAB) 389.1 (M + H)+.
R-8-Pyridinium-n-octyl-α-methyl-2-phenylacetate iodide (3).
26 (100 mg, 0.26 mmol) was dissolved in acetone (10 ml) and pyridine (0.3 ml) was added. The solution was stirred at 50 °C for three days. The solvent was evaporated and the residue dissolved in water (20 ml). The aqueous phase was washed with ether (10 ml × 3) and lyophilized to give 54 mg of pure 3 (yield 45%).
1H NMR (D2O): δ 8.80 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.09 (t, J = 6.9 Hz, 2H), 745–7.31 (m, 5H), 4.53 (t, J = 7.2 Hz, 2H), 4.19–4.05 (m, 2H), 3.88 (q, J = 6.9 Hz, 1H), 1.94–1.88 (m, 2H), 1.61–1.56 (m, 2H), 1.49–1.42 (m, 7H), 1.27–1.23 (m, 4H). MS (m/e) (positive FAB) 340.1 (M − I)+.
R-8-Pyridinium-n-butyl-α-methyl-2-phenylacetate iodide (1).
1H NMR (D2O): δ 8.67 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.07 (t, J = 6.9 Hz, 2H), 7.43–7.31 (m, 5H), 4.45 (t, J = 7.2 Hz, 2H), 4.29–4.09 (m, 2H), 3.90 (q, J = 6.9 Hz, 1H), 1.92–1.82 (m, 2H), 1.72–1.65 (m, 2H), 1.49 (d, J = 7.2 Hz, 3H). MS (m/e) (positive FAB) 284.1 (M − I)+.
R-8-Pyridinium-n-hexyl-α-methyl-2-phenylacetate iodide (2).
1H NMR (D2O) δ 8.81 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.08 (t, J = 6.9 Hz, 2H), 7.45–7.30 (m, 5H), 4.3 (t, J = 7.2 Hz, 2H), 4.21–4.12 (m, 2H), 3.89 (q, J = 6.9 Hz, 1H), 1.94–1.86 (m, 2H), 1.62–1.55 (m, 2H), 1.48 (d, J = 7.2 Hz, 3H), 1.27–1.22 (m, 4H). MS (m/e) (positive FAB) 312.1 (M − I)+.
R-8-Pyridinium-n-decyl-α-methyl-2-phenylacetate iodide (4).
1H NMR (D2O) δ 8.98–8.87 (m, 2H), 8.61–8.57 (m, 1H), 8.11–8.07 (m, 2H), 7.33–7.14 (m, 5H), 4.69–4.55 (m, 2H), 4.09–3.92 (m, 2H), 3.66 (q, J = 6.9 Hz, 1H), 2.02–1.92 (m, 2H), 1.49–1.08 (m, 17H). MS (m/e) (positive FAB) 368.3 (M − I)+.
S-8-Pyridinium-n-octyl-α-methyl-2-phenylacetate iodide (5).
1H NMR (D2O) δ 8.80 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.09 (t, J = 6.9 Hz, 2H), 7.45–7.31 (m, 5H), 4.53 (t, J = 7.2 Hz, 2H), 4.19–4.05 (m, 2H), 3.88 (q, J = 6.9 Hz, 1H), 1.94–1.88 (m, 2H), 1.61–1.56 (m, 2H), 1.49–1.42 (m, 7H), 1.27–1.23 (m, 4H). MS (m/e) (positive FAB) 340.2 (M − I)+.
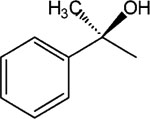
R-8-Pyridinium-n-octyl-α-hydroxy-methyl-2-phenylacetate iodide (6).
1H NMR (D2O) δ 8.84 (d, J = 6.6 Hz, 2H), 8.56 (t, J = 9 Hz, 1H), 8.08 (t, J = 6.9 Hz, 2H), 7.57–7.39 (m, 5H), 4.60 (t, J = 7.2 Hz, 2H), 4.15 (t, J = 4.2 Hz, 2H), 2.02–1.96 (m, 2H), 1.83 (s, 3H), 1.59–1.55 (m, 2H), 1.24–1.17 (m, 8H). MS (m/e) (positive FAB) 356.2 (M − I)+.
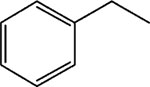
8-Pyridinium-n-octyl-propionate iodide (7).
1H NMR (D2O) δ 8.88 (d, J = 6.6 Hz, 2H), 8.66 (t, J = 9 Hz, 1H), 8.14 (t, J = 6.9 Hz, 2H), 7.47–7.38 (m, 5H), 4.55 (t, J = 7.2 Hz, 2H), 4.25 (t, J = 4.2 Hz, 2H), 3.85 (s, 2H), 2.12–1.98 (m, 2H), 1.56–1.37 (m, 4H), 1.30–1.19 (m, 6H). MS (m/e) (positive FAB) 326.2 (M − I)+.
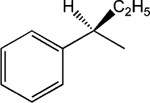
R-8-Pyridinium-n-octyl-α-ethyl-2-phenylacetate iodide (8).
1H NMR (D2O) δ 9.31 (d, J = 6.6 Hz, 2H), 8.49 (t, J = 9 Hz, 1H), 8.10 (t, J = 6.9 Hz, 2H), 7.31–7.22 (m, 5H), 4.93 (t, J = 7.2 Hz, 2H), 4.06–4.01 (m, 2H), 3.48–3.41 (m, 1H), 2.11–1.90 (m, 3H), 1.84–1.74 (m, 1H), 1.61–1.46 (m, 4H), 1.32–1.20 (m, 6H), 0.89 (t, J = 7.2 Hz, 2H). MS (m/e) (positive FAB) 355.2 (M − I)+.
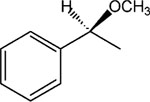
S-8-Pyridinium-n-octyl-α-methoxy-2-phenylacetate iodide (9).
1H NMR (D2O) δ 8.82 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.09 (t, J = 6.9 Hz, 2H), 7.48–7.42 (m, 5H), 4.59 (t, J = 7.2 Hz, 2H), 4.29–4.10 (m, 3H), 3.41 (s, 3H), 2.02–1.88 (m, 2H), 1.61–1.52 (m, 2H), 1.29–1.12 (m, 8H). MS (m/e) (positive FAB) 356.2 (M − I)+.
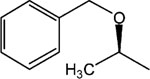
S-8-Pyridinium-n-octyl-2-benzyloxy-propionate iodide (10).
1H NMR (D2O) δ 8.81 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.07 (t, J = 6.9 Hz, 2H), 7.45–7.40 (m, 5H), 4.62–4.58 (m, 4H), 4.25 (q, J = 6.9 Hz, 1H), 4.18 (t, J = 7.2 Hz, 2H), 2.02–1.94 (m, 2H), 1.72–1.60 (m, 2H), 1.40 (d, J = 7.2 Hz, 3H), 1.38–1.26 (m, 8H). MS (m/e) (positive FAB) 370.3 (M − I)+.
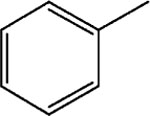
8-Pyridinium-n-octyl-benzoate iodide (11).
1H NMR (D2O) δ 8.81 (d, J = 6.6 Hz, 2H), 8.51 (t, J = 9 Hz, 1H), 8.03 (m, 4H), 7.65–7.55 (m, 1H), 7.56–7.51 (m, 2H), 4.59–4.55 (m, 2H), 4.34 (t, J = 7.2 Hz, 2H), 4.05 (q, J = 6.9 Hz, 1H), 2.02–1.94 (m, 2H), 1.78–1.74 (m, 2H), 1.62–1.26 (m, 8H). MS (m/e) (positive FAB) 312.3 (M − I)+.
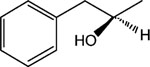
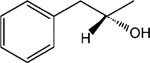
R-8-Pyridinium-n-octyl-2-hydroxy-3-phenylpropionate iodide (12) and S-8-pyridinium-n-octyl-2-hydroxy-3-phenylpropionate iodide (13).
1H NMR (D2O) δ 8.80 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.09 (t, J = 6.9 Hz, 2H), 7.38–7.22 (m, 5H), 4.65 (t, J = 7.2 Hz, 2H), 4.44 (q, J = 6.8 Hz, 1H), 4.14 (t, J = 7.2 Hz, 2H), 3.18–3.02 (m, 2H), 1.95–1.82 (m, 2H), 1.69–1.62 (m, 2H), 1.45–1.15 (m, 8H). MS (m/e) (positive FAB) 356.2 (M − I)+.
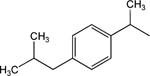
8-Pyridinium-n-octyl-α-methyl-2-[4-(2-methylpropyl)-phenyl]acetate iodide (14).
1H NMR (CD3Cl3) δ 9.35 (br s, 2H), 8.56–8.54 (m, 1H), 8.14 (br s, 1H), 7.18 (d, J = 8.1 Hz, 2H), 7.08 (d, J = 8.1 Hz, 2H), 4.98–4.81 (m, 2H), 4.05–3.98 (m, 2H), 3.94–3.62 (m, 1H), 2.43 (d, J = 7.2 Hz, 1H), 2.15 (br s, 2H), 1.88–1.78 (m, 1H), 1.59–1.18 (m, 10H), 0.88 (d, J = 6.6 Hz, 6H). MS (m/e) (positive FAB) 396.3 (M − I)+.
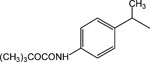
8-Pyridinium-n-octyl-α-methyl-2-[4-(N-Boc amino)-phenyl]acetate iodide (16).
1H NMR (D2O) δ 8.82 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.08 (t, J = 6.9 Hz, 2H), 7.38–7.31 (m, 4H), 4.63 (t, J = 7.2 Hz, 2H), 4.29–4.20 (m, 1H), 4.18–4.02 (m, 1H), 3.88 (q, J = 6.9 Hz, 1H), 1.98–1.88 (m, 2H), 1.61–1.56 (m, 13H), 1.27–1.23 (m, 9H). MS (m/e) (positive FAB) 455.3(M − I)+.
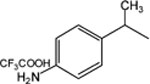
8-Pyridinium-n-octyl-α-methyl-2-[4-ammonium trifluoroacetate-phenyl]acetate iodide (17).
1H NMR (D2O) δ 8.85 (d, J = 6.6 Hz, 2H), 8.57 (t, J = 9 Hz, 1H), 8.08 (t, J = 6.9 Hz, 2H), 7.49–7.35 (m, 4H), 4.62 (t, J = 7.2 Hz, 2H), 4.14 (t, J = 7.2 Hz, 2H), 3.95 (q, J = 6.9 Hz, 1H), 1.98–1.88 (m, 2H), 1.61–1.49 (m, 5H), 1.31–1.25 (m, 8H). MS (m/e) (positive FAB) 355.3 (M − I-CF3COO)+.
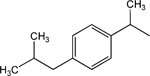
8-(3-Carboxyamido-pyridinium)-n-octyl-α-methyl-2-[4-(2-methylpropyl)phenyl]-acetate iodide (18).
1H NMR (CDCl3) d 10.21 (s, 1H), 9.16 (d, J = 9 Hz, 1H), 8.98 (d, J = 6.6 Hz, 2H), 8.58 (s, 1H), 8.19 (t, J = 6.9 Hz, 2H), 7.21–7.08 (m, 4H), 6.53 (s, 1H), 4.88 (t, J = 7.2 Hz, 2H), 4.07–4.03 (m, 2H), 3.70 (q, J = 6.8 Hz, 1H), 2.44 (d, J = 9 Hz, 2H), 2.18–2.02 (m, 2H), 1.85–1.72 (m, 5H), 1.57–1.26 (m, 9H), 0.89 (d, J = 6.6 Hz, 6H). MS (m/e) (positive FAB) 439.3 (M − I)+.
2.1.1.2. Method B.
General procedure for the synthesis of the compounds 15 and 19–22.
S,S-1,8-Di(α-methyl-2-(4-[2-methylpropyl]phenyl)acetoxy)-n-octane (22).
S-α-Methyl-2-(4-[2-methylpropyl]-benzene)acetic acid (206 mg, 1 mmol), 1,8-dibromooctane (0.184 ml) were combined. A methanolic solution of benzyltriethylammonium methoxide (453 mg, 40%) and tetrabutyl ammonium iodide (10 mg) was added. The mixture was stirred at room temperature for 3 days. The product (34, 167 mg, 0.42 mmol, 42% yield) was isolated using preparative thin layer chromatography (silica gel, eluting with hexanes:ethyl acetate, 20:1). A minor product, a dimeric molecule 22 was also isolated (34 mg, 7%).
1H NMR (CDCl3) δ 7.20 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 4.05 (t, J = 6.3 Hz, 2H), 3.70 (q, J = 6.9 Hz), 2.45 (d, J = 7.2 Hz, 2H), 1.89–1.80 (m, 1H), 1.57–1.48 (m, 5H), 1.26–1.20 (m, 4H), 0.90 (d, J = 6.6 Hz, 6H). MS (m/e) (positive FAB) 523.4 (M + H)+.
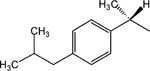
S-8-Pyridinium-n-octyl-α-methyl-2-(4-[2-methylpropyl]benzene)acetate bromide (15).
S-8-Bromo-n-octyl-α-methyl-2-(4-[2-methylpropyl]benzene)acetate (35 mg, 0.088 mmol) and pyridine (0.2 ml, 2.5 mmol) were dissolved in acetone (3 ml). Tetrabutyl ammonium iodide (10 mg) was added, and the mixture was stirred for 2 days at 50 °C. Acetone was removed in vacuo. The product (15, 16 mg, 0.035 mmol, 40% yield) was isolated using preparative thin layer chromatography (silica gel, eluting with chloroform:methanol, 5:1).
1H NMR (CD3OD) * δ 9.02 (d, J = 6.6 Hz, 2H), 8.59 (t, J = 9 Hz, 1H), 8.09 (t, J = 6.9 Hz, 2H), 7.19 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 4.62 (t, J = 7.2 Hz, 2H), 4.10–4.01 (m, 2H), 3.70 (q, J = 6.9 Hz, 1H), 2.45 (d, J = 7.2 Hz, 2H), 2.03–1.98 (m, 2H), 1.87–1.78 (m, 1H), 1.59–1.51 (m, 2H), 1.43 (d, J = 7.2 Hz. 3H), 1.33–1.25 (m, 8H), 0.90 (d, J = 6.6 Hz, 6H). MS (m/e) (positive FAB) 396.3 (M − Br)+.
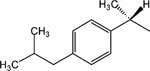
S-8-(4-n-Propylpyridinium)-n-octyl-α-methyl-2-(4-[2-methylpropyl]phenyl)acetate bromide (19).
S-8-Bromo-n-octyl-α-methyl-2-(4-[2-methylpropyl]phenyl)acetate (35 mg, 0.088 mmol) and 4-n-propylpyridine (0.2 ml) were dissolved in acetone (3 ml). Tetrabutyl ammonium iodide (5 mg) was added, and the mixture was stirred for 2 days at 50 °C. Acetone was removed in vacuo. The product (19, 16 mg, 0.028 mmol, 35% yield) was isolated using preparative thin layer chromatography (silica gel, eluting with chloroform:methanol, 5:1).
1H NMR (CD3OD) δ 9.32 (d, J = 6.6 Hz, 2H), 7.84 (d, J = 6.3 Hz, 2H), 7.19 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 4.91 (t, J = 7.2 Hz, 2H), 4.02 (t, J = 7.2 Hz, 2H) 3.70 (q, J = 6.9 Hz, 1H), 3.39–3.34 (m, 2H), 2.86 (t, J = 7.2 Hz, 2H), 2.45 (d, J = 7.2 Hz, 2H), 2.03–1.98 (m, 2H), 1.87–1.64 (m, 3H), 1.56–1.30 (m, 5H), 1.33–1.25 (m, 8H), 1.01 (t, J = 7.2 Hz, 2H), 0.90 (d, J = 6.6 Hz, 6H). MS (m/e) (positive FAB) 438.3 (M − Br)+.
S-8-[4-(2-hydroxy-ethyl)pyridinium)-n-octyl-a-methyl-2-(4-[2-methylpropyl]phenyl)acetate bromide (20).
1H NMR (CDCl3) δ 8.86 (d, J = 6.6 Hz, 2H), 7.98 (d, J = 6.3 Hz, 2H), 7.19 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 4.69 (t, J = 7.2 Hz, 2H), 4.05–3.92 (m, 3H), 4.62–4.59 (m, 1H), 3.19–3.10 (m, 2H), 2.86 (t, J = 7.2 Hz, 1H), 2.45 (d, J = 7.2 Hz, 2H), 2.03–1.98 (m, 2H), 1.49–1.25 (m, 14H), 0.90 (d, J = 6.6 Hz, 6H). MS (m/e) (positive FAB) 440.3 (M − Br)+.
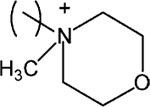
S-8-[N-Methylmorpholinium)-n-octyl-α-methyl-2-(4-[2-methylpropyl]phenyl)acetate bromide (21).
1H NMR (CDCl3) δ 7.19 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 4.13–3.98 (m, 6H), 3.89–3.78 (m, 4H), 3.71–3.55 (m, 6H), 3.40–3.24 (m, 2H), 2.45 (d, J = 7.2 Hz, 2H), 1.86–1.26 (m, 13H), 1.02 (t, J = 7.2 Hz, 2H), 0.90 (d, J = 6.6 Hz, 6H). MS (m/e) (positive FAB) 418.3 (M − Br)+.
2.2. Cells and culture methods
The CF lung epithelial cells IB-3 and AAV-[wildtype]CFTR-repaired IB-3/S9 have been previously described [23,32,33]. Both IB-3 and IB-3/S9 cells were grown in serum-free LHC-8 medium (Biofluids, Bethesda, MD, USA), formulated without gentimycin. HeLa cells were obtained from the ATCC, and cultured in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (100 mg/ml).
2.3. Default conditions and controls for screening paradigm
Initial assays for drug effects in the screening paradigm were performed in duplicate at 10, 3, 1, and 0.3 μM concentrations on IB-3 cells grown in 96-well microtiter plates. Each individual plate contained IB-3 and IB-3/S9 cells to set the boundary conditions of the assay. “Hits” in the screen were chosen on the basis of at least a 50% reduction in constitutive IL-8 secretion. Following a positive retest, candidate compounds were subjected to more detailed analysis under the same assay conditions. Many of the compounds we have screened require organic solvents such as DMSO or ethanol for initial dissolution. In most circumstances we use a maximum final solvent concentration of 0.1% DMSO or 0.1% EtOH. Higher concentrations of DMSO substantially suppress IL-8 secretion. The solvent control, which at the concentration selected minimally deviates from medium alone, is then used as a basis for 100% activity.
2.4. Detection of IL-8
The details of this assay have been described in a previous publication [22]. Briefly, IB-3 cells were grown in 96-well microtiter plates to 80% confluency. Drugs, diluted in LHC-8 medium at the given concentrations, were added, and the cells incubated for an additional 24 h. To initiate the experiment, cells were washed with fresh LHC-8 medium, and then incubated for an additional 16 h in the same medium supplemented with drug. At the end of the time period, the supernatant solutions were collected and assayed for IL-8 by an ELISA assay. Alternatively, the samples were immediately frozen at −80 °C. No quantitative differences have been noted when both paradigms were applied to the same sample handled in either manner. The IL-8 ELISA assay was assembled from bulk materials purchased from R&D (Boston, MA, USA), and performed exactly according to the manufacturer’s specifications. To detect possible drug toxicity on cells, as well as normalize to total cells per well, we measured the double stranded DNA content of cells attached to the plate after removal of supernatant solutions for IL-8 assay. Cells remaining on the plate were then fixed with 10% formalin for 30 min at room temperature before being incubated with propidium iodide (kit from Boehringer Mannheim). Propidium iodide content in each well was then measured with an automated fluorescence plate reader (FLUOstar Optima, MBG LabTechnologies, Durham, NC, USA). Final IL-8 data were calculated as a ratio of secreted IL-8 to DNA for each well. On occasion, cell proliferation assays were also carried out using the MTT assay (Roche, Basel, Switzerland).
2.5. Western blot analysis
HeLa cells, IB3-1 and IB3-1/S9 cells were pretreated with MRS compounds, as described above, and the experiments initiated by incubation with TNFα (R&D Systems) for 15 min. Cells were washed with PBS, and then collected and lysed in M2 buffer (20 mM pH 7.0 Tris, 0.5% NP-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerol phosphate, 1 mM sodium vanadate, 1 mg/ml of leupeptin). Twenty micrograms of the celllysate from each sample were fractionated by SDS-PAGE and immunoblotted. The blots were visualized with chemiluminescent substrate (Pierce).
2.6. Data analysis
IC50 values obtained in IL-8 radioimmunoassays were calculated using the GraphPad software package (GraphPad, San Diego, CA). All concentration-effect curves were repeated in at least three separate experiments, carried out in duplicate or triplicate, and the data normalized to propidium iodide binding by DNA in the adherent cells to correct for cell recovery.
3. Results
3.1. Amphiphilic pyridinium salts are inhibitors of CF IL-8 production
Employing a systematic paradigm designed for library screening, we identified an amphiphilic pyridinium salt that suppressed constitutive hypersecretion of IL-8 from CF lung epithelial IB3-1 cells. The strategy behind assembling this library was to construct binary species with molecules having history of being both biocompatible and pharmacologically active. The two species were then connected by a hydrophobic carbon chain, in an approach similar to that we have previously used to enhance affinity in muscarinic acetylcholine receptor antagonists [34]. Based on an initial “hit”, we synthesized 21 structural derivatives, hoping to increase the potency. The structures for the pyridinium derivatives 1–22 are shown in Table 1, along side their potencies in the IL-8 suppression assay. The derivatives consisted of a hydrophobic ester moiety and a hydrophilic pyridinium moiety linked through an alkyl chain of 4–10 carbons (see compounds 1–4). The chemical strategies for this set of syntheses are summarized in Fig. 1, and are labeled as schemes A–C. The structure with the currently optimum activity is compound 3, MRS2481, with an IC50 of ca. 1.8 μM.
Table 1.
Structures of pyridinium compounds prepared for testing in the IL-8 assay
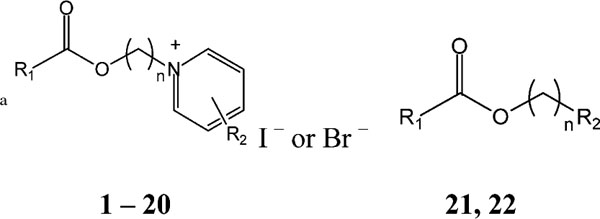
| |||
|---|---|---|---|
| R1 | Compound | n | IC50 |
| Unsubstituted pyridinium salts (R2 = H) | |||
|
1 | 4 | >30 |
| 2 | 6 | >30 | |
| 3, MRS 2481 | 8 | 1.81 ± 0.58 | |
| 4 | 10 | 2.52 ± 0.39 | |
|
5, MRS 2485 | 8 | >25 |
|
6 | 8 | >30 |
|
7 | 8 | 12 ± 0.8 |
|
8 | 8 | 3.16 ± 0.52 |
|
9 | 8 | >30 |
|
10 | 8 | >30 |
|
11 | 8 | >30 |
|
12 | 8 | >30 |
|
13 | 8 | >30 |
|
14 | 8 | Toxic at 1 μM |
|
15 | 8 | 2.2 ± 0.8 |
|
16 | 8 | 4.6 ± 0.9 |
|
17 | 8 | >30 |
| R1 | Compound | R2 | IC50 |
| Substituted pyridinium salts and other derivatives, n = 8 | |||
|
18 | 3-CONH2 | 5.56 ± 0.98 |
|
19 | p-(CH2)2CH3 | 3.3 ± 0.5 |
| 20 | p-(CH2)2-OH | 18 ± 0.9 | |
| 21 |
|
24 ± 1.0 | |
| 22 |
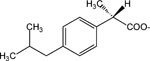
|
>30 | |
The IC50 determinations were performed in duplicate at concentrations ranging from 0.3 to 10 μM on IB-3 cells grown in 96-well microtiter plates (n = 3, or more). The solvent control containing EtOH or DMSO for initial dissolution of the compounds, which at the concentration selected minimally deviates from medium alone, is then used as a basis for 100% activity.
Fig. 1.
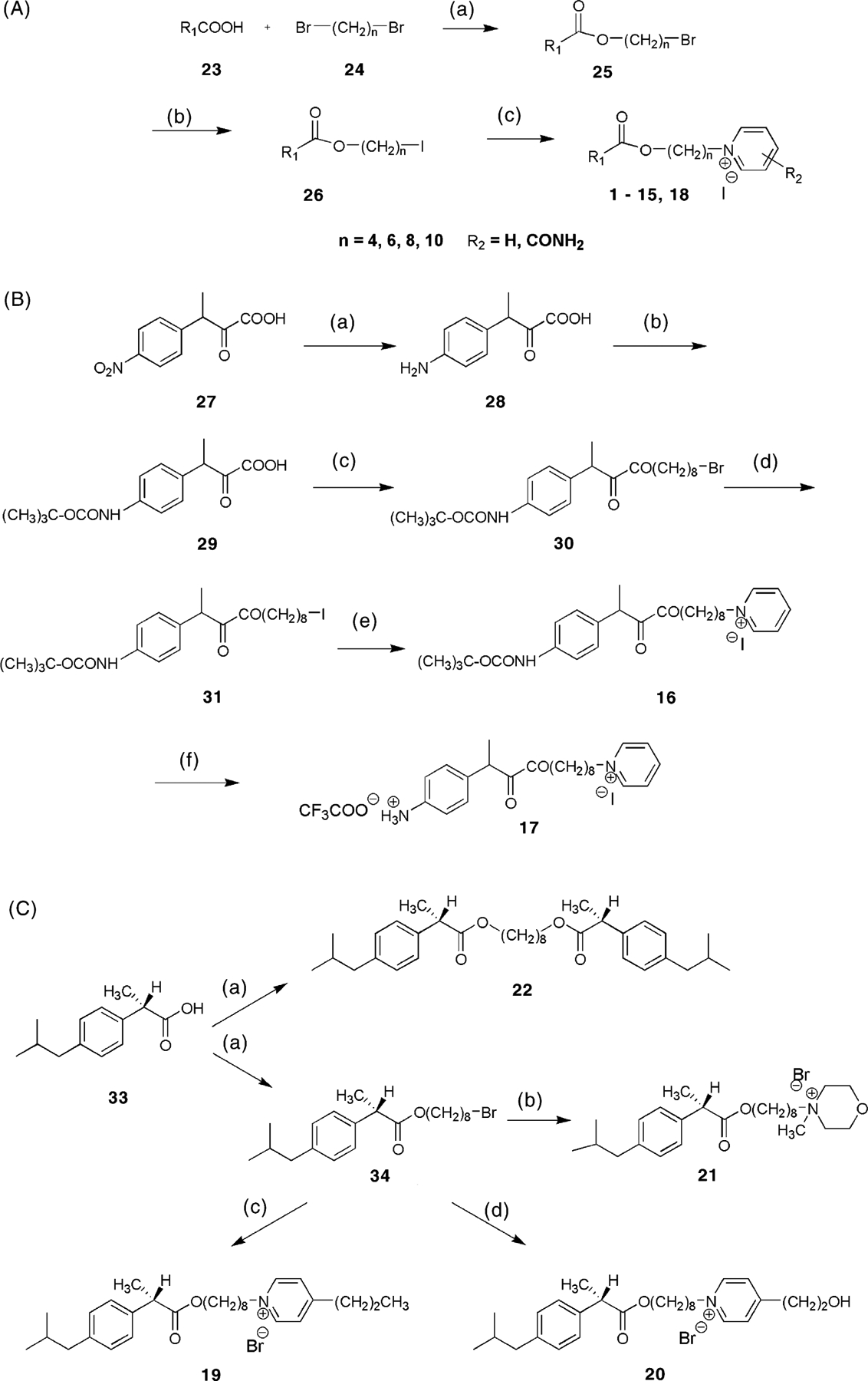
Chemical strategies for syntheses of MRS2481 and analogs. (A) Preparation of analogues of variable spacer chain lengths and variable substitution of the pyridinium moiety. For the structure of R1, refer to Table 1. Reagents: (a) benzyltrimethylammonium hydroxide, tetrabutylammonium iodide at r.t. for 2 days; (b) sodium iodide in acetone; (c) pyridine derivative in acetone at 50 °C for 3 days. (B) Preparation of compound 17, which contains a chemically reactive aryl amino group. Reagents: (a) tin, acetic acid, HCl at 100 °C for 1.5 h; (b) di-t-butyl-dicarbonate in methanol at 45 °C for 1 h; (c) benzyltrimethylammonium hydroxide, tetrabutylammonium iodide at r.t. for 2 days; (d) sodium iodide in acetone; (e) pyridine in acetone at 50 °C for 3 days; (f) trifuoroacetic acid. (C) Preparation of 4-substituted pyridinium salts 19 and 20 and non-pyridinium derivatives, including the symmetric diester 22. Reagents: (a) 1,8-dibromooctane, benzyltrimethylammonium hydroxide, tetrabutylammonium iodide at r.t. for 3 days; (b) N-methylmorpholine, tetrabutylammonium iodide in acetone at 50 °C for 2 days; (c) 4-propyl-pyridine, tetrabutylammonium iodide in acetone at 50 °C for 3 days; (d) 4-(2-hydroxyethyl)-pyridine, tetrabutylammonium iodide in acetone at 50 °C for 3 days.
Modification of the ester group of 9, leading to 10, failed to restore the activity. Removal of two carbons at the α-position of 3, to result in 11, eliminated activity of the molecule. The stereoselectivity of the effects of two enantiomeric 1-hydroxy-2-phenylethyl derivatives, i.e. 12 and 13, could not be assessed, since no activity was evident. Substitution at the p-position of the phenyl ring with a branched alkyl group resulted in the pure S-enantiomer 15, which was roughly equipotent to MRS2481. The corresponding racemic derivative 14 was toxic to cells in the relevant concentration range. Thus its effects on IL-8 could not be assessed. It is likely that the toxicity originates in the R-enantiomer, which was not prepared. The substitution of the p-position of the phenyl ring in two racemic derivatives indicated that the introduction of a urethane group in 16 was possible with retention of the activity, while the small hydrophilic p-amino group in 17 eliminated activity.
Finally, addition of a p-substituent of the pyridinium moiety of 15 led to either retention of potency (n-propyl 19) or an eight-fold loss of potency (2-hydroxyethyl 20) relative to 15. The pyridinium moiety could be replaced with an N-methylmorpholino moiety in 21, with only an 11-fold loss of potency in comparison to 15, while the replacement with an uncharged moiety identical to the ester side of the molecule to give a dimeric structure in 22 eliminated the activity. Thus, the activity is associated with the presence of a positively charged ammonium group.
3.2. The optimum chain length between pyridinium and hydrophobic moieties is eight carbons
The original decision to link the pyridinium and hydrophobic moieties by an eight-carbon chain was based on previous experience. However, we decided to test the contribution of this chain to potency by varying the chain length. As shown in Fig. 2, we tested molecules with four different chain lengths. These included 1 (n = 4); 2 (n = 6); 3 (n = 8); and 4 (n = 10). The data show that the eight-carbon species (MRS2481) is slightly more potent than the 10-carbon species 4. However, the six-carbon species 2 is much less active, and the four-carbon species 1 is even less active than the six-carbon species. We summarize these data as follows: [C8 > C10 ≫ C6 > C4]. Thus the choice of an n-octyl linker is optimal.
Fig. 2.
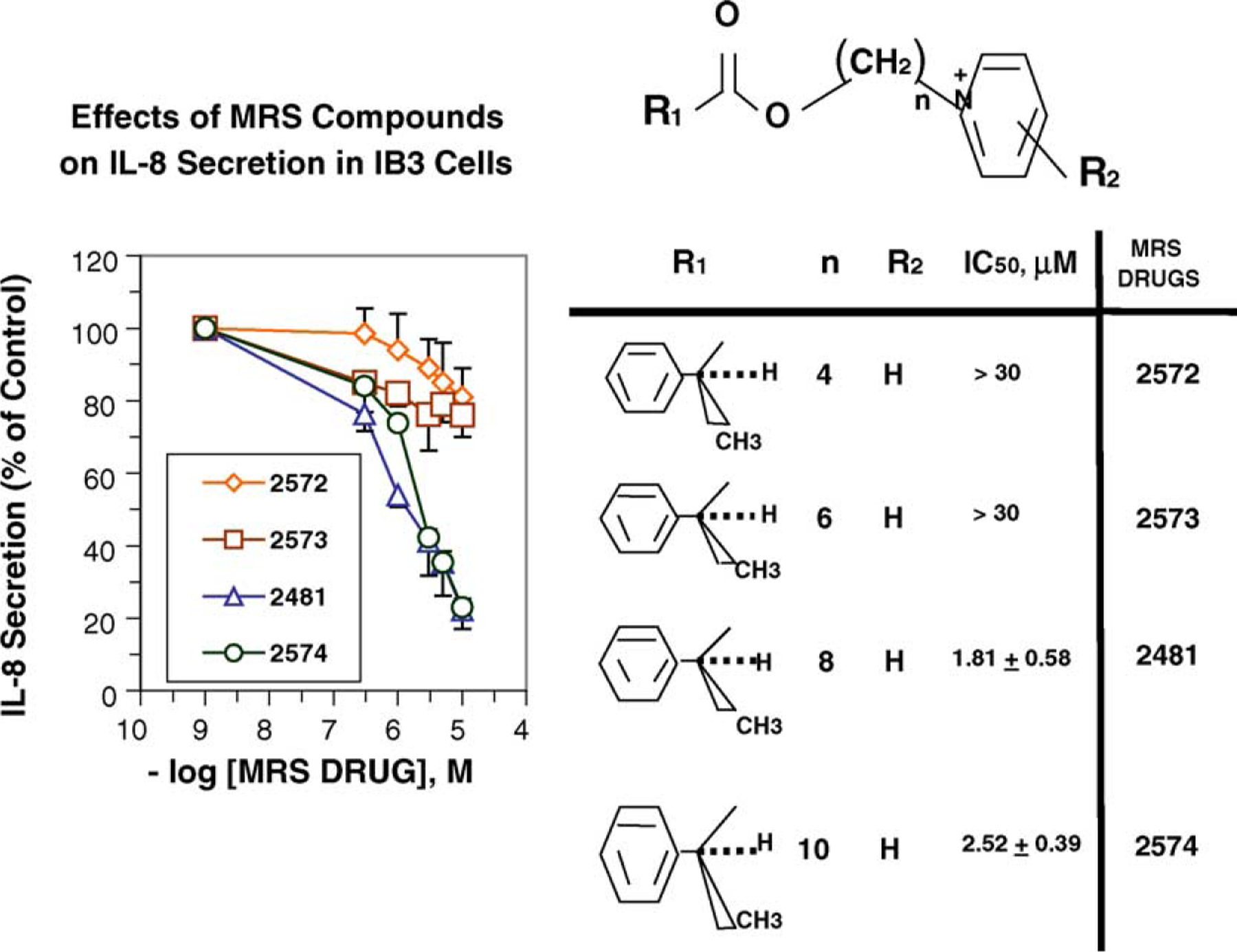
Optimum chain length between pyridinium and hydrophobic moieties of MRS2481. The carbon chain length was varied systematically between 4 and 10 carbons. Based on IC50 values, an eight-carbon chain length is optimal.
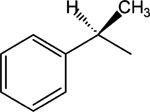
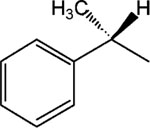
3.3. The (R)-enantiomer of MRS2481 is much more active than the (S)-enantiomer
We noted that the optimal species 3 (MRS2481) had an asymmetric carbon in the R1 moiety (i.e., an (R)-1-phenylpropionic acid ester). To test whether this asymmetric carbon might have importance for the function of the compound we changed the group to an (S)-1-phenylpropionic acid ester (see 5, MRS2485). A titration of both MRS2481 and MRS2485 is shown in Fig. 3. As summarized in Table 1, this change leads to a profound reduction in the apparent IC50, from ca. 1 μM to >25 μM. We also replaced the methyl group with an −H, thereby eliminating the chiral center. This step, as indicated by molecule 7, led to a six-fold loss of potency. We therefore conclude that this chiral group is critical for interacting with a site somewhere in the CF cell that regulates the expression of IL-8.
Fig. 3.
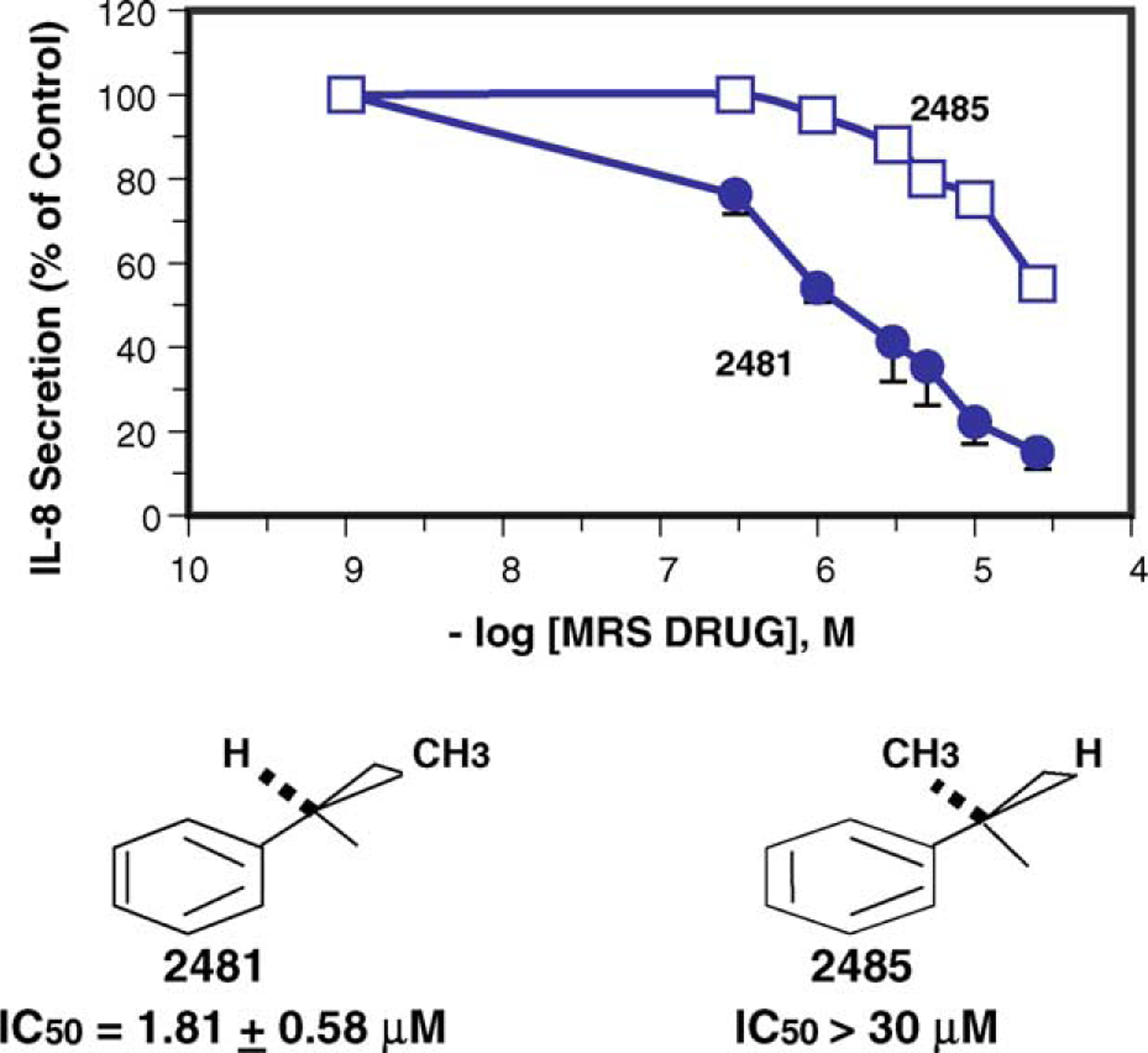
Activity comparison of (R)- and (S)-enantiomers of MRS2481. The (R)-enantiomer, MRS2481, and the (S)-enantiomer, MRS2485, were synthesized and the IL-8 suppression activity titrated.
3.4. Potency is affected by the size and polarity of the substituent on the asymmetric carbon
To further probe the significance of the structures around the asymmetric carbon, we questioned whether the size or polarity of the carbon at this site had any consequences for potency or efficacy. The optimum structure, 3, MRS2481, has a methyl group on an asymmetric carbon in the hydrophobic domain, and proceeded to substitute this methyl group with either an −OCH3 (viz, 9) or an − C2H5 (viz, 8). As shown in Fig. 4, increasing the size of the substituent from one carbon to two carbons increases the IC50 by ca. two-fold. By contrast substituting the methyl with −OCH3 profoundly reduces potency. We conclude that both the orientation and the polarity of the organic substituent on the asymmetric carbon significantly affects the potency of the compound.
Fig. 4.
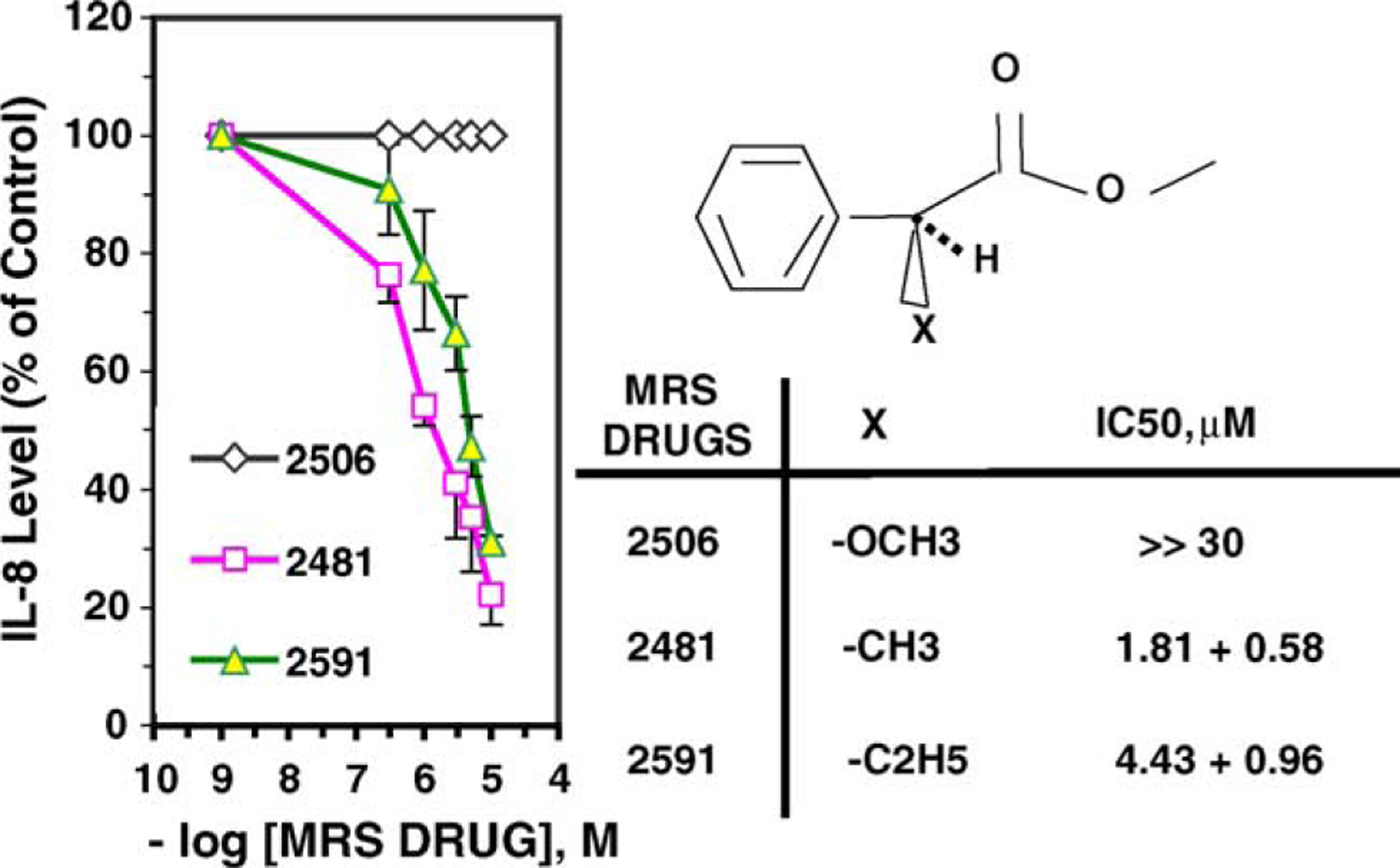
Effect of size and polarity of the substituent at the asymmetric carbon on the activity of MRS2481 and its analogues. The methyl substituent on the optically active carbon of MRS2481 was replaced with a more hydrophobic ethyl group (MRS2591) or a more polar O-Me (MRS2506). The latter substitution abolished activity
3.5. MRS2481 inhibits TNFα-dependent NFκB signaling in CF and control cells
We were much impressed by that fact that the size, polarity and orientation around the asymmetric carbon in MRS2481 had such influence on the potency of the compound. Such a fact often indicates that a compound with these properties might have a specific site in the cell upon which to bind and act. We have hypothesized in previous reports that constitutive hyperexpression of IL-8 in CF lung epithelial cells was due to a dysfunctional TNFα/NFκB signaling pathway [22]. We therefore decided to test whether MRS2481 might inhibit NFκB signaling in CF and control cells. As shown in Fig. 5a, HeLa cells express substantial amounts of IκBα. This means that NFκB is not activated under control conditions. As expected, when these cells are exposed to TNFα, the IκBα is vastly reduced, and NFκB is activated. However, when the cells are preincubated with 5 μM MRS2481 for up to 6 h, there is a modest protection when TNFα is added (see Fig. 5b). With the less active enantiomeric species, MRS2485, one can get some protection as well, except that 50 μM rather than 5 μM is needed to obtain a comparable protective effect. These data are consistent with the differential potency of these two species previously noted for suppression of IL-8 from CF cells.
Fig. 5.
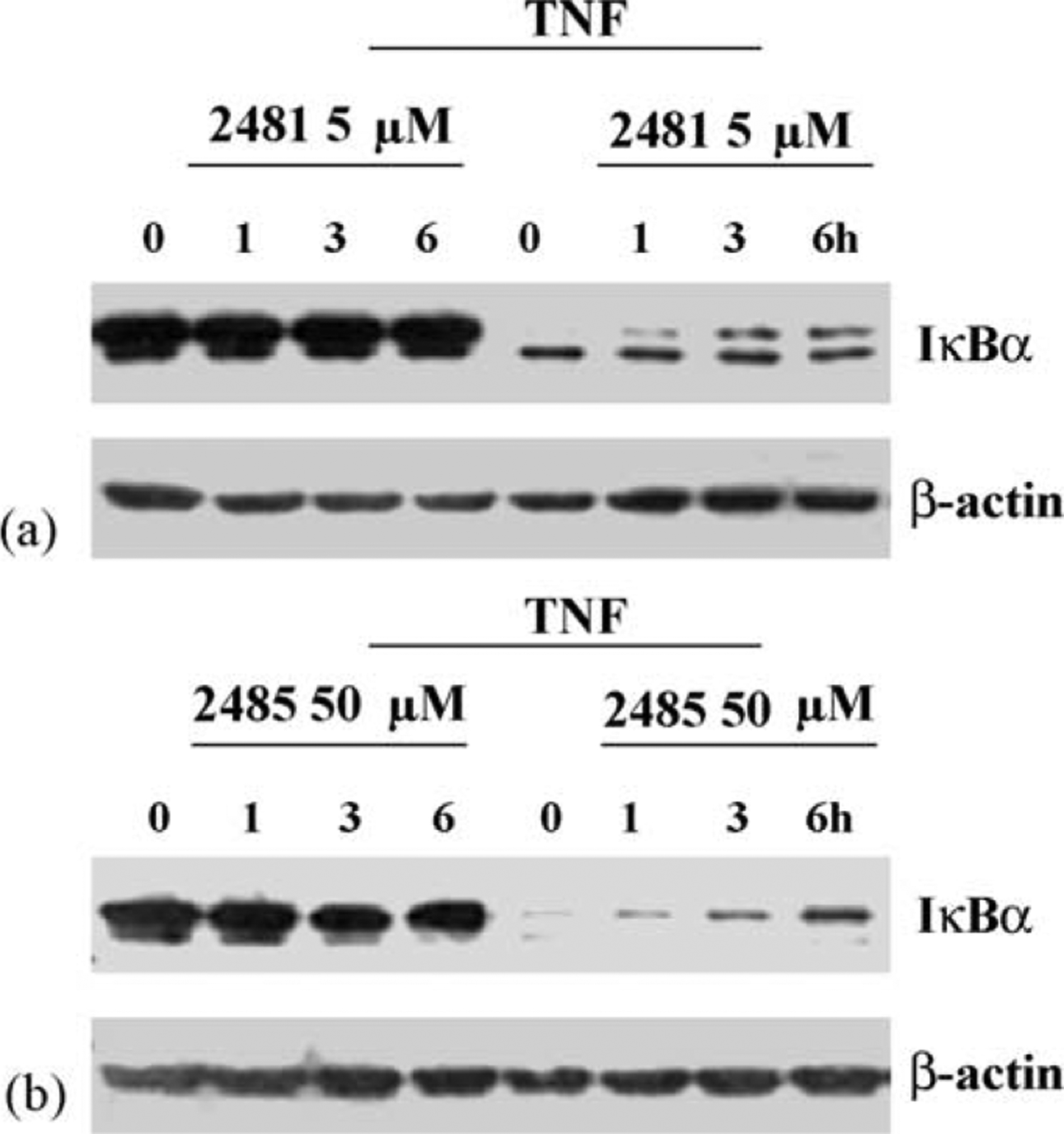
Influence of MRS2481 on TNFα-induced IκBα degradation in HeLa cells by MRS2481. (a) HeLa cells were pretreated with MRS2481 (5 μM) for 0, 1, 3, or 6 h, and then treated with 2 ng/ml TNFα for 15 min. Reaction mixtures were then subjected to polyacrylamide gel electrophoresis, blotted onto nitrocellulose, and probed with anti-IκBa antibody. (b) HeLa cells were pretreated with the less potent analogue MRS2485 (50 μM) for 0, 1, 3, or 6 h, and then treated with 2 ng/ml TNFα for 15 min. Reaction mixtures were then subjected to polyacrylamide gel electrophoresis, blotted onto nitrocellulose, and probed with anti-IκBα antibody.
A qualitatively similar differential effect of these two compounds is also seen when IB3-1 or IB3-1/S9 cells are tested in the same way. As shown in Fig. 6a, preincubation of IB3-1 cells with 5 μM MRS2481 is more effective than an equivalent amount of MRS2485 at protecting IκBα from TNFα-induced destruction. However, the distinction is lost when cells are preincubated with 10 μM of each compound. As shown in Fig. 7a, an equivalent effect is also seen in CFTR-repaired IB3-1/S9 cells when the cells are pretreated with 5 μM or 10 μM of each compound. The data with the CF and repaired cells are particularly consistent with the differential potency noted in these same cells by the two compounds.
Fig. 6.
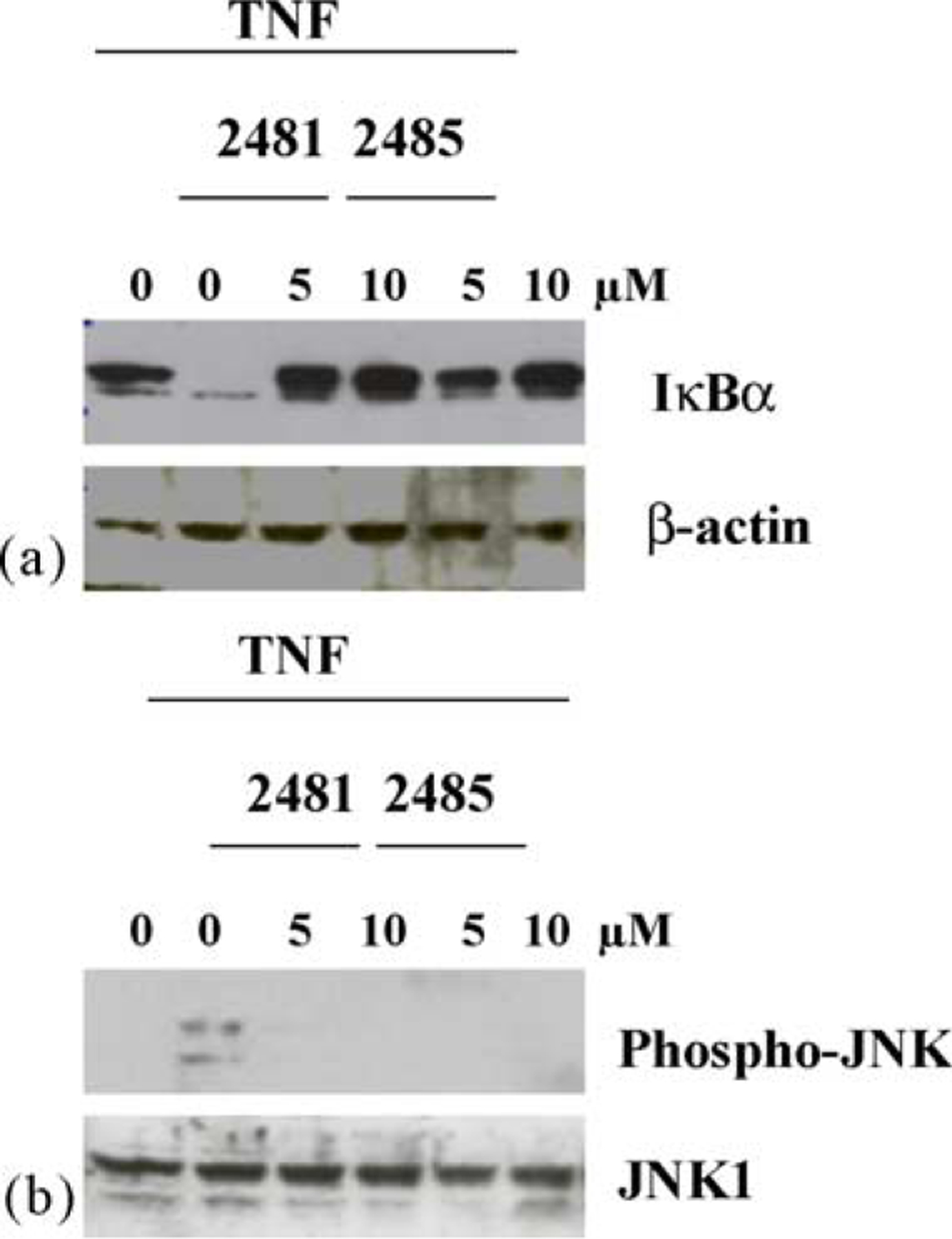
Influence of MRS2481 on TNFα-induced IκBα degradation and JNK phosphorylation in CF IB3-1 cells. (a) IB3-1 cells were pretreated with either 5 or 10 μM MRS2481, or the less potent analogue MRS2485 for 6 h. The cells were then treated with 10 ng/ml TNFα for 15 min. Reaction mixtures were then subjected to polyacrylamide gel electrophoresis, blotted onto nitrocellulose, and probed with anti-IκBα antibody. The left-most “0” is an untreated condition. The inner “0” is upon the addition of TNFα only. (b) IB3-1 cells were pretreated with either 5 or 10 μM MRS2481, or the less potent analogue MRS2485 for 6 h. The cells were then treated with 10 ng/ ml TNFα for 15 min. Reaction mixtures were then subjected to polyacrylamide gel electrophoresis, blotted onto nitrocellulose, and probed with anti-phospho-JNK or anti-JNK1 antibodies. The left-most “0” is an untreated condition. The inner “0” is upon the addition of TNFα only.
Fig. 7.
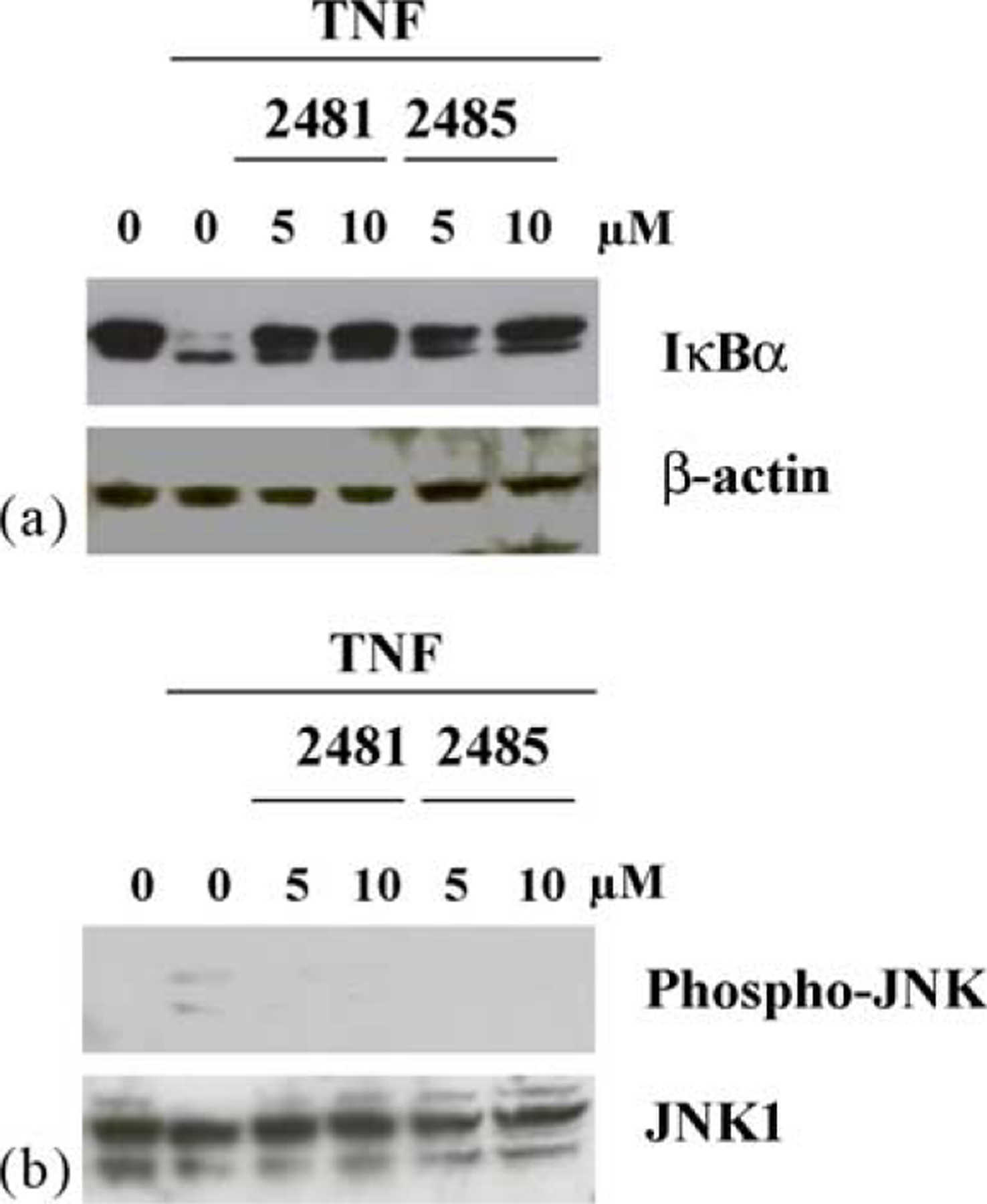
Influence of MRS2481 on TNFα-induced IκBα degradation and JNK phosphorylation in repaired IB3-1/S9 cells. (a) IB3-1/S9 cells were pretreated with either 5 or 10 μM MRS2481, or the less potent analogue MRS2485 for 6 h. The cells were then treated with 10 ng/ml TNFα for 15 min. Reaction mixtures were then subjected to polyacrylamide gel electrophoresis, blotted onto nitrocellulose, and probed with anti-IκBα antibody. The left-most “0” is an untreated condition. The inner “0” is upon the addition of TNFα only. (b) IB3-1/S9 cells were pretreated with either 5 or 10 μM MRS2481, or the less potent analogue MRS2485 for 6 h. The cells were then treated with 10 ng/ml TNFα for 15 min. Reaction mixtures were then subjected to polyacrylamide gel electrophoresis, blotted onto nitrocellulose, and probed with anti-phospho-JNK or anti-JNK1 antibodies. The left-most “0” is an untreated condition. The inner “0” is upon the addition of TNFα only.
3.6. MRS2481 inhibits TNFα-dependent activation of AP-1 signaling
The IL-8 promoter is driven by both NFκB and AP-1 signaling, and we therefore decided to test the AP-1 signaling system for sensitivity to MRS2481. The proximal step in AP-1 signaling is based on phosphorylation of Jun kinase (JNK1) and subsequent phosphorylation of c-Jun by the activated phospho-JNK. As shown in Fig. 6b, IB3-1 cells under control conditions show very little evidence of JNK phosphorylation, indicating that AP-1 signaling is not activated. However, when TNFα is added phospho-JNK is made from JNK, and the AP-1 site is activated. The data show that MRS2481 and MRS2485, both at 5 and 10 μM, suppress this reaction equivalently. Equivalent data are shown in Fig. 7b for IB3-1/S9 cells treated in the same manner. Thus the AP-1 signaling pathway appears to show less discrimination between the two enantiomeric species.
4. Discussion
We report here the discovery of a water-soluble, potent and efficacious compound, MRS2481, which has the property of suppressing the constitutitive hypersecretion of IL-8 from cystic fibrosis lung epithelial cells. MRS2481 is an amphiphilic pyridinium salt, connected by an eight-carbon chain to an (R)-1-phenylpropionic acid ester. The individual components of this compound are inactive in the assay at concentrations as high as 25–30 μM, and the eight-carbon chain connecting the two principal components is absolutely required for optimal potency. We have pioneered the concept of increasing drug potency by the functionalized congener approach [34], and pyridinium salts have historically been used to create water-soluble pro-drugs of otherwise less soluble compounds [35]. However, we find that the positive charge on the pyridinium moeity is absolutely required for activity. In addition, we also find that an enantiomer of the lead compound, MRS2485, containing an (S)-1-phenyl propionic acid ester, exhibits greatly reduces IL-8 suppression activity in CF cells. These results indicate that the more potent enantiomer, MRS2481, appears to require all aspects of the structure for its primary action, and that it very likely binds to a specific site within the cell.
4.1. Mechanism of action of MRS2481
Consistently, the mechanism of action of MRS2481 involves the inhibition of at least two signaling pathways, NFκB and AP-1. Both of these pathways are known to be potent drivers of the IL-8 promoter [12,13]. Our data indicate that in both HeLa cells and CF IB3-1 cells, MRS2481 blocks the TNFα-activated destruction of IκBα. When IκBα is phosphorylated by the IKKα,β,γ complex, the inactive cytosolic localization of the NFκB p65/NFκB p50 is lost, and the dimer translocates to the nucleus. Once in the nucleus the NFkB dimer can bind to the κB site on the IL-8 promoter, and can become a powerful driver of IL-8 expression.
Our data also indicate that MRS2481 blocks the TNFα-activated phosphorylation of JNK. Phosphorylation of JNK activates the kinase activity to phosphorylate c-JUN. Phospho-cJUN forms a dimer with phospho-c-FOS, which binds to the AP-1 site on the IL-8 promoter, thereby also driving IL-8 expression. TNFα is known to drive both NFκB and AP-1 activation. Since MRS2481 blocks both pathways, we suggest that the site of MRS2481 action mustlie upstream of or near the NFκB/AP-1 signaling bifurcation. The exact site of MRS2481 action in this pathway is still not known, but is under active investigation in our laboratory.
4.2. Classification of IL-8 suppression candidate CF drugs
There now appear to be at least two classes of candidate CF drugs that suppress the constitutive hypersecretion of IL-8 from CF lung epithelial cells. Class C (for CPX) compounds are typified by the xanthine CPX [22]. CPX, and the more potent homologue DAX, binds to the first nucleotide binding-domain of CFTR (viz, NBD1/NBF1) near the mutant ΔF508 site [19]. The folding defect is apparently rescued, thereby permitting efficient trafficking of the mutant protein to the plasma membrane. The rescued mutant CFTR now functions to both transport chloride and suppress IL-8 expression. We have hypothesized that both rescued mutant CFTR and [wildtype]CFTR suppress IL-8 expression by similar mechanisms [22]. In fact, genomic analysis has shown that both CPX and [wildtype]CFTR act by suppressing the TNFα/NFκB pathway [22]. However, it is not yet clear whether CPX only suppresses IL-8 production by virtue of its CFTR trafficking rescue property. In addition, it is not yet known to what extent other trafficking rescue/corrector compounds may also suppress IL-8 expression. These other compounds include the less active xanthines such as IBMX (ka = ca. 1 mM: Ref. [24]); flavones such as genistein (ka = ca. 50–75 μM: Ref. [25,26]); benzo[c]quinolizium (ka = ca. 50–75 μM; Ref.27); phenylbutyrate (ka = ca. 1 mM: Ref. [28]); and the benzothiazines (ka = ca. 0.5 μM: Ref. [29]).
Class D (for digitoxin) compounds are typified by digitoxin [23]. These compounds have the most modest effects, if any, on rescue of mutant CFTR trafficking. Yet digitoxin profoundly suppresses IL-8 hypersecretion from CF lung epithelial cells. Into the latter class we can now place the new candidate CF drug MRS2481. Both digitoxin and MRS2481 act by suppressing TNFα/NFκB signaling. However ongoing comparative experiments indicate that they appear to act at different sites on this complex pathway. The fact that the sites of action may be different suggests that there are multiple approaches available for cytokine suppression in CF. This is important because some of these compounds may function as site-specific mimics for CFTR suppression of inflammation without actually affecting rescue of the mutant CFTR trafficking defect per se. Details of the exact locations of action of these two compounds are presently being studied.
4.3. Conclusion
IL-8 is the most powerful known physiological attractant for inflammatory cells, and the importance of this discovery lies in the fact that the lung pathophysiology of cysticfibrosis is based substantially on an intrinsically inflammatory phenotype in the CF airway. Thus, the ability of MRS2481, and other candidate drugs with this property, to potently and efficaciously suppress constitutive hypersecretion of IL-8 secretion from the CF airway epithelial cells carries the promise of a strategy for the development of a potential CF therapeutic. Preliminary exploration of the mechanism of action of these amphiphilic pyridinium salts implicates both NFκB and AP-1 pathways, which are known to control IL-8 gene transcription. The possible usefulness of this class of compounds for other disorders of baseline IL-8 expression and resultant inflammation remain to be investigated.
Acknowledgements
Drs. Tchilibon, Zhang and Yang should be considered joint first authors on this paper. This work was supported by a grant from the Cystic Fibrosis Foundation, Bethesda, MD (to HBP), and by the NIH (RO1-DK53051, to HBP; NO1-HV-28187, to HBP). Mass spectral measurements were carried out by Dr. Victor Livengood, and NMR measurements by Wesley White (NIDDK). We thank Heng Duong (NIDDK) for proofreading the manuscript.
References
- [1].Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev 1999;79:S215–55. [DOI] [PubMed] [Google Scholar]
- [2].Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic fibrosis. In: The metabolic and molecular bases of inherited diseases (Scriver CL, Beaudet AL, Valle D, Sly WS, eds; Childs B, Kinzler KW, Vogelstein B, Assoc Eds) 2001; eighth ed. 5121–5188, McGraw-Hill, New York. [Google Scholar]
- [3].Dean TP, Dai Y, Shute JK, Church MK, Warner JO. Interleukin-8 concentrations are elevated in bronchoalveolar lavage, sputum, and sera of children with cystic fibrosis. Pediatr Res 1993;34:159–61. [DOI] [PubMed] [Google Scholar]
- [4].Richman-Eisenstat JB, Jorens PG, Hebert CA, Ueki I, Nadel JA. Interleukin-8: an important chemoattractant in sputum of patients with chronic inflammatory airway diseases. Am J Physiol 1993; 264:L413–8. [DOI] [PubMed] [Google Scholar]
- [5].Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutierrez JP, Hull J, et al. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med 1997;156:1197–204. [DOI] [PubMed] [Google Scholar]
- [6].Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995;151:1075–82. [DOI] [PubMed] [Google Scholar]
- [7].DiMango E, Ratner AJ, Bryan R, Tabibi S, Prince A. Activation of NF-kappaB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest 1998;101:2598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Care Med 1995;152:2111–8. [DOI] [PubMed] [Google Scholar]
- [9].Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol 1995;13:257–61. [DOI] [PubMed] [Google Scholar]
- [10].Tirouvanziam R, de Bentzmann S, Hubeau C, Hinnrasky J, Jacquot J, Peault B, et al. Inflammation and infection in naive human cystic fibrosis airway grafts. Am J Respir Cell Mol Biol 2000;23:121–7. [DOI] [PubMed] [Google Scholar]
- [11].Cruse JM, Lewis RE. Illustrated dictionary of immunology. Boca Raton: CRC Press, 1995, Appendix 3. [Google Scholar]
- [12].Roebuck KA. Regulation of interleukin-8 gene expression. J Interferon Cytokine Res 1999;19:429–38. [DOI] [PubMed] [Google Scholar]
- [13].Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol 2002;72: 847–55. [PubMed] [Google Scholar]
- [14].Cheng SH, Gregory RJ, Marshall J, Souza DW, White GA, O’Riordan CR, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990;63:827–34. [DOI] [PubMed] [Google Scholar]
- [15].Eidelman O, Guay-Broder C, van Galen PJM. Jacobson KA, Turner RJ, Cabantchik ZI, et al. A1-adenosine antagonists activate chloride efflux from cystic fibrosis cells. Proc Nat Acad Sci USA 1992;89:5562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guay-Broder C, Jacobson KA, BarNoy S, Cabantchik ZI, Guggino WB, Zeitlin PL, et al. Al-Receptor antagonist 8-cyclopentyl-1,3-dipropyl-xanthine (CPX) selectively activates chloride efflux from human epithelial and mouse fibroblast cell lines expressing the CFTR (ΔF508) mutation, but not the wild type CFTR. Biochemistry 1995;34:9079–87. [DOI] [PubMed] [Google Scholar]
- [17].Jacobson KA, Guay-Broder C, van Galen PJM. Gallo-Rodriguez C, Melman N, Jacobson MA, et al. Stimulation of alkylxanthines of chloride efflux from CFPAC-1 cells does not involve A1-adenosine receptors. Biochemistry 1995;34:9088–94. [DOI] [PubMed] [Google Scholar]
- [18].Haws CM, Nepomuceno I, Krause ME, Wakelen H, Law T, Xia Y, et al. ΔF508-CFTR channels: kinetics, activation by forskolin, and potentiation by xanthines. Am J Physiol 1996;270:C1544–55. [DOI] [PubMed] [Google Scholar]
- [19].Cohen BE, Lee G, Jacobson KA, Kim Y-C, Huang Z, Sorscher EJ, et al. CPX (1,3-dipropyl-8-cyclopentyl xanthine) and other alkylxanthines differentially bind to the wild type and ΔF508 mutant first nucleotide binding fold (NBF-1) domains of the cystic fibrosis transmembrane conductance regulator (CFTR). Biochemistry 1997;36: 6455–61. [DOI] [PubMed] [Google Scholar]
- [20].Arispe N, Ma J, Jacobson KA, Pollard HB. Direct activation of cystic fibrosis transmembrane conductance regulator by 8-cyclopentyl-1,3-dipropylxanthine and 1,3-diallyl-8-cyclohexylxanthine. J Biol Chem 1998;273:5724–34. [DOI] [PubMed] [Google Scholar]
- [21].McCarty NA, Standaert TA, Teresi M, Tuthill C, Launspach J, Kelley TJ, et al. A phase I randomized, multicenter trial of CPX in adult subjects with mild cystic fibrosis. Pediatr Pulmonol 2002;33:90–8. [DOI] [PubMed] [Google Scholar]
- [22].Eidelman O, Srivastava M, Zhang J, Murthy J, Heldman E, Jacobson KA, et al. Genes from the TNFαR/NFκB pathway control the proinflammatory state in cystic fibrosis epithelial cells. Mol Med 2001;7:523–34. [PMC free article] [PubMed] [Google Scholar]
- [23].Srivastava M, Eidelman O, Zhang J, Paweletz C, Caohuy H, Yang Q-F, et al. Digitoxin mimics CFTR-gene therapy, and suppresses hypersecretion of proinflammatory interleukin-8 (IL-8) from cystic fibrosis lung epithelial cells. Proc Nat Acad Sci USA 2004;101:7693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, et al. Chloride conductance expressed by ΔF508 and other mutant CFTR’s in Xenopus oocytes. Science 1991;254:1797–9. [DOI] [PubMed] [Google Scholar]
- [25].Hwang T-C, Wang F, Yang I, Reenstra WW. Genistein potentiates wild-type and ΔF508-CFTR channel activity. Am J Physiol 1997; 273:C988–98. [DOI] [PubMed] [Google Scholar]
- [26].Andersson C, Servetnyk Z, Roomans GM. Activation of CFTR by genistein in human airway epithelial cell lines. Biochem Biophys Res Commun 2003;308:518–22. [DOI] [PubMed] [Google Scholar]
- [27].Becq F, Mettey Y, Gray MA, Galietta LJ, Dormer RL, Merten M, et al. Development of substituted Benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J Biol Chem 1999;274:27415–25. [DOI] [PubMed] [Google Scholar]
- [28].Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of ΔF508-CFTR. Am J Physiol Cell Physiol 2000;278:C259–67. [DOI] [PubMed] [Google Scholar]
- [29].Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, et al. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem 2003;278:35079–85. [DOI] [PubMed] [Google Scholar]
- [30].Konstan MW, Byard PJ, Hoppel C-L, Davis PB. Effect of high dose ibuprofen in patients with cystic fibrosis. New Eng J Med 1995; 332:848–54. [DOI] [PubMed] [Google Scholar]
- [31].Accurso FJ, Sagel SD, Sontag MK, Wegener JS. Anti-inflammatory endpoints in cystic fibrosis (CF): lessons from clinical trials. Pediatr Pulmonol 2003;S25:104–5. [Google Scholar]
- [32].Zeitlin PL, Lu L, Hwang T-C, Rhim J, Cutting GR, Keiffer KA, et al. A cystic fibrosis bronchial epithelial cell line: immortalization by adeno12-SV40 infection. Am J Respir Cell Mol Biol 1991;4:313–9. [DOI] [PubMed] [Google Scholar]
- [33].Flotte TR, Afione SA, Solow R, Drumm ML, Markakis D, Guggino WB, et al. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J Biol Chem 1993;268:3781–90. [PubMed] [Google Scholar]
- [34].Karton Y, Bradbury BJ, Baumgold J, Paek R, Jacobson KA. Functionalized congener approach to muscarinic antagonists: analogues of pirenzepine. J Med Chem 1991;34:2133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Davidsen SK, Summers JB, Albert DH, Holms JH, Heyman HR, Magoc TJ, et al. N-(Acyloxyalkyl)pyridinium salts as soluble prodrugs of a potent platelet activating factor antagonist. J Med Chem 1994; 37:4423–9. [DOI] [PubMed] [Google Scholar]