

SUMMARY
The exocyst is an evolutionarily conserved protein complex that regulates vesicular trafficking and scaffolds signal transduction. Key upstream components of the exocyst include monomeric RAL GTPases, which help mount cell-autonomous responses to trophic and immunogenic signals. Here, we present a quantitative proteomics-based characterization of dynamic and signal-dependent exocyst protein interactomes. Under viral infection, an Exo84 exocyst subcomplex assembles the immune kinase Protein Kinase R (PKR) together with the Hippo kinase Macrophage Stimulating 1 (MST1). PKR phosphorylates MST1 to activate Hippo signaling and inactivate Yes Associated Protein 1 (YAP1). By contrast, a Sec5 exocyst subcomplex recruits another immune kinase, TANK binding kinase 1 (TBK1), which interacted with and activated mammalian target of rapamycin (mTOR). RALB was necessary and sufficient for induction of Hippo and mTOR signaling through parallel exocyst subcomplex engagement, supporting the cellular response to virus infection and oncogenic signaling. This study highlights RALB-exocyst signaling subcomplexes as mechanisms for the integrated engagement of Hippo and mTOR signaling in cells challenged by viral pathogens or oncogenic signaling.
Graphical abstract
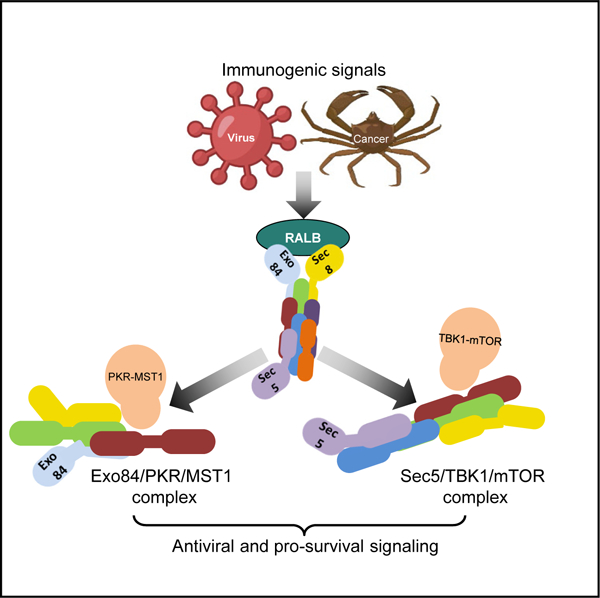
In brief
Zaman et al. demonstrate that distinct exocyst subcomplexes modulate a RALB-mediated antiviral response. The authors identify PKR/MST1 interactions in an Exo84 subcomplex and TBK1/mTOR interactions in a Sec5 subcomplex, leading to engagement of Hippo and mTOR signaling, respectively, in pathogenic contexts, such as virus infection and cancer.
INTRODUCTION
RAS Like Proto-Oncogene A and B (RALA and RALB) are RAS GTPase superfamily small G proteins (Moskalenko et al., 2002). RALA/B harbor various biological functions, including the regulation of targeting and tethering of cellular secretory vesicles via the hetero-octameric protein complex known as the exocyst (Bodemann et al., 2011; Ou et al., 2011; Chien et al., 2006; Torres et al., 2015; Ahmed et al., 2018). Distinct from regulation of membrane trafficking, RAL-exocyst signaling can contribute to stimulus-dependent regulation of growth homeostasis and host defense signaling (Bodemann et al., 2011; Ou et al., 2011; Chien et al., 2006). During trophic and immunogenic signaling, different RAL-exocyst subcomplexes act as molecular scaffolds by coordinating the local concentration of downstream effectors. The RALB/Exo84 subcomplex associates with the UNC-51-like kinase 1 (ULK1) and the Beclin1/Vps34 complex to induce autophagy (Bodemann et al., 2011). A separate RALB/Sec5 subcomplex promotes activation of the innate immune kinase TANK binding kinase 1 (TBK1) to promote cancer cell survival (Ou et al., 2011). The multi-functional nature of the exocyst during both homeostatic and pathologic cellular signaling is specified by the context-specific assembly of sub-stoichiometric signaling scaffolds (Moskalenko et al., 2002, 2003). The extent of the diversity of these subcomplexes and their mechanistic coordination is not well understood, nor is the full role of RALA/B GTPases in subcomplex assembly and function(s).
In response to appropriate trophic and immunogenic stimuli, multicellular and unicellular organisms maintain homeostasis via coordinated adjustment of mass accumulation, proliferation, and survival coupling upstream instructive signals to kinetic, spatial, and dosage regulation of effector signaling pathways (Levine and Kroemer, 2008; Zoncu et al., 2011; Yu et al., 2015). In higher metazoans, mammalian target of rapamycin (mTOR) and Yes Associated Protein 1 (YAP1), a transcription coactivator located downstream of Hippo signaling, are two sentinel signaling effectors responsive to acute perturbation of cellular nutrient and immune status (Ma and Blenis, 2009; Laplante and Sabatini, 2012; Yu et al., 2012; Wang et al., 2017; Zhang et al., 2017). However, upstream events leading to Hippo responsiveness during immunogenic signals remain elusive. Furthermore, context-dependent coordination between mTOR and Hippo signaling is yet to be fully understood.
Here, we demonstrate that activation of RALB by immunogenic stimuli promotes formation of distinct RALB/Exo84 and RALB/Sec5 subcomplexes that result in activation of Hippo pathway signaling and an mTOR-dependent interferon-β (IFN-β) response, respectively.
RESULTS
Dynamic modulation of exocyst subcomplexes in response to host defense pathway activation
Sec8 is a core exocyst subunit that copurifies with the fully assembled hetero-octameric exocyst (i.e., Sec6/8) complex and subcomplexes implicated in the regulation of mTOR, autophagy, and innate immune signaling (Grindstaff et al., 1998; Bodemann et al., 2011; Chien et al., 2006; Bhuvanakantham et al., 2010; Heider et al., 2016). An understanding of the repertoire of proteins forming the exocyst subcomplexes and the connection to subcomplex-specific biological functions, such as nutrient or pathogen response, remains incomplete.
We set out to comprehensively evaluate the control of biological state-selective exocyst complex composition. We first pursued quantitative proteomic characterization of endogenous Sec8 protein interactomes isolated under cell culture conditions that corresponded to physiologically relevant conditions of nutrient challenge or pathogen challenge (Figure S1A). To simulate nutrient-dependent biological states of cellular growth and autophagy, we incubated cells in either standard culture media or in Earle’s balanced saline solution (EBSS) lacking serum and amino acids. Amino acid deprivation in EBSS induced endogenous LC3 protein re-localization from a diffuse cytosolic pattern to a punctate pattern and accumulation of the proteolytically cleaved, active form of the lysosomal protein LC3 (Figures S1B and S1C). Amino acid deprivation induced a cellular starvation response marked by an increase in activating phosphorylation of AMP-activated protein kinase (AMPK) and a decrease in activating phosphorylation of the mTOR substrate p70-S6 kinase 1 (S6K) (Figure S1C). To simulate cellular pathogen-dependent innate immune pathway activation, we used a synthetic uncoated double-stranded RNA (dsRNA) analog, poly(I:C), a potent Toll-like receptor 3 (TLR3) and Rig-I-like receptor (RLR) ligand (Chiu et al., 2009; Kawai and Akira, 2010) or Sendai virus (SeV). RNA sequencing (RNA-seq) analysis of cells challenged with either poly(I:C) and or SeV showed activation of a TBK1 and nuclear factor κB (NF-κB) and IFN-β-mediated transcriptional response (Figures S1D–S1F). Because the poly(I:C) challenge was simple to calibrate and scale, we chose this for initial protein interactome analysis using affinity purification coupled to mass spectrometry (AP-MS) experiments.
In each of two human epithelial cell lines, HEK293T and HeLa, we performed AP multiplex tandem mass tag (TMT) MS with or without amino acid media supplementation and with or without poly(I:C) incubation. Three independent Sec8 monoclonal antibodies and two independent monoclonal control antibodies were used. The resulting candidate interactions were evaluated for selective recovery with Sec8 and altered abundance in Sec8 complexes upon amino acid starvation or poly(I:C) challenge (Figure S1G). Iterative clustering of normalized protein abundance and orthogonal detection by immunoprecipitation (IP)/western blot highlighted 14 proteins that consistently coimmunoprecipitated with Sec8 independently of culture conditions (see STAR Methods for details), which we considered as a constitutive core Sec8 interactome (Figure 1A; Figure S1I). This included a cohort of exocyst subunits (positive controls), the TBK1-Azi2 complex (Chien et al., 2006), the CDK-activating kinase (CAK) complex (CDK7, MNAT1, CCNH), and a phosphoribosyl pyrophosphate synthetase (PRPS) complex (PRPS1, PRPS2, PRPSAP1) (Figures 1A and 1B).
Figure 1. Static and dynamic exocyst complexes.
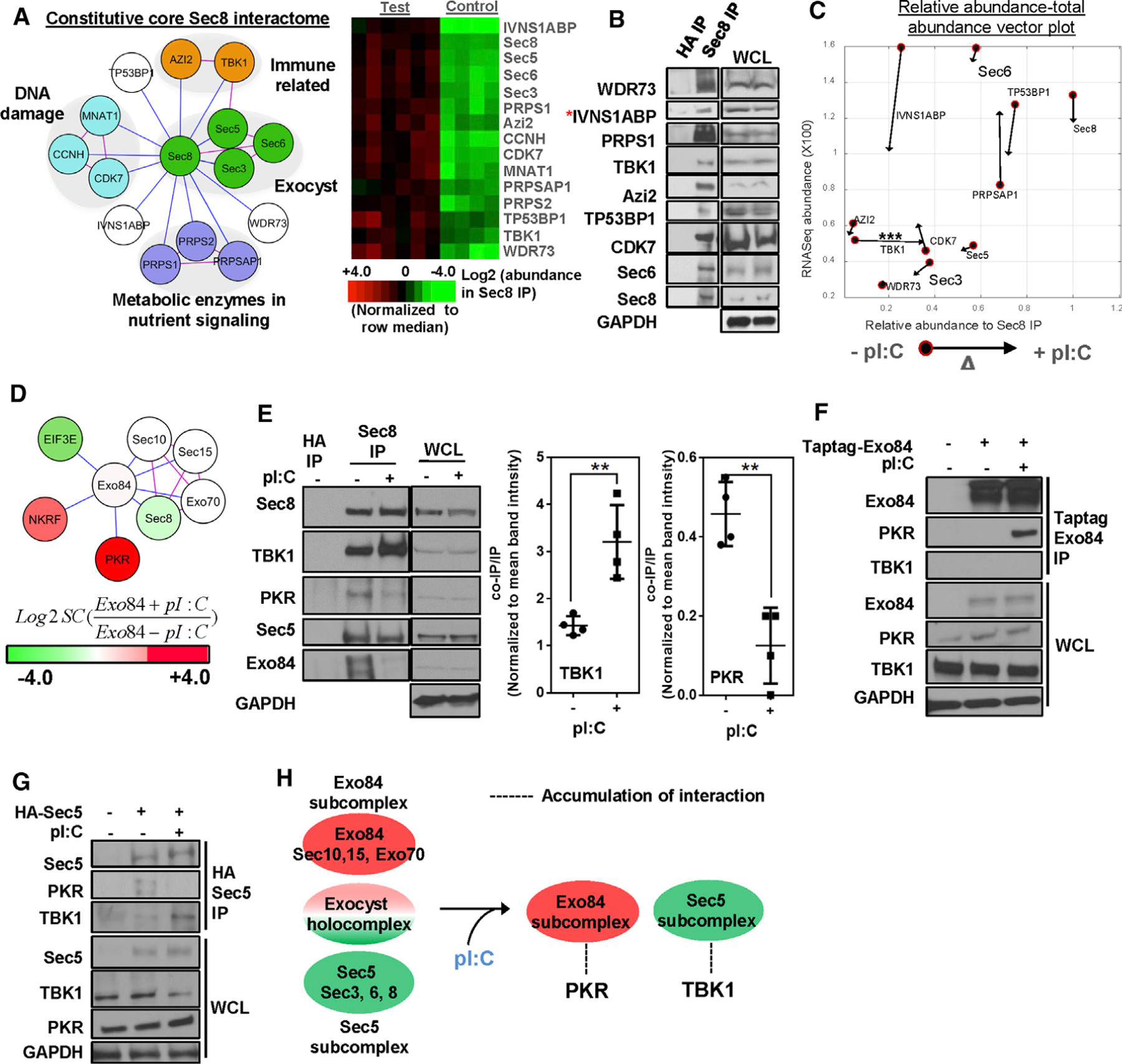
(A) Sec8 endogenous interactome. (Left) Nodes represent proteins, blue edges represent experimentally observed interactions, and pink edges represent known interaction imported from the curated BIOGRID database. Gene functions are labeled as reported in STRING and GO databases. (Right) Heatmaps represent intensity profiles (row median normalized) of the indicated proteins. Test IP: n = 20; control IP: n = 12).
(B) Western blot validation of Sec8 interactome. Asterisk indicates the presence in CRAPOME database (n = 3).
(C) Stimulus-dependent effects on Sec8 interactome. Poly(I:C) challenge-specific relative abundance versus total abundance vector plot of core Sec8 complex members is shown. Filled circles indicate pre-stimulus values, and arrowheads indicate post-stimulus values. Statistical significance for TBK1 enrichment is indicated (***p < 0.005, unpaired Student’s t test).
(D) Exo84 interactome. HEK293T cells overexpressing tagged Exo84 were immunoprecipitated with or without of poly(I:C) and were analyzed for coimmunoprecipitation of proteins using LC-MS. Node color indicates hit abundance with or without (+/−) poly(I:C), and edge color is coded as in (A) (n = 3).
(E) Sec8 interactions with immune signaling kinases PKR and TBK1. Endogenous Sec8 from HEK293T cells was immunoprecipitated with or without poly(I:C) and was evaluated by SDS-PAGE. For statistical significance (unpaired Student’s t test) for quantified band intensities, **p < 0.01 (n = 4).
(F and G) Exo84 and Sec5 interactions with PKR and TBK1. (F) Epitope-specific immunoprecipitates were evaluated for coimmunoprecipitation in Taptag-Exo84 and (G) HA-Sec5-expressing HEK293T cells with or without poly(I:C) (n = 3).
(H) Schematic summary of dynamic exocyst subcomplex partners upon poly(I:C) challenge.
The core Sec8 interactome contained a subset of the canonical exocyst subunits (Sec3, Sec5, Sec6, and Sec8) (Figure 1A; Figure S1I), consistent with previous reports (Boldt et al., 2016). Deeper proteome coverage of affinity-purified Sec8 using spectral count-based MS recovered all exocyst subunits (Figure S1J). The RAL-GTPase effectors Sec5 and Exo84 have been implicated in both pathogen and nutrient-sensing regulatory programs as components of two distinct exocyst subcomplexes (Chien et al., 2006; Bodemann et al., 2011). Evaluation of affinity-purified Sec5 and Exo84 complexes indicated relative enrichment of Sec10, Sec15, and Exo70, together with Exo84, and of Sec3, Sec6, and Sec8, together with Sec5 (Figure S1K). Most of the native Exo84 and Sec5 eluted from gel filtration columns in separate fractions of ~500 and ~700 kDa, respectively (Figures S1H and 1M). The RALB partition profile indicated coelution of a ~34-kDa modified form of RALB with Sec5, consistent with previous data and reports that a ubiquitinated form of RALB preferentially associates with Sec5 versus Exo84 (Bodemann et al., 2011; Simicek et al., 2013).
Nutrient deprivation did not significantly alter the relative abundance of any detected Sec8 partners within Sec8 complexes (Figure S1L). In contrast, poly(I:C) challenge promoted significant accumulation of TBK1/Sec8 complexes without affecting TBK1 gene expression or protein stability and Sec8’s association with other partners such as the CAK complex (Figures 1C and 1E; Figure S1N) (Chien et al., 2006). Given the low abundance of Exo84 in Sec8 complexes, we directly evaluated affinity-purified Taptag-Exo84 for differential association with protein partners upon poly(I:C) challenge. This revealed a stimulus-dependent association of Exo84 with the dsRNA-activated protein kinase, PKR (Figures 1D and 1F). TBK1 and PKR were detectable in Sec8 immunoprecipitated fractions together with Sec5 and Exo84. Poly(I:C) challenge enriched the TBK1-Sec5 complex and depleted PKR and Exo84 complexes (Figure 1E). Poly(I:C) challenge promoted formation of HA-Sec5/TBK1 complexes and disruption of HA-Sec5/PKR complexes (Figure 1G). The data suggest that an immunogenic stimulus promotes an Exo84 subcomplex interaction with PKR and a Sec5 subcomplex interaction with TBK1 (Figure 1H).
Virus infection induces assembly of Exo84-PKR-MST1 and Sec5-TBK1-mTOR complexes
To help evaluate the context and mechanistic relevance of the dynamic associations of TBK1 and PKR with the exocyst, we next examined the composition of endogenous TBK1 and PKR protein complexes with and without poly(I:C) challenge. We examined the protein complexes recovered with epitope-tagged wild-type and kinase-impaired TBK1 and PKR variants. Consistent with the exocyst subunit interactomes described above, poly(I:C) exposure promoted accumulation of TBK1/Sec5 complexes and PKR/Exo84 complexes (Figure 2A; Figures S2A and 2B). This was observed with wild-type (native and epitope tagged), but not with kinase-impaired variants of TBK1 and PKR. There was a preferential association of S6K and RAPTOR with wild-type TBK1, in a manner dependent upon TBK1 kinase activity (Figure S2C). The mTOR pathway proteins S6K and RAPTOR have been reported as TBK1 substrates before (Cooper et al., 2017; Hasan et al., 2017). In the PKR interactome, previously undescribed PKR interactions with the Hippo/YAP1 regulatory proteins MST1, NF2, and KIBRA were also identified (Hansen et al., 2015) (Figure 2A; Figures S2B and S2C) (Laplante and Sabatini, 2012; Ma and Blenis, 2009; Yu et al., 2012; Zhao et al., 2010; Varelas et al., 2010; Aragona et al., 2013; Liu et al., 2016).
Figure 2. Interaction landscape of PKR- and TBK1-containing exocyst subcomplexes under viral infection.
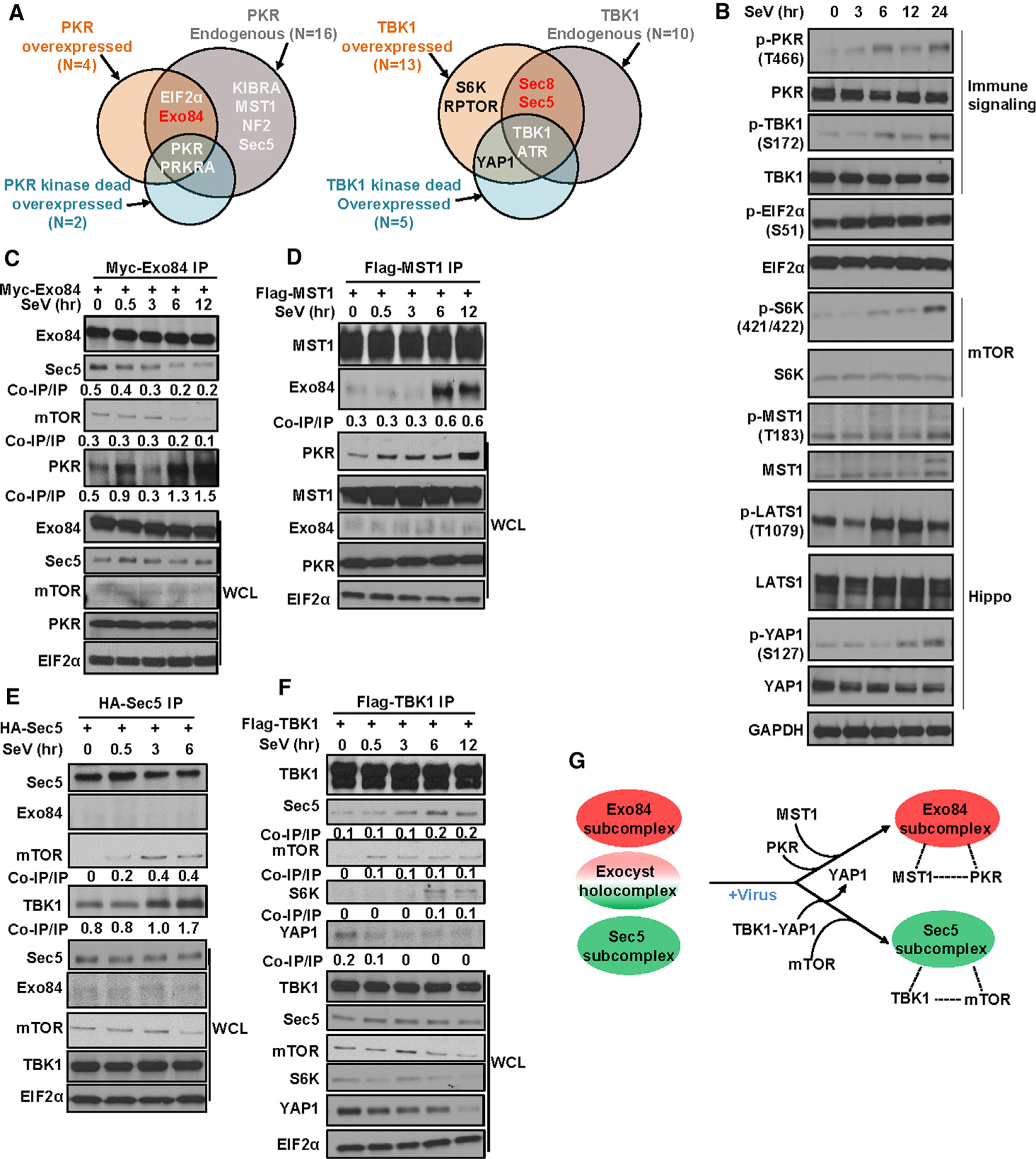
(A) PKR and TBK1 interactome identifies MST1 and mTOR as interactors. Endogenous (+/− poly(I:C)) and overexpressed PKR and TBK1 (both kinase active and kinase impaired`) were immunoprecipitated from HEK293T cells and were analyzed using LC-MS for potential interactors. Euler plot compares hits in datasets where red hits are enriched upon poly(I:C) treatment. n, number of hits identified (n = 2).
(B) Sendai virus (SeV) infection activates PKR, TBK1, Hippo, and mTOR signaling. Western blot was carried out in HEK293T cells infected with 50 HAU (hemagglutination assay unit)/mL SeV (n = 2).
(C and D) Under SeV infection, Exo84 forms a stable complex with PKR and MST1. Exo84 and MST1 immunoprecipitated from HEK293T cells transiently expressing (C) Myc Exo84 and (D) FLAG-MST1 were analyzed for interactors using SDS-PAGE (n = 2).
(E and F) Unader SeV infection, Sec5 forms a stable complex with TBK1 and mTOR. TBK1 and Sec5 immunoprecipitated from HEK293T cells transiently expressing (E) FLAG-TBK1 or (F) HA-Sec5 were analyzed for interactors using SDS-PAGE (n = 2).
(G) Schematic representation of Exo84/MST1/PKR and Sec5/mTOR/TBK1 complex formation under virus infection.
We further evaluated the consequence of pathogen exposure on exocyst subcomplexes and their relationships to mTOR and Hippo pathway activation (Figure 2B; Figures S2D and S2E). SeV and herpes simplex virus type 1 (HSV1) infection activated both TBK1 and PKR in a time-dependent manner, indicated by activating phosphorylation of TBK1 and PKR (Figure 2B; Figure S2E). This correlated with signatures of mTOR and Hippo pathway engagement, indicated by activating phosphorylation on the mTOR substrate, S6K, and on the Hippo core kinase, Large Tumor Suppressor Kinase 1/2 (LATS1/2) (Figure 2B; Figure S2E).
Because the canonical LATS kinase, MST1, was identified as a PKR complex member, we reasoned that LATS1/2 activation may be a direct consequence of MST1 activation in the PKR/MST1 complex. To test this, we first examined whether activating phosphorylation on MST1 was associated with PKR activation induced by pathogen exposure. Virus infection caused a molecular weight shift in MST1 and an increase in canonical MST1 phosphorylation (Figure 2B; Figure S2E). These data suggest that virus infection may mediate post-translational modification (PTM) of MST1.
We next investigated whether Exo84, PKR, and MST1 form a multi-protein complex upon virus exposure. Within 6 h of SeV exposure, Exo84 dissociated from Sec5 and mTOR (Figure 2C; Figure S2D) and associated with PKR and MST1 (Figures 2C and 2D; Figure S2D). The same stimulus induced the formation of a Sec5/TBK1/mTOR complex (Figure 2E; Figure S2E). There was accumulation of Sec5/TBK1/mTOR complexes concurrent with disruption of baseline TBK1/YAP1 interactions (Figure 2F; Figure S2E). Thus, pathogen exposure mediates the coordinated formation of distinct multi-protein exocyst subcomplexes.
Under identical stimuli, we performed analyses of other pathways linked to exocyst function, such as autophagy and p38 activation (Balakireva et al., 2006; Bodemann et al., 2011). p38 activity, measured by activating phosphorylation of p38 mitogen-activated protein kinase (MAPK), was not affected (Figure 2D). As for autophagy, HSV1 (dsDNA virus), but not SeV (dsRNA virus), infection induced LC3-I degradation (Figures S2F and S2G). These observations suggest that the antiviral xenophagy response is not universal across different virus classes and does not correlate with SeV-induced TBK1 and PKR activation kinetics (Tallóczy et al., 2006). HSV1, but not SeV, infection resulted in viral replication-dependent cellular toxicity (Figures S2H and S2I), marked by an increase in cleaved caspase 3 (Figures S2F and S2G). Thus, we used SeV instead of HSV1 for further mechanistic studies.
In summary, we found evidence for the dynamic assembly of distinct Exo84/PKR/MST1 and Sec5/TBK1/mTOR multi-protein complexes and concomitant activation of Hippo and mTOR pathways (Figure 2G).
PKR phosphorylates MST1 to regulate Hippo signaling
We next evaluated the functional impact of PKR and MST1 interaction with Exo84. Because both PKR and MST1 have kinase activity, we postulated the presence of a kinase cascade involving them where Exo84 acts as a scaffold. PKR acts as a pattern recognition receptor for virus-associated molecular patterns, such as dsRNA (Taylor et al., 2005). Hence we reasoned that PKR may be positioned upstream in the kinase cascade, and MST1 may be a substrate for PKR. Because recent publications have also indicated that virus infection can activate Hippo signaling (Figure 2B) (Wang et al., 2017; Zhang et al., 2017), for which MST1 is a core kinase component (Figure 3A) (Zhou et al., 2009), we hypothesized that PKR may regulate Hippo signaling under virus infection directly or indirectly (Figure 3A).
Figure 3. PKR regulates hippo signaling through MST1.
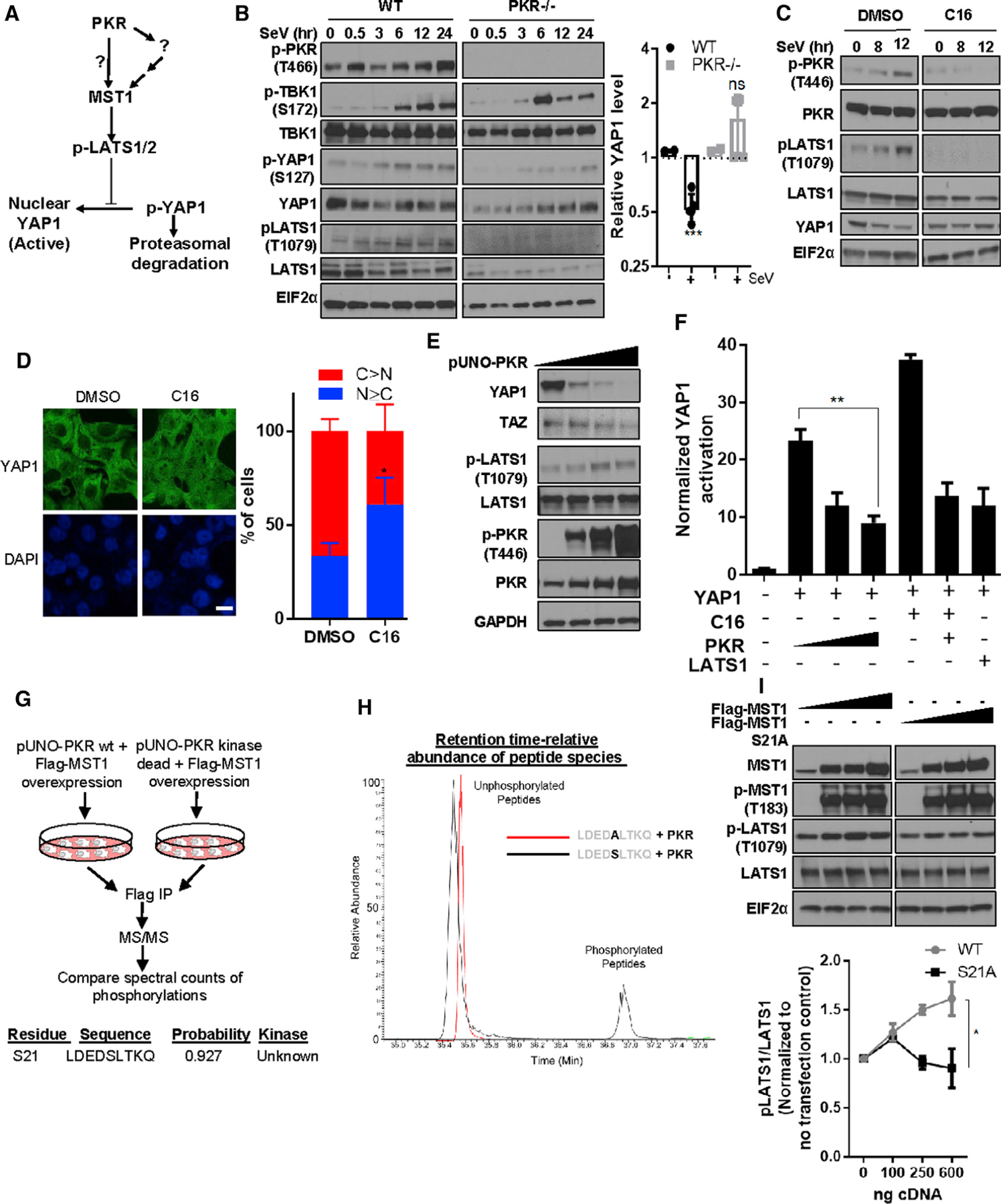
(A) Schematic diagram outlines the model for Hippo signaling and potential regulation by PKR (direct or indirect).
(B and C) PKR is required for maximal activation of hippo signaling under immune challenge. (B) (Left) Lysates from PKR null and wild-type (WT) MEFs incubated ± SeV were analyzed using SDS-PAGE. (Right) Band intensity for YAP1 (n = 2) from the ‘‘12-h’’ as well as ‘‘24-h’’ treatment cohorts were grouped and compared with that of ‘‘0-h’’ treatment cohort. Unpaired t test with equal variance, the p value was <0.005. (C) HEK293T cells either pre-treated with DMSO or 0.2 μM C16 for were treated with SeV and collected for western blot analysis.
(D) PKR inhibition causes nuclear localization of YAP1. HepG2 cells treated with DMSO or C16 were fixed and stained for YAP1 immunofluorescence. Scale bar: 10 μm. Counted number of cells for DMSO = 49 and for C16 = 42. Cells were qualitatively designated into two classifiers: C > N and N ≥ C, where N represents nuclear YAP and C represents cytosolic YAP. Unpaired t test p value was <0.05.
(E) PKR is sufficient to induce Hippo signaling. PKR cDNA was overexpressed in HEK293T cells and analyzed using SDS-PAGE (n = 2).
(F) PKR negatively regulates YAP1-mediated transcriptional output and rescues C16-mediated Hippo activation. 8XGTIIC/YAP-firefly luciferase reporter activity was measured in HEK293T cells. Cells expressing indicated cDNA or empty vector were grown for 24 h, and normalized YAP1-mediated transcriptional output was measured. Cells were treated with 0.1 μM C16 for 6 h in the presence or absence of PKR cDNA. Mean ± SD is represented as bar diagram (n = 3).
(G) PKR activity is associated with and phosphorylates MST1 on S21. MST1 was immunoprecipitated from HEK293T cells expressing FLAG-MST1 along with either pUNO-PKR WT or pUNO-PKR kinase-deficient mutant forms and was analyzed using LC-MS. Identified peptide sequence containing the MST1 S21 phosphorylation site was analyzed for kinase phosphorylation probability using the NetPhos3.1 server.
(H) In vitro phosphorylation of MST1 peptide by purified PKR. Indicated peptides were incubated with purified active PKR protein for 2 h. Purified peptides from the reaction mixture were analyzed using LC-MS, and the abundance of the peptide species phosphorylated or unphosphorylated were measured along with retention time duration.
(I) Characterization of MST1 S21A mutant: HEK293T cells were transfected with titrating amounts of MST1 WT or MST1 S21A mutant cDNA-expressing plasmids and were probed using SDS-PAGE. Band intensities were quantified and represented as mean ± SD in a trendline plot (n = 2). Two-way ANOVA significance between WT and S21 cohort where p < 0.05.
We investigated the role of PKR in Hippo pathway signaling upon virus infection using PKR-deficient mouse embryonic fibroblasts (MEFs). Hippo signaling induction is marked by an increase in activating phosphorylation in Hippo core kinases, LATS and MST, and by a decrease in YAP1 protein. Our data indicate SeV infection of wild-type MEFs lowered total YAP1 levels, whereas PKR- and MST-deficient MEFs showed higher YAP1 levels post-infection (Figure 3B; Figure S3A). Short interfering RNA (siRNA)-mediated depletion of PKR ablated SeV-induced LATS1 activation (Figure S3B). Thus, PKR is required for Hippo pathway induction by pathogen exposure. Human epithelial cell lines pre-treated with a non-toxic dose of a PKR inhibitor C16 failed to activate Hippo signaling following SeV infection, as noted by ablation of LATS phosphorylation and stabilization of YAP1 (Williams 1999; Ingrand et al., 2007; Tronel et al., 2014; Xiao et al., 2016). The PKR inhibitor lowered LATS activity even in the absence of SeV stimulus (Figure 3C), which indicated the presence of a stimulus-independent baseline regulation of Hippo signaling by PKR. Inhibition of PKR caused nuclear translocation of YAP1 (Figure 3D). PKR overexpression induced LATS phosphorylation and decreased YAP1 protein expression (Figure 3E; Figure S3C). Using a YAP/TEA Domain Transcription Factor (TEAD) reporter assay (Kim and Gumbiner, 2015), we found that ectopic PKR expression suppressed YAP/TEAD activity, and the PKR inhibitor promoted YAP/TEAD activity, which could be suppressed by ectopic PKR expression (Figure 3F). Pharmacological inhibition of a canonical PKR substrate Eukaryotic Translation Initiation Factor 2A (EIF2α) using salubrinal did not increase YAP1 reporter activity, indicating that PKR-mediated regulation of Hippo signaling was EIF2α independent (Figure S3D). Thus, PKR is a direct regulatory node in the Hippo signaling pathway that is positioned upstream of LATS and YAP1.
In HEK293T cells, SeV exposure caused a molecular weight increase in MST1 and induced a minimal increase of known phosphorylation events on MST1 (Figure 2B), and MST1/2-deficient MEFs lacked SeV-mediated YAP1 degradation (Figure S3D). Because MST1 is predominantly activated by kinase-dependent phosphorylation, we reasoned that PKR may directly phosphorylate MST1 at an unknown site to activate Hippo signaling. To test this, we expressed in HEK293T cells MST1 and either active or kinase-impaired PKR. We affinity-purified MST1 from both conditions and queried PTMs enriched in the active PKR specific cohort using liquid chromatography-tandem mass spectrometry (LC-MS/MS). We calculated the ratio of the MST1 PTM probability scores between PKR active and PKR kinase impaired cohorts and found that MST1 serine 21 had 49 times greater phosphorylation probability in PKR kinase active versus kinase-impaired samples (Figure 3G) (Trudgian et al., 2012). Sequence motif analysis indicated that a nine-residue-long peptide sequence matched six residues of the putative PKR recognition sequence identified by consensus analysis of PKR substrates (Figure S3E). We hypothesized that S21 on MST1 is phosphorylated by PKR. We incubated a synthetic peptide containing the putative phospho-site sequence with recombinant active PKR and found that the native peptide was phosphorylated on serine (Figure 3H), with a peptide where serine was replaced by alanine serving as a negative control. The MST1 S21A variant showed attenuated phosphorylation of its canonical substrate LATS1 (Figure 3I; Figure S3F), suggesting S21 phosphorylation is important for MST1-mediated LATS1 activation. These combined observations indicate a PKR-mediated PTM and activation of MST1, engaging Hippo signaling to suppress YAP1.
In HeLa cells, baseline PKR was localized mostly in the trans-Golgi compartment, and this localization oriented more toward the cis-Golgi network and lysosomes upon virus infection (Figure S3G). In response to both biologic (HSV1Δ34.5) and chemical (STATTIC, a STAT3 inhibitor) (Shen et al., 2012) modulators of PKR-dependent xenophagy (Figures S3H–S3J), HeLa and U2OS cells induced a productive autophagic response (Figures S3K–S3O), which was abrogated by loss of PKR, Exo84, and RALB, a major regulator of Exo84 (Figures S3L–S3O). Thus, PKR/Exo84 protein complex functionality may extend beyond induction of Hippo signaling and be involved in the PKR-mediated xenophagy response.
TBK1 supports mTOR signaling and regulates YAP1 through kinase-independent cytosolic sequestration
Virus exposure favors Sec5–8/TBK1/mTOR complex formation (Figures 2E and 2F; Figure S2E). TBK1 activation mediated by virus infection precedes mTOR re-activation, and TBK1 is a core kinase responsible for productive host defense signaling (Figure 2B) (Tanaka and Chen 2012). Hence we hypothesized that TBK1 may directly activate mTOR in the protein complex. YAP1 was an interacting partner of TBK1 (Figure 2A; Figure S2C). These observations were consistent with recent studies suggesting TBK1 may regulate YAP1 and the mTOR substrate S6K (Cooper et al., 2017; Wang et al., 2017; Zhang et al., 2017; Eskiocak et al., 2017; Kim et al., 2013; Antonia et al., 2019; Martin et al., 2014). Pathogen exposure formed TBK1/mTOR complexes and suppressed TBK1/YAP1 complexes (Figures 2H and 4A). Because pathogen exposure also activated TBK1, we hypothesized TBK1 bound to the Sec5 subcomplex is in an active form and TBK1 bound to YAP1 is inactive.
Figure 4. TBK1 switches from a YAP1 complex to mTOR/S6K, facilitating LATS1-mediated YAP1 suppression.
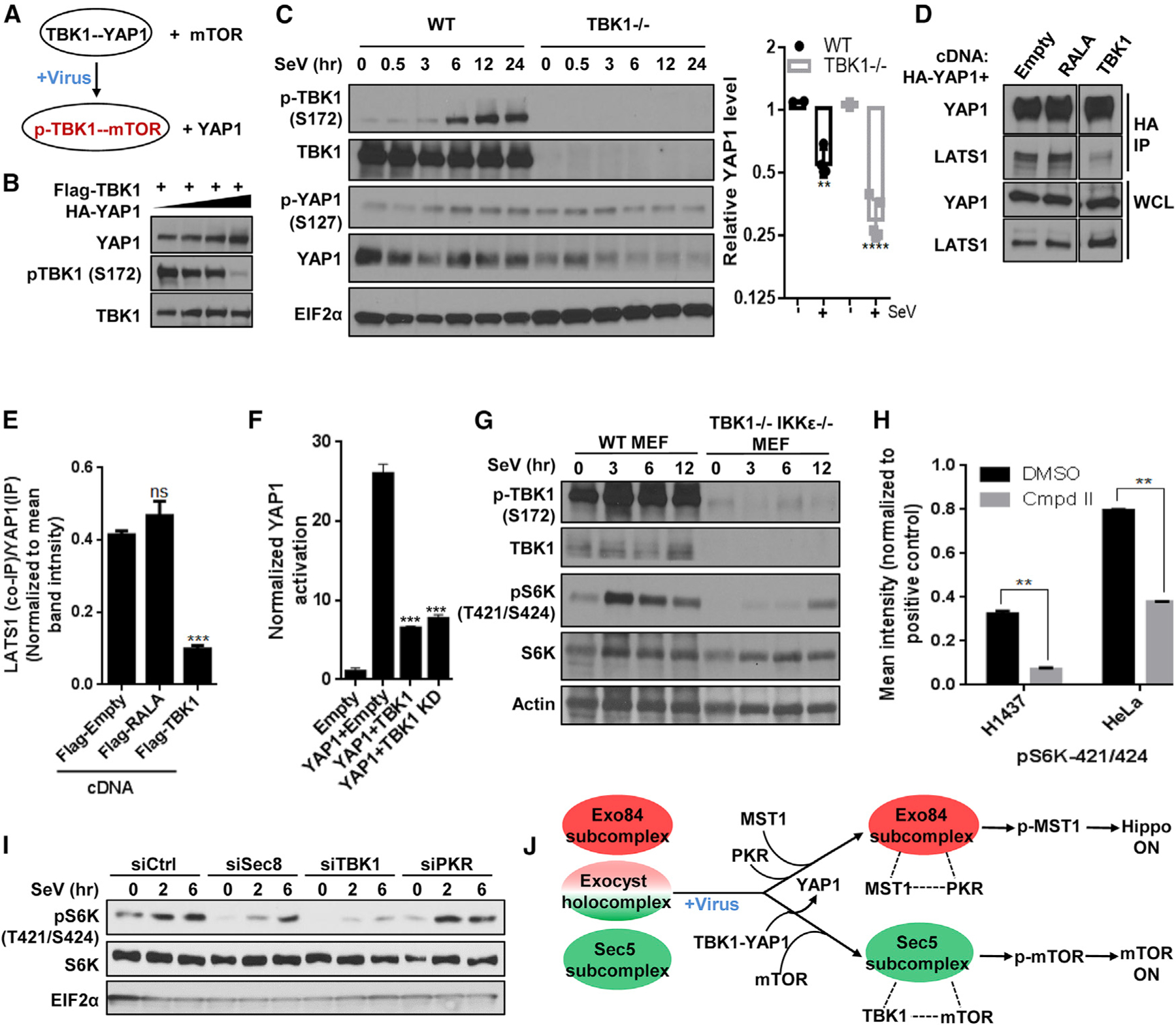
(A) Schematic diagram illustrating partner swapping event for TBK1 stable complex.
(B) YAP1 inhibits TBK1 activity. HEK293T cells expressing FLAG-TBK1 along with increasing amount of HA-YAP1 were grown and analyzed for western blot.
(C) TBK1 is protective for YAP1. (Left) TBK1 null and WT MEFs were incubated with SeV and analyzed using SDS-PAGE. (Right) YAP1 level for each time point was calculated by measuring individual band intensity. For comparison between WT and null cohort (as described in Figure 3B), unpaired t test p value was **p < 0.001 and ****p < 0.0001. Control WT cohorts were identical to the ones indicated in Figure 3B (n = 2).
(D and E) TBK1 competes with LATS1/2 for binding with YAP1. (D) Cells transiently expressing HA-YAP1 with or without plasmids co-expressing indicated cDNAs were grown to confluence, and YAP1 was affinity purified using an antibody against the HA tag and was analyzed for levels of indicated proteins using SDS-PAGE on the same gel. (E) Intensities of the bands were quantified and represented as mean ± SD in the bar graph (n = 2).
(F) TBK1 inhibits YAP1 activity. YAP reporter activity was measured in the presence of empty vector or the cDNAs indicated. Normalized fold change in Hippo signaling activation is represented as histograms representing mean ± SD. Statistical comparison between treatment cohorts was measured using a Student’s unpaired t test where p < 0.001 and n = 3.
(G) TBK1 regulates virus infection-induced S6K phosphorylation. Near-confluent culture of TBK1 WT and TBK1- and IKKε-deficient MEFs were incubated with 50 HAU/mL SeV. Lysates were collected and analyzed using SDS-PAGE (n = 2).
(H) Compound II, a specific TBK1 inhibitor, inhibits S6K phosphorylation on residues 421/424. Confluent cultures from eight cell lines from melanoma and lung cancer origin were treated with either DMSO or CmpdII (TBK1i/TBK1 inhibitor) for 30 min, and cells harvested were analyzed for phospho-protein level of 16 AKT pathway proteins by using phospho-ELISA array. Quantification of the phosphorylation of p70S6K in DMSO-versus CmpdII-treated H1437 and HeLa cell line samples in Figure S4E are represented as bar graph, and data are represented as mean ± SD. Statistical significance was calculated using Mann-Whitney U test, where p < 0.01 (n = 2).
(I) The exocyst and TBK1 support virus challenge-induced S6K phosphorylation. HEK293T cells reverse transfected with indicated short interfering RNAs (siRNAs) were grown to confluence and incubated with SeV. Cell lysates were analyzed for S6K phosphorylation using SDS-PAGE (n = 2).
(J) Summary of the virus infection stimulus-specific regulatory interaction from TBK1’s orientation.
Ectopic expression of YAP1 reduced activating phosphorylation of TBK1 and downstream IFN-β activity (Figure 4B; Figure S4A). Because YAP1 and TBK1 can interact in the cytosol (Figure 2A) (Zhang et al., 2017), we next measured the effect of reducing cytosolic YAP1 protein expression on TBK1-mediated IFN-β signaling. LATS1 expression decreased YAP1 reporter activity, as expected (Moroishi et al., 2016) (Figure 4F). Using an IFN-β reporter assay, we found IFN-β inactivation that occurred upon YAP upregulation could be overcome by LATS1 overexpression (Figure S4A), consistent with recent findings (Zhang et al., 2017).
To test whether TBK1 regulates YAP1 activity and stability, we measured Hippo signaling induction during pathogen exposure in TBK1-deficient and wild-type MEFs (Cooper et al., 2017). TBK1 deficiency promoted YAP degradation when cells were infected with SeV, compared with TBK1-proficient cells (Figure 4C). The TBK1/YAP1 complex was competitive with the canonical LATS/YAP1 interaction, as forced TBK1 overexpression dissociated the latter complex (Figures 4D and 4E).
Ectopic expression of both kinase-proficient and kinase-impaired TBK1 constructs lowered YAP/TEAD reporter activity, and treatment with the TBK1 inhibitors BX795 and Compound II neither increased nor decreased YAP activity (Figure 4F; Figure S4B) (Feldman et al., 2005). This was in agreement with previous reports in which YAP1 and TBK1 proteins interact via a defined region in YAP1, separate from YAP’s transcriptional co-activator domain (Zhang et al., 2017). We concluded that TBK1 sequesters YAP1 from LATS-mediated degradation in a kinase-independent manner to blunt YAP-mediated transcriptional output.
We examined the functional relevance of the TBK1/mTOR interaction. TBK1 and IKKε (TBK1 homolog) double-knockout MEFs and TBK1-deficient MEFs showed attenuated activation of mTOR signaling after viral infection when compared with wild-type MEFs (Figure 4G; Figure S4C). The effect was more apparent in double-knockout MEFs than in TBK1-deficient MEFs, suggesting that virus-induced mTOR activation might be regulated by both TBK1 and IKKε. Thus, the Sec5–8/TBK1 complex likely regulates a pool of mTOR activity. Previous studies showed direct regulation of the mTOR substrate S6K activity by TBK1 (Kim et al., 2013; Cooper et al., 2017). Consistent with this, the TBK1 inhibitor Compound II suppressed S6K phosphorylation on T421/S424 (Figure 4H; Figure S4D). siRNA-mediated depletion of TBK1 and the TBK1 exocyst partner Sec8 attenuated S6K phosphorylation after viral infection (Figure 4I). Loss of Sec8 resulted in selective disruption of the Sec5/mTOR/TBK1 complex, but not of the Exo84/RALB complex (Figure S4E). Thus, TBK1 supports mTOR signaling in a kinase-dependent manner and antagonizes activation of YAP1 by kinase-independent cytosolic sequestration (Figure 4J).
In HeLa cells, we found that TBK1 was detected at lysosomes and mitochondria, consistent with previous findings (Figure S4F) (Cooper et al., 2017; Sun et al., 2006). Upon virus infection, the TBK1 signalosome colocalized proximal to structures marked by LAMP1, a lyso-endosomal marker (Figure S4G). Because we detected lysosomal colocalization of both TBK1 and PKR, and because lysosomes are considered to be a vital signaling hub for mTOR and autophagy signaling, we next queried if the exocyst is localized at the lysosome. We found that Sec8 localizes at the lysosome in a RALB-dependent manner (Figures S4H–S4K). Additionally, our data demonstrated that a virally compromised cell state favors formation of Exo84/PKR protein complex puncta and Sec5/TBK1 protein complex puncta (Figure S4L).
RAL-exocyst is required for activation of TBK1, PKR, and Hippo signaling
We examined the effect of loss of exocyst subunits on mTOR and Hippo signaling activation. siRNA-mediated depletion of Sec8 perturbed not only mTOR but also TBK1 activation driven by pathogen or poly(I:C) exposure (Figures 4I and 5A; Figure S5A). Given that the TBK1 inhibitor Compound II and the mTOR inhibitor rapamycin attenuated IFN-β activity induced by TBK1 expression, we concluded mTOR signaling is necessary for this host defense IFN response (Figure S5B). An equivalent analysis of Exo84 depletion showed attenuated PKR autophosphorylation and MST1-dependent LATS1 activating phosphorylation (Figure 5B). Accordingly, selective expression of Exo84, but not Sec5, lowered YAP1 reporter activity (Figure 5C).
Figure 5. Functional requirement and sufficiency of the RAL-exocyst for TBK1, PKR, and Hippo signaling.
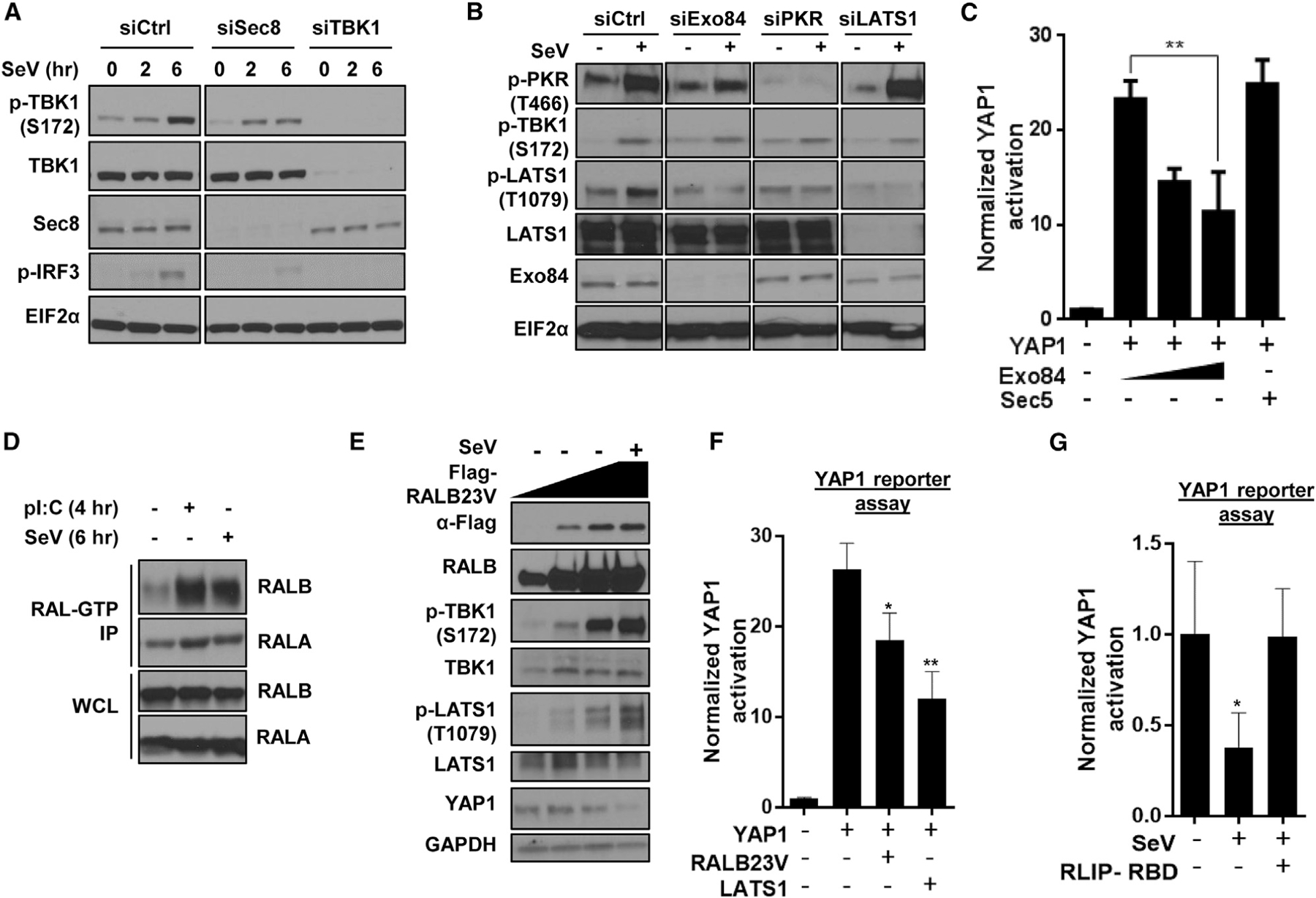
(A) Sec8 supports virus infection-dependent TBK1 activation. HEK293T cells reverse-transfected with indicated siRNAs and incubated with SeV (50 HAU/mL) were analyzed using SDS-PAGE (n = 2).
(B) Exo84 is required for virus challenge-dependent LATS1 phosphorylation. HEK293T cells reverse-transfected with indicated siRNAs and incubated with SeV (50 HAU/mL; 10 h) were analyzed using SDS-PAGE (n = 2).
(C) Exo84 overexpression inactivates YAP1 transcriptional activity. YAP reporter activity was measured in the presence of a titrated quantity of Exo84 cDNA expression. Normalized YAP activation is represented as histograms representing mean ± SD (n = 3), where Student’s unpaired t test p value was <0.01.
(D) Host defense stimulus activates RALB. HEK293T cells were incubated with poly(I:C) (2 μg/mL) for 4 h or SeV (50 HAU/mL) for 6 h, and active RAL-GTP was pulled down using the GST-RBD under each condition. Active RAL immunoprecipitated and total proteins were analyzed for RALA and RALB (n = 2).
(E) Active RALB is sufficient to induce PKR and LATS1/2 phosphorylation. U2OS cells overexpressing varying amounts of active RALB cDNA were collected and analyzed using SDS-PAGE (n = 2).
(F) RALB is sufficient to inhibit YAP1 activity. YAP reporter activity was measured in HEK293T cells, and activation was represented as histograms representing mean ± SD (n = 3; *p < 0.05 and **p < 0.01 for unpaired t test).
(G) RALB is required to inhibit YAP1 activity. In HEK293T cells, YAP reporter activity was measured in the presence of 200 HAU/mL SeV for 12 h, and activation was represented as histograms representing mean ± SD (n = 3; *p < 0.05 for unpaired t test).
Mobilization of the exocyst is a major function of the Ras-like GTPase RALB (Moskalenko et al., 2002; Chien et al., 2006; Ishikawa and Barber, 2008). Consistent with previous reports, RALB was activated by immunogenic signals, as indicated by accumulation of GTP-bound-RALB (Figure 5D). By contrast, RALA was unaffected (Figure 5D).
An immunogenic stimulus-driven assembly of Exo84 and Sec5 into distinct protein complexes was consistent with previous reports (Chien et al., 2006; Bodemann et al., 2011). Using hierarchical clustering from the FUSION database (Potts et al., 2013; McMillan et al., 2019), unbiased evaluation of pairwise functional homology within selected candidates from Hippo, mTOR, and RAL-exocyst signaling pathways suggested partitioning of RALB, RAPTOR, and MST1 into a distinct functional clade (Zaman et al., 2013) (Figure S5C).
Ectopic expression of a constitutively active RALB23V was sufficient to induce TBK1 and LATS1 activation (Figure 5E). Ectopic expression of RALB23V promoted IFN-β promoter activation and lowered YAP1-mediated transcriptional activity in human epithelial cells (Figure 5F; Figure S5D). YAP inactivation induced by RALB23V was comparable with half of the degree of inactivation imparted by LATS overexpression (Figure 5F). In agreement, expression of a dominant RAL-effector interaction interfering protein, RLIP-RBD (Bodemann et al., 2011), reduced SeV-induced IFN-β response and rescued RALB-mediated YAP1 inactivation (Figure 5G; Figure S5E). These data, along with previous reports (Chien et al., 2006), establish RALB activation as a signaling component induced by viral infection.
RALB drives MST1/PKR/Exo84 and TBK1/mTOR/Sec5 complexes
Because RALB activation is a signaling node for immunogenic regulation, we investigated if host defense stimulus-dependent partners of the exocyst are RALB responsive. We examined the composition of the endogenous RALB interactome and tagged Exo84 or Sec5 interactome in the presence of RALB23V expression (Figures S6A and S6B). Evaluation of the RALB interactome identified the mTOR regulator Ragulator complex components LAMTOR1 and LAMTOR3 as partners (Figures S6A and S6B). Active RALB favored both Sec5 and Exo84 association with the LAMTORs (Figures S6C–S6E), consistent with the recent report that RALB and LAMTOR3 form a high-molecular-weight endogenous protein complex (Havugimana et al., 2012).
Under active RALB expression, we detected mTOR and mTOR regulators RAPTOR and LAMTORs as partners of the exocyst core subunit Sec8 (Figure S6F). LAMTOR’s RALB-driven association with Sec5, Exo84, and Sec8 suggested accumulation of an exocyst holocomplex/LAMTOR complex under active RALB conditions. Additional exocyst subunits (Sec6 and Exo70) and Sec8 were detected in the RAPTOR and LAMTOR interactomes (Figures S6G and S6H). Considering LAMTOR’s canonical role in regulating mTOR signaling (Sancak et al., 2010) and because pathogen exposure accumulated mTOR/Sec5 and depleted Exo84/mTOR complexes (Figures 2C and 2F), we examined the exocyst subcomplex association with LAMTOR3 in a stimulus-dependent manner. Like mTOR, LAMTOR3 favored Sec5 versus Exo84 association under virus exposure (Figure S6I). To systematically examine baseline LAMTOR3 complexes associated with exocyst entities (i.e., octameric holocomplex, Exo84 subcomplex, and Sec5 subcomplex), we employed a holocomplex-specific RALB variant 39L and loss-of-function RALB variants that discriminate between Exo84 and Sec5 as effectors (Moskalenko et al., 2002; Bodemann et al., 2011). RALB39L engages both the exocyst holocomplex and Exo84 and Sec5 subcomplexes, whereas RALB23V-48W has 40-fold higher affinity for Sec5 and RALB23V-38R has 100-fold higher affinity for Exo84 (Moskalenko et al., 2002; Bodemann et al., 2011). Expression of F39L and 48W, but not 38R, promoted Sec5/LAMTOR3 complex accumulation (Figure S6J). LAMTOR3/Exo84 complexes were unaffected by expression of 38R but were favored in the presence of the other two mutants (Figure S6J). These observations indicated the presence of a baseline LAMTOR3/exocyst protein holocomplex. Pathogen exposure then results in LAMTOR3/Sec5 subcomplex enrichment.
Active RALB expression promoted accumulation of MST1/Exo84 and Sec5/mTOR complexes (Figure S6C; Figures 6A and 6B). We noted a shared neighborhood of Hippo signaling components MST1, NF2, and KIBRA in the RALB and PKR interactome (Figure 6A). RALB expression promoted association of the Hippo signaling components KIBRA and MST1 with the exocyst core subunit Sec8 (Figure S6F). Equivalent analysis of the RALB and TBK1 interactomes identified YAP1 as a shared partner (Figure 6B). These interactions were orthogonally validated by IP/western blot analysis (Figures S6D and S6E).
Figure 6. RALB drives formation of Exo84/PKR/MST1 and Sec5/TBK1/mTOR complexes.
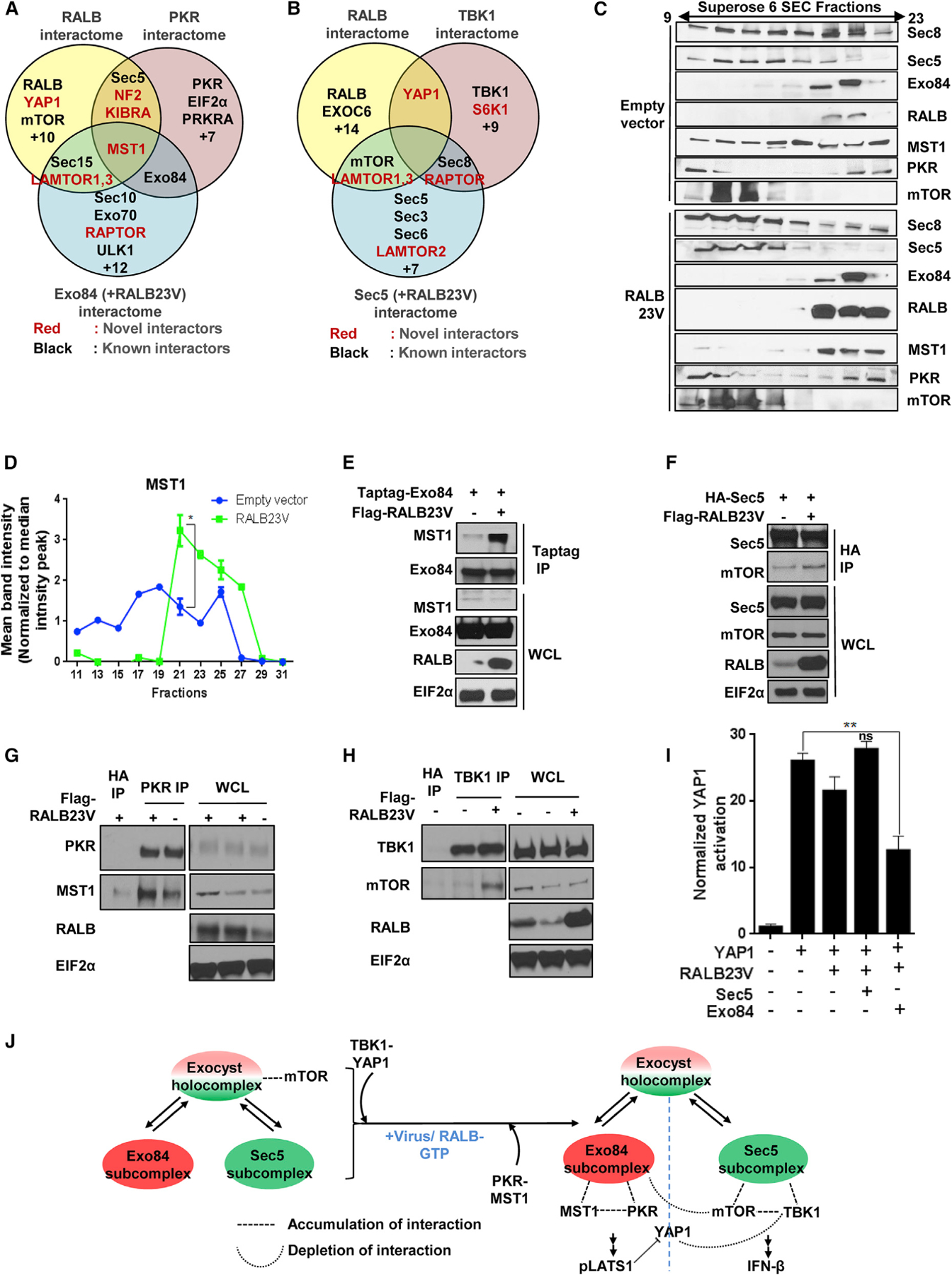
(A and B) Euler plot representation for identifying RALB-dependent exocyst interactors. Endogenous RALB, PKR, overexpressed TBK1, and overexpressed Exo84 or Sec5 in the presence or absence of RALB23V, isolated from HEK293T cells, were analyzed for coimmunoprecipitation of proteins using mass spectrometry. Euler diagram shows comparison among the PKR (described before), RALB-driven Exo84 and RALB interactome (A), and TBK1 (described before), RALB-driven Sec5 and RALB interactome (B).
(C and D) RALB23V expression is sufficient to alter endogenous high-molecular-weight complexes of MST1 and Sec5/8 subunits. (C) Size exclusion column chromatography fractions (odd serial number from 9th till 23rd) of high-molecular-weight complexes in HEK293T cells were analyzed for the partition profile using SDS-PAGE in the presence or absence of transient expression of RALB23V cDNA (n = 2). (D) Band intensity of the MST1 partition profile was represented as a line graph. Statistical comparison of fractions 19–27 between empty vector (EV) and RALB23V overexpressing cohorts was measured using a two-way ANOVA where p < 0.01 and n = 3.
(E) Active RALB promotes Exo84-MST1 interaction. Exo84 was immunoprecipitated from HEK293T cells transiently expressing Myc-Exo84 with or without FLAG-RALB and analyzed using SDS-PAGE (n = 2).
(F) Active RALB promotes Sec5-mTOR interaction. Sec5 was immunoprecipitated from HEK293T cells transiently expressing HA-Sec5 with or without FLAG-RALB and analyzed using SDS-PAGE (n = 2).
(G) RALB is sufficient to increase PKR-MST1 interaction. HEK293T cells transfected with the indicated cDNAs were grown, and endogenous PKR was immunoprecipitated and analyzed using SDS-PAGE (n = 2).
(H) RALB is sufficient to increase TBK1-mTOR interaction. HEK293T cells transfected with the indicated cDNAs were grown, and endogenous TBK1 was immunoprecipitated and analyzed using SDS-PAGE (n = 2).
(I) RALB23V-Exo84 overexpression is sufficient to inactivate YAP1 transcriptional activity. YAP reporter activity was measured, and data are represented as a histogram representing mean ± SD (n = 3). Unpaired t test p < 0.01.
(J) Model summary of virus or RALB-driven exocyst complexes and their functional significance.
RALB expression promoted a majority of Sec8 and Sec5 to cofractionate with mTOR at a molecular weight of approximately 500 kDa by size exclusion chromatography (Figure 6C). This was concomitant with a shift from a more diffuse MST1 partition profile to a more concentrated one, where it co-eluted with the majority of endogenous Exo84 and PKR at a molecular weight of ~500 kDa (Figures 6C and 6D). We isolated increased Exo84/MST1 (Figure 6E), Sec5/mTOR (Figure 6F), PKR/MST1 (Figure 6G; Figure S6K), and mTOR/TBK1 (Figure 6H) complexes under active RALB conditions.
Under viral infection there was enrichment of Exo84/MST1 complexes and depletion of Exo84/mTOR (Figure 2E). Because RALB caused Exo84 interaction with both mTOR and MST1 (Figure S6D), we hypothesized that these interactions occur in two distinct Exo84 entities: a full octameric holocomplex and an Exo84 subcomplex. Expression of MST1 reduced mTOR/Exo84 complexes (Figure S6L). mTOR expression decreased MST1/Exo84 complexes (Figure S6L). These data indicated that Exo84 may contain an identical binding determinant for MST1 and mTOR and can modulate formation of signaling foci for one arm versus the other. Co-expression of active RALB and Exo84, but not RALB and Sec5, decreased YAP1-mediated transcriptional activity, indicating a distinct contribution of the RALB-Exo84 subcomplex for Hippo signaling induction (Figure 6I). RALB and Sec5 co-expression, but not RALB and Exo84 co-expression, resulted in IFN-β promoter activation, along with a shift of the TBK1 partition profile toward Sec5 fractions (Figures S6M and S6N). These combined observations suggest that RALB mediates dynamic reorganization of the steady-state population of the exocyst holocomplex and the Exo84 or Sec5 subcomplex interactomes, resulting in engagement of both mTOR and Hippo pathways (Figure 6J).
PKR-mediated and TBK1-mediated Hippo and mTOR activation serves a pro-survival function
To further investigate the relationship between exocyst-dependent Hippo and mTOR signaling, we performed genetic epistasis experiments. LATS1/2 genetic suppression in HEK293A cells increased baseline mTOR signaling as marked by phospho-S6 levels and higher baseline TBK1 activity (Figure S7A) (Moroishi et al., 2016). To examine if this was due to potential adaptive signaling compensation, we compared other components of the pathways in a similar manner. We found PKR-deficient and TBK1-deficient MEFs also showed higher baseline TBK1 and PKR activity, respectively (Figures S7B and S7C). Inhibition of TBK1 and PKR abrogated S6K and LATS1 activity, respectively, while also activating the other protein (Figures S7D–S7F).
To examine the biological significance of PKR-mediated Hippo and TBK1-mediated mTOR activation during host defense response, we quantitively measured the host defense response against escalating dose of cytotoxic virus HSV1 (Figure S7G) in cells deficient in either YAP1, PKR, or TBK1 and expressing active RALB (or control). YAP1-deficient mouse myoblast cells and RALB23V-expressing mammalian epithelial cells both showed increased tolerance, whereas TBK1-deficient and PKR-deficient MEFs showed lower fitness, against cytotoxic HSV1 (Figures 7A–7D) (Wang et al., 2017). Mammalian cells pre-treated with the mTOR inhibitor rapamycin showed lower fitness (Figure S7H). Consistent with these observations, TBK1 and PKR deficiency accelerated HSV1-mediated apoptosis, whereas RALB23V and YAP1 expression did not, in an isogenic HEK293T cell system (Figure S7I). These observations highlight both Hippo and mTOR pathway activation as a pro-survival host defense response.
Figure 7. Concomitant PKR and TBK1 activation favors survival during virus detection and cancer.
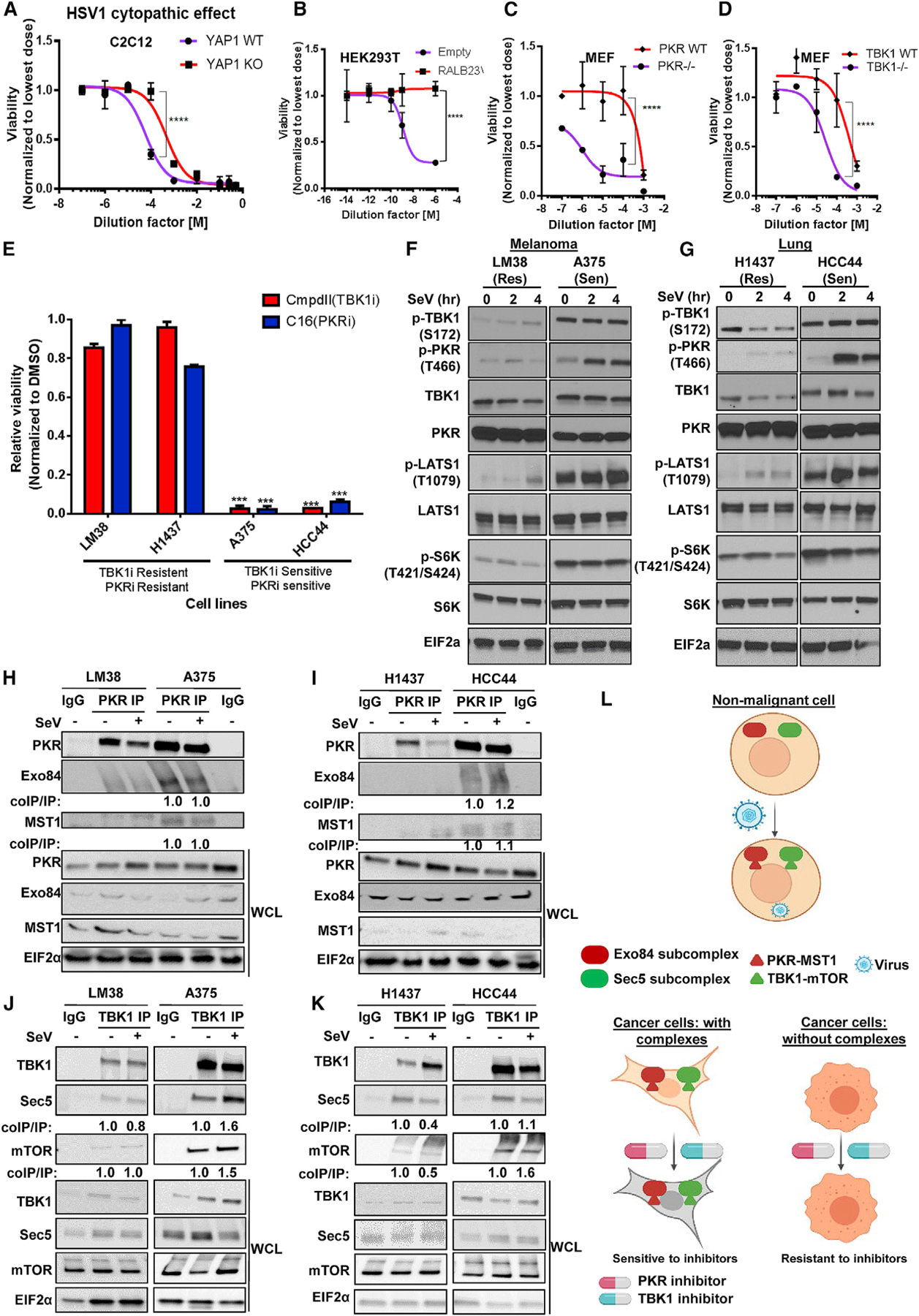
(A–D) Hippo induction, active RALB, TBK1, and PKR are immune protective. 109 plaque-forming units (PFUs) HSV1 virus stock was diluted as indicated and YAP1 WT or knockout C2C12 cell line (A), HEK293T cells stably expressing RALB23V or empty vector control (B), and MEFs WT or null for TBK1 (C) and PKR (D) were infected with the virus, and cell viability was measured via Cell Titer Glo and was plotted as 4-parameter non-linear curve; mean ± SD (n = 3). Two-way ANOVA, ****p < 0.001.
(E) Identifying PKR and TBK1 immune pathway-dependent melanoma and lung cancer cells lines. Indicated cells were treated with 2 μM TBK1i (CmpdII) and PKRi (C16) for 72 h, and cell viability was measured by Cell Titer Glo. Normalized viability is shown as a bar graph representing mean ± SD (n = 3). t test, p < 0.005 between control and treated cohorts.
(F and G) Innate immune signaling-dependent cancer cell lines with high PKR and TBK1 activity show high Hippo, S6K, and RALB activity. The indicated melanoma (F) and lung (G) cancer cell lines were incubated with SeV, and the levels of the indicated proteins were measured using SDS-PAGE (n = 2).
(H–K) TBK1/Sec5/mTOR and PKR/Exo84/MST1 form a protein complex in sensitive cells. PKR (H and I) and TBK1 (J and K) were immunoprecipitated in the presence or absence of SeV from H1437, HCC44 (lung), LM38, and A375 (melanoma) cells, which were probed for coimmunoprecipitation using SDS-PAGE (n = 2).
(L) Schematic model for the cell biology function of Exo8/PKR/MST1 and Sec5/TBK1/mTOR complexes in normal and cancer cell contexts.
Cell-autonomous innate immune/host defense signaling can be co-opted by cancer cells to support oncogenic, pro-survival signaling networks (Chien et al., 2006; Chien and White 2008; Barbie et al., 2009). To investigate how host defense-associated TBK1-dependent mTOR and PKR-dependent Hippo signaling may be used by cancer cells, we measured dose-response curves for the TBK1 inhibitor, Compound II, and the PKR inhibitor, C16, in a panel of 12 lung cancer (Figures S7J and S7K) and 10 melanoma cell lines (Figures S7L and S7M), where salubrinal, an EIF2α inhibitor, was used as a control (Figure S7N). To mitigate against the possibility of an identical mode of action for both inhibitors, we compared intracellular phospho-alterations of 35 growth and immune signaling proteins in 10 cell lines. Quantified vehicle (DMSO) normalized readouts for Compound II and C16 by unsupervised hierarchical clustering showed divergent effects, indicating different modes of action (Figure S7O).
A cohort of sensitive (IC50 < 5μM) and resistant (IC50 > 10μM) cell lines were defined from this analysis (Figures S7J–S7M). Approximately 40% of the cell lines, such as H1437 (lung) and LM38 (melanoma), were resistant to both compounds (9 of 22; ‘‘Res’’). We hypothesized that the cells selectively sensitive to either or both compounds (13 of 22; ‘‘Sen’’) would likely be under selective pressure for gain of expression of gene products belonging to immune function-specific pro-survival signaling. Gene set enrichment analysis (GSEA) using publicly available datasets and comparing samples from the former and latter cohorts indicated significant upregulation of ‘‘Inflammation pathway’’ in the ‘‘Sen’’ cohort (Eskiocak et al., 2017) (Figures S7P and S7Q). Both compounds individually showed selective activity in a sub-cohort of cells (HCC44, A375, and LOXMV1), yet mTOR activity or LATS1 activity can both be chemically modulated by each compound regardless of the sensitivity status (Figure 7E; Figure S7R), ruling out cellular drug levels as a confounder. We sought to examine if the selective sensitivity was due to a dependency of sensitive cells to TBK1- and PKR-dependent mTOR and Hippo pathway signaling (Figure S7Q). Assessment of cells with intrinsic vulnerability to both compounds (HCC44 and A375), compared with non-susceptible cells, revealed higher baseline activity of RALB, mTOR, and LATS1 (Figures S7R and S7S; Figures 7F and 7G). Genetic and pharmacological perturbation of PKR and TBK1 induced apoptosis in these cells (Figure S7T). High baseline mTOR and Hippo signaling activity in sensitive cells was associated with high baseline TBK1/mTOR/Sec5 and PKR/MST1/Exo84 complexes (Figure S7U; Figures 7H–7K). These observations indicate use of exocyst-dependent host defense signaling as a survival mechanism in certain cancer cells and a potentially targetable vulnerability (Figure 7L).
DISCUSSION
Collectively, this study establishes that RALB-exocyst effector subcomplexes comprise two proximal regulatory signaling pathways that specify a cellular host defense response and can be co-opted by certain cancer cells to promote their growth. Our findings are consistent with a model in which the RALB-exocyst signaling axis acts as a switch during host defense signaling to coordinate the amplitude and organization of mTOR and Hippo signaling outputs. RALB activation promotes dynamic association of the RALB effector duo Exo84 and Sec5 exocyst subcomplexes, with distinct kinase modules: PKR/MST1 (with Exo84) and TBK1/mTOR (with Sec5).
Functional convergence between Hippo and RAL-exocyst signaling was anticipated because of their shared participation in the regulation of cell polarity and previous reports of an interaction between the MST1 regulator, KIBRA (WWC1), and exocyst subunits (Moskalenko et al., 2002; Horikoshi et al., 2009; Bryant et al., 2010; Banks et al., 2012; Das et al., 2016; Rosse et al., 2009; Bodemann et al., 2011; Yu et al., 2015; Yoshihama et al., 2011; Zuo et al., 2019; Tay et al., 2019). Our work extends previous literature in which PKR-mediated regulation of anti-viral immunity was shown to be indirect via the regulation of protein translation, autophagy, and apoptosis (Tallóczy et al., 2002; Asakura et al., 2007; Levine and Kroemer, 2008).
Use of exocyst subcomplexes as a scaffold mechanism to couple anabolic (mTOR) and catabolic (Hippo and autophagy) signaling arms may represent an immune-surveillance strategy to facilitate gene expression of antiviral gene products, yet impede immediate propagation of infected cells (Meng et al., 2015). We report two distinct mechanisms for regulating this crosstalk between the signaling arms, calibration of: (1) the level of YAP1 bound to TBK1 or LATS1, and (2) the level of MST1 versus mTOR bound to Exo84. In a cohort of tumor cells, the level of baseline and adaptive coupling between the two signaling arms accurately predicted vulnerability to pathway perturbations. This suggests an adoption of the pathways for tumor cell survival. Selective toxicity to TBK1 depletion in KRAS mutant and BRAF mutant cancers is in alignment with our observations (Barbie et al., 2009; Eskiocak et al., 2017). Similar adaptive signaling relationships between RAL homologs A and B in tumor cell cellular proliferation (RALA) and survival (RALB) have been reported (Eskiocak et al., 2017; Cooper et al., 2013; Camonis and White, 2005). The interplay between host defense and cell survival signaling programs coordinated by RALA/B exocyst subcomplex protein networks warrants continued investigation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts Trever G. Bivona (trever.bivona@ucsf.edu)
Materials availability
All reagents generated in this study are available from the Lead Contact with proper credit.
Data and code availability
The published article includes all datasets generated or analyzed during this study. RNaseq dataset has been deposited on Gene Expression Omnibus (GEO: GSE176496).
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell line and culture condition
HEK293T, HeLa, HEBC30, U2OS, TBK1+/+ and TBK1−/− MEFs, were cultured as previously described (Stojdl et al., 2000; Chien et al., 2006; Eskiocak et al., 2017). A2058, RPMI7951, MNT1, SKMEL5, YUMAC, LM20, LM38, A375, PKR wt and PKR−/− MEFs cells, C2C12 wt and C2C12 YAP knock out cell lines were grown on DMEM supplemented with 10% fetal bovine serum (Atlanta Biologicals), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen). SKMEL5 and LOXIMVI was a gift from National Cancer Institute, Bethesda, MD; LM20 and LM38 were gifts from Monica Rodolfo Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy; YUMAC was a gift from Yale Skin Disease Research Center (Eskiocak et al., 2017). HepG2 cells were cultured on EMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. H596, H1437, H1573, HCC2450, H2887, H2347, H3255, A427, H1705, HCC44, HCC4017, H1755, H596, H125, HCC2279, HCC4019 cells (gifts from Dr. John Minna, UT Southwestern Medical Center) were cultured in RPMI supplemented with 5% fetal bovine serum (Atlanta Biologicals), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen). C2C12 wt and C2C12 YAP knock out cell lines were kindly provided by Dr. Duojia Pan (University of Texas Southwestern Medical Center), PKR wt and PKR−/− MEFs were kindly provided by Dr. John Bell (Ottawa Hospital, University of Ottawa), TBK1 wt and TBK1−/− MEFs were generated from mouse fibroblasts as previously described (Ou et al., 2011). Cell lines were fingerprinted and mycoplasma tested every 12 months.
METHOD DETAILS
Plasmids, cloning and established cell-line preparation
The mammalian expression plasmids pCDNA-FLAG-TBK1 (WT and K38M kinase deficient) were generously provided by Dr. James Chen (UT Southwestern Medical Center). Flag-MST1 expressing plasmid was generously provided by Duojia Pan (University of Texas Southwestern Medical Center). Exo84 was PCR amplified from pCMV-Myc-Exo84 vector (Bodemann et al., 2011) using primers with overhanging 5′ NOT1 and 3′ KPN1 sequences and ligated into NOT1/KPN1 digested N-terminal 3XFLAG pIRESpuro (Invitrogen). pUNO-PKR and pUNO-PKR-DN (kinase dead) expressing cDNA were purchased from invivogen. pCMV-Myc-Sec8, pCDNA3.1-HA-Sec5, pCMV-Myc-Sec5 and pCMV-Myc-exo84 expression constructs were amplified from previously generated constructs (Bodemann et al., 2011). The pBABE-RALB-Hygro plasmid was generated by restriction digestion of pBABE-RALB-puro (Bodemann et al., 2011) with EcoRI and BamHI followed by ligation of the resulting RALB cDNA into the corresponding polyclonal region of pBABE-Hygro (Advanced mammalian gene transfer: high titer retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line (Morgenstern and Land 1990).
To generate stable RALB23V expressing cell lines, retroviral expression constructs were packaged by cotransfection of phoenix cells (ATCC CRL-3213) with pBABE-RALB-Hygro using Fugene6. 48 hour and 72 hour viral supernatants were collected. HEK293T cells were plated at a density of 2.5x106 cells/10-cm dish. Two infections, one at 24 hour and another at 48 hours, were carried out with retroviral particles and polybrene (10 μg/ml). On day 3 post-infection, cells were seeded into multiple 6-well dishes at 50% confluency. 24-hour post-seeding cells were subjected to 100μg/ml hygromycin selection. RALB expression was validated by SDS-PAGE.
Antibodies and reagents
Sec8 2E12, 10C2, 5C3 epitope specific antibodies were generated as previously described (Inamdar et al., 2016); monoclonal anti-Flag M2 (Sigma Catalog#F1804) mouse anti-cMyc or mouse monoclonal anti-HA F-7 (Santa Cruz Biotechnology sc-7392). Anti-Sec5, -Sec6, -Exo84 and -RALB monoclonal antibodies were used as previously described (Bodemann et al., 2011). Protein A/G Plus-Agarose beads (sc-2003) and anti-HA antibody (sc-7392 and sc-805) were purchased from Santa Cruz. Anti-FLAG antibody (F3165) (Cat# A2220) were purchased from Sigma. ICPs and M antibodies were from Abcam; HN antibody was from Kerafast. Additional antibodies were purchased from Sigma (anti-Actin, A1978), Santa Cruz Technology (anti-IRF3, YAP1 63.7, anti-ICP0), Cell Signaling Technology (anti-PKR, 12297; S6K, 9202; p-S6K (T421/S424), 9204; S6, 2217; p-S6 (S235/236), 4858; TBK1, 3504; p-TBK1 5483S, YAP1 8418, p-YAP1 (S127), LATS1 9153, p-LATS1 (T1079) MST1 3682 p-MST1 (T183) 3681, EIF2α 5324, p-EIF2α 3398, ULK1 6439, LAMTOR3 8168 LAMTOR1 8175 LAMTOR2 8145, RAGA 4357, RAGD 4470, KIBRA 8774, GAPDH 5174, mTOR 2972, CDK7 2916), Abcam (p-PKR (T466) ab32036, Azi2 ab232654, WDR73 ab103864, PRPS1 ab137577, IVNS1ABP ab127566, anti-HNM), Fitzgerald (anti-ICP5) Bethyl laboratories (TP53BP1 A300–272A). pan-RAL antibodies were purchased from NewEast bioscience (#26913). GST-RBD was prepared as demonstrated before (Bodemann et al., 2011)
Trophic and immunogenic stimulation
HEK293T, HeLa or Human Bronchial Epithelial Cells (HBEC) cells grown on normal growth media enriched in growth nutrients (GIBCO high glucose DMEM) or grown on nutrient depleted EBSS (Earle’s basic saline solution) (Sigma# E2888). A range of different doses and duration of the stimuli were tested to determine the ultimate induction of host defense signaling and autophagic starvation signaling. Relevant molecular markers were measured by western blot, qPCR and RNaseq as appropriate.
For host defense responses, HeLa cells were treated with DMEM (+10%FBS) with or without 2ug/ml (for HeLa) or 50ug/ml (for HEK293T) pI:C; 200 HA (Hemagglutination Assay)/ml Sendai virus (Cantell strain); or 2.5 MOI HSV1 as previously described (Orvedahl et al., 2007).
Protein detection by immunoblot
Cellular proteins were solubilized with denaturing lysis buffer (40% Glycerol, 240mM Tris-HCl, pH 6.8, 8% SDS). Resulting protein concentrations were quantified with a commercial BCA kit (Thermo scientific). For SDS-PAGE, samples with equivalent total protein were denatured at 95°C in the presence of 0.04% bromophenol blue and 5% β-marcapto ethanol. Samples were separated by 4%–20% gradient gels and transferred onto PVDF membranes (BIO-RAD) using standard protocols. Membranes were blocked with BSA for 1–2 hour before overnight primary antibody incubation followed by washes and 1- to 2-hour secondary antibody incubation (peroxidase conjugated antibody from Jackson lab; fluorophore conjugated antibody from Li-Cor). Secondary antibodies were detected using the enhanced chemiluminescence peroxidase substrate reagent (Pierce) and by fluorescence intensity measured in Image Studio Lite™ software built into the LI-COR Odyssey imager.
Immunoprecipitation
For large-scale endogenous protein immunoprecipitations, five 150 cm dishes per experimental condition were grown to confluence. Cells were washed on ice with PBS and collected in non-denaturing lysis buffer (20 mM Tris-HCl pH 7.4, 137mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 10mM MgCl2, 2mM EGTA, Roche EDTA-free complete ULTRA protease inhibitor and Roche PhosphoSTOP). Cells were incubated with mild agitation for 1 hour at 4°C and cleared at 16,000 X g for 10 min at 4°C. 2mg of lysate was brought to a concentration of 1.5mg/mL by dilution with fresh lysis buffer. Immunoprecipitations were carried using 2.5mg of antibody per sample. For endogenous and single tagged proteins this was followed by 2–18 hour precipitation of antibody-antigen complexes (as optimized for each antibody) using 90μl of Protein A/G-agarose beads (Santa Cruz Biotechnology, SC-2003). Subsequently, complexes were washed in lysis buffer 4 times at 4°C and eluted with 90μl 2X SDS sample buffer (BIORAD Catalog#161–0737) at 95°C for 12 min.
For overexpression IP, 3X106 HEK293T cells were seeded, in four 10cm dishes per condition, in DMEM + 10% FBS. The following day, cells were transfected with 3μg plasmid using Fugene 6 at a ratio of 3:1 (mL Fugene 6 to mg DNA). 48 hours post transfection, cells were collected in non-denaturing lysis buffer (20 mM Tris-HCl pH 8.0, 100mM KCl, 0.1% NP-40, 0.5% sodium deoxycholate, 10mM MgCl2, 2mM EDTA) with protease and phosphatase inhibitors (Roche EDTA-free cOmplete ULTRA and PhosphoSTOP) and lysates prepared as above. Endogenous IP, for when RALB is a target, had to be performed either with 0.4mM EDTA or in presence of ectopic overexpression of RALB.
For Taptagged proteins, antibody-antigen complexes were isolated using 200ul of IgG-Sepharose (GE healthcare) followed by a 10 hour protease digestion and subsequent second degree capture with 200ul of calmodulin-Sepharose beads (GE healthcare) in calmodulin binding buffer (150mM Tris-HCl pH 8.0, 150mM NaCl, 1mM Mg-Acetate, 1mM Imidazole, 0.1% NP40, 2 mM CaCl2). Complexes were washed in rinsing buffer (10mM Tris-HCl pH 8.0, 75mM NaCl, 1mM Mg-Acetate, 1mM Imidazole, 2 mM CaCl2) 6 times at 4°C and eluted with 120μl 2X SDS sample buffer (BIORAD Catalog#161–0737) at 95°C for 12 min.
Mass spectrometry sample preparation
Samples were separated with SDS-PAGE 4%–20% gradient gels (Bio-Rad) and stained with colloidal Coomassie Gel code Blue (Thermo Fisher Scientific, San Jose, CA). For each sample, fixed immunoprecipitated proteins were separated on the gel for 15mm and were excised as a single fragment with sterile scalpel and homogenized in an Eppendorf tube with a sterile needle. Fragments were destained with 50 mM triethylammonium bicarbonate (TEAB)/acetonitrile (1:1, v/v) at 37°C for 30 minutes, and dehydrated with acetonitrile at room temperature, followed by reduction/alkylation using DTT and iodoacetamide. Samples were then rehydrated with trypsin solution (200ng/μl in 50mM TEAB) and digestion proceeded at 37°C overnight. Peptides were extracted by a 30 min incubation at 37°C with extraction buffer (Guo et al., 2014) to a final concentration of 66.7% acetonitrile and 5% trifluoroacetic acid (TFA). All steps were carried out on a thermomixer shaker (Eppendorf, NJ). Extracts were dried by vacuum centrifugation. Salts were removed using Oasis HLB micro-Elution plate (Waters, MA) before LC-MS/MS analysis.
MS/MS data were acquired with an Orbitrap Fusion LumosTM as described below and analyzed using PD2.1 for TMT-based methods (Proteome Discoverer 2.1 by Thermo-Fisher) and a previously established CPFP pipeline (Trudgian et al., 2010) for spectral count based methods. To minimize batch variability, control and biological replicates were run consecutively.
Tandem mass tag (TMT) labeling
10-plex and 6-plex isobaric tags (Thermo Scientific) were dissolved in 41 μL of acetonitrile (20 ng/μl). 10 μL of TMT reagent stock solution was mixed with digested peptides and incubated at room temperature for 2h. The reaction was quenched for 15 min with hydroxylamine (0.5% v/v). The TMT-labeled samples were mixed in equimolar ratio. Samples were then run through C18 solid-phase extraction (SPE) cartridge (Sep-Pak, Waters) and lyophilized prior to fractionation. Labeled peptides were reconstituted in 2% acetonitrile, 0.1% TFA prior to MS analysis using a Fusion Orbitrap Lumos (Thermo) mass spectrometer connected to a Dionex Ultimate 3000 UHPLC (Thermo). An Easy-Spray column (75 μm x 50 cm) packed with 2 μm C18 material was used for orthogonal peptide separation at low pH using a linear gradient from 100% mobile phase A (2% acetonitrile, 0.1% Formic acid) to 28% mobile phase B (80% acetonitrile, 0.1% Formic acid) in 90 min. Source voltage was set to 2.2 kv and capillary temperature at 275°C in the positive ion mode. The mass spectrometer was set to acquire data in data-dependent mode at MS1 level (full scan) ions within the m/z range of 400–1600 were scanned at the resolution of 120,000. In the second level of scanning (MS2), the top-10 previously selected ions were isolated and fragmented using collision-induced dissociation (CID) mode with collision energy of 35. At the MS3 level, a further fragmentation of the MS2 fragments was performed using high-energy collision-induced dissociation (HCD) mode to identify TMT labeled peptides. A multinotch MS3-based TMT method was used for analysis of the samples. Raw data acquired by the mass spectrometer was searched against a reviewed UniProt human database in Proteome Discoverer 2.2 (Thermo). Carbamidomethyl cysteine, TMT6 plex labeling at lysine and N terminus were set as static modifications and oxidation of methionine was used as dynamic modifications. False discovery rate was set to 1% and up to 3 missed cleavages were accepted. Reporter ion quantification method was used, top 3 peptides were used for area calculation and results were normalized based on total peptide amount. In total ~9173 protein groups were identified of which 7801 protein groups were successfully labeled and quantified. Ingenuity Pathway analysis (IPA) was used to analysis the identified proteins (Trudgian et al., 2010).
Data analysis strategy for quantitative TMT-based mass spectrometry
Data from the Orbitrap Fusion were processed using Proteome Discoverer Software (ver. 2.1.0.62). MS2 spectra were searched using Sequest HT and MS Amanda against UniProt database. Search parameters were specified as: trypsin enzyme, 3 missed cleavages allowed, minimum peptide length of 6, precursor mass tolerance of 10 ppm, and a fragment mass tolerance of 0.2 Daltons. Carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da), ubiquitination (+GG; +114.043 Da) and phosphorylation (+79.966 Da) was set as a dynamic modification. Peptide-spectral matches (PSMs) error rates were determined using the target-decoy strategy coupled to Percolator modeling of positive and false matches (Käll et al., 2007; Spivak et al., 2009).
Acquired quantitative reporter ion intensities were exported as a 10xN or 6XN column matrix where N number of rows represented quantitative estimates of the candidate partners and column indicated sample IDs (Table S1). These columns were subjected to following adjustments- 1) columns were separated onto specific and control cohorts and a signal to noise ratio was measured between protein intensity profiles of the two cohorts to detect background noise; 2) proteins with negative S2N score were characterized as background and discarded from the dataset 3) noise-controlled matrix was row and column normalized and were clustered 1D along the protein intensity profile axis using Euclidian distance as a metric. This clustering method clustered the protein profiles based on their intensity across the specific and control samples as well as variation within the intensity profiles within specific and control cohorts. From the generated clustered intensity profiles, a set of 50 protein profiles closest Euclidian rank to bait Sec8 was picked as a list of ‘Top50’ proteins from each of the four datasets.
Intersection of the four datasets was taken to define the Sec8 constitutive core interactome (Table S1). The inclusion criteria for the core complex were as such as Top (Ab1 Ո Ab2 Ո Ab3); Top (Run1 Ո Run2); Top (HEK293T Ո HeLa), where Top = Proteins with reporter ion intensity (specific/control) ≥ 2. These core interactome was manually curated for functional annotation using STRING and GO databases and represented as ‘hub and spoke network’ where bait Sec8 is represented as a hub and rest of the partners as spoke surrounding it. Known interactions between the proteins were imported from BIORID database. Subsequently, the core Sec8 Next, we calculated relative abundance of a protein by converting quantitative estimate for each protein’s abundance into a ratio of bait- Sec8’s. The data were then represented as a stimulus-specific ‘Relative abundance-total abundance vector plot’ where the X and Y variables were defined as follows: Relative abundance of Protein X to IP = Average reporter ion intensity (Protein X/Sec8); Abundance = Normalized transcript level quantified by RNaseq data. Statistical analysis was performed to measure the significance of the enrichment using students paired t test.
Data analysis strategy for semiquantitative spectral count-based mass spectrometry
For label-free analyses, specific interactors were characterized by quantitative enrichment of the identified protein in the test sample versus a negative control. For overexpression-based IP-MS, the negative control was the IP enrichment value of the identified protein in an empty vector transfection sample. For endogenous IP, the negative control was the IP enrichment value of the identified protein in a non-specific antibody control such as anti-HA or anti-IgG.
LC-MS/MS data were generated using the CPFP pipeline and quantified using spectral count-based, semiquantitative label-free quantification. The finalized dataset for overexpression-based IP was represented as a 2xN matrix where row indicates average intensity values of the potential partners and column indicates two cohorts: specific and controls. This dataset was further controlled for false positives using the ‘‘CRAPOME’’ database (https://reprint-apms.org) to exclude commonly identified non-specific interactors (Mellacheruvu et al., 2013). High stringency inclusion criteria cutoffs allowed proteins to pass if they were identified in less than 5% of the reports within the CRAPOME database, spectral count ≤ 1 in the negative control IP condition, and present in specific pull down with at least 2 spectral counts. Whenever a ratio was taken, empty cells in the generated matrix were replaced with a spectral count value of 0.5. Furthermore, ratio was taken of the average spectral count values of the two cohorts- with or without stimuli. Ratios were Log normalized to generate a linear scale ranging from +n to –n centered around 0 (modulus of n = maxima/minima). Candidate partners with positive value in this scale indicated partners that were enriched when exposed to stimuli whereas partners with negative values depleted upon stimuli. Partners with ‘0’ value indicate protein complex membership that is unresponsive to the stimuli.
Following are the numbers of replicates for each interactome analysis: endogenous RALB endogenous interactome, n = 4; Exo84/Sec5-pI:C, n = 2; Exo84/Sec5-RALB23V, n = 2; Exo84/Sec5+RALB23V, n = 2; active TBK1/PKR and inactive TBK1/PKR n = 2; TBK1/PKR-pI:C/SeV n = 2; TBK1/PKR+pI:C/SeV n = 2 (Tables S2–S5).
For RALB dependent Exo84 and Sec5 interactomes, separately generated interactomes were integrated and quantitative values for each protein were represented as Log2 of the ratio of the intensity values in Exo84 (+RALB23V) and Sec5 (+RALB23V) datasets. Similarly, for TBK1/PKR ± pI:C/SeV, the quantitative value for each protein was represented as Log2 of the ratio of the intensity values in TBK1/PKR-pI:C/SeV and TBK1/PKR+pI:C/SeV datasets (Tables S6 and S7).
qPCR and RNaseq
500,000 HeLa cells were plated in 6-well cell culture dishes and the following day cells were then washed with PBS. Cells were lysed, and RNA was collected via RNeasy Kit (QIAGEN). mRNA was reverse transcribed into cDNA and relative transcript abundance was measured by Taqman-qPCR probe sets (Applied Bio-Science). RNaseq for the whole genome transcripts was performed using UTSW sequencing core facility (GEO dataset: GSE176496). For background noise detection and correction, a non-parametric version of model-based background correction method which uses an extended model of robust multiarray analysis (RMA) that incorporates information from negative control beads (Xie et al., 2009). The background-corrected data were then subjected to quantile normalization to obtain identical sample distributions. To assess the differential expression between samples, we used the linear models for microarray data (LIMMA) method Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. LIMMA is specialized to minimize the standard error, for sparse sample matrices, by using an empirical Bayes method to compute the statistical significance and the fold change between two classes of samples. P values for expression changes were computed and adjusted for multiple hypothesis testing (Smyth, 2004).
Immunofluorescence
Cells were seeded in plastic-bottomed 8-well chamber slides (Corning) and grown to 90% confluency prior to treatment with indicated reagents. Cells were fixed with 3.7% formaldehyde for 15 minutes then permeabilized with 0.5% Triton-X for 15 min. Slides were washed with PBS and blocked with PBTA (PBS, 1% Tween 20, and 2% BSA). Slides were stained with primary antibody for an hour followed by appropriate secondary antibodies and DAPI. Stained slides were mounted on glass with anti-fade mounting medium (VECTASHIELD ProLong Gold). Secondary antibodies DyLight 488 donkey anti-Rabbit and DyLight 594 donkey anti-Mouse were purchased from Jackson ImmunoResearch Laboratories, inc. AlexaFluor 350 donkey anti-Mouse and Alexa Fluor 647 donkey anti-Mouse were purchased from Invitrogen.
HeLa LC3-GFP cells (Bodemann et al., 2013) were grown to 90% confluence and exposed to the indicated stimuli. Cells were fixed with 3.7% PFA, quenched with NH4Cl, and permeabilized with digitonin followed by mounting as described above.
Epifluorescence images (Figure S1C; Figure 3D) were captured using band-pass excitation/emission filter sets (GFP, AlexaFluor 488) (Rhodamine, AlexaFluor 594) with a 100X, 40X, 20X or 10X objective on a Zeiss LSM 700 laser scanning confocal microscope. Images were captured using microscope default Micro-Manager software and further analyzed with ImageJ (with FiJi opensource package).
Superose 6 FPLC
HEK293T, HeLa and MDCK cells were grown to confluence on 15 cm dishes under the indicated culture conditions. Cells were then washed 2X with PBS and collected in DHE buffer (20mM HEPES, 150mM NaCl, 0.5% NP40, and the protease inhibitors Pefabloc, Antipain, Leupeptin, and Pepstatin A). Resulting lysates were incubated on ice for 20 min, then cleared at 20,000Xg for 10 min at 4°C. The supernatant was filtered through 0.22um filter and 0.5 mL of filtered sample was injected into a Superose 6 column. The column was operated at a flow rate of 0.2 ml/min and 0.5 mL fractions were collected. The collected samples were mixed with 100ml 6X Laemmli sample buffer and boiled prior to separation by SDS-PAGE.
siRNA
Synthetic siRNA custom pools targeting LONRF1 (control), Sec8, LATS1, TBK1 and PKR were purchased from Dharmacon and used as previously described (Cooper et al., 2017; Eskiocak et al., 2017). Sense sequences of are as follows:
LONRF1#1: UCAGAGAGCUUCAUGAUUU; LONRF1#2: GACCAAGAAUGUUCCAAUA; LONRF1#3: UCACACAGCUGUUGGAAGA; LONRF1#4: GCACUGCCGACAUUGAAUA Sec8#1: GAAUUGAGCAUAAGCAUGU Sec8#2: UAACUGAGUACUUGGAUAU; Sec8#3: GCCGAGUUGUGCAGCGUAA; Sec8#4: ACUGAGUGACCUUCGACUA; LATS1#1: GAACCAAACUCUCAAACAA; LATS1#2: GCAAGU CACUCUGCUAAUU; LATS1#3: GAAAUCAAGUCGCUCAUGU; LATS1#4: GAUAAAGACACUAGGAAUA; TBK1#1: GAACGUAGAUU AGCUUAUA; TBK1#2: UGACAGAGAUUUACUAUCA; TBK1#3: UAAAGUACAUCCACGUUAU; TBK1#4: GGAUAUCGACAGCAGA UUA; PKR#1: CAAAUUAGCUGUUGAGAUA; PKR#2: GGAAAGACUUACGUUAUUA; PKR#3: GCAGAUACAUCAGAGAUAA; PKR#4: GAUCUUAAGCCAAGUAAUA
Synthetic siRNA pools targeting Exo84 and RALB were purchased from Sigma (Exo84#1: SASI_Hs01_00215827; Exo84#2: SASI_Hs01_00215828 Exo84#3: SASI_Hs01_00215829; RALB#1: SASI_Hs02_00334659; RALB#2: SASI_Hs01_00242021; RALB#3: SASI_Hs01_00242022) and used as previously described (Bodemann et al., 2011; Ou et al., 2011; Chien et al., 2006).
YAP1 Reporter assay
Cells were transiently transfected with 8XGTIIC-luc-TEAD (for YAP1) or IFN-β promoter luciferase reporter expressing firefly luciferase (~575nm), CMV promoter regulated renilla luciferase (~475nm) and empty vector or cDNAs of interest (Dupont et al., 2011). Following a 48- or 60-hour incubation, cell was lysed in reporter lysis buffer (PROMEGA). Luminescence was quantified using PHERAstar plate reader (BMG Biotech) at indicated wavelengths. Firefly by renilla ratios were calculated. They were further normalized to either a negative control transfection or a positive control transfection as indicated. Normalized hippo activation fold change was represented as histograms representing mean ± SD. Statistical comparisons between treatment cohorts were evaluated using Student’s unpaired t test.
Peptide phosphorylation assay
Peptide and purified PKR protein mixture was diluted to 1 ug/uL in 2% acetonitrile (ACN), 0.1% formic acid, and analyzed by LC/MS/MS, using an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex). Samples were injected onto a 75 μm i.d., 50-cm long EasySpray column (Thermo), and eluted with a gradient from 1%–28% buffer B over 60 min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 2.2 kV and an ion transfer tube temperature of 275°C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Phosphorylated peptides of interest were observed by generating an extracted ion chromatogram corresponding to a 0.01 Da window around the theoretical m/z of the +3 charged monoisotopic mass of the singly phosphorylated peptide of interest.
Small Molecule Cell Viability Assays
For dose response analyses, cells were seeded in 96-well plates at a density of 1–5K/well. Twenty-four hours post seeding, compounds solubilized in vehicle or vehicle alone (equal volume) were added to achieve the required final concentrations. Cell viability was measured with Cell Titer Glo (Promega), 72–96 hours post compound exposure. Luminescence values were normalized using vehicle control treated cells. Response curves, from biological triplicates, were modeled using a nonlinear regression curve fit with a three-parameter dose response using GraphPad Prism 6.
HSV1 cytotoxic assay
Cells were seeded in 96-well plates at a density of 1–5K/well in 90 μL growth media. Twenty-four hours post seeding 109 PFU HSV1 virus stock were serially diluted in Eppendorf tubes using growth media. 10μl of the virus stock dilution was added each well. 72–96 hours post virus exposure, cell viability was measured with Cell Titer Glo (Promega). Data were normalized to the lowest non-toxic dose. Response curves, from biological triplicates, were modeled using a nonlinear regression curve fit with a three-parameter dose response using GraphPad Prism 6.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analyses were performed using GraphPad Prism6. A two-tailed unpaired Student’s t test was used to determine significance when two conditions were compared unless otherwise specified. For comparison between two groups in multivariate data, a two-way ANOVA was used. In both cases values of p < 0.05 were considered as significant. p values are represented in Figure legends and on figures as asterisks where * indicates p < 0.05, ** indicates p < 0.01, ***indicates p < 0.005 and ****indicates p < 0.001. Data are shown as the mean ± SD (standard deviation from the mean) unless otherwise specified. SEM stands for standard error of the mean. Number of replicates (n) for each experiments are denoted in the individual figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Flag M2 | Sigma-Aldrich | F1804: RRID:AB_262044 |
| Flag | Sigma-Aldrich | F3165: RRID:AB_259529 |
| c-Myc | Santa Cruz | sc-40: RRID:AB_2857941 |
| HA | Santa Cruz | sc-7392: RRID:AB_627809 |
| HA | Santa Cruz | sc-805: RRID:AB_631618 |
| Actin | Sigma-Aldrich | A1978: RRID:AB_476692 |
| PKR | Cell Signaling Technology | 12297: RRID:AB_2665515 |
| S6K | Cell Signaling Technology | 9202: RRID:AB_331676 |
| p-S6K (T421/S424) | Cell Signaling Technology | 9204: RRID:AB_2265913 |
| S6 | Cell Signaling Technology | 2217: RRID:AB_331355 |
| p-S6 (S235/236) | Cell Signaling Technology | 4858: RRID:AB_916156 |
| TBK1 | Cell Signaling Technology | 3504: RRID:AB_2255663 |
| p-TBK1 | Cell Signaling Technology | 5483: RRID:AB_10693472 |
| YAP1 | Cell Signaling Technology | 8418: RRID:AB_10950494 |
| p-YAP1 (S127) | Cell Signaling Technology | 13008: RRID:AB_2650553 |
| LATS1 | Cell Signaling Technology | 9153: RRID:AB_2296754 |
| p-LATS1 (T1079) | Cell Signaling Technology | 8654: RRID:AB_10971635 |
| MST1 | Cell Signaling Technology | 3682: RRID:AB_2144632 |
| p-MST1 (T183) | Cell Signaling Technology | 3681: RRID:AB_330269 |
| EIF2α | Cell Signaling Technology | 5324: RRID:AB_10692650 |
| p-EIF2α (S51) | Cell Signaling Technology | 3398: RRID:AB_2096481 |
| LAMTOR3 | Cell Signaling Technology | 8168: RRID:AB_10949501 |
| LAMTOR1 | Cell Signaling Technology | 8175: RRID:AB_11178807 |
| LAMTOR2 | Cell Signaling Technology | 8145: RRID:AB_10971636 |
| GAPDH | Cell Signaling Technology | 5174: RRID:AB_10622025 |
| mTOR | Cell Signaling Technology | 2972: RRID:AB_330978 |
| CDK7 | Cell Signaling Technology | 2916: RRID:AB_2077142 |
| p-PKR (T466) | Abcam | ab32036: RRID:AB_777310 |
| Azi2 | Abcam | ab232654 |
| WDR73 | Abcam | ab103864: RRID:AB_10716295 |
| PRPS1 | Abcam | ab137577 |
| IVNS1ABP | Abcam | ab127566:RRID:AB_11140332 |
| TP53BP1 | Bethyl laboratories | A300–272A: RRID:AB_185520 |
| RALB | Gift from Dr. Larry Feig | NA |
| ICP5 | Abcam | ab6508: RRID:AB_305530 |
| ICP0 | Abcam | ab6513: RRID:AB_305536 |
| HN | Kerafast | EMS016 |
| M | Abcam | ab34752: RRID:AB_777075 |
| Sec6 | Inamdar et al., 2016 | NA |
| Sec5 | This paper | NA |
| Sec8 Ab1 (2E12) | This paper | NA |
| Sec8 Ab2 (5C1) | This paper | NA |
| Sec8 Ab3 (10C2) | This paper | NA |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| 5-alpha Competent E. coli | New England Biolabs | C2987H |
| Sendai virus (Cantell strain) | ATCC | VR-907 |
| Herpes simplex virus Δ34.5 (HSV1Δ34.5) | Orvedahl et al., 2007 | NA |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Trypsin | GE Healthcare | SH30042.02 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15070063 |
| FBS | Atlanta Biologicals | S11150 |
| Rapamycin | Selleck Chemicals | S1039–1ml |
| C16 | Tocris | 5382 |
| polyI:C | Invivogen | tlrl-picw |
| EBSS | Thermo Fisher Scientific | 24010043 |
| Lipofectamine RNAiMAX Transfection Reagent | Life Technologies | 13778150 |
| Protein A/G Plus-Agarose beads | Santa Cruz Biotechnology | sc-2003 |
| Fugene6 | Promega | E2692 |
| Puromycin | Sigma-Aldrich | P7255 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Cell titer glo | Promega | G7573 |
| Caspase glo | Promega | G8090 |
| Bright glo luciferase assay | Promega | E2620 |
| TMT reagent (6 plex and 10 plex) | Thermo Fisher Scientific | 90402, 90110 |
| RNeasy kit | QIAGEN | 74106 |
|
| ||
| Deposited data | ||
|
| ||
| RNaseq | Gene Expression Omnibus (GEO) | GEO: GSE176496 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HeLa | ATCC | CCL-2 |
| HEK293T | ATCC | CRL-3216 |
| U2OS | ATCC | HTB-96 |
| C2C12 | ATCC | CRL-1772 |
| MNT1 | ATCC | CRL-3450 |
| A2058 | ATCC | CRL-11147 |
| SKMEl5 | ATCC | HTB-70 |
| HepG2 | ATCC | HB-8065 |
| Phoenix cells | ATCC | CRL-3213 |
| HBEC30 | Gift from Dr. John Minna | NA |
| TBK1 +/+ and TBK1 −/− MEFs | Ou et al., 2011 | NA |
| PKR wt and PKR−/−MEFs | Gift from Dr. John Bell | NA |
| RPMI7951 | ATCC | HTB-66 |
| LOXIMVI | Millipore sigma | SCC201 |
| YUMAC | Gift from Yale Skin Disease Research Center | NA |
| LM20 | Gift from Dr. Monica Rodolfo Fondazione | NA |
| LM38 | Gift from Dr. Monica Rodolfo Fondazione | NA |
| A375 | ATCC | CRL-1619 |
| C2C12 wt and C2C12 YAP knock out cells | Gift from Dr. Duojia Pan | NA |
| LATS KO 293A cells | Gift from Dr. Duojia Pan | NA |
| TBK1 KO 293T cells | Gift from Dr. Zhijian ‘James’ Chen | NA |
| PKR KO 293T cells | This paper | |
| H596 | Gift from Dr. John Minna | NA |
| H1437 | Gift from Dr. John Minna | NA |
| H1573 | Gift from Dr. John Minna | NA |
| HCC2450 | Gift from Dr. John Minna | NA |
| H2887 | Gift from Dr. John Minna | NA |
| H2347 | Gift from Dr. John Minna | NA |
| H3255 | Gift from Dr. John Minna | NA |
| A427 | Gift from Dr. John Minna | NA |
| H1705 | Gift from Dr. John Minna | NA |
| HCC44 | Gift from Dr. John Minna | NA |
| HCC4017 | Gift from Dr. John Minna | NA |
| H1755 | Gift from Dr. John Minna | NA |
| H596 | Gift from Dr. John Minna | NA |
| H125 | Gift from Dr. John Minna | NA |
| HCC2279 | Gift from Dr. John Minna | NA |
| HCC4019 | Gift from Dr. John Minna | NA |
|
| ||
| Oligonucleotides | ||
|
| ||
| Exo84#1 | Sigma Aldrich | SASI_Hs01_00215827 |
| Exo84#2 | Sigma Aldrich | SASI_Hs01_00215828 |
| Exo84#3 | Sigma Aldrich | SASI_Hs01_00215829 |
| RALB#1 | Sigma Aldrich | SASI_Hs02_00334659 |
| RALB#2 | Sigma Aldrich | SASI_Hs01_00242021 |
| RALB#3 | Sigma Aldrich | SASI_Hs01_00242022 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pIRESpuro3 | Clontech | 631619 |
| pUNO-PKR | Invivogen | puno1-hpkr |
| pUNO-PKR DN (kinase dead) | Invivogen | puno1-hpkr-dn |
| V5-PKR | This paper | NA |
| 8XGTIIC-luc-TEAD | Addgene | 34615 |
| pCDNA-FLAG-TBK1 WT and K38M kinase deficient | Seth et al., 2005 | NA |
| Flag-MST1 | Gift from Dr. Duojia Pan | NA |
| Flag-MST1-S21A | This paper | NA |
| Flag-MST1-S21E | This paper | NA |
| Myc-Exo84 | Moskalenko et al., 2003 | NA |
| Taptag-Exo84 | This paper | NA |
| HA-Sec5 | Moskalenko et al., 2002 | NA |
| pIRESpuro-Exo84 | This paper | NA |
| Flag-RalB | Cascone et al., 2008 | NA |
| Flag-RalB(G23V) | Cascone et al., 2008 | NA |
| Flag-RalB(G23V, E38R) | Cascone et al., 2008 | NA |
| Flag-RalB(G23V, A48W) | Cascone et al., 2008 | NA |
| Flag-RalB(E38R) | Cascone et al., 2008 | NA |
| Flag-RalB(A48W) | Cascone et al., 2008 | NA |
| Myc-Rlip(RBD) | Moskalenko et al., 2002 | NA |
| pBABE-puro | Morgenstern and Land, 1990 | Addgene; 1764 |
| pBABE-hygro | Morgenstern and Land, 1990 | Addgene, 1765 |
| Software and algorithms | ||
| BioRender | BioRender | https://biorender.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 6 | GraphPad | RRID:SCR_002798 |
Highlights.
Exocyst protein subcomplexes impart phenotypic specificity
Exo84 exocyst contains PKR/MST1, activating Hippo signaling
Sec5 exocyst contains TBK1/mTOR, activating mTOR signaling
Cancer cells can coopt these subcomplexes for cellular survival
ACKNOWLEDGMENTS
We thank Drs. John Minna, Adi Gazdar, Rolf Brekken, Zhijian ‘‘James’’ Chen, Duojia Pan, Lawrence Lum, and Sandra Schmid for several of the reagents and helpful discussions. This research project was conducted with support from the National Cancer Institute (NCI), Cancer Prevention and Research Institute of Texas (CPRIT) immunity and cancer grant, Welch Foundation, National Institutes of Health (R01CA231300, U54CA224081, R01CA204302, R01CA211052, and R01CA169338), and the Pew and Stewart Foundations.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109491.
DECLARATION OF INTERESTS
T.G.B. is an advisor to Novartis, AstraZeneca, Revolution Medicines, Array, Springworks, Strategia, Relay, Jazz, and Rain and receives research funding from Novartis and Revolution Medicines and Strategia. M.A.W. is the current Chief Scientific Officer for Samumed LLC and former Chief Scientific Officer and Vice President of Tumor Cell Biology at Pfizer, Inc. E.A.M. is currently Principal Scientist at Pfizer, Inc.
REFERENCES
- Ahmed SM, Nishida-Fukuda H, Li Y, McDonald WH, Gradinaru CC, and Macara IG (2018). Exocyst dynamics during vesicle tethering and fusion. Nat. Commun 9, 5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonia RJ, Castillo J, Herring LE, Serafin DS, Liu P, Graves LM, Baldwin AS, and Hagan RS (2019). TBK1 Limits mTORC1 by Promoting Phosphorylation of Raptor Ser877. Sci. Rep 9, 13470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, and Piccolo S (2013). A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059. [DOI] [PubMed] [Google Scholar]
- Asakura Y, Fujiwara Y, Kato N, Sato Y, and Komatsu T (2007). Serine/threonine kinase PKR: a sentinel kinase that discriminates a signaling pathway mediated by TLR4 from those mediated by TLR3 and TLR9. Am. J. Hematol 82, 640–642. [DOI] [PubMed] [Google Scholar]
- Balakireva M, Rossé C, Langevin J, Chien YC, Gho M, Gonzy-Treboul G, Voegeling-Lemaire S, Aresta S, Lepesant JA, Bellaiche Y, et al. (2006). The Ral/exocyst effector complex counters c-Jun N-terminal kinase-dependent apoptosis in Drosophila melanogaster. Mol. Cell. Biol 26, 8953–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks L, Pim D, and Thomas M (2012). Human tumour viruses and the deregulation of cell polarity in cancer. Nat. Rev. Cancer 12, 877–886. [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuvanakantham R, Li J, Tan TT, and Ng ML (2010). Human Sec3 protein is a novel transcriptional and translational repressor of flavivirus. Cell. Microbiol 12, 453–472. [DOI] [PubMed] [Google Scholar]
- Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, Formstecher E, Maiti M, Hazelett CC, Wauson EM, Balakireva M, et al. (2011). RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen TM, Texier Y, van Beersum SE, Horn N, Willer JR, Mans DA, et al. ; UK10K Rare Diseases Group (2016). An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun 7, 11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Datta A, Rodríguez-Fraticelli AE, Peränen J, Martín-Belmonte F, and Mostov KE (2010). A molecular network for de novo generation of the apical surface and lumen. Nat. Cell Biol 12, 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camonis JH, and White MA (2005). Ral GTPases: corrupting the exocyst in cancer cells. Trends Cell Biol 15, 327–332. [DOI] [PubMed] [Google Scholar]
- Cascone I, Selimoglu R, Ozdemir C, Del Nery E, Yeaman C, White M, and Camonis J (2008). Distinct roles of RalA and RalB in the progression of cytokinesis are supported by distinct RalGEFs. EMBO J 27, 2375–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, and White MA (2008). Characterization of RalB-Sec5-TBK1 function in human oncogenesis. Methods Enzymol 438, 321–329. [DOI] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Bala-kireva MG, Romeo Y, Kopelovich L, Gale M Jr., et al. (2006). RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127, 157–170. [DOI] [PubMed] [Google Scholar]
- Chiu YH, Macmillan JB, and Chen ZJ (2009). RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JM, Bodemann BO, and White MA (2013). The RalGEF/Ral pathway: evaluating an intervention opportunity for Ras cancers. Enzymes 34 (Pt B), 137–156. [DOI] [PubMed] [Google Scholar]
- Cooper JM, Ou YH, McMillan EA, Vaden RM, Zaman A, Bodemann BO, Makkar G, Posner BA, and White MA (2017). TBK1 Provides Context-Selective Support of the Activated AKT/mTOR Pathway in Lung Cancer. Cancer Res 77, 5077–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Fischer RS, Pan D, and Waterman CM (2016). YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-dependent, Myosin II- and Phospho-YAP-independent Pathway during Extracellular Matrix Mechanosensing. J. Biol. Chem 291, 6096–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- Eskiocak B, McMillan EA, Mendiratta S, Kollipara RK, Zhang H, Humphries CG, Wang C, Garcia-Rodriguez J, Ding M, Zaman A, et al. (2017). Biomarker Accessible and Chemically Addressable Mechanistic Sub-types of BRAF Melanoma. Cancer Discov 7, 832–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D, Biroc SL, Alicke B, Bryant J, Yuan S, et al. (2005). Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem 280, 19867–19874. [DOI] [PubMed] [Google Scholar]
- Grindstaff KK, Yeaman C, Anandasabapathy N, Hsu SC, Rodriguez-Boulan E, Scheller RH, and Nelson WJ (1998). Sec6/8 complex is recruited to cell-cell contacts and specifies transport vesicle delivery to the basal-lateral membrane in epithelial cells. Cell 93, 731–740. [DOI] [PubMed] [Google Scholar]
- Guo X, Trudgian DC, Lemoff A, Yadavalli S, and Mirzaei H (2014). Confetti: a multiprotease map of the HeLa proteome for comprehensive proteomics. Mol. Cell. Proteomics 13, 1573–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Moroishi T, and Guan KL (2015). YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol 25, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M, Gonugunta VK, Dobbs N, Ali A, Palchik G, Calvaruso MA, DeBerardinis RJ, and Yan N (2017). Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc. Natl. Acad. Sci. USA 114, 746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. (2012). A census of human soluble protein complexes. Cell 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider MR, Gu M, Duffy CM, Mirza AM, Marcotte LL, Walls AC, Farrall N, Hakhverdyan Z, Field MC, Rout MP, et al. (2016). Subunit connectivity, assembly determinants and architecture of the yeast exocyst complex. Nat. Struct. Mol. Biol 23, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi Y, Suzuki A, Yamanaka T, Sasaki K, Mizuno K, Sawada H, Yonemura S, and Ohno S (2009). Interaction between PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain development of epithelial cells. J. Cell Sci 122, 1595–1606. [DOI] [PubMed] [Google Scholar]
- Inamdar SM, Hsu SC, and Yeaman C (2016). Probing Functional Changes in Exocyst Configuration with Monoclonal Antibodies. Front. Cell Dev. Biol 4, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingrand S, Barrier L, Lafay-Chebassier C, Fauconneau B, Page G, and Hugon J (2007). The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. FEBS Lett 581, 4473–4478. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Canterbury JD, Weston J, Noble WS, and MacCoss MJ (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925. [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Kim NG, and Gumbiner BM (2015). Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol 210, 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Welsh EA, Oguz U, Fang B, Bai Y, Kinose F, Bronk C, Remsing Rix LL, Beg AA, Rix U, et al. (2013). Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl. Acad. Sci. USA 110, 12414–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, and Sabatini DM (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, and Kroemer G (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zheng Y, Yin F, Yu J, Silverman N, and Pan D (2016). Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila. Cell 164, 406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, and Blenis J (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol 10, 307–318. [DOI] [PubMed] [Google Scholar]
- Martin TD, Chen XW, Kaplan RE, Saltiel AR, Walker CL, Reiner DJ, and Der CJ (2014). Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol. Cell 53, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan EA, Kwon G, Clemenceau JR, Fisher KW, Vaden RM, Shaikh AF, Neilsen BK, Kelly D, Potts MB, Sung YJ, et al. (2019). A Genome-wide Functional Signature Ontology Map and Applications to Natural Product Mechanism of Action Discovery. Cell Chem. Biol 26, 1380–1392.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, Park HW, Mo JS, Lu W, Lu S, et al. (2015). MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun 6, 8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, and Land H (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hayashi T, Pan WW, Fujita Y, Holt MV, Qin J, Carson DA, and Guan KL (2016). The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 167, 1525–1539.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, and White MA (2002). The exocyst is a Ral effector complex. Nat. Cell Biol 4, 66–72. [DOI] [PubMed] [Google Scholar]
- Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, Camonis J, and White MA (2003). Ral GTPases regulate exocyst assembly through dual subunit interactions. J. Biol. Chem 278, 51743–51748. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, and Levine B (2007). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35. [DOI] [PubMed] [Google Scholar]
- Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, Brekken R, Wurz R, Tasker A, Polverino T, et al. (2011). TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol. Cell 41, 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Kim HS, Fisher KW, Hu Y, Carrasco YP, Bulut GB, Ou YH, Herrera-Herrera ML, Cubillos F, Mendiratta S, et al. (2013). Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci. Signal 6, ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse C, Formstecher E, Boeckeler K, Zhao Y, Kremerskothen J, White MD, Camonis JH, and Parker PJ (2009). An aPKC-exocyst complex controls paxillin phosphorylation and migration through localised JNK1 activation. PLoS Biol 7, e1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, and Sabatini DM (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, and Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. [DOI] [PubMed] [Google Scholar]
- Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Mariño G, Lachkar S, Senovilla L, et al. (2012). Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 48, 667–680. [DOI] [PubMed] [Google Scholar]
- Simicek M, Lievens S, Laga M, Guzenko D, Aushev VN, Kalev P, Baietti MF, Strelkov SV, Gevaert K, Tavernier J, and Sablina AA (2013). The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol 15, 1220–1230. [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol 3, Article3. [DOI] [PubMed] [Google Scholar]
- Spivak M, Weston J, Bottou L, Käll L, and Noble WS (2009). Improvements to the percolator algorithm for Peptide identification from shotgun proteomics data sets. J. Proteome Res 8, 3737–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Abraham N, Knowles S, Marius R, Brasey A, Lichty BD, Brown EG, Sonenberg N, and Bell JC (2000). The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol 74, 9580–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, and Chen ZJ (2006). The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24, 633–642. [DOI] [PubMed] [Google Scholar]
- Tallóczy Z, Jiang W, Virgin HW 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, and Levine B (2002). Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 99, 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallóczy Z, Virgin HW 4th, and Levine B (2006). PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2, 24–29. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, and Chen ZJ (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay YD, Leda M, Spanos C, Rappsilber J, Goryachev AB, and Sawin KE (2019). Fission Yeast NDR/LATS Kinase Orb6 Regulates Exocytosis via Phosphorylation of the Exocyst Complex. Cell Rep 26, 1654–1667.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Haste NM, and Ghosh G (2005). PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking. Cell 122, 823–825. [DOI] [PubMed] [Google Scholar]
- Torres MJ, Pandita RK, Kulak O, Kumar R, Formstecher E, Horikoshi N, Mujoo K, Hunt CR, Zhao Y, Lum L, Zaman A, Yeaman C, White MA, and Pandita TK (2015. November). Role of the Exocyst Complex Component Sec6/8 in Genomic Stability. Mol. Cell Biol 35, 3633–3645. 10.1128/MCB.00768-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronel C, Page G, Bodard S, Chalon S, and Antier D (2014). The specific PKR inhibitor C16 prevents apoptosis and IL-1β production in an acute excitotoxic rat model with a neuroinflammatory component. Neurochem. Int 64, 73–83. [DOI] [PubMed] [Google Scholar]
- Trudgian DC, Thomas B, McGowan SJ, Kessler BM, Salek M, and Acuto O (2010). CPFP: a central proteomics facilities pipeline. Bioinformatics 26, 1131–1132. [DOI] [PubMed] [Google Scholar]
- Trudgian DC, Singleton R, Cockman ME, Ratcliffe PJ, and Kessler BM (2012). ModLS: Post-Translational Modification Localization Scoring with Automatic Specificity Expansion. J. Proteomics Bioinform 5, 283–289. [Google Scholar]
- Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, and Wrana JL (2010). The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell 19, 831–844. [DOI] [PubMed] [Google Scholar]
- Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T, Gao L, Wang L, Ling L, Jia J, et al. (2017). YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKε-mediated phosphorylation. Nat. Immunol 18, 733–743. [DOI] [PubMed] [Google Scholar]
- Williams BR (1999). PKR; a sentinel kinase for cellular stress. Oncogene 18, 6112–6120. [DOI] [PubMed] [Google Scholar]
- Xiao J, Tan Y, Li Y, and Luo Y (2016). The Specific Protein Kinase R (PKR) Inhibitor C16 Protects Neonatal Hypoxia-Ischemia Brain Damages by Inhibiting Neuroinflammation in a Neonatal Rat Model. Med. Sci. Monit 22, 5074–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Wang X, and Story M (2009). Statistical methods of background correction for Illumina BeadArray data. Bioinformatics 25, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihama Y, Sasaki K, Horikoshi Y, Suzuki A, Ohtsuka T, Hakuno F, Takahashi S, Ohno S, and Chida K (2011). KIBRA suppresses apical exocytosis through inhibition of aPKC kinase activity in epithelial cells. Curr. Biol 21, 705–711. [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al. (2012). Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, and Guan KL (2015). Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman A, Rahaman MH, and Razzaque S (2013). Kaposi’s sarcoma: a computational approach through protein-protein interaction and gene regulatory networks analysis. Virus Genes 46, 242–254. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S, Li X, Zhou R, Wang J, Zhao B, et al. (2017). Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol 19, 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, and Guan KL (2010). The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 24, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, and Bardeesy N (2009). Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, and Sabatini DM (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol 12, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Lobo G, Fulmer D, Guo L, Dang Y, Su Y, Ilatovskaya DV, Nihalani D, Rohrer B, Body SC, et al. (2019). The exocyst acting through the primary cilium is necessary for renal ciliogenesis, cystogenesis, and tubulogenesis. J. Biol. Chem 294, 6710–6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. RNaseq dataset has been deposited on Gene Expression Omnibus (GEO: GSE176496).
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request