

Abstract
Immunodeficiency often coincides with hyperactive immune disorders such as autoimmunity, lymphoproliferation, or atopy, but this is rarely understood molecularly. We describe four families with immunodeficiency coupled with atopy, lymphoproliferation, and cytokine overproduction with mutations in NCKAP1L, encoding the hematopoietic-specific HEM1 protein. These mutations cause loss of the HEM1 protein and the WAVE regulatory complex (WRC) or disrupt binding to the WRC regulator, Arf1, thereby impairing actin polymerization, synapse formation, and immune cell migration. Diminished cortical actin networks caused by WRC loss led to uncontrolled cytokine release and immune hyperresponsiveness. HEM1 loss also blocked mTORC2-dependent AKT phosphorylation, T cell proliferation, and selected effector functions causing immunodeficiency. Thus, the evolutionarily conserved HEM1 protein simultaneously regulates F-actin and mTORC2 signaling to achieve equipoise in immune responses.
Keywords: T cells, immunodeficiency, hyper-immunity, AKT, RICTOR, mTOR, NCKAP1L
One sentence summary:
A novel inborn immunodysregulatory disease reveals a role for HEM1 in independent regulation of F-actin and mTORC2 signaling.
Inborn errors of immunity (IEI) can affect global cellular regulatory systems (1). The mechanistic targets of rapamycin complex 1 (mTORC1) and mTORC2 are global regulators of metabolism and cell signaling. mTORC2, comprised of the mTOR, RICTOR, mSIN1, mLST8, PROTOR1/2, and DEPTOR proteins, activates AGC kinases downstream of phosphoinositide 3-kinase (PI3K) to promote T cell survival, proliferation, and differentiation (2–5). Similarly, actin is a global regulator of cellular behavior and immune synapse (IS) formation (6, 7). Signals activating the WAVE Regulatory Complex (WRC), containing CYFIP1/2, HEM1/2, ABI1/2/3, HSPC300, and WAVE1/2/3, control the dynamics of Arp2/3-mediated branched filamentous actin (F-actin) nucleation and polymerization. In the WRC, HEM1/2 and CYFIP1/2 form a membrane-associated scaffold supporting the ABI1/2, HSPC300, and WAVE1/2/3 proteins and is directly activated by the small GTPases Rac1/Arf1, although the Arf1 binding site is uncertain (8) (9) (Fig. 1). Whether the WRC regulates the cortical actin network (CAcN) is unknown (6, 7, 10, 11) (8, 9, 12). Mutations affecting actin regulatory proteins underlie immunodeficiencies (table S1), but none are yet reported for WRC components (13).
Figure 1. Immunodysregulatory disorder due to genetic HEM1 deficiency.
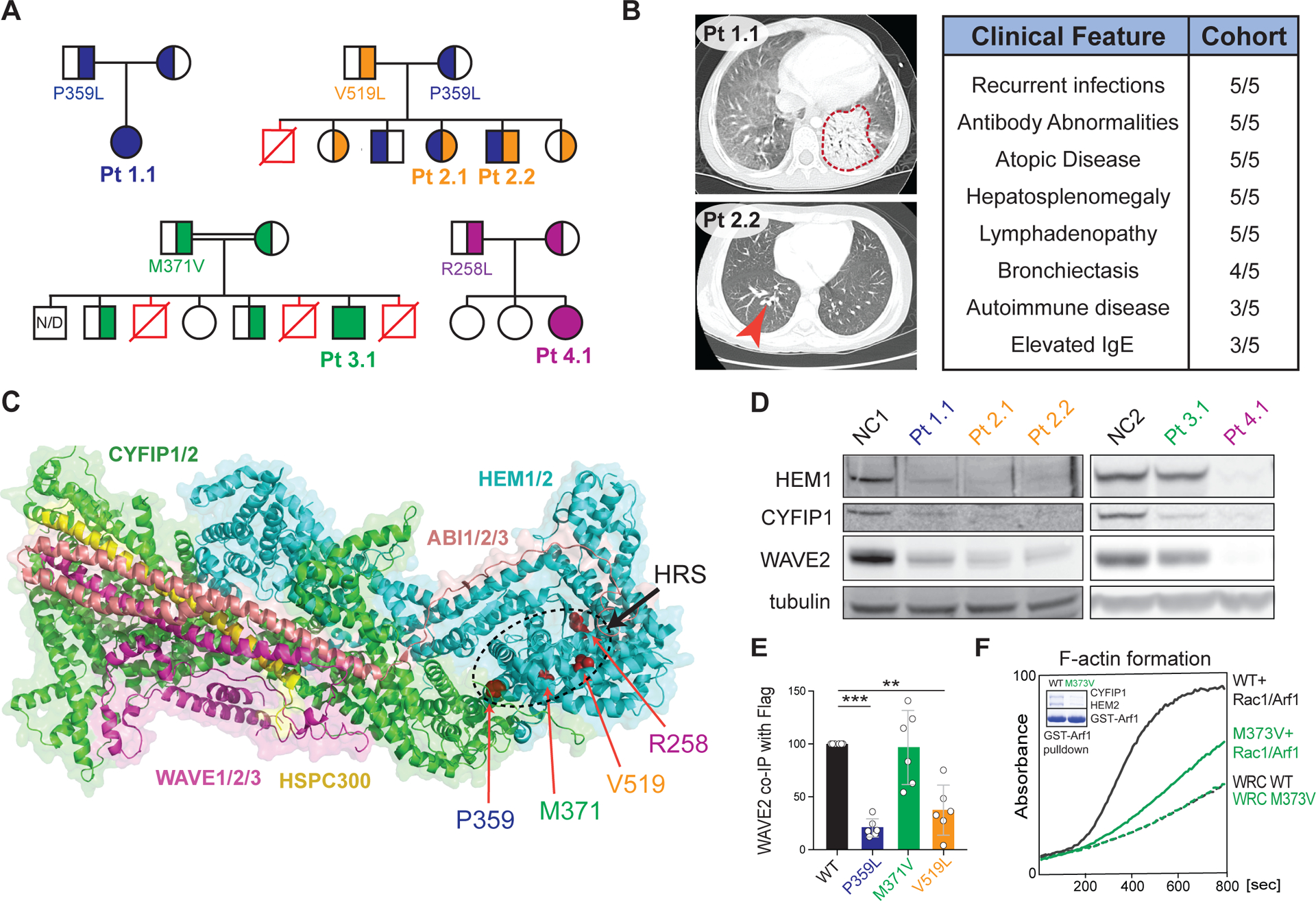
(A) Patient (Pt) pedigrees showing recessive inheritance of disease and HEM1 amino acid substitutions. Red symbols: deceased affected siblings, unknown genotype; N/D: not determined. (B) Chest CT scans showing ground glass opacity and pneumonia (red outline) in Pt 1.1 (upper left), bronchiectasis (red arrow) in Pt. 2.2 (bottom left). Key shared clinical features (right). (C) Structural location of patient variants in HEM1 in the WRC (PDB 3P8C, PMID 21107423). HRS: HEM1 regulatory site. (D) Immunoblot of WRC components in lysates derived from Pt and normal control (NC) CD4+ (left) and CD8+ (right) T cell blasts. (E) Quantification of WAVE2 co-immunoprecipitated by WT or mutant HEM1-Flag constructs in six independent experiments. Statistical analysis was performed using a one-sample t-test. (F) Pyrene-actin polymerization assay with WRC230VCA containing HEM2 WT or M373V, with or without activation by a Rac1-Arf1 heterodimer pre-loaded with GMPPNP. Inset: Coomassie blue-stained gel showing GST-Arf1 pull-down of WRC230VCA containing HEM2 WT or M373V and Rac1 (Q61L/P29S). Data are representative of four independent experiments. [**P ≤ 0.01, ***P ≤ 0.001.]
We investigated five patients from four unrelated families with recurrent bacterial and viral skin infections and severe respiratory tract infections leading to pneumonia and bronchiectasis (Fig. 1A and B left panels and fig. S1A), and poor specific antibody responses (Fig. 1B right panel and table S2). Paradoxically, the patients also exhibited atopic and inflammatory disease alongside chronic hepatosplenomegaly and lymphadenopathy, sometimes with elevated IgE or IgG and autoimmune manifestations (Fig. 1B, fig. S1,B and C, and tables S2 and S3). FoxP3+ T regulatory cells were normal (fig. S1D). All five patients harbored bi-allelic NCKAP1L mutations encoding missense variants in HEM1, the hematopoietic-specific member of the WRC (Fig. 1,A and C, and table S4). The amino acid substitutions affected conserved residues that were not homozygous in gnomAD or internal databases and were bioinformatically predicted to be damaging (fig. S2,A and B) (14). The altered residues clustered within the quaternary structure of the WRC near the distal Rac1-binding site located on CYFIP1/2 (fig. S2A–D), which we call the “HEM1 regulatory site” (HRS). The human immune phenotype differs from the lymphopenia, neutrophilia, or bone marrow failure observed in HEM1-deficient mice (10, 15, 16).
Biochemical analyses showed that Patients 1.1, 2.1, 2.2, and 4.1 lost HEM1, CYFIP1, and WAVE2 indicating WRC destabilization, whereas Patient 3.1 expressed normal WRC proteins (Fig. 1D and S3A–C). Moreover, the HEM1 substitutions in Patient 1.1 and 2.1, but not Patient 3.1, reduced affinity for WAVE2 (Fig. 1E and S3D). The WRC could be restored in HEM1 CRISPR/Cas9 knockout (KO) Jurkat cells by normal and Patient 3.1 HEM1 but less so by the Patient 1.1 and 2.1 variants (fig. S3E). Immunoprecipitation/mass spectrometry (IP/MS) showed that the M371V HEM1 variant (Patient 3.1) maintained association with HEM1–WAVE2 interacting proteins (fig. S4 and table S5). We therefore hypothesized that M371V disrupted the activation by either Rac1 or Arf1, small GTPases that activate the WRC. By reconstituting the WRC in vitro with recombinant proteins (containing HEM2 with the equivalent M373V substitution, not HEM1 which had insufficient yield), we observed that the HEM2-M373V protein interacted poorly with GST-Arf1 and could not promote F-actin polymerization upon stimulation with an Arf1-Rac1 dimer (Fig. 1F and fig. S3, F–H). Thus, the M371/373 residues located in the HRS of HEM1/2 are crucial for Arf1 binding and WRC activation, analogous to binding of Rac1 to CYFIP1/2 (8). Therefore, the patient HEM1 mutants either destabilize the WRC or disrupt its Arf1-mediated activation (Fig. S2E).
We observed that IL-2 stimulation caused patient and HEM1-knockdown cells to hypersecrete perforin and granzymes and hyperproliferate in response to IL-2 (Fig. 2,A and B, and fig. S5,A, B, and E). Proximal IL-2 signaling was normal implying that downstream processes, such as CAcN control of granule release, might be affected (fig. S6) (17) (18, 19). We found evidence for an abnormal CAcN because patient cells displayed reduced cortical F-actin and aberrant membrane spikes and puncta due to unregulated formin and WASp, respectively (Fig. 2C and movie S1) (20). Also, we observed defective cell spreading and lamellipodia. (fig. S5C). Patient T cells expressed higher levels of surface CD107a/LAMP1, showing increased granule membrane fusion, especially following phorbol myristate acetate and ionomycin (PMA/I)-induced degranulation (Fig. 2D and fig. S5D). Experimental reduction of HEM1 in primary T cells and NKL cells also increased release of cytokines and granule contents, CD107a expression, or both (fig. S5E–H). Three-dimensional imaging revealed a diminished CAcN and a dramatic accumulation of lytic granules at the IS of patient cells (Fig. 2E). Latrunculin A (LatA), which depolymerizes F-actin, increased exocytosis-based CD107a surface expression and Gzm A and B release in a dose-dependent manner (Fig. 2D bottom and fig. S5,I and J). Thus, HEM1/WRC enables the CAcN to restrain excessive degranulation and granule release by T cells. Constitutive cytokine release was blocked by a Jak inhibitor (fig. S5E).
Figure 2. HEM1 is essential for regulating cortical actin and granule release.
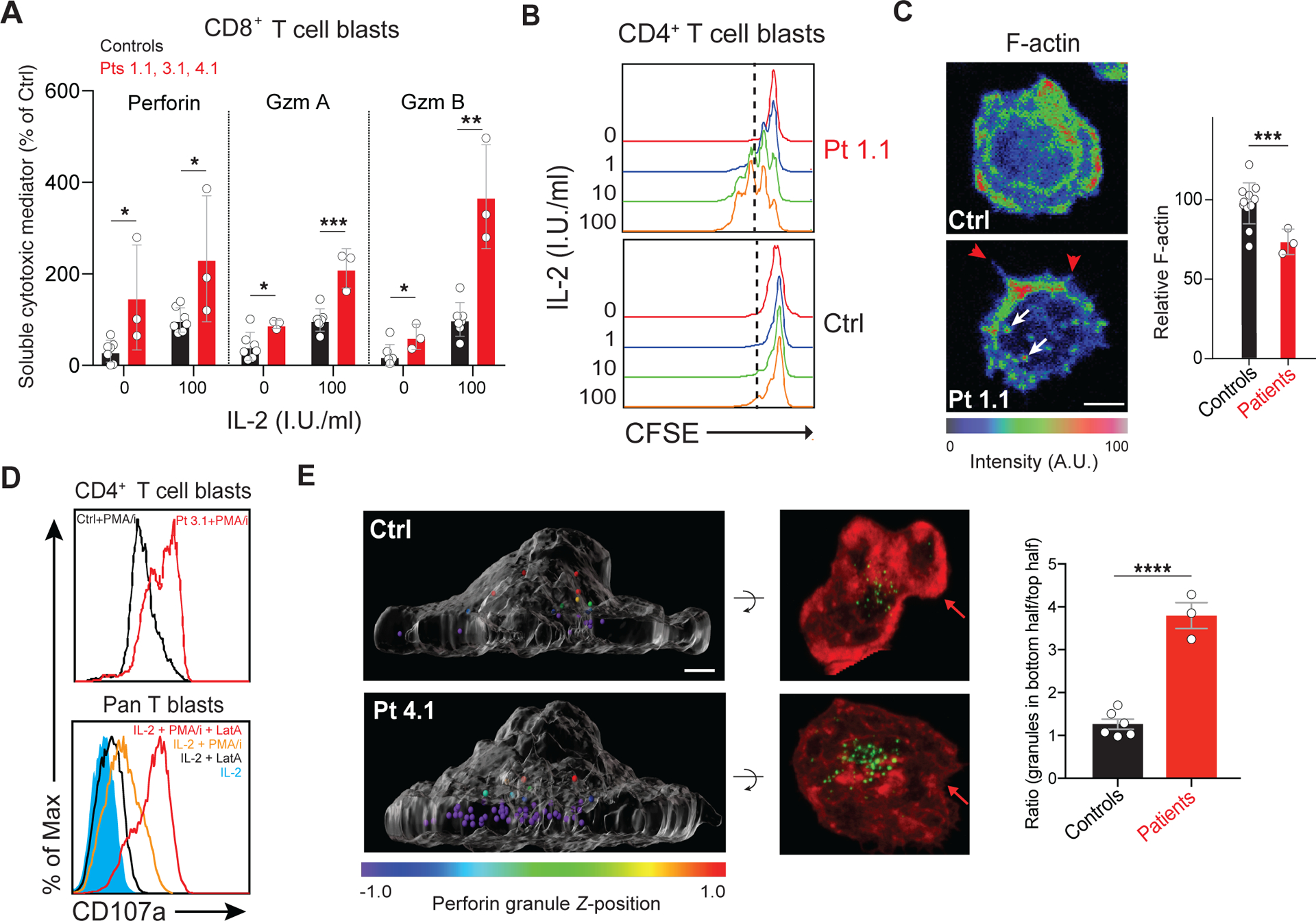
(A) Release of granzymes (Gzm) A and B or perforin from Pt or control (Ctrl) CD8+ T cell blasts following 18-hours of IL-2 stimulation in international units (I.U.) in three independent experiments. (B) Flow cytometric histograms measuring proliferation of rested CD4+ T cell blasts from a normal control (Ctrl) or Patient 1.1 (Pt 1.1) measured by carboxyfluorescein succinimidyl ester (CFSE) dilution after IL-2 restimulation for 96 hours. (C) Ctrl or Pt 1.1 CD4+ T cell blasts spreading on coverslips coated with anti-CD3, anti-CD28, and ICAM-1 (1 μg/ml each), stained with phalloidin, and pseudo-colored for F-actin (left). F-actin was quantified in three experiments (right). Red arrows: formin-mediated spikes; white arrows: WASp-mediated actin puncta. Scale bar: 4 μm. (D) Surface CD107a on Ctrl and Pt 3.1 CD4 T cell blasts following 1-hour phorbol myristate acetate (PMA)/ionomycin (I) stimulation (top) or stimulated pan T cells with 1 μM latrunculin A (LatA) (bottom). (E) Side view of perforin granules pseudo-colored by Z-position relative to the cell center in Ctrl or Pt 1.1 CD8+ T cell blasts (left). Corresponding 90° forward rotation top views of F-actin (red) and perforin (green) (middle). Red arrows: lamellipodial F-actin density. Scale bar: 2 μm. Mean ratios of granules in the bottom half to top half of the cell, quantified in at least 30 cells per sample (right). Bars represent mean ± SEM (control n = 6, patient n = 3). Data represent at least three repeat experiments. Statistical analyses for (A), (C), and (E) were performed using a t-test without assuming equal variance. [P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.]
We next explored other F-actin functions using live cell imaging of the T cell IS and found that patient cells and HEM1-KO Jurkat cells reconstituted with mutant HEM1 alleles, cannot form symmetrical and stable synaptic contacts with the coverslip (movies S1 and S2) (20). We also observed abnormal formin spikes, WASp-mediated puncta, and defective lamellipodia. Since lamellipodia guide cell migration, we evaluated spontaneous T cell and neutrophil migration (21, 22). We found that patient T cells exhibited defective membrane ruffling, loss of lamellipodial extensions, decreased F-actin density at the leading edge with abnormal puncta, spikes, and blebs, and reduced migratory velocity (fig. S7,A and B, and movie S3). Similarly, patient neutrophils migrating in chemokine gradients exhibited reduced velocity and directional persistence, unusual elongation, and misdirected competing leading edges (Fig. 3A and B, movie S4, and fig. S7C). In patients for whom we had sufficient samples, we found decreased NK cell abundance along with defective F-actin accumulation at the IS and a corresponding defect in conjugate formation (Fig. 3C and D and fig. S7E). Additionally, HEM1-KO NKL clones displayed reduced effector function after stimulation (fig. S7,F and G).
Figure 3. HEM1 loss causes immunodeficiency by abnormal immune cell behavior and activation.
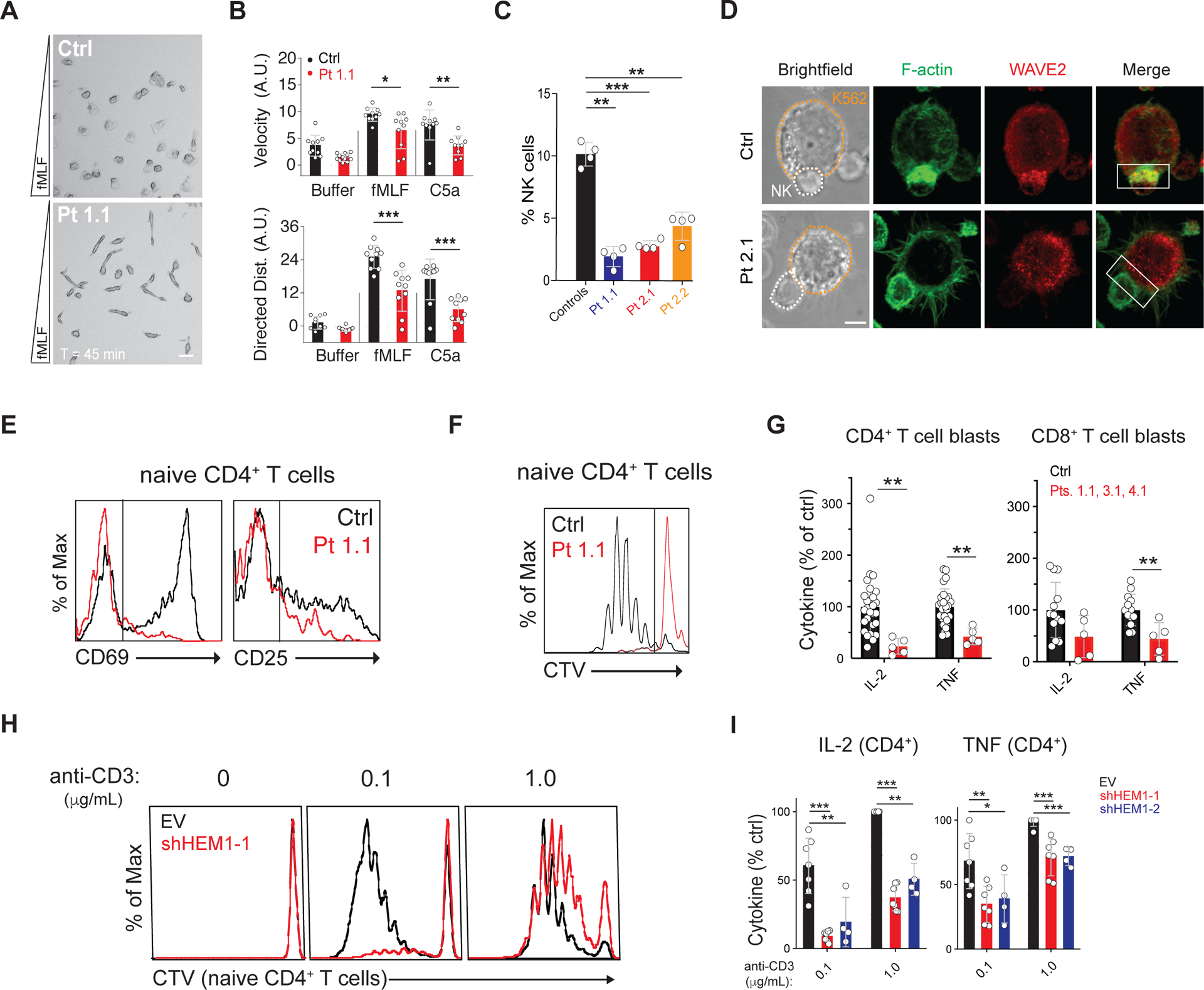
(A) Single frame from movie S4 showing healthy control (Ctrl) or Pt 1.1 neutrophils migrating in a gradient (bottom = greatest concentration) of N-formyl-L-methionyl-L-leucyl-L-phenylalanine (fMLF). Scale bar: 20 μm. (B) Displacement velocity (top) and net directed distance (Dist.)(bottom) in arbitrary units (A.U.) of ten randomly selected Ctrl or Pt 1.1 neutrophils migrating in chemoattractant gradients. (C) Percentage of NK cells in four peripheral blood samples. (D) Photomicrographs of immunological synapses between K562 target cells (orange outline) and NK cells (white outline) stained with phalloidin for F-actin and WAVE2 antibody. White box: area of synapse. Scale bar: 5 μm. (E) CD69 and CD25 upregulation on Ctrl or Pt 1.1 naïve CD4+ T cells after stimulation with immobilized anti-CD3/28 (1 μg/ml each). (F) Cell Trace Violet (CTV) proliferation plots of cells as in (E) stimulated for 5 days. (G) IL-2 and TNF secretion by CD4+ or CD8+ T cell blasts after restimulation for 36 hours with immobilized anti-CD3/28 and ICAM-1 (1 μg/ml each) in three independent experiments. (H) CTV plots of naive CD4+ T cells transduced with empty vector (EV) or small hairpin RNA against HEM1 (sh-HEM1) stimulated on immobilized ICAM-1/anti-CD28 (1 μg/ml each) and the indicated dose of anti-CD3. (I) IL-2 and TNF secretion by CD4+ T cell blasts transduced with empty vector (EV) or shRNAs targeting HEM1 (sh-HEM1–1 and sh-HEM1–2) and stimulated as in (H). Neutrophil migration was analyzed for two independent donors, otherwise data represent at least four independent trials of each assay. Statistical analyses for (B), (C), and (G) was performed using a t-test without assuming equal variance. Statistical analysis for (I) was performed using a Wilcoxon matched-pairs signed-rank test. [*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.]
We also found abnormal T cell activation manifested by reduced CD69 and CD25 expression, blunted proliferation, and decreased IL-2 and TNF production (Fig. 3E–G and fig. S9,A and B). Additionally, patient T cells had defective integrin activation with lower soluble ICAM-1 binding, though adherence to immobilized ICAM-1 was largely intact, indicating abnormal integrin affinity maturation. Interestingly, we found CD8+ T cell cytotoxicity and release of granzyme A, granzyme B, and perforin were normal (fig. S8,A–C and fig. S9,C and D). (21, 23, 24). Proliferation and cytokine defects were recapitulated by shRNA knockdown of HEM1 (Fig. 3H and I).
Despite the IS abnormalities, proximal T cell receptor (TCR) signaling events in HEM1-deficient patient cells were normal (fig. S10A–C) (21). Nevertheless, we found that both patient and HEM1 knockdown T cells showed defective TCR-induced phosphorylation of a well-known substrate of the mTORC2 complex, AKT (Protein kinase B), at serine (Ser)473 (Fig. 4A and fig. S11,A–C) (25). Phosphorylation of mTORC2-independent targets, including AKT Threonine (Thr)308 and ribosomal protein S6 Ser235/236 and Ser240/244, was moderately reduced in patient cells, but these defects were not recapitulated by HEM1 knockdown (Fig. 4B and fig. S11,C and D). Immunoblotting showed decreased phosphorylation of AKT Ser473 as well as decreased Ser21 of the downstream AKT substrate, glycogen synthase kinase (GSK) 3a (Fig. 4C). TCR-induced AKT Ser473 in T cells could be blocked by an mTOR catalytic inhibitor (Ku0063794), whereas inhibition of the actin network by LatA moderately enhanced AKT Ser473 phosphorylation. Thus, the AKT Ser473 phosphorylation defect appeared to be independent of HEM1 regulation of the CaCN (fig. S11E and F).
Figure 4. HEM1 associates with RICTOR and governs mTORC2 activation.
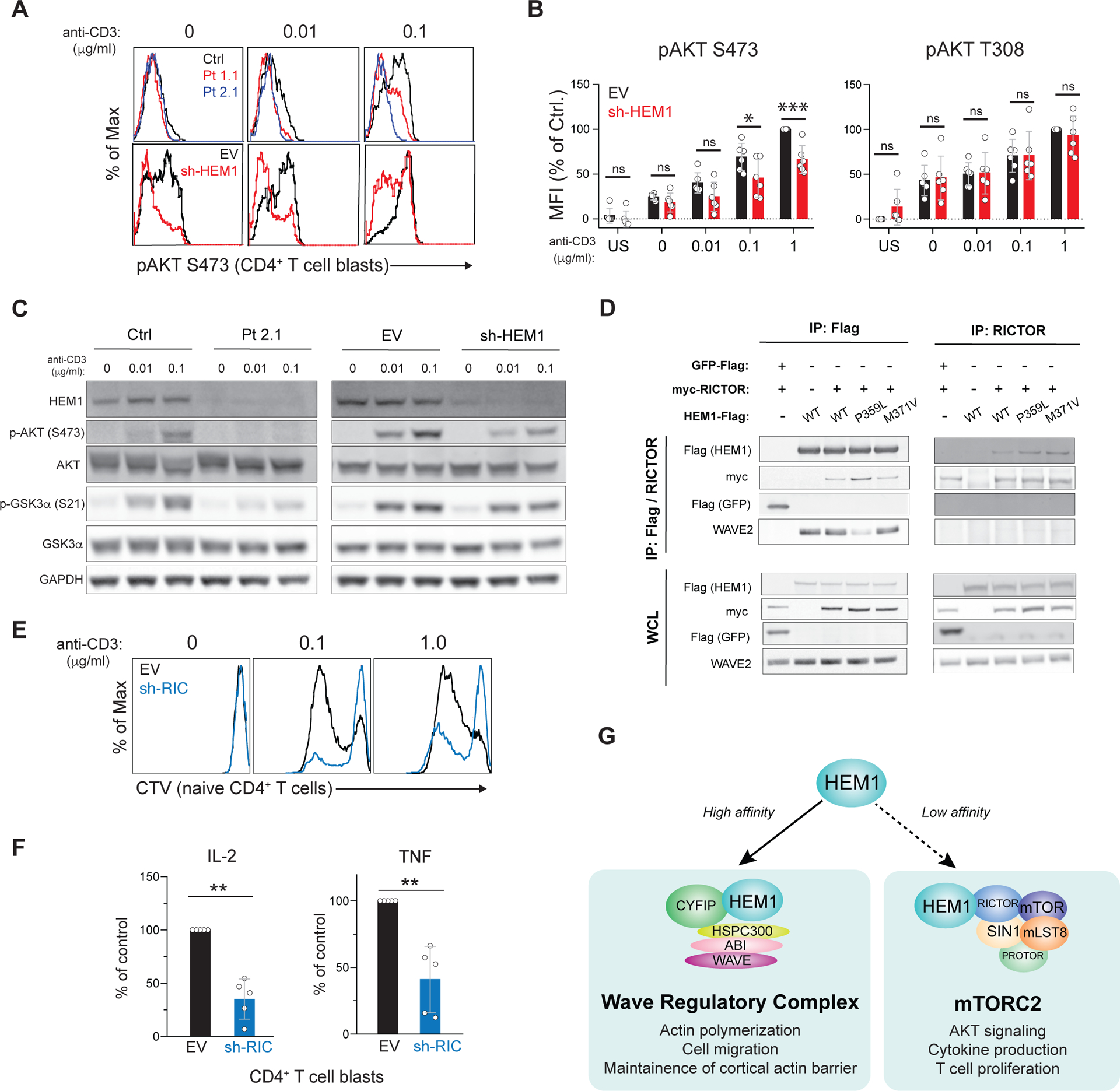
(A) Phospho-flow cytometry of purified CD4+ T cell blasts from control (ctrl) or patient (Pt) (top row) or empty vector (EV) transduced or sh-HEM1 knockdown cells (bottom row) for AKT phosphorylated on Ser473 (pAKT S473). Cells were stimulated for 10 min with anti-CD28/ICOS (1 μg/ml each) and the indicated dose of anti-CD3. (B) Mean fluorescence intensity (MFI) of pAKT S473 or AKT pAKT T308 in EV or sh-HEM1 CD4+ T cell blasts stimulated as in (A) in 6 independent experiments. US: unstimulated; ns: not significant. (C) Immunoblot of Ctrl or Pt CD4 T cell blasts, or healthy CD4 T cell blasts cells transduced with the empty vector (EV) or shRNA directed against HEM1 (sh-HEM1). Cells were rested and restimulated with ICAM-1/anti-CD28 (1 μg/ml each) and the indicated dose of anti-CD3. (D) Flag and RICTOR IP from 293T cells transduced with myc-RICTOR and either Flag-tagged GFP or Flag-tagged HEM1 (WT or mutant) and blotted. (E) Cell Trace Violet (CTV) proliferation plots of naïve CD4+ T cells transduced with empty vector (EV) or shRNA directed against RICTOR (sh-RIC). (F) Cytokine secretion by control and RICTOR knockdown (sh-RIC) CD4+ T cell blasts following 18-hour restimulation in five independent experiments. (G) Provisional model of HEM1 independently regulating WRC- and mTORC2-mediated functions. Statistical analyses for (B) and (F) were performed using a Wilcoxon matched-pairs signed-rank test. [*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.]
To investigate how HEM1 regulates the phosphoinositide 3-kinase (PI3K)/AKT/mTORC2 pathway, we searched our IP/MS datasets and found that several mTORC2 components, including mTOR and RICTOR, the key scaffolding protein of mTORC2, were precipitated by HEM1 but not WAVE2 (fig. S4, fig. S12A, and table S5). This observation suggested the existence of a pool of HEM1 outside of the WRC that interacts with, and regulates, mTORC2 (22). Testing HEM1-Flag (WT/P359L/M371V), Flag-GFP, myc- RICTOR, or WAVE2 from 293T cells, we observed that HEM1, but not WAVE2, specifically co-immunoprecipitated with RICTOR (Fig. 4D and fig. S12,A and B). Notably, the P359L HEM1 strongly associated with RICTOR, suggesting that the interaction occurs when HEM1 is not in complex with the WRC. Knockdown of RICTOR in CD4+ T cells impaired proliferation and cytokine secretion (Fig. 4,E and F and fig. S12D). Chemical inhibitors of the PI3K, AKT, or mTOR kinases also abrogated T cell proliferation and IL-2 and TNF secretion (fig. S12,E and F). Critically, specific inhibition of mTORC1 with rapamycin had little effect compared to inhibition of both complexes with an mTOR catalytic inhibitor, suggesting that mTORC2, but not mTORC1, is required. However, mTOR inhibition had little effect on perforin and granzymes secretion or CD69/CD25 upregulation, essentially phenocopying the defects observed in patient cells (fig. S12,G–I). Thus, HEM1 plays an additional role in human T cells outside of the WRC as an upstream regulator of mTORC2 enzymatic activity (Fig. 4G).
Previous studies showed that individual WRC components can have non-canonical roles in cellular processes beyond actin filament nucleation and that HEM1/2 exists, and likely functions, outside of the WRC complex (11, 22). We now show in human patients with immunodeficiency and immune hyperactivation, that loss-of-function mutations in NCKAP1L, the gene encoding HEM1, disrupt WRC-mediated actin polymerization and abrogate mTORC2 activation of AKT. The resulting autosomal recessive IEI affects multiple hematopoietic lineages and leads to bacterial and viral infections, atopic disease, autoimmunity, cytokine overproduction, and lymphoproliferative disease. We demonstrate that HEM1 and the WRC maintain the CAcN, which restricts cytokine secretion and lytic granule release. We also show that HEM1 plays a key binding role in Arf1-mediated WRC activation. Our findings suggest a broader effect of genetic HEM1 deficiency on the cytokine repertoire and cellular effector function that should be addressed in future work. Finally, we identified an interaction between HEM1 and RICTOR essential for mTORC2 regulation. HEM1 may have escaped detection in previous RICTOR precipitation experiments because the interaction appears to be weak and because commonly used 293T cells do not express the hematopoietically restricted HEM1. We posit that HEM1 independently coordinates WRC-mediated actin nucleation and mTORC2 catalytic activity in response to signals that activate both protein complexes, such as PI3K, Arf1, and Rac1, during T cell activation and possibly during B and NK cell activation. These data could explain how mTORC2 is activated downstream of actin-generated membrane tension and can negatively regulate the WRC (26). Because mTORC2 exerts similar roles in all lymphocytes, and since their activation involves actin-dependent regulation, it is likely that B and NK cell abnormalities contribute to immunopathology in the HEM1-deficient patients (27–29). In sum, our study elucidates a new human congenital disorder caused by loss of HEM1 and highlights new potential routes for immunological therapy.
Supplementary Material
Acknowledgments
The authors thank the patients and family members for participating in this study and making this research possible. We thank H.Su for scientific input, discussions, careful reading of the manuscript. The authors would like to thank members of the Bioinformatics and Computational Biosciences Branch (BCBB), NIAID for bioinformatics support and the Office of Cyber Infrastructure and Computational Biology (OCICB), NIAID for high-performance computing support and the Laboratory of Immune System Biology Flow Cytometry Core, NIAID for cell analysis. Finally, we thank the staff of the Advanced Mass Spectrometry Core, NIDDK.
Funding:
This work was supported by The Jeffrey Modell Foundation Translational Research Award to IKC, NIH-NIAID R01 AI120989 to JSO, NIH-NIGMS R35 GM128786 to BC, and NIH-NHGRI UM1 HG006542 to the Baylor-Hopkins Center for Mendelian Genomics, and the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. Additional support came from the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, the Division of Intramural Research, National Institute of Diabetes, Digestive, and Kidney Diseases (NIDDK), and the Deputy Director of Intramural Research, National Institutes of Health, through the Clinical Center Genomics Opportunity. This work was also funded by a fellowship grant (1-16-PDF-025, to W.A.C) from the American Diabetes Association, a F12 postdoctoral fellowship (1FI2GM119979-01, to W.A.C.) from the National Institute of General Medical Sciences, NIGMS, NIH, and M.C.P. was supported by Fondecyt#11181222. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and Materials Availability: Whole-exome sequencing (WES) data for the kindred of Pt 1.1 was submitted to dbGaP (accession: phs001561). WES data for the kindred of Pts 2.1 and 2.2 were submitted to dbGaP (accession: phs000711).
References
- 1.Cunningham-Rundles C, Ponda PP, Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol 5, 880–892 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Saxton RA, Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Guertin DA et al. , Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11, 859–871 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Lee K et al. , Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van de Velde LA, Murray PJ, Proliferating Helper T Cells Require Rictor/mTORC2 Complex to Integrate Signals from Limiting Environmental Amino Acids. J Biol Chem 291, 25815–25822 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen B, Padrick SB, Henry L, Rosen MK, Biochemical reconstitution of the WAVE regulatory complex. Methods Enzymol 540, 55–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stradal TE et al. , Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol 14, 303–311 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Chen B et al. , Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z et al. , Structure and control of the actin regulatory WAVE complex. Nature 468, 533–538 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shao L et al. , The Wave2 scaffold Hem-1 is required for transition of fetal liver hematopoiesis to bone marrow. Nat Commun 9, 2377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Litschko C et al. , Differential functions of WAVE regulatory complex subunits in the regulation of actin-driven processes. Eur J Cell Biol 96, 715–727 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Lebensohn AM, Kirschner MW, Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell 36, 512–524 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns SO, Zarafov A, Thrasher AJ, Primary immunodeficiencies due to abnormalities of the actin cytoskeleton. Curr Opin Hematol 24, 16–22 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Kircher M et al. , A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park H et al. , A point mutation in the murine Hem1 gene reveals an essential role for Hematopoietic protein 1 in lymphopoiesis and innate immunity. J Exp Med 205, 2899–2913 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leithner A et al. , Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nat Cell Biol 18, 1253–1259 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Basquin C et al. , Membrane protrusion powers clathrin-independent endocytosis of interleukin-2 receptor. EMBO J 34, 2147–2161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gil-Krzewska A et al. , An actin cytoskeletal barrier inhibits lytic granule release from natural killer cells in patients with Chediak-Higashi syndrome. J Allergy Clin Immunol 142, 914–927 e916 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carisey AF, Mace EM, Saeed MB, Davis DM, Orange JS, Nanoscale Dynamism of Actin Enables Secretory Function in Cytolytic Cells. Curr Biol 28, 489–502 e489 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murugesan S et al. , Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J Cell Biol 215, 383–399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nolz JC et al. , The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol 16, 24–34 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiner OD et al. , Hem-1 complexes are essential for Rac activation, actin polymerization, and myosin regulation during neutrophil chemotaxis. PLoS Biol 4, e38 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nolz JC et al. , WAVE2 regulates high-affinity integrin binding by recruiting vinculin and talin to the immunological synapse. Mol Cell Biol 27, 5986–6000 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nolz JC et al. , The WAVE2 complex regulates T cell receptor signaling to integrins via Abl- and CrkL-C3G-mediated activation of Rap1. J Cell Biol 182, 1231–1244 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM, Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Diz-Munoz A et al. , Membrane Tension Acts Through PLD2 and mTORC2 to Limit Actin Network Assembly During Neutrophil Migration. PLoS Biol 14, e1002474 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Treanor B et al. , The membrane skeleton controls diffusion dynamics and signaling through the B cell receptor. Immunity 32, 187–199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwata TN, Ramirez-Komo JA, Park H, Iritani BM, Control of B lymphocyte development and functions by the mTOR signaling pathways. Cytokine Growth Factor Rev 35, 47–62 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang C et al. , mTORC1 and mTORC2 differentially promote natural killer cell development. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paila U, Chapman BA, Kirchner R, Quinlan AR, GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol 9, e1003153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lek M et al. , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanborn KB et al. , Myosin IIA associates with NK cell lytic granules to enable their interaction with F-actin and function at the immunological synapse. J Immunol 182, 6969–6984 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanborn KB, Rak GD, Mentlik AN, Banerjee PP, Orange JS, Analysis of the NK cell immunological synapse. Methods Mol Biol 612, 127–148 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Banerjee PP, Orange JS, Quantitative measurement of F-actin accumulation at the NK cell immunological synapse. J Immunol Methods 355, 1–13 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindelin J et al. , Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen B, Padrick SB, Henry L, Rosen MK, Biochemical reconstitution of the WAVE regulatory complex. Methods Enzymol 540, 55–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen B et al. , Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Shea EK, Lumb KJ, Kim PS, Peptide ‘Velcro’: design of a heterodimeric coiled coil. Curr Biol 3, 658–667 (1993). [DOI] [PubMed] [Google Scholar]
- 39.Keilhauer EC, Hein MY, Mann M, Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol Cell Proteomics 14, 120–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ, Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4, 484–494 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Lin Y et al. , Sodium laurate, a novel protease- and mass spectrometry-compatible detergent for mass spectrometry-based membrane proteomics. PLoS One 8, e59779 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rappsilber J, Mann M, Ishihama Y, Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Perrin C, Ohta B, Kuperman J, Beta-deuterium isotope effects on amine basicity, “inductive” and stereochemical. J Am Chem Soc 125, 15008–9. (2003) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.