

Abstract
Glioblastoma multiforme (GBM) is the most common and aggressive type of brain tumor in adulthood. Epigenetic mechanisms are known to play a key role in GBM although the involvement of histone methyltransferase KMT5B and its mark H4K20me2 has remained largely unexplored. The present study shows that DNA hypermethylation and loss of DNA hydroxymethylation is associated with KMT5B downregulation and genome-wide reduction of H4K20me2 levels in a set of human GBM samples and cell lines as compared with non-tumoral specimens. Ectopic overexpression of KMT5B induced tumor suppressor-like features in vitro and in a mouse tumor xenograft model, as well as changes in the expression of several glioblastoma-related genes. H4K20me2 enrichment was found immediately upstream of the promoter regions of a subset of deregulated genes, thus suggesting a possible role for KMT5B in GBM through the epigenetic modulation of key target cancer genes.
Keywords: epigenetics, glioblastoma, histone methyltransferase, histone posttranslational modification, tumor suppressor
Introduction
Glioblastoma multiforme (GBM) is a malignant grade IV glioma which represents the most common and aggressive primary brain tumor among adults. The first line of treatment consists of surgical resection followed by radiotherapy and/or chemotherapy with the alkylating agent temozolomide (TMZ). Nevertheless, the median overall survival after treatment is only 14 months (Weller et al., 2015). Hence there is a critical need to gain a deeper understanding of this frequently fatal and heterogeneous brain cancer, and search for new therapeutic targets.
During tumorigenesis, epigenetic alterations affect different types of cancer-related genes involved in cell cycle control, DNA repair, apoptosis, or cell signaling (Feinberg et al., 2016), among others. Moreover, they can also occur at epigenetic regulator genes, thereby triggering a chain of genome-wide massive epigenetic alterations. As examples of the latter are, on the one hand, epigenetic disruption through the downregulation of different micro RNAs that modulate histone deacetylases (HDACs) expression has been reported in several cancers, such as hepatocellular carcinoma (Yuan et al., 2011), lymphoplasmacytic lymphoma (Roccaro et al., 2010), tongue squamous cell carcinoma (Kai et al., 2014), and breast cancer (Wu et al., 2014). On the other hand, disruption of the methylation signature at the promoter of genes encoding histone deacetylases HDAC4, HDAC5, and HDAC9 has been described in papillary thyroid carcinoma (White et al., 2016). In the context of neuroblastoma and glioma, epigenetic inactivation via hypermethylation at promoter CpG island (CpGi) of the histone methyltransferase gene NSD1 has been defined as the mechanism responsible for the altered histone methylation landscape observed in both types of tumor (Berdasco et al., 2009). In the same vein, it has been shown that DNA methyltransferases (DNMTs) encoded by DNMT1 and DNMT3B are overexpressed in glioma due to, respectively, an aberrant histone code or an aberrant methylation pattern at their promoters (Rajendran et al., 2011). Recently, our laboratory has demonstrated that the expression of the epigenetic enzyme TET3 is aberrantly downregulated in GBM through epigenetic mechanisms, leading to a genome-wide reduction of 5-hydroxymethylcytosine (5hmC) levels (Carella et al., 2020).
Thus, besides genetic features (Brennan et al., 2013), DNA methylation/hydroxymethylation and histone modifications are major epigenetic mechanisms regulating gene expression and genomic stability whose alterations have been widely described in GBM (Hegi et al., 2005; Fernandez et al., 2018; García et al., 2018; Carella et al., 2020). In addition, the brain has a unique DNA methylation/hydroxymethylation landscape, characterized by the highest levels of 5-methylcytosine (5mC) and 5hmC of all human tissues (Nestor et al., 2012). In general terms, 5mC in promoters contributes to create a repressive chromatin environment and decreases gene expression, while 5hmC located within the body of genes may activate transcription (Chen et al., 2016). The balance between the two marks modulates a plethora of biological processes and has been extensively reported to be dysregulated in GBM (Johnson et al., 2016; López et al., 2017; Fernandez et al., 2018; García et al., 2018; Carella et al., 2020). In addition to the epigenetic marks of DNA, many studies have addressed how the modulation of chromatin structure via histone modifications is affected in the context of GBM (Liu et al., 2010). They have mostly focused on the function of histone modifying enzymes, specifically histone lysine demethylases (KDMs) and histone deacetylases (HDACs), and the use of different inhibitors (Singh et al., 2015).
However, to date little attention has been paid to the role of histone lysine methyltransferases (KMTs) in GBM (Gursoy-Yuzugullu et al., 2017). Methylation at lysine 20 of histone H4 (H4K20me) is frequently altered in cancer (Behbahani et al., 2012). However, the contribution of the epigenetic enzymes regulating these histone posttranslational modifications is still not fully understood. A preliminary genome-wide screening of candidate genes altered in GBM revealed that the gene encoding the histone methyltransferase KMT5B (alias SUV420H1) is frequently hypermethylated and hypo-hydroxymethylated in this tumor type. KMT5B employs H4K20me1 as a substrate, giving rise to H4K20me2 (Yang et al., 2008). In the present study we investigated the molecular basis of the deregulation of this epigenetic enzyme as well as its functional consequences and its possible tumoral role in GBM.
Materials and Methods
Human Brain and Glioblastoma Samples
Non-tumoral human brain (n = 28) and GBM samples (n = 37) were obtained from the Tumor Bank of the Institute of Oncology of Asturias (Asturias, Spain), the Neurological Tissue Bank of the Clinic Hospital (IDIBAPS, Barcelona, Spain), and the Biobank of the Virgen de la Salud Hospital (BioB-HVS, Toledo, Spain). Informed written consent was obtained from all patients involved in this study. Tissue collection and all analyses were conducted in accordance with the Declaration of Helsinki and approved by the Clinical Research Ethics Committee of the Principality of Asturias (Spain) (date of approval 14/10/2013, project identification code: 116/13).
Culture of Human Glioblastoma Cell Lines and Drug Treatments
The human glioblastoma cell lines LN-18 (RRID: CVCL_0392), LN-229 (RRID: CVCL_0393), U-87MG ATCC (RRID: CVCL_0022), and T98G (RRID: CVCL_0556) were obtained and cultured according to American Type Culture Collection (ATCC) recommendations. Recently (November, 2020), all cell lines were authenticated by short tandem repeat (STR) profiling of an extracted DNA sample using AmpFLSTRTM IdentifilerTM Plus PCR Amplification Kit (Applied Biosystems, A26182) at the Scientific and Technological Resources Unit (University of Oviedo, Asturias, Spain). Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, 41965) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, F6178), 2% penicillin/streptomycin (Gibco, 15070), and 1% Amphotericin B (Gibco, 15290) at 37°C in a humidified incubator containing 5% CO2. Cell cultures were regularly tested and verified to be mycoplasma negative with the Mycoplasma Gel Detection kit from Biotools (Biotools, 90.022-4544), and all experiments were performed with mycoplasma-free cells. For the selection of LN-229 clones overexpressing KMT5B, medium was prepared that contained DMEM, 10% FBS, MEM non-essential aminoacids (Sigma-Aldrich, M7145), and Geneticin (G-418, Sigma-Aldrich, A1720) as the selection antibiotic. For the 5-aza-2′-deoxycytidine (5-AZA-dC) and vitamin C treatments, 2 × 106 LN-229 cells were seeded onto P-100 plates and supplemented for 72 h with either 4 μM 5-AZA-dC (Sigma-Aldrich, A3656) alone or in combination with 50 μg/mL vitamin C (Sigma-Aldrich, A7506). Control wells contained the solvent dimethyl sulfoxide (DMSO, Sigma-Aldrich, D5879). For the KMT5B inhibitor A-196 treatment (Sigma-Aldrich, SML1565), 2 × 103 stably-transfected LN-229 clone cells were seeded onto 8-well Lab-Tek chamber slides (Thermo Scientific, 177445) and grown for 48 h in selective medium before adding the drug (dissolved in DMSO) at 10 μM for another 48 h, with DMSO alone used in control wells.
DNA Methylation Analysis With High-Density Array and Whole Genome Bisulfite Sequencing
Data corresponding to the oxBS and BS conversion of brain or GBM samples were obtained from our previously reported HumanMethylation 450K array (E-MTAB-6003) (Fernandez et al., 2018). Processed WGBS data were obtained from Arrayexpress (E-MTAB-5171) (Raiber et al., 2017). Both datasets were filtered to highlight the CpG coverage along the KMT5B gene. Estimations of 5mC and 5hmC levels were calculated with the R/bioconductor package ENmix (version 1.12.3) using the oxBS.MLE method (Xu et al., 2016).
DNA Extraction
DNA was extracted from freshly frozen GBM, control brains and human GBM cell lines with standard phenol–chloroform-isoamyl alcohol protocol. Samples were stored at −20°C until further analysis.
Bisulfite Pyrosequencing
Bisulfite modification of DNA was performed with the EZ DNA methylation-gold kit (Zymo Research) following the manufacturer’s instructions. The set of primers for PCR amplification and sequencing was designed using the specific software PyroMark assay design (version 2.0.01.15) (Supplementary Table 1). After PCR amplification, pyrosequencing was performed using Pyro-Mark Q24 reagents, vacuum prep workstation, equipment, and software (Qiagen).
5-Hydroxymethylcytosine Immunoprecipitation and Quantitative Real-Time Polymerase Chain Reaction Assay
EpiQuik Hydroxymethylated DNA Immunoprecipitation Kit (hMeDIP, Epigentek) was used for immunoprecipitation of 5hmC in 11 samples (corresponding to 5 brain samples, 5 GBM samples and LN-229 GBM cell line), according to supplier’s protocol. Input, non-specific IgG and 5hmC-enriched fractions were amplified by Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) in a StepOnePlusTM Real-Time PCR machine (Applied Biosystems) with SYBR Green 2X PCR Master Mix (Applied Biosystems, 4309155) and oligonucleotides for the CpGs in the promoter of KMT5B listed in Supplementary Table 1. Relative 5hmC enrichment was calculated as a Fold Change relative to Input Ct Mean.
Quantitative Real-Time Reverse Transcriptase Polymerase Chain Reaction
Total RNA was isolated from human samples and GBM cell lines using TRIzol Reagent (Life Technologies, Ref. 15596) according to the manufacturer’s instructions. To remove any possible residual genomic DNA, total RNA was treated with DNase I (Turbo DNA-free kit, Ambion-Life Technologies, Ref. AM1906). RNA was quantified both before and after DNase treatment with Nanodrop (ThermoScientific) checking purity as A260/280 ratio. cDNA was synthesized from total RNA using a SuperScriptTM III Reverse Transcriptase kit (Invitrogen, Ref. 18080) and following the manufacturer’s instructions. Quantitative PCR was carried out in triplicate for each sample, using SYBR Green 2X PCR Master Mix (Applied Biosystems, 4309155) and specific primers detailed in Supplementary Table 1. qRT-PCR was performed using the StepOnePlusTM Real-Time PCR System (Applied Biosystems). Gene expression was normalized using GAPDH as endogenous control and analyzed by the comparative threshold (ΔΔCt) method.
KMT5B Transfection
LN-229 cells were stably transfected with either expression vectors encoding human KMT5B (pEGFP-KMT5B) or with empty vector (pEGFP-C1), kindly provided by Professor Miki Hieda (Ehime Prefectural University of Health Sciences, Matsuyama, Japan) and constructed as described previously (Yokoyama et al., 2014), using Lipofectamine 3000 (Invitrogen, L3000015) according to the manufacturer’s instructions. Geneticin-resistant cells were selected 72 h after transfection with 1 mg/mL G-418 (Sigma-Aldrich, A-1720), after which sub-cloning was carried out by limiting dilution, and several clones overexpressing KMT5B were obtained. KMT5B expression was tested in positive clones by qRT-PCR using specific primers for KMT5B transcription variant 1 detection (Supplementary Table 1).
Chromatin Immunoprecipitation Assays and Real-Time PCR Quantification (qChIP)
For real-time PCR quantification of H4K20me2-enriched genomic regions (qChIP), LN-229 cells (empty vector, KMT5B #3, and KMT5B #7) were freshly processed using the SimpleChIP Enzymatic Chromatin IP Kit with magnetic beads (Cell Signaling, 9003). Immunoprecipitations were performed using antibodies against H4K20me2 (ab9052, Abcam), total histone H3 (Abcam, ab1791) as positive control, and IgG antiserum (Abcam, ab46540) as negative control (see Supplementary Table 2). DNA was purified and used for quantitative real-time PCR with SYBR Green 2X PCR Master Mix (Applied Biosystems) and the primers for the gene loci listed in Supplementary Table 1. Input chromatin DNA was used to create a standard curve and determine the efficiency of amplification for each primer set in a StepOnePlusTM Real-Time PCR machine (Applied Biosystems). All samples were measured in triplicate. IgG was used as negative control. ChIP data were analyzed and are shown in the results as the percentage relative to the input DNA amount by the equation:
Immunohistochemistry
The immunohistochemical analyses of KMT5B protein levels in three GBM samples and paired-normal brain tissue were performed using the EnVision FLEX Mini Kit (DAKO, K8024) and Dako Autostainer system. Briefly, paraffin embedded tissue (3–5 μm) was deparaffinized, rehydrated, and then epitopes were retrieved by heat induction (HIER) at 95°C for 20 min at pH 6.0 (DAKO, GV805) in the Pre-Treatment Module, PT-LINK (DAKO). Sections were incubated with anti-KMT5B antibody (sc-169462, Santa Cruz) at 1:100 dilution in EnVisionTM FLEX Antibody Diluent (DAKO, K8006) for 60 min after blocking endogenous peroxidase with EnVisionTM FLEX Peroxidase-Blocking Reagent (DAKO, DM821). Signal was detected using diaminobenzidine chromogen as substrate after incubation with Dako EnVisionTM FLEX/HRP (DAKO, DM822). Sections were counterstained with hematoxylin. Appropriate negative and positive controls were also tested. After the complete process, sections were dehydrated and mounted with permanent medium (Dako mounting medium, CS703). Images were captured using a Nikon Eclipse Ci microscope equipped with a 20x objective and a camera (Nikon Instruments Europe B. V.).
Immunofluorescence
For immunofluorescence experiments 2 × 103 cells were seeded onto 8-well Lab-Tek chamber slides (Thermo Scientific, 177445) and grown for 48 h. Samples were fixed with 4% formaldehyde (252931 Panreac) for 15 min at RT, and permeabilized with 1 × PBS/0.1% Triton X-100 (Sigma-Aldrich) for 20 min at RT. Blocking was performed with 1 × PBS/10% BSA (A7906 Sigma-Aldrich) at RT for 1 h, followed by incubation with the corresponding primary antibody _ rabbit anti-H4K20me1 (ab9051, Abcam) or rabbit anti-H4K20me2 (ab9052, Abcam)_ at 1:1,000 dilution (see Supplementary Table 2) in antibody diluent (EnVision FLEX DM830 Dako), for 1 h at 4°C. Secondary antibody chicken anti-rabbit IgG Alexa Fluor 488 (A-21441 Invitrogen) at 1:500 dilution was incubated for 1 h at RT protected from light. Finally, slides were mounted using EverBrite mounting medium with DAPI (23002 Biotium). Immunofluorescence images were acquired with a Zeiss microscope, equipped with a 63X/1.4 NA immersion objective and an AxioCam MRm camera (Carl Zeiss). Fluorescence intensity measurements were performed using the ZEN lite software (ZEN lite 2.3 SP1, Carl Zeiss).
Cell Proliferation Assay
Cell proliferation rates were measured using xCELLigence Real Time Cell Analyzer (RTCA, ACEA Biosciences). Quadruplicates of mock or stably-transfected LN-229 clone cells (15 × 103) were seeded onto analyzer specific plates. Cell impedance was measured every 2 h for eight consecutive days through micro-electric biosensors located at the base of the plate wells. Cell proliferation was represented by Cell Index, slope, and doubling time parameters.
Cell Viability Assay
Cell viability of LN-229 clones stably-transfected with KMT5B or empty vectors was determined by 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay. Briefly, ten replicates of 2 × 103 cells per condition and time point (up to 72 h) were seeded onto 96-well plates. MTT (Sigma-Aldrich, M5655) was added to the corresponding wells (every 24 h for 3 days) to a final concentration of 500 μg/mL, and incubated at 37°C, 5% CO2 for 3 h. The MTT was then removed and the formazan crystals formed were dissolved in DMSO (Sigma-Aldrich-Aldrich, D5879). Absorbance at 570 nm was measured with an automated microtiter plate reader (Synergy HT, BioTek).
Colony Formation Assay
Colony formation assays of LN-229 clones stably-transfected with KMT5B or empty vectors were conducted as described by Franken et al. (2006), with minor modifications. Briefly, cells were seeded at two different densities (100 and 200 cells) onto P-6 wells. After incubation for 14 days, stable colonies were fixed and stained with a mixture of 6.0% glutaraldehyde (Sigma-Aldrich, 340855) and 0.5% crystal violet (Sigma-Aldrich, C3886). After washing, the number of colonies was counted manually. The results were expressed as mean Plating Efficiency (PE), a parameter which represents the ratio between the number of grown colonies versus the number of cells seeded at each different density.
Cell Cycle Analysis
Cell cycle analysis of LN-229 clones stably-transfected with KMT5B or empty vectors was conducted by flow cytometry using propidium iodide (PI, Sigma-Aldrich, P4170). Briefly, cells were trypsinized, washed with PBS, and fixed with cold absolute ethanol overnight at −20°C. Cells were then washed twice with PBS and dyed with PI, and RNase A for 30 min at 37°C in the dark. The suspension was filtered with a nylon mesh filter and analyzed using a BD FACS Aria Ilu cytometer (BD Biosciences) and FlowJoV10 software.
Tumor Xenografts
For the xenograft experiments, 5-weeks old NU/NU female mice (n = 4) were purchased from Charles River Japan Inc. (Kanagawa, Japan). Animal housing and experimental procedures were approved by the Animal Ethics Committee of University of Oviedo (Asturias, Spain) on 21/06/2018 with the project identification code PROAE 18/2018. Mice were subcutaneously injected in each flank with 1 × 106 LN-229 cells (mock and KMT5B stably-transfected clones) suspended in culture medium 1:1 with Matrigel (Corning, Ref 354248). Tumor volumes were measured with a calliper twice a week and calculated using the formula V = 4/3π (Rr)2, were R is the maximum diameter and r is the minimum diameter. After animal sacrifice, the tumors were excised and weighed.
RNAseq
For RNA-seq, total RNA from three replicates of mock and KMT5B stably-transfected clones was isolated using the RNeasy Mini Kit (Cat. No. 74104, Qiagen) according to the manufacturer’s instructions, submitted to GeneWiz (Germany) for next-generation sequencing purposes, and to the European Nucleotide Archive (ENA) database (accession number PRJEB39613). Library preparation was performed using the Illumina Poly(A) selection method according to GeneWiz pipeline for standard RNA sequencing. Samples were sequenced using the Illumina HiSeq platform using 2 × 150 bp configuration mode. Adaptor removal was performed using fastp (v. 0.20.1), and the filtered reads were aligned to the human reference genome GRCh38 using the ultra-fast selective-alignment provided by SALMON (v.1.2.1). A prior step was introduced to generate the gentrome files from the concatenated GENCODE transcriptome and GRCh38 genome primary assembly (V29). The sequencing coverage and quality statistics for each sample are summarized in Supplementary Table 6. Differential gene expression analyses were conducted using the DESeq2 R/Bioconductor package (v.1.22.2) using as input the read count matrices obtained from SALMON. Genes with an absolute Log2 fold change = 1 and FDR < 0.05 were considered to be significant for downstream purposes. Subsequent enrichment analyses were performed using the R/Bioconductor package clusterProfiler (v.3.16.0). Significant gene-disease associations were represented using the R/Bioconductor package enrichplot (v.1.8.1).
Statistical Analysis
All statistical analyses were conducted using the R statistical programming language (version 3.4.0). Specific analyses are described in the corresponding section.
Results
KMT5B Shows Altered 5mC/5hmC Pattern in GBM
On the basis of data from our previously reported 450 K Illumina methylation array of 11 GBM and 5 non-tumoral brain samples (Fernandez et al., 2018), we performed a genome-wide screening of candidate genes which are altered in GBM. We found that KMT5B, a gene encoding a histone methyltransferase, had altered DNA methylation and hydroxymethylation patterns. Specifically, compared to healthy brain tissue, GBM showed higher levels of 5mC and greatly reduced levels of 5hmC in the CpGi at the promoter and several CpG sites within the KMT5B gene body (Figure 1A and Supplementary Table 3). Comparison with publicly available whole genome bisulfite sequencing (WGBS) data, with a deeper CpG coverage along the KMT5B locus (Raiber et al., 2017), confirmed a 5mC/5hmC imbalance in GBM as compared to non-tumoral brain (Figure 1B and Supplementary Table 4).
FIGURE 1.

5mC and 5hmC profiling along the KMT5B locus in brain and glioblastoma multiforme (GBM) samples and analysis of KMT5B expression in brain, GBM and GBM cell lines. (A) Line plots represent percentage of average 5mC (upper panel) and 5hmC values (lower panel) corresponding to 25 CpG positions in the KMT5B gene for five control brain and eleven GBM samples, depicted in blue and violet, respectively. Data were obtained from a genome-wide Illumina HumanMethylation 450 K array (E-MTAB-6003). Welch t-tests were applied for each CpG and those that were significant are depicted in red (q-value < 0.05). (B) WGBS dataset with high content profiling of 5mC and 5hmC (accession number E-MTAB-5171) confirmed the altered 5mC/5hmC pattern of KMT5B. CpGs depicted in red represent those with changes = 15%. Lower diagrams in panels (A,B) represent the relative location of the analyzed CpG sites along KMT5B locus. (C,D) Biological validation of the methylation and hydroxymethylation values in five KMT5B promoter CpGs using, respectively, bisulfite pyrosequencing and hMeDIP. The gray dotted line in panel (D) indicates 5hmC levels in GBM cell line LN-229. Wilcoxon rank sum tests were applied (*p < 0.05; **p < 0.01; ***p < 0.001). (E) Box plot represents KMT5B mRNA relative expression in 16 white matter and 7 gray matter control brains, 13 GBM samples and 4 GBM cell lines, measured by qRT-PCR in relation to GAPDH and represented as Fold Change relative to white-matter control brains. Wilcoxon rank sum tests were applied (*p < 0.05; **p < 0.01; ***p < 0.001). (F) Representative images of KMT5B protein levels detected in GBM samples and their paired non-tumoral brain tissue by immunohistochemistry (IHC), shown at 20× magnification. Only non-tumoral brain sections showed positive KMT5B nuclear staining.
We validated 5mC array data by means of the bisulfite pyrosequencing of five representative CpG sites at KMT5B promoter in the same specimens included in the array, as well as in a larger set of samples consisting of: 27 GBM, 4 non-tumoral brains, and 4 human GBM cell lines. Pyrosequencing results confirmed the 5mC pattern observed in the array (Figure 1C and Supplementary Table 5). With respect to 5hmC array data, we validated those results using a different technique that relied not on oxidative bisulfite conversion, but on DNA immunoprecipitation with an antibody against 5hmC. We used an hMeDIP Kit (see “Materials and Methods” section) with 5 GBM, 5 non-tumoral brain samples and the human GBM cell line LN-229. qRT-PCR was used to analyse the samples and the results confirmed the extensive loss of 5hmC affecting the KMT5B locus in GBM (Wilcoxon rank sum test, ∗∗p < 0.01; Figure 1D).
KMT5B Expression Is Downregulated in a Subset of Human GBM Samples and in GBM Cell Lines
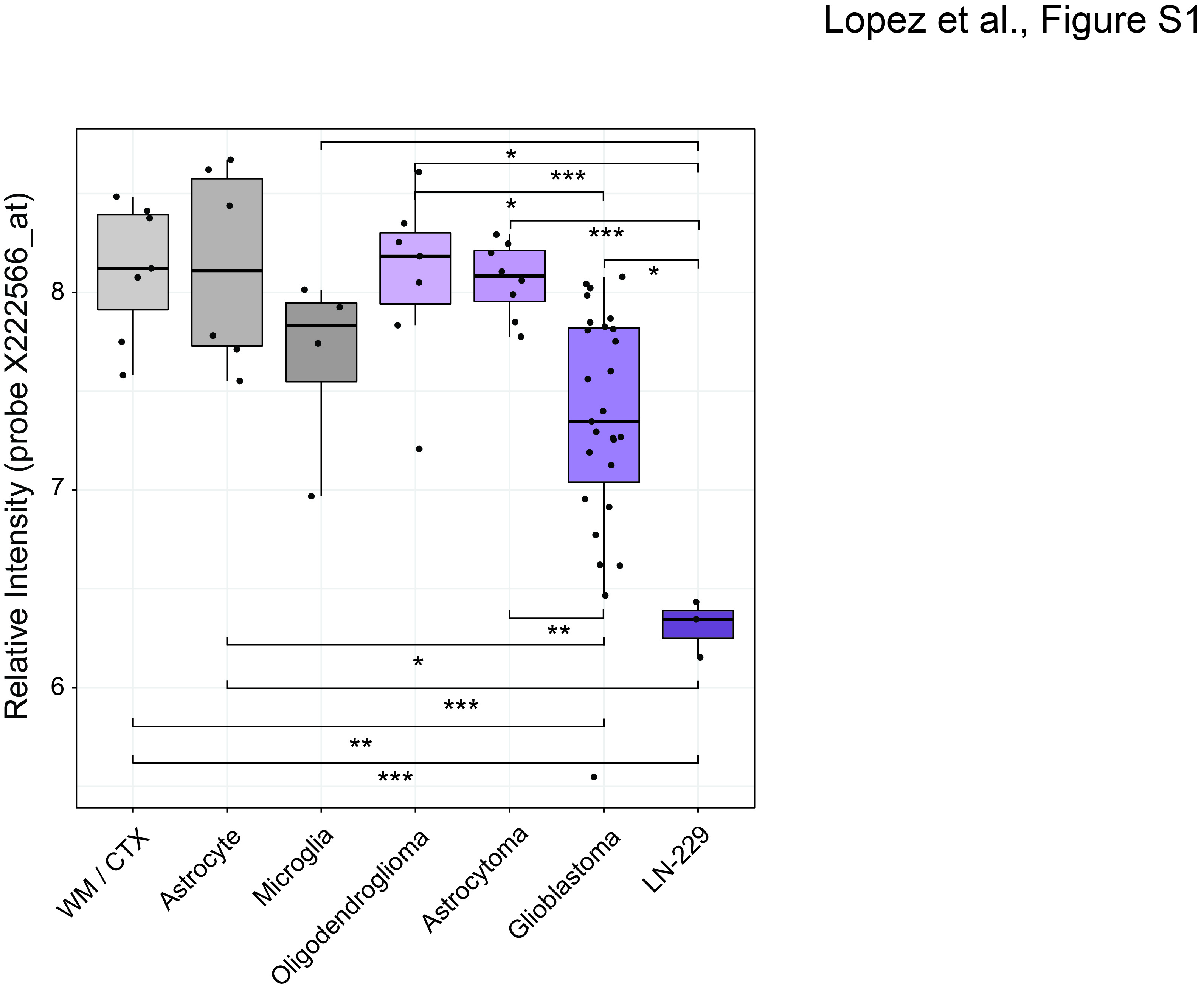
Next, we wanted to investigate whether the aberrant methylation landscape of KMT5B could have an effect at the transcriptional regulation level. In order to determine the expression status of KMT5B in GBM, we measured its mRNA levels by means of qRT-PCR in 13 GBM samples, four human GBM cell lines, and 23 non-tumoral control brains (seven corresponding to gray matter and 16 to white matter samples). The results showed that KMT5B mRNA expression was significantly downregulated in a subset of GBM samples compared with non-tumoral brains (Figure 1E). Comparison with gene expression array data obtained from different brain datasets in ArrayExpress (Günther et al., 2008; Sim et al., 2009; Grzmil et al., 2011) confirmed that downregulation of KMT5B affects a subset of GBMs (Supplementary Figure 1, Supplementary Table 8, and Supplementary Methods).
To validate our observations at the protein level, we subjected three GBM samples and their paired non-tumoral brain tissue to immunohistochemistry (IHC). The results were in line with the mRNA expression data and revealed that KMT5B protein expression was high in non-tumoral brain tissues and low to absent in GBM (Figure 1F).
These findings point to a potential epigenetic silencing of KMT5B through the methylation of the promoter and hypo-hydroxymethylation of the gene body in GBM.
Correlation Between Epigenetic Regulation and KMT5B Gene Expression
To further explore the possible functional link between the epigenetic alterations described above and KMT5B repression, we performed a pharmacological restoration of 5mC/5hmC levels in human GBM cell line LN-229. To this end, we incubated LN-229 cells with 5-aza-2′-deoxycytidine (5-AZA-dC) either alone or together with vitamin C. 5-AZA-dC is an epigenetic drug, widely used in clinical practice, that decreases 5mC levels through DNA methyl-transferases (DNMTs) inhibition, and increases 5hmC levels through upregulating the expression of Ten-Eleven-Translocation proteins (TETs) (Sajadian et al., 2015). Vitamin C, a TET cofactor, has been shown to increase intragenic 5hmC levels and to act synergistically with 5-AZA-dC in vitro (Sajadian et al., 2016). The treatments carried out here caused a significant decrease in 5mC and an increase in 5hmC levels, as confirmed at two intragenic CpG sites (CpG in the array CG27086672 and the next downstream CpG position) by means of bisulfite pyrosequencing (Wilcoxon rank sum tests, ∗p < 0.05; ∗∗p < 0.01; Figure 2A and Supplementary Table 5). At the mRNA level, KMT5B expression was also upregulated, especially when both drugs were present (Welch t-test, ∗∗p < 0.01; Figure 2B).
FIGURE 2.

Pharmacological restoration of 5mC/5hmC levels in human GBM cell line LN-229. Cells were treated with demethylating agent 5-AZA-dC, either alone or in combination with vitamin C (Vit C). The effects on 5mC/5hmC levels and on KMT5B expression were assessed by (A) bisulfite pyrosequencing at two intragenic CpG positions (Wilcoxon rank sum test; *p < 0.05; **p < 0.01) and (B) KMT5B mRNA relative expression by means of qRT-PCR. The data are the average ±SEM of three independent experiments (Welch t-test; *p < 0.05; **p < 0.01).
Taken together, these data indicate that aberrant epigenetic regulation contributes to KMT5B repression in GBM.
Ectopic Expression of KMT5B Reduces the Tumorigenic Potential of Glioblastoma Cells in vitro
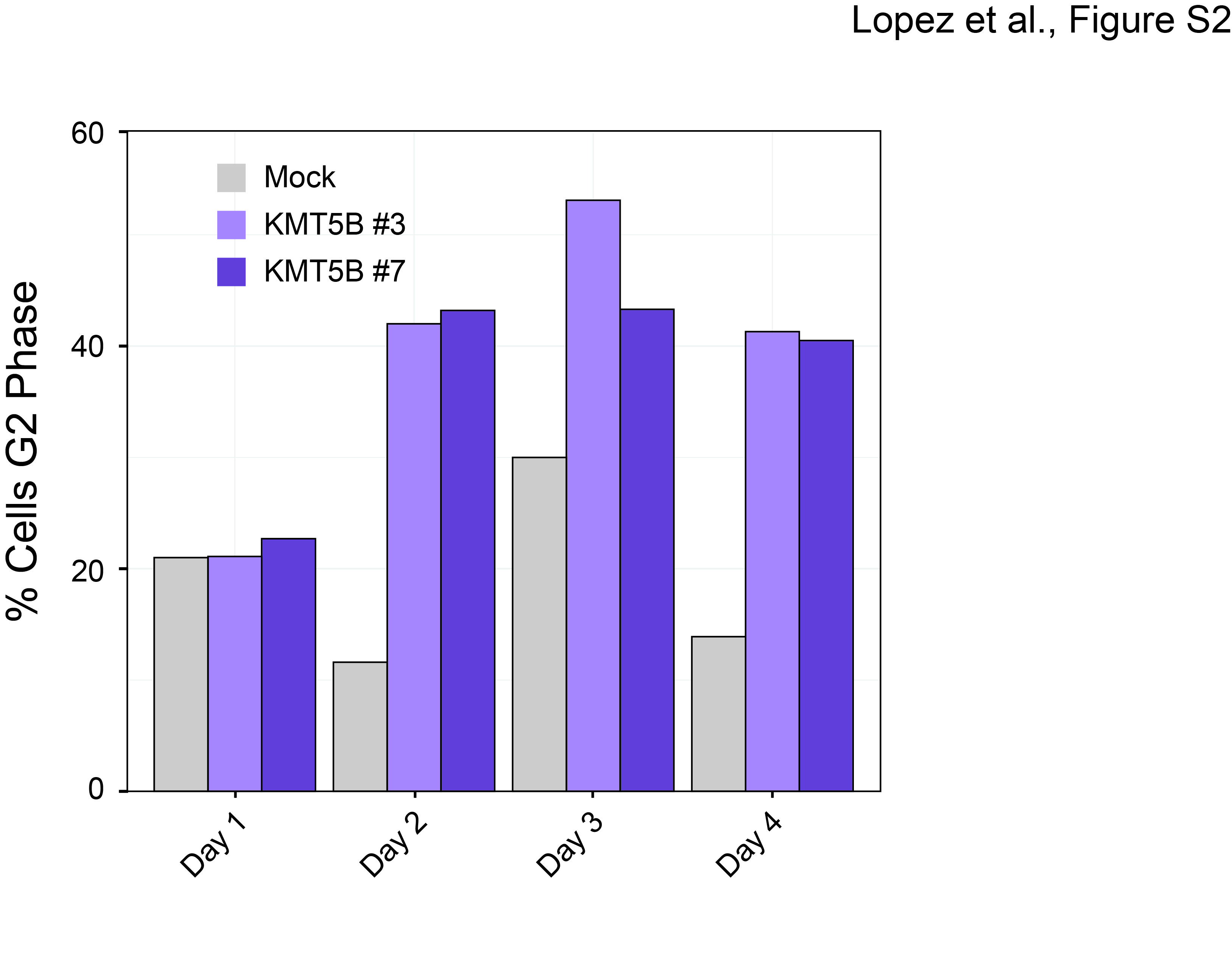
To delve into the possible role of KMT5B epigenetic repression in GBM, we stably transfected either a KMT5B expression plasmid or an empty vector (mock) into the human GBM cell line LN-229, which exhibits hypo-hydroxymethylation and downregulation of KMT5B. Two clones, namely KMT5B #3 and KMT5B #7, were selected among those that were drug resistant, and the expression of KMT5B was confirmed by qRT-PCR (Figure 3A). Impedance-based proliferation assays showed that KMT5B-transfected clones grew less than control cells (Figure 3B). Overexpression of KMT5B also reduced the clonogenicity of the clones compared with the mock transfected cells (Figure 3C). In addition, an MTT assay demonstrated that KMT5B overexpression caused a significant decrease in cell viability (Figure 3D). Cell cycle analysis confirmed that KMT5B overexpression induced G2/M arrest, as was demonstrated previously by Evertts et al. (2013), thus explaining the decreased cell proliferation and viability of the overexpressing clones (Supplementary Figure 2).
FIGURE 3.

In vitro effect of KMT5B ectopic expression. Human GBM cell line LN-229 was transfected with an empty (control) or a KMT5B expression vector. (A) Stable transfectants were obtained after selection with 1 mg/mL G-418, and named mock, KMT5B #3, and KMT5B #7. KMT5B expression was confirmed by qRT-PCR (the data shown are the average ±SEM of three independent experiments. Welch t-test; **p < 0.01; ***p < 0.001). (B) The proliferation rate of mock and the two KMT5B stably-transfected clones was measured by an impedance-based assay and represented as cell index units [General linear model (GLM) ***p-value < 0.001], slope and doubling time for three to four technical replicates (Wilcoxon rank sum tests; *p-value < 0.05). (C) On the left, representative image of plates showing colony formation assay. Experiments were done in triplicate for each clone and seeding density. Results are represented in the box plot on the right as plate efficiency (Wilcoxon rank sum tests; ***p-value < 0.001). (D) Cell viability was determined by MTT assay on control and KMT5B transfected cells. Box plot represents the average absorbance of ten replicates at several time points, relative to the absorbance at time 0 h for each clone (Wilcoxon rank sum test; *p < 0.05; **p < 0.01; ***p < 0.001).
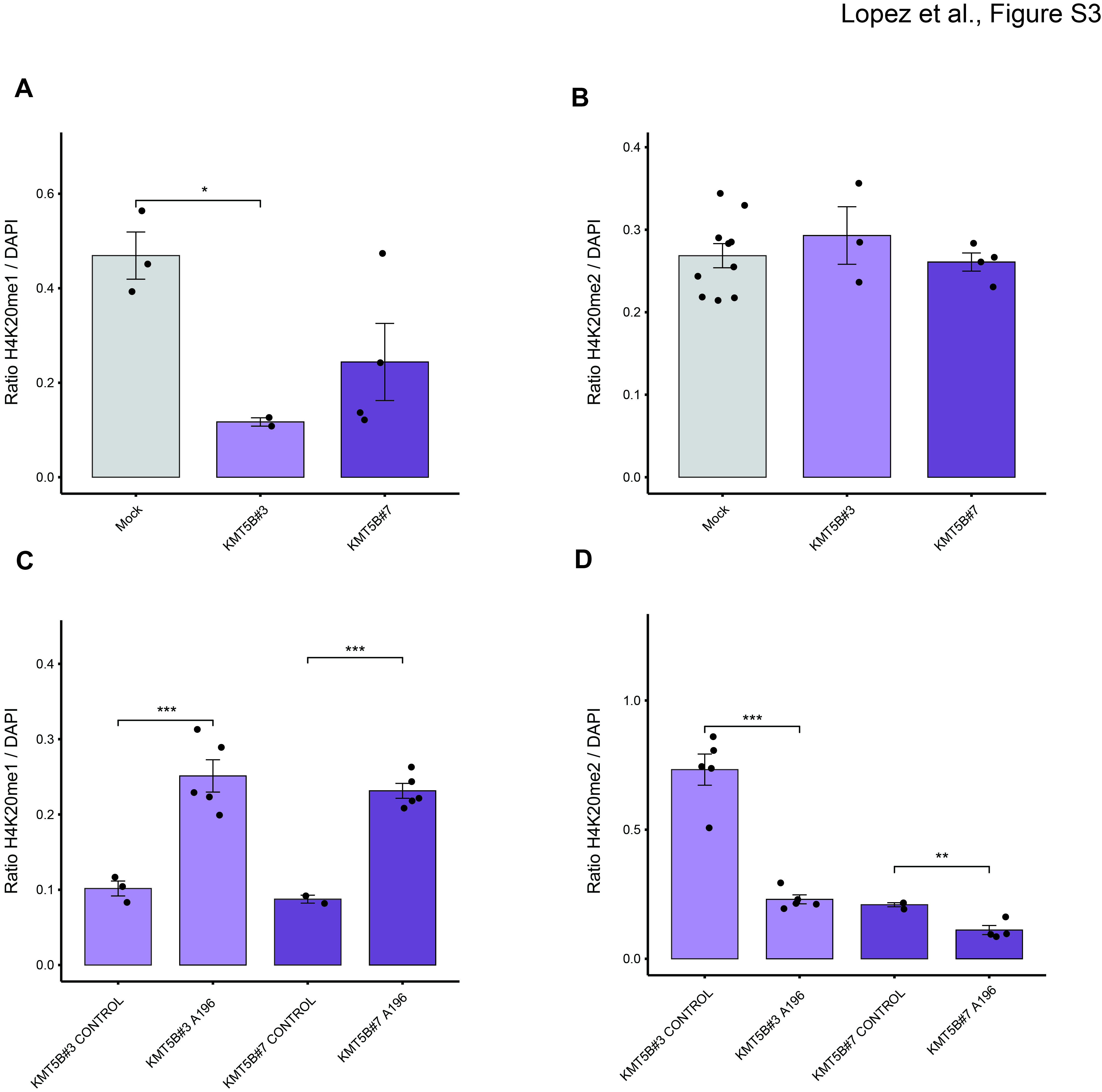
As expected, the increased expression of KMT5B elevated the levels of its histone mark H4K20me2, concomitantly reducing those of H4K20me1, as seen by immunofluorescence (IF) in stably-transfected clones versus mock (Figures 4A,B and Supplementary Figures 3A,B). Conversely, inhibition of KMT5B enzymatic activity with the selective drug A-196 (Bromberg et al., 2017) increased monomethylated and decreased dimethylated forms of H4K20 in stably-transfected clones, thus recapitulating the histone H4K20 methylation landscape of LN-229 GBM cell line (Figures 4C,D and Supplementary Figures 3C,D).
FIGURE 4.

Immunofluorescence analysis of mono- and dimethylation of H4K20. Representative images of mock and KMT5B-transfected clones immunostained with (A) anti-H4K20me1 or (B) anti-H4K20me2 antibodies (green). In panels (C,D) representative images of KMT5B-transfected clones immunostained with (C) anti-H4K20me1 or (D) anti-H4K20me2 antibodies (green) after treating the cells with KMT5B inhibitor A-196 for 48 h. Nuclei were counterstained with DAPI (blue). Bars: 10 μm.
KMT5B Overexpression Reduces Tumor Growth in Nude Mice
In light of the above data, we sought to further characterize the role of KMT5B expression in GBM in vivo. To this end, we subcutaneously injected stably-transfected KMT5B or mock LN-229 cells into nude mice. Tumor volume was notably smaller in mice flanks receiving KMT5B-overexpressing LN-229 clones compared to those injected with empty vector transfected cells (General linear model, ∗∗∗p-value < 0.001; Figure 5A). At the time of sacrifice, 56 days after tumor-xenograft implantation, the average weight of tumors generated by KMT5B-overexpressing clones was significantly lower than those from control cells (Wilcoxon rank sum tests; ∗p < 0.05; ∗∗p < 0.01; Figure 5B). The tumor xenografts thereby revealed that KMT5B overexpression also slows tumorigenesis in vivo, suggesting that KMT5B could play a possible role as a putative tumor suppressor in GBM.
FIGURE 5.

In vivo effect of KMT5B overexpression. (A) Mock or KMT5B-transfected LN-229 cells were subcutaneously injected into the flanks or the neck base of 4 nude mice. An image of the tumors excised after animal sacrifice is shown on the left. Tumor growth was monitored for 56 days [General linear model (GLM) ***p-value < 0.001]. (B) Box plot represents average tumor weights for the tumors generated by each clone (Wilcoxon rank sum tests; *p < 0.05; **p < 0.01).
Overexpression of KMT5B Affects Genome-Wide Gene Expression in GBM Cells
In an attempt to shed light on the molecular pathways and mechanisms by which KMT5B decreases GBM tumorigenesis, we subjected our stably-transfected overexpressing or mock LN-229 clones to RNAseq (Supplementary Table 6). KMT5B has been proposed to impact transcriptional regulation by means of its product H4K20me2 (Miao and Natarajan, 2005; Vakoc et al., 2006). Our results showed that overexpression of KMT5B changed the transcriptome of GBM cells through up- or downregulation of hundreds of genes (Figure 6 and Supplementary Table 7). Specifically, for clone KMT5B#3 a total of 725 genes were significantly affected, corresponding to 333 upregulated and 392 downregulated ones. Regarding KMT5B#7, expression changes implicated a total of 962 genes of which 484 were upregulated and 478 downregulated. The Venn diagram in Figure 6B represents the overlap of differentially expressed genes in KMT5B clones as compared to the LN-229 control cells (mock). The two clones shared a total of 315 differentially expressed genes. Among the upregulated ones, CDH11 (cadherin-11) has been associated with blocking the invasion of GBM cells in vitro (Delic et al., 2012). Interestingly, several of those genes had been previously described to be overexpressed and implicated in GBM tumorigenesis, including: CA9 (carbonic anhydrase 9) (Boyd et al., 2017), PDL1 (Programmed Death Ligand 1, also known as CD274) (Jacobs et al., 2009), and IL13RA2 (Interleukin 13 Receptor Subunit Alpha 2). We focused on IL13RA2 as this gene encodes a monomeric IL4-independent and high grade glioma-associated IL13 receptor (Mintz et al., 2002). IL13RA2 is overexpressed in most patients with GBM but not in normal brain (Brown et al., 2013). As expected, the gene-disease associations platform DisGeNET related the above-mentioned genes with malignant glioma. Interestingly, it also highlighted the association of certain other genes with both malignant glioma and kidney diseases, thus pointing to a yet-undescribed hypothetical role of KMT5B in such renal pathologies (Figure 6C).
FIGURE 6.

Overexpression of KMT5B alters the expression of several genes, including IL13RA2. (A) Principal component analysis of the three replicates of each clone subjected to RNAseq (B) Venn diagram showing differentially expressed genes in KMT5B-expressing clones KMT5B#3 and KMT5B#7. Both comparisons were made independently against the expression profile of the LN-229 control cells (mock). (C) Network diagram showing the functional relationships between the genes differentially expressed in the RNAseq analysis and disease ontologies (glioblastoma and renal diseases) obtained from DisGeNET. To facilitate the interpretation of the data, the relationships of genes and ontologies common to both conditions are indicated. Information on the expression levels of the different genes compared to the control (mock) is indicated by the colour scale. (D) Enrichment of H4K20me2 upstream of the IL13RA2 and CDH11 transcription start sites due to KMT5B overxpression. Chromatin immunoprecipitation (ChIP) assays were performed in mock and KMT5B stably-transfected LN-229 cells using either anti-H4K20me2 polyclonal antibody or control IgG (see Supplementary Table 2). DNA eluted from the ChIP assay was amplified by qRT-PCR with primer sets mapping to IL13RA2 or CDH11 promoters (listed in Supplementary Table 1). The enrichment of H4K20me2 at the indicated regions in KMT5B clones was compared to the same regions in mock control cells (Welch t-tests; *p < 0.05; **p < 0.01; ***p < 0.001).
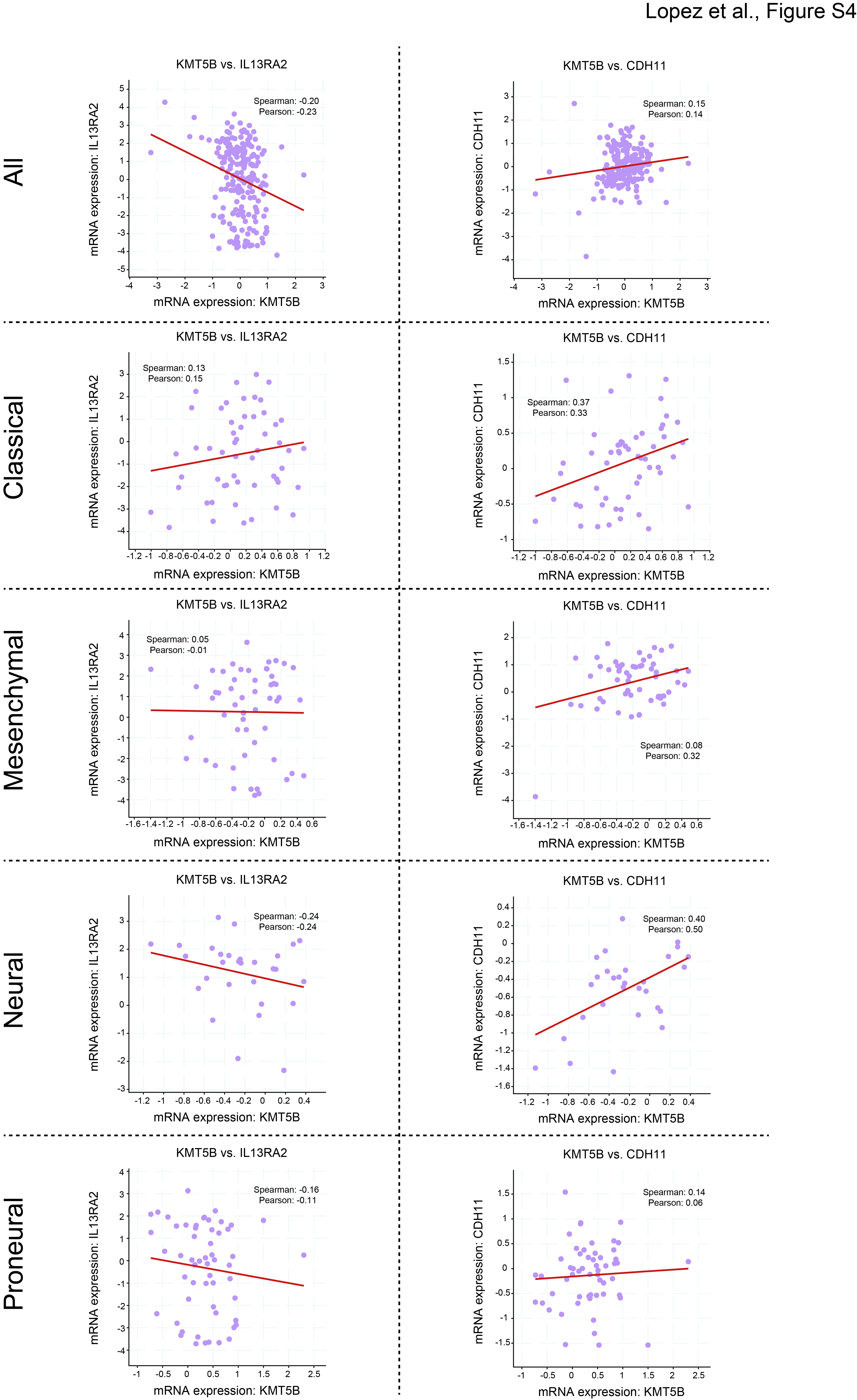
To verify the RNAseq results and assess the involvement of KMT5B and H4K20 methylation in the regulation of IL13RA2 and CDH11 expression, we performed a ChIP assay for H4K20me2 in our clones. The analysis of immunoprecipitated DNA by qRT-PCR demonstrated that H4K20me2 is enriched immediately upstream of the IL13RA2 and CDH11 transcription start sites in the KMT5B-overexpressing clones, but not in mock-transfected GBM cells (Figure 6D). Additionally, we conducted pairwise correlation analyses between KMT5B and candidate genes using the cBioPortal platform (Cerami et al., 2012) with microarray data obtained from the comprehensive characterization of glioblastoma performed by the TCGA consortium (Cancer Genome Atlas Research Network, 2008). Considering all the samples from the GBM dataset (n = 206), KMT5B mRNA expression showed inverse correlation with IL13RA2 and direct correlation with CDH11, which is in line with our RNAseq and qChIP results (Supplementary Figure 4, upper panels). When grouping the samples by their corresponding GBM gene expression profile (Classical, Mesenchymal, Neural, and Proneural), we observed a similar trend in correlations across all subtypes, and they were particularly significant in the neural GBM subtype (Supplementary Figure 4). Altogether, these data suggest that IL13RA2 and CDH11 could represent direct targets of KMT5B-mediated gene expression regulation.
Discussion
The involvement of KMTs in GBM pathogenesis has not as yet been fully elucidated. Referring back to our data from a 450K Infinium Illumina methylation array reported in 2018 (Fernandez et al., 2018), we found that the KMT5B gene shows an altered DNA methylation and hydroxymethylation profile in GBM as compared to non-tumoral brain samples. The present study aimed to investigate the contribution of this chromatin modifying gene to the etiology of GBM. We obtained evidence that it could exert an anti-tumoral effect, likely through its histone mark H4K20me2.
Expression analyses revealed that KMT5B hypermethylation and hypo-hydroxymethylation correlated with its decreased expression in some of our GBM specimens in comparison with normal brain tissue strongly indicating that the epigenetic downregulation of KMT5B might be relevant for a subset of GBM tumors. In fact, gene inactivation by DNA hypermethylation of CpGis at gene promoters is a well-established epigenetic hallmark in cancer (Koch et al., 2018). As for DNA hydroxymethylation, its presence at gene bodies is positively correlated with gene expression (Jin et al., 2011b), while its loss has been described in several cancers, including GBM (Jin et al., 2011a). This was reinforced in this work through the pharmacological reestablishment of 5mC and 5hmC levels, using a DNMTs inhibitor and a TETs cofactor, which successfully rescued KMT5B expression in our human GBM cell line LN-229. Nevertheless, unlike Sajadian et al. (2016), we did not find differences in the magnitude of changes in 5mC and 5hmC levels using 5-AZA-dC alone or when in combination with vitamin C. This apparent discrepancy might be explained by the fact that we specifically evaluated the methylation/hydroxymethylation state of two intragenic CpGs rather than considering global levels of those marks as in the study cited.
At the functional level, overexpression KMT5B reduced cell proliferation, cell viability and clonogenic potential in vitro, and tumor growth in vivo in mice xenografts. Interestingly, the loss of proliferative capacity induced by KMT5B overexpression in our clones was not related to apoptotic mechanisms, but rather to a G2/M cell cycle blockade. This is in agreement with a previous study showing that KMT5B overexpression causes G2 arrest (Evertts et al., 2013), and is consistent with the widely described role of methylated forms of H4K20 in cell cycle progression (Pesavento et al., 2008).
Regarding the histone mark deposited by KMT5B, H4K20me2, its levels were confirmed to be diminished in mock-transfected GBM cells, and to recover upon KMT5B expression in stably-transfected clones, as demonstrated through immunohistochemical analyses. Concurrently, levels of the histone mark H4K20me1, which serves as KMT5B substrate, varied according to the expression of this enzyme, i.e., they were very low in KMT5B-expressing clones and higher in mock-transfected ones. We further confirmed the H4K20 patterns by treating the cells with A-196, a drug which causes KMT5B enzymatic inhibition. Indeed, the treatment reverted the levels of both marks in KMT5B stably-transfected clones, increasing H4K20me1 and decreasing H4K20me2 to a comparable extent as seen in LN-229 GBM cells.
Furthermore, ectopic expression of KMT5B was associated with transcriptional changes of several GBM-related genes, as seen by the results of RNAseq analyses. The role of H4K20 monomethylation in gene expression has been controversial for some time due to studies associating this mark with both transcriptional repression (Nishioka et al., 2002) and activation (Talasz et al., 2005; Vakoc et al., 2006). However, more recent works and all the genome-wide ChIP analyses to date have rendered compelling results linking this mark with transcription activation (Barski et al., 2007; Li et al., 2011) and, indeed, it is one of the histone modifications that is most strongly correlated with active transcription (Barski et al., 2007; Karliæ et al., 2010; Veloso et al., 2014). By contrast, to date, the function of H4K20 dimethylation in gene expression has scarcely been addressed. That said, studies carried out in human cells suggest that H4K20me2 could be related to gene silencing (Miao and Natarajan, 2005; Vakoc et al., 2006; Gwak et al., 2016). The results presented here highlight the importance of KMT5B epigenetic downregulation in GBM, which leads to a distorted H4K20 methylation pattern. This is in line with the roles for H4K20me1 and H4K20me2 in gene expression mentioned above, and tie in with the concept of epigenetic de-repression of oncogenes. While it is well-known that the epigenetic silencing of tumor suppressor genes occurs during tumorogenesis, the role of epigenetic de-repression of oncogenes due to the loss of repressive chromatin marks has remained partially unexplored (Baylin and Jones, 2016). Our experiments showed that the epigenetic downregulation of KMT5B impairs the balance between H4K20me species in human GBM cell line LN-229. In turn, this may cause global transcriptomic changes, given our finding that overexpression of KMT5B restored H4K20me2 patterns and decreased the expression of several oncogenes, such as IL13RA2, a tumor-restricted receptor (Mintz et al., 2002). IL13 is a pleiotropic Th2-derived cytokine implicated in inflammation and immunomodulation, and is known to induce apoptosis in different cell types, including GBM (Hsi et al., 2011). IL13RA2 binds IL13 with a higher affinity than the physiological IL13RA1/IL4RA receptor but prevents apoptosis as it does not transduce the signal for STAT6 pathway activation (Kawakami et al., 2001). Although the functional significance of IL13RA2 expression is not fully understood, it is considered an inhibitory or decoy receptor in GBM (Rahaman et al., 2002) that contributes to tumor growth (Hsi et al., 2011) and several studies have found that its overexpression is associated with higher glioma grades and poor patient prognosis (Brown et al., 2013; Han and Puri, 2018). Investigations in pancreatic, colorectal, and ovarian cancer have shown that IL13RA2 overexpression promotes tumor migration and invasion (Fujisawa et al., 2009, 2012; Barderas et al., 2012). More recent studies in GBM have also pointed to a role for IL13RA2 in stimulating cell growth and metastasis (Tu et al., 2016). As such, IL13RA2 has become an attractive therapeutic target in GBM, even though the regulatory mechanisms of its expression are currently unknown (Sharma and Debinski, 2018). By means of RNAseq and ChIP experiments, our study suggests that KMT5B and its mark H4K20me2 might be involved in IL13RA2 regulation since overexpression of KMT5B in our clones caused H4K20me2 deposition upstream of the IL13RA2 promoter and a concomitant downregulation of IL13RA2 mRNA.
Notwithstanding, we observed that some genes were upregulated upon KMT5B overexpression. Among them CDH11, which encodes Cadherin 11, has been previously reported to block GBM cell invasion in vitro (Delic et al., 2012). One plausible explanation is that H4K20me2 might function as both transcriptional activator or repressor depending on the cellular context, in concert with other associated histone modifications and readers. That dual role on gene expression modulation would fit with the abundance and ubiquitous distribution of this mark on chromatin (Pesavento et al., 2008; Schotta et al., 2008). Future research is guaranteed in order to elucidate the context dependant function of H4K20me2. Consequently, it is possible that the anti-tumoral effect of KMT5B observed in our study would not be due to the overexpression of this epigenetic modifier itself, but rather might be mediated by the re-establishment of levels of its product H4K20me2 in the promoter region of key GBM-related genes, such as IL13RA2 and CDH11.
The dynamic and reversible nature of H4K20me2 opens the possibility of targeting its demethylase (KDM), via small-molecule inhibitors, to restore this histone mark in GBM. Indeed, in recent years, histone modifiers, such as KDMs, are envisaged as attractive druggable targets for GBM therapy (Jones et al., 2016). Although H4K20me2 is the most abundant form of H4K20 in eukaryotic cells (Pesavento et al., 2008), only a H4K20me2 demethylase has been described so far. It is LSD1n, a neuron-specific splicing variant of the flavin-dependent monoamine oxidase LSD1 with acquired new specificity against H4K20me2 and H4K20me1 (Wang et al., 2015). However, caution is needed when considering LSD1n as a potential therapeutic target using a hypothetical specific KDM inhibitor. On the one hand, there is increasing evidence that histone modifications do not act alone, but influence one another. Hence, modulation of H4K20me2 levels via a LSD1n inhibitor could potentially affect the modification status of other histone residues on the vicinity with unknown effects. On the other hand, non-histone targets of LSD1n should also be taken into account. Moreover, it has been reported that LSD1n promotes neuronal activity-regulated gene expression and plays a critical role in neurite morphogenesis, spatial learning, long-term memory formation, and modulation of emotional behavior (Zibetti et al., 2010; Wang et al., 2015; Rusconi et al., 2016). All of the above reveals the challenging pleiotropic consequences and the complexity of the epigenetic regulation via KDM inhibitors within CNS. In the future, a deeper understanding of the roles and LSD1n targets other than histones, as well as the transcriptional networks regulated by H4K20me2, or even the discovery of other yet unknown specific demethylases, could boost the development of novel specific KDM inhibitors and inform novel strategies in cancer therapy.
Taken together, our results suggest that when KMT5B expression and its histone mark H4K20me2 are perturbed in GBM through deregulated epigenetic mechanisms, this process has a genome-wide effect on gene transcription and may contribute to malignant transformation.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: ENA, PRJEB39613.
Ethics Statement
The studies involving human participants were reviewed and approved by Clinical Research Ethics Committee of the Principality of Asturias (Spain). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Animal Ethics Committee of University of Oviedo (Asturias, Spain).
Author Contributions
MF and AF: conceptualization, study supervision, and funding acquisition. VL, JT, AC, MG, PS-O, RP, CM, RU, AiA, and DN: development of methodology. VL, MC-T, AuA, MM, and BM: acquisition of data. VL, JT, AF, and MF: analysis and interpretation of data, and writing–review and editing. JT and RP: software analysis. VL: writing–original draft preparation. MF: final approval. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We sincerely apologize to all colleagues whose work could not be cited because of space constraints. We would like to thank Miki Hieda (Ehime Prefectural University of Health Sciences, Matsuyama, Japan) for kindly providing the plasmids used in this study. Ronnie Lendrum for editorial assistance. Laura Santos and the Technical Services of University of Oviedo for technical support. All the members of Cancer Epigenetics and Nanomedicine laboratory (FINBA-ISPA, IUOPA, and CINN-CSIC) for their positive feedback and helpful discussions.
Abbreviations
- 5-AZA-dC
5-aza-2 ′-deoxycytidine
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
- ATCC
American Type Culture Collection
- CFA
colony formation assay
- ChIP
chromatin immunoprecipitation
- CpGi
CpG island
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethyl sulfoxide
- DNMTs
DNA methyltransferases
- ENA
European Nucleotide Archive
- FBS
fetal bovine serum
- GBM
glioblastoma multiforme
- GLM
general linear model
- H4K20me
methylation at lysine 20 of histone H4
- HDACs
histone deacetylases
- hMeDIP
5-hydroxymethylcytosine immunoprecipitation
- KDMs
histone lysine demethylases
- KMTs
histone lysine methyltransferases
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PCR
polymerase chain reaction
- PE
platting efficiency
- PI
propidium iodide
- RTCA
real time cell analysis system
- qRT-PCR
quantitative real-time reverse transcriptase polymerase chain reaction
- SAHA
suberoylanilide hydroxamic acid
- STR
short tandem repeat
- TET
ten–eleven translocation
- TMZ
temozolomide
- Vit C
vitamin C
- WGBS
whole genome bisulfite sequencing.
Footnotes
Funding. This research was funded by the Health Institute Carlos III (Plan Nacional de I+D+I) cofounding FEDER (PI15/00892 and PI18/01527 to MF and AF); the Government of the Principality of Asturias PCTI-Plan de Ciencia, Tecnología e Innovación de Asturias co-funding 2018–2022/FEDER (IDI/2018/146 to MF); AECC (PROYE18061FERN to MF); FGCSIC (0348_CIE_6_E to MF); Severo Ochoa Program BP17-165 to PS-O and BP17-114 to RP); the Ministry of Economy and Competitiveness of Spain (VL, Juan de la Cierva fellowship IJCI-2015-23316; JT, Juan de la Cierva fellowship FJCI-2015-26965); FICYT (AC and MG); FINBA-ISPA (VL); and IUOPA (VL and CM). The IUOPA is supported by the Obra Social Cajastur-Liberbank, Spain.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.671838/full#supplementary-material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Barderas R., Bartolomé R. A., Fernandez-Aceñero M. J., Torres S., Casal J. I. (2012). High expression of IL-13 receptor α2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 72 2780–2790. 10.1158/0008-5472.CAN-11-4090 [DOI] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T.-Y., Schones D. E., Wang Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129 823–837. 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- Baylin S. B., Jones P. A. (2016). Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 8:a019505. 10.1101/cshperspect.a019505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behbahani T. E., Kahl P., von, der Gathen J., Heukamp L. C., Baumann C., et al. (2012). Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urol. 12:5. 10.1186/1471-2490-12-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco M., Ropero S., Setien F., Fraga M. F., Lapunzina P., Losson R., et al. (2009). Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl. Acad. Sci. U. S. A. 106 21830–21835. 10.1073/pnas.0906831106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd N. H., Walker K., Fried J., Hackney J. R., McDonald P. C., Benavides G. A., et al. (2017). Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2:92928. 10.1172/jci.insight.92928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C. W., Verhaak R. G. W., McKenna A., Campos B., Noushmehr H., Salama S. R., et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155 462–477. 10.1016/j.cell.2013.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg K. D., Mitchell T. R. H., Upadhyay A. K., Jakob C. G., Jhala M. A., Comess K. M., et al. (2017). The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat. Chem. Biol. 13 317–324. 10.1038/nchembio.2282 [DOI] [PubMed] [Google Scholar]
- Brown C. E., Warden C. D., Starr R., Deng X., Badie B., Yuan Y.-C., et al. (2013). Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoSOne 8:e77769. 10.1371/journal.pone.0077769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455 1061–1068. 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carella A., Tejedor J. R., García M. G., Urdinguio R. G., Bayón G. F., Sierra M., et al. (2020). Epigenetic downregulation of TET3 reduces genome-wide 5hmC levels and promotes glioblastoma tumorigenesis. Int. J. Cancer 146 373–387. 10.1002/ijc.32520 [DOI] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2 401–404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Zhao B. S., He C. (2016). Nucleic acid modifications in regulation of gene expression. Cell Chem. Biol. 23 74–85. 10.1016/j.chembiol.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delic S., Lottmann N., Jetschke K., Reifenberger G., Riemenschneider M. J. (2012). Identification and functional validation of CDH11, PCSK6 and SH3GL3 as novel glioma invasion-associated candidate genes. Neuropathol. Appl. Neurobiol. 38 201–212. 10.1111/j.1365-2990.2011.01207.x [DOI] [PubMed] [Google Scholar]
- Evertts A. G., Manning A. L., Wang X., Dyson N. J., Garcia B. A., Coller H. A. (2013). H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 24 3025–3037. 10.1091/mbc.E12-07-0529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A. P., Koldobskiy M. A., Göndör A. (2016). Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 17 284–299. 10.1038/nrg.2016.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A. F., Bayón G. F., Sierra M. I., Urdinguio R. G., Toraño E. G., García M. G., et al. (2018). Loss of 5hmC identifies a new type of aberrant DNA hypermethylation in glioma. Hum. Mol. Genet. 27 3046–3059. 10.1093/hmg/ddy214 [DOI] [PubMed] [Google Scholar]
- Franken N. A. P., Rodermond H. M., Stap J., Haveman J., van Bree C. (2006). Clonogenic Assay of Cells in Vitro. Nat. Protoc. 1 2315–2319. 10.1038/nprot.2006.339 [DOI] [PubMed] [Google Scholar]
- Fujisawa T., Joshi B., Nakajima A., Puri R. K. (2009). A novel role of interleukin-13 receptor alpha2 in pancreatic cancer invasion and metastasis. Cancer Res. 69 8678–8685. 10.1158/0008-5472.CAN-09-2100 [DOI] [PubMed] [Google Scholar]
- Fujisawa T., Joshi B. H., Puri R. K. (2012). IL-13 regulates cancer invasion and metastasis through IL-13Rα2 via ERK/AP-1 pathway in mouse model of human ovarian cancer. Int. J. Cancer 131 344–356. 10.1002/ijc.26366 [DOI] [PubMed] [Google Scholar]
- García M. G., Carella A., Urdinguio R. G., Bayón G. F., Lopez V., Tejedor J. R., et al. (2018). Epigenetic dysregulation of TET2 in human glioblastoma. Oncotarget 9 25922–25934. 10.18632/oncotarget.25406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzmil M., Morin P., Lino M. M., Merlo A., Frank S., Wang Y., et al. (2011). MAP kinase-interacting kinase 1 regulates SMAD2-dependent TGF-β signaling pathway in human glioblastoma. Cancer Res. 71 2392–2402. 10.1158/0008-5472.CAN-10-3112 [DOI] [PubMed] [Google Scholar]
- Günther H. S., Schmidt N. O., Phillips H. S., Kemming D., Kharbanda S., Soriano R., et al. (2008). Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 27 2897–2909. 10.1038/sj.onc.1210949 [DOI] [PubMed] [Google Scholar]
- Gursoy-Yuzugullu O., Carman C., Serafim R. B., Myronakis M., Valente V., Price B. D. (2017). Epigenetic therapy with inhibitors of histone methylation suppresses DNA damage signaling and increases glioma cell radiosensitivity. Oncotarget 8 24518–24532. 10.18632/oncotarget.15543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak J., Shin J. Y., Lee K., Hong S. K., Oh S., Goh S.-H., et al. (2016). SFMBT2 (Scm-like with four mbt domains 2) negatively regulates cell migration and invasion in prostate cancer cells. Oncotarget 7 48250–48264. 10.18632/oncotarget.10198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Puri R. K. (2018). Analysis of the cancer genome atlas (TCGA) database identifies an inverse relationship between interleukin-13 receptor α1 and α2 gene expression and poor prognosis and drug resistance in subjects with glioblastoma multiforme. J. Neurooncol. 136 463–474. 10.1007/s11060-017-2680-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegi M. E., Diserens A.-C., Gorlia T., Hamou M.-F., de Tribolet N., Weller M., et al. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352 997–1003. 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- Hsi L. C., Kundu S., Palomo J., Xu B., Ficco R., Vogelbaum M. A., et al. (2011). Silencing IL-13Rα2 promotes glioblastoma cell death via endogenous signaling. Mol. Cancer Ther. 10 1149–1160. 10.1158/1535-7163.MCT-10-1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J. F. M., Idema A. J., Bol K. F., Nierkens S., Grauer O. M., Wesseling P., et al. (2009). Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro-Oncol. 11 394–402. 10.1215/15228517-2008-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.-G., Jiang Y., Qiu R., Rauch T. A., Wang Y., Schackert G., et al. (2011a). 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 71 7360–7365. 10.1158/0008-5472.CAN-11-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.-G., Wu X., Li A. X., Pfeifer G. P. (2011b). Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 39 5015–5024. 10.1093/nar/gkr120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. C., Houseman E. A., King J. E., von Herrmann K. M., Fadul C. E., Christensen B. C. (2016). 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat. Commun. 7:13177. 10.1038/ncomms13177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. A., Issa J.-P. J., Baylin S. (2016). Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17 630–641. 10.1038/nrg.2016.93 [DOI] [PubMed] [Google Scholar]
- Kai Y., Peng W., Ling W., Jiebing H., Zhuan B. (2014). Reciprocal effects between microRNA-140-5p and ADAM10 suppress migration and invasion of human tongue cancer cells. Biochem. Biophys. Res. Commun. 448 308–314. 10.1016/j.bbrc.2014.02.032 [DOI] [PubMed] [Google Scholar]
- Karlić R., Chung H.-R., Lasserre J., Vlahovicek K., Vingron M. (2010). Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. U. S. A. 107 2926–2931. 10.1073/pnas.0909344107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K., Taguchi J., Murata T., Puri R. K. (2001). The interleukin-13 receptor alpha2 chain: an essential component for binding and internalization but not for interleukin-13-induced signal transduction through the STAT6 pathway. Blood 97 2673–2679. 10.1182/blood.v97.9.2673 [DOI] [PubMed] [Google Scholar]
- Koch A., Joosten S. C., Feng Z., de Ruijter T. C., Draht M. X., Melotte V., et al. (2018). Analysis of DNA methylation in cancer: location revisited. Nat. Rev. Clin. Oncol. 15 459–466. 10.1038/s41571-018-0004-4 [DOI] [PubMed] [Google Scholar]
- Li Z., Nie F., Wang S., Li L. (2011). Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc. Natl. Acad. Sci. U. S. A. 108 3116–3123. 10.1073/pnas.1009353108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Cheng J., Zhang X., Wang R., Zhang W., Lin H., et al. (2010). Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol. Biomark. Prev. 19 2888–2896. 10.1158/1055-9965.EPI-10-0454 [DOI] [PubMed] [Google Scholar]
- López V., Fernández A. F., Fraga M. F. (2017). The role of 5-hydroxymethylcytosine in development, aging and age-related diseases. Ageing Res. Rev. 37 28–38. 10.1016/j.arr.2017.05.002 [DOI] [PubMed] [Google Scholar]
- Miao F., Natarajan R. (2005). Mapping global histone methylation patterns in the coding regions of human genes. Mol. Cell. Biol. 25 4650–4661. 10.1128/MCB.25.11.4650-4661.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz A., Gibo D. M., Slagle-Webb B., Christensen N. D., Debinski W. (2002). IL-13Ralpha2 is a glioma-restricted receptor for interleukin-13. Neoplasia N. Y. N 4 388–399. 10.1038/sj.neo.7900234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor C. E., Ottaviano R., Reddington J., Sproul D., Reinhardt D., Dunican D., et al. (2012). Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 22 467–477. 10.1101/gr.126417.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K., Rice J. C., Sarma K., Erdjument-Bromage H., Werner J., Wang Y., et al. (2002). PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9 1201–1213. 10.1016/s1097-2765(02)00548-8 [DOI] [PubMed] [Google Scholar]
- Pesavento J. J., Yang H., Kelleher N. L., Mizzen C. A. (2008). Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 28 468–486. 10.1128/MCB.01517-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaman S. O., Sharma P., Harbor P. C., Aman M. J., Vogelbaum M. A., Haque S. J. (2002). IL-13R(alpha)2, a decoy receptor for IL-13 acts as an inhibitor of IL-4-dependent signal transduction in glioblastoma cells. Cancer Res. 62 1103–1109. 10.1093/intimm/10.8.1103 [DOI] [PubMed] [Google Scholar]
- Raiber E.-A., Beraldi D., Martínez Cuesta S., McInroy G. R., Kingsbury Z., Becq J., et al. (2017). Base resolution maps reveal the importance of 5-hydroxymethylcytosine in a human glioblastoma. NPJ Genomic Med. 2:6. 10.1038/s41525-017-0007-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran G., Shanmuganandam K., Bendre A., Muzumdar D., Mujumdar D., Goel A., et al. (2011). Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J. Neurooncol. 104 483–494. 10.1007/s11060-010-0520-2 [DOI] [PubMed] [Google Scholar]
- Roccaro A. M., Sacco A., Jia X., Azab A. K., Maiso P., Ngo H. T., et al. (2010). microRNA-dependent modulation of histone acetylation in Waldenstrom macroglobulinemia. Blood 116 1506–1514. 10.1182/blood-2010-01-265686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi F., Grillo B., Ponzoni L., Bassani S., Toffolo E., Paganini L., et al. (2016). LSD1 modulates stress-evoked transcription of immediate early genes and emotional behavior. Proc. Natl. Acad. Sci. U. S. A. 113 3651–3656. 10.1073/pnas.1511974113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadian S. O., Ehnert S., Vakilian H., Koutsouraki E., Damm G., Seehofer D., et al. (2015). Induction of active demethylation and 5hmC formation by 5-azacytidine is TET2 dependent and suggests new treatment strategies against hepatocellular carcinoma. Clin. Epigenetics 7:98. 10.1186/s13148-015-0133-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadian S. O., Tripura C., Samani F. S., Ruoss M., Dooley S., Baharvand H., et al. (2016). Vitamin C enhances epigenetic modifications induced by 5-azacytidine and cell cycle arrest in the hepatocellular carcinoma cell lines HLE and Huh7. Clin. Epigenetics 8:46. 10.1186/s13148-016-0213-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G., Sengupta R., Kubicek S., Malin S., Kauer M., Callén E., et al. (2008). A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 22 2048–2061. 10.1101/gad.476008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Debinski W. (2018). Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 19:9113326. 10.3390/ijms19113326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim F. J., Windrem M. S., Goldman S. A. (2009). Fate determination of adult human glial progenitor cells. Neuron Glia Biol. 5 45–55. 10.1017/S1740925X09990317 [DOI] [PubMed] [Google Scholar]
- Singh M. M., Johnson B., Venkatarayan A., Flores E. R., Zhang J., Su X., et al. (2015). Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro-Oncol. 17 1463–1473. 10.1093/neuonc/nov041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talasz H., Lindner H. H., Sarg B., Helliger W. (2005). Histone H4-lysine 20 monomethylation is increased in promoter and coding regions of active genes and correlates with hyperacetylation. J. Biol. Chem. 280 38814–38822. 10.1074/jbc.M505563200 [DOI] [PubMed] [Google Scholar]
- Tu M., Wange W., Cai L., Zhu P., Gao Z., Zheng W. (2016). IL-13 receptor α2 stimulates human glioma cell growth and metastasis through the Src/PI3K/Akt/mTOR signaling pathway. Tumour Biol. 37 14701–14709. 10.1007/s13277-016-5346-x [DOI] [PubMed] [Google Scholar]
- Vakoc C. R., Sachdeva M. M., Wang H., Blobel G. A. (2006). Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell. Biol. 26 9185–9195. 10.1128/MCB.01529-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veloso A., Kirkconnell K. S., Magnuson B., Biewen B., Paulsen M. T., Wilson T. E., et al. (2014). Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 24 896–905. 10.1101/gr.171405.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Telese F., Tan Y., Li W., Jin C., He X., et al. (2015). LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 18 1256–1264. 10.1038/nn.4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M., Wick W., Aldape K., Brada M., Berger M., Pfister S. M., et al. (2015). Glioma. Nat. Rev. Dis. Primer 1:15017. 10.1038/nrdp.2015.17 [DOI] [PubMed] [Google Scholar]
- White M. G., Nagar S., Aschebrook-Kilfoy B., Jasmine F., Kibriya M. G., Ahsan H., et al. (2016). Epigenetic Alterations and Canonical Pathway Disruption in Papillary Thyroid Cancer: A Genome-wide Methylation Analysis. Ann. Surg. Oncol. 23 2302–2309. 10.1245/s10434-016-5185-4 [DOI] [PubMed] [Google Scholar]
- Wu M.-Y., Fu J., Xiao X., Wu J., Wu R.-C. (2014). MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett. 354 311–319. 10.1016/j.canlet.2014.08.031 [DOI] [PubMed] [Google Scholar]
- Xu Z., Taylor J. A., Leung Y.-K., Ho S.-M., Niu L. (2016). oxBS-MLE: an efficient method to estimate 5-methylcytosine and 5-hydroxymethylcytosine in paired bisulfite and oxidative bisulfite treated DNA. Bioinforma. Oxf. Engl. 32 3667–3669. 10.1093/bioinformatics/btw527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Pesavento J. J., Starnes T. W., Cryderman D. E., Wallrath L. L., Kelleher N. L., et al. (2008). Preferential dimethylation of histone H4 lysine 20 by Suv4-20. J. Biol. Chem. 283 12085–12092. 10.1074/jbc.M707974200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama Y., Matsumoto A., Hieda M., Shinchi Y., Ogihara E., Hamada M., et al. (2014). Loss of histone H4K20 trimethylation predicts poor prognosis in breast cancer and is associated with invasive activity. Breast Cancer Res. BCR 16:R66. 10.1186/bcr3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Yang F., Chen B., Lu Z., Huo X., Zhou W., et al. (2011). The histone deacetylase 4/SP1/microrna-200a regulatory network contributes to aberrant histone acetylation in hepatocellular carcinoma. Hepatol. Baltim. Md 54 2025–2035. 10.1002/hep.24606 [DOI] [PubMed] [Google Scholar]
- Zibetti C., Adamo A., Binda C., Forneris F., Toffolo E., Verpelli C., et al. (2010). Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J. Neurosci. Off. J. Soc. Neurosci. 30 2521–2532. 10.1523/JNEUROSCI.5500-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: ENA, PRJEB39613.