

Abstract
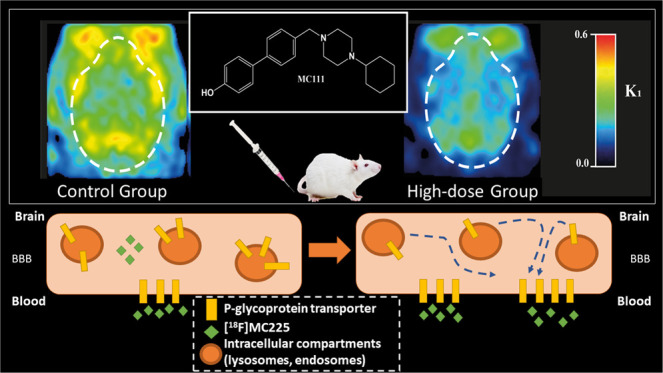
P-Glycoprotein (P-gp) is an efflux pump located at the blood–brain barrier (BBB) that contributes to the protection of the central nervous system by transporting neurotoxic compounds out of the brain. A decline in P-gp function has been related to the pathogenesis of neurodegenerative diseases. P-gp inducers can increase the P-gp function and are considered as potential candidates for the treatment of such disorders. The P-gp inducer MC111 increased P-gp expression and function in SW480 human colon adenocarcinoma and colo-320 cells, respectively. Our study aims to evaluate the P-gp inducing effect of MC111 in the whole brain in vivo, using the P-gp tracer [18F]MC225 and positron emission tomography (PET). Eighteen Wistar rats were treated with either vehicle solution, 4.5 mg/kg of MC111 (low-dose group), or 6 mg/kg of MC111 (high-dose group). Animals underwent a 60 min dynamic PET scan with arterial-blood sampling, 24 h after treatment with the inducer. Data were analyzed using the 1-tissue-compartment model and metabolite-corrected plasma as the input function. Model parameters such as the influx constant (K1) and volume of distribution (VT) were calculated, which reflect the in vivo P-gp function. P-gp and pregnane xenobiotic receptor (PXR) expression levels of the whole brain were assessed using western blot. The administration of MC111 decreased K1 and VT of [18F]MC225 in the whole brain and all of the selected brain regions. In the high-dose group, whole-brain K1 was decreased by 34% (K1-high-dose = 0.20 ± 0.02 vs K1-control = 0.30 ± 0.02; p < 0.001) and in the low-dose group by 7% (K1-low-dose = 0.28 ± 0.02 vs K1-control = 0.30 ± 0.02; p = 0.42) compared to controls. Whole-brain VT was decreased by 25% in the high-dose group (VT-high-dose = 5.92 ± 0.41 vs VT-control = 7.82 ± 0.38; p < 0.001) and by 6% in the low-dose group (VT-low-dose = 7.35 ± 0.38 vs VT-control = 7.82 ± 0.37; p = 0.38) compared to controls. k2 values did not vary after treatment. The treatment did not affect the metabolism of [18F]MC225. Western blot studies using the whole-brain tissue did not detect changes in the P-gp expression, however, preliminary results using isolated brain capillaries found an increasing trend up to 37% in treated rats. The decrease in K1 and VT values after treatment with the inducer indicates an increase in the P-gp functionality at the BBB of treated rats. Moreover, preliminary results using brain endothelial cells also sustained the increase in the P-gp expression. In conclusion, the results verify that MC111 induces P-gp expression and function at the BBB in rats. An increasing trend regarding the P-gp expression levels is found using western blot and an increased P-gp function is confirmed with [18F]MC225 and PET.
Keywords: ABC transporters, brain Imaging, efflux transporters, P-glycoprotein, P-gp inducers
Introduction
P-glycoprotein (P-gp) is an efflux transporter that belongs to the ATP-binding cassette (ABC) transporter family and is located at the luminal side of the endothelial cells at the blood–brain barrier (BBB) and other blood-tissue barriers.1 P-gp pumps a wide variety of compounds, including endogenous and exogenous substances from the brain to the circulation, by adenosine triphosphate (ATP)-driven transport.2 Thereby, P-gp located at the BBB contributes to the protection of the brain from harmful substances and maintains the homeostasis needed for the appropriate neural function.3 However, several factors can affect P-gp function or expression such as inflammatory responses, stress, therapeutic drugs, and diet.4
A decline in the P-gp function at the BBB can increase the concentration of neurotoxic compounds inside the brain, which can be related to the onset and progression of several neurodegenerative diseases such as Alzheimer’s (AD). It has been reported that P-gp is involved in the clearance of Aβ peptides since the inhibition of P-gp decreased extracellular levels of Aβ secretions in vitro.5 Moreover, an inverse correlation between Aβ deposition and expression of P-gp was found in the BBB of elderly nondemented subjects.6 Also, positron emission tomography (PET) imaging studies performed in AD patients found an increase of [11C]verapamil uptake inside the brain compared to healthy volunteers, which indicates a decreased P-gp function.7
Several studies have already reported the use of P-gp inducers to restore the function of P-gp in neurodegenerative diseases.8,9 P-gp induction does not occur via direct binding to the P-gp transporter, and P-gp expression and function can be regulated by different mechanisms including the activation of several transcription factors and post-transcriptional and post-translational mechanisms.10−12 Recently, it has been proposed that an increase in P-gp expression at the cell surface can be caused by relocating intracellular P-gp protein to the plasma membrane.10,11 Among the transcription factors, the nuclear receptors pregnane xenobiotic receptor (PXR) and constitutive androstane receptor (CAR) have been identified as key factors in the P-gp upregulation.13 The PXR and CAR can be activated by endogenous or exogenous compounds.14 PXR is activated not only by steroids, chemotherapeutics, HIV protease inhibitors, glucocorticoids, and anticonvulsants but also by dietary compounds and natural steroids such as pregnenolone and progesterone.15 CAR is activated by endogenous ligands such as bile acids and also by drugs, dietary compounds, and pollutants.16
Since P-gp inducers can play an important role in the treatment of neurodegenerative diseases,8,9 several preclinical studies have already reported the efficacy of such drugs. A study performed in a transgenic mouse model of human AD found a reduced P-gp function in these animals compared to wild-type mice. After treatment with pregnenolone-16α-carbonitrile for 7 days, PXR was activated, and consequently, the P-gp function was restored resulting in an increase of Aβ clearance from the brain.17 1,25-Dihydroxyvitamin D3-enriched diet also induced the P-gp function and reduced the Aβ deposition in mice models of AD. In contrast, AD model mice fed with a vitamin D-deficient diet showed a reduction in P-gp expression and increased Aβ plaques in the brain as well as increased cognitive deficits.18 The administration of St. John’s Wort, which is a herbal drug used in the treatment of depression, to transgenic AD mice enhanced the cerebral P-gp function and reduced Aβ accumulation.19
Recently, a new promising compound, 4′-((4-cyclohexylpiperazin-1-yl)methyl)-[1,1′-biphenyl]-4-ol (MC111) has been identified as a P-gp inducer by in vitro studies. MC111 activity was assessed in colo-320 cells. After treatment with MC111, an increase in [3H]verapamil efflux was observed, which indicates an increase in the P-gp function. Western blot studies also indicated that MC111 increases P-gp expression in SW480 human colon adenocarcinoma cells.20 Furthermore, this new compound also activates the breast cancer resistance protein (BCRP) function and expression in colo-320 and MCF7 cells.20 The dual activity of MC111 may be beneficial for the treatment of several neurodegenerative diseases where not only an impaired P-gp function and expression has been observed but also alterations in the BCRP and other ABC transporters.21,22
This study aims to demonstrate the P-gp induction effect of MC111 in vivo at the BBB of healthy rats. For this purpose, we used positron emission tomography (PET) imaging, which is a noninvasive imaging technique that quantifies the biochemical process in vivo, and the tracer [18F]MC225 for measuring the P-gp function. [18F]MC225 is considered as a weak P-gp substrate and therefore, it shows relatively high uptake in the brain at baseline conditions when the P-gp transporter is completely functional. This characteristic of [18F]MC225 could be beneficial for imaging increased P-gp function upon treatment with an inducer when the tracer uptake in the brain is expected to decrease.23,24
In the present study, we will evaluate the effect of an inducer for the first time in vivo, after its administration to living subjects. To this aim, a dynamic PET scan will be made in eighteen healthy male rats. The kinetics of [18F]MC225 will be assessed by the 1-tissue compartment model (1-TCM) and changes in the P-gp function will be reflected by the influx constant (K1) and volume of distribution (VT). Moreover, the P-gp expression level in brain tissue will be evaluated postmortem using western blots. The results may yield insight into the potential therapeutic application of MC111 in diseases where the P-gp function/expression is decreased.
Experimental Section
Chemicals
6,7-Dimethoxy-2-(3-(5-(2-((methylsulfonyl)oxy)ethoxy)-1,2,3,4-tetrahydronaphthalen-1-yl)propyl)-1,2,3,4-tetrahydroisoquinolin-2-ium methanesulfonate salt (mesylate [18F]MC225 precursor) was purchased from Syncom (Syncom, Groningen, the Netherlands). The inducer MC111 (4′-((4-cyclohexylpiperazin-1-yl)methyl)-[1,1′-biphenyl]-4-ol) was supplied by the University of Bari, Italy. Chemicals were purchased from Sigma-Aldrich (Sigma-Aldrich, St.Louis, MO) and isoflurane from Pharmachemie (Pharmachemie, Haarlem, the Netherlands).
Tracer Production
[18F]MC225 was synthesized by a one-step reaction.25 Briefly, the radiosynthesis was automated and performed in Synthera modules (IBA RadioPharma Solutions, Ottignies-Louvain-la-Neuve, Belgium). After drying the [18F]Fluoride, the mesylate [18F]MC225 precursor dissolved in dimethylformamide (DMF) was added to the vial and the mixture was heated at 140 °C for 30 min. After purification on a semipreparative high-performance liquid chromatography (HPLC) system, the radiotracer was reformulated to 15% ethanol in sterile 0.9% NaCl solution. Quality control procedures were performed in an ultraperformance liquid chromatography (UPLC) system, as previously described.24
Animals
Eighteen healthy male Wistar rats (RjHan: WI) were obtained from Janvier Labs (Janvier Labs, Le Genest-Saint-Isle, France) (281 ± 9 g). Before starting the experiments, rats were acclimatized to the new environment for at least 7 days. Rats were housed in groups of 2 in a temperature-controlled room (21 ± 2 °C) with 12 h dark/12 h light cycles. Water and standard laboratory chow were available ad libitum. All animal experiments were performed in compliance with Dutch and EU regulations. The experimental protocol was approved by the National Committee on Animal Experiments of the Netherlands (CCD, the Hague) and the Institutional Animal Care and Use Committee of the University of Groningen (CCD license number: AVD105002015166, IvD protocol: 15166-01-002 and CCD license number: AVD1050020198648 IvD protocol: 198648-01-001).
Rats were randomly divided into three groups treated with either vehicle solution (control group, n = 6), the MC111 (Figure 1) inducer at a dose of 4.5 mg/kg (low-dose group, n = 6), or MC111 inducer at a dose of 6 mg/kg (high-dose group, n = 6). The injection of MC111 was performed 24 h before the PET scan, intravenously through a tail vein under isoflurane anesthesia (12 ± 9 min). First, MC111 was dissolved in dimethyl sulfoxide (DMSO) (5% v/v) (42 μL) and poly(ethylene glycol) (PEG) 300 (10%) (84 μL), and subsequently, sterile water (85%) was added to the mixture, resulting in a total volume of 0.85 mL (injection volume). An extra group of healthy male Wistar rats (RjHan: WI) (Janvier Labs) (315 ± 15 g) were used to evaluate the P-gp expression level in the isolated brain capillaries by means of western blot. Animals were divided into two groups. One group of animals (n = 6) was injected with 6 mg/kg of MC111 and the other group (n = 5) with vehicle solution. Twenty-four hours after the injection of the treatment, animals were sacrificed, and the brain tissue was collected. Afterward, the brain endothelial cells were isolated, as described elsewhere.26,27
Figure 1.

Molecular structures of the P-gp tracer [18F]MC225 (A) and the P-gp inducer MC111 (B).
PET Imaging
One day after the treatment with MC111, animals were transported to the microPET facility. Before the PET scans, rats underwent surgery to place a cannula in a side branch of the femoral artery, which allows blood sampling during the scan. After the surgery, anesthetized rats were placed in the bed of the PET scanner (Focus 220 microPET, Siemens Medical Solutions Inc., Malvern, PA) with their head placed in the center of the field of view. First, a transmission scan was acquired using a 57Co point source. Then, a 60 min dynamic PET scan was started simultaneously with the tracer injection. Rats were injected with [18F]MC225 (28.4 ± 10.7 MBq in a total volume of 1 mL) via the tail vein at a speed of 1 mL/min. After the administration of [18F]MC225 (Figure 1), 15 blood samples were taken from the arterial cannula. These blood samples were used for the determination of the radiotracer concentration in blood and plasma and to measure the percentage of [18F]MC225 and its radioactive metabolites in plasma. Rats were anesthetized with isoflurane in 95% oxygen (induction 5% and maintenance 1.5–2%) during the surgery, the tracer administration, and the PET scan. The body temperature was maintained close to normal values and blood oxygenation and the heart rate were continuously monitored during the scan, using pulse oximeters. After the scan, rats were perfused with cold phosphate-buffered saline (PBS) from the left ventricle under the deep isoflurane anesthesia and then sacrificed by removal of the heart. Brain tissues were immediately collected and stored at −80 °C after flash-freezing in liquid nitrogen.
Arterial-Blood Sampling and Metabolism
Immediately after the injection of the tracer, blood samples were collected at the following time points: 10, 20, 30, 40, and 50 s, and 1, 1.3, 2, 3, 5, 7.3, 10, 15, 30, and 60 min. The drawn volume of blood (0.1 mL) was replaced by a warm saline solution containing 0.1% heparin. From each blood sample, a 25 μl sample of whole blood was pipetted into a different tube. The remaining blood was centrifuged for 5 min at 3700 rpm (Mikro 20, Hettich, Germany), and 25 μl of plasma was transferred to another tube. Radioactivity in whole blood and plasma was measured in a γ counter (LKB Wallac, Turku, Finland) and values were corrected for decay from the time of tracer injection.
The [18F]MC225 parent fraction and radiometabolites in plasma were assessed by thin-layer chromatography (TLC) (F-254 silica gel plates, Sigma-Aldrich) as follows.24 Plasma samples acquired at 10 s and 1, 3, 5, 7.3, 10, 15, 30, and 60 min were used for metabolite analysis. Fifty microliters of acetonitrile (ACN) was added to the plasma sample to precipitate the proteins. Then, plasma samples were vortexed and centrifuged for 5 min at 6000 rpm (Hettich Mikro 20, Hettich Zentrifugen, Germany). The protein-free supernatant (2.5 μL) was spotted onto a silica gel TLC plate. When the sample spots were dried, the samples on TLC plates were eluted with 10% of MeOH in EtOAc. The TLC plates were exposed to activated phosphor storage plates (PerkinElmer, Waltham, MA) overnight. The following day, the storage plates were read using a GE Amersham Typhoon molecular imager (GE Healthcare, Life Sciences, MA). OptiQuant 03.00 software (PerkinElmer) was used to calculate the fractions of the parent tracer and radioactive metabolites.
To calculate the percentage of the radioactive metabolites in plasma, a single exponential equation was fitted to the values obtained from the metabolite analysis, using an iterative nonlinear least-squares approach and GraphPad software (GraphPad Prism version 7.02, San Diego, CA): Y = (Y0 – plateau)*exp(−Ke*X) + plateau,28 where Y is the percentage of the parent fraction at different time points; Y0, the intercept which was fixed to 100% (percentage of the parent fraction at the beginning of the scan); Ke, the elimination constant; and X, the time.
PET Image Reconstruction and Analysis
Data from the transmission scan were used for attenuation and scatter corrections. The list-mode data of the emission scan was reconstructed into 21 frames (6 × 10; 4 × 30; 2 × 60; 1 × 120; 1 × 180; 4 × 300; 3 × 600) using OSEM 2D, 4 iterations, and 16 subsets. The PET data were normalized and corrected for scattering, attenuation, and radioactivity decay.
PMOD v3.8 software (PMOD Technologies, Zürich, Switzerland) was used to automatically and individually register the PET images to an [18F]MC225 specific template29,30 by rigid transformation.
The amygdala, cerebellum, corpus callosum, medial geniculate, mesencephalic, septum, superior colliculus, striatum, cortex, hippocampus, hypothalamus, midbrain, brainstem, thalamus, basal ganglia, and a whole-brain region were selected as volumes of interest (VOI) for this study. The radioactivity concentration in each VOI (KBq/mL) was calculated and corrected for the injected dose (KBq) and the bodyweight of the animal (g) and thus, it was expressed as the standardized uptake value (SUV).
Input Function Analysis
Time-activity curves (TAC) of whole blood, plasma, and plasma-corrected for metabolites were calculated using the blood measurement and expressed in SUV. The metabolite-corrected plasma TAC–SUV was obtained by multiplying the SUV values of the plasma samples by the percentage of the parent fraction in each time-point.
The first-order rate constant of elimination (Ke) was calculated by fitting a single exponential curve to the metabolite-corrected plasma TAC–SUV of each subject, by an iterative nonlinear least-squares approach using GraphPad software: Y = Y0 × exp(−Ke × X), where Y is the metabolite-corrected SUV value in plasma; Y0 is the Y-intercept; Ke is the first-order rate constant of elimination, and X is the time.31
Pharmacokinetic Modeling
Measured radioactivity in whole blood and plasma as well as the percentage of the [18F]MC225 parent tracer were used as input functions for the pharmacokinetic analysis (PMOD Technologies).
The 1-tissue compartmental model (1-TCM) has been selected as the model of choice to analyze the [18F]MC225 data in rats and the cerebral volume of blood was fixed to 5%.24,29 The TACs were corrected for blood delay. First, the blood delay of the whole-brain region TAC was estimated and then this value was fixed for the rest of the brain regions.
The influx rate constant K1 is considered as the best parameter to measure the P-gp at the BBB,24,32,33 thus the study evaluated the changes in K1 caused by the administration of the treatment. Since the volume of distribution (VT) can also be used to assess changes in the P-gp function, this parameter was also calculated.24 Some studies reported changes in the efflux constant k2 due to the P-gp inhibition,34,35 thus the effect of the treatment on k2 values was also evaluated.
Parametric Images
Parametric images representing K1, VT, and k2 values of three rats from different groups (control, low-dose, and high-dose group) were used for illustrative purposes. To this aim, the metabolite-corrected plasma TAC of each subject was used as the input function for l-TCM analysis (vB = 5%), using PMOD.
Western Blot Analysis
P-glycoprotein (P-gp) and pregnane X receptor (PXR) expression levels in whole-brain tissue were quantified by means of western blot.
Protein extraction was carried out by adding cold RIPA buffer (Sigma-Aldrich) with the protease inhibitor cocktail (Sigma-Aldrich) to brain slices in an approximated 10:1 proportion (volume/weight), and tissue disruption was performed in a TissueLyser II (Qiagen, Hombrechtikon, Switzerland). Then, tissue lysates were centrifuged for 30 min at 20 000g, and supernatants were collected to perform western blot. Protein concentration in the lysates was quantified using a micro BCA Protein assay kit (Thermo Fisher, Waltham, MA).
The necessary volume of cell lysate containing a total amount of 25 μg of protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) in 4–15% Criterion TGX precast midi protein gel (BioRad, Hercules, CA) using a constant voltage of 140 V. Then, proteins were transferred onto a poly(vinylidene difluoride) (PVDF) membrane (Millipore, Cork, Ireland) using a Trans-Blot semidry system (BioRad) with a limited voltage of 25 V and 180 mA for 2 h. After the blotting step, membranes were blocked for 1 h with a 3% bovine serum albumin (BSA) solution (in Tris-Chloride Buffer with a 0.1% of Tween 20 (TBST)). Once blocked, target proteins were detected through incubation with primary antibodies against P-gp (Rabbit monoclonal to P-glycoprotein ab170904, Abcam, Cambridge, U.K.), PXR (Rabbit polyclonal to PXR ab192579, Abcam), β-Actin (Mouse monoclonal to β-Actin ab8226, Abcam), and histone H3 (Rabbit polyclonal to H3, ab1791, Abcam) diluted 1:1000, 1:1000, 1:5000, and 1:1000, respectively, in TBS with 3% of BSA. Primary antibodies were incubated overnight at room temperature under agitation, and then membranes were washed three times in TBST to remove the excess of primary antibodies and avoid unspecific signaling. As secondary antibodies, an HRP-conjugated Goat anti-Rabbit IgG (P044801-2, Dako, Denmark) and HRP-conjugated Rabbit anti-Mouse IgG (P026002-2) were used. They were diluted to 1:5000 in TBS with 3% BSA and incubated for 1 h at room temperature under agitation. Eventually, after washing, the HRP activity was revealed with a Pierce _ECL western blotting substrate (Thermo Fisher) and detected in a Chemi Doc MP imaging system (BioRad). WB results were analyzed by measuring the mean gray value of protein bands delimited in ROIs using ImageJ software (Rasband, WS). The relative expression of P-gp to β-Actin and PXR to histone H3 was calculated for each sample, and the average of samples for each group was normalized to the control.
As previously mentioned, additional experiments were performed to evaluate the changes in the P-gp expression level in isolated brain capillaries. Animals were sacrificed, and brains were immediately collected in ice-cold Dulbecco′s phosphate-buffered saline (DPBS) (Cytiva, Global Life Sciences Solutions, Marlborough, MA). The isolation of the capillaries was performed, as previously described by Hartz et al.26,27 Briefly, meninges were carefully removed, and the olfactory bulb, cerebellum, and white matter structures were separated from the cortex. The cortex was homogenized in DPBS with Ficoll PM400 (Cytiva, Global Life Sciences Solutions) and centrifuged at 5800g for 20 min at 4 °C. The supernatant was discarded, and the pellet was resuspended in 1% BSA–DPBS buffer. The suspension was filtered through a 200 μm mesh and after several washing steps, the filtrate was centrifuged at 1500g for 3 min at 4 °C. Afterward, the pellet was flash-frozen and stored at −80 °C. The amount of protein in each sample was also quantified using a micro BCA protein assay kit, and western blot studies were performed loading 40 μg of the protein in each well. Western blots using the brain isolated endothelial cells were carried out following the previously described protocol.
Statistical Analysis
Descriptive data are presented as mean ± standard deviation (SD), whereas the results of the statistical analysis are shown as mean ± standard error (SE) unless otherwise indicated. IBM SPSS 23 (Chicago, IL) was used for the statistical analysis. Generalized estimated equation (GEE) analysis with the independent matrix36 was used to assess the differences in the SUV–TAC of the blood and brain. Generalized linear models (GLM) were used to evaluate the differences in SUV40–60 min, pharmacokinetic parameters (VT, K1, k2), and blood kinetics (Ke) among the groups, independently for each region. P-values < 0.05 were considered statistically significant. Relative changes among the groups were calculated, using the control as the reference.
Results
Tracer Production
The final product was obtained in 5.6 ± 2.9% yield (mean ± SD) (n = 9) (decay-corrected) in an average synthesis time of 121 ± 24 min (mean ± SD). The radiochemical purity of [18F]MC225 was 95.3 ± 2.7% (mean ± SD) and the molar activity higher than 80 TBq/mmol (mean ± SD).
Plasma Kinetics and Metabolism of [18F]MC225
No significant differences between the control and the treated animals were found concerning the parent fraction in plasma. However, statistical analysis revealed that the (metabolite-corrected) SUV–TAC of [18F]MC225 in plasma was higher in the high-dose group than in the other two groups. The SUV–TAC of the tracer was 44% (p = 0.042) higher in the high-dose group than in the control group (high-dose group = 0.97 ± 0.10, low-dose group = 0.67 ± 0.06, control group = 0.70 ± 0.10) (Figure 2).
Figure 2.

Metabolite-corrected SUV–TACs for plasma of the control (blue), low-dose (green), and high-dose groups (red) (A) and the percentage of plasma radioactivity representing [18F]MC225 as a function of time (B).
The statistical analysis found a significant increase in the Ke in the 6 mg/kg treated group. The Ke from the high-dose group increased by 25% compared to the control group (Ke high-dose group = 0.048 ± 0.003 s–1 vs Ke control group = 0.039 ± 0.003 s–1, p = 0.014). However, no significant differences were found between the control group and the low-dose group (Ke low-dose group = 0.035 ± 0.003 s–1 vs Ke control group = 0.039 ± 0.002 s–1, p = 0.35).
Brain Kinetics of [18F]MC225
SUV–TAC of the Whole Brain
SUV–TACs of the whole brain were calculated. In all groups, [18F]MC225 showed an initially high uptake followed by a subsequent decrease (Figure 3). In all groups, the peak was observed at 3.3 ± 2.4 min (mean ± SD) after injection of the tracer. Statistical analysis did not find any significant differences in the SUV–TAC of the whole brain among the groups.
Figure 3.

SUV–TACs of the whole brain in the control (blue color), low-dose (4.5 mg/kg) (green color), and high-dose groups (6 mg/kg) (red color).
SUV40–60 min of the Whole-Brain Region
Since the last part of the PET scans shows more stable SUV values, a mean SUV value from 40 to 60 min was calculated for all of the animals (SUV40–60 min). The SUV40–60 min calculated using the last 20 min of the scan decreased in the high-dose group with regard to the control group, but this difference was not statistically significant (SUV40–60 min control = 0.45 ± 0.02; SUV40–60 min low-dose= 0.45 ± 0.02; SUV40–60 min high-dose = 0.40 ± 0.02).
Pharmacokinetic Modeling
A low dose of 4.5 mg/kg MC111 decreased the VT and K1 values compared to the control group, however, the decrease was not statistically significant. The higher dose (6 mg/kg) caused a significant reduction in the K1 and VT values in all of the brain regions compared to the control group. The analysis also found significant differences in K1 and VT between the high-dose group and the low-dose group. Figure 4 shows the VT, K1, and k2 values of all of the brain regions in all of the experimental groups.
Figure 4.

Mean ± SE values of K1 (A), VT (B), and k2 (C) in the control (blue), low-dose (4.5 mg/kg) (green), and high-dose (6 mg/kg) (red) groups.
The whole-brain region K1 significantly decreased by 34% (p < 0.001) after the high-dose treatment compared to the control group, and the whole-brain region VT also decreased by 24% (p < 0.001). A similar trend was observed in all brain regions. The largest change in the K1 was observed in the septum, the superior colliculus, and the cortex region with a decrease of 39, 38, and 36% compared to the control group, respectively. In the case of VT, the largest changes were found in the brainstem, the thalamus, and the superior colliculus with reductions of 30, 27, and 26%, respectively (more information in Table 1). The analysis did not find significant differences in k2 values between the low-dose group and the control in any of the brain regions. However, k2 values decreased in the high-dose group compared to the control.
Table 1. Mean ± SE K1 and VT Values of All Selected Brain Regions in All Experimental Groups, Percentages of Change Relative to the Control Group, and p-Values.
|
K1 (mL/(mL min)) |
VT (mL/mL) |
||||||
|---|---|---|---|---|---|---|---|
| region | group | mean ± SE | changes relative to control (%) | p-value | mean ± SE | changes relative to control (%) | p-value |
| amygdala | control | 0.27 ± 0.02 | 7.38 ± 0.47 | ||||
| 4.5 mg/kg | 0.27 ± 0.02 | –0.94 | 0.92 | 6.94 ± 0.47 | –6.03 | 0.50 | |
| 6 mg/kg | 0.19 ± 0.02 | –30.62 | 0.00 | 5.41 ± 0.51 | –26.81 | 0.00 | |
| basal ganglia | control | 0.27 ± 0.02 | 6.23 ± 0.33 | ||||
| 4.5 mg/kg | 0.26 ± 0.02 | –4.63 | 0.62 | 5.89 ± 0.33 | –5.53 | 0.46 | |
| 6 mg/kg | 0.19 ± 0.02 | –30.43 | 0.00 | 4.6 ± 0.36 | –26.19 | 0.00 | |
| brainstem | control | 0.41 ± 0.03 | 8.66 ± 0.43 | ||||
| 4.5 mg/kg | 0.37 ± 0.03 | –8.80 | 0.31 | 7.88 ± 0.43 | –9.02 | 0.20 | |
| 6 mg/kg | 0.29 ± 0.03 | –29.33 | 0.00 | 6.1 ± 0.48 | –29.63 | 0.00 | |
| cerebellum | control | 0.38 ± 0.02 | 8.7 ± 0.49 | ||||
| 4.5 mg/kg | 0.36 ± 0.02 | –4.77 | 0.58 | 8.18 ± 0.49 | –6.01 | 0.45 | |
| 6 mg/kg | 0.26 ± 0.03 | –30.60 | 0.00 | 6.41 ± 0.54 | –26.37 | 0.00 | |
| corpus callosum | control | 0.26 ± 0.02 | 7.25 ± 0.34 | ||||
| 4.5 mg/kg | 0.23 ± 0.02 | –8.00 | 0.43 | 7.04 ± 0.34 | –2.80 | 0.68 | |
| 6 mg/kg | 0.17 ± 0.02 | –34.47 | 0.00 | 5.65 ± 0.37 | –22.08 | 0.00 | |
| cortex | control | 0.27 ± 0.02 | 8.57 ± 0.41 | ||||
| 4.5 mg/kg | 0.24 ± 0.02 | –9.65 | 0.29 | 8.07 ± 0.41 | –5.89 | 0.38 | |
| 6 mg/kg | 0.17 ± 0.02 | –36.39 | 0.00 | 6.76 ± 0.45 | –21.19 | 0.00 | |
| hippocampus | control | 0.27 ± 0.02 | 6.8 ± 0.35 | ||||
| 4.5 mg/kg | 0.26 ± 0.02 | –1.33 | 0.90 | 6.5 ± 0.35 | –4.48 | 0.54 | |
| 6 mg/kg | 0.18 ± 0.02 | –33.77 | 0.00 | 5.24 ± 0.38 | –22.94 | 0.00 | |
| hypothalamus | control | 0.35 ± 0.03 | 7.57 ± 0.37 | ||||
| 4.5 mg/kg | 0.33 ± 0.03 | –3.30 | 0.75 | 7.27 ± 0.37 | –3.96 | 0.57 | |
| 6 mg/kg | 0.24 ± 0.03 | –30.33 | 0.00 | 6.08 ± 0.41 | –19.61 | 0.01 | |
| medial geniculate | control | 0.32 ± 0.03 | 6.06 ± 0.29 | ||||
| 4.5 mg/kg | 0.3 ± 0.03 | –5.13 | 0.72 | 5.85 ± 0.29 | –3.59 | 0.59 | |
| 6 mg/kg | 0.2 ± 0.04 | –35.96 | 0.02 | 4.65 ± 0.32 | –23.34 | 0.00 | |
| mesencephalic region | control | 0.35 ± 0.02 | 7.14 ± 0.33 | ||||
| 4.5 mg/kg | 0.34 ± 0.02 | –1.86 | 0.83 | 6.65 ± 0.33 | –6.75 | 0.31 | |
| 6 mg/kg | 0.26 ± 0.02 | –26.91 | 0.00 | 5.49 ± 0.37 | –23.00 | 0.00 | |
| midbrain | control | 0.35 ± 0.02 | 7.07 ± 0.34 | ||||
| 4.5 mg/kg | 0.33 ± 0.02 | –5.43 | 0.52 | 6.92 ± 0.34 | –2.13 | 0.75 | |
| 6 mg/kg | 0.26 ± 0.02 | –25.27 | 0.00 | 5.58 ± 0.37 | –21.06 | 0.00 | |
| septum | control | 0.27 ± 0.02 | 6.37 ± 0.35 | ||||
| 4.5 mg/kg | 0.26 ± 0.02 | –5.95 | 0.60 | 5.91 ± 0.35 | –7.23 | 0.35 | |
| 6 mg/kg | 0.17 ± 0.02 | –38.90 | 0.00 | 4.89 ± 0.38 | –23.28 | 0.00 | |
| striatum | control | 0.26 ± 0.02 | 6.36 ± 0.32 | ||||
| 4.5 mg/kg | 0.24 ± 0.02 | –9.46 | 0.33 | 6.05 ± 0.32 | –5.00 | 0.48 | |
| 6 mg/kg | 0.17 ± 0.02 | –35.30 | 0.00 | 4.95 ± 0.35 | –22.18 | 0.00 | |
| superior colliculus | control | 0.39 ± 0.03 | 6.98 ± 0.33 | ||||
| 4.5 mg/kg | 0.36 ± 0.03 | –9.66 | 0.30 | 6.34 ± 0.33 | –9.21 | 0.17 | |
| 6 mg/kg | 0.25 ± 0.03 | –37.67 | 0.00 | 5.13 ± 0.36 | –26.42 | 0.00 | |
| thalamus | control | 0.3 ± 0.02 | 6.19 ± 0.31 | ||||
| 4.5 mg/kg | 0.29 ± 0.02 | –3.23 | 0.76 | 5.89 ± 0.31 | –4.82 | 0.49 | |
| 6 mg/kg | 0.2 ± 0.03 | –33.61 | 0.00 | 4.54 ± 0.34 | –26.70 | 0.00 | |
| whole-brain | control | 0.3 ± 0.02 | 7.82 ± 0.38 | ||||
| 4.5 mg/kg | 0.28 ± 0.02 | –7.18 | 0.42 | 7.35 ± 0.38 | –5.99 | 0.38 | |
| 6 mg/kg | 0.2 ± 0.02 | –33.68 | 0.00 | 5.92 ± 0.41 | –24.29 | 0.00 | |
Parametric Images
Figure 5 shows parametric maps of K1, VT, and k2 in three representative subjects of each group using 1-TCM.
Figure 5.

Parametric images calculated using 1-TCM and 60 min scan duration of three representative subjects of each group: control, low-dose (4.5 mg/kg), and high-dose (6 mg/kg).
Western Blot
Western blot analysis using the whole brain showed no statistical differences between the control and treated groups. There was a noticeable intragroup variability regarding P-gp analysis, but no significant differences were found (Figure 6). In PXR analysis, there was a tendency to lower levels of PXR in the high-dose group (Figure 7 and 8).
Figure 6.

P-gp expression levels and western blot bands corresponding to P-gp (band close to 160 kDa predicted molecular weight) and ß-actin as the load control (band close to 42 kDa predicted molecular weight).
Figure 7.

PXR expression levels and western blot bands corresponding to PXR (two bands close to 50 kDa predicted molecular weight) and histone H3 as the load control (band close to 17 kDa predicted molecular weight). Notice that in our analysis only the 50 kDa band was quantified.
Figure 8.

P-gp expression levels in isolated brain endothelial cells and western blot bands corresponding to P-gp (band close to 160 kDa predicted molecular weight) and ß-actin as the load control (band close to 42 kDa predicted molecular weight).
Preliminary results using western blot analysis in isolated brain endothelial cells showed an increasing trend up to 37% (p = 0.087) in treated rats compared to control rats.
Discussion
The present study was designed to determine the effect of MC111 on the P-gp function in vivo using the P-gp tracer [18F]MC225 and PET. Our analysis highlights the induction effect of MC111 on the P-gp function at the BBB of healthy rats. This finding was in line with the results that were previously observed in vitro.20 Moreover, the results demonstrate that [18F]MC225 can be used to measure increases in the P-gp function reflected in decreases in K1 and VT values of [18F]MC225 in all brain regions. Western blot analyses performed using the isolated capillaries cells of the rats found an increasing trend regarding the P-gp expression levels in rats treated with the inducer.
The blood analysis revealed that the treatment with MC111 did not alter the metabolism of the tracer since the analysis did not find significant differences in the percentages of radiometabolites among the groups. However, the treatment with 6 mg/kg dose of MC111 increased the (metabolite-corrected) concentration of the tracer in plasma compared to the control group and low-dose group. However, the elimination of the tracer from plasma (Ke) was also higher in the high-dose group than in the other two groups, which means that the washout of the tracer from the blood was faster in the high-dose group. Therefore, it seems that although the concentration of the tracer in plasma was higher in the high-dose group, the washout of the tracer was also faster. One rat (from the high-dose group) showed very high concentration levels of [18F]MC225 in blood reaching SUV values of 6 in blood and plasma. This value had been considered as an outlier and removed from the analysis.
Analysis of the whole-brain SUV–TACs did not find significant differences among the experimental groups. The SUV calculated using the last 20 min of the scan showed a reduction of 10% in the high-dose group compared to the control, however, this reduction in the SUV40–60 min was not statistically significant. These results refute the use of SUV for quantification of P-gp induction and support the use of pharmacokinetic parameters.
Following previous recommendations, the kinetics of [18F]MC225 was analyzed using 1-TCM.24 The statistical analysis found a significant decrease in the K1 and VT values in animals treated with a high-dose of MC111 compared to the control and low-dose groups, which indicates an increase in the P-gp function. The whole-brain region K1 in the high-dose group decreased by 34% (p < 0.001) compared to the control group and by 29% (p = 0.005) compared to the low-dose group. The VT of the whole-brain region in the high-dose group decreased by 24% (p = 0.001) compared to the control and by 19% (p = 0.0.11) compared to the low-dose group. Similar changes were observed in all of the selected brain regions. As was also observed in previous studies, changes in K1 are larger than the change in VT. For this reason, K1 has been identified as a more sensitive parameter to measure the P-gp function at the BBB in rats and in nonhuman primates,24,37 however, VT can also be used as a surrogate parameter to estimate the P-gp function.24
Many therapeutic drugs have shown an induction effect on the P-gp transporter.38 Subchronic morphine administrations induced P-gp expression in brain microvessels of rats.39 In the mentioned study, rats were treated with increasing doses of morphine by intraperitoneal injections (ip) twice a day for 5 consecutive days. Six hours after the last dose of morphine, the levels of P-gp were not higher but after 24 h of abstinence, the P-gp expression increased 1.5 times compared to control animals.39 Long-term exposure (60 days) to phenobarbital, phenytoin, carbamazepine, and valproic acid (antiepileptic drugs) increased the P-gp expression levels and P-gp function in rat brain microvascular endothelial cells in vitro.40 P-gp expression in brain capillaries of mice was also increased 24 h after the administration of rifampin (50 mg/kg). That study also observed an increase of P-gp expression and function in vitro using isolated brain capillaries after the exposure to rifampicin and hyperforin, two PXR-ligands.41 After the treatment with the corticosteroid dexamethasone for 4 days at dose 25 mg/kg ip, brain capillaries of rats showed an increase in the P-gp expression.42 The previously described studies evaluated the P-gp function in vitro using endothelial cells acquired from brain postmortem. However, our study confirms the P-gp induction effect of MC111 in vivo using PET imaging. This compound has been selected from an extensive P-gp/BCRP library and in vitro studies have already shown that MC111 increases the P-gp and BCRP function and expression.20 Several studies have suggested a synergetic role between the two efflux transporters.10 Moreover, the expression of these two transporters is usually increased in various neurodegenerative diseases.22 Therefore, it is expected that the dual action of MC111 may be beneficial for the treatment of AD.
Since P-gp function can be altered after several treatments, it is important to monitor such alterations to prevent decreases in drug efficacy or drug toxicities. Moreover, since P-gp inducers have the ability to restore the P-gp function and thus can be a potential treatment for neurodegenerative diseases where the P-gp function is decreased, PET imaging with P-gp tracers may allow the evaluation of the effect of promising inducer drug candidates.
Tracers for imaging P-gp function such as [11C]verapamil and [11C]-N-desmethyl-loperamide show very low uptake inside the brain at baseline conditions due to their strong affinity toward the transporter protein, thus hampering the evaluation of P-gp induction.43−46 However, weak P-gp substrates such as [18F]MC225 show higher baseline brain uptake levels than [11C]verapamil24 and thus have been proposed as adequate tracers to measure P-gp induction. The results of our studies confirm the ability of [18F]MC225 to measure increases in the P-gp function because the treatment with the P-gp inducer MC111 significantly decreased the K1 and VT values of [18F]MC225 in the high-dose group compared to the control group, indicating an increase in the P-gp function in these rats.
Increases in the P-gp function do not always mean increases in the P-gp expression, for this reason, it is important to assess simultaneously the P-gp expression level and function to identify the mechanism of action of a drug. For instance, western blot and flow cytometry analysis indicated that colchicine increased the P-gp expression in caco-2 cells without increasing the P-gp function.47 In the present study, at first, the P-gp expression level was assessed postmortem using the whole-brain tissue of the rats, however, these results did not find any change in the P-gp expression among the groups. To adequately evaluate the P-gp expression level at the BBB of rats and to not overestimate the P-gp levels, the brain endothelial cells must be isolated because the P-gp transporter is also expressed at low levels by nonendothelial cell types and because the volume of the brain capillaries represents approximately 1% of the total brain volume.27,48,49 Thus, additional western blot experiments were performed to determine the P-gp expression levels in isolated brain endothelial cells of the rats. The preliminary results have shown a tendency to higher P-gp expression levels in rats treated with MC111 compared to control rats, however, this increase was not statistically significant. Additional data is needed to confirm these results and to examine the mechanism of action of this novel compound. Moreover, P-gp transporters are also expressed in intracellular compartments, whereas only the transporter molecules on the cell surface are functional.50,51 Therefore, it is important to use antibodies that specifically recognize an external epitope of the P-gp transporter to avoid the detection of P-gp stored in intracellular vesicles.27,52,53 For this reason, more research is required to distinguish the expression level of the P-gp transporter at the membrane of the cell, which will be more related to the functionality of the transporter, from the intracellular P-gp. Our study also evaluated the PXR expression level in the whole-brain tissue since we have hypothesized that MC111 may increase the P-gp expression by the activation of the PXR. However, the analysis did not detect any increase in the expression levels of this nuclear receptor. Interestingly, in the PXR western blot, a band between 37 and 50 kDa was also found in addition to the expected PXR 50 kDa band. This could be due to the presence of alternative splicing variants. Isoforms 1b and 2b of 43 and 39 kDa, respectively, were reported in uniport database but further analysis is needed to elucidate this issue.54 Notice that in our analysis only the 50 kDa band quantification is shown. In future studies, the PXR expression levels will be also determined in isolated brain cells to find out if MC111 is also a PXR ligand.
Conclusions
In conclusion, this study shows the ability of MC111 to induce the P-gp expression and function at the BBB of rats. An increasing trend regarding the P-gp expression levels in isolated brain capillaries is found using western blot. Moreover, an increased P-gp function is confirmed with [18F]MC225 and PET. This study demonstrates that the administration of a 6 mg/kg dose of MC111 can be safely administered to rats to induce the P-gp expression and function at the BBB. Moreover, the results also verify the ability of [18F]MC225 to measure increases in the P-gp function of rats. [18F]MC225 may be the first radiofluorinated tracer able to monitor increases and decreases of the P-gp function at the mammalian BBB. Moreover, this technique may be used to screen potential P-gp inducers that can be applied as experimental treatments for several neurodegenerative diseases.
Acknowledgments
The authors would like to thank Jürgen W.A. Sijbesma and Anna Schildt for their assistance during the preclinical studies.
Author Contributions
L.G.-V. performed the synthesis of the tracer and the preclinical studies, analyzed the data, conducted the pharmacokinetic analysis, and wrote the manuscript with help from all authors. M.R.-P. performed the western blot studies and commented on the final manuscript. A.C. helped in the isolation of the brain capillaries and the western blot studies. R.M.-A. provided assistance during the in vivo studies. N.A.C. contributed to the design of the experiment, provided the P-gp inducer (MC111), and commented on the final manuscript. P.A. and T.S. helped in the design of the western blot studies and commented on the final manuscript. A.v.W. contributed to the design of the experiment and commented on the final manuscript. RAJOD and PEH commented on the final manuscript. G.L. contributed to the design of the experiment, helped in the interpretation of the data, and in the preparation of the manuscript.
The authors declare no competing financial interest.
Notes
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
References
- Mahringer A.; Fricker G. ABC Transporters at the Blood–Brain Barrier. Expert Opin. Drug Metab. Toxicol. 2016, 12, 499–508. 10.1517/17425255.2016.1168804. [DOI] [PubMed] [Google Scholar]
- Locher K. P. Mechanistic Diversity in ATP-Binding Cassette (ABC) Transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. 10.1038/nsmb.3216. [DOI] [PubMed] [Google Scholar]
- Löscher W.; Potschka H. Role of Drug Efflux Transporters in the Brain for Drug Disposition and Treatment of Brain Diseases. Prog. Neurobiol. 2005, 76, 22–76. 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Miller D. S. Regulation of P-Glycoprotein and Other ABC Drug Transporters at the Blood-Brain Barrier. Trends Pharmacol. Sci. 2010, 31, 246–254. 10.1016/j.tips.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam F. C.; Liu R.; Lu P.; Shapiro A. B.; Renoir J.-M.; Sharom F. J.; Reiner P. B. β-Amyloid Efflux Mediated by p-Glycoprotein. J. Neurochem. 2001, 76, 1121–1128. 10.1046/j.1471-4159.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- Vogelgesang S.; Cascorbi I.; Schroeder E.; Pahnke J.; Kroemer H. K.; Siegmund W.; Kunert-Keil C.; Walker L. C.; Warzok R. W. Deposition of Alzheimer’s Beta-Amyloid Is Inversely Correlated with P-Glycoprotein Expression in the Brains of Elderly Non-Demented Humans. Pharmacogenetics 2002, 12, 535–541. 10.1097/00008571-200210000-00005. [DOI] [PubMed] [Google Scholar]
- van Assema D. M. E.; Lubberink M.; Bauer M.; van der Flier W. M.; Schuit R. C.; Windhorst A. D.; Comans E. F. I.; Hoetjes N. J.; Tolboom N.; Langer O.; Müller M.; Scheltens P.; Lammertsma A. A.; van Berckel B. N. M. Blood–Brain Barrier P-Glycoprotein Function in Alzheimer’s Disease. Brain 2012, 135, 181–189. 10.1093/brain/awr298. [DOI] [PubMed] [Google Scholar]
- Colabufo N. A.; Berardi F.; Cantore M.; Contino M.; Inglese C.; Niso M.; Perrone R. Perspectives of P-Glycoprotein Modulating Agents in Oncology and Neurodegenerative Diseases: Pharmaceutical, Biological, and Diagnostic Potentials. J. Med. Chem. 2010, 53, 1883–1897. 10.1021/jm900743c. [DOI] [PubMed] [Google Scholar]
- Miller D. S.; Bauer B.; Hart A. M. S. Modulation of P-Glycoprotein at the Blood-Brain Barrier: Opportunities to Improve CNS Pharmacotherapy. Pharmacol Rev. 2008, 60, 196–209. 10.1124/pr.107.07109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W.; Gericke B. Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain?. Pharmaceutics 2020, 12, 966 10.3390/pharmaceutics12100966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. Regulation of ABC Transporters at the Blood-Brain Barrier. Clin. Pharmacol. Ther. 2015, 97, 395–403. 10.1002/cpt.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gameiro M.; Silva R.; Rocha-Pereira C.; Carmo H.; Carvalho F.; Bastos M. D. L.; Remião F. Cellular Models and In Vitro Assays for the Screening of Modulators of P-Gp, MRP1 and BCRP. Molecules 2017, 22, 600 10.3390/molecules22040600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmeliegy M.; Vourvahis M.; Guo C.; Wang D. D. Effect of P-Glycoprotein (P-Gp) Inducers on Exposure of P-Gp Substrates: Review of Clinical Drug–Drug Interaction Studies. Clin. Pharmacokinet. 2020, 59, 699–714. 10.1007/s40262-020-00867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley L. A.; Horsburgh B. C.; Ross J.; Scheer N.; Roland Wolf C. PXR and CAR: Nuclear Receptors Which Play a Pivotal Role in Drug Disposition and Chemical Toxicity. Drug Metab. Rev. 2006, 38, 515–597. 10.1080/03602530600786232. [DOI] [PubMed] [Google Scholar]
- Miller D. S.; Bauer B.; Hartz A. M. S. Modulation of P-Glycoprotein at the Blood-Brain Barrier: Opportunities to Improve Central Nervous System Pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. 10.1124/pr.107.07109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Sykes D. B.; Miller D. S. Constitutive Androstane Receptor-Mediated Up-Regulation of ATP-Driven Xenobiotic Efflux Transporters at the Blood-Brain Barrier. Mol. Pharmacol. 2010, 78, 376–383. 10.1124/mol.110.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz A. M. S.; Miller D. S.; Bauer B. Restoring Blood-Brain Barrier P-Glycoprotein Reduces Brain Amyloid-β in a Mouse Model of Alzheimer’s Disease. Mol. Pharmacol. 2010, 77, 715–723. 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durk M. R.; Han K.; Chow E. C. Y.; Ahrens R.; Henderson J. T.; Fraser P. E.; Pang K. S. 1, 25-Dihydroxyvitamin D3 Reduces Cerebral Amyloid- Accumulation and Improves Cognition in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2014, 34, 7091–7101. 10.1523/JNEUROSCI.2711-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenn A.; Grube M.; Jedlitschky G.; Fischer A.; Strohmeier B.; Eiden M.; Keller M.; Groschup M. H.; Vogelgesang S. St. John’s Wort Reduces Beta-Amyloid Accumulation in a Double Transgenic Alzheimer’s Disease Mouse Model - Role of P-Glycoprotein. Brain Pathol. 2014, 24, 18–24. 10.1111/bpa.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colabufo N. A.; Contino M.; Cantore M.; Berardi F.; Perrone R.; Tonazzi A.; Console L.; Panaro M. A.; Savolainen H.; Luurtsema G. An Innovative Small Molecule for Promoting Neuroreparative Strategies. RSC Adv. 2018, 8, 5451–5458. 10.1039/C7RA11812K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qosa H.; Miller D. S.; Pasinelli P.; Trotti D. Regulation of ABC Efflux Transporters at Blood-Brain Barrier in Health and Neurological Disorders. Brain Res. 2015, 1628, 298–316. 10.1016/j.brainres.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Martins E.; Barbosa D. J.; Silva V.; Remião F.; Silva R. Dysfunction of ABC Transporters at the Blood-Brain Barrier: Role in Neurological Disorders. Pharmacol. Ther. 2020, 213, 107554 10.1016/j.pharmthera.2020.107554. [DOI] [PubMed] [Google Scholar]
- Savolainen H.; Cantore M.; Colabufo N. A.; Elsinga P. H.; Windhorst A. D.; Luurtsema G. Synthesis and Preclinical Evaluation of Three Novel Fluorine-18 Labeled Radiopharmaceuticals for P-Glycoprotein PET Imaging at the Blood-Brain Barrier. Mol. Pharmaceutics 2015, 12, 2265–2275. 10.1021/mp5008103. [DOI] [PubMed] [Google Scholar]
- Savolainen H.; Windhorst A. D.; Elsinga P. H.; Cantore M.; Colabufo N. A.; Willemsen A. T.; Luurtsema G. Evaluation of [18F]MC225 as a PET Radiotracer for Measuring P-Glycoprotein Function at the Blood-Brain Barrier in Rats: Kinetics, Metabolism, and Selectivity. J. Cereb. Blood Flow Metab. 2017, 37, 1286–1298. 10.1177/0271678X16654493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Varela L.; Attia K.; Antunes I.; Kwizera C.; Niezink A.; Zijlma R.; Visser T.; Dierckx R.; Elsinga P.; Luurtsema G. One-step synthesis of [18F]MC225 intended for GMP compliant productions. J. Labelled Compd. Radiopharm. 2019, 62, S80. [Google Scholar]
- Hartz A. M. S.; Schulz J. A.; Sokola B. S.; Edelmann S. E.; Shen A. N.; Rempe R. G.; Zhong Y.; El Seblani N.; Bauer B. Isolation of Cerebral Capillaries from Fresh Human Brain Tissue. J. Vis. Exp. 2018, 2018, 57346 10.3791/57346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz A. M. S.; Pekcec A.; Soldner E. L. B.; Zhong Y.; Schlichtiger J.; Bauer B. P-Gp Protein Expression and Transport Activity in Rodent Seizure Models and Human Epilepsy. Mol. Pharmaceutics 2017, 14, 999–1011. 10.1021/acs.molpharmaceut.6b00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn R. N.; Summerfield S. G.; Salinas C. A.; Read K. D.; Guo Q.; Searle G. E.; Parker C. A.; Jeffrey P.; Laruelle M. Combining PET Biodistribution and Equilibrium Dialysis Assays to Assess the Free Brain Concentration and BBB Transport of CNS Drugs. J. Cereb. Blood Flow Metab. 2012, 32, 874–883. 10.1038/jcbfm.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Varela L.; Vállez García D.; Rodríguez-Pérez M.; van Waarde A.; Sijbesma J. W. A.; Schildt A.; Kwizera C.; Aguiar P.; Sobrino T.; Dierckx R. A. J. O.; Elsinga P. H.; Luurtsema G. Test–Retest Repeatability of [18 F]MC225-PET in Rodents: A Tracer for Imaging of P-Gp Function. ACS Chem. Neurosci. 2020, 11, 648–658. 10.1021/acschemneuro.9b00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vállez Garcia D.; Casteels C.; Schwarz A. J.; Dierckx R. A. J. O.; Koole M.; Doorduin J. A Standardized Method for the Construction of Tracer Specific PET and SPECT Rat Brain Templates: Validation and Implementation of a Toolbox. PLoS One 2015, 10, e0122363 10.1371/journal.pone.0122363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; de Lannoy I. A. M. Pharmacokinetics. Biochem. Pharmacol. 2014, 87, 93–120. 10.1016/j.bcp.2013.09.007. [DOI] [PubMed] [Google Scholar]
- Muzi M.; Mankoff D. A.; Link J. M.; Shoner S.; Collier A. C.; Sasongko L.; Unadkat J. D. Imaging of Cyclosporine Inhibition of P-Glycoprotein Activity Using 11C-Verapamil in the Brain: Studies of Healthy Humans. J. Nucl. Med. 2009, 50, 1267–1275. 10.2967/jnumed.108.059162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberink M. Kinetic Models for Measuring P-Glycoprotein Function at the Blood-Brain Barrier with Positron Emission Tomography. Curr. Pharm. Des. 2016, 22, 5786–5792. 10.2174/1381612822666160804093852. [DOI] [PubMed] [Google Scholar]
- Pottier G.; Marie S.; Goutal S.; Auvity S.; Peyronneau M.-A.; Stute S.; Boisgard R.; Dolle F.; Buvat I.; Caille F.; Tournier N. Imaging the Impact of the P-Glycoprotein (ABCB1) Function on the Brain Kinetics of Metoclopramide. J. Nucl. Med. 2016, 57, 309–314. 10.2967/jnumed.115.164350. [DOI] [PubMed] [Google Scholar]
- Auvity S.; Caillé F.; Marie S.; Wimberley C.; Bauer M.; Langer O.; Buvat I.; Goutal S.; Tournier N. P-Glycoprotein (ABCB1) Inhibits the Influx and Increases the Efflux of 11 C-Metoclopramide across the Blood-Brain Barrier: A PET Study on Non-Human Primates. J. Nucl. Med. 2018, 59, 1609–1615. 10.2967/jnumed.118.210104. [DOI] [PubMed] [Google Scholar]
- Liang K.-Y.; Zeger S. L. Longitudinal Data Analysis Using Generalized Linear Models. Biometrika 1986, 73, 13–22. 10.1093/biomet/73.1.13. [DOI] [Google Scholar]
- García-Varela L.; Arif W. M.; Vállez García D.; Kakiuchi T.; Ohba H.; Harada N.; Tago T.; Elsinga P. H.; Tsukada H.; Colabufo N. A.; Dierckx R. A. J. O.; van Waarde A.; Toyohara J.; Boellaard R.; Luurtsema G. Pharmacokinetic Modeling of [18 F]MC225 for Quantification of the P-Glycoprotein Function at the Blood–Brain Barrier in Non-Human Primates with PET. Mol. Pharmaceutics 2020, 17, 3477–3486. 10.1021/acs.molpharmaceut.0c00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R.; Vilas-Boas V.; Carmo H.; Dinis-Oliveira R. J.; Carvalho F.; de Lourdes Bastos M.; Remião F. Modulation of P-Glycoprotein Efflux Pump: Induction and Activation as a Therapeutic Strategy. Pharmacol. Ther. 2015, 149, 1–123. 10.1016/j.pharmthera.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Yousif S.; Chaves C.; Potin S.; Margaill I.; Scherrmann J.-M.; Declèves X. Induction of P-Glycoprotein and Bcrp at the Rat Blood-Brain Barrier Following a Subchronic Morphine Treatment Is Mediated through NMDA/COX-2 Activation. J. Neurochem. 2012, 123, 491–503. 10.1111/j.1471-4159.2012.07890.x. [DOI] [PubMed] [Google Scholar]
- Yang H.; Liu H.; Liu X.; Zhang D.; Liu Y.; Liu X.; Wang G.; Xie L. Increased P-Glycoprotein Function and Level after Long-Term Exposure of Four Antiepileptic Drugs to Rat Brain Microvascular Endothelial Cells in Vitro. Neurosci. Lett. 2008, 434, 299–303. 10.1016/j.neulet.2008.01.071. [DOI] [PubMed] [Google Scholar]
- Bauer B.; Yang X.; Hartz A. M. S.; Olson E. R.; Zhao R.; Kalvass J. C.; Pollack G. M.; Miller D. S. In Vivo Activation of Human Pregnane X Receptor Tightens the Blood-Brain Barrier to Methadone through P-Glycoprotein Up-Regulation. Mol. Pharmacol. 2006, 70, 1212–1219. 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- Wen J.; Shen Y.; Zhang M.; Wang C.; Xiang Y.; Cai H.; Fang P.; Li H. Dexamethasone Changes the Pharmacokinetics of Amitriptyline and Reduces Its Accumulation in Rat Brain: The Roles of P-Gp and Cyp3a2. J. Pharmacol. Sci. 2019, 140, 54–61. 10.1016/j.jphs.2019.04.007. [DOI] [PubMed] [Google Scholar]
- Luurtsema G.; Molthoff C. F. M.; Schuit R. C.; Windhorst A. D.; Lammertsma A. A.; Franssen E. J. F. Evaluation of (R)-[11C]Verapamil as PET Tracer of P-Glycoprotein Function in the Blood-Brain Barrier: Kinetics and Metabolism in the Rat. Nucl. Med. Biol. 2005, 32, 87–93. 10.1016/j.nucmedbio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Zoghbi S. S.; Liow J.-S.; Yasuno F.; Hong J.; Tuan E.; Lazarova N.; Gladding R. L.; Pike V. W.; Innis R. B. 11C-Loperamide and Its N-Desmethyl Radiometabolite Are Avid Substrates for Brain Permeability-Glycoprotein Efflux. J. Nucl. Med. 2008, 49, 649–656. 10.2967/jnumed.107.047308. [DOI] [PubMed] [Google Scholar]
- Wanek T.; Mairinger S.; Langer O. Radioligands Targeting P-Glycoprotein and Other Drug Efflux Proteins at the Blood-Brain Barrier. J. Labelled Compd. Radiopharm. 2013, 56, 68–77. 10.1002/jlcr.2993. [DOI] [PubMed] [Google Scholar]
- Luurtsema G.; Elsinga P.; Dierckx R.; Boellaard R.; Waarde A. PET Tracers for Imaging of ABC Transporters at the Blood-Brain Barrier: Principles and Strategies. Curr. Pharm. Des. 2016, 22, 5779–5785. 10.2174/1381612822666160810123634. [DOI] [PubMed] [Google Scholar]
- Silva R.; Carmo H.; Vilas-Boas V.; Barbosa D. J.; Palmeira A.; Sousa E.; Carvalho F.; Bastos M.; de L.; Remião F. Colchicine Effect on P-Glycoprotein Expression and Activity: In Silico and in Vitro Studies. Chem. Biol. Interact. 2014, 218, 50–62. 10.1016/j.cbi.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Sharom F. J. The P-Glycoprotein Multidrug Transporter. Essays Biochem. 2011, 50, 161–178. 10.1042/bse0500161. [DOI] [PubMed] [Google Scholar]
- Pardridge W. M. The Isolated Brain Microvessel: A Versatile Experimental Model of the Blood-Brain Barrier. Front. Physiol. 2020, 11, 1–27. 10.3389/fphys.2020.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg K.; Thomas J.; Ashford D.; Cartwright J.; Coldwell R.; Weston D. J.; Pillmoor J.; Surry D.; O’Toole P. Quantification of Proteins by Flow Cytometry: Quantification of Human Hepatic Transporter P-Gp and OATP1B1 Using Flow Cytometry and Mass Spectrometry. Methods 2015, 82, 38–46. 10.1016/j.ymeth.2015.03.030. [DOI] [PubMed] [Google Scholar]
- Zhu H.-J.; Liu G.-Q. Glutamate Up-Regulates P-Glycoprotein Expression in Rat Brain Microvessel Endothelial Cells by an NMDA Receptor-Mediated Mechanism. Life Sci. 2004, 75, 1313–1322. 10.1016/j.lfs.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Vahedi S.; Lusvarghi S.; Pluchino K.; Shafrir Y.; Durell S. R.; Gottesman M. M.; Ambudkar S. V. Mapping Discontinuous Epitopes for MRK-16, UIC2 and 4E3 Antibodies to Extracellular Loops 1 and 4 of Human P-Glycoprotein. Sci. Rep. 2018, 8, 12716 10.1038/s41598-018-30984-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi E.; Iwahashi T.; Tsuruo T. Monoclonal Antibodies Specific for P-Glycoprotein. Leukemia 1997, 11, 1119–1123. 10.1038/sj.leu.2400658. [DOI] [PubMed] [Google Scholar]
- Nuclear Receptor Subfamily 1 Group I Member 2. https://www.uniprot.org/uniprot/O75469 (accessed January 20, 2021).