

Abstract
Introduction
Head injuries (HI) are a risk factor for dementia, but the underlying etiology is not fully known. Understanding whether tau might mediate this relationship is important.
Methods
Cognition and tau deposition were compared between 752 individuals with (impaired, n = 302) or without cognitive impairment (CN, n = 450) with amyloid and [18F]flortaucipir positron emission tomography, HI history information, and cognitive testing from the Alzheimer's Disease Neuroimaging Initiative and the Indiana Memory and Aging Study.
Results
Sixty‐three (38 CN, 25 impaired) reported a history of HI. Higher neuropsychiatric scores and poorer memory were observed in those with a history of HI. Tau was higher in individuals with a history of HI, especially those who experienced a loss of consciousness (LOC). Results were driven by impaired individuals, especially amyloid beta‐positive individuals with history of HI with LOC.
Discussion
These findings suggest biological changes, such as greater tau, are associated with HI in individuals with cognitive impairment. Small effect sizes were observed; thus, further studies should replicate and extend these results.
Keywords: Alzheimer's disease, head injury, mild cognitive impairment, tau, traumatic brain injury, [18F]flortaucipir positron emission tomography (PET)
1. INTRODUCTION
Alzheimer's disease (AD) affects more than 5.7 million people in the United States.1 Mild cognitive impairment (MCI) is considered a prodromal stage of AD.2 Pathological AD is defined by the presence of amyloid beta (Aβ) plaques and tau neurofibrillary tangles (NFT).1
Traumatic brain injury (TBI) associated with head injury (HI) is a risk factor for dementia, including AD and other neurodegenerative conditions.3, 4, 5, 6 Chronic traumatic encephalopathy (CTE) is associated with repeated TBI and characterized by deposition of hyperphosphorylated tau at the sulcal depths, along with TDP‐43 and Aβ in some cases, and neuronal loss.7, 8 Neuroimaging studies of individuals with TBI have shown abnormalities in white matter microstructure9, 10 and gray and white matter atrophy in symptomatic individuals thought to have CTE.11, 12, 13 In addition, a history of a HI was associated with cortical thinning in AD‐like regions in preclinical AD.14 Amyloid positron emission tomography (PET) studies in individuals with HI due to sports concussions and/or military‐related injuries have been mixed, with some studies showing increased amyloid deposition,9, 15 especially in those with cognitive impairment,16 and others showing no increased amyloid positivity.17, 18, 19 Studies with [18F]flortaucipir tau PET in small samples of suspected CTE and/or others with repetitive HI due to sports or military exposure (often case reports or small cohort studies) have indicated increased [18F]flortaucipir binding in subcortical regions and the frontal, occipital, and temporal lobes.11, 12, 17, 18, 20, 21, 22, 23, 24 However, a neuropathological study using autoradiography in five cases of CTE indicated minimal sensitivity of [18F]flortaucipir to CTE tau pathology.25
The goal of this study was to assess tau deposition with [18F]flortaucipir PET in older adults with and without a history of HI in a large sample of older individuals from the Alzheimer's Disease Neuroimaging Initiative (ADNI) and the Indiana Memory and Aging Studies (IMAS). We sought to determine whether tau deposition in patients at risk for or with clinical AD would be exacerbated by HI. We first evaluated differences in tau deposition on a regional level between older adults with and without a history of HI. Then, we evaluated regional tau differences between those with and without HI with loss of consciousness (LOC). We evaluated differences in tau deposition using voxel‐wise analysis. Finally, we assessed the interaction of Aβ deposition and HI on tau deposition on regional and voxel‐wise analyses.
2. METHODS
2.1. ADNI participants
Data used in the preparation of this article were obtained from ADNI (http://adni.loni.usc.edu). See supporting information, http://adni.loni.usc.edu, and previous reports26, 27, 28, 29, 30, 31, 32, 33, 34 for more details. Informed consent was obtained according to the Declaration of Helsinki. Self‐reported medical history was manually reviewed for the presence of a reported HI with or without LOC. In ADNI, no direct question was asked about HI and data were only acquired through self‐initiated report. All included individuals were screened for the presence of a reported HI with or without LOC. These incidents were often a fall (“fall ‐ hit head on curb, unconscious for 4‐5 h”), sports participation (“Three helmeted hits resulting in LOC for a few seconds in High School football”), accident (“Head injury [no LOC] from MVA [MRI negative]”), or other event (“Concussion”). These described events are only selected examples. Participants were divided by diagnosis and history of HI with or without LOC. A second analysis divided participants based on diagnosis and history of HI with LOC (any amount of time). Diagnosis was assessed as described previously26, 34 at http://adni.loni.usc.edu, and in the supporting information. Due to small sample sizes, participants were pooled into two diagnostic categories, including cognitively normal (CN) and cognitively impaired.
2.2. IMAS participants
IMAS is a longitudinal observational study of older adults at risk for and with clinical AD. For more information, see supporting information. Informed consent was obtained according to the Declaration of Helsinki. The participants were asked about history of HI, along with presence and length of LOC. As in ADNI, participants were divided based on diagnosis and HI history, with or without LOC first and then based on diagnosis and HI with LOC (any amount of time).
2.3. Amyloid PET imaging
ADNI [18F]florbetapir and [18F]florbetaben scans were downloaded and processed with standard techniques (see supporting information). Standardized uptake value ratio (SUVR) images were converted to Centiloid (CL) units as previously described.35, 36 However, processing was done in SPM12, so new formulas were created. Specifically, SUVR from the CL cortical region of interest (ROI) for [18F]florbetapir scans was adjusted using the formula ([181.04*SUVR]‐192.1), while the cortical SUVR from [18F]florbetaben scans were adjusted using the formula ([158.47*SUVR]‐162.9). See Figure S1 in supporting information for confirmation of successful CL processing.
IMAS amyloid PET scans were done with [18F]florbetapir (Eli Lilly and Co.) or [18F]florbetaben (Life Molecular Imaging) as described previously36, 37 and in the supporting information. The resulting SUVR images were converted to CL units as described above.
Global amyloid in CL units was extracted using the CL cortical ROI. CL≥20.76 was considered Aβ positive (Aβ+), as this cut‐off best predicted the SUVR cut‐offs produced by UC Berkeley (SUVR > 1.11 for [18F]florbetapir and SUVR > 1.08 for [18F]florbetaben, data not shown).
2.4. [18F]Flortaucipir PET imaging
Pre‐processed ADNI [18F]flortaucipir scans were downloaded (http://adni.loni.usc.edu) and processed using standard techniques in SPM12. In IMAS, [18F]flortaucipir scans were collected as described previously36, 37 and in the supporting information. [18F]Flortaucipir SUVR was extracted from regions from subject‐specific parcellations using FreeSurfer v6. Specifically, bilateral mean SUVR was extracted from the medial temporal lobe (MTL), inferior parietal, precuneus, and frontal lobe.
2.5. Statistical analyses
Demographic and neuropsychological variables were compared between groups using a Chi‐square (for dichotomous variables [i.e., sex]) or a two‐way analysis of covariance (ANCOVA). Age, sex, and years of education were included as covariates where appropriate. A P‐value < .05 was considered significant for all comparisons.
Mean [18F]flortaucipir SUVR in all regions was not normally distributed. Thus, we performed a rank‐based normal transformation and performed target analyses with transformed variables. Both the raw and transformed SUVR values had consistent results. We present the transformed [18F]flortaucipir SUVR values in the body of the manuscript and the raw [18F]flortaucipir SUVR in the supporting information figures. Transformed and raw [18F]flortaucipir SUVR was compared between groups (diagnosis by history of HI) using a two‐way ANCOVA model, covaried for age, sex, global cortical amyloid, and race/ethnicity (all analyses), as well as cohort (raw values only). Other covariates (education, apolipoprotein E [APOE] ε4 carrier status) were tested for inclusion, but were non‐significant and did not change the pattern of results. Both of these analyses were repeated for HI with LOC. Finally, Cohen's d was calculated to estimate effect size using a transformation of η2 for the main effect of diagnosis and HI ([(2*[SQRT(η2/1‐ η2])]) and for the interaction using an adjustment for intermediate group variability ([2*(SQRT[η2/1‐ η2])*(3*[k‐1]/[k+1])]), where k is number of groups (k = 4).38
RESEARCH IN CONTEXT
Systematic Review: To investigate our research question of the impact of history of head injury on tau deposition and cognition, we searched PubMed for: “head injury,” “Alzheimer's,” “tau,” “[18F]flortaucipir,” “traumatic brain injury (TBI),” and “chronic traumatic encephalopathy (CTE).” We then combined the returned articles to generate a summary of previous reports of associations between tau and history of head injury.
Interpretation: Our results provide new evidence that Alzheimer's disease–related tau deposition in cognitively impaired individuals may be exacerbated by a history of head injury, particularly one with loss of consciousness. These findings provide evidence that changes in tau may be important underlying biology linking history of head injury with dementia risk.
Future directions: To confirm the current findings, additional analyses with larger and more diverse samples and longitudinal studies would be beneficial. In addition, longitudinal prospective studies of older adults with impaired cognition and head injury with tau positron emission tomography are warranted.
Exploratory voxel‐wise analyses were completed to evaluate the relationship of diagnosis and HI with [18F]flortaucipir SUVR across the whole brain. Two‐way ANCOVA models (diagnosis by HI), masked for gray and white matter, were calculated in SPM12 to evaluate tau deposition across all participants and in impaired individuals only, covaried for age, sex, global cortical amyloid, cohort, and race/ethnicity. Separate models were calculated for those with a history of any HI and for those with only HI with LOC. Voxel‐wise results were displayed at cluster‐level P< .05 (family‐wise error [FWE] correction for multiple comparisons).
We also sought to determine whether Aβ positivity and HI interact to influence tau deposition. Thus, transformed and raw [18F]flortaucipir SUVR were compared between groups (Aβ positivity by history of HI) using a two‐way ANCOVA model, covaried for age, sex, diagnosis, and race/ethnicity (all), as well as cohort (raw only). Other covariates (years of education, APOE ε4 carrier status) were tested for inclusion into the model but were non‐significant and did not change the pattern of results. Cohen's d effect sizes were calculated as described above. The association of Aβ positivity by history of HI with LOC with tau deposition was also evaluated on a voxel‐wise level using a two‐way ANCOVA, masked for gray and white matter and covaried for age, sex, global cortical amyloid, cohort, and race/ethnicity. Voxel‐wise results were displayed at cluster‐level P < .05 (FWE correction for multiple comparisons). All analyses were repeated for HI with LOC.
3. RESULTS
3.1. Demographics, clinical measures, and cognitive performance
Of 450 CN, 38 (8.4%) self‐reported a history of HI with or without a LOC, as did 25 (8.3%) of 302 impaired participants. In addition, 18 (4.0%) of 450 CN reported a HI with LOC, as did 13 (4.3%) of 302 impaired participants. Demographic, clinical, and cognitive measures are displayed in Table 1. Cohort distribution was different across groups, with IMAS being a greater percentage of those reporting HI relative to ADNI. Approximately 25% of IMAS participants reported a history of HI versus only 6% of ADNI participants. No difference in MCI/AD distribution was observed between those with and without HI. Age was greater in impaired individuals than CN. Education was higher in those with HI relative to those without. Sex was significantly different by group, which was driven by diagnosis rather than HI. Global amyloid and amyloid positivity were not associated with history of HI. However, global amyloid and amyloid positivity were associated with diagnostic status, such that amyloid was significantly greater in impaired individuals than CN. No significant interaction effect of history of HI and diagnosis on amyloid deposition was observed. Impaired individuals demonstrated cognitive impairment relative to CN on all measures. Interestingly, individuals with HI showed higher levels of neuropsychiatric symptoms as reported on the Neuropsychiatric Inventory–Questionnaire (NPI‐Q). This was primarily driven by increased levels of agitation/aggression, frontal symptoms (elation/euphoria, apathy/indifference, disinhibition, irritability/lability), and delusion/hallucination scores rather than mood disturbances, grouped as in Trzepacz et al. (data not shown, all P < .05).39 Alternatively, grouped according to Apostolova et al.,40 history of HI was associated with increased levels of distress/tension and psychotic behaviors, rather than affective symptoms (data not shown, all P < .05). Finally, individuals with HI showed lower performance on delayed recall and a trend toward poorer performance on Trail Making B (Table 1), even when covaried for amyloid positivity. When adjusted for multiple testing, only the effect of HI on NPI‐Q remained significant. All effects of diagnosis also remained significant.
TABLE 1.
Demographics and neuropsychological performance
| Cognitive Normals | Impaired | DX P‐value | HI P‐value | DX by HI P‐value | |||
|---|---|---|---|---|---|---|---|
| No head injury | Head injury | No head injury | Head injury | ||||
| n | 412 | 38 | 277 | 25 | n/a | n/a | n/a |
| Cohort distribution (ADNI, IMAS) | 375, 37 | 24, 14 | 250, 27 | 19, 6 | P < .001 | ||
| Age (y) | 72.47 (7.38) | 72.21 (7.52) | 74.84 (8.52) | 75.52 (8.17) | .007 | .844 | .658 |
| Education (y) | 16.61 (2.46) | 17.34 (2.11) | 16.19 (2.56) | 16.92 (2.53) | .209 | .029 | .995 |
| Sex (M, F)* | 164, 248 | 14, 24 | 156, 121 | 18, 7 | <.001 | ||
| Ethnicity/Race (% Non‐Hispanic White)* | 87.38% | 86.84% | 89.53% | 84.00% | .880 | ||
| APOE Genotype (% ε4 positive)a , * | 36.83% | 37.14% | 40.38% | 54.54% | .363 | ||
| Diagnosis (% MCI, % AD)* | n/a | n/a | 74.73%, 25.27% | 68.00%, 32.00% | .763 | ||
| CDR – Memoryb | 0.02 (0.11) | 0.04 (0.14) | 0.68 (0.45) | 0.76 (0.48) | <.001 | .232 | .410 |
| CDR – Globalb | 0.02 (0.11) | 0.05 (0.16) | 0.56 (0.32) | 0.62 (0.39) | <.001 | .121 | .569 |
| CDR – Sum of Boxesb | 0.07 (0.23) | 0.13 (0.41) | 2.33 (2.35) | 2.70 (2.42) | <.001 | .293 | .452 |
| GDS Totalc | 0.88 (1.28) | 1.25 (1.56) | 2.11 (2.18) | 2.35 (2.33) | <.001 | .204 | .772 |
| FAQ Totald | 0.18 (0.80) | 0.27 (0.77) | 6.00 (7.29) | 7.11 (7.11) | <.001 | .370 | .448 |
| NPI‐Q Totale | 1.12 (2.70) | 3.34 (9.13) | 5.34 (7.63) | 6.80 (8.79) | <.001 | .016 | .720 |
| MoCA Total Scoref, l | 26.29 (2.66) | 26.42 (2.09) | 21.35 (4.88) | 20.67 (4.37) | <.001 | .578 | .405 |
| RAVLT – Immediate Recallg, l | 46.35 (10.39) | 44.56 (8.87) | 32.99 (11.35) | 30.78 (7.46) | <.001 | .163 | .883 |
| RAVLT – Delayed Recallg, l | 8.35 (4.30) | 7.29 (3.59) | 3.66 (4.23) | 2.37 (2.65) | <.001 | .039 | .838 |
| Trail Making A (seconds)h, l | 30.33 (9.06) | 32.03 (9.96) | 41.27 (20.94) | 37.98 (14.18) | <.001 | .696 | .223 |
| Trail Making B (seconds)h, l | 73.08 (33.67) | 77.85 (30.48) | 122.08 (78.38) | 146.91 (82.83) | <.001 | .058 | .197 |
| Animal Fluency Scorei, l | 21.89 (5.58) | 22.26 (4.16) | 16.71 (6.06) | 14.95 (4.82) | <.001 | .353 | .159 |
| Self ECog – Memory Scorej | 1.65 (0.53) | 1.71 (0.56) | 2.38 (0.78) | 2.36 (0.77) | <.001 | .833 | .679 |
| Self ECog – Total Scorej | 1.38 (0.35) | 1.41 (0.36) | 1.9 (0.62) | 1.82 (0.45) | <.001 | .708 | .437 |
| Informant ECog – Memory Scorek | 1.34 (0.44) | 1.46 (0.5) | 2.55 (0.93) | 2.62 (0.92) | <.001 | .323 | .770 |
| Informant ECog – Total Scorek | 1.18 (0.27) | 1.28 (0.3) | 2.06 (0.77) | 2.18 (0.80) | <.001 | .149 | .914 |
| Mean Global Cortical Amyloid Centiloidm | 20.60 (2.10) | 11.24 (6.97) | 46.84 (2.56) | 50.95 (8.51) | <.001 | .115 | .714 |
| Amyloid Positivity (% positive)* | 32.38% | 28.95% | 58.12% | 64.00% | <.001 | ||
77 participants missing (59 CN‐no HI, 3 CN‐HI, 12 IMP‐no HI, 3 IMP‐HI).
5 participants missing (3 CN‐no HI, 2 IMP‐no HI).
12 participants missing (3 CN‐no HI, 2 CN‐HI, 5 IMP‐no HI, 2 IMP‐HI).
39 participants missing (8 CN‐no HI, 1 CN‐HI, 24 IMP‐no HI, 6 IMP‐HI).
12 participants missing (5 CN‐no HI, 7 IMP‐no HI).
24 participants missing (11 CN‐no HI, 13 IMP‐no HI).
20 participants missing (10 CN‐no HI, 1 CN‐HI, 8 IMP‐no HI, 1 IMP‐HI).
24 participants missing (3 CN‐no HI, 1 CN‐HI, 17 IMP‐no HI, 3 IMP‐HI).
4 participants missing (1 CN‐no HI, 3 IMP‐no HI).
53 participants missing (13 CN‐no HI, 3 CN‐HI, 32 IMP‐no HI, 5 IMP‐HI).
90 participants missing (31 CN‐no HI, 8 CN‐HI, 47 IMP‐no HI, 4 IMP‐HI).
Covaried for age, sex, and years of education.
Covaried for age, sex, cohort, and race/ethnicity.
Chi‐square test.
Abbreviations: ADNI, Alzheimer's Disease Neuroimaging Initiative; APOE, apolipoprotein E; CDR, Clinical Dementia Rating; CN, cognitively normal; HI, head injury; DX, diagnosis; HI, head injury; IMAS, Indiana Memory and Aging Study; IMP, impairment; MCI, mild cognitive impairment; MoCA, Montreal Cognitive Assessment; NPI‐Q, Neuropsychiatric Inventory–Questionnaire; RAVLT, Rey Auditory Verbal Learning Test.
BOLD p‐values represent those meeting statistical significance after multiple comparison correction; Italicized BOLD p‐values represent those meeting statistical significance without multiple comparison correction; Italicized p‐values (non‐BOLD) represent those with trend‐level significance.
3.2. Regional tau deposition by diagnosis and history of HI
A main effect of HI was observed where participants with HI showed greater tau deposition in the MTL, inferior parietal lobe, precuneus, and frontal lobe relative to those without using both transformed (Figure 1A‐D; all P < .05) and raw values (Figure S2 in supporting information; all P < .05). The main effect of HI had a Cohen's d value of 0.1 to 0.2, representing a small effect size. No interaction between diagnosis and HI was observed using the transformed values, but an interaction was observed when evaluating the raw values (Figure S2; all P < .05, Cohen's d = 0.25–0.3). A significant main effect of HI with LOC on tau deposition in all assessed regions was observed in with the transformed values (Figure 2A‐D; all P < .05, Cohen's d = 0.1–0.2) and the raw values (Figure S3 in supporting information; all P < .05, Cohen's d = 0.2–0.25). A trend for an interaction between diagnosis and HI with LOC was observed using the transformed values (Figure 2; P ≤ .10, Cohen's d = 0.2–0.25) and was significant using the raw values (Figure S3; all P < .05, Cohen's d = 0.4–0.45). Again, increased tau in impaired participants with HI relative to those without was observed, but not in CNs. As expected, significant effects of diagnosis were also observed (all P < .05, Cohen's d = 0.3–0.45).
FIGURE 1.
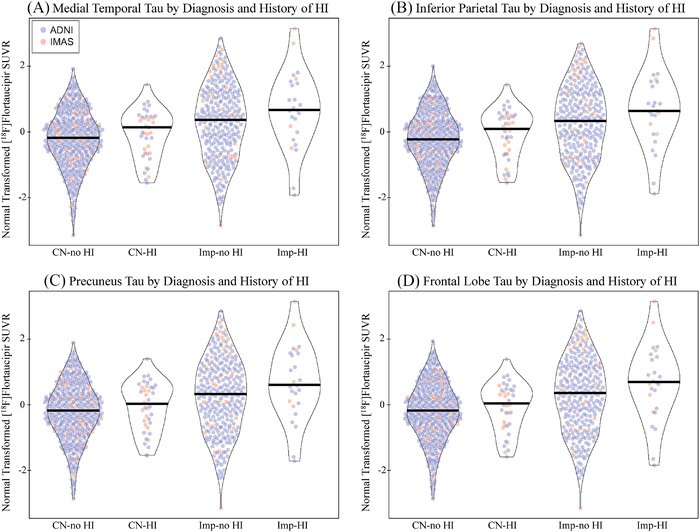
Tau deposition by diagnosis and history of head injury. Individuals with a history of head injury (HI) with or without a loss of consciousness (LOC) show greater normal transformed [18F]flortaucipir SUVR in the (A) medial temporal lobe (DX: P < .001, d = 0.313; HI: P = .034, d = 0.156; DX by HI: P > .1), (B) inferior parietal lobe (DX: P < .001, d = 0.307; HI: P = .024, d = 0.166; DX by HI: P > .1), (C) precuneus (DX: P < .001, d = 0.302; HI: P = .025, d = 0.165; DX by HI: P > .1), and (D) frontal lobe (DX: P < .001, d = 0.309; HI: P = .038, d = 0.153; DX by HI: P > .1). This effect appears to be driven by the impaired participants in the study. Age, sex, mean global amyloid, and race/ethnicity were included as covariates. Note: Participants include 412 CN without history of HI, 38 CN with history of HI, 277 impaired without history of HI, 25 impaired with history of head injury. Abbreviations: ADNI, Alzheimer's Disease Neuroimaging Initiative; CN, cognitively normal; d, Cohen's d; DX, diagnosis; HI, head injury; IMAS, Indiana Memory and Aging Study; LOC, loss of consciousness; SUVR, standardized uptake value ratio
FIGURE 2.
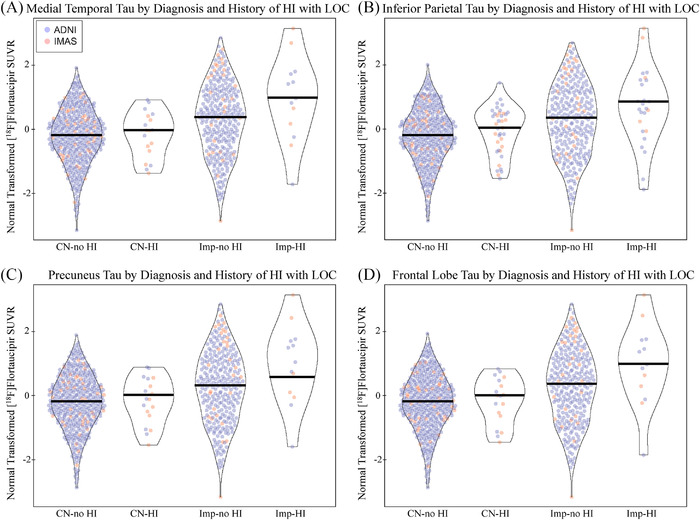
Tau deposition by diagnosis and history of head injury with loss of consciousness. Individuals with a history of head injury (HI) with a loss of consciousness (LOC) show significantly greater normal transformed [18F]flortaucipir SUVR in the (A) medial temporal lobe (DX: P < .001, d = 0.296; HI with LOC: P = .049, d = 0.144; DX by HI with LOC: P = .104, d = 0.215), (B) inferior parietal lobe (DX: P < .001, d = 0.295; HI with LOC: P = .025, d = 0.164; DX by HI with LOC: P = .096, d = 0.220), (C) precuneus (DX: P < .001, d = 0.294; HI with LOC: P = .036, d = 0.154; DX by HI with LOC: P = .083, d = 0.230), and (D) frontal lobe (DX: P < .001, d = 0.296; HI with LOC: P = .034, d = 0.149; DX by HI with LOC: P = .086, d = 0.227). This effect appears to be due to increased tau in impaired participants with HI with LOC. Age, sex, mean global amyloid, and cohort were included as covariates. Note: Participants include 432 CN without history of HI with LOC, 18 CN with history of HI with LOC, 289 impaired without history of HI with LOC, 13 impaired with history of head injury with LOC. Abbreviations: ADNI, Alzheimer's Disease Neuroimaging Initiative; CN, cognitively normal; d, Cohen's d; DX, diagnosis; HI, head injury; IMAS, Indiana Memory and Aging Study; LOC, loss of consciousness; SUVR, standardized uptake value ratio
3.3. Voxel‐wise analysis
A significant effect of HI (with or without LOC) was observed, with higher [18F]flortaucipir SUVR in bilateral superior and medial parietal lobes and frontal lobe on voxel‐wise analysis (Figure 3A). In addition, a significant interaction between diagnosis and HI was observed in the bilateral superior lateral and medial parietal lobes (Figure 3B). In impaired participants only, significantly higher [18F]flortaucipir SUVR was observed in the bilateral superior frontal lobes and superior and medial parietal lobes (Figure S4A in supporting information) in those with HI relative to those without. No effect of HI was observed in CN only, suggesting the findings are primarily driven by the impaired individuals.
FIGURE 3.
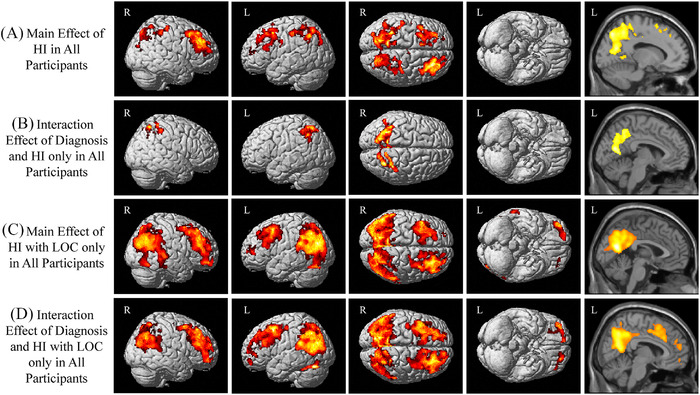
Voxel‐wise effect of diagnosis and history of head injury on tau deposition. (A) A significant main effect of history of head injury (HI) with or without a loss of consciousness (LOC) was observed, with significant clusters indicating greater [18F]flortaucipir SUVR in the parietal and frontal lobes. (B) In addition, an interaction between diagnosis and history of HI was observed, with significant clusters in the medial and lateral parietal lobes. (C) A similar but more extensive pattern of the frontal and parietal lobes shows a significant main effect of TBI with LOC. (D) An interaction between diagnosis and history of HI with LOC was also observed, with significant clusters in the frontal and parietal lobes. Significant effects of diagnosis were also observed (data not shown). All results shown at a cluster‐wise threshold of P < 0.05 (family‐wise error correction for multiple comparisons) and covaried for age, sex, global cortical amyloid, and cohort. Note: For (A) and (B), participants include 412 CN without history of HI, 38 CN with history of HI, 277 impaired without history of HI, 25 impaired with history of head injury; for (C) and (D), participants include 432 CN without history of HI with LOC, 18 CN with history of HI with LOC, 289 impaired without history of HI with LOC, 13 impaired with history of head injury with LOC. Abbreviations: CN, cognitively normal; HI, head injury; L, left; LOC, loss of consciousness; R, right; TBI, traumatic brain injury
A significant effect of HI with LOC was also observed, with higher [18F]flortaucipir SUVR in the bilateral superior and medial parietal lobes and superior frontal lobes (Figure 3C). In addition, the interaction of diagnosis and HI with LOC was significant in widespread bilateral regions of the parietal and frontal lobes (Figure 3D). Again, in impaired individuals only, higher [18F]flortaucipir SUVR was observed in bilateral medial and lateral parietal lobes and frontal regions (Figure S4B) in those with HI with LOC relative to those without. No effect of HI with LOC was seen in CN only.
3.4. Regional tau deposition by amyloid positivity and history of HI with LOC
A main effect of HI with LOC was observed in the MTL, inferior parietal lobe, precuneus, and frontal lobe using both transformed (Figure 4; all P < .05, Cohen's d = 0.14–0.2) and raw values (Figure S5 in supporting information; all P < .05, Cohen's d = 0.2–0.25) with small effect sizes as indicated by Cohen's d. Aβ+ individuals with HI with LOC had higher tau than all other groups. No interaction of amyloid positivity and HI with LOC was observed for the transformed values, but interaction was significant with the raw values (Figure S5; all P < .05, Cohen's d = 0.45–0.5).
FIGURE 4.
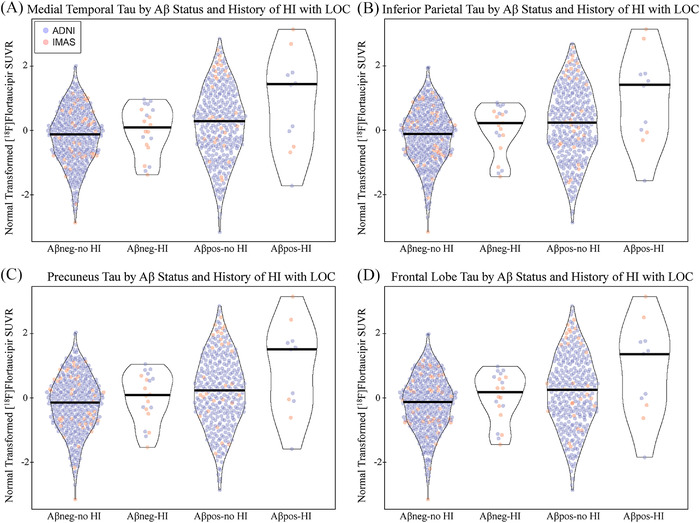
Tau deposition by amyloid positivity and history of head injury with loss of consciousness. A main effect for history of head injury (HI) with a loss of consciousness (LOC) was observed in the (A) medial temporal lobe (Aβ status: P < .001, d = 0.348; HI with LOC: P = .046, d = 0.146; Aβ status by HI with LOC: P > .1), (B) inferior parietal lobe (Aβ status: P < .001, d = 0.362; HI with LOC: P = .020, d = 0.171; Aβ Status by HI with LOC: P > .1), (C) precuneus (Aβ status: P < .001, d = 0.343; HI with LOC: P = .035, d = 0.155; Aβ status by HI with LOC: P > .1), and (D) frontal lobe (Aβ status: P < .001, d = 0.344; HI with LOC: P = .044, d = 0.148; Aβ status by HI with LOC: P > .1). Although not significant, a trend toward an interaction was observed, such that Aβ positive participants with a history of HI with LOC showed the highest normal transformed [18F]flortaucipir SUVR as a group. Age, sex, mean global amyloid, race/ethnicity, and diagnostic group were included as covariates. Note: Participants include 411 Aβ negative participants without history of HI, 20 Aβ negative participants with history of HI, 310 Aβ positive participants without history of HI, 11 Aβ positive participants with history of head injury. Abbreviations: Aβ, amyloid beta; ADNI, Alzheimer's Disease Neuroimaging Initiative; d, Cohen's d; DX, diagnosis; HI, head injury; IMAS, Indiana Memory and Aging Study; LOC, loss of consciousness
Voxel‐wise analyses indicated that the interaction between Aβ positivity and HI was significant with greater tau deposition in the bilateral parietal and frontal lobes (Figure S6A in supporting information). In impaired individuals only, a significant interaction of HI with LOC and Aβ positivity was observed in the bilateral parietal and frontal lobes was observed (Figure S6B). No effect of HI or Aβ positivity was observed in CNs (data not shown).
4. DISCUSSION
In this sample, we observed that 8.4% of cognitively normal individuals and 8.3% of impaired participants self‐report a history of HI with or without a LOC. Further, 4.0% of cognitive normal and 4.3% of impaired participants reported a HI with LOC. Greater tau deposition was observed in older adults with a history of HI, with or without LOC, in the frontal, temporal, and parietal lobes. This effect was also observed in those with HI with LOC only. These results were similar in voxel‐wise analysis. Observed effects were driven by individuals with cognitive impairment. No significant effects of HI were observed in CN. No significant effect of HI on amyloid was observed. We also observed significantly higher neuropsychiatric symptoms and poorer cognition in older adults with HI. Finally, we observed an interaction between amyloid positivity and HI with LOC. Aβ+ individuals with HI with LOC had the highest group levels of tau, which were driven by impaired individuals. Of note, the effects of HI on tau showed only small to medium effect sizes in the full sample. However, in IMAS only, which directly asked participants about history of HI, the effect sizes were medium to high (Cohen's d = 0.6–0.7).
This aritcle is the first to our knowledge to evaluate the association between history of HI and tau deposition on PET in an aging cohort who were not specifically recruited for a history of HI nor were from high‐risk groups (i.e., military or sport). However, our findings are similar to those observed previously in individuals with repeated HI due to sports and military exposure, specifically increased tau deposition on PET in those with repeated HI.11, 12, 17, 18, 20, 21, 22, 23, 24, 36, 41 Further, our analyses showed relatively small effect sizes for the impact of HI on tau, similar to previous reports. However, given the recent report about the lack of sensitivity of [18F]flortaucipir to CTE‐related tau on autoradiography, it seems unlikely that the higher tau observed in the present study is reflective of CTE.25 Further, the anatomical locations of the higher tau deposition in those with HI overlap with those known to be higher in patients with MCI and AD. Thus, it is plausible that the increased tau observed in the present study is an exacerbation of “AD‐like” tau rather than CTE in these individuals. Thus, we hypothesize that a history of HI may be accelerating or exacerbating the deposition of tau in individuals with AD pathology rather than causing co‐occurring CTE pathology. Although CTE and AD tau are similar in structure and contain both 3R and 4R tau, a recent cryomicroscopy study showed slight differences in the protein conformation of CTE‐related tau and AD‐related tau.42 However, because we only have PET studies and not neuropathology, at the current time we cannot discern whether the higher tau is due to co‐occurring mild CTE, exacerbation of the AD tau, or some other type of tau associated with HI in these cases. Future longitudinal studies in larger samples and potentially with PET tracers specific to different tau species and conformations, or with neuropathological confirmation, are needed to fully explore the nature of the higher tau we observed in those with a history of HI.
One limitation of our study is that all HIs were self‐reported and thus, may have been underestimated, overestimated, or biased in reporting. In IMAS, history of HI was asked directly along with presence/absence and length of LOC. The rate of HI in IMAS (≈25%) is similar to previously reported rates of TBI‐related emergency department visits, hospitalizations, and deaths in older individuals.43 However, in ADNI, HI was only determined as part of a general medical history interview if the participant volunteered the information. As only 6% of ADNI participants reported HI, we are likely underestimating the HI rate in ADNI. When the analyses of tau deposition associations with HI are done in IMAS only, we observe a similar pattern to the overall results with less power due to the smaller sample size.
Our study has a few other notable limitations. Although the largest study to date with [18F]flortaucipir in individuals with and without a history of HI, the results rest on only 25 impaired participants with HI. Much larger samples are needed for replication and expansion of these findings. In addition, because the self‐reported HIs were not the focus of ADNI and IMAS, we had incomplete information about LOC, age of injury, or other important factors. Direct questioning about history of HI likely better captures the actual rate and effect of HI in older adults. Finally, we considered the reports as a history of HI rather than a TBI history as reporting of concussive sequelae was incomplete. Future studies with more direct questioning and complete capture of HI information, as well as well‐designed prospective studies of HI and dementia are needed.
In summary, older adults with a history of HI, particularly those with cognitive impairment, have greater tau deposition than those without a history of HI. These findings support the importance of HI in cerebral tau pathology in dementia and suggest that HI should be considered a possible risk factor for AD pathophysiology. These findings suggest that TBI‐associated increased risk for cognitive decline and dementia may be mediated by tau‐related pathways.
CONFLICTS OF INTEREST
Dr. Risacher has previously consulted for Biogen. Dr. Apostolova has previously consulted for Eli Lilly, Biogen, and Two Labs and received honoraria for participating in independent data safety monitoring boards and providing educational CME lectures and programs. Dr. Jagust has previously consulted for Biogen, Bioclinica, and Genentech. Dr. Landau has served on the scientific advisory board for KeifeRx. Dr. Weiner has served on advisory boards for Eli Lilly, Cerecin/Accera, Roche, Alzheon, Inc., Merck Sharp & Dohme Corp., Nestle/Nestec, PCORI/PPRN, Dolby Family Ventures, National Institute on Aging (NIA), Brain Health Registry, and ADNI; Editorial Boards for Alzheimer's & Dementia, TMRI, and MRI; has provided consulting for and/or acted as a speaker/lecturer to Cerecin/Accera, Inc., BioClinica, Nestle/Nestec, Roche, Genentech, NIH, The Buck Institute for Research on Aging, FUJIFILM‐Toyama Chemical (Japan), Garfield Weston, Baird Equity Capital, University of Southern California (USC), Cytox, and Japanese Organization for Medical Device Development, Inc. (JOMDD) and T3D Therapeutics; and holds stock options with Alzheon, Inc., Alzeca, and Anven. Dr. Saykin has received support from Avid Radiopharmaceuticals, a subsidiary of Eli Lilly (in kind contribution of PET tracer precursor), Bayer Oncology (Scientific Advisory Board), and Springer‐Nature Publishing (Editorial Office Support as Editor‐in‐Chief, Brain Imaging and Behavior). Mr. West, Ms. Deardorff, and Drs. Brosch, Farlow, Gao, McAllister, and Wu have no relevant disclosures.
Supporting information
Supporting material
Supporting material
Supporting material
Supporting material
Supporting material
Supporting material
Supporting material
ACKNOWLEDGMENTS
The authors thank Mary G. Austrom, Michelle Beal, Steve Brown, David Clark, Danai Chasioti, Bradley Glazier, Kala Hall, Lili Kyurkchiyska, Evan Finley, James W. Fletcher, Tatiana M. Foroud, Yolanda Graham‐Dotson, Jenna Groh, Gary D. Hutchins, Nenette M. Jessup, Brenna McDonald, Heather Polson, T.J. Olivares, Mary Kate Starner, Wendy Territo, Fredrick Unverzagt, Sophia Wang, Donna Wert, and Karmen K. Yoder for their contributions to this work. We thank Avid Radiopharmaceuticals, a subsidiary of Eli Lilly, for provision of the precursor for [18F]flortaucipir and permission to cross‐reference their IND for AV1451 tracer manufacturing, which was performed by PETNET of Indiana in partnership with the Indiana University Department of Radiology and Imaging Sciences.
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.;Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Additional support for this work is from the National Institute on Aging (K01 AG049050, R01 AG061788, R01 AG19771, and P30 AG10133) and Donor's Cure Foundation. Part of this research was also supported in part by Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute, and in part by the Indiana METACyt Initiative. The Indiana METACyt Initiative at IU was also supported in part by Lilly Endowment, Inc. This material is based upon work supported by the National Science Foundation under Grant No. CNS‐0521433.
Risacher SL, WestJD,Deardorff R, et al., for the Alzheimer's Disease Neuroimaging Initiative . Head injury is associated with tau deposition on PET in MCI and AD patients. Alzheimer's Dement. 2021;13:e12230. 10.1002/dad2.12230
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
REFERENCES
- 1.Alzheimer's Association. 2019 Alzheimer's disease facts and figures. Alzheimer's & Dementia. 2019;15:321‐387. [DOI] [PubMed] [Google Scholar]
- 2.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183‐194. [DOI] [PubMed] [Google Scholar]
- 3.Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K. Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA Neurol. 2018;75:1055‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fann JR, Ribe AR, Pedersen HS, et al. Long‐term risk of dementia among people with traumatic brain injury in Denmark: a population‐based observational cohort study. Lancet Psychiatry. 2018;5:424‐431. [DOI] [PubMed] [Google Scholar]
- 5.Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, Yaffe K. Mild TBI and risk of Parkinson disease: a chronic effects of neurotrauma Consortium study. Neurology. 2018;90:e1771‐e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendez MF. What is the relationship of traumatic brain injury to dementia?. J Alzheimers Dis. 2017;57:667‐681. [DOI] [PubMed] [Google Scholar]
- 7.McKee AC, Abdolmohammadi B, Stein TD. The neuropathology of chronic traumatic encephalopathy. Handb Clin Neurol. 2018;158:297‐307. [DOI] [PubMed] [Google Scholar]
- 8.McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015;25:350‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott G, Ramlackhansingh AF, Edison P, et al. Amyloid pathology and axonal injury after brain trauma. Neurology. 2016;86:821‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taber KH, Hurley RA, Haswell CC, et al. White matter compromise in veterans exposed to primary blast forces. J Head Trauma Rehabil. 2015;30:E15‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickstein DL, Pullman MY, Fernandez C, Short JA, Kostakoglu L, Knesaurek K, Soleimani L, Jordan BD, Gordon WA, Dams‐O'Connor K, Delman BN, Wong E, Tang CY, DeKosky ST, Stone JR, Cantu RC, Sano M, Hof PR, Gandy S. Cerebral [18 F]T807/AV1451 retention pattern in clinically probable CTE resembles pathognomonic distribution of CTE tauopathy. Translational Psychiatry. 2016;6: 9:e900–e900. 10.1038/tp.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorgoraptis N, Li LM., Whittington A, Zimmerman KA, Maclean LM, McLeod C, Ross E, Heslegrave A, Zetterberg H, Passchier J, Matthews PM, Gunn RN, McMillan TM, Sharp DJ. In vivo detection of cerebral tau pathology in long‐term survivors of traumatic brain injury. Science Translational Medicine. 2019;11: 508:eaaw1993. 10.1126/scitranslmed.aaw1993. [DOI] [PubMed] [Google Scholar]
- 13.Koerte IK, Lin AP, Willems A, et al. A review of neuroimaging findings in repetitive brain trauma. Brain Pathol. 2015;25:318‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang ML, Wei XE, Yu MM, Li PY, Li WB. Alzheimer's Disease Neuroimaging I. Self‐reported traumatic brain injury and in vivo measure of AD‐vulnerable cortical thickness and AD‐related biomarkers in the ADNI cohort. Neurosci Lett. 2017;655:115‐120. [DOI] [PubMed] [Google Scholar]
- 15.Hong YT, Veenith T, Dewar D, et al. Amyloid imaging with carbon 11‐labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014;71:23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mielke MM, Savica R, Wiste HJ, et al. Head trauma and in vivo measures of amyloid and neurodegeneration in a population‐based study. Neurology. 2014;82:70‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lesman‐Segev OH, La Joie R, Stephens ML, et al. Tau PET and multimodal brain imaging in patients at risk for chronic traumatic encephalopathy. Neuroimage Clin. 2019;24:102025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stern RA, Adler CH, Chen K, et al. Tau positron‐emission tomography in former national football league players. N Engl J Med. 2019;380:1716‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiner MW, Harvey D, Hayes J, et al. Effects of traumatic brain injury and posttraumatic stress disorder on development of Alzheimer's disease in Vietnam Veterans using the Alzheimer's Disease Neuroimaging Initiative: preliminary Report. Alzheimers Dement (N Y). 2017;3:177‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitsis EM, Riggio S, Kostakoglu L, et al. Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatry. 2014;4:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohamed AZ, Cumming P, Gotz J, Nasrallah F, Department of Defense Alzheimer's Disease Neuroimaging Initiative. Tauopathy in veterans with long‐term posttraumatic stress disorder and traumatic brain injury. Eur J Nucl Med Mol Imaging. 2019;46:1139‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okonkwo DO, Puffer RC, Minhas DS, et al. [(18)F]FDG, [(11)C]PiB, and [(18)F]AV‐1451 PET imaging of neurodegeneration in two subjects with a history of repetitive trauma and cognitive decline. Front Neurol. 2019;10:831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson ME, McKee AC, Salat DH, et al. Positron emission tomography of tau in Iraq and Afghanistan Veterans with blast neurotrauma. Neuroimage Clin. 2019;21:101651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wooten DW, Ortiz‐Teran L, Zubcevik N, et al. Multi‐modal signatures of tau pathology, neuronal fiber integrity, and functional connectivity in traumatic brain injury. J Neurotrauma. 2019;36:3233‐3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marquié M, Agüero C, Amaral AC, et al. [18F]‐AV‐1451 binding profile in chronic traumatic encephalopathy: a postmortem case series. Acta Neuropathol Commun. 2019;7:164. 10.1186/s40478-019-0808-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aisen PS, Petersen RC, Donohue M, Weiner MW, Alzheimer's Disease Neuroimaging Initiative . Alzheimer's Disease Neuroimaging Initiative 2 Clinical Core: progress and plans. Alzheimers Dement. 2015;11:734‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beckett LA, Donohue MC, Wang C, et al. The Alzheimer's Disease Neuroimaging Initiative phase 2: increasing the length, breadth, and depth of our understanding. Alzheimers Dement. 2015;11:823‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franklin EE, Perrin RJ, Vincent B, et al. Brain collection, standardized neuropathologic assessment, and comorbidity in Alzheimer's Disease Neuroimaging Initiative 2 participants. Alzheimers Dement. 2015;11:815‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jack CR Jr, Barnes J, Bernstein MA, et al. Magnetic resonance imaging in Alzheimer's Disease Neuroimaging Initiative 2. Alzheimers Dement. 2015;11:740‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jagust WJ, Landau SM, Koeppe RA, et al. The Alzheimer's Disease Neuroimaging Initiative 2 PET Core: 2015. Alzheimers Dement. 2015;11:757‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang JH, Korecka M, Figurski MJ, et al. The Alzheimer's Disease Neuroimaging Initiative 2 Biomarker Core: a review of progress and plans. Alzheimers Dement. 2015;11:772‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saykin AJ, Shen L, Yao X, et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: progress, opportunities, and plans. Alzheimers Dement. 2015;11:792‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toga AW, Crawford KL. The Alzheimer's Disease Neuroimaging Initiative informatics core: a decade in review. Alzheimers Dement. 2015;11:832‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiner MW, Veitch DP, Aisen PS, et al. 2014 Update of the Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2015;11:e1‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1‐15 e1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Risacher SL, Tallman EF, West JD, et al. Olfactory identification in subjective cognitive decline and mild cognitive impairment: association with tau but not amyloid positron emission tomography. Alzheimers Dement (Amst). 2017;9:57‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Risacher SL, Fandos N, Romero J, et al. Plasma amyloid beta levels are associated with cerebral amyloid and tau deposition. Alzheimers Dement (Amst). 2019;11:510‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. New Jersey: Lawrence Erlbaum Associates, Inc.; 1988. [Google Scholar]
- 39.Trzepacz PT, Saykin A, Yu P, et al. Subscale validation of the neuropsychiatric inventory questionnaire: comparison of Alzheimer's disease neuroimaging initiative and national Alzheimer's coordinating center cohorts. Am J Geriatr Psychiatry. 2013;21:607‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Apostolova LG, Di LJ, Duffy EL, et al. Risk factors for behavioral abnormalities in mild cognitive impairment and mild Alzheimer's disease. Dement Geriatr Cogn Disord. 2014;37:315‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montenigro PH, Alosco ML, Martin BM, et al. Cumulative head impact exposure predicts later‐life depression, apathy, executive dysfunction, and cognitive impairment in former high school and college football players. J Neurotrauma. 2017;34:328‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. 2019;568:420‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic, brain injury‐related emergency department visits, hospitalizations and deaths ‐ United States, 2007 and 2013. MMWR Surveill Summ. 2017;66:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting material
Supporting material
Supporting material
Supporting material
Supporting material
Supporting material
Supporting material