

SUMMARY
The pathogenic mechanism by which dominant mutations in VCP cause multisystem proteinopathy (MSP), a rare neurodegenerative disease that presents as fronto-temporal lobar degeneration with TDP-43 inclusions (FTLD-TDP), remains unclear. To explore this, we inactivate VCP in murine postnatal forebrain neurons (VCP conditional knockout [cKO]). VCP cKO mice have cortical brain atrophy, neuronal loss, autophago-lysosomal dysfunction, and TDP-43 inclusions resembling FTLD-TDP pathology. Conditional expression of a single disease-associated mutation, VCP-R155C, in a VCP null background similarly recapitulates features of VCP inactivation and FTLD-TDP, suggesting that this MSP mutation is hypomorphic. Comparison of transcriptomic and proteomic datasets from genetically defined patients with FTLD-TDP reveal that progranulin deficiency and VCP insufficiency result in similar profiles. These data identify a loss of VCP-dependent functions as a mediator of FTLD-TDP and reveal an unexpected biochemical similarity with progranulin deficiency.
Graphical abstract
In brief
Wani et al. demonstrate that loss of neuronal VCP recapitulates FTLD-TDP pathology in mice. Conditional expression of a VCP-R155C mutation, associated with MSP1, also recapitulates FTLD-TDP consistent with its hypomorphic function. A transcriptomic and proteomic signature of VCP inactivation resembles a transcriptomic signature in brains of patients with GRN insufficiency.
INTRODUCTION
Fronto-temporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) is associated with dominantly inherited mutations in C9orf72, progranulin (GRN), and valosin-containing protein (VCP) (Neumann and Mackenzie, 2019). VCP disease-associated mutations cause other degenerative phenotypes in addition to FTLD-TDP. These diseases include inclusion body myopathy (IBM), Paget’s disease of the bone (PDB), and amyotrophic lateral sclerosis (ALS), often in the same patient or family (Al-Obeidi et al., 2018). This has led to the nomenclature, multisystem proteinopathy (MSP), as a means of describing VCP-associated disease and related syndromes, since degenerative phenotypes such as dementia, PDB, IBM, ALS, parkinsonism, and axonal neuropathy can co-occur in patients with MSP (Taylor, 2015). Although affecting disparate tissues, MSP pathology is unified by cellular degeneration, ubiquitinated aggregates, and TDP-43 inclusions in post-mitotic and terminally differentiated tissue such as muscle and neurons (Weihl et al., 2009).
VCP is an essential and abundant AAA+ protein that participates in multiple cellular processes, including cell cycle regulation, organelle formation, vesicular sorting, and protein homeostasis, via the ubiquitin-proteasome system (UPS) and autophago-lysosomal systems (Meyer and Weihl, 2014). Germline knockout of VCP in mice is embryonic lethal, with death occurring prior to embryonic day 4.5 (Müller et al., 2007). The essential role of VCP in non-rapidly dividing or differentiated tissues such as neurons is less clear. VCP knockdown in primary cultured hippocampal neurons decreases dendritic spine formation due to a decrease in endoplasmic reticulum (ER)-associated protein synthesis (Shih and Hsueh, 2016; Wang et al., 2011). In Drosophila, strong inactivation of ter94, the Drosophila homolog of VCP, in neurons is cell lethal while mild inhibition impairs dendritic pruning (Rumpf et al., 2011). These studies support a developmental role for VCP in spinogenesis and dendritic pruning. However, the role of VCP in terminally differentiated neurons and the consequences of VCP inactivation in these cells remain unclear.
More than 60 different VCP mutations are identified as causing disease in patients with MSP (Al-Obeidi et al., 2018). Most of these structurally reside at the interface between the N and D1 domains and may affect N domain engagement with adaptors during the ATPase hydrolysis cycle (Meyer and Weihl, 2014). Most VCP disease mutations increase ATPase activity and subsequent substrate processing, suggesting that MSP mutations behave in a gain-of-function manner (Blythe et al., 2017; Halawani et al., 2009; Niwa et al., 2012; Weihl et al., 2006). This is bolstered by the finding that VCP chemical inhibitors can suppress MSP mutant phenotypes in Drosophila (Zhang et al., 2017). In contrast, some studies suggest that MSP-associated mutants fail to engage selected adaptor proteins essential for VCP function indicative of a loss-of-function mechanism of disease (Papadopoulos et al., 2017; Ritz et al., 2011). For example, quantitative proteomics demonstrates that when expressed in cells, MSP mutant VCP fails to interact with UBXD1, an essential VCP adaptor needed for endolysosomal function (Ritz et al., 2011). A more recent study identified a unique VCP mutation in a family with FTLD-tau that, in contrast to previous MSP-associated variants, reduced ATPase and unfoldase activity, supporting the loss of VCP function hypothesis (Darwich et al., 2020). Distinguishing between gain- and loss-of-function mechanisms of pathogenesis is critical to future therapeutic strategies.
Several mouse models of VCP-associated MSP have been generated using both knockin and transgenic approaches (Clemen et al., 2018; Custer et al., 2010; Nalbandian et al., 2013; Weihl et al., 2007). These models recapitulate some pathologic features of MSP such as muscle weakness and lytic bone lesions, albeit at advanced ages (Custer et al., 2010; Nalbandian et al., 2013; Weihl et al., 2007). Notably, neuronal pathology has been minimally characterized in these models with TDP-43 pathology observed between 13 and 15 months and a reduction in motor neurons after 20 months (Custer et al., 2010; Nalbandian et al., 2013; Yin et al., 2012). MSP pathology in non-neuronal tissues such as muscle derived from patients with VCP-associated myopathy accumulates ubiquitinated inclusions, autophagicdebris, and endolysosomal remnants (Ju et al., 2009; Papadopoulos et al., 2017; Ritz et al., 2011; Weihl et al., 2008). The pathologic features of MSP-associated neurodegeneration also include ubiquitin- and sequestosome 1 (SQSTM1)-positive aggregates along with TDP-43 inclusions in cortical neurons and/or motor neurons when ALS is present (Forman et al., 2006; Neumann et al., 2007). Some cases of VCP-associated MSP have also demonstrated other pathologies that include α-synuclein, tau, and amyloid-β, suggesting that VCP dysfunction is related to other forms of neurodegeneration (Darwich et al.,2020; Kaleem et al., 2007; Shamirian et al., 2015; Spina et al., 2013).
RESULTS
Neuronal inactivation of VCP leads to cortical brain atrophy and behavioral changes
We crossed mice carrying two LoxP insertions within the murine VCP gene (VCPFL/FL) with CaMKIIα-Cre mice that express Cre re-combinase under the CaMKIIα (calcium/calmodulin-dependent protein kinase II alpha) promoter (VCPFL/FL:CamkCre, termed VCP conditional knockout [cKO] hereafter) (Figure 1A). Deletion of VCP from the cortex at 6 weeks of age was confirmed via PCR amplification through the deleted region (Figure 1B). There was no significant decrease in total VCP protein levels in VCP cKO brain lysates due to the presence of VCP in other brain regions and non-neuronal cells (Figure 1C). In contrast, there was a decrease in both nuclear and cytosolic VCP in neurons from the CA1 region of the hippocampus and cortex in VCP cKO mouse brains at 1 month compared with matched controls when immunostained with an anti-VCP antibody (Figure 1D). Brain mass and cortical brain volume were reduced in 2-month-old VCP cKO mice compared with control mice (Figures 1E and 1F). Cresyl violet staining of coronal brain sections confirmed the loss of brain volume was principally due to a reduction in cortical thickness (Figures 1G and 1H). VCP cKO mice were born at a normal Mendelian ratio and appeared grossly normal.
Figure 1. Loss of VCP in forebrain neurons causes cortical brain atrophy.
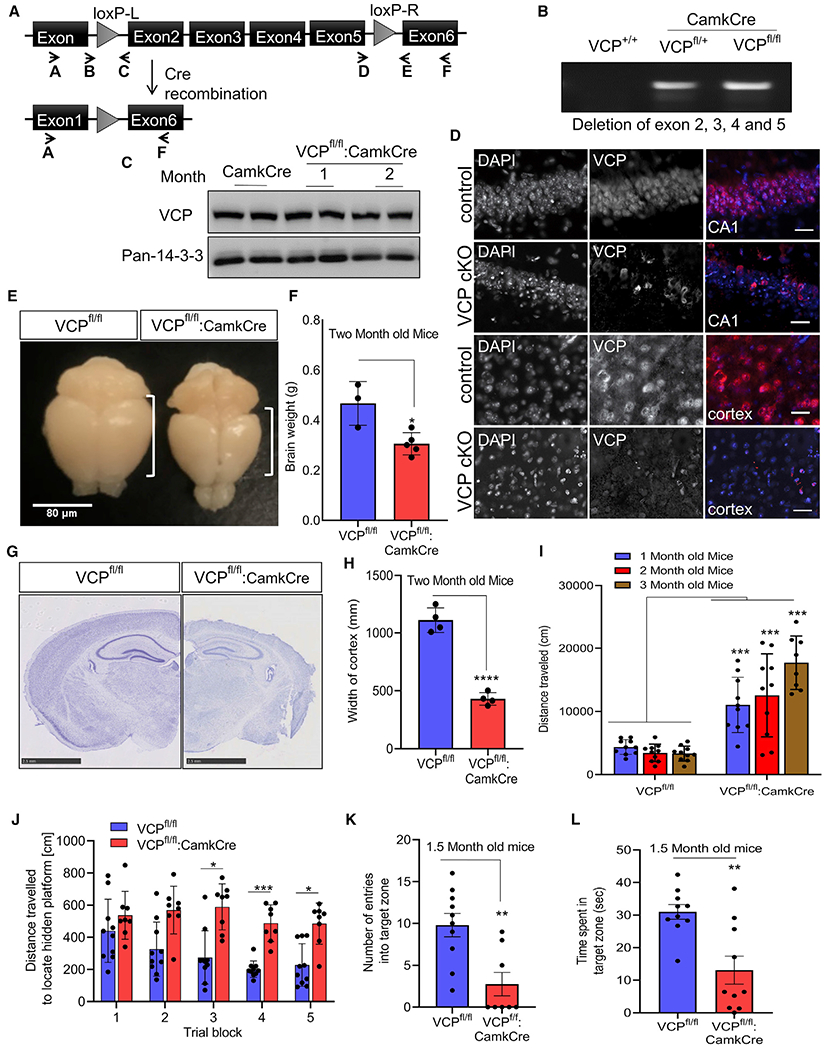
(A) VCP inactivation strategy.
(B) Agarose gel of cortical brain genomic DNA PCR amplification using primers A and F from (A), confirming LoxP recombination in VCPFL/WT or VCPFL/FL crossed to CaMKIIα-Cre mice (VCP cKO).
(C) Immunoblot for VCP and 14-3-3 (loading control) of cortical brain lysates from control (CamkCre) or VCP cKO mice at 1 and 2 months.
(D) VCP (red) immunofluorescence and DAPI nuclear (blue) fluorescence of coronal sections through the CA1 region of the hippocampus or cortex of 1-month-old VCP cKO mice and 2-month-old control mice (representative images of four control and six VCP cKO mice). Scale bars, 25 μm.
(E) Gross brain image of 2-month-old control (VCPFL/FL) or VCP cKO mice. Scale bar, 80 μm.
(F) Bar graph of brain weights from 2-month-old control or VCP cKO mice. Data represent mean ± SD (error bars) (n = 3 and 5 animals, respectively). *p < 0.05 by unpaired t test.
(G) Micrograph of 2-month-old control or VCP cKO coronal mouse brain sections stained with cresyl violet. Scale bars, 2.5 mm.
(H) Bar graph of cortical width from 2-month-old control of VCP cKO mice. ****p < 0.0001 by unpaired t test. Data represent mean ± SD (error bars) (n = 3 slices from four mice per group).
(I) Locomotor activity levels of VCP cKO mice (RM ANOVA interaction between genotype and age, F(1,52) = 103.6, p < 0.0001; ***p < 0.001 for each comparison within age). Data represent mean ± SEM (error bars) (n = 10 mice/group).
(J) Distance traveled to locate hidden platform (RM ANOVA) (F(4,80) = 3.187.6, p = 0.0176). Main effect of genotype p < 0.0001, with post hoc comparisons showing a shorter distance between genotypes by trial blocks 3 (*p < 0.05), 4 (***p < 0.001), and 5 (*p < 0.05). Data represent mean ± SEM (error bars) (n = 10 mice/group).
(K) Entries into the target zone during the probe trial compared to controls (**p < 0.01 by t test).
(L) Time searching the quadrant where platform is hidden (**p < 0.01 by t test). Data represent mean ± SEM (error bars) (n = 10 mice/group).
VCP cKO mice did not show differences in strength on the inverted screen test or time to turn and climb to the top of a 90° screen as a measure of general motor function (Figures S1A and S1B). In contrast, VCP cKO mice show significantly higher levels of locomotor activity (Figure 1I) and rearing behavior (Figure S1C) compared to controls. VCP cKO mice became progressively more hyperactive with repeated testing at 2 and 3 months of age (Figure 1I). On memory tasks, 1.5-month-old VCP cKO mice did not significantly improve their path length to locate the submerged (hidden) platform in the spatial learning trials, while control mice showed significantly shorter path lengths over trials, indicating impaired spatial learning in VCP cKO mice (Figure 1J). Following the spatial learning trials, a probe trial was conducted to assess memory for the location of the escape platform. Control mice show significantly more entries in the target zone and more time in the target quadrant as compared to VCP cKO mice, suggesting impaired spatial memory (Figures 1K and 1L).
Neuronal inactivation of VCP leads to progressive neuronal loss, gliosis, and TDP-43 pathology
VCP cKO mouse brains were sectioned and immunostained with anti-NeuN antibody to visualize the nuclei of neurons in the cortex and hippocampus. The area of NeuN staining in the cortex and CA1 region of the hippocampus from VCP cKO mice progressively decreased from 1 to 4 months (Figures 2A and 2B). To explore other markers of degeneration, we immunostained similar sections with an anti-GFAP antibody to define astrogliosis. GFAP staining of 1-month-old VCP cKO mouse brains was similar to controls (Figures 2C and 2D). In contrast to NeuN staining, an increase in GFAP immunostaining in 2- and 4-month-old VCP cKO mouse brains was present as compared to control brains (Figures 2C and 2D). Consistent with neuronal loss and astrogliosis, anti-IBA1 staining, a marker of activated microglia, was increased beginning in 1-month-old VCP cKO mouse brains (Figures 2E and 2F).
Figure 2. Loss of VCP in forebrain neurons causes neurodegeneration and TDP-43 pathology.
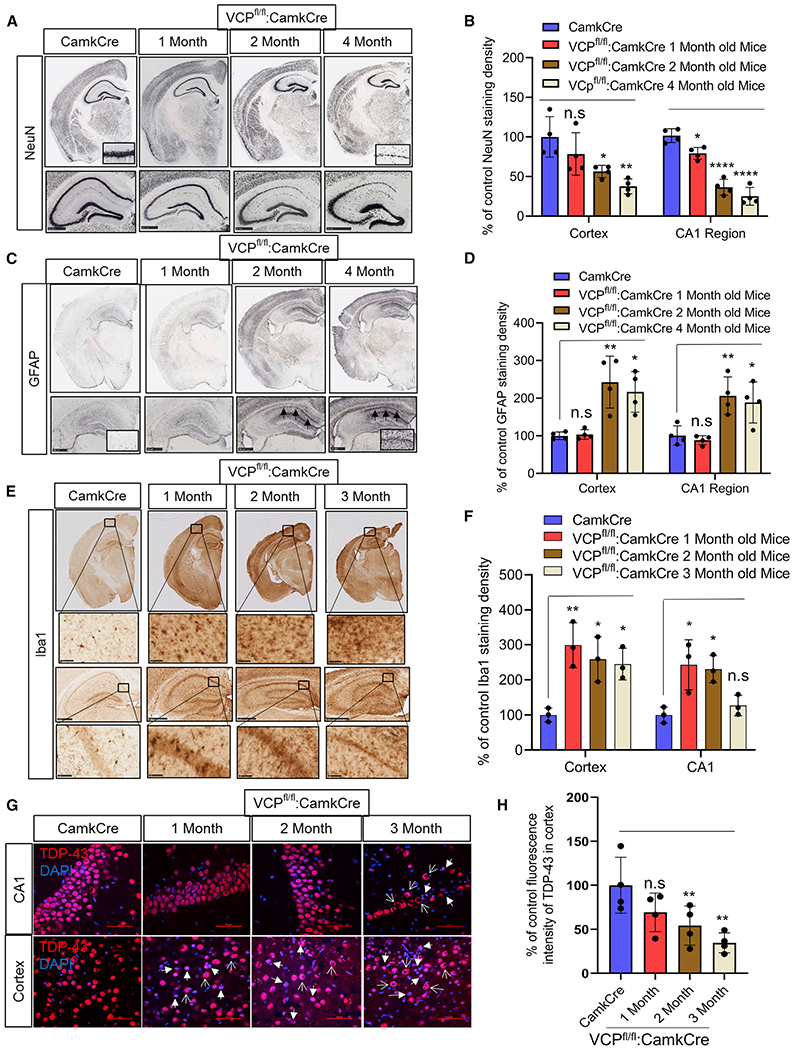
(A) NeuN staining of coronal sections through the cortex and hippocampus of control (CamKcre) or 1-, 2-, and 4-month-old VCP cKO mice. Insets denote CA1 region of the hippocampus. Scale bars, 500 μm.
(B) Quantitation of NeuN. Data represent mean ± SD (error bars) (data points represent mice) (n = 4 slices from four mice per group). A one-way ANOVA was used for statistical testing, treating four slices from four mice as an independent sample; cortex is F(3,12) = 57.34, p = 0.0001 and CA1 region is F(3,12) = 7.689, p = 0.0040. Post hoc comparisons using Tukey’s test, *p < 0.02.
(C) GFAP staining of coronal sections through the cortex and hippocampus of control (Camkcre) or 1-, 2-, and 4-month-old VCP cKO mice. Insets and arrows denote CA1 region of the hippocampus. Scale bars, 500 μm.
(D) Quantitation of GFAP staining. Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing, treating four slices from four mice as an independent sample; cortex is F(3,12) = 11, p = 0.0009 and CA1 region is F(3,12) = 9, p = 0.0019. Post hoc comparisons using a Tukey’s test, *p < 0.02.
(E) Iba1 staining of coronal sections through the cortex and hippocampus of control (Camkcre) or 1-, 2-, and 4-month-old VCP cKO mice. Scale bars, 2.5 mm. Insets denote CA1 region of the hippocampus. Scale bars, 500 μm.
(F) Quantitation of Iba1 staining. Data represent mean ± SD (error bars) (n = 2 slices from four mice per group). The quantification was done by ImageJ software. Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing treating two slices from three mice as an independent sample; cortex is F(3,8) = 8.4, p = 0.0073 and CA1 region is F(3,8) = 7.8, p = 0.0091. Post hoc comparisons using Tukey’s test, *p < 0.02.
(G) TDP-43 (red) immunofluorescence and DAPI nuclear (blue) fluorescence of the cortex and CA1 region of the hippocampus from control (CamkCre) and 1-, 2-, and 3-month old VCP cKO mice. Closed arrows indicate the loss of TDP-43 from the nucleus, and open arrows indicate cytosolic TDP-43. Scale bars, 50 μm.
(H) Quantitation of TDP-43 fluorescence intensity. Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing, treating three slices from four mice as an independent sample; F(3,12) = 5, p = 0.0111. Post hoc comparisons using Tukey’s test, *p < 0.02.
In contrast to control mouse brain sections where TDP-43 was nuclear, TDP-43 was lost from the nucleus and accumulated within the cytoplasm of cortical neurons of VCP cKO mouse brains at 1 month of age (Figures 2G and 2H). This pattern persisted at 2 and 3 months of age and was also apparent in CA1 hippocampal neurons when neuronal loss became evident in VCP cKO mouse brains (Figures 2G and 2H). Moreover, TDP-43 immunoblotting of brain homogenates from control or VCP cKO mice subjected to differential centrifugation to generate a soluble and insoluble fraction found an increase in insoluble TDP-43 and TDP-43 phosphorylated at serines 409/410 (pTDP-43) at 1 month of age (Figures 3A and 3B). The pattern of pTDP-43 immunofluorescence was similar to that of total TDP-43 but diminished in intensity with age, suggesting that cytosolic TDP-43 inclusions were primarily non-phosphorylated (Figure S1C). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive nuclei appeared in the cortex and CA1 region of the hippocampus at 2 months of age and disappeared, consistent with their death after 3 months (Figures 3C and 3D). Further evidence of apoptotic activity in the VCP cKO mouse was demonstrated by an increase in Bcl-2-associated X protein (BAX) on immunoblot (Figures 3C and 3D).
Figure 3. Loss of VCP in forebrain neurons leads to neuronal death and autophago-lysosomal dysfunction.
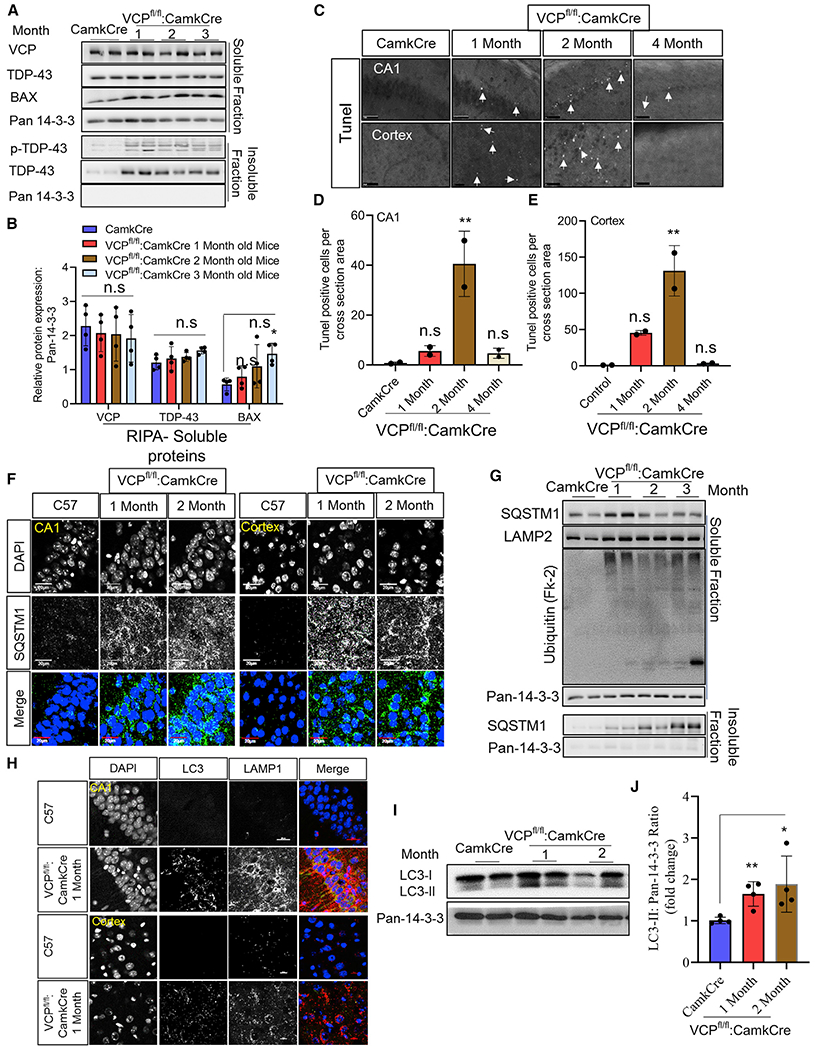
(A) Soluble and insoluble proteins from brain lysates of control and 1-, 2-, and 3-month-old VCP cKO mice immunoblotted with antibodies to VCP, TDP-43, pTDP-43, BAX, and 14-3-3 (as a loading control).
(B) Quantitation of band intensities of VCP, TDP-43, and BAX relative to 14-3-3 (n = 4 animals per group). The quantification was done by ImageJ software. Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing treating each brain as an independent sample; VCP is F(3,12) = 0, p = 0.8863, TDP-43 is F(3,12) = 1, p = 0.2082, and BAX is F(3,12) = 3, p = 0.0408. Post hoc comparisons using a Bonferroni test, *p < 0.02.
(C) TUNEL staining of the cortex and CA1 region of the hippocampus from control (CamkCre) and 1-, 2-, and 4-month-old VCP cKO mice. Closed arrows mark TUNEL-positive nuclei. Scale bars, 500 μm.
(D and E) Quantitation of TUNEL-positive cells. Data represent mean ± SD (error bars) (n = 2 slices from two animals per group). Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing, treating two slices from two mice as an independent sample; cortex is F(3,4) = 24, p = 0.0050 and for the CA1 region is F(3, 4) = 15, p = 0.0118. Post hoc comparisons using Tukey’s test, **p < 0.01.
(F) SQSTM1 (green) immunofluorescence and DAPI nuclear (blue) fluorescence of the cortex and CA1 region of the hippocampus from control (C57) and 1- or 2-month-old VCP cKO mice. Scale bars, 20 μm (n = 3 slices from four mice per group).
(G) Soluble and insoluble proteins from brain lysates of control (CamkCre) and 1-, 2-, and 3-month-old VCP cKO mice immunoblotted with antibodies to SQSTM1, Lamp2, ubiquitin-conjugates (FK2), and 14-3-3 (as a loading control) (n = 4 animals per group).
(H) LC3 (green), Lamp1 (red) immunofluorescence, and DAPI nuclear (blue) fluorescence of the cortex and CA1 region of the hippocampus from control (C57) and 1-month-old VCP cKO mice. Scale bars, 20 μm (n = 3 slices from four mice per group).
(I) Immunoblot for LC3 and 14-3-3 (loading control) of brain lysates from control (CamkCre) or 1-month-old VCP cKO mice.
(J) Quantitation of the ratio of the band intensities of LC3II:14-3-3. Data represent mean ± SD (error bars) (n = 4 animals per group). Unpaired t test, **p < 0.01; n.s., not significant.
Autophago-lysosomal dysfunction precedes neuronal degeneration in VCP-deficient neurons
SQSTM1-positive puncta were present by immunofluorescence at 1 and 2 months of age in the CA1 hippocampal region and cortex of VCP cKO mice as compared with control mouse brains (Figure 3F). Immunoblotting for ubiquitin and SQSTM1 demonstrated an increase in ubiquitinated proteins and total SQSTM1 at 1 month of age in VCP cKO mouse cortex (Figure 3G). Moreover, SQSTM1 moved from the soluble to insoluble fraction after 2 months of age in VCP cKO mouse brains (Figure 3G). Similarly, co-immunofluorescent staining for LC3 and Lamp1 demonstrated an increase in LC3-positive puncta that occasionally co-localized with Lamp1 (Figure 3H). The accumulation of LC3-positive puncta was not seen in control mouse brains (Figure 3H). Immunoblots for LC3 from control and 1- and 2-month-old VCP cKO mouse brains further demonstrated a selective increase in LC3II protein levels (Figures 3I and 3J). Co-immunofluorescence with NeuN demonstrated that the increase in Lamp1 puncta occurred in neurons (Figure S1D). Inactivation of the essential autophagy protein ATG5 in CaMKIIα-expressing neurons, generated by crossing ATG5 floxed mice with CaMKIIα-Cre mice (ATG5FL/FL:CamkCre, termed ATG5 cKO hereafter), showed SQSTM1 accumulation with no neuronal loss at 2 and 4 months of age, consistent with previous studies (Figures S2A–S2E) (Inoue et al., 2012).
Early proteomic changes in VCP cKO mouse brains
We performed quantitative mass spectrometry on cortical lysates from control and 1- and 2-month-old VCP cKO mice (three mice per time point). Proteomic analysis reflects changes in neuronal and non-neuronal cells within the cortex of these mice. At 1 month of age, only 35 proteins were significantly upregulated and 14 proteins significantly downregulated by >2-fold in VCP cKO mice (Figure 4A; Table S2). Of these, all three C1q components of complement and several lysosomal proteases were increased (Figure 4A). Pathway analysis on all 299 proteins that were significantly changed at 1 month of age identified protein ubiquitination as the most enriched pathway (Figure 4B). Similar analysis utilizing 2-month-old VCP cKO mouse brains identified an enrichment of 248 proteins upregulated by >2-fold, many of which included lysosomal proteins, lysosomal proteases, and lysosomal enzymes (Figure 4A). Additionally, there was a further increase in complement proteins and glial-derived proteins (Figure 4A). C1q immunostaining of control and 1- and 2-month-old VCP cKO brains demonstrated an increase above control mice at 2 months of age, consistent with proteomic data (Figures S3A and S3B). In contrast to 1-month data, 126 proteins were downregulated by >2-fold, including synaptic proteins such as GRIN1 and GRIN2A/B (Figure 4A; Table S2). Pathway analysis on all 1,091 proteins that were significantly changed at 2 months identified synaptogenesis as the most enriched pathway (Figure 4B).
Figure 4. Proteomic and transcriptomic signatures of VCP inactivation in the brain.
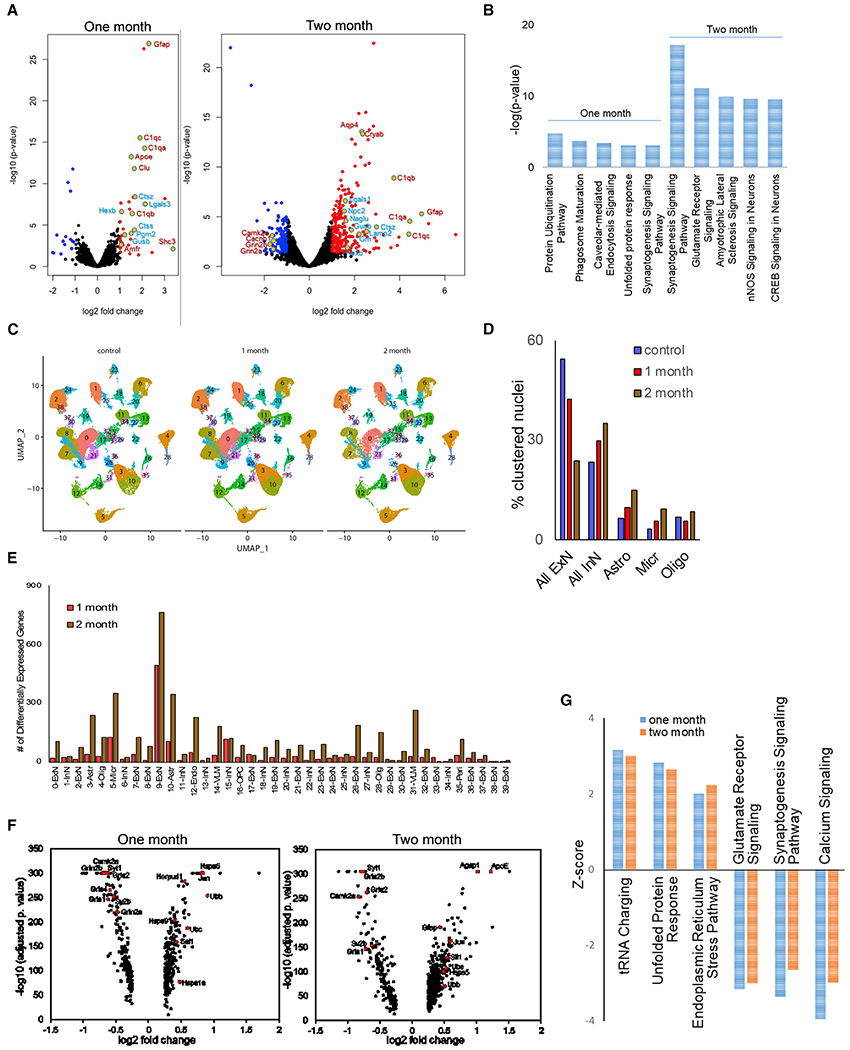
(A) Volcano plots of differentially abundant proteins (DAPs) in 1- and 2-month-old VCP cKO cortices as compared with age-matched controls.
(B) Graph of the most significantly enriched pathways following Ingenuity Pathway Analysis (IPA) of DAPs at 1 and 2 months.
(C) UMAP of nuclear clusters from control and 1- or 2-month-old VCP cKO cortices.
(D) Graph of the percentage of nuclei clustered within the categories of excitatory neuron (ExN), inhibitory neuron (InN), astrocytes (Astro), microglia (Micro), and oligodendrocytes (Oligo) in control and 1- or 2-month-old VCP cKO cortices.
(E) Graph of the number of differentially expressed genes (DEGs) in each nuclear cluster from 1- and 2-month-old VCP cKO cortices. VLM, vascular leptomeningeal cell; Peri, pericyte; OPC, oligodendroglia precursor cell; Endo, endothelial cell.
(F) Volcano plots of DEGs from cluster 9 in 1- and 2-month-old VCP cKO cortices as compared with age-matched controls.
(G) Graph of the three most increased and decreased pathways according to Z score following IPA of DEGs at 1 and 2 months.
Early transcriptomic changes in VCP knockout cortical neurons
Since VCP inactivation was performed in a specific subpopulation of neurons, we reasoned that performing single nuclei RNA sequencing on control, 1-month old VCP cKO, and 2-month-old VCP cKO mouse cortices (three mice per group) would reveal changes specific to VCP inactivation since these time points were before and during neuronal loss. A total of 160,127 individual nuclei were arranged by uniform manifold approximation and projection (UMAP) in two dimensions for visualization. Unsupervised clustering revealed a total of 39 distinct clusters across all samples (Figure 4C). These clusters were manually identified on the basis of the expression of known cell type-specific markers as excitatory neurons, inhibitory neurons, oligodendrocytes, astrocytes, vascular leptomeningeal cells, microglia, oligodendrocyte precursor cells, pericytes, and endothelial cells (Figure 4C). For all three groups, all clusters were similarly represented in each of the three mice analyzed. As expected, the percentage of nuclei from excitatory neuron clusters decreased from control to 1 month and further in 2-month VCP cKO mouse brains (Figure 4D). In contrast, the relative proportions of nuclei in clusters from inhibitory neurons, astrocytes, and microglia increased during this same time period (Figure 4D). Notably one excitatory neuron cluster had the greatest number of significant differentially expressed genes (DEGs) at both 1 month and 2 months in VCP cKO cortices (Figures 4E and 4F; Table S3). Pathway analysis from this cluster revealed that the unfolded protein response (UPR) and ER stress pathways were significantly upregulated, and glutamate receptor signaling and synaptogenesis were significantly decreased (Figure 4G).
To validate these transcriptomic findings, we immunoblotted brain lysates from control and 1-, 2-, and 3-month-old VCP cKO mice for UPR and ER stress markers. There was an increase in BIP, phosphorylated (phospho-)PERK, and phospho-IRE1 (Figures S3C and S3D). Immunofluorescence staining with the pre- and post-synaptic marker synapsin and PSD-95 was performed on coronal brain sections of control and 1- and 2-month-old VCP cKO mice, and Synaptic Evaluation and Quantification by Imaging of Nanostructure (SEQUIN) quantitated synaptic loci (Figure S3E) (Sauerbeck et al., 2020). Quantitation from the CA1 region of the hippocampus demonstrated a 25% reduction in synaptic loci in 2-month-old VCP cKO mice, consistent with our proteomic and transcriptomic studies (Figure S3F).
VCP disease mutations are hypomorphic
To understand the pathogenic nature of VCP disease mutations, we characterized and generated several additional mouse models. Two previous studies have described VCP knockin mouse lines carrying a VCP-R155H or a VCP-R155C disease mutation (Clemen et al., 2018; Nalbandian et al., 2013). We evaluated CNS pathology in both mouse lines at 6 months and 13 months of age as compared with C57BL/6 (C57) littermate controls (Figure S4). Notably, there was no neuronal loss, TDP-43 pathology, SQSTM1 inclusions, ubiquitin accumulation, or autophago-lysosomal dysfunction as measured by immunoblot or immunostaining at these ages (Figure S4). Homozygous VCP-R155H mice have reduced embryonic viability, growth retardation, and lethality by 21 days (Nalbandian et al., 2012), and homozygous VCP-R155C mice are embryonic lethal (Clemen et al., 2018). We crossed VCP cKO (VCPFL/FL:CamkCre) to heterozygous VCP-R155C (VCPRC/WT) knockin mice to generate mice that expressed only the VCP-R155C (VCPRC/FL:CamkCre) in CaMKIIα-expressing neurons, referred to as VCP cRC mice (Figure S5A). VCP cRC mice developed progressive neuronal loss and degenerative pathology between 6 and 12 months of age as evaluated by NeuN and IBA1 (Figures 5A–5D). Notably, while total TDP-43 immunostaining intensity decreased in the cortex, there was an increase in pTDP-43 immunostaining intensity (Figures 5E–5H). VCP cRC mice show significantly higher levels of locomotor activity at 10 months (Figure 6A) compared to VCPRC/FL controls. VCP cRC mice spend less time in the target quadrant as compared with age-matched mice that maintained the floxed allele, suggesting development of spatial memory impairment with age (Figure 6B). Similar to VCP cKO mice, VCP cRC mouse brains had accumulation of SQSTM1-, LAMP1/2-, and LC3-positive structures in neurons from the cortex and CA1 region of the hippocampus (Figure 6C; Figure S5B). These pathologic features were also seen via immunoblot and included the accumulation of insoluble TDP-43, soluble and insoluble SQSTM1, ubiquitin conjugates, Bax, and ER stress markers (Figures 6E–6G). These pathologies were not due to the loss of one VCP-wild-type (WT) allele (i.e., haploinsufficiency) since VCPWT/FL:CamkCre mice that express 50% of VCP in CaMKIIα-expressing neurons had no pathologic changes, accumulation of TDP-43, or autophagic markers at 6 and 12 months of age (Figure S6). This is consistent with previous studies suggesting that VCP haploinsufficient mice have no measurable phenotype (Clemen et al., 2015).
Figure 5. Re-expression of a VCP-R155C mutant allele in VCP cKO mouse neurons recapitulates FTLD-TDP pathology.
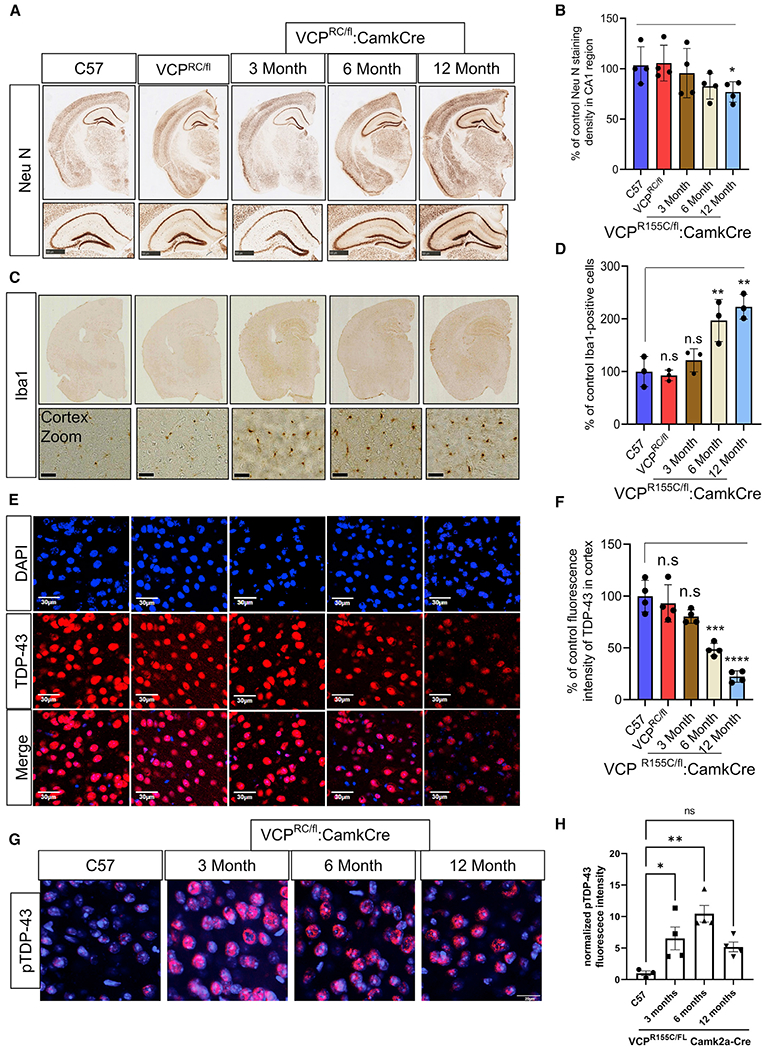
(A) NeuN staining of coronal sections through the cortex and hippocampus of 9-month-old control (C57), 9-month-old VCP-R155C heterozygous (VCPRC/FL), or VCP cRC (VCPRC/FL:CamkCre) mice at 3, 6, and 12 months of age. Scale bars, 500 μm in bottom panel.
(B) Quantitation of NeuN. Data represent mean ± SD (error bars) (n = 3 slices from four mice per group). *p < 0.05 with paired t test against C57 control.
(C) Iba1 staining of coronal sections through the cortex and hippocampus of 9-month-old control (C57), 9-month old VCP-R155C heterozygous (VCPRC/FL), or VCP cRC (VCPRC/FL:CamkCre) mice at 3, 6, and 12 months of age. Scale bars, 30 μm in bottom panel.
(D) Quantitation of Iba1 staining. Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing, treating three slices from three mice as an independent sample; F(4,10) = 14.83, p = 0.0003. Post hoc comparisons using Tukey’s test, **p < 0.01.
(E) TDP-43 (red) immunofluorescence and DAPI nuclear (blue) fluorescence of the cortex from 9-month-old control (C57), 9-month-old VCP-R155C heterozygous VCPRC/FL), or VCP cRC (VCPRC/FL:CamkCre) mice at 3, 6, and 12 months of age. Scale bars, 30 μm.
(F) Quantitation of TDP-43 fluorescence intensity. Data represent mean ± SD (error bars) (n = 3 slices from four mice per group). Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing, treating three slices from four mice as an independent sample; F(4,15) = 32, p < 0.0001. Post hoc comparisons using a Bonferroni test, ***p < 0.0001.
(G) Phospho-TDP-43 (red) immunofluorescence and DAPI nuclear (blue) fluorescence of the cortex from 9-month-old control (C57) or VCP cRC (VCPRC/FL:CamkCre) mice at 3, 6, and 12 months of age. Scale bar, 20 μm.
(H) Quantitation of pTDP-43 fluorescent intensity. Data represent mean ± SD (error bars) (n = 3 slices from four mice per group). F(3,11) = 8.469, p = 0.0034 followed by Tukey’s test was used for comparisons.
Figure 6. VCP-R155C is a hypomorphic allele and resembles VCP loss of function in mouse neurons.
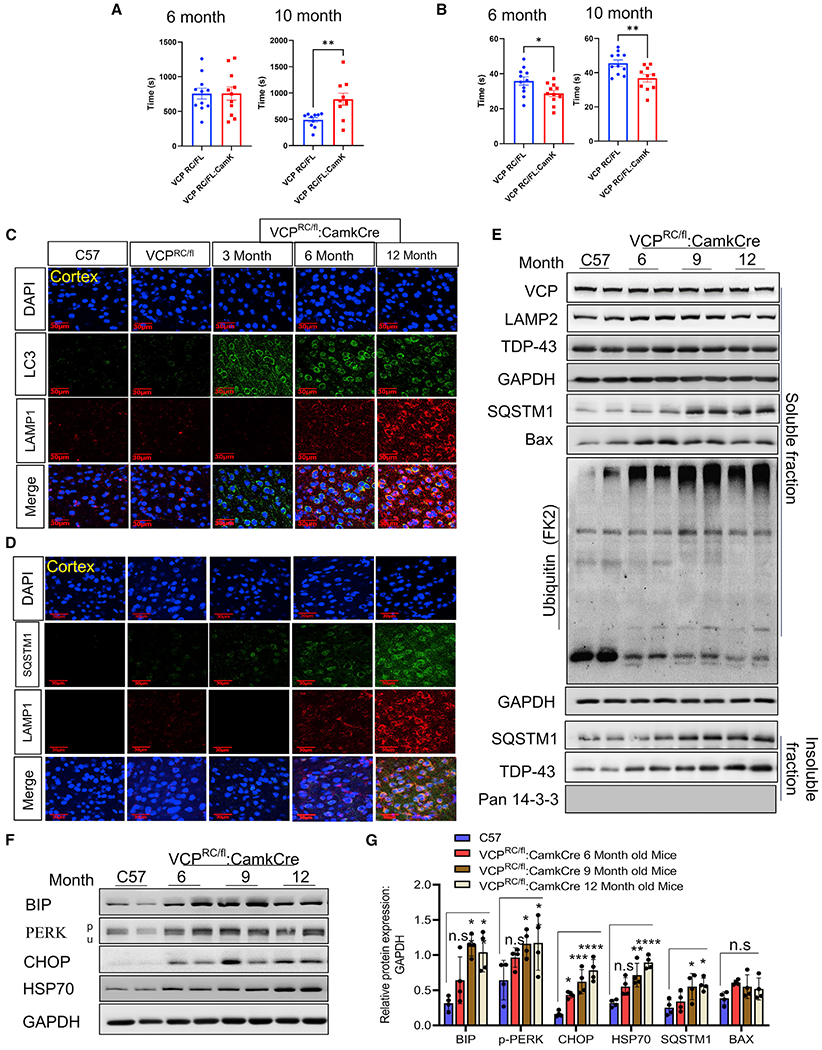
(A) Locomotor activity of VCP cRC mice and control VCPRC/FL mice at 6 and 10 months of age; paired t test between genotypes, **p < 0.01. Data represent mean ± SEM (error bars) (n = 10 mice/group).
(B) Time spent searching the quadrant where the hidden platform at both VCP cRC at 6 months (paired t test, *p < 0.05) and at 10 months (**p < 0.01) compared to control VCPRC/FL mice at each age. Data represent mean ± SEM (error bars) (n = 10 mice/group).
(C) LC3 (green), Lamp1 (red) immunofluorescence, and DAPI nuclear (blue) fluorescence of the cortex from 9-month-old control (C57), 9-month-old VCP-R155C heterozygous (VCPRC/FL), or VCP cRC (VCPRC/FL:CamkCre) mice at 3, 6 and 12 months of age. Scale bars, 30 μm.
(D) SQSTM1 (green), Lamp1 (red) immunofluorescence, and DAPI nuclear (blue) fluorescence of the cortex from 9-month-old control (C57), 9-month-old VCPR155C heterozygous (VCPRC/FL), or VCP cRC (VCPRC/FL:CamkCre) mice at 3, 6, and 12 months of age. Scale bars, 30 μm (n = 3 slices from four mice per group).
(E) Soluble and insoluble proteins from brain lysates of 9-month-old control (C57) or VCP cRC (VCPRC/FL:CamkCre) mice at 6, 9, and 12 months of age were immunoblotted with antibodies to VCP, Lamp2, TDP-43, SQSTM1, BAX, ubiquitin conjugates (FK2), GAPDH (loading control), and 14-3-3 (loading control).
(F) Immunoblot for BIP, PERK, CHOP, HSP70, and GAPDH (loading control) of brain lysates from 9-month-old control (C57) or VCP cRC (VCPRC/FL:CamkCre) mice at 6, 9, and 12 months of age.
(G) Quantitation band intensities of BIP, PERK, CHOP, HSP70, SQSTM1, and BAX relative to GAPDH (n = 4 animals per group). Quantification was determined by ImageJ software. Data represent mean ± SD (error bars) (data points represent mice). A one-way ANOVA was used for statistical testing, treating mice brains as an independent sample; for BIP is F(3,12) = 10.32, p < 0.0012; for p-PERK is F(3,12) = 6, p = 0.0062; for CHOP is F(3,12) = 19.19, p = 0.0001; for HSP70 is F(3,12) = 17.19, p = 0.0001; for SQSTM1 is F(3,12) = 5, p = 0.0127; for BAX is F(3,12) = 2.297, p = 0.1296. Post hoc comparisons using a Bonferroni test, *p < 0.05.
Proteomic and transcriptomic signatures in VCP cKO mice resemble GRN carriers
We reasoned that the early proteomic and transcriptomic changes seen in VCP cKO mouse brains may reveal insights into the pathogenesis of other genetically defined human FTLD-TDPs caused by pathogenic mutations in GRN and repeat expansions in C9orf72. To test this hypothesis, we evaluated the extent of overlap and directionality of differentially expressed proteins (DEPs) and DEGs from 1- and 2-month-old VCP cKO brains with transcriptomic profiles altered in the parietal cortex of GRN (n = 5) and C9orf72 (n = 5) carriers compared to neuropathology-free controls (n = 16). We identified significant overlap between the DEPs and DEGs from VCP cKO mice and the GRN carriers (Figures 7A and 7B; Figure S7; Table S4). Our analyses showed that overlap was more remarkable with VCP cKO at 2 months (Figure S7) compared to 1-month samples (Figures 7A and 7B). In contrast, we did not identify a significant overlap (p > 0.05) when we analyzed the transcriptomic profiles altered in carriers of the C9orf72 repeat expansions (Figures 7A and 7B). Similarly, we observed that only GRN carriers recapitulated the signatures of VCP cKO mice when the analyses were restricted to those genes in the human cortex that show the same direction of effect of DEPs and DEGs from the VCP cKO model. To visualize the VCP-deficient transcriptomic signature within individual control, GRN, and C9orf72 participants, we generated a heat map for all overlapped genes from 1- and 2-month VCP cKO mouse cortex (Figures 7C and 7D; Figure S7). We did not have any transcriptomic data from the brains of patients with VCP-associated MSP1 for this study.
Figure 7. The proteomic and transcriptomic signatures of VCP deficiency are present in GRN carriers.
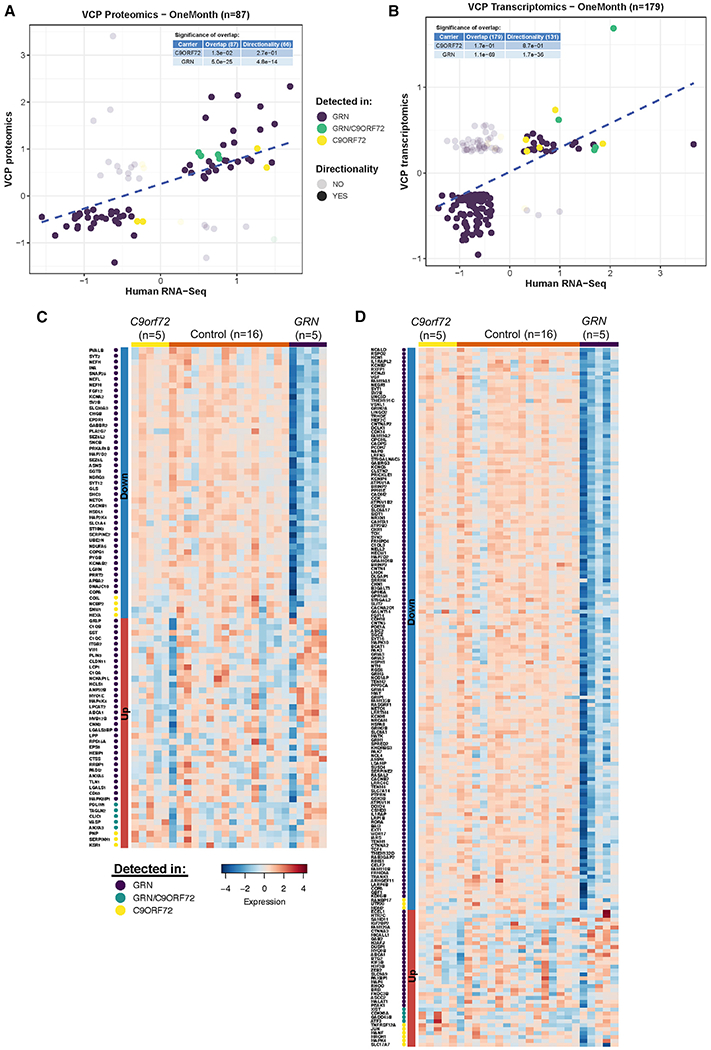
(A and B) The effect sizes of significantly different DEPs (A) and DEGs found in neurons from cluster 9 (B) from 1-month-old VCP cKO mice are compared to those effects from transcriptomic profiles of human parietal cortices with FTLD-TDP. The effects of significant differential gene expression products by comparing five GRN carriers (purple) versus neuropathology-free controls are shown (75 DEPs and 166 neuronal DEGs, respectively). Similarly, the effect of the comparison between the five carriers of the repeat expansion in C9orf72 (yellow) versus neuropathology-free controls is shown (seven DEPs and nine neuronal DEGs, respectively). The five DEPs and four neuronal DEGs identified for both genetic strata are shown in green. The tables embedded in each plot report the p value (hypergeometric test) product of comparing the extent of overlap (all) and directionality (same direction of effect) between genes detected in VCP cKO mice and human brains.
(C) A heatmap representing the gene expression (TPM) for significantly upregulated/downregulated genes overlapping between VCP cKO DEPs and FTLD-TDP GRN and C9orf72 carriers indicated in (A). The genes are ordered by their genetic strata and log fold change (FC).
(D) Same as (C), but showing the genes overlapping between VCP cKO neuronal DEGs and brain transcriptomics from FTLD-TDP GRN and C9orf72 reported in (B).
DISCUSSION
Most VCP disease mutant mouse models have focused on phenotypes other than FTLD. Transgenic expression of either an R155H or A232E mutant VCP in mice recapitulates muscle weakness and myopathology consistent with IBM (Custer et al., 2010; Weihl et al., 2007). Similarly, VCP-R155H knockin mice have muscle weakness and IBM-like pathology (Nalbandian et al., 2013). CNS pathology, if present, develops later, often after 18 months, and is less robust (Custer et al., 2010; Nalbandian et al., 2013). Indeed, even dedicated neuronal overexpression of mutant VCP-A232E does not lead to cortical neuronal loss until 20 months (Rodriguez-Ortiz et al., 2013). The present study further supports previous studies demonstrating that two different VCP knockin mouse models, VCPRH/WT and VCPRC/WT, have limited CNS pathology at 1 year of age (Clemen et al., 2018; Nalbandian et al., 2013). In contrast, young VCP cKO and VCP cRC mice develop the full spectrum of FTLD-TDP with neuronal degeneration, behavioral changes, TDP-43 inclusions, and lysosomal pathology. Other mouse models of FTLD-TDP pathology have been generated. These include GRN knockout and C9orf72 expanded repeat expression (Ahmed et al., 2010; Ghoshal et al., 2012; O’Rourke et al., 2015; Peters et al., 2015; Solomon et al., 2019). Similar to previous VCP mouse models, they develop modest neuropathology after 18 months. Other approaches have included transgenic expression of WT or mutant TDP-43 in the CNS or forebrain cortical neurons (Igaz et al., 2011; Porta et al., 2018). These mouse models develop robust TDP-43 pathology and neurodegeneration, suggesting that TDP-43 represents the final toxic pathway in FTLD-TDP.
The pathogenic mechanism by which VCP disease mutations lead to TDP-43 pathology and ultimately neurodegeneration is not clear. In our study, the earliest feature of VCP inactivation is autophago-lysosomal dysfunction as demonstrated by the accumulation of autophagosomes and endolysosomes. Concomitantly, TDP-43 moves from a soluble to insoluble fraction consistent with its loss from the nuclei of cortical neurons. Whether VCP-associated lysosomal dysfunction leads to TDP-43 pathology and ultimately neuronal death or, alternatively, VCP directly protects against TDP-43 proteotoxicity and the lysosomal pathology is mediated by TDP-43 is unresolved. The co-occurrence of autophago-lysosomal pathology and TDP-43 is a common feature seen in other FTLD-TDP model systems such as GRN knockout and even TDP-43 overexpression models (Elia et al., 2019; Werner et al., 2020; Xia et al., 2016).
Our data support that the mechanism of VCP-associated MSP1 mutant pathogenesis is due to a loss of VCP function. We demonstrate that VCP inactivation in cortical neurons leads to the hallmark pathologies seen in FTLD-TDP with neuronal loss, endolysosomal dysfunction, and TDP-43 inclusions. While heterozygous VCP mutant mouse models modestly recapitulate pathology at very late ages, our hemizygous VCP-R155C mutant neuronal-expressing cRC mice have similar pathologic changes to VCP knockout neurons, albeit at later onset and with slower progression, consistent with hypomorphic function. Pathway enrichment analysis of proteomic and transcriptomic changes from VCP cKO mouse brains demonstrates disruption of the UPS, ERAD, autophagic, and endolysosomal systems. These same pathways are disrupted in VCP cRC mouse brains, suggesting a similar loss of VCP activity with VCP-R155C expression. VCP is able to mediate cellular processes ranging from proteasome degradation, organelle reformation, endocytosis, and lysosomal function via its ability to interact with distinct adaptor proteins that ultimately dictate VCP’s function in ubiquitin-dependent processes (Meyer and Weihl, 2014).
Our previous studies using cell culture systems suggested that VCP disease mutations disrupt only a subset of VCP functions that are dependent on the VCP adaptors UBXD1 and PLAA (Ritz et al., 2011). Indeed, immunoprecipitation of VCP disease mutations failed to identify UBXD1 and PLAA in the precipitates (Ritz et al., 2011). UBXD1 and PLAA are necessary for VCP’s function within the endolysosomal pathway and, in particular, ubiquitin-mediated endolysosomal degradation of caveolin and the recognition/clearance of damaged late endosomes via lysophagy (Papadopoulos et al., 2017; Ritz et al., 2011). Notably, both of these processes are disrupted in muscle tissue of patients with MSP1 (Papadopoulos et al., 2017; Ritz et al., 2011). Recessive loss of function mutations in PLAA cause a rare infantile onset leukoencephalopathy with delayed myelination and a thin corpus callosum (Falik Zaccai et al., 2017; Hall et al.,2017). PLAA deficiency is embryonic lethal in mice (Falik Zaccai et al., 2017; Hall et al., 2017). Mice homozygous for disease mutations are viable and have impairment in ubiquitin-mediated endolysosomal degradation of synaptic proteins (Hall et al., 2017). Notably, proteasome activity and autophagic flux are unaffected, and the transcriptional signatures of the UPR and ERAD are unaltered in PLAA knockout cells (Hall et al., 2017). These data would suggest that VCP disease mutations lead to a loss of function beyond those associated with UBXD1/PLAA.
Our demonstration that VCP disease mutations are hypomorphic contrasts with studies evaluating VCP mutant function in cell-free systems. Most VCP disease mutations have an increase in basal ATPase activity, including the VCP-R155H and VCP-R155C used in this study (Blythe et al., 2017; Halawani et al., 2009; Niwa et al., 2012; Weihl et al.,2006). In addition, VCP disease mutations have an increased affinity for the adaptors UFD1/NPL4, resulting in an increase in unfoldase activity (Blythe et al., 2019). One principal function of VCP is to hydrolyze ATP and unfold substrates, facilitating their dissociation from a molecular complex or enhancing proteasomal degradation. This process and its enhanced activity with disease mutants are inhibited by VCP ATPase-specific inhibitors such as CB-5083 that are currently in clinical trials for various malignancies (Blythe et al., 2019; Blythe et al., 2017). These inhibitors have been shown to improve VCP mutant-associated mitochondrial dysfunction in patient fibroblasts and Drosophila (Zhang et al., 2017). However, no studies have evaluated whether there is an increase in VCP unfoldase activity in vivo. More importantly, our data would suggest that therapeutic inhibition of VCP in patients with MSP1 would not be beneficial and potentially exacerbates disease.
An increase in in vitro VCP ATPase activity does not correlate with disease phenotype or in vivo dysfunction (Al-Obeidi et al.,2018). In fact, two disease-associated mutations, VCP-I27V and VCP-E185K, do not have elevated ATPase activity, yet they both disrupt autophagosome maturation similar to all other MSP1 mutations, clearly demonstrating a discordance between in vitro activity and cellular function (Gonzalez et al., 2014; Weihl et al., 2015). Recently, dominant VCP disease mutations were identified in an FTD family with a vacuolar tauopathy (Darwich et al., 2020). This mutation, VCP-D395G, had a reduction in basal ATPase activity as compared with WT VCP and failed to block the aggregation of tau fibrils, suggesting that it was hypomorphic (Darwich et al., 2020). Although, the neuropathology of the vacuolar tau/VCP-D395G patient did not have TDP-43 inclusions and was dominated by phosphorylated tau-positive NFTs, note that patients with MSP1 have been reported to have both NFTs and α-synuclein-positive inclusions (Forman et al., 2006; Shamirian et al., 2015; Spina et al., 2013). These findings further support that dominant VCP disease mutations are hypomorphic and may lead to accumulation of aggregate-prone proteins associated with neurodegeneration.
Our studies do not preclude the possibility that VCP disease mutations are hypomorphic and also behave in a dominant negative manner. The WT VCP protein is not required for pathogenesis since our VCP cRC mice have only a single copy of the VCP-R155C allele. This is similar to human genetic studies in which a patient with a homozygous VCP-R159H variant was identified with IBM and PDB manifesting at age 31 (De Ridder et al., 2020). WT VCP forms stable hexamers, and VCP disease mutations stably incorporate into these complexes (Blythe et al., 2019). Mixing studies demonstrate that VCP dysfunction increases as the ratio of mutant to WT VCP increases (Blythe et al., 2019). How many mutant subunits are needed and what the hexameric orientation of the mutant subunits are that leads to VCP dysfunction are unclear. Our data would support a model in which VCP mutant subunits incorporate with WT subunits leading to a greater than 50% reduction in VCP function, resulting in disease.
Loss-of-function mutations in GRN are associated with a dominant FTLD-TDP (Baker et al., 2006; Cruts et al., 2006). The mechanism of action is presumed to be haploinsufficiency. Patients homozygous for GRN mutations have a syndrome ranging from adolescent onset neuronal ceroid lipofuscinosis (NCL) to early onset FTD (Huin et al., 2020; Smith et al., 2012). This spectrum of pathology may correlate with GRN protein levels, and augmenting GRN levels may be therapeutic (Huin et al., 2020). GRN is localized to the lysosome, and GRN knockout in cells and mice disrupts lysosomal function (Ahmed et al.,2010; Hu et al., 2010). Lysosomal dysfunction leads to features of NCL such as lipofuscin deposits and enlarged lysosomes (Ahmed et al., 2010; Wils et al., 2012). In addition, GRN knockout cells and mice accumulate ubiquitinated proteins and SQSTM1 inclusions and have a reduction in autophagic flux similar to that seen in our VCP cKO and VCP cRC mouse models (Ahmed et al., 2010; Chang et al., 2017; Tanaka et al., 2014; Wils et al., 2012). Our finding that a transcriptional signature of VCP insufficiency is consistent that seen in GRN carriers suggests a further pathogenic overlap with MSP1.
Our study supports therapies that increase VCP activity or function in patients with MSP1. More importantly, these data suggest that enhancing VCP function may also be therapeutic in other forms of FTLD-TDP and proteinopathies. This is consistent with studies demonstrating that VCP overexpression is protective in fly models of TDP-43 dysfunction and polyglutamine toxicity in C. elegans (Kushimura et al., 2018; Yamanaka et al., 2004). In mammalian systems, selective cardiac overexpression of VCP corrects pressure-induced left ventricular hypertrophy, and selective neuronal expression improves protein synthesis deficits in a model of neurofibromatosis (Shih et al., 2020; Zhou et al., 2017). The mechanism for improvement in these systems relates to VCP’s participation in the regulation of mTORC1 activity (Ching et al., 2013). We have previously demonstrated that loss of VCP or mutant VCP expression in skeletal muscle leads to endolysosomal dysfunction, impaired mTORC1 activation, and a persistent activation of TFEB (Arhzaouy et al., 2019; Ching et al., 2013). VCP’s overlapping roles in protein degradation via the UPS and autophago-lysosomal pathway coupled with its regulation of protein synthesis in terminally differentiated cells place VCP at a critical position for targeted therapies aimed at restoring protein homeostasis for many neurodegenerative diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Conrad C. Weihl (weihlc@wustl.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Data for figures in the paper is available in supplemental tables. The mass spectrometry proteomics data related to Figure 4 has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD026685 and 10.6019/PXD026685. Single nuclei RNA sequence data related to Figure 4 has been deposited in Gene expression omnibus (GEO) as GSE178257. RNA sequence analysis and phenotypic data of patients and brain samples of patients with dementia that is related to Figures 7 and S7 and Table S4 has been deposited in Synapse portal (handle syn25882533) at https://www.synapse.org/#!Synapse:syn25882533/. Notably, one of the 26 subjects included the study did not consent to share data, and by Human Research Protection Office at Washington University cannot share data for that single subject. An archived version of record source code used in Figures 7 and S7 and Table S4 analysis is available at https://zenodo.org/record/4957857.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J wild-type mice and CaMKIIα-Cre mice were purchased from Jackson laboratory. VCP floxed mice which were generated by our laboratory as described previously (Arhzaouy et al., 2019), and Atg5 floxed mice were a gift from Noburu Mizushima (University of Tokyo). VCP and ATG5 floxed mice were crossed with CaMKIIα-Cre mice. Progeny of interest were VCPFL/FL:CamkCre. VCP-R155C mice were previously reported (Clemen et al., 2018) and were generated by crossed VCP cKO (VCPFL/FL:CamkCre) to heterozygous VCP-R155C (VCPRC/WT) knockin mice that expressed only the VCP-R155C (VCPRC/FL:CamkCre) in CaMKIIα expressing neurons. All genotypes were determined by PCR on genomic DNA. Primers used for genotyping are listed in Key resource table. Unless otherwise noted, all experiments were performed on mixed sex cohorts of VCPFL/FL:CamkCre at one month upto 4-months of age, VCPRC/FL:CamkCre at 6 month upto 12 months of age. All mice were housed in a pathogen-free environment under controlled environmental conditions with 12 h light-dark cycles. Age and number of mice used for each experiment are detailed in figure legends. All the animal protocols used in this study were approved by the Institutional Animal Studies Committee at Washington University School of Medicine.
Human study subjects
In this work, we studied single-cell transcriptome profiles of parietal lobe tissue of patients with GRN (Age 71.41 ± 12.58), C9ORF872 (Age 74.74 ± 8.86) and unaffected control (Age 87.21 ± 10.82) of the post-mortem brain from The Charles F. and Joanne Knight Alzheimer Disease Research Center (Knight ADRC) brain bank was obtained with informed consent for research. Washington University in St. Louis institutional review board approved this study. The clinical status of participants was neuropathologically confirmed (Mirra et al., 1991). Clinical dementia rating (CDR) scores were obtained during regular visits throughout the study before the subject’s decease (Morris, 1997).
METHOD DETAILS
Tissue preparation of mouse brains
Mice were anesthetized with isoflurane and perfused with ice-cold PBS. Half brain was fixed overnight at 4°C in 4% paraformaldehyde, rinsed with PBS and incubated overnight at 4°C in 30% sucrose. The floating brains were transected coronally on a microtome with 40 μm brain section thickness into cryoprotectant (30% ethylene glycol, 0.2M PO4 buffer, 30% sucrose, water). Other half-brain was carefully dissected out and flash frozen for biochemical analysis.
Cresyl violet staining
The free-floating brain sections were washed 5 times with PBS and mounted on glass slides overnight at room temperature. The samples were rinsed with PBS and stained with 1% cresyl violet for 6 min at room temperature. The samples were washed 6 times with distilled water and dehydrated in ethanol in a row (70%, 95% and 100% for 4 min each) followed by xylene for 2 mins. The slides were coverslipped with cytoseal as a mounting media. Images were acquired using NanoZoomer 2.0-HT slide scanner. Images were processed and cortical width was quantified from stereological identical images of the cortex of the stained sections (3 slice per animal from 4 mice per group) using NDP.view2 (Hamamatsu), ImageJ version 2.0.0 (National Institutes of Health).
Fractionation of lysates and immunoblotting
The frozen brains were homogenized in radioimmunoprecipitation assay (RIPA) buffer containing 100 mM PMSF and 10X protease inhibitor cocktail. The samples were sonicated for 5 min and ultracentrifuged at 40,000 rcf. for 30 min at 4°C. The supernatant was taken as a RIPA soluble fraction, and the protein estimation was done by the bicinchoninic acid (BCA) method. The pellet was sonicated into RIPA buffer for 5 min and ultracentrifuged again for 30 min. Supernatant was discarded and the pellet was dissolved into 7 M urea followed by sonication. The 3X loading buffer (SDS, 1M Tris pH 8.0, glycerol, Bromophenol Blue, Water, β-mercaptoethanol) were added into each sample and denatured at 100°C for 10 min. The samples were subjected to SDS-PAGE and were transferred into nitrocellulose membrane for 2 h at 4°C. The protein membranes were blocked into 5% skimmed milk and were incubated with primary antibodies overnight at 4°C. After washing with PBST (PBS-5% tween 20), horseradish peroxidase (HRP) secondary antibodies were probed into each membrane for 1 h at room temperature. The proteins were detected by Amersham ECL chemiluminescent HRP substrate were visualized with G:Box Chemi XT4, Genesys version 1.1.2.0 (Syngene, Cambridge, UK). The quantification of western blots was done by ImageJ version 2.0.0 (National Institutes of Health).
Immunohistochemistry
Free floating brain sections from the cryoprotectant were washed 3 times with PBS and incubated with 0.3% hydrogen peroxide for 30 min at room temperature for endogenous quenching of peroxidases. The sections were washed 3 times with PBS for 15 min. The sections were permeabilized with 0.25% Triton X-100 for 15 minutes and washed with PBS for 10 min. Next, the samples were blocked in 5% normal goat serum in 0.1% Triton X-100 for 1 hour at room temperature. Primary antibodies were incubated into each section overnight at 4°C followed by PBS washing for 15 mins. The sections were further incubated with biotinylated secondary antibody for 2 hours at room temperature. Before 30 min of termination of the secondary antibody 1:250 ABC elite solution was added into each sample followed by PBS washing at room temperature. DAB (3,3′-Diaminobenzidine) mixture was added for 30 min into each sample for the detection of the antibody-bound complex. The reaction was stopped by adding distilled water and was further washed 3 times with PBS. Samples were further dried into the slides and dehydrated with an increasing concentration of ethanol (70%, 95%, 100% for 4 minute each) followed by xylene for 2 minutes. The slides were coverslipped with cytoseal as a mounting media. Images were acquired using NanoZoomer 2.0-HT slide scanner with 20x objective. Images were processed and quantified using NDP.view2 (Hamamatsu), ImageJ version 2.0.0 (National Institutes of Health). All acquisition settings were set equally for sections of all groups within each immunostaining. Images were quantified by measuring the mean intensity in defined region of interests (ROI).
TUNEL assay
TUNEL Staining were performed with Apoptag Fluorescein in situ apoptosis kit (Millipore, S7110) as per manufacturer instruction. Briefly, the brain sections were washed 3 times with PBS and mounted on glass slides overnight at room temperature. The samples were rinsed with PBS and fixed with 4% paraformaldehyde in PBS pH 7.4 for 10 min at room temperature. Each sample were washed 3 times with PBS for 5 min each wash and post-fix in pre-cooled ethanol: acetic acid in a ration of 2:1 for 5 min at −20°C. The samples were again washed 2 times with PBS followed by applying 75 μL/5 cm2 of equilibration buffer for 20 s at room temperature. Apply working strength TdT enzyme 75 μL/5 cm2 and incubate in a humidified chamber for 1 hour at 37°C. The reaction was stopped by incubating with working strength wash buffer for 10 min at room temperature. After washing 3 times with PBS for 1 min each, samples were incubated with 65 μL/5 cm2 of anti-digoxigenin conjugate for 30 min at room temperature followed with PBS wash for 4 times. The sections were mounted with mounting medium containing 1.0 μg/mL of DAPI. Images were acquired using NanoZoomer 2.0-HT slide scanner with 20x objective. Images were processed and quantified using NDP.view2 (Hamamatsu), ImageJ version 2.0.0 (National Institutes of Health). All acquisition settings were set equally for sections of all groups within each immunostaining. The Images were quantified by manually counting the number of TUNEL+ cells from stereological identical images of the CA1 region of the hippocampus and cortex of the stained sections (Two slices from 2 animals per group).
Immunofluorescence
Mice were anesthetized with isoflurane, perfused with PBS and the brains were fixed overnight at 4°C into 4% paraformaldehyde followed by incubation with 30% sucrose solution. The floating brains were transected coronally on a microtome with 40 μm brain section thickness into cryoprotectant (30% ethylene glycol, 0.2M PO4 buffer, 30% sucrose, water). The brain slices were washed extensively with tris buffer saline (TBS) and blocked with a blocking buffer containing 2% normal goat serum and 0.25% Triton X-100 in TBS. These sections were incubated with primary antibodies like TDP-43, LC3, SQSTM1 and LAMP1 overnight at 4°C followed by TBS washing and corresponding incubation with secondary antibody for 2 hours in dark at room temperature. After 3 washes with TBS, DAPI at a concentration of 1:1000 were added into each sample for staining the nuclei. The brain slices were further mounted into the slides and were incubated True black (70% in ethanol) for 5 min at room temperature followed by TBS washing until the excessive true black will be removed. The samples were dried and covered with coverslips using Prolong Gold Anti-Fade mounting media. All the images were captured with Olympus Flu and Nikon microscope with FV10-ASW 4.2 viewer and NIS-Elements Viewer 4.20 software respectively.
Synaptic Evaluation and Quantification by Imaging of Nanostructure (SEQUIN)
Tissue was processed, imaged, and analyzed for SEQUIN as described (Sauerbeck et al., 2020). Briefly, 50μm free floating tissue sections were rinsed in PBS (Coring Cat# 46-013-CM) and blocked for one hour in 20% normal goat serum (NGS, Vector Cat# S-1000, RRID:AB_2336615) in PBS. Sections were stained in 10% NGS/0.3% Triton X-100 in PBS overnight at room temperature with primary antibodies against synapsin 1/2 (1:1000, Synaptic Systems Cat# 106004 Lot# 2-27; RRID: AB_1106784) and PSD-95 (1:200, Invitrogen Cat# 51-6900; RRID: AB_2533914). Sections were then rinsed in PBS and incubated with Goat anti-Guinea Pig IgG-Alexa 488 (1:200, Invitrogen Cat# A11073, RRID:AB_2534117) and Goat anti-Rabbit IgG-Alexa 594 (1:200, Invitrogen Cat# A11037; RRID: AB_2534095) for four hours at room temperature using the same labeling buffer. Sections were counterstained with DAPI, rinsed in PBS, and mounted on charged slides. Sections were allowed to adhere to slides by drying, briefly rinsed in distilled water, dried again, and coverslipped with a 9:1 mixture of Mowiol 488 (Electron Microscopy Services Cat# 17977-150) to Antifade 300 (Electron Microscopy Services Cat# 17977-25) and 1.5 H coverglass (Marienfeld Cat# 0107242). After seven days of mountant curing, sections were imaged on Zeiss LSM 880 Fast Airyscan microscope (Zeiss, Oberkochen, Germany) with a 1.6NA 63x Oil Objective (Zeiss, Cat# 420782-9900-000). Four fields were acquired in the hippocampal CA1 stratum radiatum on both the left and right sides of each animal. Images were Airyscan Processed with Strength setting 6. Pre- and postsynaptic puncta were detected using Imaris 9.5 (Bitplane RRID:SCR_007370) with a spot size of 0.2 μm XY x 0.6 μm Z. SEQUIN analysis was performed using custom scripts (https://github.com/KummerLab/SEQUIN) isolating the top 20% brightest spots for each marker and quantifying the number of PSD-synapsin puncta pairs within 400 nm after performing reflect subtraction to remove noise (Sauerbeck et al., 2020).
Behavior tests
Behavior tests and analyses were conducted through the Animal Behavior Core at Washington University. Two separate cohorts of male and female VCPfl/fl CaMKIIα-Cre+ and Cre− (control) littermate mice were used for phenotypical analysis. The first cohort (n = 5 per genotype) were tested first in a sensorimotor battery, 1 hour locomotor activity, water maze, novel object and novel location recognition beginning at 1 month of age. Locomotor activity tests were repeated at 2 and 3 months of age. In a second cohort (n = 5 per genotype) was evaluated on the same behavioral tests, in the same order as the first. All behavior was conducted during the light phase by a female experimenter blind to the genotypes of each mouse. Mice were moved to the behavior room and allowed to acclimate for at least 30 minutes before testing began each day. All equipment was cleaned with 2% chlorhexidine diacetate or 70% ethanol between animals.
One-hour locomotor activity and open-field behavior test
To evaluate general activity levels and possible alterations in emotionality, mice in cohorts 1 and 2 were evaluated over a 1-h period in transparent (47.6 × 25.4 × 20.6 cm high) polystyrene enclosures. Each cage was surrounded by a frame containing a 4 × 8 matrix of photocell pairs, the output of which was fed to an on-line computer (Hamilton-Kinder, LLC, Poway, CA). The system software (Hamilton-Kinder, LLC) was used to define a 33 × 11 cm central zone and a peripheral or surrounding zone that was 5.5 cm wide with the sides of the cage being the outermost boundary. This peripheral area extended along the entire perimeter of the cage. Variables that were analyzed included the total number of ambulations and rearing on hindlimbs, as well as the number of entries, the time spent, and the distance traveled in the center area as well as the distance traveled in the periphery surrounding the center.
Sensorimotor battery
Walking initiation, ledge, platform, pole, and inclined and inverted screen tests were performed as previously described (Wozniak et al., 2004). Time in each task was manually recorded. The average for two trials was used for analyses. Test duration was 60 s, except for the pole test, which was extended to 120 s. For walking initiation, time for an animal to leave a 21x21cm square on a flat surface was recorded. For ledge and platform tests, the time the animal was able to balance on an acrylic ledge (0.75cm wide and 30cm high), and on a wooden platform (1.0cm thick, 3.0cm in diameter and elevated 47cm) was recorded, respectively. The pole test was used to evaluate fine motor coordination by quantifying time to turn 180° and climb down a vertical pole. The screen tests assessed a combination of coordination and strength by quantifying time to climb up or hang onto a mesh wire grid measuring 16 squares per 10cm, elevated 47cm and inclined (60° or 90°) or inverted.
Morris water maze (MWM)
MWM was conducted as previously described (Wozniak et al., 2004). Briefly, cued, place and probe trials were conducted in a galvanized steel pool, measuring 120cm in diameter, and filled with opaque water (diluted nontoxic white tempera paint). The PVC escape platform measured 11.5cm in diameter. A digital video camera connected to a PC computer and the computer software program ANY-maze (Stoelting Co., Wood Dale, IL) tracked the swimming pathway of the mouse to the escape platform and quantified path length, latency to find escape platform, and swimming speeds. On two consecutive days, animals received four cued trials, separated by 1h, during which a red tennis ball atop a rod was attached to the escape platform and served as a visual cue. To prevent spatial learning, the escape platform was moved to a different quadrant location for each trial. The mouse was released from the quadrant opposite to the platform location and allowed 60 s to locate the platform. Once the mouse found the platform, it was allowed to remain there for 10 s before being returned to its home cage. Three days following visible platform testing, the cue was removed from the platform and it was submerged 1cm under the water for the hidden platform tests. Animals received two blocks of two consecutive trials on five consecutive days, with an inter-trial interval between 30-90 s and approximately 2h separating trial blocks. The escape platform remained in the same quadrant location for all trials and distal cues were placed on the walls of the room to support spatial learning. The mouse was released from a different location for each trial on each day. The mouse was allowed 60 s to find the escape platform and allowed to sit on it for 10 s before being returned to its home cage. Cued and hidden platform trials were combined into blocks of two or four trials for analyses, respectively. Following completion of hidden platform trials on the 5th day of training, the escape platform was removed from the pool and one 60 s probe trial was conducted to assess memory retention for the location of the platform.
Proteomic sample preparation
Mouse cortex samples were lysed using the lysis buffer from Thermo EasyPep Mini MS Sample Prep Kit (REF A4006). The Bradford (cat# 5000006) assay were used to determine protein concentration, and 100 μg of proteins were digested and desalted by following the instruction of the same MS Prep Kit. Then the samples were resuspended in 0.1% FA solution, and the peptide concentrations were measured through Pierce Quantitative Fluorometric Peptide Assay (cat# 23290).
Mass spectrometry analysis
The LC-MS/MS experiments were performed using an EASY-nLC 1000 (ThermoFisher Scientific, San Jose, CA) connected to an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA). 0.5 μg sample was loaded onto an Aurora UHPLC Columns (25 cm x 75 μm, 1.6 μm C18, AUR2-25075C18A, Ion Opticks) and separated over 136 min at a flow rate of 0.35 μL/min with the following gradient: 2%–6% Solvent B (7.5 min), 6%–25% B (82.5 min), 25%–40% B (30 min), 40%–98% B (1 min), and 98% B (15 min). Solvent A consisted of 97.9% H2O, 2% ACN, and 0.1% formic acid, and solvent B consisted of 19.9% H2O, 80% ACN, and 0.1% formic acid. MS1 scan was acquired in the Orbitrap at 120,000 resolution with a scan range of 350-1500 m/z, the AGC target was 4 × 105, and the maximum injection time was 50 ms. Dynamic exclusion was set to exclude features after 1 time for 60 s with a 10 ppm mass tolerance. Higher-energy collisional dissociation (HCD) fragmentation was performed with 35% collision energy after quadruple isolation of features using 1.6 m/z isolation window, 1 × 105 AGC target, and a 35 ms maximum injection time. MS2 scans were then acquired by the IonTrap. Ion source settings were as follows: ion source type, NSI; spray voltage, 2400 V; ion transfer tube temperature, 275°C. System control and data collection were performed by Xcalibur software.
Proteomics data analysis
The proteomic data processing was performed through Proteome Discoverer 2.4 (Thermo Scientific) using Uniprot mouse database and the Byonic search algorithm (Protein Metrics). The search allowed for a precursor mass tolerance of 10 ppm, a minimum peptide length of 6, and a minimum peptide sequences number of 1. Carbamidomethyl (C) was set as fixed modification; oxidation (M) was set as dynamic common modification; acetyl (protein N-term), Met-loss (Protein N-term M) and Met-loss+acetyl (Protein N-term M) were set as dynamic rare modifications. Percolator FDRs were set at 0.001 (strict) and 0.01 (relaxed). Normalization was performed on the total peptide amount. Raw data is included Table S2.
Single nuclei sample preparation
Frozen cortex was isolated from three mice per experimental group. Tissue was homogenized using a dounce homogenizer in lysis buffer (10 mM Tris-HCl, pH = 7.4; 10 mM NaCl, 3 mM MgCl2; 0.005% NP40; and 0.2 U/μl RNase Inhibitor in nuclease free water at 4°C) and incubated on ice for 15 min. A 30 μm MACS SmartStrainer was used to remove cell debris and large clumps, followed by centrifugation at 500 x g for 5 min at 4°C. After carefully removing the supernatant, the nuclei pellet was resuspended with Nuclei wash and resuspension buffer (1% BSA and 0.2 U/μl RNase Inhibitor in 1 X PBS). Then cell debris removal step, centrifugation, and resuspension were repeated 2 times. Only 500 μl of Nuclei wash and resuspension buffer was added into the last resuspension step. This solution was mixed with 900 μl of Sucrose Cushion Buffer I (2.7 mL Nuclei Pure 2M Sucrose Cushion Solution with 300 μl Nuclei Pure Sucrose Cushion Solution and then carefully layered to the top of 500 μl Sucrose Cushion Buffer I in a 2 mL Eppendorf tube. This sucrose gradient was centrifuged at 13,000 x g for 45 min at 4°C. After centrifugation, the nuclear pellet was resuspended by 500 μl Nuclei Wash and Resuspension Buffer. The nuclei concentration was determined using a Countess with pre-DAPI stain. Finally nuclei concentration was adjust to ~1200 nuclei/μl using Nuclei Wash and Resuspension Buffer followed by proceeding to the 10 x Genomics protocol.
Single-nucleus RNA sequencing
Isolated nuclei were processed for droplet-based 3′ end massively parallel single-cell RNA sequencing using Chromium Single Cell 3′ Reagent Kits (10x Genomics) following manufacturer’s instructions. The libraries were sequenced at the Genome Technology Access Center (GTAC) using an Illumina HiSeq3000 (Illumina). Sample demultiplexing, barcode processing, and single-cell 3′ counting was performed using the Cell Ranger Single-Cell Software Suite (10x Genomics). Cellranger count was used to align samples to a pre-mRNA reference package (mm10), quantify reads, and filter reads with a quality score below 30.
Processing data with Seurat package
The Seurat v3.1 R package was used for subsequent analysis (Butler et al., 2018). Nuclei with mitochondrial content > 5% or total UMI < 200 or > 5000 were removed in Seurat. For each group, the % of mitochondrial genome genes was regressed out as a nuisance variable, gene counts were normalized and variable features identified using the SCTransform function in Seurat. The top 3000 variable genes were used to integrate experimental groups using the PrepSCTIntegration, FindIntegrationAnchors, and IntegrateData commands in Seurat. Principal component analysis (PCA) was performed on the integrated dataset and the first 20 PCs were selected for downstream analysis using FindNeighbors. Clusters were identified using the FindClusters function with a granularity ranging from 0.1 to 1.2. Final clustering was performed using a resolution of 0.6. The first 20 PCs were passed into UMAP using the RunUMAP command with default parameters. Differential gene expression between each cell cluster and all other clusters was performed on SCT data to identify marker genes for each individual cell cluster. Clusters containing high mitochondrial-genome content or marker genes for more than one broad cell type (i.e., microglia and excitatory neurons) were removed and data reclustered using the first 20 PCs and a resolution of 0.6. In total, 160,127 nuclei with a median UMI of 3194 and median gene number of 1716 across all 3 experimental groups were used in the final analysis. Differential gene expression to identify marker genes was again performed using a Wilcoxan Rank-Sum test and broad cell types identified based on known cell-type specific markers. Differential gene expression analysis based on cluster was performed between one-month-old or two-month-old and controls groups for each cluster using a Wilcoxan Rank-Sum test.
Pathway enrichment analysis
For pathway analysis, molecules from the dataset that are associated with Ingenuity’s Knowledge Base are considered for the analysis. The significance of the association and between the dataset and the specific pathways of interest is determined in two ways: i) Fisher’s exact test is used to calculate a p value determining the probability that the association between the genes in the dataset and the pathway of interest can be explained by chance alone (p value) and ii) z-score algorithm to identify biological functions that are expected to be more active or less active in the experimental dataset versus control dataset (positive or negative z-score) according to our proteomic or RNaseq data. In order to enhance the stringency of our analysis, we considered only functions with a z-score > 2 or < −2.
Human brain sample RNA sequencing
Parietal lobe tissue of the post-mortem brain from The Charles F. and Joanne Knight Alzheimer Disease Research Center (Knight ADRC) brain bank was obtained with informed consent for research. A range of other pathological measurements were collected during the autopsy, as previously described (Braak and Braak, 1995). Table S1 summarizes the demographic characteristics of the samples included in this study.
RNA was extracted from frozen brain tissues using Tissue Lyser LT and RNeasy Mini Kit (QIAGEN, Hilden, Germany) following the manufacturer’s instruction. RIN (RNA integrity) and DV200 were measured with RNA 6000 Pico Assay using Bioanalyzer 2100 (Agilent Technologies). Bioanalyzer determined the using the entire electrophoretic trace of the RNA, including the presence or absence of degradation products. The DV200 value is defined as the percentage of nucleotides greater than 200nt. RIN and DV200 for all the samples can be found in Table S1. Each sample’s yield was determined by the Quant-iT RNA Assay (Life Technologies) on the Qubit Fluorimeter (Fisher Scientific). The cDNA library was prepared with the TruSeq Stranded Total RNA Sample Prep with Ribo-Zero Gold kit (Illumina) and then sequenced by HiSeq 4000 (Illumina) using 2 × 150 paired-end reads at McDonnell Genome Institute, Washington University in St. Louis with a mean of 58.14 ± 8.62 million reads. The number of reads and other QC metrics can be found in Table S1. Raw sequening data is deposited at https://www.synapse.org/#!Synapse:syn25882533/.
RNA-seq QC, alignment, and gene expression quantification
FastQC was initially used to perform a quality check on various aspects of RNA-seq libraries . Sequences were aligned to human GRCh37 primary assembly using STAR (version 2.5.2b) (Dobin et al., 2013). We used the primary assembly and aligned reads to the assembled chromosomes, un-localized and unplaced scaffolds, and discarded alternative haploid sequences. Sequencing metrics, including coverage, distribution of reads in the genome, ribosomal and mitochondrial contents, and alignment quality, were further obtained by applying Picard CollectRnaSeqMetrics (version 2.8.2) to detect sample deviation. Additional QC metrics can be found in Table S1. Aligned and sorted bam files were loaded into IGV (Robinson et al., 2011) to perform a visual inspection of target variants. In addition, variants were called from RNA-seq following BWA/GATK pipeline (Li and Durbin, 2009; McKenna et al., 2010). The samples’ identity was later verified by performing IBD analysis against genomic typing from GWAS chipsets and/or whole genome/exome sequences as reported elsewhere (Dube et al., 2019). C9orf72 repeat expansion repeat quantification is described in detail in (Harms et al., 2013).
We applied Salmon transcript expression quantification (version 0.7.2) (Patro et al., 2017) to infer the gene expression. We quantified the coding transcripts of Homo sapiens included in the GENCODE reference genome (GRCh37.75).
Differential expression
Differential expression (DE) analysis was performed to compare subject carriers of the pathogenic variant 9AD in GRN (n = 5), repeat expansion C9orf72 (n = 5) compare to neuropathology-free controls subjects (n = 16) using DESeq2 version 1.22.2 (Love et al., 2014) to test read counts quantified by the Salmon method. The statistical model corrected from confounding factors, including RNA integrity number (RIN), gender, and age of death (AOD) as covariates. We also performed a t test to determine whether the AOD was significantly different between all subject carriers (n = 21) and the control subjects. Given the significant difference of the AOD (p = 1.3 × 10−04), we evaluated a second model in which AOD was not included. All genes with a p value < 0.05 were considered as differentially expressed genes and used in downstream analysis.
Identify common genes between VCP cKO and transcriptomic profiles from patients with FTD
We evaluated whether the transcriptome of subjects with FTD carrying a pathogenic variant in GRN and the repeat expansion C9orf72 recapitulated the one- and two-month-old VCP cKO proteomic and neuronal signatures. To do so, we determined the extent of overlap among the DEG and DEP from the VCP cKO and the differentially expressed genes between GRN/C9orf72 carriers compare to controls. We employed the hypergeometric distribution to perform a Fisher’s exact test and compute the significance of the overlap between the data of each time point and mice omics versus human genetic strata. In total, we performed eight tests (for example, we compared mice VCP cKO proteomics collected at one month versus genes DE in GRN carriers). Scatterplots and heatmaps showing the effect size and expression profiles (Figures 7 and S7) were generated using R packages ggplot2 (version 3.3.2) and gplots (version 3.04). All source code used in this analysis is available at https://zenodo.org/record/4957857.
QUANTIFICATION AND STATISTICAL ANALYSIS
Please refer to the legend of the figures for sample size and statistical test performed. All statistical analyses were performed using Graphpad Prism software (v.8). The data were analyzed with one way ANOVA followed by Tukey’s (Figures 2B, 2D, 2F, 3B, 3D, 3F, 3G, 6D, 6F, 6H, 7G, S5B, S6B, S7B,S8E, S8F, S8G, S8H, S11E, and S11F) multiple comparisons test, t test (Figures 1F, 1G, 4F, 6B, 7A, 7B, S1C, S4B, S11G, and S11H). Prior to analyses, all data were screened for fit of distributions with assumptions underlying univariate analyses, which included the Shapiro-Wilk test and q-q plot investigations for normality. Means and standard errors were computed for each measure. Analysis of variance (ANOVA), including repeated-measures, and independent samples t tests were used to analyze behavioral data. With a statistically significant interaction between main factors, simple main effects were calculated to provide clarification of statistically significant between-subjects and within-subjects differences. Where appropriate, the Huynh-Feldt adjustment was used to protect against violations of sphericity, the Bonferroni correction was applied to multiple pairwise comparisons, and Tukey’s HSD method was used as the post hoc test. Statistical results were confirmed with two-tailed non-parametric testing, when available, for any datasets with violations of the univariate assumptions. Probability value for all analyses was p < 0.05, unless otherwise stated. P values can be found in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| VCP | Abcam | Cat# ab11433 |
| VCP | Fitzgerald | Cat# 10R-P104A |
| Pan-14-3-3 | Santa Cruz | Cat# Sc1657 |
| NEUN | CST | Cat# 12943S |
| Gfap | Abcam | Cat# Ab7260 |
| Iba1 | Walko Chem | Cat# 019-19741 |
| C1Q | Abcam | Cat# ab182451 |
| TDP-43 | Proteintech | Cat# 10782-2-AP |
| p-TDP-43 | BioLegend | Cat# 829901 |
| Ubiquitin | Abcam | Cat# ab134953 |
| SQSTM1/p62 | Sigma | Cat# P0067 |
| LAMP1 | BD Transduction Labs | Cat# 553792 |
| LC3 | Sigma | Cat# L7543 |
| LAMP2 | Thermo/Invitrogen | Cat# PA1-655 |
| p-IRE1 | Invitrogen | Cat# PA116927 |
| GAPDH | CST | Cat# 2118 |
| IRE1 | Abcam | Cat# ab37073 |
| HSP70 | Enzo | Cat# ADI-SPA-812 |
| PERK | CST | Cat# 3192 |
| BIP/Grp78 | BD Transduction Labs | Cat# 610979 |
| BIP/Grp78 | Millipore | Cat# 4392 |
| ATG5 | CST | Cat# 12994 |
| FK2 (mono & poly ubiquitin) | Enzo | Cat# pw8810-050 |
| TOM20 | Santa Cruz | Cat# sz1415 |
| PSD-95 | Invitrogen | Cat# 516900 |
| Synapsin 1/2 | Synaptic Systems | Cat# 10600 |
| PSD-95 | Invitrogen | Cat# 51-6900 |
| Bax | CST | Cat# 14796s |
| CHOP | CST | Cat# 2895S |
| LC3 | Nanotools | Cat# 0231-100 |
| Goat anti-Guinea Pig IgG-Alexa 488 | Invitrogen | Cat# A11073 |
| Goat anti-Rabbit IgG-Alexa 594 | Invitrogen | Cat# A11037 |
| Chemicals, peptides, and recombinant proteins | ||
| ProLong Diamond DAPI mounting media | Invitrogen | Cat# P36971 |
| Critical commercial assays | ||
| Thermo EasyPep Mini MS Sample Prep Kit | Thermo Fischer | REF A4006 |
| ApopTag® Fluorescein In Situ Apoptosis Detection Kit | Millipore | REF S7110 |
| Tissue Lyser LT and RNeasy Mini Kit | QIAGEN | N/A |
| Deposited data | ||
| Raw proteomic data related to Figure 4 and Table S2 | This Paper | Data are available via ProteomeXchange with identifier PXD026685 |
| Code for analysis of transcriptomic and proteomic overlay related to Figures 7 and S7 | This Paper | Code available via Github https://zenodo.org/record/4957857 |
| Raw single nuclei RNA sequence data from cVCP KO mice cortex related to Figure 4 and Table S3 | This paper | GEO: GSE178257 |
| RNA sequencing and phenotypic data of patients and brain samples from patients with dementia related to Figures 7 and S7 and Table S4 | This paper | Data deposited in Synapse portal (handle syn25882533) at https://www.synapse.org/#!Synapse:syn25882533/. |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Jackson Labs | Stock No: 000664 |
| CamKIIa-cre | Jackson Labs | Stock No: 005359 |
| B6;129S-Vcptm1Itl/J (VCP R155H knockin) | Jackson Labs | Stock No. 021968 |
| VCP R155C CaMKIIα-Cre | This Paper | N/A |
| ATG5fl/fl | University of Tokyo | Noburu Mizushima |
| VCPfl/fl | Washington University in saint louis | Chris Weihl |
| VCPfl/fl CaMKIIα-Cre | This paper | N/A |
| ATG5fl/fl CaMKIIα-Cre | This paper | N/A |
| Oligonucleotides | ||
| Primers for LoxP site #1 in VCP floxed mice Forward 5′-GGTCTTGTTGTAGAGCCCTGTTCTGTAG-3′ | This paper | N/A |
| Primers for LoxP site #1 in VCP floxed mice Reverse 3′-CCTGGGACAAGGACACCACGCTGTTATC-5′ | This paper | N/A |
| Primers for LoxP site #2 in VCP floxed mice Forward 5′-GTGAGGGGGAGCCAATCAAGCGAGAGG-3′ | This paper | N/A |
| Primers for LoxP site #2 in VCP floxed mice Reverse 3′-CAATATTCCCACCTCCACCTCCTAAGC-5′ | This paper | N/A |
| Primers for Cre Forward 5′-GCATTACCGGTCGATGCAACGAGTGATGAG-3′ | This paper | N/A |
| Primers for Cre Reverse 3′-GAGTGAACGAACCTGGTCGAAATCAGTGCG-5′ | This paper | N/A |
| Primers for VCP-R155C allele Forward 5′-CAGTTCTCATGCCTCTCTGAAGGATAATGT-3′ | This paper | N/A |
| Primers for VCP-R155C allele Reverse 3′-TCTACAACTTTGAACTCCACAGCACGC-5′ | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH open access | https://imagej.nih.gov/ij/ |
| NIS Elements Viewer | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| FV10-ASW 4.2 viewer | Olympus | https://www.olympus-lifescience.com/en/support/downloads/fv10i_vw_license/ |
| Seurat V3.1 R package | Butler et al., 2018 | |
| Ingenuity Pathway Analysis | QIAGEN | |
| GraphPad Prism 7.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| IMARIS 9.5 | Oxford Instruments | https://imaris.oxinst.com/versions/9-5 |
| Hamilton-Kinder | ||
| ANY-maze | Stoelting Co | https://www.any-maze.com |
| Xcalibur | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30965#/OPTON-30965 |
| Cell Ranger Single-Cell Software Suite | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| R packages ggplot2 (version 3.3.2) | N/A | |
| gplots (version 3.04) | N/A | |
Highlights.
Loss of VCP in cortical neurons leads to neurodegeneration resembling FTLD-TDP
Conditional expression of an MSP disease mutation similarly leads to FTLD-TDP pathology
MSP-associated VCP mutations are hypomorphic in cortical neurons
VCP LOF has an expression signature resembling GRN deficiency in patients with FTLD
ACKNOWLEDGMENTS
This work was supported by NIH grants K24AR073317 and R01AG031867 (to C.C.W.); I01BX005204 to (T.T.K.); R01AG044546, P01AG003991, P30AG066444, RF1AG053303, RF1AG058501, and U01AG058922 (to C.C.); R01AG057777 (to O.H.); R01NS118146 (to B.A.B.); and R01NS102279 (to T.F.C. and C.C.W.). O.H. is supported as an Archer Foundation Research Scientist.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109399.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, et al. (2010). Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol 177, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Obeidi E, Al-Tahan S, Surampalli A, Goyal N, Wang AK, Hermann A, Omizo M, Smith C, Mozaffar T, and Kimonis V (2018). Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin. Genet 93, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhzaouy K, Papadopoulos C, Schulze N, Pittman SK, Meyer H, and Weihl CC (2019). VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy 15, 1082–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. (2006). Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. [DOI] [PubMed] [Google Scholar]
- Blythe EE, Olson KC, Chau V, and Deshaies RJ (2017). Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. USA 114, E4380–E4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe EE, Gates SN, Deshaies RJ, and Martin A (2019). Multisystem proteinopathy mutations in VCP/p97 increase NPLOC4.UFD1L binding and substrate processing. Structure 27, 1820–1829.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, and Braak E (1995). Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–278, discussion 278–284. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MC, Srinivasan K, Friedman BA, Suto E, Modrusan Z, Lee WP, Kaminker JS, Hansen DV, and Sheng M (2017). Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med 214, 2611–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching JK, Elizabeth SV, Ju JS, Lusk C, Pittman SK, and Weihl CC (2013). mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum. Mol. Genet 22, 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemen CS, Marko M, Strucksberg KH, Behrens J, Wittig I, Gärtner L, Winter L, Chevessier F, Matthias J, Türk M, et al. (2015). VCP and PSMF1: Antagonistic regulators of proteasome activity. Biochem. Biophys. Res. Commun 463, 1210–1217. [DOI] [PubMed] [Google Scholar]
- Clemen CS, Winter L, Strucksberg KH, Berwanger C, Türk M, Kornblum C, Florin A, Aguilar-Pimentel JA, Amarie OV, Becker L, et al. (2018). The heterozygous R155C VCP mutation: Toxic in humans! Harmless in mice? Biochem. Biophys. Res. Commun 503, 2770–2777. [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, et al. (2006). Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. [DOI] [PubMed] [Google Scholar]
- Custer SK, Neumann M, Lu H, Wright AC, and Taylor JP (2010).Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet 19, 1741–1755. [DOI] [PubMed] [Google Scholar]
- Darwich NF, Phan JM, Kim B, Suh E, Papatriantafyllou JD, Changolkar L, Nguyen AT, O’Rourke CM, He Z, Porta S, et al. (2020).Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science 370, eaay8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ridder W, Azmi A, Clemen CS, Eichinger L, Hofmann A, Schröder R, Johnson K, Töpf A, Straub V, De Jonghe P, et al. (2020). Multisystem proteinopathy due to a homozygous p.Arg159His VCP mutation: A tale of the unexpected. Neurology 94, e785–e796. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube U, Del-Aguila JL, Li Z, Budde JP, Jiang S, Hsu S, Ibanez L, Fernandez MV, Farias F, Norton J, et al. ; Dominantly Inherited Alzheimer Network (DIAN) (2019). An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci 22, 1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia LP, Mason AR, Alijagic A, and Finkbeiner S (2019). Genetic regulation of neuronal progranulin reveals a critical role for the autophagy-lysosome pathway. J. Neurosci 39, 3332–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falik Zaccai TC, Savitzki D, Zivony-Elboum Y, Vilboux T, Fitts EC, Shoval Y, Kalfon L, Samra N, Keren Z, Gross B, et al. (2017). Phospholipase A2-activating protein is associated with a novel form of leukoencephalopathy. Brain 140, 370–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, et al. (2006). Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J. Neuropathol. Exp. Neurol 65, 571–581. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, Dearborn JT, Wozniak DF, and Cairns NJ (2012). Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol. Dis 45, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MA, Feely SM, Speziani F, Strickland AV, Danzi M, Bacon C, Lee Y, Chou TF, Blanton SH, Weihl CC, et al. (2014). A novel mutation in VCP causes Charcot-Marie-Tooth type 2 disease. Brain 137, 2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halawani D, LeBlanc AC, Rouiller I, Michnick SW, Servant MJ, and Latterich M (2009). Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol. Cell. Biol 29, 4484–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall EA, Nahorski MS, Murray LM, Shaheen R, Perkins E, Dissanayake KN, Kristaryanto Y, Jones RA, Vogt J, Rivagorda M, et al. (2017). PLAA mutations cause a lethal infantile epileptic encephalopathy by disrupting ubiquitin-mediated endolysosomal degradation of synaptic proteins. Am. J. Hum. Genet 100, 706–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M, Benitez BA, Cairns N, Cooper B, Cooper P, Mayo K, Carrell D, Faber K, Williamson J, Bird T, et al. ; NIA-LOAD/NCRAD Family Study Consortium (2013). C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol. 70, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, and Strittmatter SM (2010). Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huin V, Barbier M, Bottani A, Lobrinus JA, Clot F, Lamari F, Chat L, Rucheton B, Fluchère F, Auvin S, et al. (2020). Homozygous GRN mutations: New phenotypes and new insights into pathological and molecular mechanisms. Brain 143, 303–319. [DOI] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ, and Lee VM (2011). Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest 121, 726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, Komatsu M, and Abeliovich A (2012). Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol. Neurodegener 7, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, and Weihl CC (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol 187, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaleem M, Zhao A, Hamshere M, and Myers AJ (2007). Identification of a novel valosin-containing protein polymorphism in late-onset Alzheimer’s disease. Neurodegener. Dis 4, 376–381. [DOI] [PubMed] [Google Scholar]
- Kushimura Y, Tokuda T, Azuma Y, Yamamoto I, Mizuta I, Mizuno T, Nakagawa M, Ueyama M, Nagai Y, Yoshida H, and Yamaguchi M (2018). Overexpression of ter94, Drosophila VCP, improves motor neuron degeneration induced by knockdown of TBPH, Drosophila TDP-43. Am. J. Neurodegener. Dis 7, 11–31. [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, and Weihl CC (2014). The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci 127, 3877–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, and Berg L (1991). The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- Morris JC (1997). Clinical dementia rating: A reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int. Psychogeriatr 9 (Suppl 1), 173–176, discussion 177–178. [DOI] [PubMed] [Google Scholar]
- Müller JM, Deinhardt K, Rosewell I, Warren G, and Shima DT (2007). Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem. Biophys. Res. Commun 354, 459–465. [DOI] [PubMed] [Google Scholar]
- Nalbandian A, Llewellyn KJ, Kitazawa M, Yin HZ, Badadani M, Khanlou N, Edwards R, Nguyen C, Mukherjee J, Mozaffar T, et al. (2012). The homozygote VCPR155H/R155H mouse model exhibits accelerated human VCP-associated disease pathology. PLoS ONE 7, e46308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A, Llewellyn KJ, Badadani M, Yin HZ, Nguyen C, Katheria V, Watts G, Mukherjee J, Vesa J, Caiozzo V, et al. (2013). A progressive translational mouse model of human valosin-containing protein disease: The VCPR155H/+ mouse. Muscle Nerve 47, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, and Mackenzie IRA (2019). Review: Neuropathology of nontau frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol 45, 19–40. [DOI] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, and Forman MS (2007). TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol 66, 152–157. [DOI] [PubMed] [Google Scholar]
- Niwa H, Ewens CA, Tsang C, Yeung HO, Zhang X, and Freemont PS (2012). The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J. Biol. Chem 287, 8561–8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke JG, Bogdanik L, Muhammad AKMG, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, et al. (2015). C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 88, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, et al. (2017). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, et al. (2015). Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88, 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, Lee EB, Trojanowski JQ, and Lee VM (2018). Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun 9, 4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz D, Vuk M, Kirchner P, Bug M, Schütz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, et al. (2011). Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat. Cell Biol 13,1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ortiz CJ, Hoshino H, Cheng D, Liu-Yescevitz L, Blurton-Jones M, Wolozin B, LaFerla FM, and Kitazawa M (2013). Neuronal-specific overexpression of a mutant valosin-containing protein associated with IBMPFD promotes aberrant ubiquitin and TDP-43 accumulation and cognitive dysfunction in transgenic mice. Am. J. Pathol 183, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf S, Lee SB, Jan LY, and Jan YN (2011). Neuronal remodeling and apoptosis require VCP-dependent degradation of the apoptosis inhibitor DIAP1. Development 138, 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbeck AD, Gangolli M, Reitz SJ, Salyards MH, Kim SH, Hemingway C, Gratuze M, Makkapati T, Kerschensteiner M, Holtzman DM, et al. (2020). SEQUIN multiscale imaging of mammalian central synapses reveals loss of synaptic connectivity resulting from diffuse traumatic brain injury. Neuron 107, 257–273.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamirian S, Nalbandian A, Khare M, Castellani R, Kim R, and Kimonis VE (2015). Early-onset Alzheimers and cortical vision impairment in a woman with valosin-containing protein disease associated with 2 APOEε4/APOEε4 genotype. Alzheimer Dis. Assoc. Disord 29, 90–93. [DOI] [PubMed] [Google Scholar]
- Shih YT, and Hsueh YP (2016). VCP and ATL1 regulate endoplasmic reticulum and protein synthesis for dendritic spine formation. Nat. Commun 7, 11020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih YT, Huang TN, Hu HT, Yen TL, and Hsueh YP (2020). Vcp overexpression and leucine supplementation increase protein synthesis and improve fear memory and social interaction of Nf1 mutant mice. Cell Rep. 31, 107835. [DOI] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, et al. (2012). Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet 90, 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Mitchell JC, Salcher-Konrad MT, Vance CA, and Mizielinska S (2019). Review: Modelling the pathology and behavior of frontotemporal dementia. Neuropathol. Appl. Neurobiol 45, 58–80. [DOI] [PubMed] [Google Scholar]
- Spina S, Van Laar AD, Murrell JR, Hamilton RL, Kofler JK, Epperson F, Farlow MR, Lopez OL, Quinlan J, DeKosky ST, and Ghetti B (2013). Phenotypic variability in three families with valosin-containing protein mutation. Eur. J. Neurol 20, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, and Nishihara M (2014). Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol. Commun 2, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP (2015). Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology 85, 658–660. [DOI] [PubMed] [Google Scholar]
- Wang HF, Shih YT, Chen CY, Chao HW, Lee MJ, and Hsueh YP (2011). Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Invest 121, 4820–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Dalal S, Pestronk A, and Hanson PI (2006). Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet 15, 189–199. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Miller SE, Hanson PI, and Pestronk A (2007). Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum. Mol. Genet 16, 919–928. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, Hanson PI, Kimonis V, and Pestronk A (2008). TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 79, 1186–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Pestronk A, and Kimonis VE (2009). Valosin-containing protein disease: Inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul. Disord 19, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl CC, Baloh RH, Lee Y, Chou TF, Pittman SK, Lopate G, Allred P, Jockel-Balsarotti J, Pestronk A, and Harms MB (2015). Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul. Disord 25, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner G, Damme M, Schludi M, Gnörich J, Wind K, Fellerer K, Wefers B, Wurst W, Edbauer D, Brendel M, et al. (2020). Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep. 21,e50241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, Cuijt I, Joris G, De Deyn PP, Haass C, et al. (2012). Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J. Pathol 228, 67–76. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, and Muglia LJ (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis 17, 403–414. [DOI] [PubMed] [Google Scholar]
- Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, and Wang G (2016). TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 35, 121–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Okubo Y, Suzaki T, and Ogura T (2004). Analysis of the two p97/VCP/Cdc48p proteins of Caenorhabditis elegans and their suppression of polyglutamine-induced protein aggregation. J. Struct. Biol 146, 242–250. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Nalbandian A, Hsu CI, Li S, Llewellyn KJ, Mozaffar T, Kimonis VE, and Weiss JH (2012). Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 3, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Mishra P, Hay BA, Chan D, and Guo M (2017). Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. eLife 6, e17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N, Ma B, Stoll S, Hays TT, and Qiu H (2017). The valosin-containing protein is a novel repressor of cardiomyocyte hypertrophy induced by pressure overload. Aging Cell 16, 1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for figures in the paper is available in supplemental tables. The mass spectrometry proteomics data related to Figure 4 has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD026685 and 10.6019/PXD026685. Single nuclei RNA sequence data related to Figure 4 has been deposited in Gene expression omnibus (GEO) as GSE178257. RNA sequence analysis and phenotypic data of patients and brain samples of patients with dementia that is related to Figures 7 and S7 and Table S4 has been deposited in Synapse portal (handle syn25882533) at https://www.synapse.org/#!Synapse:syn25882533/. Notably, one of the 26 subjects included the study did not consent to share data, and by Human Research Protection Office at Washington University cannot share data for that single subject. An archived version of record source code used in Figures 7 and S7 and Table S4 analysis is available at https://zenodo.org/record/4957857.