

Abstract
Ynamides are among the most powerful building blocks in organic synthesis and have become invaluable starting materials for the construction of multifunctional compounds and challenging architectures that would be difficult to prepare otherwise. The rapidly growing popularity originates from the unique reactivity and ease of manipulation of the polarized ynamide triple bond, the advance of practical methods for making them, and the simplicity of storage and handling. These attractive features and the demonstration of numerous synthetic applications have spurred the development of intriguing asymmetric reaction strategies during the last decade. An impressive variety of chemo-, regio- and stereoselective carbon-carbon and carbon-heteroatom bond forming reactions with ynamides have been developed and now significantly enrich the toolbox of synthetic chemists. This review provides a comprehensive overview of asymmetric ynamide chemistry since 2010 with a focus on the general scope, current limitations, stereochemical reaction control and mechanistic aspects.
1. Introduction
The chemical properties and reactivity of ynamides are largely defined by the interaction between the triple bond and the adjacent electron-donating nitrogen atom. The distinctive polarity of the aminoalkyne functionality coincides with exceptional synthetic opportunities that extend far beyond traditional alkyne chemistry. The introduction of straightforward methods for the preparation of ynamines and ynamides together with a well-developed understanding of the diverse reactivity patterns of this unique class of compounds have set the stage for intriguing synthetic applications. Ynamines are considerably more reactive and difficult to handle than ynamides which carry a sulfonyl, alkoxycarbonyl, acyl or another electron-withdrawing group to regulate the polarization of the alkyne bond by the amine moiety (Figure 1). Ynamides therefore are more practical to work with and provide better reaction control which has been particularly advantageous in efforts aimed at asymmetric catalysis development.
Figure 1.
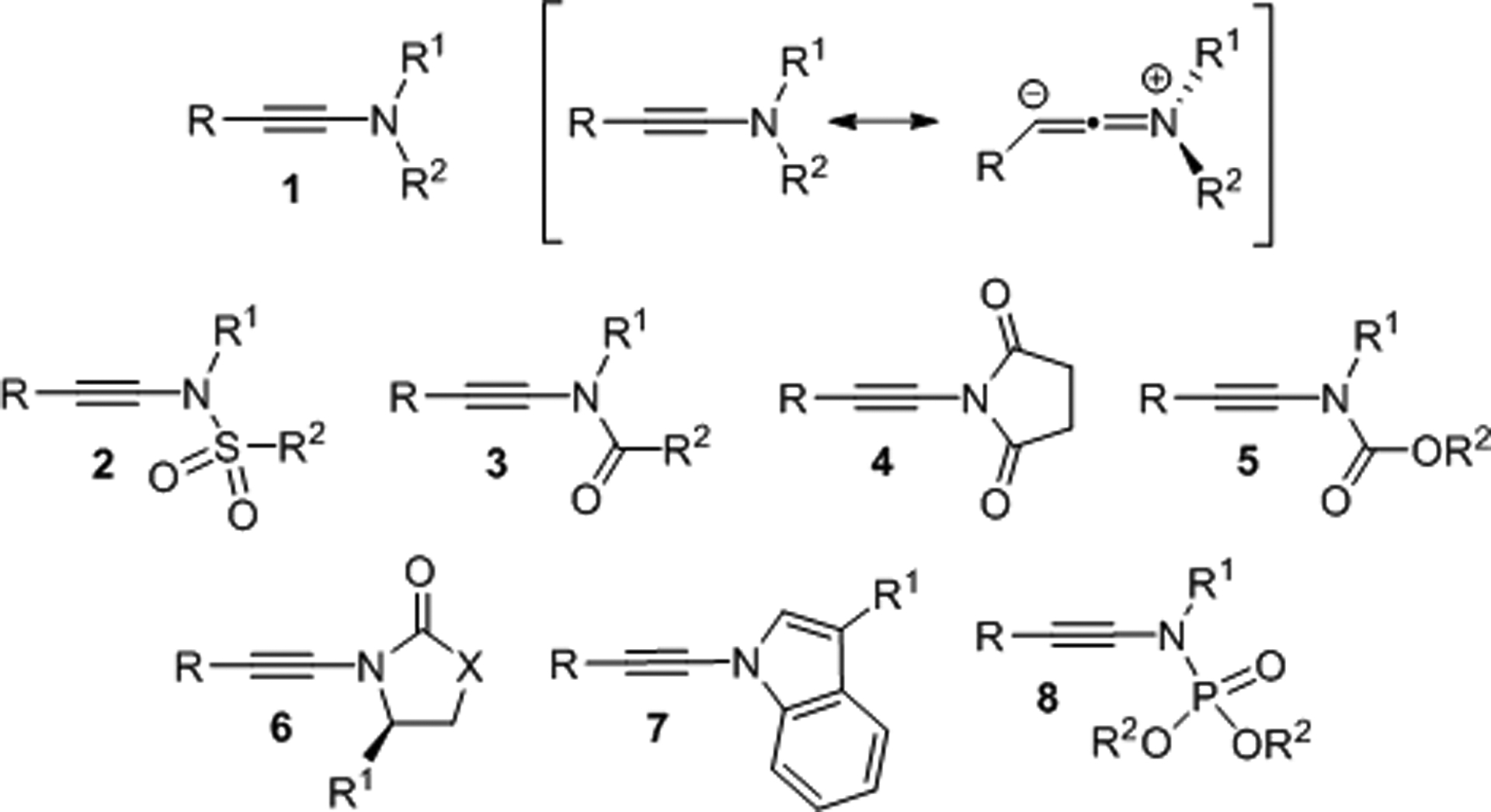
Structures and polarization of ynamines and ynamides.
The staggering diversity of reactions of ynamines and in particular of ynamides was covered with great detail in authoritative reviews by Hsung and Evano in 2010.1,2 Since then, this field has grown tremendously and remarkable progress with asymmetric methods that produce structurally diverse, multifunctional compounds often with excellent stereocontrol has been made. During the last ten years, a personal perspective and several excellent reviews focused on various selected ynamide topics have appeared in the literature.3,4 Four reports about cyclizations and cycloisomerizations collectively underscore the general importance of ynamides for these type of transformations.5–8 Hsung highlighted the properties and use of structural analogues of ynamides9 and Evano evaluated the anionic chemistry of aminoalkynes.10 In 2015, our group reviewed the synthesis and chemistry of terminal ynamides.11 Cariou examined the progress with the conversion of ynamides to ketenimines and the use of these building blocks for the production of nitrogen-containing scaffolds12 while others elaborated on specific applications in peptide synthesis13 or Ugi and Passerini reactions.14 Recently, Blanchard covered Diels-Alder cycloadditions15 and Kerwin discussed the chemistry of N-alkynyl azoles.16 Finally, Ye provided a very intriguing discussion of strategies that enable control of regioselectivity aspects of ynamide chemistry.17 To the best of our knowledge, a review that focuses on the immense utility of ynamides for the asymmetric synthesis of chiral compounds has not appeared to date.
This review covers the advance of enantioselective synthesis with ynamides during the last decade by providing a comprehensive overview that evaluates the general usefulness and promise of this rapidly emerging field (Figure 2). Both stoichiometric reactions and catalytic processes are discussed and, when available, key mechanistic insights are included to explain the success and sense of asymmetric induction. Considerable emphasis is generally laid on individual substrate scope and limitations by showing carefully selected examples to provide useful guidance for the interested reader. Reports describing the synthesis of cis/trans-enamides,18 achiral N-heterocyclic derivatives,19 E/Z-olefins,20 or nonstereoselective formation of chiral products21 are outside the scope of this review. To help the reader with navigating through the diversity of asymmetric ynamide transformations we classified the reactions into a few major categories: Aldol and Mannich Reactions, Cycloadditions, Rearrangements and Cycloisomerizations, Functionalization of Terminal Ynamides, and Miscellaneous Addition Reactions. We realize that this classification is not unambiguous and some of the transformations discussed herein are actually combinations of two or more than two of these reaction types. We hope, however, that the organization of this review provides the readers with a comprehensive overview of the impressive developments since 2010 and at the same time facilitates the search for specific aspects of asymmetric ynamide chemistry. The structures of frequently used protecting groups are summarized in Figure 3.
Figure 2.

Structural diversity of asymmetric synthesis with ynamides.
Figure 3.

Structures and abbreviations of protecting groups commonly used in this review.
2. Aldol and Mannich Reactions
Asymmetric aldol and Mannich reactions are among the most important transformations in organic chemistry and countless examples with all types of nucleophiles including terminal alkynes exist in the literature. During the past decade, remarkable progress that complements existing synthetic methodologies and conquers new chemical space has been made with internal ynamides. A wide variety of multifunctional products have been prepared through careful reaction development enabling C–C bond formation with high asymmetric induction while the ynamide moiety is transformed to a new functionality, typically an enamide or amide group. In some cases, this was accomplished with chiral ynamides but catalytic enantioselective methods that use achiral starting materials have been introduced as well. It is important to note that the use of terminal ynamides in aldol-type reactions allows asymmetric C–C bond formation without concomitant loss of the N-alkynyl motif, which is discussed separately in Section 5.
In 2010, the Walsh group presented a multi-step reaction sequence that furnishes chiral β-hydroxy (E)-enamines via hydroboration of N-tosyl ynamides 2 followed by boron-to-zinc transmetalation and morpholino isoborneol (MIB)-catalyzed enantioselective addition of the intermediate alkenylzinc species 10 to aldehydes 11 (Scheme 1).22 This procedure is applicable to aliphatic and aromatic aldehydes and provides efficient access to the enamides 12 with 67–86% yield and 54–98% ee. While N-tosylated ynamides gave excellent results trifluoroacetyl and imide analogues did not work. The introduction of a Simmons-Smith cyclopropanation step to essentially the same protocol generated a series of aminocyclopropyl carbinols 13 carrying three contiguous stereocenters (Scheme 2). These challenging building blocks were obtained with moderate to high yields, up to 94% ee and diastereomeric ratios greater than 20:1. The reaction was sluggish, however, with internal ynamides and cyclopropanation of the β-hydroxy enamides derived from aromatic aldehydes or aryl-substituted ynamides did not occur. The cheletropic reaction is believed to proceed via transition state A.
Scheme 1.

Asymmetric synthesis of β-hydroxy enamides.
Scheme 2.

Walsh’s stereoselective one-pot synthesis of aminocyclopropyl carbinols.
Chiral ynamides are also excellent starting materials for the synthesis of highly substituted aldol compounds.23 To achieve this task with oxazolidinone derived ketene N,O-acetals, Marek’s group explored two different pathways (Scheme 3). In path A, sequential addition of a Gilman cuprate, R22CuLi·SMe2, to the ynamide 6 followed by treatment with tert-butyl hydroperoxide and trimethylsilyl chloride gives the O-silyl ketene acetal 14. Alternatively, this can be achieved with the organocopper complexes, R2Cu·SMe2, and a similar work-up. Interestingly, it was found that the latter approach provides higher yields. The ketene N,O-acetals then react with aliphatic and aromatic aldehydes in the presence of titanium tetrachloride to the corresponding Mukaiyama aldol products 15 in good yields and with high diastereoselectivity. The stereochemical outcome is consistent with Zimmerman-Traxler transition states.
Scheme 3.

Marek’s one-pot preparation of ketene N,O-acetals and conversion to the corresponding aldol products.
A similar method for Mannich and aldol type reactions with aldehydes and sulfonylimines, respectively, was introduced by the same group (Scheme 4).24 This chemistry eliminates the silylation step and the final products 16 and 17 exhibiting two adjacent chiral centers were obtained in moderate yields but with high diastereomeric ratios. Alternatively, quenching of the reaction with an alcohol, amino alcohol or another protic additive at low temperature provides the α-substituted imides 18 and SN2 reactions with activated electrophiles is also possible as discussed in more detail in Section 6.25
Scheme 4.
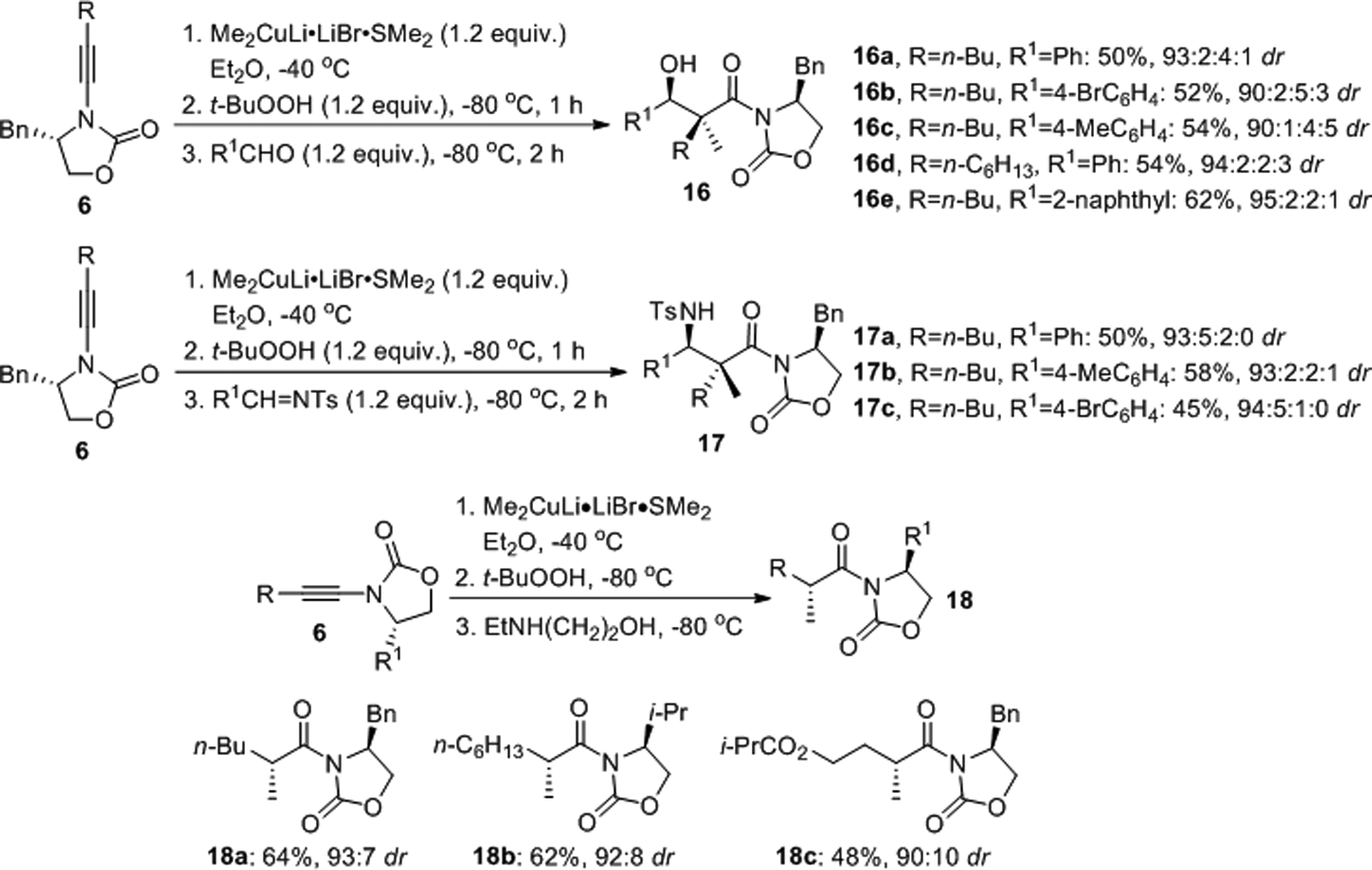
Diastereoselective formation of aldol and Mannich reaction products (top) and α-alkyl imides (bottom).
Gaunt and coworkers developed a scandium(III) catalyzed stereoselective anti-aldol reaction with aliphatic and aromatic aldehydes 11 (Scheme 5).26 The triphenylsilanol derived anti-aldol products 19 were produced in 43–80% yield and with dr values ranging from 67:33 to greater than 95:5. The authors also showed that these ynamides can undergo [3,3] rearrangements upon addition of allylic alcohols 20 under acidic conditions at room temperature, thus forming the α,β-disubstituted imides 21 in 54–97% yields and in some cases greater than 95:5 dr.
Scheme 5.

Acid catalyzed stereoselective C–C bond formation with aldehydes and allylic alcohols by Gaunt et al.
Diastereoselective nickel-catalyzed reductive coupling of chiral oxazolidinone-derived ynamides and aldehydes was reported by Sato et al.27 In the presence of 10 mol% of a nickel catalyst derived from SIMes·HBF4 L-1 and excess of triethylsilane, Et3SiH, the oxazolidinone-derived ynamides 6 reacted with aromatic and aliphatic aldehydes 11 to the γ-siloxyenamides 22 which were obtained in 73–98% yield and 66–99% de (Scheme 6). The authors rationalized that the rate-limiting step in the reductive coupling mechanism is the preferential formation of oxanickelacycle B via oxidative cycloaddition from the tetracoordinate complex A exhibiting minimized steric interactions between the NHC ligand and the approaching aldehyde. The reduction of B then results in the predominant formation of the homochiral diastereomer 22.
Scheme 6.

Sato’s asymmetric synthesis of γ-siloxyenamides by nickel catalyzed reductive coupling of ynamides and aldehydes.
Liu described a stereoselective hydrative aldol reaction between enynamides 2, water and aromatic aldehydes 23 that allows synthesis of the branched compounds 24 with tunable anti/syn-selectivity.28 In the presence of 5 mol% of zinc(II) triflate and 2 equivalents of water, the anti-aldol products were produced with diastereomeric ratios greater than 20:1 at room temperature (Scheme 7). The authors observed that incorporation of (S)-α-methylbenzyl into the N-sulfonamide moiety favors formation of syn-products such as 24d under the same reaction conditions. Lin and coworkers further extended this chemistry by carrying out a solvent-free scandium(III) triflate catalyzed aldol reaction with ketones 25 employed in considerable excess and obtained the diastereomerically pure anti-aldol products 26 .29
Scheme 7.
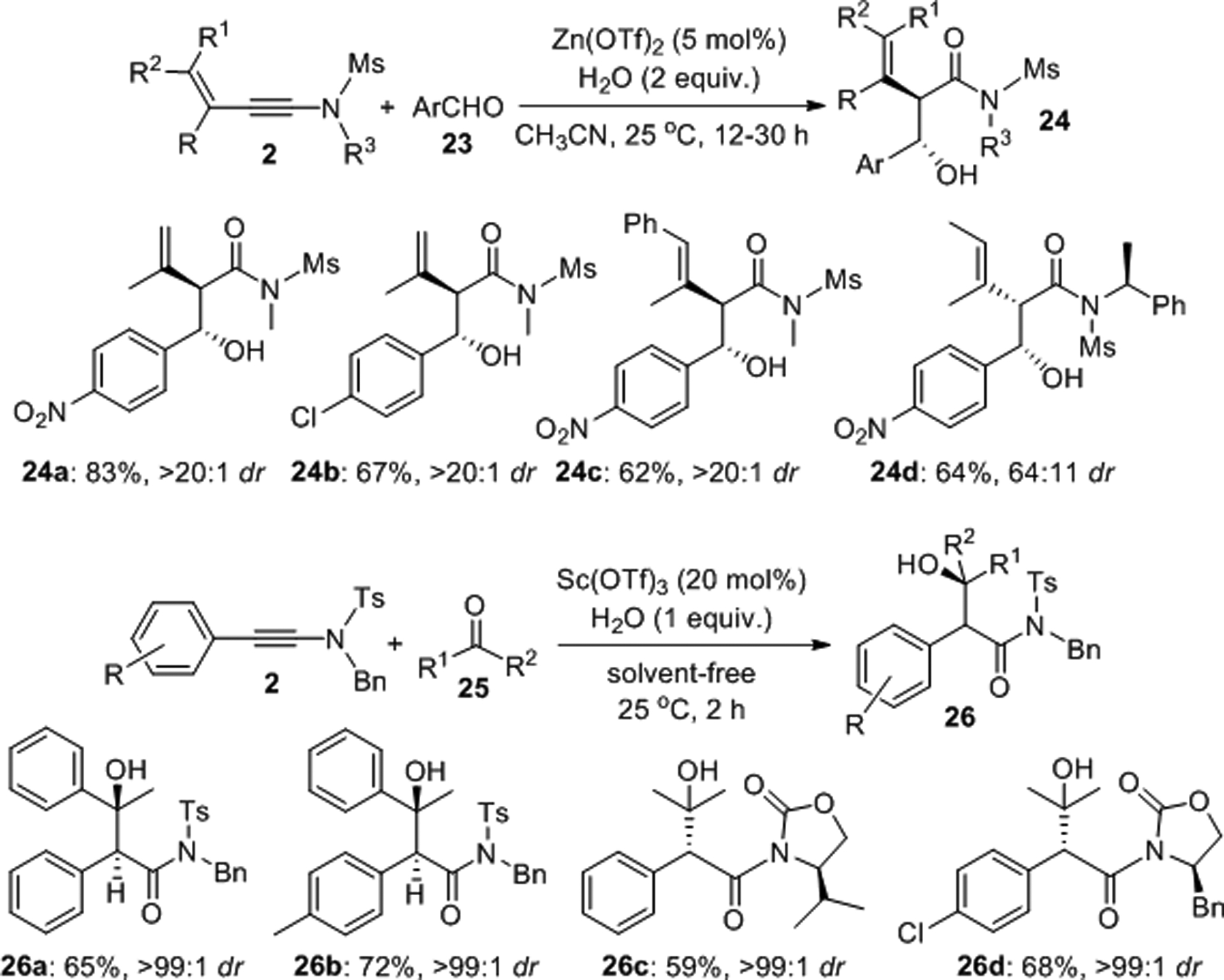
Diastereoselective hydrative aldol reactions with aldehydes and ketones.
3. Cycloadditions
Ynamides have been extremely useful substrates in a variety of cycloadditions that may be truly pericyclic in nature or following a stepwise, nonconcerted mechanism. The exceptional utility of these transformations is apparent from the diversity of challenging cyclic scaffolds that have been prepared with excellent regio- and stereocontrol, in particular via transition metal catalysis. In several cases, cycloadditions are exploited in tandem with isomerization or sigmatropic rearrangement pathways which underscores the unique reactivity and ease of functional group manipulation of ynamides.
Following Ficini’s seminal work with ynamines such as N,N-diethylprop-1-yn-1-amine, 1, in 1976,30 several asymmetric [2+2] cycloadditions with more easily controlled ynamide analogues and a variety of electron-deficient electrophiles have appeared in the literature (Scheme 8).31 In 2010, Hsung prepared cyclobutenamines 28 from sulfonynamides 2 and cyclohexenone, 27, using catalytic amounts of CuCl2 and AgSbF6 at 0 °C.32 This protocol was also applied to other α,β-unsaturated carbonyl compounds. Two years later, Lam extended this chemistry to rhodium catalyzed additions with trans-nitroalkenes 29.33 Liu employed cyclic isoimidium salts 31 in the Ficini reaction and obtained syn-cyclobutenamides 32 in high yields under catalyst-free conditions.34
Scheme 8.

Comparison of asymmetric [2+2] cycloadditions with ynamines and ynamides.
In 2011, Mikami examined catalytic asymmetric [2+2] cycloadditions of alkynes and ynamides with ethyltrifluoropyruvate (Scheme 9).35 They found that the dicationic (S)-BINAP derived palladium(II) complex Cat-2 catalyzes the formal cycloaddition between 2 and the ketoester 33 to oxetene 34 in quantitative yield and literally perfect enantiomeric excess. Despite this impressive result, the synthesis of other chiral analogues of 34 was not reported.
Scheme 9.

Chiral oxetene synthesis based on a formal [2+2] cycloaddition.
Hsung et al. presented a diastereoselective Staudinger-type [2+2] cycloaddition with the ketenimine A generated in situ via aza-Claisen rearangement from the N-phosphoryl ynamide 8 (Scheme 10).36 The reaction was observed upon heating to 125 °C and the transient cumulene A was successfully trapped with the aldimine 35 as the azetidin-2-imine 36 which was obtained in 73% yield and 4:1 dr.
Scheme 10.
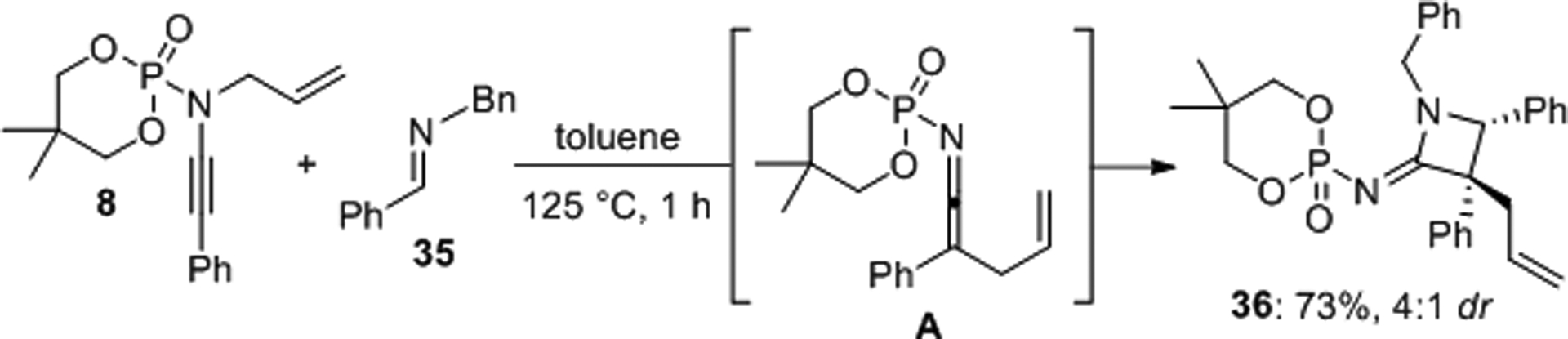
Staudinger-type ketenimine-imine [2+2] cycloaddition of an N-phosphoryl ynamide.
The same group discovered that palladium-catalyzed diastereoselective [2+2] cycloadditions of propargylic allyl ethers exhibiting 1,1- or 1,2-disubstituted alkene moieties affords structurally diverse bicyclic ketimines.37 The intramolecular cycloaddition coincides with an intriguing N-to-C allyl transfer and depending on the alkene geometry goes either through transition state A or B affording compounds 37 and 38, respectively (Scheme 11). The cycloadducts were produced in good yields and with high diastereoselectivities using 5 mol% of readily available tetrakis(triphenylphosphine)palladium(0) at 70 °C.
Scheme 11.

Diastereoselective [2+2] cycloadditions with propargylic allyl ethers.
Schotes and Mezzetti employed various sulfonynamides in the catalytic enantioselective Ficini reaction with tert-butyl 5-oxocyclopent-1-ene-1-carboxylate, 39.38–39,40,41 The stepwise [2+2] cycloaddition was achieved using catalytic amounts of the ruthenium/PNNP complex Cat-3 and excess of Et3O(PF6) at 55 °C in dichloromethane in a sealed tube, providing a series of the strained unsaturated [3.2.0] bicyclic structures 40 with a tetrasubstituted carbon center in good to high yields and enantioselectivities (Scheme 12). When the ethyl ester derivative of 40 was employed in the same protocol lower ee values were obtained, confirming the necessity of a sterically encumbered ester group for high asymmetric induction. Cyclohexenone ethyl esters, however, performed on par with the five-membered ethyl ester analogues of 39, indicating additional potential for enantioselective sulfonamidocyclobutene synthesis by means of a Ficini reaction.
Scheme 12.
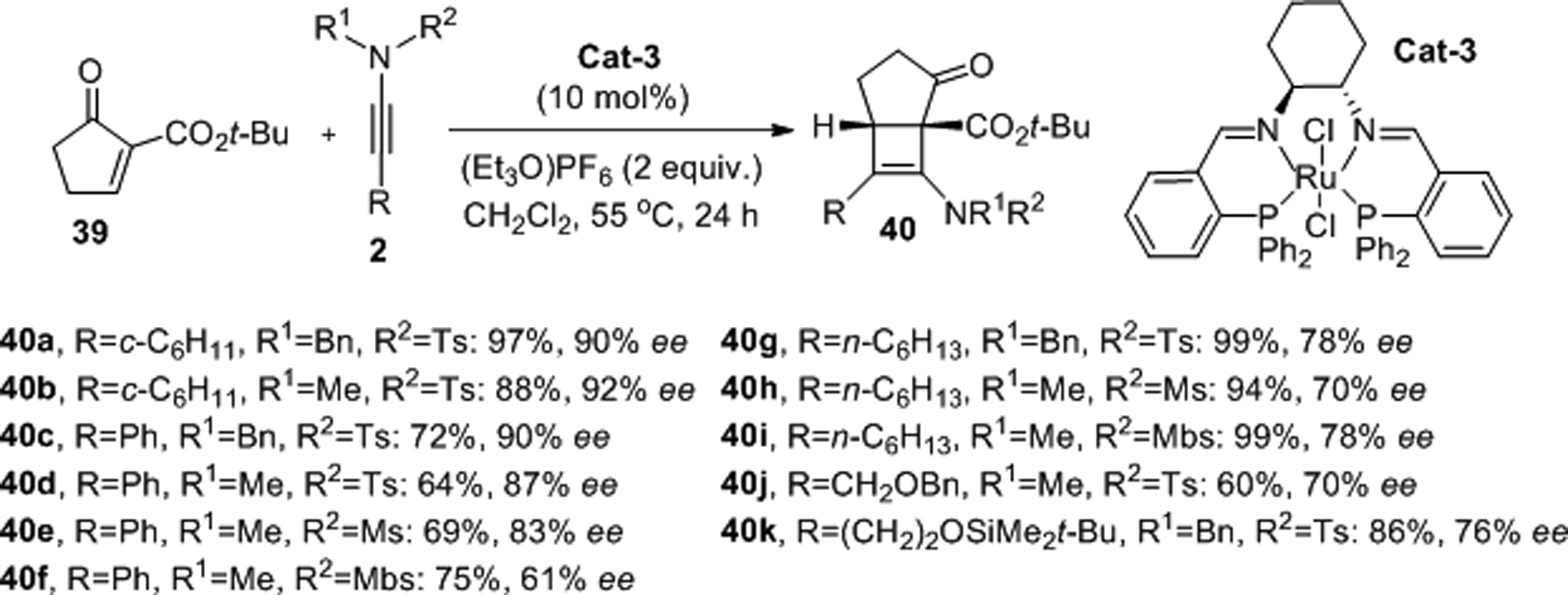
Catalytic Ficini reactions with β-ketoester 39 and ynamides 2.
Catalytic enantioselective [2+2] cycloadditions with cyclic α-alkylidene β-oxo imides 41 were reported by the Nakada group (Scheme 13).42 In contrast to the ruthenium catalyzed [2+2] cycloadditions reported by Mezzetti,43 the authors employed chiral Cu(bisoxazoline) complexes at low temperatures to produce multifunctional cyclobutene-fused lactones 42 as well as their ketone analogues. Excellent yields and ee’s were observed when ynamides carrying small substituents on the carbon terminus were used. It was proposed that the reaction proceeds via the conformationally restricted Cu(II) complex A which is stabilized by intramolecular hydrogen bonding between the imide and carbonyl moieties in the substrate.
Scheme 13.

Nakada’s catalytic asymmetric [2+2] cycloaddition with cyclic α-alkylidene β-oxo imides.
Chang developed a torquoselective Lewis acid catalyzed annulation method with o-quinone methides 43 and terminally substituted ynamides 2 (Scheme 14).44 This reaction produces chiral 4-amino-2H-chromenes 44 displaying central and axial chirality with excellent stereocontrol through a sequence of three pericyclic steps. Initially, [2+2] cycloaddition with the exocyclic double bond gives the strained spirane intermediate A, which then undergoes 4π electrocyclic ring opening to 1-oxatriene B. Lastly, a 6π electrocyclic ring closure generates chromenes 44. Overall, this reaction course establishes two chirality elements with high diastereoselectivity.
Scheme 14.

Chang’s annulation of o-quinone methides.
Danheiser et al. developed a benzannulation/ring closing metathesis route to access highly substituted benzofused nitrogen heterocycles (Scheme 15).45 This strategy was applied to the formal total synthesis of the anticancer agents (+)-FR66979 and (+)-FR900482. The key chiral ynamide intermediate 49 was obtained in six steps from (S)-valinol and used together with cyclobutenone 50 in a pericyclic cascade involving a 4π electrocyclic ring opening, a regioselective [2+2] cycloaddition, a second 4π electrocyclic ring opening and finally a 6π electrocyclization to construct aniline 51. Benzylation and ring closing metathesis gave the benzazocine scaffold 53, the precursor to (+)-FR66979, in 92% yield. The synthesis of a close analogue provided access to (+)-FR900482.
Scheme 15.
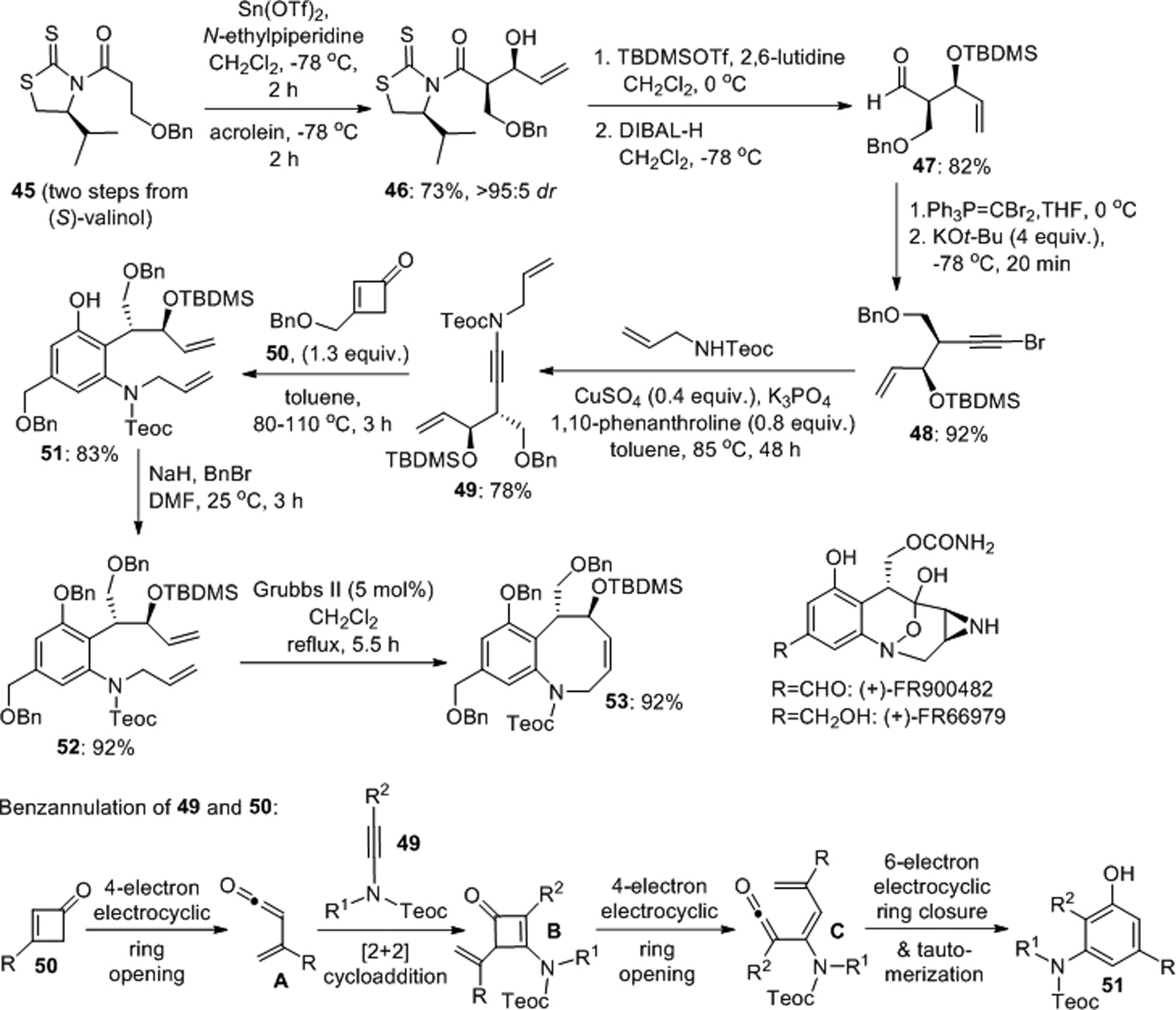
Enantioselective synthesis of benzazocines via benzannulation/ring-closing metathesis.
Buono developed a palladium-catalyzed [2+1] cycloaddition between norbornene derivatives 54 and ynamides such as 2 (Scheme 16).46 A variety of functionalized bicyclo[2.2.1]hept-2-enes 55 were produced in moderate to good yields albeit with low diastereoselectivity. For example, 55a and 55b were obtained in 2:1 and 1.3:1 dr, respectively. This work is remarkable because it constitutes an unusual example of a cycloaddition with terminal ynamides. Furthermore, an enantioselective variation of this reaction seems possible. Compound 55c was obtained in 21% ee when the chiral phosphapalladacycle Cat-5 was used. For additional asymmetric reactions with terminal ynamides, see Section 5.
Scheme 16.

Palladium-catalyzed [2+1] cycloaddition of norbornenes and terminal sulfonynamides.
Zhang et al. prepared bicyclic aza compounds through a gold(I) catalyzed one-pot stereoselective ynamide isomerization/[4+2] cycloaddition sequence (Scheme 17).47 Initial isomerization of the triple bond to an electron-rich diene sets the stage for either intra- or intermolecular Diels-Alder reaction with electron-poor dienophiles producing racemic 56 and 58 in 51 and 78% yield, respectively.
Scheme 17.
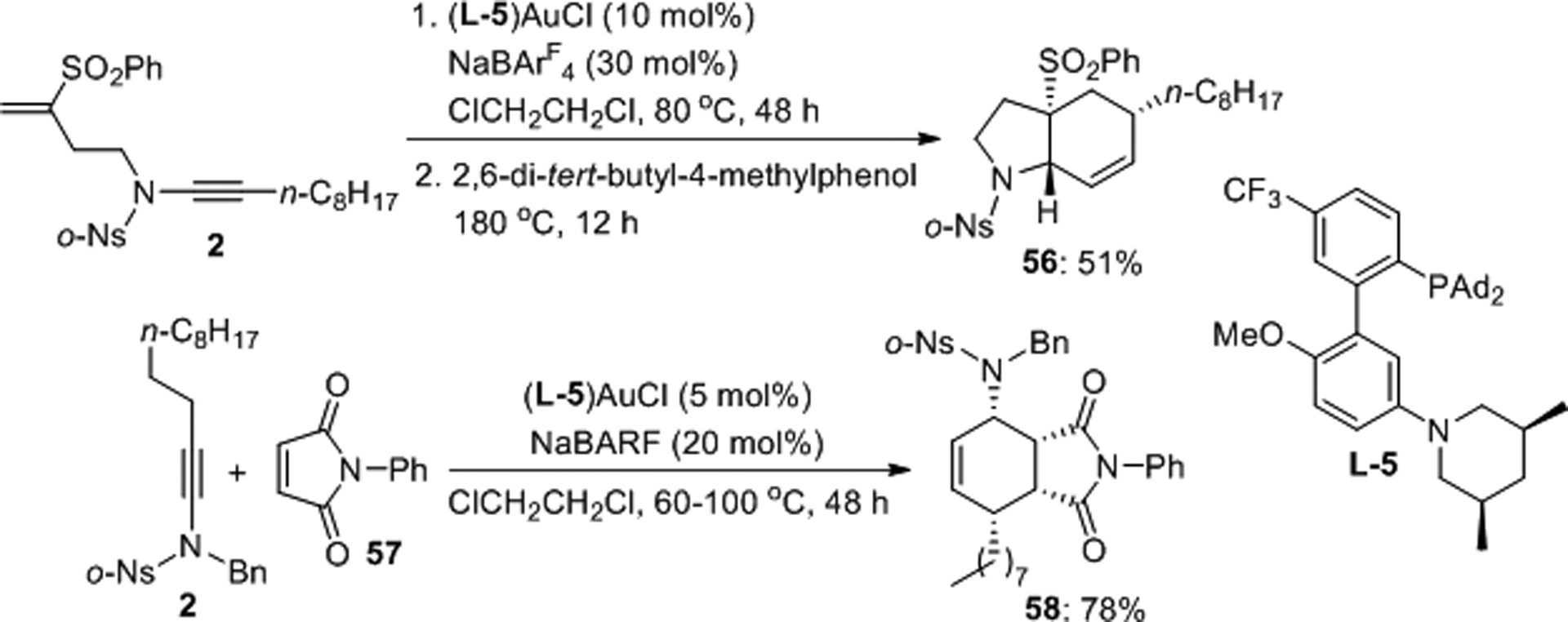
Zhang’s gold catalyzed isomerization/[4+2] cycloaddition sequence.
Liu et al. applied sulfonynamides in gold-catalyzed diastereoselective [4+2] and [2+2+2] cycloadditions (Scheme 18).48 The formal intermolecular [4+2] cycloaddition of the sulfonynamide 2 with the enolether 59 or the electron-rich alkene 60 in the presence of 5 mol% of [(L-6)AuCl]/AgNTf2 in 1,2-dichloroethane at 25 °C gave 61 and 62 in 61 and 91% yield, respectively. The authors also found that terminal ynamides 2 undergo rapid [2+2+2] cycloaddition with enolethers toward multisubstituted cyclohexenes 64 in moderate to good yields using [(L-7)AuCl]/AgNTf2 as catalyst. The reaction is complete within five minutes and diastereomeric ratios greater than 20:1 were achieved at room temperature. The stereochemical outcome was attributed to a stepwise mechanism involving the initial formation of the cyclopropyl gold-carbenoid complex A which is attacked by a second equivalent of 63a. The resulting axial oxonium B then undergoes cyclization to C which eliminates the [Au] complex, affording trans-64a with high diastereoselectivity. In situ ynamide formation has also been successfully exploited for diastereoselective Diels-Alder reactions.49
Scheme 18.

Gold catalyzed [4+2] and [2+2+2] cycloadditions with enol ethers.
Sato recognized the potential of ynamides for the total synthesis of herbindoles (Scheme 19).50 The indole 65 was prepared in 97% yield by intramolecular [2+2+2] cycloaddition of the sulfonynamide derived precursor 2 using RhCl3(PPh3)3 as catalyst at 50 °C. This set the stage for the synthesis of (−)-herbindole A, 66, in four steps with an overall yield of 87%.
Scheme 19.
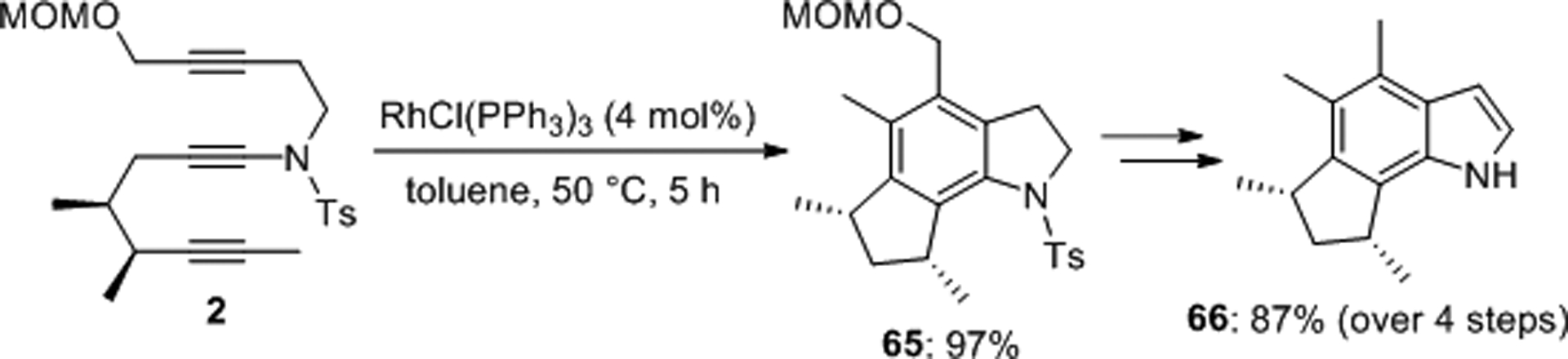
Total synthesis of (−)-herbindole A.
Gold catalyzed formal [4+3] cycloadditions of N-methyl-N-(phenylethynyl)methanesulfonamide, 2, and chiral epoxides 67 were reported by Liu et al. who prepared several benzene-fused seven-membered oxacycles 68 with moderate to high enantioselectivity (Scheme 20).51 The proposed mechanism for this transformation involves ynamide activation by the gold catalyst via ketenimine A. The subsequent reaction with (R)-2-phenyloxirane 67 results in the formation of oxonium B, which undergoes SN2-type attack by the neighboring benzene ring to cyclohexadienyl cation C. Deauration and proton transfer then produces (S)-68c.
Scheme 20.
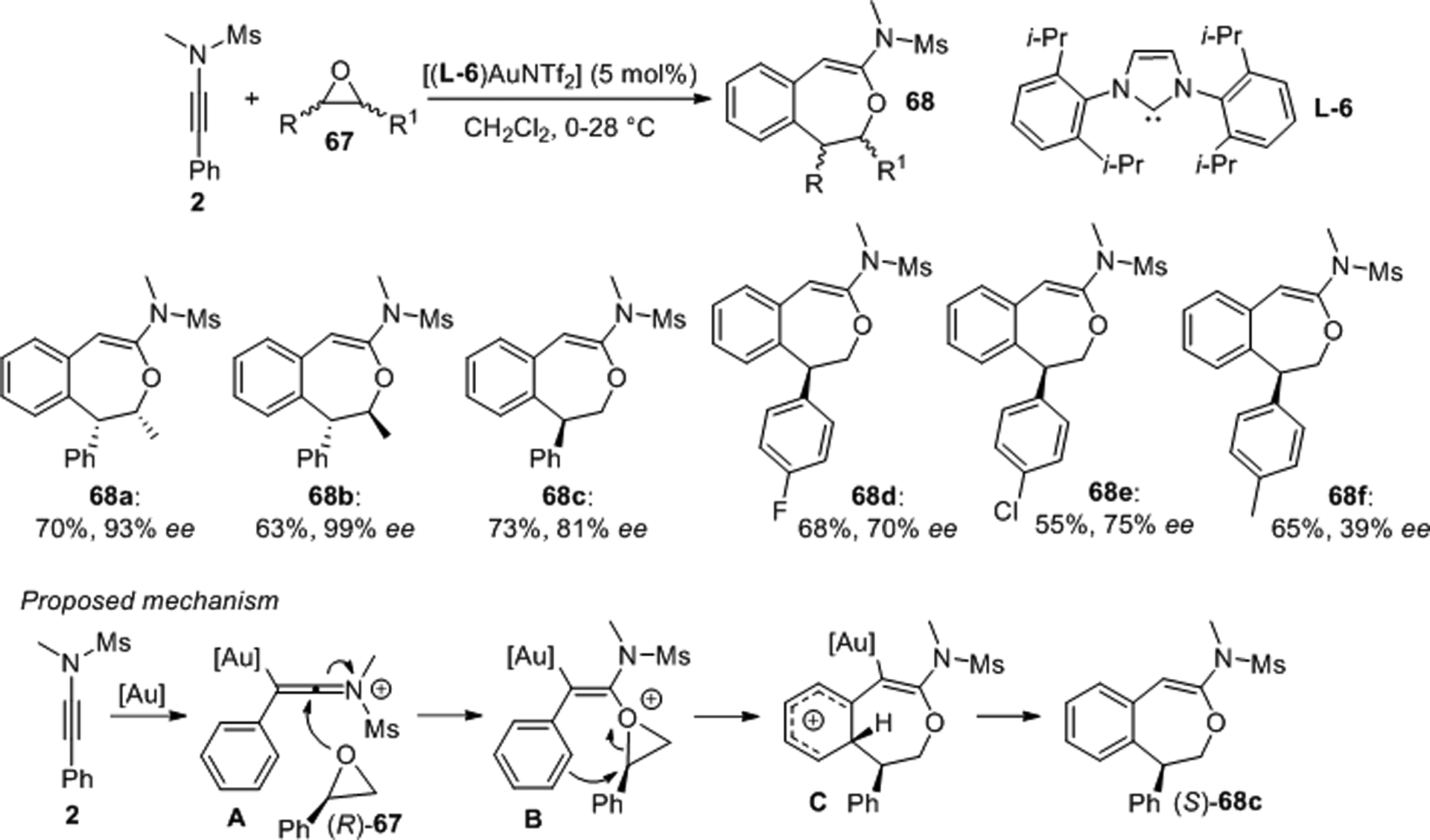
Asymmetric gold-catalyzed [4+3] cycloaddition of ynamides and chiral epoxides.
Recently, Ye achieved asymmetric copper-catalyzed formal [3+2] cycloadditions with alkenyl N-propargyl ynamides 2 and styrene, 69, which gives access to the interesting pyrrole-fused [2.2.1] scaffolds 70 (Scheme 21).52 High yields and stereoselectivities were obtained using catalytic amounts of Cu(CH3CN)4PF6, (S)-DTBM-SEGPHOS L-8 and sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF4) at slightly elevated temperatures. With this protocol in hand, over forty-five chiral [2.2.1] adducts were produced in 57–90% yield, 52–99% ee and diastereomeric ratios greater than 50:1. Detailed reaction analysis suggests that two possibly competing pathways may be operative. In the common first cyclization step, the ynamide 2 attacks the [Cu] activated C–C triple bond affording the copper carbene intermediate A. A [1,4]-H shift can then afford intermediate B which forms the second ring structure D via nucleophilic attack of the alkene moiety on the carbene carbon center. Alternatively, A may undergo metallahexatriene-type cyclopentannulation and subsequent [1,4]-H migration to produce D via C. Finally, highly regio- and diastereoselective [3+2] cycloaddition of D with a styrene dipolarophile furnishes the observed cycloaddition product.
Scheme 21.
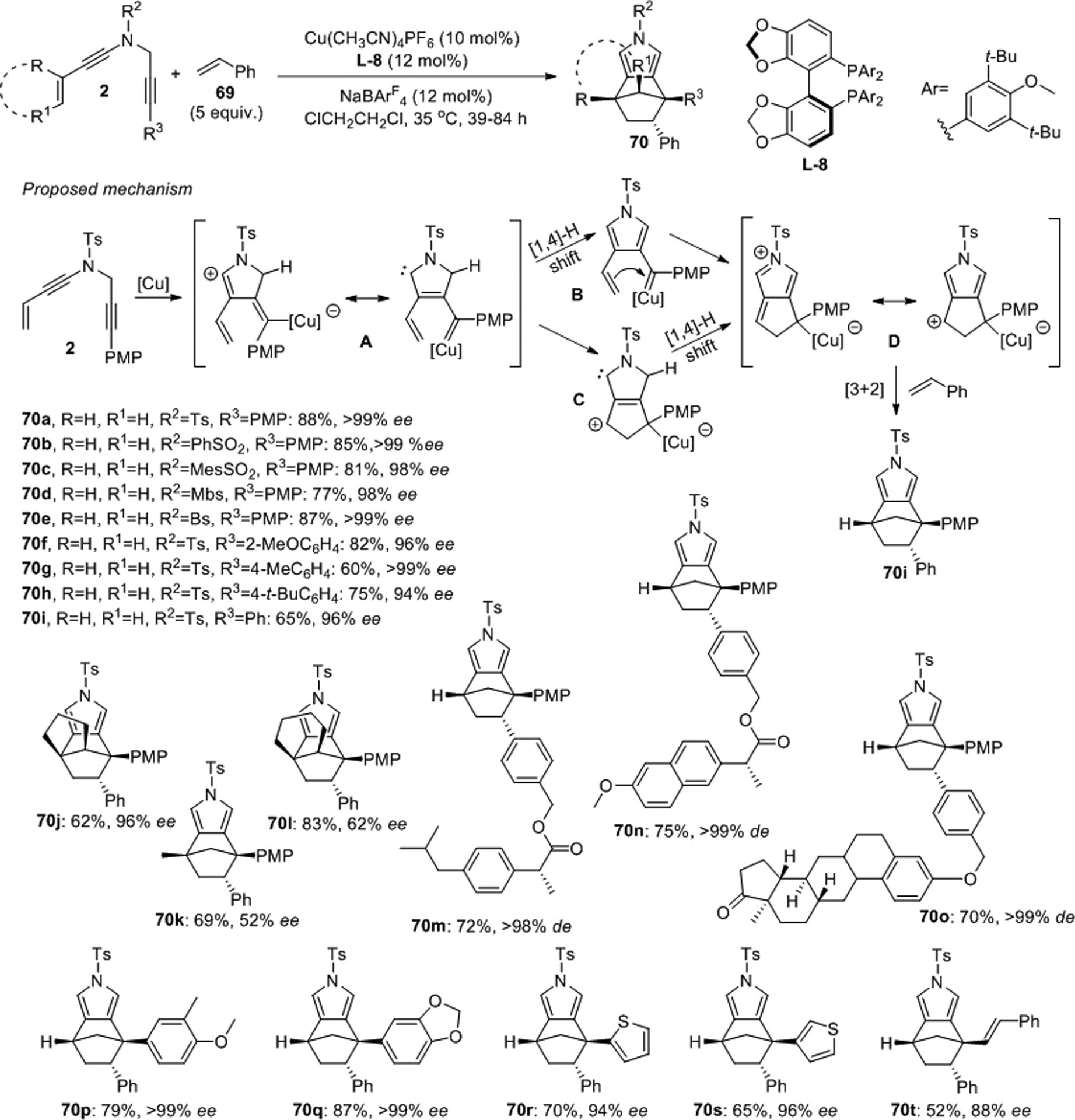
Ye’s catalytic asymmetric formal [3+2] cycloaddition between N-propargyl ynamides and styrenes.
An intramolecular variation of this chemistry enables synthesis of polycyclic pyrroles through copper-catalyzed enantioselective cascade cyclization of N-propargyl ynamide diynes.53 The tetracyclic pyrroles 71 were produced from the vinyl derivatives 2 in up to 94% ee and high dr’s, greater than 50:1, with catalytic amounts of Cu(CH3CN)4PF6 and the chiral BOX ligand L-9 (Scheme 22). Alternatively, tricyclic products 72 can be obtained at slightly lower temperatures under otherwise fairly similar conditions with SEGPHOS L-10 as the ligand using 2 devoid of the vinyl group as starting material. Again, triple bond activation is followed by sequential cyclization and a [1,4]-hydride shift of the copper carbene A. The resulting key intermediate B can further react with the vinyl moiety to form 71 or insert into the adjacent aryl C–H bond to generate 72. Asymmetric [3+2] cycloadditions between ynamides and 1,3-dipoles generated in situ through cyclopropane ring opening have also been reported.54
Scheme 22.
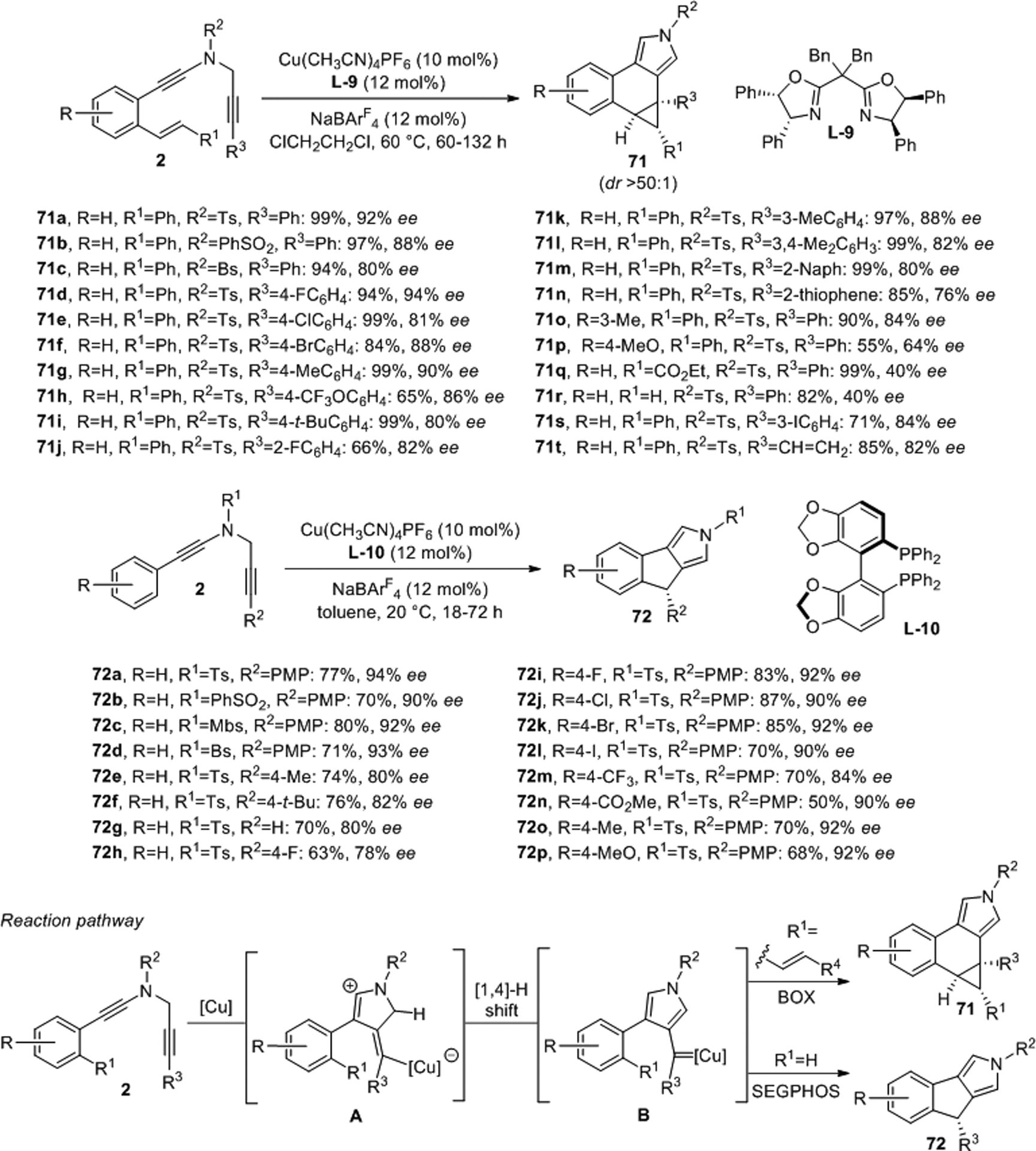
Enantio- and diastereoselective copper catalyzed cascade cyclization of N-propargyl ynamides.
A three component formal [5+2+1] cycloaddition of ynamides 2, isoxazoles 73 and water toward bicyclic tetrahydro-1,4-oxazepines was disclosed by Wan et al.55 In the presence of 15 mol% of trifluoromethanesulfonimide and one equivalent of water compound 74 was obtained in 48% yield and 5:1 dr (Scheme 23). The authors verified that the water molecule serves as the source of the bridging oxygen.
Scheme 23.

Diastereoselective [5+2+1] cycloaddition between an ynamide, isoxazole and water.
Blanchard and colleagues reported the synthesis of polycyclic 4-aminopyridines by intramolecular inverse electron-demand [4+2] cycloaddition of ynamidyl pyrimidines 6 (Scheme 24).56,57 The reaction was carried out in sulfolane under microwave irradiation at 210 °C furnishing 75a and 75b in 72 and 78% yields, respectively. This transformation involves formation of intermediate B via transition state A and subsequent retro Diels-Alder reaction eliminating HCN to produce the final 4-aminopyridine structure.
Scheme 24.
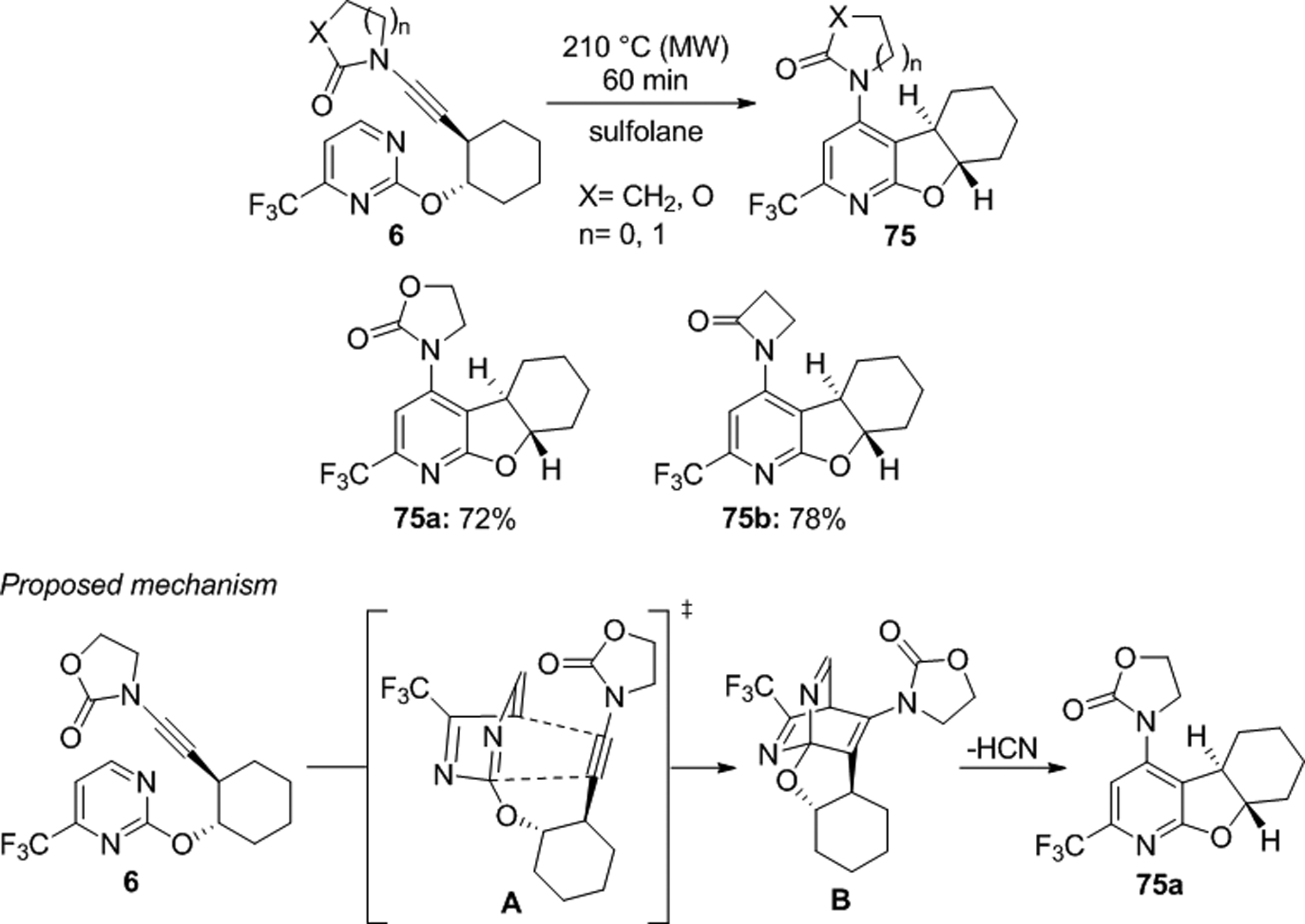
Inverse electron-demand [4+2] cycloaddition of ynamidyl pyrimidines.
4. Rearrangements and Cycloisomerizations
Ynamides are attractive starting materials for rearrangement and cycloisomerization pathways and inspiring asymmetric transition metal catalyzed and organocatalytic variants of these type of reactions have been introduced during the last 10 years. Without a doubt, this area has seen tremendous progress and practical access to many complicated polycyclic architectures and multifunctional scaffolds, often exhibiting more than one chirality center, that are difficult to prepare through other means has been established.
In contrast to intermolecular ring construction strategies, cyclization reactions of appropriately functionalized starting materials often have the advantage of increased stereocontrol and may thus produce superior stereoselectivities. Following this strategy, Hayashi and colleagues developed highly enantioselective cycloisomerizations of 1,6-ene-ynamides using [RhCl(L-11)]2 as catalyst at ambient temperatures (Scheme 25).58 This work establishes up to three chirality centers in the cyclopropane ring and typically furnishes the 3-aza- and oxabicyclo[4.1.0]heptenes 76, which are different from the bicycloheptene scaffolds described by Buono as shown in Scheme 16,59 with high yields and enantioselectivities.
Scheme 25.

Hayashi’s asymmetric rhodium catalyzed cycloisomerization of 1,6-ene-ynamides.
Malacria, Fensterbank and Aubert presented a tandem cycloisomerization/Diels-Alder reaction sequence with allenynamides such as 2 (Scheme 26).60 In the presence of catalytic amounts of silver triflate, the allenynamide cycloisomerization generates a reactive diene A that can be isolated or trapped with the dienophilic N-phenyl maleimide 57 to form the tricyclic structure 77. Alternatively, asymmetyric cycloisomerization/cycloaddition cascades can be achieved with alkenyl ynamides formed in situ.61
Scheme 26.

A silver catalyzed tandem cycloisomerization/Diels-Alder reaction with an allenynamide.
Following work from Carbery,62 Meyer and Cossy developed an asymmetric method that affords allenamides by [3,3] Claisen rearrangement of propargylic N-Boc glycinates 2 (Scheme 27).63 In a two-step protocol, enolate formation triggers the sigmatropic rearrangement to the allenamides 78 which are isolated as the corresponding methyl esters. The reaction proceeds with high stereocontrol and conservation of chirality which was demonstrated with the synthesis of allenamide 78a in high ee and dr from an enantiomerically pure glycinate ynamide. Silver catalyzed ring closure provides access to multifunctional 3-pyrroline 79a with minor erosion of enantiomeric purity.
Scheme 27.

[3,3] Claisen rearrangement of propargylic N-Boc glycinates.
Maulide and coworkers introduced chiral sulfoxides to a powerful [3,3] sigmatropic rearrangement course exhibiting a highly uncommon 1,4-chirality transfer from sulfur to a carbon atom.64,65 This strategy provides efficient access to α-arylamides with good to high yields and enantioselectivities under mild conditions (Scheme 28). Initially, the authors studied the reaction between 3-(3-methylbut-1-yn-1-yl)oxazolidin-2-one, 6, and (R)-cyclohexyl p-tolyl sulfoxide, 80, in the presence of 50 mol% of trifluoromethanesulfonic acid in CH2Cl2 which gave (R)-81a in 71% yield and 73:27 er. Further optimization showed that the enantioselectivity can be improved to 93:7 er when the reaction is carried out in 1,2-dichloroethane with 50 mol% of trifluoromethanesulfonimide, Tf2NH. A variety of cyclic and acyclic arylalkyl sulfoxides react with ynecarbamate or ynesulfonamides under these conditions, however, superior ee’s were obtained with cyclic sulfoxides and oxazolidinone-derived ynamides carrying bulky groups at the C-terminus. Quantum chemical calculations and experimental evidence are in agreement with a stereospecific chirality transfer mechanism that involves formation of intermediate A and transition state B. If desired, compounds 81 undergo amide hydrolysis and reductive desulfurization via Raney nickel hydrogenolysis with high yields and without loss of enantiopurity.
Scheme 28.
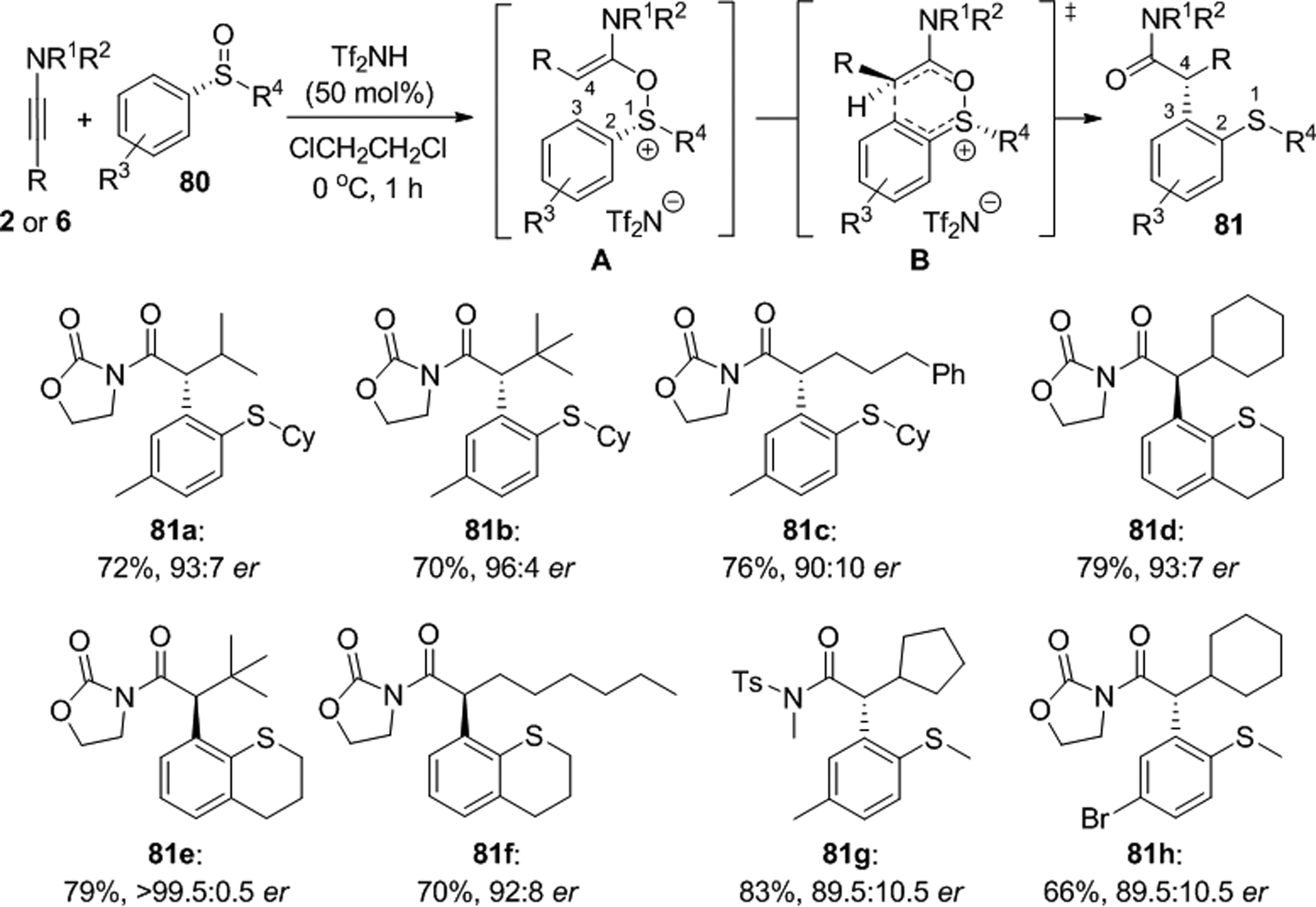
Representative examples of enantiospecific [3,3] sigmatropic rearrangements with chiral sulfoxides.
The same group further extended the [3,3] sulfonium rearrangement chemistry to alkenylsulfoxides which undergo traceless stereodivergent Brønsted acid catalyzed C–C bond formation toward pharmaceutically important 1,4-dicarbonyl motifs with moderate to high yields, excellent enantioselectivities and often high dr values (Scheme 29).66 The chirality at the sulfur atom controls the absolute configuration, and the relative stereochemistry in the product is governed by the E/Z geometry of the vinylsulfoxide double bond. (E)-Vinylsulfoxides produce syn-dicarbonyls while (Z)-vinylsulfoxides lead to the formation of anti-dicarbonyls which was attributed to Zimmerman-Traxler like transition states. The stereochemical outcome is mostly controlled by the chiral sulfoxide but when chiral ynamides were used, the diastereomeric products 83l and 83l’ were obtained with 10:1 and 3:1 dr indicating the significance of matched and mismatched double stereodifferentiation. The general utility of this chemistry was highlighted by the synthesis of a precursor of an important MMP inhibitor via high yielding aldehyde oxidation and amino acid coupling.
Scheme 29.

Maulide’s asymmetric synthesis of syn- and anti-1,4-dicarbonyl motifs from chiral vinylsulfoxides.
An unusual ynamide oxoarylation which produces α-aryl amides via Claisen rearrangement in modest yields but with high asymmetric induction was observed with the chiral sulfoxide 88 (Scheme 30).67 The Maulide group discovered that the acid promoted addition to 6 affords the regioisomers 89 and 90 in 37 and 14% yield, respectively, which was rationalized with two competing reaction pathways. Accordingly, ESI-MS and computational studies suggest that product 89 is formed via intermediate A after rearomatization, while the formation of 90 occurs via intermediate B which undergoes a [1,2] shift to B’ and subsequent deprotonation to regenerate the aromatic ring.
Scheme 30.
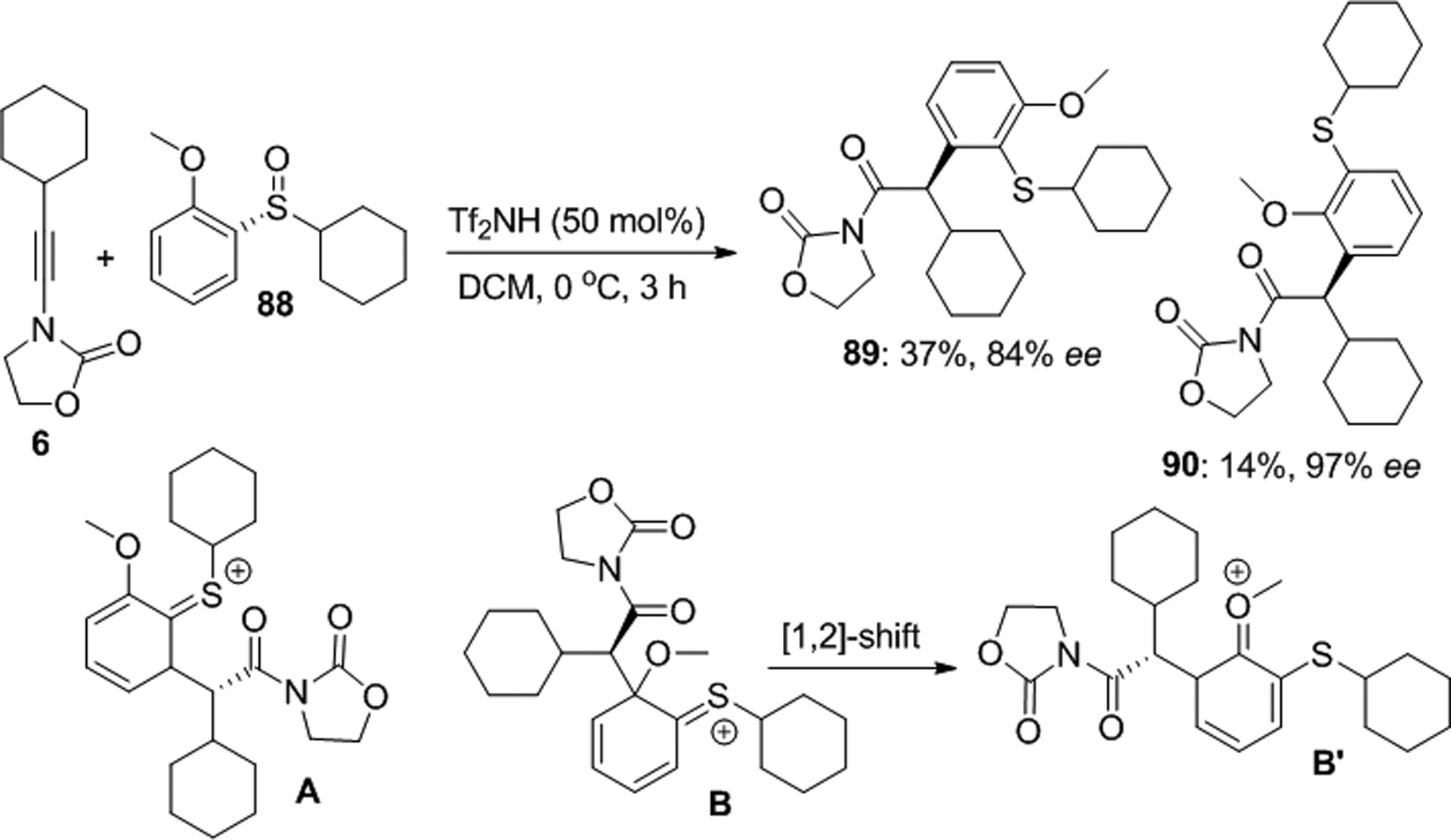
Asymmetric ynamide oxoarylation with a chiral sulfoxide.
A [Au(picolinate)Cl2] catalyzed C–H insertion/cyclization cascade of N-allyl enynamides 2 was used by Davies for the synthesis of the tetracyclic compounds 91 which were produced with moderate diastereoselectivity in most cases (Scheme 31).68 The reaction probably occurs via gold keteniminium A which first undergoes [1,5] hydride transfer to generate benzylic cation B. A 4π electrocyclic ring closure (4π ERC) may then form the gold complex C before cyclopropanation completes the cascade.
Scheme 31.
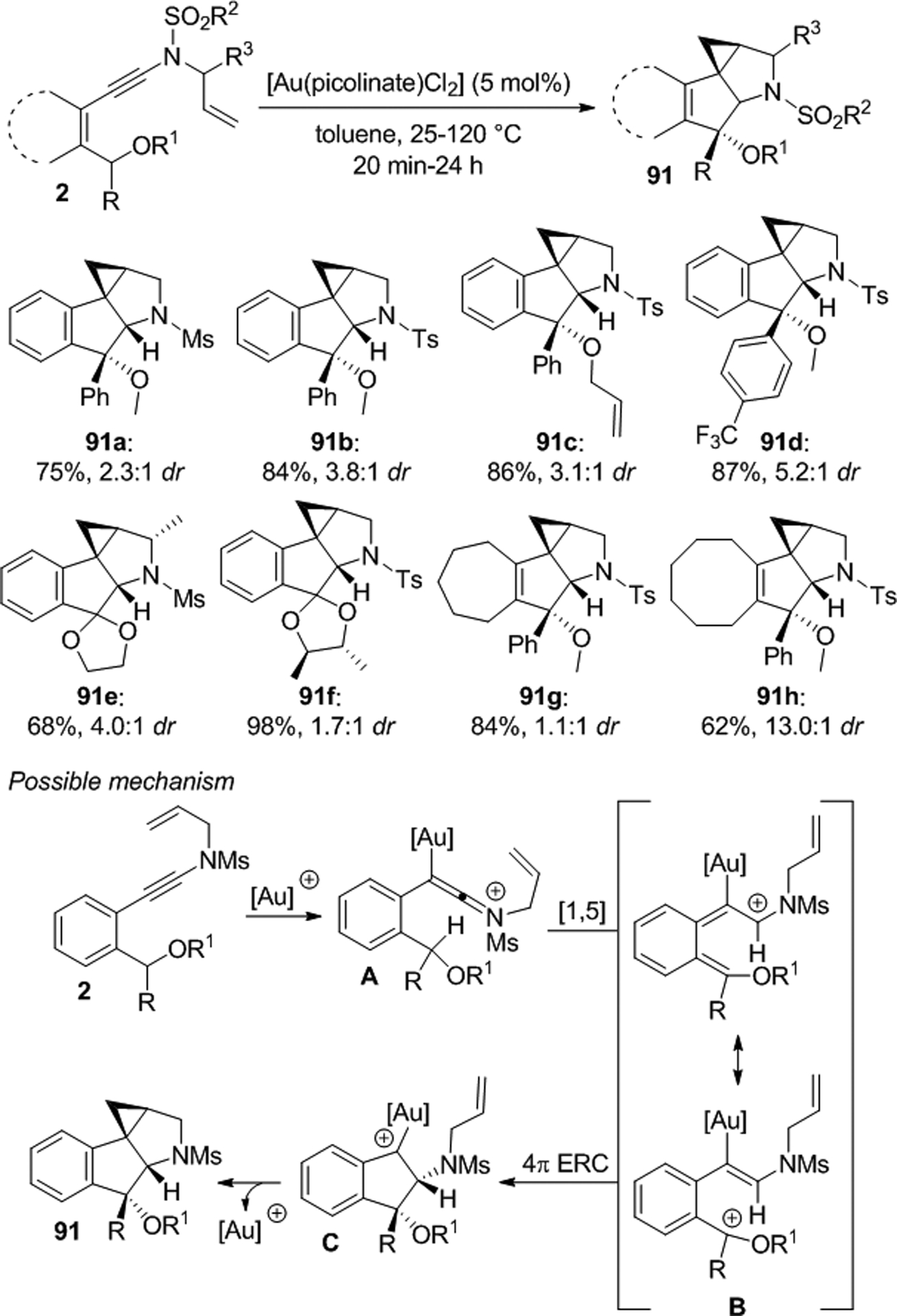
Gold catalyzed C–H insertion/cyclization cascade of N-allyl ynamides.
Yang and coworkers prepared spirocyclic pyrrolidinoindolines via gold catalyzed intramolecular annulation of indole ynamides 2 at room temperature (Scheme 32).69 This reaction probably involves an intermediate keteniminium A which can undergo 5-endo-dig cyclization to B and then 6-exo-trig nucleophilic attack to afford the tetracyclic N,O-acetals 92.
Scheme 32.

Yang’s diastereoselective synthesis of spirocyclic pyrrolidinoindolines from indole ynamides.
Liu et al. coupled the cycloisomerization of tryptamine-ynamides 2 toward intermediate spiroindolenium scaffolds with the nucleophilic addition of indoles 93 (Scheme 33).70 Their silver triflate/N-fluorobenzenesulfonimide (NFSI) catalyzed protocol produces spiro(indoline-3,4’-piperidine) derivatives 94 as a single diastereomer in up to 99% yield.
Scheme 33.

Cycloisomerization and indole trapping with tryptamine-tethered ynamides.
Rational rhodium catalyst design and ligand optimization studies paved the way toward a powerful method that produces chiral unsaturated [5.3.0] azabicyclic structures via enantio- and diastereoselective [5+2] cycloisomerization of the vinylcyclopropanes 2.71 Guided by invaluable theoretical and experimental mechanistic insights, the Anderson group systematically developed an efficient procedure that uses [RhCl(C2H4)2]2, the phosphoramidite ligand L-12 and NaBArF4 for selective formation of 95 or 96 in good to high yields, up to 99% ee and, in some cases, greater than 20:1 dr (Scheme 34). An attractive feature of this method is that the stereochemical outcome of the reaction is predominantly controlled by the chiral Rh complex which allows successful double stereodifferentiation with both matched (95i–k) and mismatched (96a–c) catalyst/substrate combinations. It is noteworthy that other multisubstituted chiral pyrroles and piperidines can be prepared with high stereo- and regioselectivity via palladium or ruthenium catalyzed cycloisomerization of alkenyl ynamides.72
Scheme 34.

Rhodium-catalyzed enantio- and diastereoselective [5+2] cycloisomerizations developed by the Anderson group.
Pharmaceutically important 3-isochromanones were prepared through acid catalyzed ring closure of allyl ynamide ethers 6.73 The Ye group showed that high asymmetric induction can be achieved when a chiral oxazolidinone auxiliary is used, producing esters 97a–d in up to 95% ee (Scheme 35). A plausible mechanism begins with ynamide activation due to protonation by bis(trifluoromethane)sulfonimide. The corresponding keteniminium B reacts with the ether moiety to form the oxonium intermediate C, which rearranges to oxonium D. Addition of water generates the orthoester E and subsequent elimination of the oxazolidinone auxiliary produces 97a. A neat feature of this method is that a small amount of water in the reaction mixture cleaves the chiral auxiliary from the isochromanone scaffold, a task that often requires elaborate work-up or several additional steps. The observed sense of asymmetric induction is in agreement with the proposed transition state A.
Scheme 35.

Metal-free intramolecular alkoxylation initiated cascade reaction of ether tethered allylic ynamides.
This Brønsted acid catalyzed cyclization protocol gave also rise to indole-fused bridged [4.2.1] lactones 98 and 99 that were obtained in most cases with dr’s greater than 20:1 and with high ee’s when a chiral oxazolidinone ynamide was used as the traceless chiral auxiliary (Scheme 36).74 It is noteworthy that a wide variety of functional groups is tolerated by this reaction.
Scheme 36.

Intramolecular alkoxylation of indolyl ynamides.
Evano et al. developed a Brønsted acid promoted ring closure protocol that starts with N-benzyl ynesulfonamides 2 or ynecarbamates 5 (Scheme 37).75,76 The cyclization is very fast and gives rise to tetracyclic compounds 100 with greater than 19:1 dr in most cases. Activation of ynamide 2 produces keteniminium ion A, which undergoes a [1,5] sigmatropic hydrogen shift to intermediate B. This is followed by Nazarov 4π conrotatory electrocyclization to carbocation C and intramolecular Friedel-Crafts alkylation forming cis-100b.
Scheme 37.
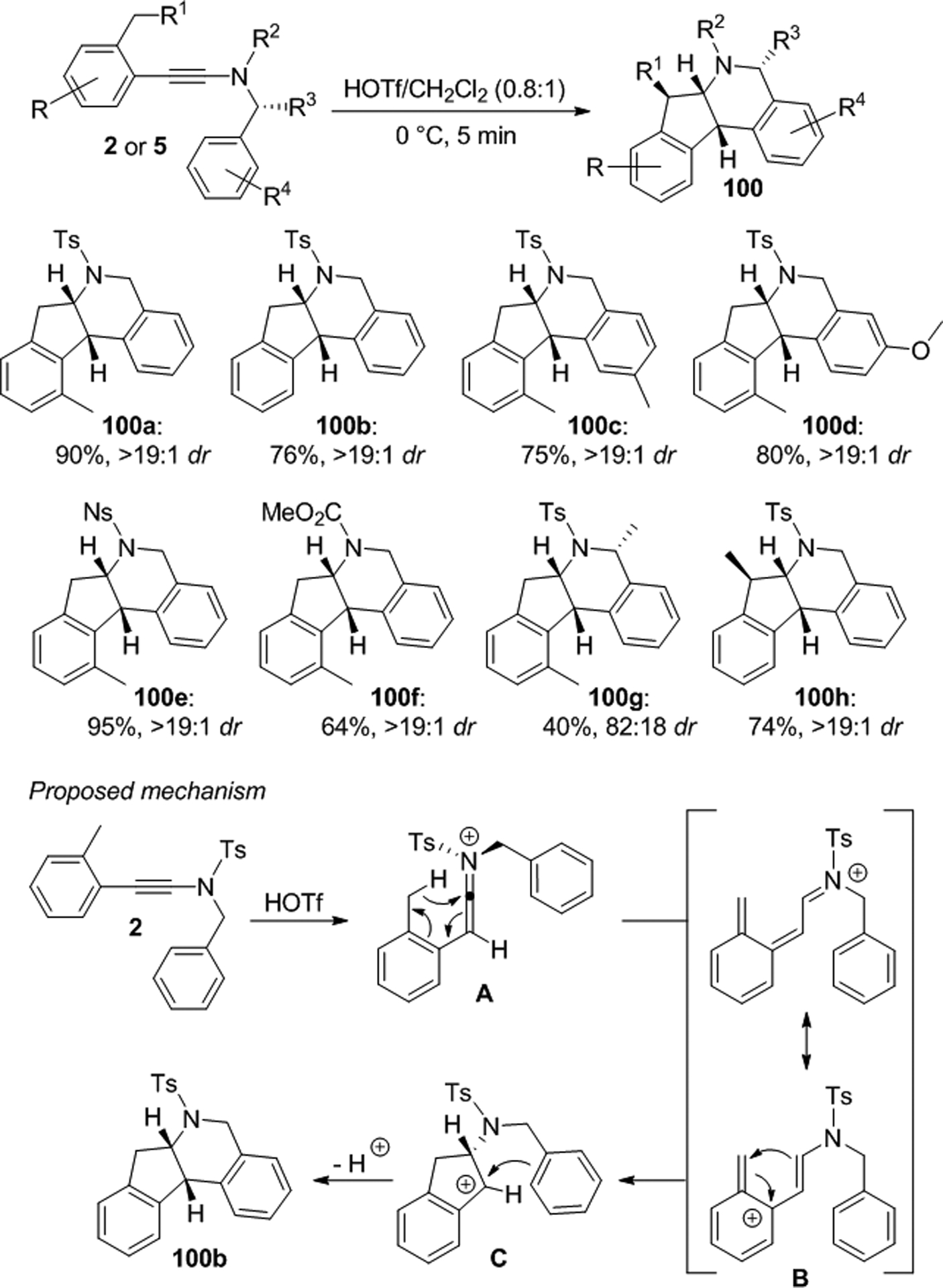
Brønsted acid promoted asymmetric ring closure with N-benzyl ynamides.
Ye et al. developed an yttrium triflate catalyzed hydroalkoxylation/Claisen rearrangement sequence with the Morita-Baylis-Hillman alcohols 101 (Scheme 38).77 This protocol allows diastereoselective synthesis of γ,δ-unsaturated amides 102 with 50–75% yields, excellent E/Z stereocontrol and in diastereomeric ratios often greater than 10:1. Addition of the alcohol to the sulfonynamide probably generates the transient ketene N,O-acetal A which is poised to form 102 via Claisen rearrangement.
Scheme 38.

Yttrium catalyzed tandem hydroalkoxylation/Claisen rearrangement.
In 2019, the same group synthesized a series of eight-membered lactams 103 from the chiral benzylic alcohols 2 through an intramolecular hydroalkoxylation/[1,3] rearrangement sequence (Scheme 39).78 The cyclization is highly diastereoselective and can be initiated by very small amounts of a strong Brønsted acid upon heating to 80 °C. It was proposed that ynamide protonation allows the alcohol group to add to the initially formed keteniminium A. Deprotonation of the oxonium B and subsequent O-to-C rearrangement can then furnish the lactam scaffold. This methodology was further refined into a powerful enantioselective version and several lactams were produced in good to high enantiomeric excess using the chiral phosphoramide catalyst Cat-7 under milder conditions. The same group developed a similar yttrium catalyzed intramolecular hydroalkoxylation/Claisen rearrangement method which was used for the synthesis of the diastereomerically pure lactam 104 and other benzazocinones.79
Scheme 39.
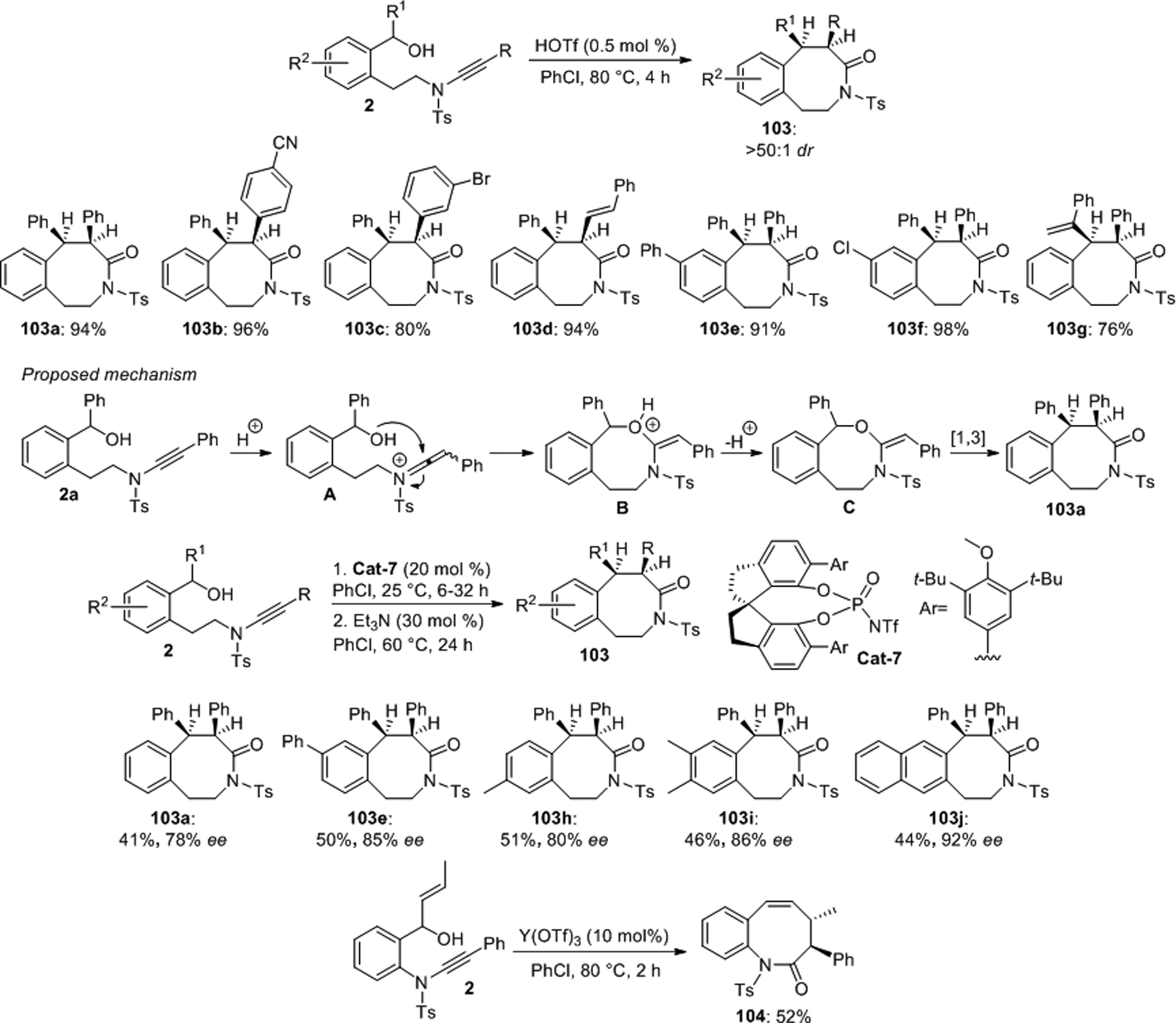
Ye’s enantio- and diastereoselective preparation of eight-membered lactams.
Hsung’s laboratory found that epoxidation of the chiral oxazolidinone derived ynamides 6 with dimethyldioxirane generates oxirenes that spontaneously rearrange to the push-pull carbenes A prone to intramolecular cyclopropanation (Scheme 40).80 Depending on the length of the tether between the triple and double bonds, 5-membered and 6-membered ring structures 105 and 106 can be obtained by this method.
Scheme 40.
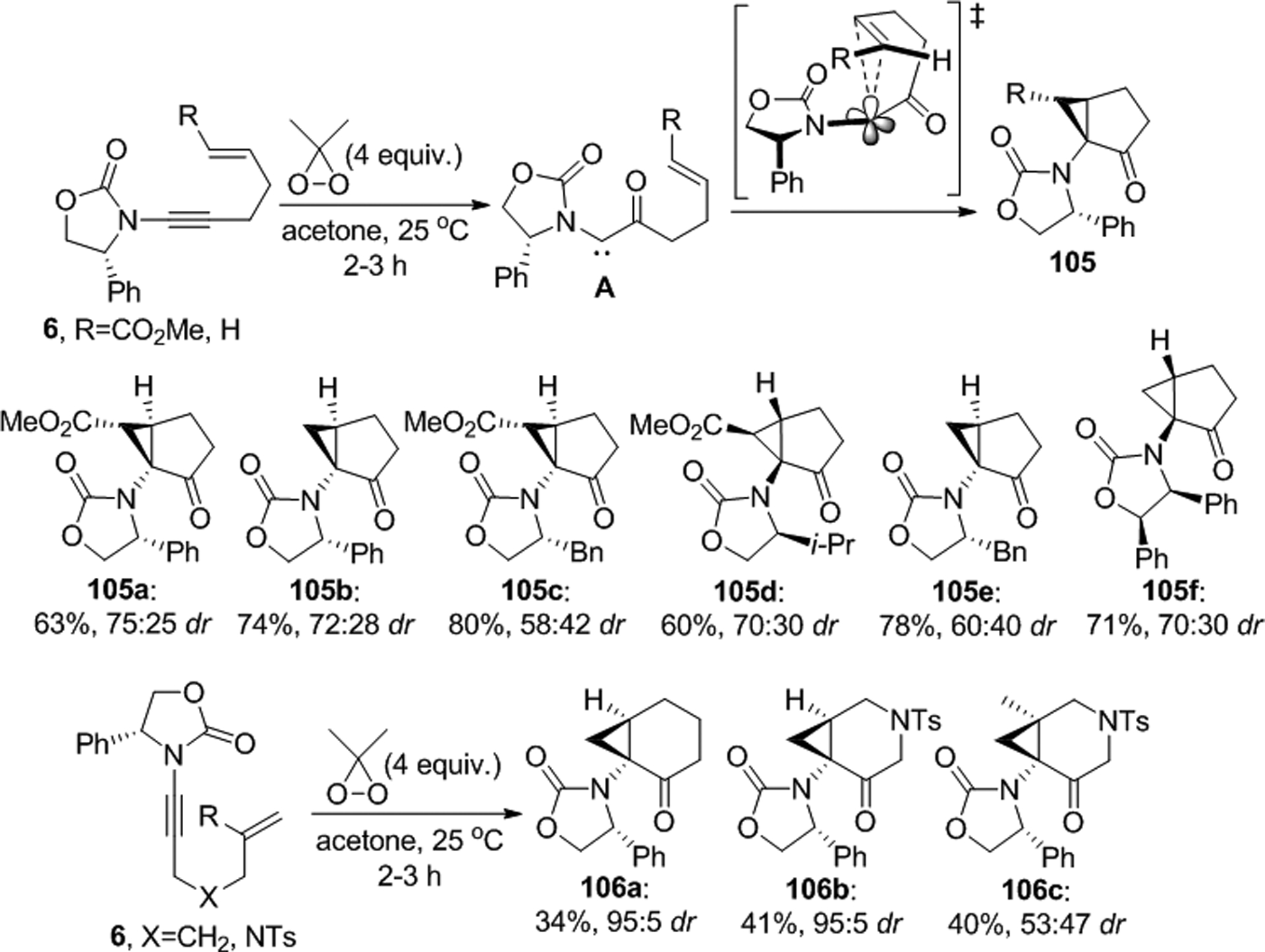
Diastereoselective epoxidation and subsequent cyclopropanation of chiral oxazolidinone derived ynamides.
A gold catalyzed dearomative ynamide spirocyclization course that affords 2-azaspiro[4.5]decan-3-ones 107 in moderate to good yields was discovered by Shibata and coworkers.81 Scheme 41 shows a plausible mechanism involving the formation of gold carbene species A upon oxidation of the ynamide unit in 2 and subsequent spirocyclization to B. Hydrolysis and protodeauration then furnishes the spirane 107b. A similar protocol was developed by Huang et al. for the diastereoselective synthesis of tetracyclic spiroindolines from tryptamine derived enynamides and with pyridine N-oxide as the oxidant.82
Scheme 41.

Comparison of Shibata’s oxidative spirocyclization with ynamides (top) and Huang’s spiroindoline synthesis from enynamides (bottom).
Voituriez developed a method for gold catalyzed enantioselective hydrative cyclizations (Scheme 42).83 Following previous reports of an analogous achiral variant,84 asymmetric ring closure of N-propargyl ynamides 2 was carried out with p-toluenesulfonic acid monohydrate and a SEGPHOS L-13 derived Au(I) complex, which gave several N-tosyl 3-aryl-3,6-dihydropyridin-2(1H)-ones 109 with moderate to good enantioselectivities. It is assumed that the gold catalyst activates the alkyne triple bond while the sulfonic acid generates a ketene N,O-acetal moiety in the intermediate A. This is followed by 6-endo-dig cyclization, sulfonate hydrolysis and protodeauration toward dihydropyridinones 109.
Scheme 42.

Gold catalyzed hydrative cyclizations of N-propargyl sulfonynamides.
An organocatalytic enantioselective Conia-ene-type cyclization of ynamides that produces chiral morphans and normorphan isomers was developed by Ye.85 Stereoselectivities up to 97% ee were achieved using the cyclohexanone derived sulfonynamides 2 and 20 mol% of the chiral secondary amine Cat-8 (Scheme 43). The reaction outcome can be steered by careful selection of the ynamide protection group and the solvent. A mechanism that explains the formation of morphans 110 and normorphans 111 based on alternative cyclization paths of the enamine intermediate B was proposed. Apparently, nucleophilic attack at the β-carbon is favored with arylsulfonyl protected ynamides, resulting in the formation of the 6-endo-dig cyclization intermediate C which finally gives rise to 110 carrying an endocyclic double bond. Ms protection and the use of polar solvents promotes nucleophilic attack at the α-carbon toward the 5-exo-dig intermediate C’ which subsequently hydrolyses to 111 with an exocyclic double bond.
Scheme 43.

Conia-ene-type cyclization of ynamide cyclohexanones.
The same group also found that transition-metal free oxidative cyclization of chiral N-propargyl ynamides with catalytic amounts of NaBArF4 and slight excess of 2-bromopyridine N-oxide 112 gives access to an array of diverse polycyclic lactams 113 at elevated temperatures (Scheme 44).86 It was proposed that the N-oxide does not only serve as the oxidant but is also required to initiate the catalysis. First, it generates the strong Lewis acid BArF3 in situ which then activates the ynamide triple bond as the zwitterion A. This enables reaction with another N-oxide molecule toward B. Cleavage of the N–O bond produces the resonance-stabilized carbocation C, which subsequently undergoes cyclization affording D. Migration of the boron moiety gives E and this sets the stage for a second cyclization step. Aromatization of F and elimination of the BArF3 catalyst gives rise to the thermodynamically favored diastereomer 113d. This chemistry was further extended to indolyl N-propargyl ynamides 2, which form the tetracyclic N-heterocycles 115. In all cases, high dr’s greater than 20:1 were obtained. Asymmetric rhodium catalyzed oxidative cycloisomerization of alkenyl ynamides toward azabicyclo[3.1.0]hexanes is also possible.87
Scheme 44.

Transition-metal free oxidative cyclization of chiral N-propargyl ynamides.
The Ye laboratory also reported the synthesis of medium-sized lactams with this type of chemistry (Scheme 45).88 Diastereomeric oxidative cyclization of the chiral indolyl ynamide 2 was achieved with 2,6-dichloropyridine N-oxide, 114, as the oxidant in the presence of catalytic amounts of Zn(OTf)2 and NaBArF4. This protocol furnished lactam 116 in 75% yield and 2.5:1 dr.
Scheme 45.
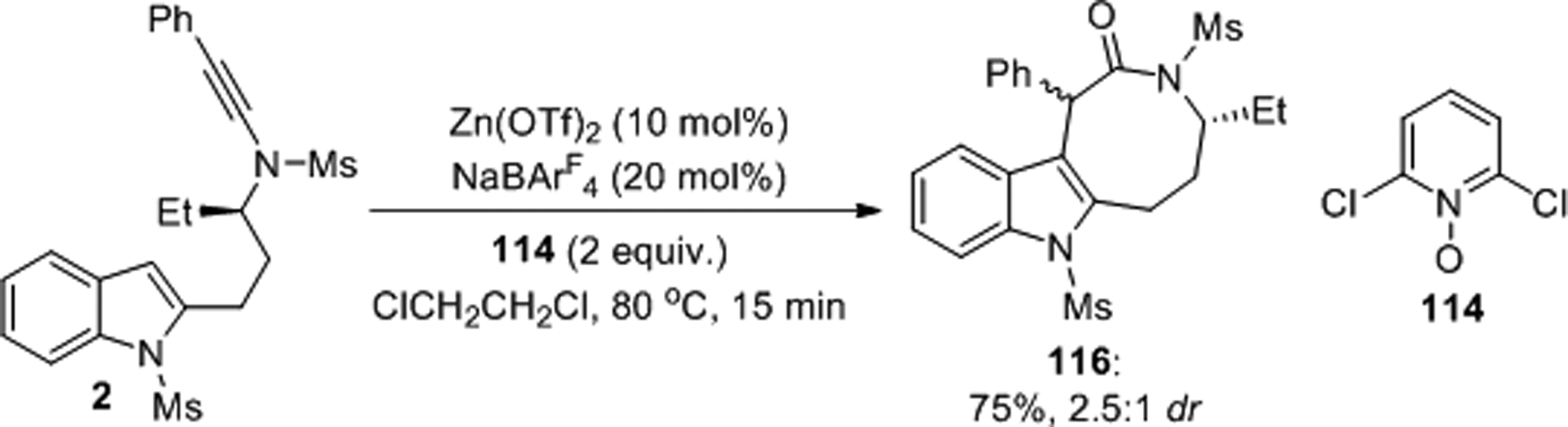
Oxidative cyclization of a chiral indolyl ynamides.
Flynn and colleagues exploited the Nazarov reaction for the production of multisubstituted cyclopentanoids 120 from oxazolidinone-derived ynamides.89,90 This was achieved with a tandem transition metal catalysis strategy setting the stage for the final ring closure which proceeds under acidic conditions (Scheme 46). At the onset, stereoselective Pd-mediated syn-hydrostannylation of ynamides 6 affords alkenes 117 which then undergo copper-catalyzed Stille coupling with tigloyl chlorides 118 to the divinyl ketones 119. Addition of excess of MeSO3H then furnishes the cyclopentanoids 120 in high dr’s by means of an asymmetric Nazarov cyclization. Alternatively, vinylstannanes or organotrifluoroboronate salts together with carbon monoxide can be employed in carbonylative Stille or Suzuki cross-coupling with oxazolidinone (Ox) derived alkenylbromide intermediates to generate compounds 119. The chiral auxiliary provides exceptional stereocontrol in the final step by directing the torquoselectivity of the electrocyclic reaction. The sense of asymmetric induction was explained with a “coupled-torque” mechanism involving a transition state that is stabilized by interactions between the oxazolidinone nitrogen lone electron pair and the nascent pentadienyl cation while the chiral auxiliary steers the unidirectional conrotatory motion of the pentadienyl cation termini during ring formation. The authors showed that the oxazolidinone moiety can be fully removed and recovered using either lithium naphthalenide or SmI2, or reduced to the corresponding amine derivatives by hydrogenation.
Scheme 46.

Torquoselective Nazarov cyclizations reported from the Flynn laboratory.
The same group further extended the scope of diastereoselective ynamide Nazarov cyclizations to the synthesis of complicated cyclopentane structures carrying several quaternary chirality centers (Scheme 47).91 In analogy to their previous work, a stepwise protocol toward the central divinyl ketone motif was used. The treatment of 6 with either a Grignard reagent or Et2Zn in the presence of CuBr·SMe2 or Rh(acac)COD, Cat-9, respectively, first generates intermediate enamides 121. Two alternative methods can then be followed to make the divinyl ketones 119. This is either accomplished by a carbonylative Stille cross-coupling reaction with a vinylstannane and carbon monoxide in the presence of catalytic amounts of [1,1′-bis(di-tert-butylphosphino)ferrocene]dichloropalladium(II) Cat-10 and copper iodide or by addition of 121 to vinyl or aryl aldehydes, followed by oxidation with Dess-Martin periodinane (DMP). Compounds 119 are finally transformed to 122 via Nazarov cyclization with a Lewis or Brønsted acid. The chiral oxazolidinone auxiliary again controls the torquoselectivity in this reaction affording asymmetric cyclopentanoid products with high diastereoselectivity. It is also possible to trap the oxyallyl cation intermediates of the Nazarov cyclization with suitable nucleophiles. For example, the addition of 10 equivalents of N-methyl indole 93 produced compound 123 in greater than 20:1 dr and intramolecular trapping with 119b gave 124 containing three contiguous quaternary chirality centers as a single isomer.
Scheme 47.

Synthesis of cyclopentanoids by asymmetric electrocyclization.
Lin developed a diastereoselective method providing access to multifunctional pyrido- and pyrrolo[1,2-c][1,3]oxazin-1-ones via a one-pot nucleophilic addition-cyclization process with ynamides and chiral N,O-acetals.92 The coupling of N,O-acetals 125 with ynesulfonamides 2 or cyclic and acyclic ynecarbamates 5 and 6, respectively, in the presence of BF3·Et2O at low temperatures afforded the bicyclic unsaturated bicycles 126 in moderate to high yields and with dr’s greater than 99:1 (Scheme 48). A plausible mechanism for this reaction involves Lewis acid promoted formation of iminium ion A that is attacked by the nucleophilic ynamide. The 1,2-asymmetric induction is controlled by the bulky TBDMS ether moiety. Removal of the Boc group and subsequent intramolecular cyclization then furnishes compounds 126.
Scheme 48.
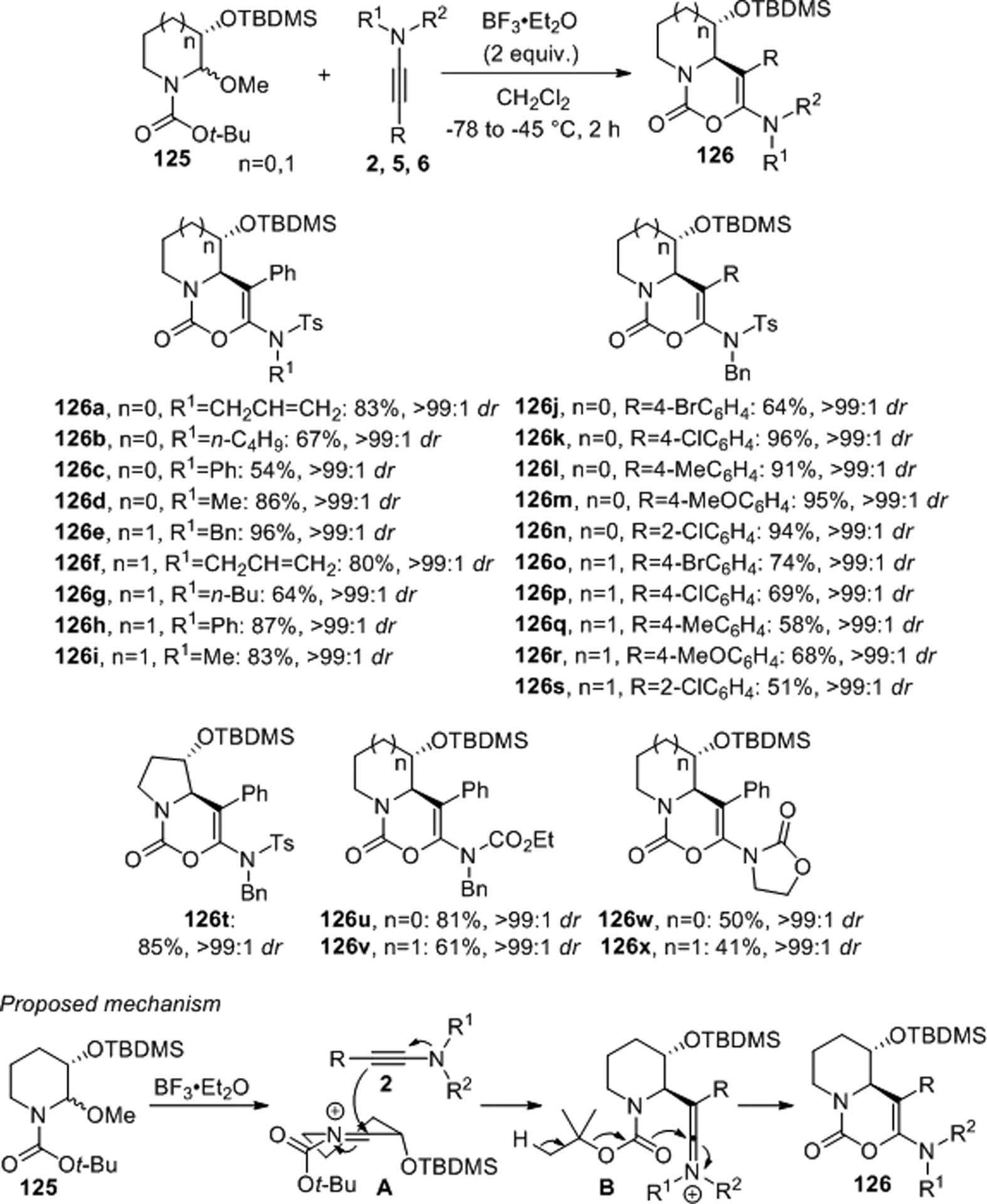
Nucleophilic addition-cyclization of ynamides with N,O-acetals.
5. Functionalization of Terminal Ynamides
The vast majority of asymmetric reactions has been accomplished with internal ynamides through conversion of the nitrogen substituted triple bond into a new scaffold, for example during a cycloaddition or sigmatropic rearrangement. The presence of a terminal CH bond generates additional opportunities for carbon-carbon bond formation that leave the ynamide moiety intact and available for further modifications if desired. The possibility of CH deprotonation pathways and the inherent enamide-like nucleophilicity of terminal ynamides have been explored in several 1,2-addition reactions with carbonyl and imine electrophiles. Typically, these transformations proceed with excellent stereoselectivities. Other activation modes discussed above for internal ynamides, for example the formation of transient N,O-ketene acetals, are of course also available and occur with concurrent triple bond derivatization.
The diastereoselective addition of litihiated ynesulfonamides 2 or ynecarbamates 6 to the Ellman-Davis N-tert-butylsulfinyl imines 127 was reported by Hsung and coworkers in 2013 (Scheme 49).93,94 Initially, relatively low diastereoselectivities were observed when the reaction was conducted at −78 °C. Further optimization, however, revealed that the (SS,S)-γ-aminoynamides 128 are formed in moderate to high yields with up to 25:1 dr when the temperature is increased to −40 °C. The sense of asymmetric induction, i.e. the formation of homochiral products, is in agreement with the Zimmerman-Traxler type transition state A. Interestingly, addition of boron trifluoride diethyl etherate resulted in reversed stereoselectivity. The preferential formation of (SS,R)-γ-aminoynamides under these conditions was attributed to acyclic synclinal and antiperiplanar transition states B and C, respectively. It is noteworthy that this procedure generated the heterochiral adducts in high yields and diastereoselectivities from a variety of N-sulfonyl-, carbamoyl-, phosphoryl-, and oxazolidinone derived ynamides. The incorporation of a chiral oxazolidinone ring into the ynamide structure did show minor effects on the selectivity which apparently is overwhelmingly controlled by the chiral N-tert-butylsulfinyl moiety.
Scheme 49.

Hsung’s diastereoselective synthesis of γ-aminoynamides.
In 2014, our group reported the first catalytic enantioselective nucleophilic 1,2-additions with terminal ynamides.95 Extensive screening of reaction variables revealed that the indole-derived ynamides 7 react with aldehydes 11 to the propargylic alcohols 129 in high yields and ee’s in the presence of Hünig’s base and catalytic amounts of zinc triflate and N-methyl ephedrine (Scheme 50). Careful reaction analysis during optimization uncovered that the ynamide addition is reversible. As a result of competing racemization, high conversions were initially only achieved at the expense of the enantioselectivity which significantly decreased over time. This issue was successfully addressed by resorting to apolar solvents to affect precipitation of the reaction product. Under these conditions, the racemization course was efficiently suppressed and the reaction between 3-benzoylindolyl ynamide 7 and 4-bromobenzaldehyde, gave 129a in 97% yield and 93% ee. Similar results were produced with a total of 15 electron-rich and electron-deficient aromatic aldehydes as well as with aliphatic substrates.
Scheme 50.

The first catalytic enantioselective nucleophilic 1,2-addition with terminal ynamides.
Two years later, we presented a catalytic procedure that enables enantioselective addition of N-ethynyl-N-butylbenzenesulfonamide, 2, to aromatic trifluoromethyl ketones 25 (Scheme 51).96 Using 10 mol% of Zn(OTf)2 and the bis(prolinol)phenol ligand L-14 together with triethyl phosphate to accelerate catalyst turnover, a series of tertiary β-hydroxy-β-trifluoromethyl ynamides 130 were produced in 91–99% yield and with 85–96% ee. Interestingly, this reaction is ligand-accelerated and does not require a base. Terminal alkynes are not consumed under the same conditions which accentuates the remarkable difference in the chemistry of nonpolarized triple bonds and ynamides. The addition products were shown to undergo regioselective hydration, diastereoselective reductions and hydroacyloxylation without erosion of the enantiomeric purity, thus providing access to synthetically useful trifluoromethylated chiral alcohols carrying adjacent (Z)- and (E)-enamide, amide and N,O-ketene acetal functionalities.
Scheme 51.
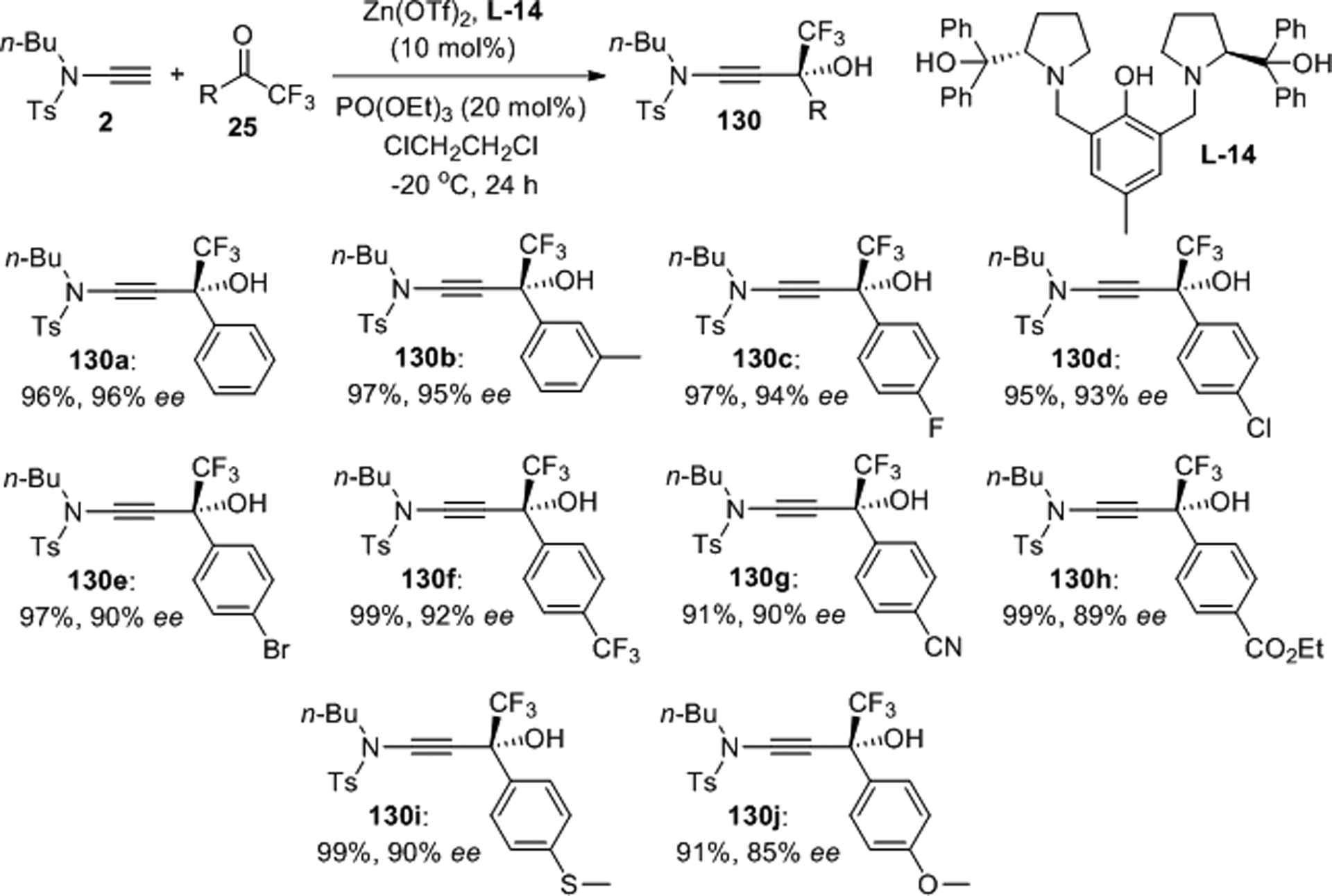
Catalytic enantioselective preparation of tertiary β-hydroxy-β-trifluoromethyl ynamides.
Catalytic enantioselective ynamide additions do not only produce versatile chiral building blocks but also provide streamlined access to important natural products.97 This was demonstrated by our group with the copper(bisoxazolidine L-15) catalyzed addition of sulfonynamides 2 to isatins 131 as shown in Scheme 52. This procedure has several remarkable features that are noteworthy. The 1,2-addition is ligand-accelerated, occurs under mild base-free conditions, is scalable, it has a broad substrate scope that includes unprotected isatins, and it affords highly sought-after 3-hydroxyoxindoles 132 carrying a tetrasubstituted chirality center with excellent yields and enantioselectivities. Again, alkynes do not react under the same conditions while ynamides display enamide-like nucleophilicity and are therefore able to react without prior deprotonation. A mechanism that is in agreement with a direct, base-free addition of the ynamide to the Lewis acid activated isatin electrophile and the formation of a keteniminium intermediate that undergoes rapid tautomerization to the final product was proposed. Exhaustive triple bond hydrogenation and the development of a reductive sulfonamide bond cleavage protocol allowed efficient asymmetric chimonamidine alkaloid synthesis. Alternatively, the addition products can be transformed into multifunctional 3-hydroxyindolinone derivatives by hydration, partial hydrogenation or hydroxyacyloxylation of the ynamide moiety at room temperature.
Scheme 52.
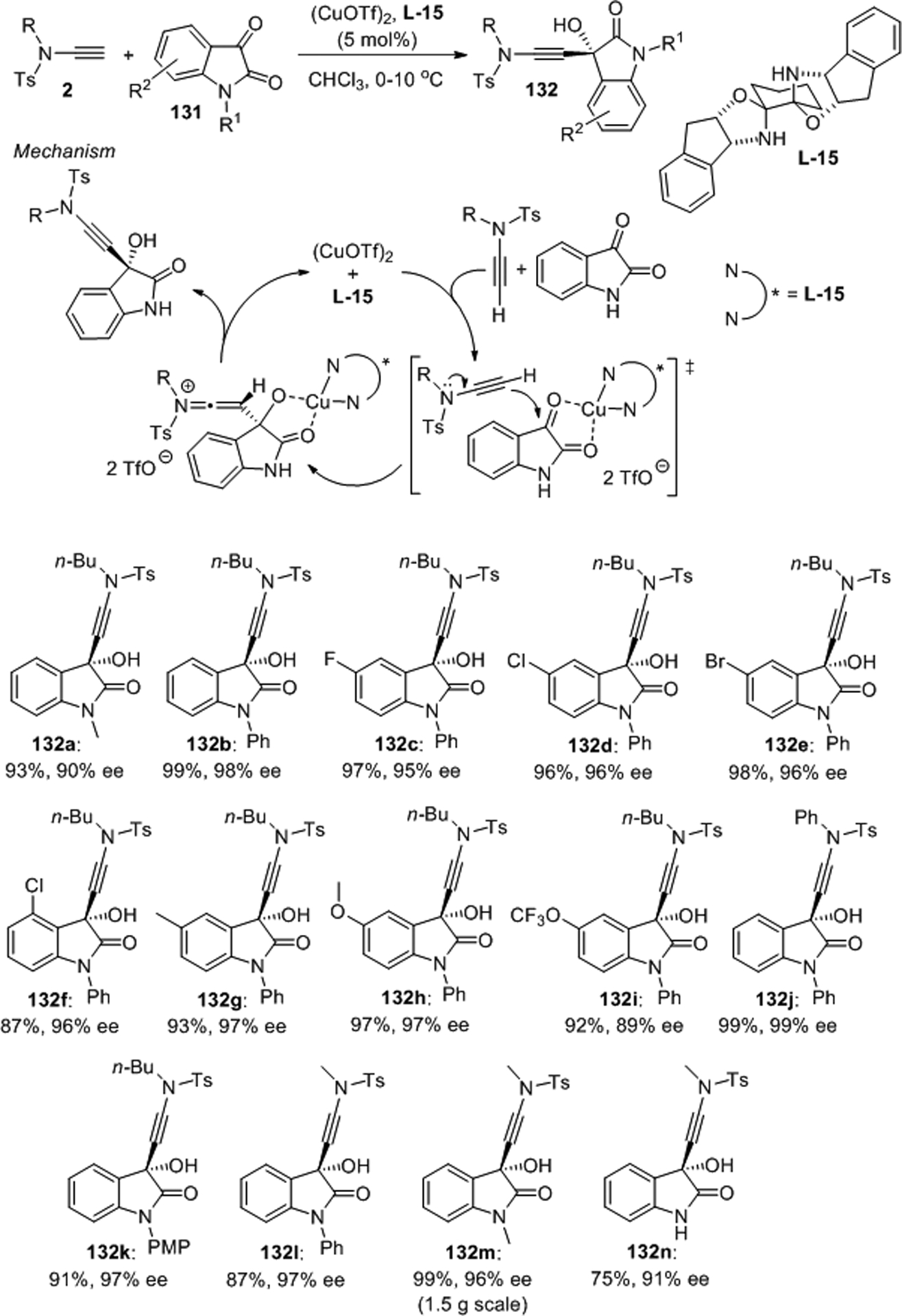
Copper catalyzed enantioselective additions to isatins.
Recently, Feng and colleagues developed a catalytic asymmetric three-component reaction with ynamides 2, carboxylic acids 133 and ortho-hydroxybenzyl alcohols 134 (Scheme 53).98 The reaction proceeds in the presence of catalytic amounts of chiral N,N’-dioxide L-16 and Sc(OTf)3 and gives chiral α-acyloxyenamides 135 in up to 99% ee under relatively mild conditions. Mechanistic studies are in agreement with a scandium(III) promoted dehydration of 134a to the transient Michael acceptor ortho-quinone methide B. In parallel, hydroacyloxylation of 2 generates the acyloxyenamide C which then undergoes 1,4-conjugate addition to B. The resulting zwitterion D finally affords the α-acyloxyenamide 135a and thereby regenerates the catalyst. It is noteworthy that α-acyloxyenamides have also been prepared from ynamides and chiral Boc-protected lactams.99
Scheme 53.

Feng’s catalytic asymmetric three-component hydroacyloxylation/1,4-conjugate addition sequence.
Akai et al. found that chiral allenes can be prepared from 2 and propargyl tertiary alcohols 136 under mild conditions by a one-pot procedure (Scheme 54).100 In the presence of 1 mol% of silver triflate the intermediates 137 are formed and undergo [3,3] sigmatropic rearrangement to allenes 138 in moderate to good yields and with excellent central-to-axial chirality transfer. This work paves the way for enantiospecific synthesis of multifunctional allenes from chiral propargylic alcohols which are readily available from terminal alkynes. It thus serves as an intriguing example that merges the rapidly increasing space of asymmetric ynamide chemistry with traditional transformations of the parental alkyne functionality.
Scheme 54.

Enantiospecific synthesis of tetrasubstituted allenes via chirality transfer with a propargylic alcohol.
6. Miscellaneous Addition Reactions
Several groups have developed powerful triple bond addition strategies that exploit the inherent ynamide polarity in distinctive ways to set the stage for asymmetric bond construction. While some of these examples are mechanistically related to the reaction types discussed in the preceding sections, they are discussed here separately to highlight the general potential of the underlying ynamide manipulation tactics.
Evano reported the asymmetric synthesis of α-halogenated imides 139 using excess of TMSCl, an N-halosuccinimide derivative, NXS, and water (Scheme 55).101 The reaction is probably initiated by ynamide hydrochlorination generating the transient chloroenamide B. Electrophilic halogenation then furnishes the chloroiminium C albeit with relatively low asymmetric induction. In the final step, the iminium hydrolyzes to the α-halogenated imide 139.
Scheme 55.

Asymmetric synthesis of α-halogenated imides from a chiral oxazolidinone derived ynamide.
Marek and coworkers prepared α,α-disubstituted N-acyl oxazolidinones with good yields and diastereoselectivites through tandem syn-addition of organocuprates to chiral ynamides, oxidation and SN2 reaction with allyl bromides (Scheme 56).102–104 Stereo- and regioselective carbometalation of 6 with organocuprates gave the exhaustively substituted enolate 140 which upon oxidation with tert-butylperoxylithium afforded 141. Carbon-carbon bond formation with allyl bromides 142 then produced the N-acyl oxazolidinones 143. Compared to allyl bromide or allyl iodide, the reaction proceeded smoothly and with higher diastereoselectivity when more electrophilic bromomethyl acrylates were employed. The authors achieved successful removal of the chiral oxazolidinone moiety to the corresponding thioesters without erosion of the stereoisomeric purity.
Scheme 56.
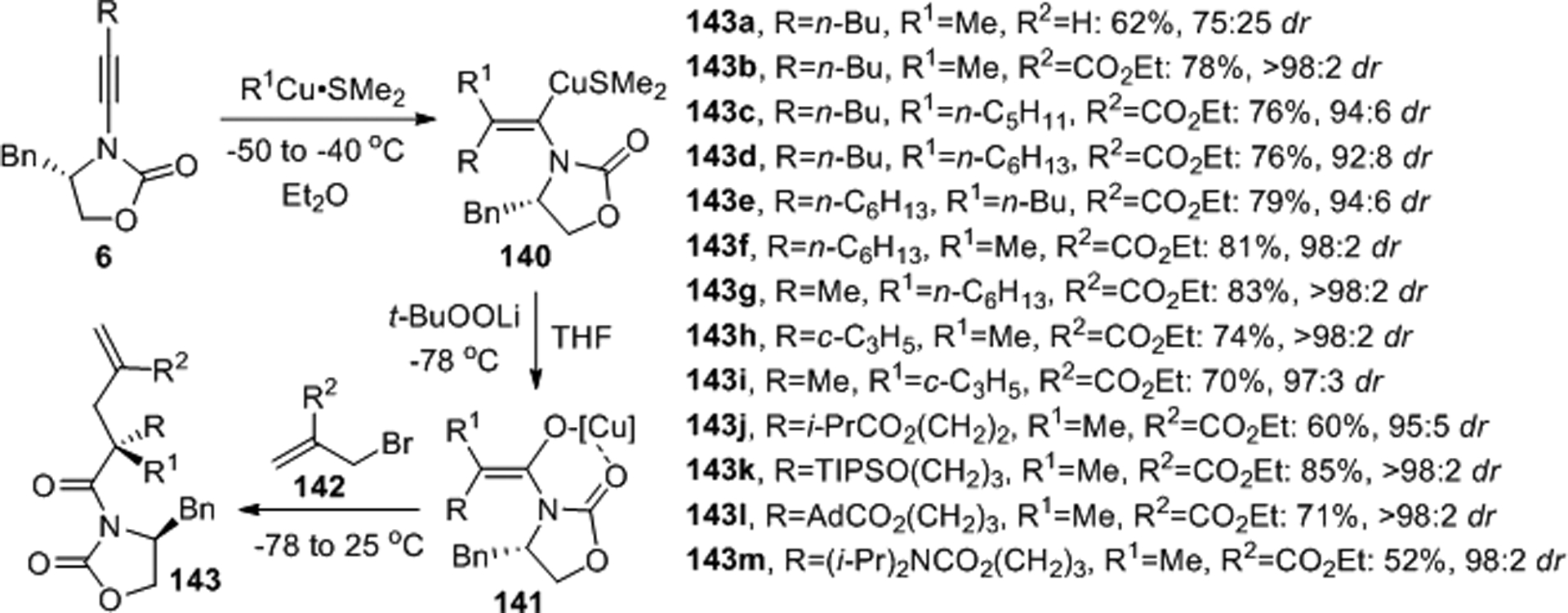
Diastereoselective tandem syn-addition of organocuprates to chiral ynamides, oxidation and SN2 reaction with allyl bromides.
The Shin group developed complementary oxidation routes that transform ynamides either into chiral α,α-diaryl amides or into α-acyl amides that undergo asymmetric rearrangement into ketones bearing an adjacent N,O-acetal group.105–107 In contrast to related work from the Maulide laboratory,108 the asymmetric conversion of sulfonynamides 2 into α-aryl amides was achieved with excess of the N,N’-dioxide (P)-144 as oxidant and 20 mol% of HNTf2 at slightly lower temperatures (Scheme 57). The authors rationalized that activation of a sulfonynamide 2 by the acid first yields the keteniminium A which reacts with the N,N’-dioxide 144. The corresponding enolonium B then undergoes a stereocontrolled Friedel-Crafts reaction with an indole or another suitable aromatic compound toward α,α-diaryl sulfonamides such as 145a. A transition state C that is stabilized by π-stacking interactions and favoring a Re-face approach of the indole through an SN2’-type reaction course was proposed. A series of over forty α,α-diaryl amides were produced from indoles, pyrroles, phenols and naphthols using this redox arylation procedure, often with excellent yields and ee’s.
Scheme 57.
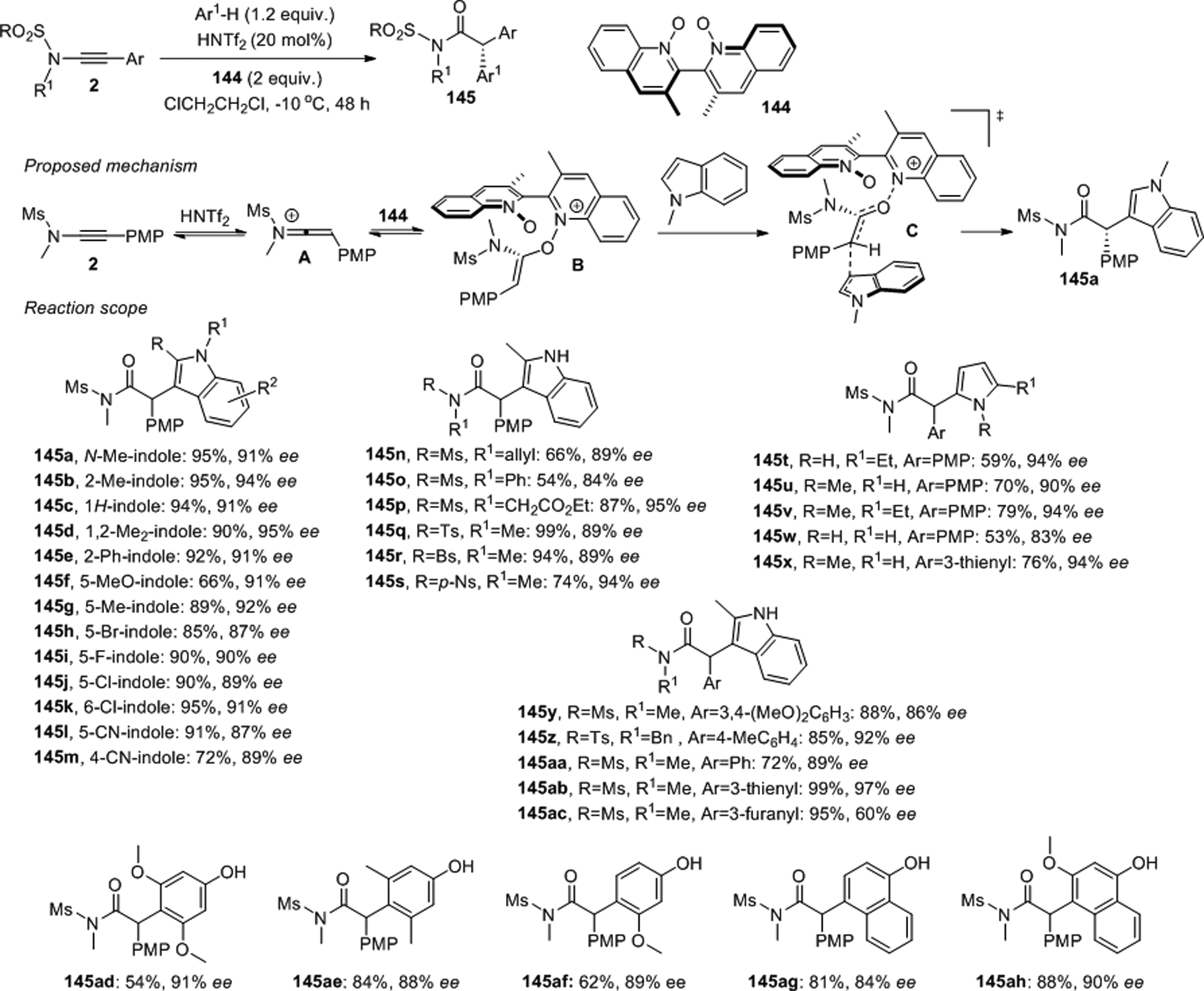
Enantioselective redox arylation of sulfonynamides with an N,N’-dioxide.
Alternatively, regioselective β-oxidation of sulfonynamides with meta-chloroperoxybenzoic acid (m-CPBA) at room temperature results in the formation of racemic N,O-acetals 146 (Scheme 58). Activation of 146 by the chiral Brønsted acid Cat-11 at 80 °C promotes asymmetric transacetalization with the diaryl alcohol 147, giving rise to synthetically useful N,O-acetals 148 that were obtained in up to 90% ee.
Scheme 58.

Shin’s β-oxidation and enantioselective transacetalization procedure.
Zhu introduced p-tolylsulfenyl chloride 149 to a DMSO mediated diastereoselective oxothiolation reaction with oxazolidinone derived ynamides 6 (Scheme 59).109 This method is unique because the N-protecting group is not only a simple spectator but actively participates in the transformation. The reaction requires relatively harsh conditions and probably involves the transient ion pair A which rearranges to oxazolidine-2,4-diones 150 carrying an arylthio-substituted quaternary carbon center.
Scheme 59.

Oxothiolation with p-tolylsulfenyl chloride and DMSO.
7. Conclusions
As we have illustrated in this review, increasingly powerful asymmetric transformations with ynamides have been developed in the last 10 years. The staggering diversity of reaction products that have become accessible through these efforts originates without a doubt to a large extent from the unique reactivity of the polarized triple bond which provides synthetic chemists with an array of opportunities that are distinctly different from traditional alkyne chemistry. To date, many groups have recognized the general utility of readily available, benchstable ynamide derivatives and successfully tailored carbon-carbon and carbon-heteroatom bond formation strategies that use these attractive building blocks for the construction of multifunctional scaffolds often displaying several chirality centers with excellent regio- and stereocontrol. The prevalence of pericyclic mechanisms, in particular cycloadditions and sigmatropic rearrangements, stands out but many other reaction types including cycloisomerizations, organometallic transformations, oxidative mechanisms, ring annulations as well as cascade-type combinations thereof add to the striking synthetic diversity.
The advance with diastereoselective methods that utilize chiral ynamides or reagents has been matched by many elegant Brønsted acid or transition metal complex catalyzed enantioselective variations. The trend toward asymmetric catalysis and diversity oriented ynamide transformations with impressive chemo-, regio- and stereocontrol is expected to continue as ample guidance for such developments already exists based on the tremendous amount of mechanistic insights uncovered during the last decade. We hope that the fascinating chemical diversity and the unique reactivity patterns highlighted in this review will encourage many synthetic chemists to further explore new directions and applications of ynamide chemistry. Despite recent achievements and the steady advance of this field, many challenges and opportunities for future discoveries remain. To this end, the scarcity of asymmetric reactions with terminal ynamides in comparison to their alkyne analogues is noticeable and points out one of several important directions. In several cases improvements of currently unsatisfactory yields and stereoselectivities need to be demonstrated to fully capitalize on the undeniable promise of asymmetric synthesis with ynamides. In fact, many reactions generate racemic products or proceed with suboptimal asymmetric induction which requires additional method optimization or if unsuccessful revised reaction strategies that address these limitations. At the same time, more practical procedures that allow further manipulation or removal of the electron-withdrawing sulfonyl, alkoxycarbonyl or acyl group typically attached to the ynamide nitrogen atom are needed. This would extend the current focus on synthetic methodology developments toward applications in natural product synthesis or drug development programs.
Acknowledgements.
We gratefully acknowledge financial support from NIH (GM106260).
8. References
- 1.DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y and Hsung RP, Chem. Rev, 2010, 110, 5064–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evano G, Coste A and Jouvin K, Angew. Chem. Int. Ed, 2010, 49, 2840–2859. [DOI] [PubMed] [Google Scholar]
- 3.Evano G, Blanchard N, Compain G, Coste A, Demmer CS, Gati W, Guissart C, Heimburger J, Henry N, Jouvin K, Karthikeyan G, Laouiti A, Lecomte M, Martin-Mingot A, Métayer B, Michelet B, Nitelet A, Theunissen C, Thibaudeau S, Wang J, Zarca M and Zhang C, Chem. Lett, 2016, 45, 574–585. [Google Scholar]
- 4.Saito N, Sato Y, J. Synth. Org. Chem. Jpn, 2018, 76, 699–709. [Google Scholar]
- 5.Wang X-N, Yeom H-S, Fang L-C, He S, Ma Z-X, Kedrowski BL and Hsung RP, Acc. Chem. Res, 2014, 47, 560–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nayak S, Prabagar B and Sahoo AK, Org. Biomol. Chem, 2016, 14, 803–807. [DOI] [PubMed] [Google Scholar]
- 7.Prabagar B, Ghosh N and Sahoo AK, Synlett, 2017, 28, 2539–2555. [Google Scholar]
- 8.Pan F, Shua C and Ye L-W, Org. Biomol. Chem, 2016, 14, 9456–9465. [DOI] [PubMed] [Google Scholar]
- 9.Lu T and Hsung RP, ARKIVOC, 2014. (i) 127–141. [PMC free article] [PubMed] [Google Scholar]
- 10.Evano G, Michelet B and Zhang C, C. R. Chimie, 2017, 20, 648–664. [Google Scholar]
- 11.Cook AM and Wolf C Tetrahedron Lett, 2015, 56, 2377–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodd RH and Cariou K, Chem. Eur. J, 2018, 24, 2297–2304. [DOI] [PubMed] [Google Scholar]
- 13.Hu L and Zhao J, Synlett, 2017, 28, 1663–1670. [Google Scholar]
- 14.Huang B and Cui S, Drug Discov. Today, 2018, 29, 43–49. [DOI] [PubMed] [Google Scholar]
- 15.Duret G, Le Fouler V, Bisseret P, Bizet V and Blanchard N, Eur. J. Org. Chem, 2017, 46, 6816–6830. [Google Scholar]
- 16.Reinus B and Kerwin SM, Molecules, 2019, 24, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou B, Tan T-D, Zhu X-Q, Shang M and Ye L-W, ACS Catal, 2019, 9, 6393–6406. [Google Scholar]
- 18.a) Selected examples:Fadel A, Legrand F, Evano G and Rabasso N, Adv. Synth. Catal, 2011, 353, 263–267. [Google Scholar]; b) Saito N, Saito K, Shiro M and Sato Y, Org. Lett, 2011, 13, 2718–2721. [DOI] [PubMed] [Google Scholar]; c) Barbazanges M, Meyer C, Cossy J and Turner P, Chem. Eur. J, 2011, 17, 4480–4495. [DOI] [PubMed] [Google Scholar]; d) Banerjee B, Litvinov DN, Kang J, Bettale JD and Castle SL, Org. Lett, 2010, 12, 2650–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shindoh N, Kitaura K, Takemoto Y and Takasu K, J. Am. Chem. Soc, 2011, 133, 8470–8473. [DOI] [PubMed] [Google Scholar]; f) Smith DL, Goundry WRF and Lam HW, Chem. Commun, 2012, 48, 1505–1507. [DOI] [PubMed] [Google Scholar]; g) Compain G, Jouvin K, Martin-Mingot A, Evano G, Marrot J and Thibaudeau S, Chem. Commun, 2012, 48, 5196–5198. [DOI] [PubMed] [Google Scholar]; h) Yang Y, Wang L, Zhang F and Zhu G, J. Org. Chem, 2014, 79, 9319–9324. [DOI] [PubMed] [Google Scholar]; i) Pan F, Shu C, Ping Y-F, Pan Y-F, Ruan P-P, Fei Q-R and Ye L-W, J. Org. Chem, 2015, 80, 10009–10015. [DOI] [PubMed] [Google Scholar]; j) Vercruysse S, Jouvin K, Riant O and Evano G, Synthesis, 2016, 48, 3373–3381. [Google Scholar]; k) Shu C, Shen C-H, Wang Y-H, Li L, Li T, Lu X and Ye L-W, Org. Lett, 2016, 18, 4630–4633. [DOI] [PubMed] [Google Scholar]; l) Chen L, Yu L, Deng Y, Cui Y, Bian G and Cao J, Org. Biomol. Chem, 2016, 14, 564–569. [DOI] [PubMed] [Google Scholar]; m) Saito N, Abdullah I, Hayashi K, Hamada K, Koyoma M and Sato Y, Org. Biomol. Chem, 2016, 14, 10080–10089. [DOI] [PubMed] [Google Scholar]; n) Zeng X, Lu Z, Liu S, Hammond GB and Xu B, J. Org. Chem, 2017, 82, 13179–13187. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Kim SW, Um T-W and Shin S, Chem. Commun, 2017, 53, 2733–2736. [DOI] [PubMed] [Google Scholar]; p) Baldassari LL, de la Torre A, Li J, Lüdtke DS and Maulide N, Angew. Chem. Int. Ed, 2017, 56, 15723–15727. [DOI] [PubMed] [Google Scholar]; q) Yu J, Xu G, Tang S, Shao J and Sun J, Org. Lett, 2019, 21, 9076–9079. [DOI] [PubMed] [Google Scholar]
- 19.For example:; a) Willumstad TP, Haze O, Mak XY, Lam TY, Wang Y-P and Danheiser RL, J. Org. Chem, 2013, 78, 11450–11469. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu J, Chen M, Zhang L and Liu Y, Chem. Eur. J, 2015, 21, 1009–1013. [DOI] [PubMed] [Google Scholar]; c) Willumstad TP, Boudreau PD, and Danheiser RL, J. Org. Chem, 2015, 80, 11794–11805. [DOI] [PubMed] [Google Scholar]; d) Gillie AD, Reddy RJ and Davies PW, Adv. Synth. Catal, 2016, 358, 226–239. [Google Scholar]; e) Forneris CC, Wang Y-P, Mamaliga G, Willumstad TP and Danheiser RL, Org. Lett, 2018, 20, 6318–6322. [DOI] [PubMed] [Google Scholar]; f) Zhou B, Tan T-D, Zhu X-Q, Shang M and Ye L-W, ACS Catal, 2019, 9, 6393–6406. [Google Scholar]
- 20.Selected examples; a) Davies PW, Cremonesi A and Martin N, Chem. Commun, 2011, 47, 379–381. [DOI] [PubMed] [Google Scholar]; b) Kramer S, Friis SD, Xin Z, Odabachian Y and Skrydstrup T, Org. Lett, 2011, 13, 1750–1753. [DOI] [PubMed] [Google Scholar]; c) Ding R, Li Y, Tao C, Cheng B and Zhai H, Org. Lett, 2015, 17, 3994–3997. [DOI] [PubMed] [Google Scholar]; d) Li C and Zhang L, Org. Lett, 2011, 13, 1738–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ao C, Yang X, Jia S, Xu X, Yuan Y, Zhang D and Hu W, J. Org. Chem, 2019, 84. 15331–15342. [DOI] [PubMed] [Google Scholar]
- 21.Selected examples:; a) Chemala F, Dulong F, Ferreira F, Nüllen MP and Pérez-Luna A, Synthesis, 2011, 9, 1347–1360. [Google Scholar]; b) Li L, Zhou B, Wang Y-H, Shu C, Pan Y-F, Lu X and Ye L-W, Angew. Chem. Int. Ed, 2015, 54, 8245–8249. [DOI] [PubMed] [Google Scholar]; c) Wang Z-S, Chen Y-B, Zhang H-W, Sun Z, Zhu C and Ye L-W, J. Am. Chem. Soc, 2020, 142, 3636–3644. [DOI] [PubMed] [Google Scholar]; d) Pinto A, Kaiser D, Maryasin B, Di Mauro G, González L and Maulide N, Chem. Eur. J, 2018, 24, 2515–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tlais SF and Danheiser RL, J. Am. Chem. Soc, 2014, 136, 15489–15492. [DOI] [PubMed] [Google Scholar]; f) Graux LV, Clavier H and Buono G, ChemCatChem, 2014, 6, 2544–2548. [Google Scholar]; g) Tona V, Ruider SA, Berger M, Shaaban S, Padmanaban M, Xie L-G, Gonzalez L and Maulide N, Chem. Sci, 2016, 7, 6032–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Jadhav AM, Huple DB, Singh RR and Liu RS, Adv. Synth. Catal, 2016, 358, 1017–1022. [Google Scholar]; i) Andna L and Miesch L, Org. Lett, 2018, 20, 3430–3433. [DOI] [PubMed] [Google Scholar]; j) Chen R, Zeng L, Huang B, Shen Y and Cui S, Org. Lett, 2018, 20, 3377–3380. [DOI] [PubMed] [Google Scholar]
- 22.Valenta P, Carroll PJ and Walsh PJ, J. Am. Chem. Soc, 2010, 132, 14179–14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nairoukh Z and Marek I, Angew. Chem. Int. Ed, 2015, 54, 14393–14397. [DOI] [PubMed] [Google Scholar]
- 24.Minko Y, Pasco M, Lercher L and Marek I, Nat. Protoc, 2013, 8, 749–754. [DOI] [PubMed] [Google Scholar]
- 25.Minko Y, Pasco M, Lercher L, Botoshansky M and Marek I, Nature, 2012, 490, 522–526. [DOI] [PubMed] [Google Scholar]
- 26.Grimster NP, Wilton DAA, Chan LKM, Godfrey CRA, Green C, Owen DR and Gaunt M, J. Tetrahedron, 2010, 66, 6429–6436. [Google Scholar]
- 27.Saito N, Katayama T and Sato Y, Heterocycles, 2010, 82, 1181–1187. [Google Scholar]
- 28.Jadhav AM, Pagar VV, Huple DB and Liu RS, Angew. Chemie - Int. Ed, 2015, 54, 3812–3816. [DOI] [PubMed] [Google Scholar]
- 29.Liu YW, Mao ZY, Nie X-D, Si CM, Wei BG and Lin GQ, J. Org. Chem, 2019, 84, 16254–16261. [DOI] [PubMed] [Google Scholar]
- 30.Ficini J, Tetrahedron, 1976, 32, 1448–1486. [Google Scholar]
- 31.Riddell N, Villeneuve K and Tam W, Org. Lett, 2005, 7, 3681–3684. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Hsung RP, DeKorver KA and Wei Y, Org. Lett., 2010, 12, 3780–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith DL, Chidipudi SR, Goundry WR and Lam HW, Org. Lett, 2012, 14, 4934–4937. [DOI] [PubMed] [Google Scholar]
- 34.Yuan Y, Bai L, Nan J, Liu J and Luan X, Org. Lett, 2014, 16, 4316–4319. [DOI] [PubMed] [Google Scholar]
- 35.Aikawa K, Hioki Y, Shimizu N and Mikami K, J. Am. Chem. Soc, 2011, 133, 20092–20095. [DOI] [PubMed] [Google Scholar]
- 36.DeKorver KA, Walton MC, North TD and Hsung RP, Org. Lett, 2011, 13, 4862–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dekorver KA, Hsung RP, Song WZ, Wang XN and Walton MC, Org. Lett, 2012, 14, 3214–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schotes C and Mezzetti A, J. Org. Chem, 2011, 76, 5862–5866. [DOI] [PubMed] [Google Scholar]
- 39.Schotes C and Mezzetti A, Angew. Chem. Int. Ed, 2011, 50, 3072–3074. [DOI] [PubMed] [Google Scholar]
- 40.Schotes C, Bigler R and Mezzetti A, Synthesis, 2012, 44, 513–526. [Google Scholar]
- 41.Schotes C, Althaus M, Aardoom R and Mezzetti A, J. Am. Chem. Soc, 2012, 134, 1331–1343. [DOI] [PubMed] [Google Scholar]
- 42.Enomoto K, Oyama H and Nakada M, Chem. Eur. J, 2015, 21, 2798–2802. [DOI] [PubMed] [Google Scholar]
- 43.Schotes C and Mezzetti A, Angew. Chem. Int. Ed, 2011, 50, 3072–3074. [DOI] [PubMed] [Google Scholar]
- 44.Yang Y, Liu H, Peng C, Wu J, Zhang J, Qiao Y, Wang X-N and Chang J, Org. Lett, 2016, 18, 5022–5025. [DOI] [PubMed] [Google Scholar]
- 45.Mak XY, Crombie AL and Danheiser RL, J. Org. Chem, 2011, 76, 1852–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clavier H, Lepronier A, Bengobesse-Minsta N, Gatineau D, Pellissier H, Giordano L, Tenaglia A and Buono G, Adv. Synth. Catal, 2013, 355, 403–408. [Google Scholar]
- 47.Li X, Wang Z, Ma X, Liu PN and Zhang L, Org. Lett, 2017, 19, 5744–5747. [DOI] [PubMed] [Google Scholar]
- 48.Dateer RB, Shaibu BS and Liu R-S, Angew. Chem. Int. Ed, 2012, 53, 113–117. [DOI] [PubMed] [Google Scholar]
- 49.Jiang C, Yu P-P, Zhang Q, Xu H-D and Shen M-H, Chin. Chem. Lett, 2019, 30, 266–268. [Google Scholar]
- 50.Saito N, Ichimaru T and Sato Y, Org. Lett, 2012, 14, 1914–1917. [DOI] [PubMed] [Google Scholar]
- 51.Karad SN, Bhunia S and Liu R-S, Angew. Chem., Int. Ed, 2012, 51, 8722–8726. [DOI] [PubMed] [Google Scholar]
- 52.Hong F-L, Chen Y-B, Ye S-H. Zhu G-Y, Zhu X-Q, Lu X, Liu R-S and Ye L-W, J. Am. Chem. Soc, 2020, 142, 7618–7626. [DOI] [PubMed] [Google Scholar]
- 53.Hong F-L, Wang Z-S, Wei D-D, Zhai T-Y, Deng G-C, Lu X, Liu R-S and Ye L-W, J. Am. Chem. Soc, 2019, 141, 16961–16970. [DOI] [PubMed] [Google Scholar]
- 54.Macka WD, Fistikci M, Carris RM and Johnson JS, Org. Lett, 2014, 16, 1626–1629. [DOI] [PubMed] [Google Scholar]
- 55.Zhao Y, Wang C, Hu Y and Wan B, Chem. Commun, 2018, 54, 3963–3966. [DOI] [PubMed] [Google Scholar]
- 56.Duret G, Quinlan R, Martin RE, Bisseret P, Neuburger M, Gandon V and Blanchard N, Org. Lett 2016, 18, 1610–1613. [DOI] [PubMed] [Google Scholar]
- 57.Duret G, Quinlan R, Yin B, Martin RE, Bisseret P, Neuburger M, Gandon V and Blanchard N, J. Org. Chem 2017, 82, 1726–1742. [DOI] [PubMed] [Google Scholar]
- 58.Nishimura T, Takiguchi Y, Maeda Y and Hayashi T, Adv. Synth. Catal, 2013, 355, 1374–1382. [Google Scholar]
- 59.Clavier H, Lepronier A, Bengobesse-Minsta N, Gatineau D, Pellissier H, Giordano L, Tenaglia A and Buono G, Adv. Synth. Catal, 2013, 355, 403–408. [Google Scholar]
- 60.Garcia P, Harrak Y, Diab L, Cordier P, Ollivier C, Gandon V, Malacria M, Fensterbank L and Aubert C, Org. Lett, 2011, 13, 2952–2955. [DOI] [PubMed] [Google Scholar]
- 61.Marien N, Reddy BN, De Vleeschouwer F, Goderis S, Van Hecke K and Verniest G, Angew. Chem. Int. Ed, 2018, 57, 5660–5664. [DOI] [PubMed] [Google Scholar]
- 62.Heffernan SJ and Carbery DR, Tetrahedron Lett, 2012, 53, 5180–5182. [Google Scholar]
- 63.Brioche J, Meyer C and Cossy J, Org. Lett, 2013, 15, 1626–1629. [DOI] [PubMed] [Google Scholar]
- 64.Peng B, Huang X, Xie L-G and Maulide N, Angew. Chem. Int. Ed, 2014, 53, 8718–8721. [DOI] [PubMed] [Google Scholar]
- 65.Kaldre D, Maryasin B, Kaiser D, Gajsek O, González L and Maulide N, Angew. Chem. Int. Ed, 2017, 56, 2212–2215. [DOI] [PubMed] [Google Scholar]
- 66.Kaldre D, Klose I and Maulide N, Science, 2018, 361, 664–667. [DOI] [PubMed] [Google Scholar]
- 67.Maryasin B, Kaldre D, Galaverna R, Klose I, Ruider S, Drescher M, Kählig H, González L, Eberlin MN, Jurberg ID and Maulide N, Chem. Sci, 2018, 9, 4124–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adcock HV, Chatzopoulou E and Davies PW, Angew. Chem., Int. Ed, 2015, 54, 15525–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng N, Chang YY, Zhang LJ, Gong JX and Yang Z, Chem. - An Asian J, 2016, 11, 371–375. [DOI] [PubMed] [Google Scholar]
- 70.Liang G, Ji Y, Liu H, Pang Y, Zhou B, Cheng M, Liu Y, Lin B and Liu Y, Adv. Synth. Catal, 2020, 362, 192–205. [Google Scholar]
- 71.Straker RN, Peng Q, Mekareeya A, Paton RS and Anderson EA, Nat. Commun, 2016, 7, 10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Craig RW, Campbell D, Suleman A, Carr G and Anderson EA, Angew. Chem. Int. Ed, 2013, 52, 9139–9143. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y-Q, Zhu X-Q, Xu Y, Bu H-Z, Wang J-L. Zhai T-Y, Zhou J-M and Ye L-W, Green Chem, 2019, 21, 3023–3028. [Google Scholar]
- 74.Li L, Zhu X-Q, Zhang Y-Q, Bu H-Z, Yuan P, Chen J, Su J, Deng X and Ye L-W, Chem. Sci, 2019, 10, 3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Theunissen C, Métayer B, Henry N, Compain G, Marrot J, Martin-Mingot A, Thibaudeau S and Evano G, J. Am. Chem. Soc, 2014, 136, 12528–12531. [DOI] [PubMed] [Google Scholar]
- 76.Theunissen C, Métayer B, Lecomte M, Henry N, Chan HC, Compain G, Gérard P, Bachmann C, Mokhtari N, Marrot J, Martin-Mingot A, Thibaudeau S and Evano G, Org. Biomol. Chem, 2017, 15, 4399–4416. [DOI] [PubMed] [Google Scholar]
- 77.Zhou B, Li L, Liu X, Tan T-D, Liu J and Ye L-W, J. Org. Chem, 2017, 82, 10149–10157. [DOI] [PubMed] [Google Scholar]
- 78.Zhou B, Zhang Y-Q, Zhang K, Yang M-Y, Chen Y-B, Li Y, Peng Q, Zhu S-F, Zhou Q-L and Ye L-W, Nat. Commun, 2019, 10, 3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou B, Li L, Zhu X-Q, Yan J-Z, Guo Y-L and Ye L-W, Angew. Chem. Int. Ed, 2017, 56, 4015–4019. [DOI] [PubMed] [Google Scholar]
- 80.Li H, Antoline JE, Yang JH, Al-Rashid ZF and Hsung RP, New J. Chem, 2010, 34, 1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ito M, Kawasaki R, Kanyiva KS and Shibata T, Chem. Eur. J, 2018, 24, 3721–3724. [DOI] [PubMed] [Google Scholar]
- 82.Lin M, Zhu L, Xia J, Yu Y, Chen J, Mao Z and Huang X, Adv. Synth. Catal, 2018, 360, 2280–2284. [Google Scholar]
- 83.Febvay J, Sanogo Y, Retailleau P, Gogoi MP, Sahoo AK, Marinetti A and Voituriez A, Org. Lett, 2019, 21, 9281–9285. [DOI] [PubMed] [Google Scholar]
- 84.Ghosh N, Nayak S and Sahoo AK, Chem. Eur. J, 2013, 19, 9428–9433. [DOI] [PubMed] [Google Scholar]
- 85.Xu Y, Sun Q, Tan TD, Yang MY, Yuan P, Wu SQ, Lu W, Hong X and Ye LW, Angew. Chem. Int. Ed, 2019, 58, 1652–1659. [DOI] [PubMed] [Google Scholar]
- 86.Wang CM, Qi LJ, Sun Q, Zhou B, Zhang ZX, Shi ZF, Lin SC, Lu X, Gong L and Ye LW, Green Chem, 2018, 20, 3271–3278. [Google Scholar]
- 87.Liu R, Winston-McPherson GN, Yang ZY, Zhou X, Song W, Guzei IA, Xu X and Tang J Am. Chem. Soc, 2013, 135, 8201–8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li HH, Ye SH, Chen YB, Luo WF, Qian PC and Ye LW, Chin. J. Chem, 2020, 38, 263–268. [Google Scholar]
- 89.Kerr DJ, Miletic M, Chaplin JH, White JM and Flynn BJ, Org. Lett, 2012, 14, 1732–1735. [DOI] [PubMed] [Google Scholar]
- 90.Manchala N, Law HYL, Kerr DJ, Volpe R, Lepage RJ, White JM, Krenske EH and Flynn BJ, J. Org. Chem, 2017, 82, 6511–6527. [DOI] [PubMed] [Google Scholar]
- 91.Volpe R, Lepage RJ, White JM, Krenske EH, and Flynn BJ, Chem. Sci, 2018, 9, 4644–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han P, Mao ZY, Si CM, Zhou Z, Wei BG and Lin GQ, J. Org. Chem, 2019, 84, 914–923. [DOI] [PubMed] [Google Scholar]
- 93.Wang XN, Hsung RP, Fox SK, Lv MC and Qi R, Heterocycles, 2014, 8, 1233–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang XN, Hsung RP, Qi R, Fox SK and Lv MC, Org. Lett, 2013, 15, 2514–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cook AM, Wolf C, Chem. Commun, 2014, 50, 3151–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cook AM, Wolf C, Angew. Chem. Int. Ed, 2016, 55, 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moskowitz M, Wolf C, Angew. Chem. Int. Ed, 2019, 58, 3402–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li X, Jiang M, Zhan T, Cao W and Feng X, Chem. Asian J, 2020, 15, 1953–1956. [DOI] [PubMed] [Google Scholar]
- 99.Han P, Mao ZY, Li M, Si CM, Wei BG and Lin GQ, J. Org. Chem, 2020, 85, 4740–4752. [DOI] [PubMed] [Google Scholar]
- 100.Egi M, Shimizu K, Kamiya M and Ota Y, Chem. Commun, 2015, 51, 380–383. [DOI] [PubMed] [Google Scholar]
- 101.Thilmany P and Evano G, Angew. Chem. Int. Ed, 2020, 59, 242–246. [DOI] [PubMed] [Google Scholar]
- 102.Minko Y, Pasco M, Chechik H and Marek I, Beilstein J. Org. Chem 2013, 9, 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nairoukh Z, Narayana Kumar GGKS, Minko Y and Marek I, Chem. Sci, 2016, 8, 627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nairoukh Z, Narayana Kumar GGKS, Minko Y and Marek I, Chem. Sci, 2017, 8, 627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Patil DV, Kim SW, Nguyen QH, Kim H, Wang S, Hoang T and Shin S, Angew. Chem. Int. Ed, 2017, 56, 3670–3674. [DOI] [PubMed] [Google Scholar]
- 106.Um TW, Lee G and Shin S, Org. Lett, 2020, 22, 1985–1990. [DOI] [PubMed] [Google Scholar]
- 107.Nguyen NH, Nguyen QH, Biswas S, Patil DV and Shin S, Org. Lett, 2019, 21, 9009–9013. [DOI] [PubMed] [Google Scholar]
- 108.Kaldre D, Maryasin B, Kaiser D, Gajsek O, González L and Maulide N, Angew. Chem. Int. Ed, 2017, 56, 2212–2215. [DOI] [PubMed] [Google Scholar]
- 109.Huang H, Fan J, He G, Yang Z, Jin X, Liu Q and Zhu H, Chem. Eur. J, 2016, 22, 2532–2538. [DOI] [PubMed] [Google Scholar]