

Abstract
High-throughput sequencing is widely used for strain detection and characterization of antibiotic resistance in microbial metagenomic samples. Current analytical tools use curated antibiotic resistance gene (ARG) databases to classify individual sequencing reads or assembled contigs. However, identifying ARGs from raw read data can be time consuming (especially if assembly or alignment is required) and challenging, due to genome rearrangements and mutations. Here, we present the k-mer-based antibiotic gene resistance analyzer (KARGA), a multi-platform Java toolkit for identifying ARGs from metagenomic short read data. KARGA does not perform alignment; it uses an efficient double-lookup strategy, statistical filtering on false positives, and provides individual read classification as well as covering of the database resistome. On simulated data, KARGA’s antibiotic resistance class recall is 99.89% for error/mutation rates within 10%, and of 83.37% for error/mutation rates between 10% and 25%, while it is 99.92% on ARGs with rearrangements. On empirical data, KARGA provides higher hit score (≥1.5-fold) than AMRPlusPlus, DeepARG, and MetaMARC. KARGA has also faster runtimes than all other tools (2x faster than AMRPlusPlus, 7x than DeepARG, and over 100x than MetaMARC). KARGA is available under the MIT license at https://github.com/DataIntellSystLab/KARGA.
Keywords: bioinformatics, high-throughput sequencing, metagenomics, antibiotic resistance, k-mer, ontology, classification
I. Introduction
Antibiotic resistance is a worldwide public health and environmental health concern, because it reduces therapeutic options in present-day infections and in future outbreaks caused by resistant strains [1]. High-throughput sequencing is widely used for strain detection and characterization of antimicrobial resistance in microbial metagenomic samples [2], [3]. Current analytical tools use curated antibiotic resistance gene (ARG) databases to classify individual sequencing reads or assembled contigs [4]. However, identifying ARGs from raw read data can be time consuming (especially if assembly or alignment is required) and challenging, due to genome rearrangements and mutations [5]. The translational utility –in terms of clinical point-of-care or veterinary employment– of ARG classification tools for high-throughput sequencing relies on comprehensive databases, regularly updated to include new resistance genes, and on accurate, fast turnaround reporting.
A. Curated, Reference ARG Databases
MEGARes (v.2.0) [6] is one of the largest (~8,000 gene entries), manually-curated ARG databases currently maintained. MEGARes utilizes a tree-like ontology to annotate ARGs, with a four-level hierarchy of antibiotic resistance (type, class, mechanism, and group). Other ARG databases include the Comprehensive Antibiotic Resistance Database (CARD) [7], which employs a different, non-tree-like ontology (~4,500 entries), and the Antibiotic Resistance Genes Database (ARDB) [8], which is no longer maintained and has been later incorporated into CARD.
B. ARG Classification Tools
The tools that are able to analyze directly short read data, without requiring first an assembly step, include AMRPlusPlus (v.2.0) [9], DeepARG [10], KmerResistance [11], and Meta-MARC [12]. AMRPlusPlus aligns reads to MEGARes using BWA and then aggregates results for each ARG, providing detailed coverage, depth, and rarefaction support statistics. DeepARG also is alignment-based, but uses the CARD ontology (plus other ancillary data sources), translates reads into proteins via PRODIGAL, aligns them with DIAMOND, and then predicts ARGs using a deep learning classifier. KmerResistance is instead alignment-free, uses ARDB, and maps the ARDB resistome through k-mer (short sequences of fixed k length) matching. Meta-MARC uses a hidden Markov model (HMM) approach on ARG clusters derived from MEGARes.
C. Our Contribution
We present the k-mer-based antibiotic gene resistance analyzer (KARGA), a multi-platform Java toolkit for identifying ARGs from metagenomics short read data. KARGA is alignment-free, utilizes hash-based k-mer mapping, providing faster runtimes and more ARG classification robustness with respect to genome rearrangements compared to alignment-based or HMM methods. Differently from KmerResistance, which is k-mer-based, KARGA employs an efficient double-lookup strategy, a statistical test for handling false positives upon the choice of k, a weighting of ambiguous k-mers, and a much larger, up-to-date, maintained reference ARG database.
II. METHODS
A. Strategy for K-mer Matching and ARG Annotation
KARGA extracts all distinct k-mers (both in forward- and reverse-strand) from the MEGARes database and places them into lookup tables as keys, storing their ARG annotations (gene identifier and type/class/group/mechanism information) in a one-to-many relation. Complementarily, each ARG entry is placed in another one-to-many lookup relation where now all k-mers and their frequencies are stored with respect to ARG keys. KARGA processes reads individually and extracts k-mers, mapping them first on to the k-mer-ARG lookup relation. If a k-mer maps on to multiple ARGs, that hit is weighted as a fraction of the number of ARGs found. After mapping all k-mers, KARGA assigns the type/class/group/mechanism of a read on the basis of the most frequent, weighted ARG hits. Note that this voting is independent at each level of the ontology hierarchy. After classifying all reads, KARGA summarizes the nucleotide coverage and the median k-mer depth for each ARG entry using the complementary lookup ARG-k-mer relation. Of note, KARGA does not provide classification for housekeeping genes where single-point mutations are responsible for resistance; these genes are removed from the MEGARes database before creation of lookup tables.
B. Statistical Assessment of K-mer Hits
Since the probability that a random k-mer matches a given gene is not null, there can be cases when reads that do not belong to ARGs are mistakenly mapped because of random matches. In order to avoid this, KARGA calculates the empirical count distribution D of randomly generated k-mers within the reference ARG database, approximating the theoretical Markovian distribution [13]. We then set a threshold (99th percentile) to filter out reads whose k-mer hit number is below the expected count in D.
C. Experimental Setup and Performance Assessment
We validate and test KARGA on synthetic, semi-synthetic and real-world experimental datasets. The synthetic metagenomic data designed for validation include: (i) reads drawn from MEGARes genes with mutations/errors up to a 25% rate; (ii) reads from MEGARes with a two-point transposition/transversion of half-read length size; (iii) non-AMR reads, randomly generated. We evaluate KARGA for different values of k, since k can influence the false positive rate with respect to non-ARG sequences and the false negative rate with respect to ARG sequences that bear mutations, gene rearrangements, or sequencing errors.
The semi-synthetic includes bacterial genomes from the Pathosystems Resource Integration Center (PATRIC) [14], for which an available antibiogram test is available, and thus information on antibiotic resistance at the molecular level (which can be mapped to the MEGARes ontology). We select both antibiotic-resistant (at least one molecule in the class) and antibiotic-susceptible genomes and simulate high-throughput sequencing data. The in silico datasets are generated using InSilicoSeq [15] parameterized for NovaSeq (Illumina, Inc.). The real-world datasets are functional metagenomics experiments of cultured bacteria that survived on an antibiotic-laden medium, named ‘Pediatric’ and ‘Soil’ from the sample sources (Genbank accessions PRJNA244044 and PRJNA215106) [16], [17]. Note that for both datasets it is not assured that an ARG is present in each read, so the resistance is known only at the whole sample level. Also, not all antibiotic classes, mechanisms or groups are represented.
On both the semi-synthetic and real-world datasets, we compare KARGA with state-of-the-art tools –AMRPlusPlus (v.2.0), DeepARG, and Meta-MARC– in terms of recall, hit rate, difference between resistance and susceptible hits, and runtime.
D. Implementation and Availability
KARGA is implemented in Java™ (www.java.com) and compiled using Oracle Corporation’s Java Development Kit v.15, 64-bit. MEGARes (v.2.0) is used by default, but any other database consistent with the MegaRES ontology and in FASTA format can be used. A HashMap indexes all distinct k-mers found across all ARGs, and each value points to an ArrayList of ARG identifiers where that k-mer is found. Complementarily, another HashMap links ARG identifiers with a complex object that stores one HashMap of distinct k-mers found in that ARG with their frequency, and another HashMap that is to be filled with potential hits from the read set.
The read file is parsed sequentially as a plain or a gzipped FASTQ, and each read is checked for consistency, flagging all non-ACGT characters as ‘N’. A preliminary scan of the first 50,000 reads assesses average read length and calculates the frequency threshold of k-mer hits for random reads. Then, each read is divided into k-mers and each k-mer is queried against the ARG HashMaps. If the total number of k-mer hits is above the false positive threshold, the program outputs the ARG classification for each hierarchy level. The level-specific k-mer hits and weights are stored in other read-specific HashMaps. If the resistome mapper module is invoked, then the program outputs the percentage of gene length spanned by mapping reads and the median k-mer coverage for each ARG.
The software and the source code are available under the MIT license at https://github.com/DataIntellSystLab/KARGA.
III. Results
The empirical count distribution analysis of k-mer hits for NovaSeq-like data (average read length of 151) on MEGARes (v.2.0) indicates that k values of 11 and above provide very low probability of finding a random k-mer once or more in the database. Fig. 1 plots the count distribution percentiles calculated over 250,000 random strings; the curve that divides the top from the bottom distribution corresponds to k=11.
Fig. 1.
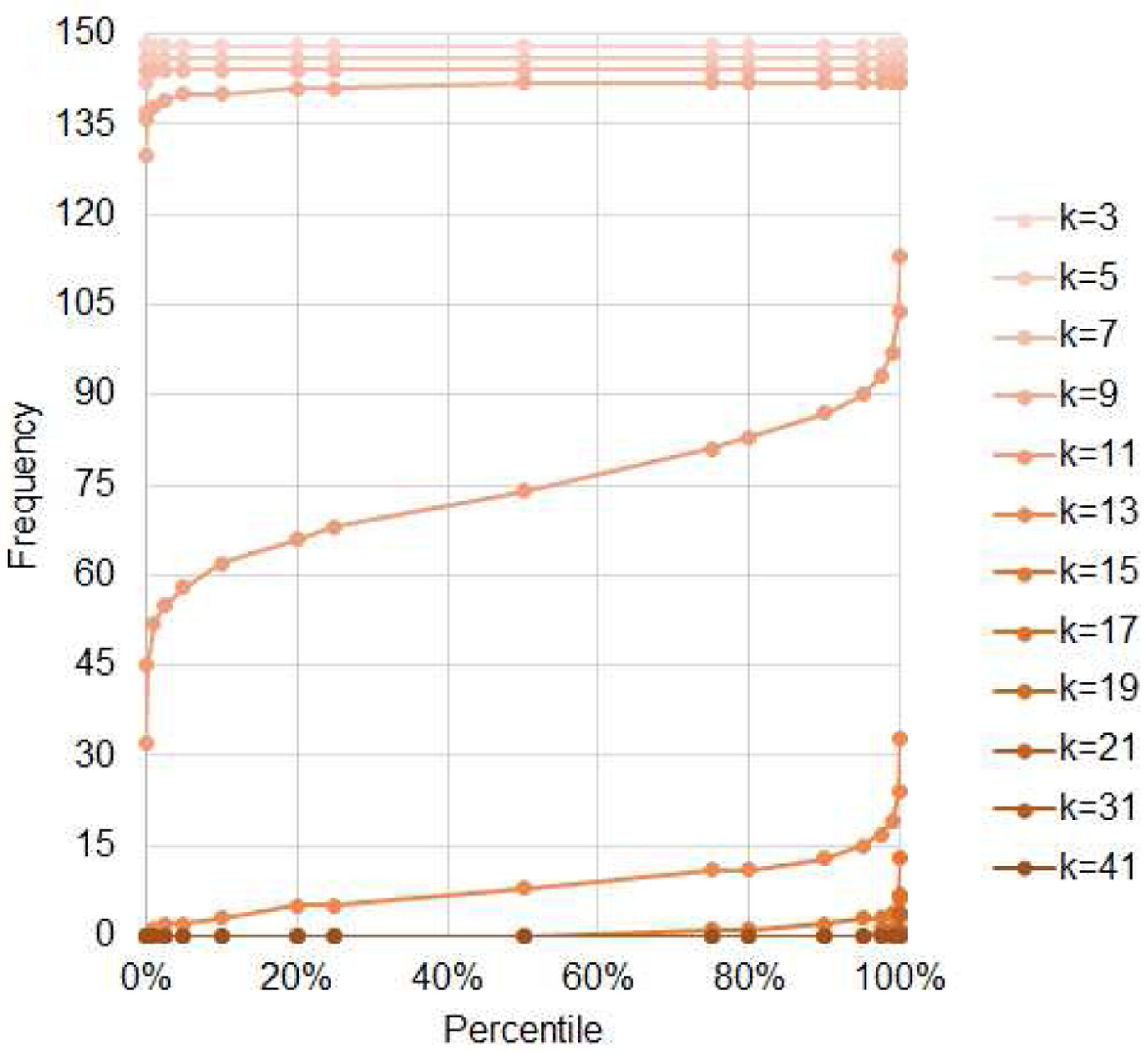
Empirical distribution of counts for random k-mers (k=3…41) found in the MEGARes database, estimated on 200,000 randomizations.
The synthetic validation dataset includes 200,000 reads (each 151 nucleotides long), of which 10% are non-ARG (random), 90% are ARGs with mutations/errors, and the remaining 10% are ARGs with gene rearrangement. The statistical test to discard non-ARG reads works as expected. Out of 10,000 randomly simulated reads, less than 1% are mistakenly assigned. The false positive rate decreases when k increases, and no false positives are found for any k larger than 19. Classification results for the ARG reads with mutations/errors are shown in Fig. 2, stratified by k value and antibiotic resistance hierarchy. Overall, there is a nonlinear, sigmoid-like decrease in performance when the error rate increases. The best k is 13 (relative to a read length of 151), with an average class recall of 99.89% for error/mutation rates within 10%, and of 83.37% for error/mutation rates between 10% and 25%. However, any value between 13 and 21 yields results above the average across all k values being tested, i.e. 11 to 41. On the gene-rearranged ARG reads, KARGA shows high robustness: on average, 99.92%, 99.66%, and 98.52% of the reads are assigned to the correct class, group, and mechanism, respectively. These performance values are stable across all k values, with no evidence of slope due to k.
Fig. 2.
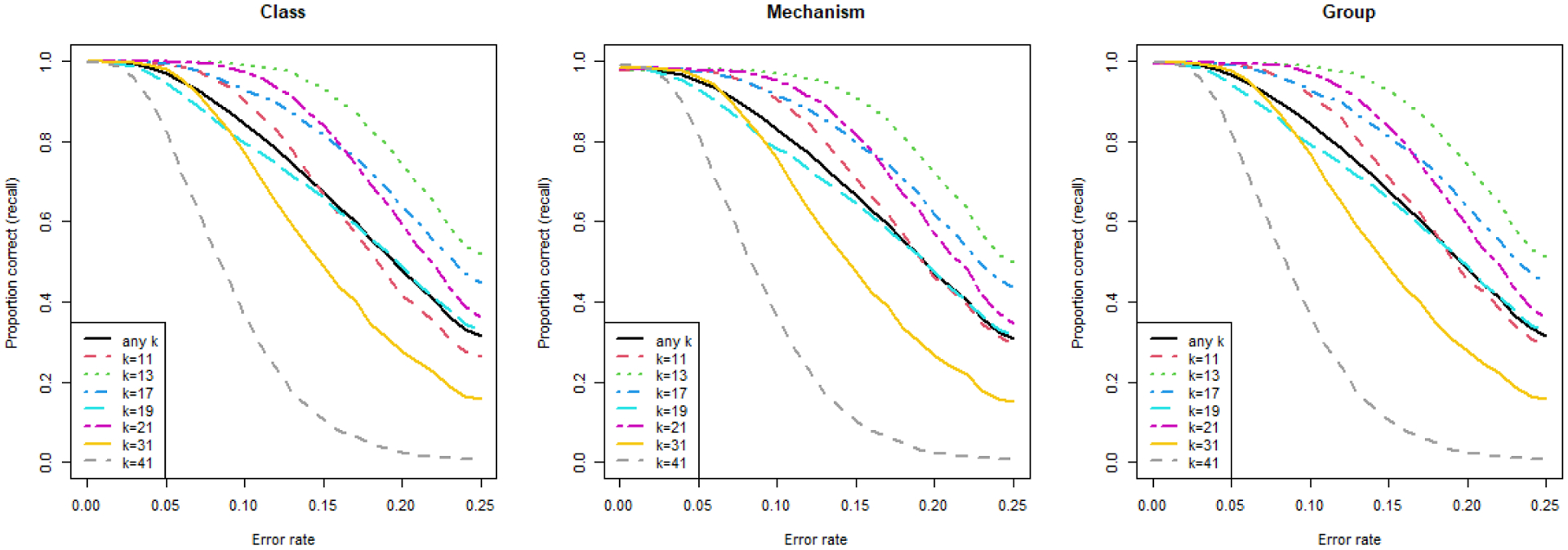
Recall performance of KARGA on synthetic short read metagenomics data (200,000 reads, 151 nucleotides) containing ARGs with mutations/sequencing errors up to 25%. Results are stratified by MEGARes annotation hierarchy (class/mechanism/group) and by value of k.
On the semi-synthetic PATRIC dataset (Fig.3), the method with the highest hit score (resistance hits minus susceptible hits) is KARGA, with a median (IQR) of 415 (193–672), followed by MetaMARC with 284 (71–454), DeepARG with 219 (39–454), and AMRPlusPlus with 46 (−262–209). On the experimental Pediatric and Soil datasets, KARGA’s average hit rate is 5.1% whereas AMRPlusPlus yields 4.2%, DeepARG 3.7%, and MetaMARC 6.4%. Of note, MetaMARC has been calibrated on the Pediatric/Soil data in its original paper, so its performance cannot be considered external validation as with the other tools.
Fig. 3.
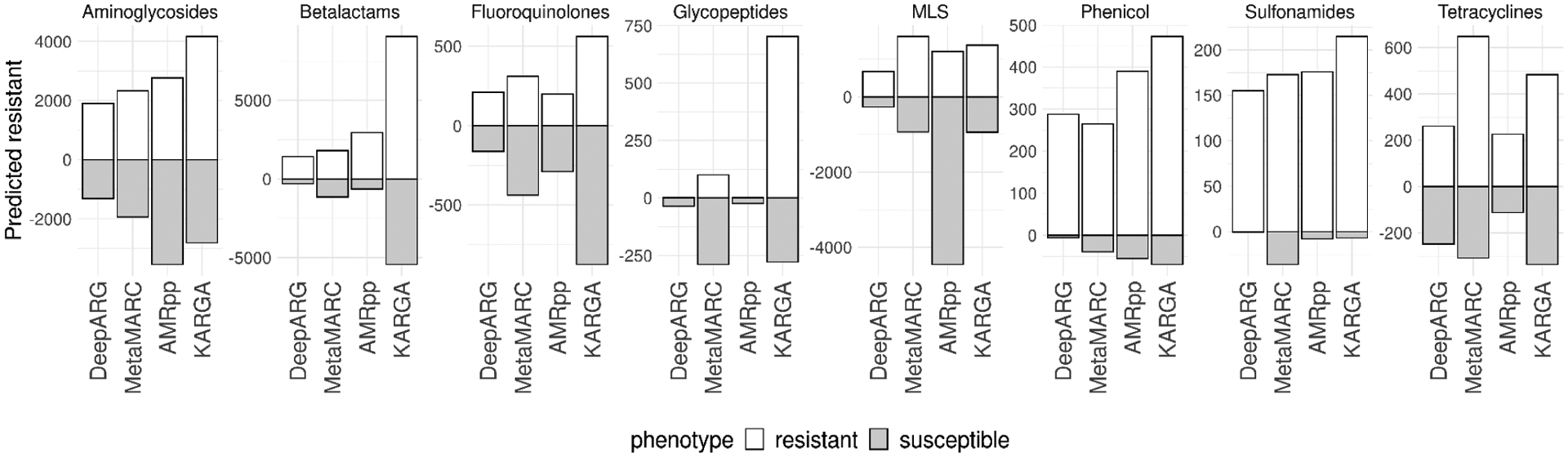
Resistance and susceptibility hit rate of KARGA, AMRPlusPlus, DeepARG and MetaMARC on semi-synthetic short read metagenomics data (~250,000 reads, 151 nucleotides) drawn from the PATRIC repository, stratified by antibiotic class.
In terms of runtimes, we tested the software on a 4-cores AMD Opteron 6378, 2.4GHz, 32GB of RAM, and KARGA is faster than all other software across all file sizes, as shown in Table 1, being on average 2x faster than AMRPlusPlus, 7x faster than DeepARG, and over 100x faster than MetaMARC.
TABLE I.
Runtime benchmarks (hh:mm:ss) comparing KARGA with AMRPlusPlus, DeepARG, and MetaMARC.
| File size | Reads | KARGA | AMR PlusPlus | DeepARG | MetaMARC |
|---|---|---|---|---|---|
| 1GB | 1.6M | 0:08:27 | 00:21:19 | 00:53:01 | 16:26:27 |
| 2GB | 3.1M | 0:15:32 | 00:49:40 | 01:38:55 | >24h |
| 5GB | 7.9M | 0:38:11 | 01:35:47 | 03:41:06 | >24h |
| 10GB | 15.8M | 1:38:20 | 2:48:43 | 11:43:16 | >24h |
IV. Discussion
KARGA demonstrates high recall and hit rate in identifying ARGs from high-throughput, short read metagenomics data. It is highly robust with gene rearrangement and with genetic divergence at lower k values, yet maintaining a low false positive rate thanks to the statistical filtering. Its hit rate on semi-synthetic and empirical data is higher than all other methods. Only in one real-world dataset KARGA is second to MetaMARC; however that dataset was used for calibration, and on large experimental data MetaMARC is practically unusable due to extremely long execution times. In addition to be the most accurate, KARGA is the fastest of all off-the-shelf ARG classification software tested here.
This work has some limitations. First, we do not classify antibiotic resistance in housekeeping genes, since resistance in these genes is determined by single nucleotide polymorphisms (SNPs). However, such feature could be added by requiring always the presence of k-mers containing the SNPs in addition to non-SNP-containing k-mer hits. Second, the data structures we implement are standard and can have high memory overhead. We use String types for k-mers, but they can be transformed, processed and stored in much more efficient data structures (e.g. using bit maps, Bloom filters and minimizers) [18]. Finally, KARGA could be parallelized: each read can be processed independently from the others and the k-mer-ARG HashMap is read-only; however, the ARG-k-mer HashMap would need to be updated concurrently.
In conclusion, KARGA is a fast, reliable and flexible ARG classifier that can be employed in multiple contexts; its multi-platform implementation makes it also ideal for mobile bioinformatics applications, e.g. Oxford’s Nanopore sequencing data generated by MinION (with the Mk1C), and the smartphone-pluggable SmidgION [19].
Acknowledgments
This work has been supported in part by National Science Foundation (NSF) grant #2013998 and National Institutes of Health - National Institute of Allergy and Infectious Diseases (NIH/NIAID) grant #R01AI141810.
Contributor Information
Mattia Prosperi, Data Intelligence Systems Lab, Department of Epidemiology, College of Public Health and Health Professions, University of Florida, Gainesville, FL, USA.
Simone Marini, Data Intelligence Systems Lab, Department of Epidemiology, College of Public Health and Health Professions, University of Florida, Gainesville, FL, USA.
References
- [1].Christaki E, Marcou M, and Tofarides A, “Antimicrobial Resistance in Bacteria: Mechanisms, Evolution, and Persistence,” J. Mol. Evol, vol. 88, no. 1, pp. 26–40, Jan. 2020, doi: 10.1007/s00239-019-09914-3. [DOI] [PubMed] [Google Scholar]
- [2].Crofts TS, Gasparrini AJ, and Dantas G, “Next-generation approaches to understand and combat the antibiotic resistome,” Nat. Rev. Microbiol, vol. 15, no. 7, pp. 422–434, Jul. 2017, doi: 10.1038/nrmicro.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gu W, Miller S, and Chiu CY, “Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection,” Annu. Rev. Pathol, vol. 14, pp. 319–338, Jan. 2019, doi: 10.1146/annurev-pathmechdis-012418-012751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hendriksen RS, Bortolaia V, Tate H, Tyson GH, Aarestrup FM, and McDermott PF, “Using Genomics to Track Global Antimicrobial Resistance,” Front. Public Health, vol. 7, p. 242, 2019, doi: 10.3389/fpubh.2019.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Doyle RM et al. , “Discordant bioinformatic predictions of antimicrobial resistance from whole-genome sequencing data of bacterial isolates: an inter-laboratory study,” Microb. Genomics, vol. 6, no. 2, Feb. 2020, doi: 10.1099/mgen.0.000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Doster E et al. , “MEGARes 2.0: a database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data,” Nucleic Acids Res, vol. 48, no. D1, pp. D561–D569, Jan. 2020, doi: 10.1093/nar/gkz1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Alcock BP et al. , “CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database,” Nucleic Acids Res, vol. 48, no. D1, pp. D517–D525, Jan. 2020, doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu B and Pop M, “ARDB--Antibiotic Resistance Genes Database,” Nucleic Acids Res, vol. 37, no. Database issue, pp. D443–447, Jan. 2009, doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lakin SM et al. , “MEGARes: an antimicrobial resistance database for high throughput sequencing,” Nucleic Acids Res, vol. 45, no. Database issue, pp. D574–D580, Jan. 2017, doi: 10.1093/nar/gkw1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Arango-Argoty G, Garner E, Pruden A, Heath LS, Vikesland P, and Zhang L, “DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data,” Microbiome, vol. 6, Feb. 2018, doi: 10.1186/s40168-018-0401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Clausen PTLC, Zankari E, Aarestrup FM, and Lund O, “Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data,” J. Antimicrob. Chemother, vol. 71, no. 9, pp. 2484–2488, Sep. 2016, doi: 10.1093/jac/dkw184. [DOI] [PubMed] [Google Scholar]
- [12].Lakin SM et al. , “Hierarchical Hidden Markov models enable accurate and diverse detection of antimicrobial resistance sequences,” Commun. Biol, vol. 2, Aug. 2019, doi: 10.1038/s42003-019-0545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Prosperi MCF, Prosperi L, Gray RR, and Salemi M, “On counting the frequency distribution of string motifs in molecular sequences,” Int. J. Biomath, vol. 05, no. 06, p. 1250055, May 2012, doi: 10.1142/S1793524512500556. [DOI] [Google Scholar]
- [14].Davis JJ et al. , “The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities,” Nucleic Acids Res, vol. 48, no. D1, pp. D606–D612, Jan. 2020, doi: 10.1093/nar/gkz943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gourlé H, Karlsson-Lindsjö O, Hayer J, and Bongcam-Rudloff E, “Simulating Illumina metagenomic data with InSilicoSeq,” Bioinformatics, vol. 35, no. 3, pp. 521–522, Feb. 2019, doi: 10.1093/bioinformatics/bty630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moore AM et al. , “Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes,” PloS One, vol. 8, no. 11, p. e78822, 2013, doi: 10.1371/journal.pone.0078822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Forsberg KJ et al. , “Bacterial phylogeny structures soil resistomes across habitats,” Nature, vol. 509, no. 7502, Art. no. 7502, May 2014, doi: 10.1038/nature13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Manekar SC and Sathe SR, “A benchmark study of k-mer counting methods for high-throughput sequencing,” GigaScience, vol. 7, no. giy125, Dec. 2018, doi: 10.1093/gigascience/giy125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oliva M, Milicchio F, King K, Benson G, Boucher C, and Prosperi M, “Portable nanopore analytics: are we there yet?,” Bioinformatics, vol. 36, no. 16, pp. 4399–4405, Aug. 2020, doi: 10.1093/bioinformatics/btaa237. [DOI] [PMC free article] [PubMed] [Google Scholar]