

Abstract
Purpose
Previously, we identified a G661R mutation of ADAMTS10 (a disintegrin-like and metalloprotease with thrombospondin type 1 motif 10) as being disease causative in a colony of Beagles with inherited primary open-angle glaucoma (POAG). Mutations in ADAMTS10 are known to cause Weill–Marchesani syndrome (WMS), which is also caused by mutations in the fibrillin-1 gene (FBN1), suggesting functional linkage between ADAMTS10 and fibrillin-1, the principal component of microfibrils. Here, we established a mouse line with the G661R mutation of Adamts10 (Adamts10G661R/G661R) to determine if they develop features of WMS and alterations of ocular fibrillin microfibrils.
Methods
Intraocular pressure (IOP) was measured using a TonoLab rebound tonometer. Central cornea thickness (CCT), anterior chamber depth (ACD) and axial length (AL) of the eye were examined by spectral-domain optical coherence tomography. Sagittal eye sections from mice at postnatal day 10 (P10) and at 3 and 24 months of age were stained with antibodies against fibrillin-1, fibrillin-2, and ADAMTS10.
Results
IOP was not elevated in Adamts10G661R/G661R mice. Adamts10G661R/G661R mice had smaller bodies, thicker CCT, and shallower ACD compared to wild-type mice but normal AL. Adamts10G661R/G661R mice displayed persistent fibrillin-2 and enhanced fibrillin-1 immunofluorescence in the lens zonules and in the hyaloid vasculature and its remnants in the vitreous.
Conclusions
Adamts10G661R/G661R mice recapitulate the short stature and ocular phenotypes of WMS. The altered fibrillin-1 and fibrillin-2 immunoactivity in Adamts10G661R/G661R mice suggests that the G661R mutation of Adamts10 perturbs regulation of the fibrillin isotype composition of microfibrils in the mouse eye.
Keywords: Adamts10, fibrillin, microfibrils, glaucoma, Weill–Marchesani syndrome
ADAMTS10 belongs to a family of secreted proteins that includes 19 ADAMTS matrix metalloproteases and seven ADAMTS-like (ADAMTSL) proteins.1 We previously identified the glaucoma-causing ADAMTS mutation, which was a G661R mutation of ADAMTS10 in a research colony of Beagles with autosomal recessive primary open-angle glaucoma (POAG).2,3 These dogs develop elevated intraocular pressure (IOP) with open iridocorneal angles followed by development of optic disc cupping, a defining feature of glaucoma.4 Subsequently, another mutation in ADAMTS10, A387T, was reported to cause POAG in Norwegian Elkhounds.5 Evidence for ADAMTS family involvement in glaucoma was further extended by identification of mutations in a closely related gene, ADAMTS17, as being causative for glaucoma in other dog breeds.6–8 In the context of human glaucoma, an ADAMTS8 locus was found to be associated with a vertical cup-to-disc ratio and IOP, important glaucoma endophenotypes.9,10 Together, these findings implicate ADAMTS genes in glaucoma pathogenesis.
In humans, mutations in ADAMTS10 are known to cause autosomal recessive Weill–Marchesani syndrome (WMS),11–13 a systemic connective tissue disorder characterized by short stature, as well as abnormalities of the musculoskeletal, ocular, and cardiovascular systems. Ocular features of WMS include dislocated lens, microspherophakia, and glaucoma. Certain mutations in the fibrillin-1 gene (FBN1) cause autosomal dominant WMS,14,15 which is clinically indistinguishable from the autosomal recessive form,16 suggesting overlapping functions for ADAMTS10 and FBN1. There are three highly homologous and evolutionarily conserved fibrillin isoforms: fibrillin-1, fibrillin-2, and fibrillin-3 (although mice lack fibrillin-3).17 Fibrillin-1 dominates in postnatal life, whereas fibrillin-2 and fibrillin-3 are primarily expressed during development, with a transition to fibrillin-1 dominancy occurring from the late gestational period into the juvenile period.18–21
Fibrillin-1 and fibrillin-2 are the main constituents of microfibrils, which are fibrillar structures in the extracellular matrix of many tissues.22,23 Microfibrils contribute to tissue biomechanics and are key regulators of transforming growth factor beta (TGF-β) and bone morphogenic protein (BMP) signaling.24 Microfibrils are required for the formation of elastic fibers, and they form sheaths that surround the elastin core of all mature elastic fibers, contributing to their mechanical properties.25 Microfibrils also exist independent of elastic fibers, such as in the lens zonules, where they are the principal structural component.26 Altered tissue biomechanics and TGF-β signaling are thought to be major factors in glaucoma pathogenesis27,28 and are known to contribute to the pathology of diseases caused by microfibril deficiencies, such as Marfan syndrome and WMS.15
In addition to genetic evidence, several studies have shown evidence of functional interactions between ADAMTS10 and fibrillin-1.15,29,30 ADAMTS10 has been shown to bind fibrilin-1 with high affinity, co-localize with microfibrils in the human dermis and lens zonule, and accelerate formation of microfibrils in cell cultures.15,31–33 Cultured skin fibroblasts from patients with WMS with ADAMTS10 mutations have been shown to have reduced formation of extracellular microfibrils.32 Based on these and other findings, ADAMTS10 is thought to be a microfibril-associated protein that plays a role in the proper formation of fibrillin microfibrils.29,30 Although ADAMTS10 is resistant to furin cleavage of its propeptide, a necessary step for activation of its metalloproteinase activity, it can cleave fibrillin-1 or fibrillin-2 after optimization of its furin recognition sequence.32,34 A relation between ADAMTS10 and fibrillin-2 was recently reported by two recent studies that showed ocular persistence of fibrillin-2 in mice with Adamts10 null mutations.33,34
For the present study, we introduced the G661R mutation of Adamts10 into the C57BL/6J mouse strain to compare phenotypes of mice homozygous for the mutation (Adamts10G661R/G661R) with contemporary wild-type (WT) littermate controls. Adamts10G661R/G661R mice recapitulate some features of WMS, such as smaller bodies and thickened cornea. Although normally fibrillin-2 is the dominant isoform through early gestation, replaced by dominance of fibrillin-1 in adults, we found that predominant fibrillin-2 immunofluorescence persisted into adulthood in the lens zonules and in the vitreous associated with remnants of the hyaloid vasculature. Additionally, fibrillin-1 fluorescence was enhanced in those structures as compared with WT mice. These findings suggest that ADAMTS10 plays an important role in determining the fibrillin isoform composition of fibrillin microfibrils and show that the G661R mutation of Adamts10 interferes with this function.
Methods
Animals
All animal studies were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of Vanderbilt University. Males and females heterozygous for the G661R mutation of Adamts10 (see below) were bred to produce cohorts of experimental animals homozygous for the G661R mutation of Adamts10, hereafter referred to as Adamts10G661R/G661R, and control animals homozygous for wild-type Adamts10, hereafter referred to as WT. The genotype of each mouse was determined at weaning and confirmed after sacrificing. Animals were housed in a facility operated by the Vanderbilt University Division of Animal Care, with 12/12-hour light/dark cycle and ad libidum access to food and water.
Introduction of the G661R Mutation of Mouse Adamts10 by CRISPR/Cas9 Genome Editing
The CRISPR protospacer targeting the desired locus of Adamts10 and having the sequence CGAGCAGCAGCTGTGGTGGA (Fig. 1) was used to design a single-guide RNA (sgRNA) that was obtained through a commercial vendor (MilliporeSigma, Burlington, MA, USA). A 120-nucleotide, single-stranded oligodeoxynucleotide (ssODN) homology-directed repair (HDR) template was designed to introduce the single base pair change resulting in the glycine-to-arginine amino acid substitution at amino acid 661 of the ADAMTS10 protein, as well as two silent base changes that introduced a diagnostic SalI restriction site (Fig. 1). The sequence of the ssODN HDR template corresponding to the bottom strand relative to the Adamts10 reading frame was TCTACCTTGCACTCGCCGCT-GACACAAATGTCCACCGTGTCAGGGCGGCAGGGTGTTCT-GTCGACCACAGCTGCTGCTCGCTCCGTATAAAAGTTGAAG-CCTTCTGCTAGGCAAGTCAGC. Cas9 mRNA, sgRNA, and HDR oligo were co-injected into C57BL/6J zygotes by the Vanderbilt Genome Editing Resource. Tail DNA samples from 60 weanlings were screened by PCR using primers F3 (GAAACTTCGCTGTTCCCTCT) and R5 (ATTTGTCCTCTCGGAGATCAG). As shown in Figure 1, the presence of the correctly edited allele was verified by SalI digestion of PCR products and Sanger sequencing (Genewiz, South Plainfield, NJ, USA). Eighteen pups were verified to carry a correctly edited allele generated through HDR with only the three desired base changes (Fig. 1). Founder animal 4 was bred to a WT C57BL6/J mouse. Germline transmission of the edited allele was confirmed in approximately 50% of progeny. Potential off-target sites in the mouse genome (mm10/GRCm38) were identified using COSMID.35 No other identical sites or single-mismatch sites were found. Eleven potential off-target sites were identified that had either two mismatches or one mismatch and one single-base indel. PCR assays were developed for four of these potential off-target sites, which were used to amplify and sequence DNA from 11 founder pups carrying the on-target G661R CRISPR edit, including founder pup 4, which was used for further breeding. No off-target mutations were detected.
Figure 1.

Introduction of the G661R mutation of mouse Adamts10 by CRISPR/Cas9 genome editing. (A) The 20-nucleotide protospacer targeting the G661R locus of Adamts10 (PS) and the reverse complement of a portion of the HDR template (ssODN) aligned with the WT genomic sequence (Genomic) is shown in relation to the protein structure of ADAMTS10. The protospacer-adjacent motif (PAM sequence) is indicated in blue font and the Cas9 cleavage site by a black triangle. In addition to the G-to-A nucleotide substitution resulting in the desired amino acid substitution (green font), two other bases (underlined) were changed in the HDR template to introduce a diagnostic SalI restriction site (orange triangle). (B) Representative diagnostic PCR using primers flanking the target site followed by SalI digestion resulted in a band pattern as expected: 401-bp band for WT and overlapping 210-bp and 191-bp bands for homozygous mutant mice and the combined set of bands for heterozygous mice. (C) Sanger sequencing of PCR reaction using flanking primers confirmed the expected nucleotide changes resulting from incorporation of the HDR template into genomic DNA. HM Mut, homozygous for the G661R mutation of Adamts10.
Intraocular Pressure
Diurnal IOP was measured at ∼10 AM at 3, 6, and 24 months of age, and nocturnal IOP was measured at ∼11 PM for mice at 24 months of age using a rebound tonometer designed for use in rodents (iCare TONOLAB; iCare Finland, Helsinki, Finland). Mice were anesthetized with 2.5% isoflurane in 95% O2/5% CO2 delivered at 1.5 L/min. The IOP of one eye of each mouse was taken within 2 minutes of loss of consciousness and was determined as the average of three readouts, each based on six consecutive measurements, following the manufacturer's recommendations.
Body Size
The body length of mice at 3 and 24 months of age was measured from nose to anus using a ruler. Body weight was determined by a digital scale.
Spectral-Domain Optical Coherence Tomography
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (7 mg/kg) in saline. Anterior segments of one eye from each mouse were visualized using the Bioptigen Envisu R2200 spectral-domain optical coherence tomography (SD-OCT) system (Leica Microsystems, Wetzlar, Germany) with a 12-mm telecentric bore lens and a rectangular scanning pattern consisting of 100 B-scans, each composed of 1000 A-scans. The anterior chamber depth (ACD) was defined as the distance from the inner surface of the central cornea to the anterior central surface of the lens determined by the digital caliper function of the Diver Analysis Software (Leica). For axial length (AL) measurement, pupils were dilated using 1% tropicamide (Bausch & Lomb, Laval, QC, Canada) and imaging was performed using a “mouse retina” lens (Leica Microsystems). AL was determined by the acquisition of a series of three images. A posterior image was used to determine the distance from the outer retinal pigment epithelium to the posterior surface of the lens (vitreous + retina); an anterior image was used to determine the distance from the outer corneal surface to the anterior surface of the lens (central cornea thickness [CCT] + ACD); and a third image in which the lens was optically folded in half was used to determine half of the axial lens diameter (1/2 lens). AL was defined as equal to (vitreous + retina) + (CCT + ACD) + 2 × 1/2 lens. Upon completion of imaging, the mice were injected with atipamezole (1 mg/kg; Patterson Veterinary, Greeley, CO, USA) to prevent xylazine-induced corneal damage.36
Immunofluorescence Staining and Microscopy
At P10 and 3 and 24 months of age, mice were sacrificed by CO2 inhalation then cardiac perfused with PBS followed by 4% paraformaldehyde in PBS. Eyes were enucleated and post-fixed in 4% paraformaldehyde in PBS, paraffin embedded, and sectioned at 7-µm thickness. Central sagittal eye sections were deparaffinized, rehydrated, and then subjected to antigen retrieval with 20 µg/mL proteinase K (Macherey-Nagel, Düren, Germany) in buffer (50-mM Tris-HCl; 1-mM EDTA; 0.5% Triton X-100, pH 8.0) for 5 minutes at room temperature. Sections were blocked with 5% normal donkey serum (MilliporeSigma) for 2 hours at room temperature in a humid chamber, then incubated with rabbit anti-fibrillin-1 diluted 1:200 (pAb 9543; kindly provided by Lynn Sakai),37 rabbit anti-fibrillin-2 diluted 1:200 (pAb 0868; kindly provided by Lynn Sakai),37 or goat anti-ADAMTS10 diluted 1:25 (sc-21505, Santa Cruz Biotechnology, Dallas, TX, USA) in blocking buffer overnight at 4°C. Sections were washed in 0.1% Tween-20 in PBS and then incubated with either donkey anti-rabbit or donkey anti-goat IgG conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) diluted 1:1000 for 2 hours, protected from light. Sections were washed and then coverslipped with ProLong Gold Antifade Mountant with DAPI (P36935; Thermo Fisher Scientific). Images were acquired under identical settings (laser power, digital offset, and gain) for each set of eye sections using a confocal microscope equipped with a 20 × 0.5-NA objective (FV1000; Olympus, Tokyo, Japan). Stacks of optical sections were visualized as maximum-intensity z-projections using ImageJ (National Institutes of Health, Bethesda, MD, USA). Negative control immunostaining omitting primary antibodies was performed and showed no immunofluorescence. Fibrillin-1 and fibrillin-2 immunofluorescence of the zonular fibers was quantified in ImageJ. For each experiment, thresholding of the 8-bit images was identical for all samples. Zonular fiber fragments were outlined and registered as regions of interest in the ImageJ ROI Manager function to measure the total intensity and total area. Mean fluorescence intensity was calculated as the total fluorescent intensity divided by the total area.
Experimental Design and Statistical Analysis
All experiments were conducted in a masked fashion. Data were analyzed using Student's two-tailed t-test or one-way analysis of variance (ANOVA) as indicated in the figure legends. Results were considered significant for P < 0.05.
Results
For this study, a line of mice carrying the G661R mutation of Adamts10 on the C57BL/6J background was created using CRISPR technology with template-directed homology-mediated repair as described in Methods (Fig. 1). Experimental animals were generated by crossing mice heterozygous for the mutation as determined by PCR followed by restriction digestion (Fig. 1B) or Sanger sequencing (Fig. 1C). Litters were of normal size with expected Mendelian inheritance. Out of 485 pups genotyped, 26.4% were homozygous WT, and 49.9% were heterozygous and 23.7% were homozygous for the mutation (Adamts10G661R/G661R). Homozygous mutant mice had normal life spans up to 2 years of age. These results indicate that the G661R mutation does not affect gestation or survival. Hematoxylin and eosin staining of sagittal eye sections from mice at 3, 6, and 24 months of age showed intact retinal layers and did not reveal apparent abnormalities in Adamts10G661R/G661R mice, including in the cornea and ciliary body (data not shown).
Intraocular Pressure
IOP elevation in the Beagle colony with the G661R mutation of ADAMTS10 begins at 8 to 16 months of age in homozygous dogs.4 To determine if IOP was elevated in the Adamts10G661R/G661R mice, diurnal IOP at 3, 6, and 24 months of age was measured by rebound tonometer at ∼10 AM (Fig. 2A). Diurnal IOP significantly decreased from 3 months to 24 months of age for both WT and Adamts10G661R/G661R mice (P < 0.0001). However, diurnal IOP at 3, 6, and 24 months of age did not differ significantly between Adamts10G661R/G661R mice and WT mice (P > 0.10). Because differences in IOP may be enhanced at night,38 nocturnal IOP measured at ∼11 PM was also determined; however, the nocturnal IOP of Adamts10G661R/G661R mice also did not differ from that of WT mice at 24 months of age (P = 0.55) (Fig. 2B). Unlike Beagle dogs, the G661R mutation of Adamts10 did not result in elevated IOP in mice on a C57BL/6J background.
Figure 2.
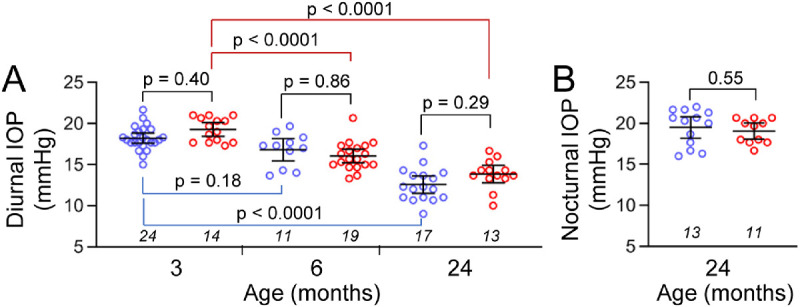
IOP of Adamts10G661R/G661R mice at 3, 6, and 24 months of age. (A) Diurnal IOP of the Adamts10G661R/G661R mice was not elevated compared with WT at all three time points. (B) Nocturnal IOP of the Adamts10G661R/G661R mice was not affected compared with WT mice at 24 months of age. Blue symbols, individual WT mice; red symbols, individual Adamts10G661R/G661R mice; lines, mean/95% confidence interval (CI). Numbers of mice are indicated in italics below each group, and P values from one-way ANOVA are indicated above brackets.
Body Size
By gross examination, Adamts10G661R/G661R mice appeared to have smaller bodies compared with WT mice (Fig. 3A), with the difference becoming more pronounced with age. Quantitative analysis showed age-dependent increases in body length and weight in both male and female mice, as expected (all P < 0.0001) (Figs. 3B, 3C). However, the body lengths of male Adamts10G661R/G661R mice were 3.8% and 7.2% shorter compared with WT mice at 3 and 24 months of age, respectively (P = 0.003 and P < 0.0001, respectively) (Fig. 3B). Female Adamts10G661R/G661R mice were 5.4% and 8.3% shorter at 3 and 24 months of age, respectively (both P < 0.0001) (Fig. 3B). Although body weight was not different at 3 months of age for either sex (P > 0.5), at 24 months of age male Adamts10G661R/G661R mice were 9.1% lighter (P = 0.003), and female Adamts10G661R/G661R mice were 17.7% lighter (P < 0.0001) compared with WT mice (Fig. 3C). The small body size phenotype of Adamts10G661R/G661R mice is consistent with the short-stature feature of human WMS.
Figure 3.

Smaller body size of Adamts10G661R/G661R mice. (A) Representative image comparing the body size of an Adamts10G661R/G661R mouse with a WT mouse. (B) Body length was shorter for Adamts10G661R/G661R mice (red symbols) compared with WT mice (blue symbols) at 3 and 24 months of age for both males and females. (C) Body weight was lower for Adamts10G661R/G661R mice (red symbols) compared with WT mice (blue symbols) at 24 months of age, but not at 3 months of age, for both males and females. Error bars represent mean ± 95% CI. Numbers of mice are indicated in italics below each group, and P values from one-way ANOVA are indicated above brackets (B and C).
SD-OCT Imaging
Patients with WMS have eye anomalies that include thickened cornea and dislocated lenses.16,39 To test if Adamts10G661R/G661R mice have similar ocular anomalies, CCT, ACD, and AL were determined by SD-OCT imaging (Fig. 4A). CCT did not significantly change from 3 to 24 months of age for either WT or Adamts10G661R/G661R mice (P > 0.4) (Fig. 4B). However, compared with WT mice, at 3 months of age the CCT of Adamts10G661R/G661R mice (0.113 ± 0.006 mm) was 5.4% thicker than that of WT mice (0.107 ± 0.004 mm; P = 0.01). At 24 months of age, the CCT was 8.5% thicker (0.114 ± 0.011 mm) than that of WT mice (0.105 ± 0.012; P = 0.02). Thicker CCT in Adamts10G661R/G661R mice is consistent with ocular features of human WMS.
Figure 4.

Thick CCT and shallow ACD with normal AL in Adamts10G661R/G661R mice. SD-OCT imaging of the anterior segment (A, left) revealed thicker CCT (B) and shallower ACD (C) for Adamts10G661R/G661R mice (red symbols) compared with WT mice (blue symbols) at 3 and 24 months of age. AL was calculated from the SD-OCT images (A, right) of three measurements: the summation of the distance from the outer surface of the central cornea to the anterior central surface of the lens (CCT + ACD), the lens thickness (2 × 1/2 lens), and the distance from the posterior central surface of the lens to the central retinal pigment epithelium layer (vitreous + retina). There was no difference in AL determined by SD-OCT between Adamts10G661R/G661R mice (red symbols) and WT mice (blue symbols) at 3 and 24 months of age (D). Scale bar: 0.05 mm (A, yellow). Error bars represent mean ± 95% CI. Numbers of mice are indicated in italics below each group, and P values from two-tailed Student's t-test are indicated above brackets (B–D).
An age-dependent increase in ACD (Fig. 4C) was seen for WT and Adamts10G661R/G661R mice (P = 6.5 × 10−13 and P = 7.9 × 10−19, respectively). In addition, at 3 months of age, the ACD of Adamts10G661R/G661R mice (0.384 ± 0.012 mm) was 5.4% shallower compared with that of WT mice (0.406 ± 0.019 mm; P = 0.002). Similarly, at 24 months of age, the ACD of Adamts10G661R/G661R mice (0.468 ± 0.017 mm) was 5.0% shallower than that of WT mice (0.498 ± 0.017 mm; P = 4.7 × 10−7). AL (Fig. 4D) significantly increased with age for WT and Adamts10G661R/G661R mice (P = 8.0 × 10−6 and P = 1.8 × 10−8, respectively), suggesting that the age-dependent deepening of the ACD is due to overall growth of the eye. However, there were no significant differences in AL between WT and Adamts10G661R/G661R mice at 6 or 24 months of age (P > 0.8) (Fig. 4D), indicating that the shallower ACD was not due to reduced eye size. Because thickening of the CCT is considerably less than the reduction of ACD in Adamts10G661R/G661R mice, anterior displacement of the lens likely contributes to reduction of the ACD.
Enhanced Immunofluorescent Signal for ADAMTS10 on Adamts10G661R/G661R Zonules
Anterior displacement of the lens indicates weakened zonules, which are primarily composed of fibrillin-1 microfibrils. Therefore, we investigated ADAMTS10 expression in the zonules by immunohistochemistry on sagittal sections of eyes from WT and Adamts10G661R/G661R mice at P10 and 3 and 24 months of age. In WT mice, the ADAMTS10 immunofluorescence signal of the zonules was barely or not detectable at all ages (Figs. 5A–5C). For Adamts10G661R/G661R mice, ADAMTS10 immunofluorescence of the zonules was dim at P10 (Fig. 5D) but became increasingly strong at 3 and 24 months of age (Figs. 5E, 5F). Overall, the ADAMTS10 immunofluorescent signal on the Adamts10G661R/G661R zonules was stronger than for WT mice at all three age points (compare Figs. 5D–5F with Figs. 5A–5C). These results indicate that the G661R mutation of Adamts10 results in greater abundance of ADAMTS10 on the zonules.
Figure 5.

Enhanced ADAMTS10 immunofluorescence on the Adamts10G661R/G661R zonules. (A–C) ADAMTS10 fluorescence (green) was barely detected in the WT zonules at P10 and 3 and 24 months. (D–F) Enhanced ADAMTS10 immunofluorescent staining (green) was detected on the Adamts10G661R/G661R zonules at P10 and 3 and 24 months. Blue indicates DAPI staining. Scale bars: 100 µm. L, lens; CB, ciliary body; *zonule.
Enhanced Immunofluorescent Signals for Fibrillin-2 and Fibrillin-1 on Adamts10G661R/G661R Zonules
Zonular fibers are principally composed of fibrillin-1 and fibrillin-2 microfibrils. In WT mice, an age-dependent switch in immunofluorescence from fibrillin-2 dominant at P10 to fibrilin-1 dominant in zonules of adults 3 and 24 months of age was observed (compare Figs. 6A–6C with Figs. 6G–6I). Fibrillin-2 fluorescence signals on the zonules of WT mice were detected at P10 (Fig. 6A) but decreased with age to barely detectable levels at 3 and 24 months of age (Figs. 6B, 6C; quantification in Fig. 6M). Conversely, zonular fibrillin-1 immunofluorescence in WT mice was barely detectable at P10 but was subsequently strongly expressed at 3 and 24 months of age (Figs. 6G–6I). However, for Adamts10G661R/G661R mice, rather than a switch in predominance, fibrillin-2 immunofluorescence maintained strong expression from P10 through 24 months of age (Figs. 6D–6F), and fibrilin-1 immunofluorescence was detectable at P10 and increased at 3 and 24 months of age to levels higher than those found in WT mice at 3 months and similar to WT mice at 24 months of age (compare Figs. 6J–6L with Figs. 6G–6I; quantitation in Fig. 6M). These findings suggest that the G661R mutation of Adamts10 perturbs the normal development of zonule composition.
Figure 6.

Enhanced fibrillin-1 and fibrillin-2 immunofluorescence in the Adamts10G661R/G661R zonules. Fibrillin-2 immunofluorescence staining (green) was stronger in the Adamts10G661R/G661R zonules (D–F) compared with the WT mice (A–C) at P10 and 3 and 24 months of age. Fibrillin-1 staining (green) was stronger in the Adamts10G661R/G661R zonules at P10 and 3 months (J, K) compared with WT mice (G, H) but was comparable at 24 months (I, L). Blue indicates DAPI staining. Scale bars: 150 µm. Quantification of fibrillin-2 (M, right) and fibrillin-1 (M, left) immunofluorescence showed increased mean intensity for Adamts10G661R/G661R zonules. Error bars represent mean ± SD. Numbers of mice are indicated in italics below each group, and P values from two-tailed Student's t-test are indicated above brackets (B–D).
Persistence of Fibrillin-2 Microfibrils in the Adamts10G661R/G661R Vitreous
The hyaloid vasculature is a transient vascular bed in the vitreous that regresses concurrently with formation of the retinal vasculature.40 For mice, regression of the hyaloid vasculature is complete by P21.41 In the vitreous of WT and Adamts10G661R/G661R mice, cell nuclei were detected at P10 (Figs. 7A, 7D, 7G, 7J) but not at 3 or 24 months of age (Figs. 7B, 7C, 7E, 7F, 7H, 7I, 7K, 7L), indicating that the hyaloid vessels underwent normal regression in both genotypes of mice. However, compared with WT mice, Adamts10G661R/G661R mice displayed a dense network of relatively strong fibrillin-2 immunostaining of the hyaloid vasculature at P10 (compare Fig. 7D with 7A). The dense network of fibrillin-2 fluorescence signal persisted in the remnants of the hyaloid vasculature at 3 months of age (Fig. 7E) and remained detectable at 24 months of age (Fig. 7F) in contrast to WT mice, which lacked fibrillin-2 immunoreactivity at those ages (Figs. 7B, 7C). A broader distribution of fibrillin-1 immunofluorescence remained for Adamts10G661R/G661R mice compared with WT mice at 3 and 24 months of age (compare Figs. 7K and 7L with Figs. 7H and 7I). These findings are consistent with a role for ADAMTS10 in determining the fibrillin isoform composition and regression of the hyaloid vasculature, a function that is perturbed by the G661R mutation.
Figure 7.

Persistent postnatal fibrillin-2 and fibrillin-1 immunofluorescence in the vitreous of Adamts10G661R/G661R mice. Fibrillin-2 immunofluorescence staining (green) was stronger in the Adamts10G661R/G661R vitreous at P10 compared with WT mice (D compared with A), and it persisted into 3 and 24 months of age in Adamts10G661R/G661R mice (E, F), with barely detectable fibrillin-2 fluorescence in WT mice (B, C). Fibrillin-1 staining (green) was enhanced in the Adamts10G661R/G661R vitreous at P10 (J, G), with a broader distribution at 3 and 24 months of age (K, L) compared with WT mice (H, I). DAPI staining (blue) shows intravitreal nuclei at P10 (A, D, G, J), but not at 3 and 24 months of age (B, C, E, F, H, I, K, L). Images are representative of staining from two independent experiments. Scale bars: 150 µm. V, vitreous; R, retina.
Discussion
Beagle dogs homozygous for the G661R mutation of ADAMTS10 develop POAG, characterized by decreased facility of aqueous humor outflow with open iridocorneal angles followed by elevated IOP and subsequent development of glaucomatous optic disc cupping.4 In this study, elevation of IOP was not observed in mice on the C57BL/6J background with the G661R mutation. This result likely indicates species-specific differences in the regulation of aqueous humor outflow and the role of ADAMTS10 in this process. The Adamts10G661R/G661R mice do have optic nerve phenotypes that may be relevant to glaucoma which will be presented in a subsequent manuscript.
The shorter and lighter body of Adamts10G661R/G661R mice found in the present study is similar to that observed by Mularczyk et al.33 in mice with a human WMS-associated truncation mutation of Adamts10 (Adamts10S236X/S236X). The authors of that study found that Adamts10S236X/S236X mice have altered chondrocyte differentiation, which could contribute to their shorter long bones.33 In another work by Wang et al.,34 in which exon 5 of Adamts10 was disrupted by a β-galactosidase reporter, the resulting Adamts10−/− mice were shown to have lower body weight, although they lacked skeletal abnormalities. WMS can also be caused by mutations in ADAMTS17, a gene that is structurally and functionally closely related to ADAMTS1030 and has been linked to height variation in humans.42,43 Study of a conditional knockout of Adamts17 in mice (Adamts17−/−) revealed that Adamts17−/− mice recapitulate the short stature phenotype of WMS and suggested that ADAMTS17 is involved in bone development through regulation of the BMP–Smad1/5/8 pathway.44 The molecular mechanism whereby ADAMTS10 regulates skeletal growth remains unclear, although our finding of smaller body size indicates that the G661R mutation of Adamts10 interferes with this function in mice. In addition to small body size, we found that Adamts10G661R/G661R mice had shallow anterior chambers in the context of normal AL, suggestive of lens dislocation, as well as significantly thicker than normal corneas, both common features of WMS.16,39 Therefore, the single amino acid change of the Adamts10G661R/G661R mice results in several features of WMS, similar to mice with null alleles of Adamts10.
Dogs homozygous for the G661R mutation of ADAMTS10 develop glaucoma and dislocated lenses,4 which are features of human WMS.16 However, body size reduction in dogs homozygous for G661R has not been reported, although this feature may not be obvious and would require careful study of a large number of dogs; for example, a study has shown that dogs homozygous for glaucoma-causing variants in ADAMTS17 are 4% to 10% shorter at the withers.45
The G661R mutation is within the highly conserved cysteine-rich region of ADAMTS102 and therefore was expected to have detrimental effects on ADAMTS10 function. ADAMTS10 has been shown to bind fibrillin-1 with high affinity and was shown to enhance or accelerate formation of fibrillin microfibrils in cell culture.32 Although Adamts10 deficiency might be expected to reduce fibrillin microfibrils, we found enhanced immunofluorescent signal for fibrillin-2 in the zonule fibers of Adamts10G661R/G661R mice. A similar enhancement of zonular fibrillin-2 immunoreactivity has also been found in the mouse strains with inactivated Adamts10, Adamts10S236X/S236X, and Adamts10−/−.33,34 ADAMTS10 is synthesized as a zymogen, although it is innately resistant to furin cleavage and activation due to its suboptimal furin processing site.32 Work from Apte's group showed that enabling ADAMTS10 activation by optimizing the furin processing site resulted in cleavage of fibrillin-1 and fibrillin-2 by ADAMTS10, suggesting that fibrillin-1 and fibrillin-2 can be substrates for ADAMTS10.32,34 Differences in fibrillin-2 immunostaining in Adamts10G661R/G661R mice may result from reduced fibrillin-2 cleavage by ADAMTS10. The positive immunoreactivity for ADAMTS10 in the zonules that we found, and which was also reported by Mularczyk et al.,33 would be consistent with this. However, determining whether and how ADAMTS10 is catalytically activated in vivo remains elusive.30
Alternatively, differences in fibrillin-2 immunostaining in ADAMTS10-deficient mice could result from increased fibrillin-2 expression rather than abnormal fibrillin-2 cleavage. ADAMTS10 could function similarly to ADAMTS17 in the suppression of fibrillin-2 incorporation into microfibrils.46 ADAMTS17 binds selectively to fibrillin-2 but does not cleave either fibrillin isoform, although it was shown to transcriptionally downregulate fibrillin-2 mRNA expression in mouse embryonic fibroblasts.46 However, enhanced fibrillin-2 immunostaining without an effect on fibrillin-2 mRNA expression has been observed in the perichondrial extracellular matrix (ECM) of the growth plate of mice lacking ADAMTS17 and in the ECM of the lungs of mice lacking ADAMTSL2.44,47 It is interesting to note that ADAMTSL2 lacks proteolytic activity and therefore appears to increase fibrillin-2 immunostaining through neither reduced proteolysis nor downregulation of Fbn2 mRNA. It seems that several ADAMTS family proteins participate in a complex regulation of the fibrillin isoform composition of microfibrils using a variety of mechanisms in a tissue-specific manner.
Contrary to previously reported normal fibrillin-1 immunofluorescent staining on the zonules of Adamts10S236X/S236X mice and possibly reduced staining in Adamts10−/− mice,33,34 we found that Adamts10G661R/G661R mice have enhanced fibrillin-1 immunofluorescence on the zonules. This would be consistent with the G661R mutation curtailing the proteolysis of fibrillin-1 by ADAMTS10. Another possibility would be that the G661R mutation of Adamts10 enhances its normal function of facilitating fibrillin-1 microfibril assembly. Other ADAMTS/ADAMTSL proteins such as ADAMTS6, ADAMTSL2, ADAMTSL4, ADAMTSL5, and ADAMTSL6 have also been shown to interact with fibrillin-1 and accelerate its assembly.47–51
An age-dependent decrease in fibrillin-2 fluorescent signal was seen in the WT and Adamts10G661R/G661R zonules. Although this could have resulted from fibrillin-2 proteolysis, another possibility is that the fibrillin-2 epitope becomes masked by fibrillin-1.52 It is well established that fibrillin-2 mRNA dominates the embryonic and juvenile development periods of mice, and its expression dramatically decreases as fibrillin-1 mRNA expression increases in the adult stage.18,20 Microfibrils incorporate fibrillin isoforms in a nonselective manner26; therefore, the isoform composition of microfibrils is partially determined by the bioavailability of the fibrillin isoforms. Using super resolution imaging, Shi et al.53 revealed that fibrillin-1 expression was restricted to the outer layer of the zonular fibers, whereas fibrillin-2 was present throughout the fiber, implying a laminar growth pattern of the zonule in which newly synthesized components are added onto the surface of the existing fibers, possibly masking fibrillin-2 epitopes.
Adamts10G661R/G661R mice developed shallow anterior chambers with normal AL. Although thickening of the cornea could contribute to reduction of the ACD, the magnitude of the CCT increase (6 µm at 3 months and 9 µm at 24 months) was smaller than the magnitude of the decreased ACD (22 µm at 3 months and 25 µm at 24 months). This suggests that the bulk of the reduction in ACD was due to dislocation of the lens anteriorly. Lens dislocation is consistent with human WMS and indicative of weakened zonular fibers that stabilize the lens within the eye.
The biological consequences of altered fibrillin isoform composition of microfibrils have been investigated in mice with fibrillin-1 or fibrillin-2 deficiencies.54,55 Conditional knockout of FBN1 in the mouse eye has been shown to result in small zonular fibers with reduced tensile strength and a deepened anterior chamber due to posterior lens dislocation.55 FBN2 knockout mice mainly develop iris abnormalities with changes in lens size rather than lens dislocation.54 An appropriate balance of fibrillin isoform composition of microfibrils seems to be highly controlled and required for proper development of the lens zonules. Abnormal fibrillin isoform composition of microfibrils of the Adamts10G661R/G661R zonule may alter their tensile strength, thus affecting zonule stability leading to lens dislocation.
In Adamts10G661R/G661R mice, fibrillin-2 immunofluorescence was detected in the vitreous, associated with the hyaloid vasculature at P10 and with likely remnants of the supporting ECM of hyaloid vessels at 3 and 24 months of age when there were no 4′,6-diamidino-2-phenylindole (DAPI)-positive nuclei in the vitreous. A similar observation was made by Wang et al.34 for Adamts10−/− mice.34 Additionally, we observed broader distribution of fibrillin-1 immunofluorescence in the Adamts10G661R/G661R vitreous, a feature that was not detected in Adamts10−/− mice. ADAMTS10 is expressed in the retinal nerve fiber layer,33 which could be the source of soluble ADAMTS10 available to interact with microfibrils in the vitreous and proteolyzing fibrillin-2 while accelerating fibrillin-1 microfibril assembly. Similar to our findings in the zonules, the G661R mutation of Adamts10 results in enhanced fibrillin-2 and fibrillin-1 immunoreactivity in the hyaloid vasculature and its remnants. Retention of the hyaloid vasculature remnants could affect visual function. However, we found no differences in electroretinograms of dark-adapted 3-month-old mice or in optomoter responses at 6 months and 2 years of age (data not shown), indicating that retention of the remnants of hyaloid vasculature did not significantly affect visual function of the Adamts10−/− mice.
There were some observed differences between our knock-in model introducing a single amino acid substitution and the knockout models that introduced premature stop codons near the 5′ end of Adamts10, as may be expected. Unlike the Adamts10S236X/S236X mice of Mularczyk et al.,33 we did not observe smaller ciliary bodies in Adamts10G661R/G661R mice (data not shown). In contrast to the Adamts10 knockout model of Wang et al.,34 the viability of Adamts10G661R/G661R mice was normal, with mating of heterozygous mice producing Adamts10G661R/G661R mice at the expected Mendelian ratio.
In summary, we investigated the effect of the glaucoma-causative G661R mutation of Adamts10 on body and fibrillin phenotypes of Adamts10G661R/G661R mice. The small body size, thickened cornea, and lens dislocation phenotypes of Adamts10G661R/G661R mice recapitulate features of human WMS. Enhanced immunofluorescent signal of fibrillin-2 and fibrillin-1 of the lens zonules and of the hyaloid vasculature and its remnants in the vitreous of Adamts10G661R/G661R mice indicate that ADAMTS10 is involved in regulating the fibrillin isoform composition of microfibrils in the mouse eye and that the G661R mutation of Adamts10 perturbs that function.
Acknowledgments
The authors thank Lynn Sakai, PhD (Oregon Health and Science University, Portland, OR, USA) for kindly providing the anti-fibrillin-1 antibodies pAb 9543 and pAb 0868 for this study. We also thank Abudi Nashabi for optic nerve histology and Samyukta Reddy for mouse colony management and genotyping.
Supported by a grant from the National Eye Institute, National Institutes of Health (EY027746 to JK), by a Vanderbilt Vision Research Center core grant (P30EY008126), and by a Departmental Unrestricted Award from Research to Prevent Blindness. Imaging was performed through the use of the Vanderbilt Cell Imaging Shared Resource, which is supported by grants from the National Institutes of Health (CA68485, DK20593, DK58404, DK59637, and S10 RR023901). The Vanderbilt Genome Editing Resource (RRID:SCR_018826) was supported by grants from the Vanderbilt Cancer Center (CA68485) and Vanderbilt Diabetes Research and Training Center (DK020593), and by the Center for Stem Cell Biology.
Disclosure: H.-J. Wu, None; D.P. Mortlock, None; R.W. Kuchtey, None; J. Kuchtey, None
References
- 1.Apte SS.A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009; 284(46): 31493–31497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuchtey J, Olson LM, Rinkoski T, et al.. Mapping of the disease locus and identification of ADAMTS10 as a candidate gene in a canine model of primary open angle glaucoma. PLoS Genet. 2011; 7(2): e1001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuchtey J, Kunkel J, Esson D, et al.. Screening ADAMTS10 in dog populations supports Gly661Arg as the glaucoma-causing variant in beagles. Invest Ophthalmol Vis Sci. 2013; 54(3): 1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gelatt KN, Peiffer RL, Gwin RM, Gum GG, Williams LW.. Clinical manifestations of inherited glaucoma in the beagle. Invest Ophthalmol Vis Sci. 1977; 16(12): 1135–1142. [PubMed] [Google Scholar]
- 5.Ahonen SJ, Kaukonen M, Nussdorfer FD, Harman CD, Komáromy AM, Lohi H.. A novel missense mutation in ADAMTS10 in Norwegian Elkhound primary glaucoma. PLoS One. 2014; 9(11): e111941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oliver JAC, Rustidge S, Pettitt L, et al.. Evaluation of ADAMTS17 in Chinese Shar-Pei with primary open-angle glaucoma, primary lens luxation, or both. Am J Vet Res. 2018; 79(1): 98–106. [DOI] [PubMed] [Google Scholar]
- 7.Oliver JAC, Forman OP, Pettitt L, Mellersh CS.. Two independent mutations in ADAMTS17 are associated with primary open angle glaucoma in the Basset Hound and Basset Fauve de Bretagne breeds of dog. PLoS One. 2015; 10(10): e0140436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forman OP, Pettitt L, Komáromy AM, Bedford P, Mellersh C.. A novel genome-wide association study approach using genotyping by exome sequencing leads to the identification of a primary open angle glaucoma associated inversion disrupting ADAMTS17. PLoS One. 2015; 10(12): e0143546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Springelkamp H, Höhn R, Mishra A, et al.. Meta-analysis of genome-wide association studies identifies novel loci that influence cupping and the glaucomatous process. Nat Commun. 2014; 5: 4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Springelkamp H, Iglesias AI, Mishra A, et al.. New insights into the genetics of primary open-angle glaucoma based on meta-analyses of intraocular pressure and optic disc characteristics. Hum Mol Genet. 2017; 26(2): 438–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dagoneau N, Benoist-Lasselin C, Huber C, et al.. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004; 75(5): 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morales J, Al-Sharif L, Khalil DS, et al.. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009; 85(5): 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kutz WE, Wang LW, Dagoneau N, et al.. Functional analysis of an ADAMTS10 signal peptide mutation in Weill-Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme. Hum Mutat. 2008; 29(12): 1425–1434. [DOI] [PubMed] [Google Scholar]
- 14.Faivre L, Gorlin RJ, Wirtz MK, et al.. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003; 40(1): 34–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sengle G, Tsutsui K, Keene DR, et al.. Microenvironmental regulation by fibrillin-1. PLoS Genet. 2012; 8(1): e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faivre L, Dollfus H, Lyonnet S, et al.. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am J Med Genet A. 2003; 123A(2): 204–207. [DOI] [PubMed] [Google Scholar]
- 17.Zeyer KA, Reinhardt DP.. Fibrillin-containing microfibrils are key signal relay stations for cell function. J Cell Commun Signal. 2015; 9(4): 309–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mariencheck MC, Davis EC, Zhang H, et al.. Fibrillin-1 and fibrillin-2 show temporal and tissue-specific regulation of expression in developing elastic tissues. Connect Tissue Res. 1995; 31(2): 87–97. [DOI] [PubMed] [Google Scholar]
- 19.Sabatier L, Miosge N, Hubmacher D, Lin G, Davis EC, Reinhardt DP.. Fibrillin-3 expression in human development. Matrix Biol. 2011; 30(1): 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Hu W, Ramirez F.. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol. 1995; 129(4): 1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corson GM, Charbonneau NL, Keene DR, Sakai LY.. Differential expression of fibrillin-3 adds to microfibril variety in human and avian, but not rodent, connective tissues. Genomics. 2004; 83(3): 461–472. [DOI] [PubMed] [Google Scholar]
- 22.Sakai LY, Keene DR, Engvall E.. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986; 103(6): 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramirez F, Sakai LY.. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010; 339(1): 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramirez F, Rifkin DB.. Extracellular microfibrils: contextual platforms for TGFβ and BMP signaling. Curr Opin Cell Biol. 2009; 21(5): 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baldwin AK, Simpson A, Steer R, Cain SA, Kielty CM.. Elastic fibres in health and disease. Expert Rev Mol Med. 2013; 15: e8. [DOI] [PubMed] [Google Scholar]
- 26.Beene LC, Wang LW, Hubmacher D, et al.. Nonselective assembly of fibrillin 1 and fibrillin 2 in the rodent ocular zonule and in cultured cells: implications for Marfan syndrome. Invest Ophthalmol Vis Sci. 2013; 54(13): 8337–8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgoyne CF, Crawford Downs J, Bellezza AJ, Francis Suh J-K, Hart RT. The optic nerve head as a biomechanical structure: a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog Retin Eye Res. 2005; 24(1): 39–73. [DOI] [PubMed] [Google Scholar]
- 28.Fuchshofer R, Tamm ER.. The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012; 347(1): 279–290. [DOI] [PubMed] [Google Scholar]
- 29.Hubmacher D, Apte SS.. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015; 47: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karoulias SZ, Taye N, Stanley S, Hubmacher D.. The ADAMTS/fibrillin connection: insights into the biological functions of ADAMTS10 and ADAMTS17 and their respective sister proteases. Biomolecules. 2020; 10(4): 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Somerville RPT, Jungers KA, Apte SS.. Discovery and characterization of a novel, widely expressed metalloprotease, ADAMTS10, and its proteolytic activation. J Biol Chem. 2004; 279(49): 51208–51217. [DOI] [PubMed] [Google Scholar]
- 32.Kutz WE, Wang LW, Bader HL, et al.. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J Biol Chem. 2011; 286(19): 17156–17167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mularczyk EJ, Singh M, Godwin ARF, et al.. ADAMTS10-mediated tissue disruption in Weill-Marchesani syndrome. Hum Mol Genet. 2018; 27(21): 3675–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang LW, Kutz WE, Mead TJ, et al.. Adamts10 inactivation in mice leads to persistence of ocular microfibrils subsequent to reduced fibrillin-2 cleavage. Matrix Biol. 2019; 77: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G.. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol Ther Nucleic Acids. 2014; 3(12): e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janssen CF, Maiello P, Wright MJ, Kracinovsky KB, Newsome JT. Comparison of atipamezole with yohimbine for antagonism of xylazine in mice anesthetized with ketamine and xylazine. J Am Assoc Lab Anim Sci. 2017; 56(2): 142–147. [PMC free article] [PubMed] [Google Scholar]
- 37.Charbonneau NL, Dzamba BJ, Ono RN, et al.. Fibrillins can co-assemble in fibrils, but fibrillin fibril composition displays cell-specific differences. J Biol Chem. 2003; 278(4): 2740–2749. [DOI] [PubMed] [Google Scholar]
- 38.Aihara M, Lindsey JD, Weinreb RN.. Twenty-four-hour pattern of mouse intraocular pressure. Exp Eye Res. 2003; 77(6): 681–686. [DOI] [PubMed] [Google Scholar]
- 39.Razeghinejad MR, Safavian H.. Central corneal thickness in patients with Weill-Marchesani syndrome. Am J Ophthalmol. 2006; 142(3): 507–508. [DOI] [PubMed] [Google Scholar]
- 40.Lutty GA, McLeod DS.. Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog Retin Eye Res. 2018; 62: 58–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Liu C-H, Huang S, Chen J.. Assessment and characterization of hyaloid vessels in mice. J Vis Exp. 2019;(147): 10.3791/59222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Duyvenvoorde HA, Lui JC, Kant SG, et al.. Copy number variants in patients with short stature. Eur J Hum Genet. 2014; 22(5): 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lango Allen H, Estrada K, Lettre G, et al.. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010; 467(7317): 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oichi T, Taniguchi Y, Soma K, et al.. Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway. Cell Mol Life Sci. 2019; 76(23): 4795–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeanes EC, Oliver JAC, Ricketts SL, Gould DJ, Mellersh CS.. Glaucoma-causing ADAMTS17 mutations are also reproducibly associated with height in two domestic dog breeds: selection for short stature may have contributed to increased prevalence of glaucoma. Canine Genet Epidemiol. 2019; 6(1): 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hubmacher D, Schneider M, Berardinelli SJ, et al.. Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci Rep. 2017; 7: 41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hubmacher D, Wang LW, Mecham RP, Reinhardt DP, Apte SS.. Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia–a novel mouse model providing insights into geleophysic dysplasia. Dis Model Mech. 2015; 8(5): 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saito M, Kurokawa M, Oda M, et al.. ADAMTSL6β protein rescues fibrillin-1 microfibril disorder in a Marfan syndrome mouse model through the promotion of fibrillin-1 assembly. J Biol Chem. 2011; 286(44): 38602–38613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabriel LAR, Wang LW, Bader H, et al.. ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Invest Ophthalmol Vis Sci. 2012; 53(1): 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bader HL, Wang LW, Ho JC, et al.. A disintegrin-like and metalloprotease domain containing thrombospondin type 1 motif-like 5 (ADAMTSL5) is a novel fibrillin-1-, fibrillin-2-, and heparin-binding member of the ADAMTS superfamily containing a netrin-like module. Matrix Biol. 2012; 31(7): 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsutsui K, Manabe R, Yamada T, et al.. ADAMTSL-6 is a novel extracellular matrix protein that binds to fibrillin-1 and promotes fibrillin-1 fibril formation. J Biol Chem. 2010; 285(7): 4870–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Charbonneau NL, Jordan CD, Keene DR, et al.. Microfibril structure masks fibrillin-2 in postnatal tissues. J Biol Chem. 2010; 285(26): 20242–20251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi Y, Jones W, Beatty W, et al.. Latent-transforming growth factor beta-binding protein-2 (LTBP-2) is required for longevity but not for development of zonular fibers. Matrix Biol. 2021; 95: 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi Y, Tu Y, Mecham RP, Bassnett S.. Ocular phenotype of Fbn2-null mice. Invest Ophthalmol Vis Sci. 2013; 54(12): 7163–7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones W, Rodriguez J, Bassnett S.. Targeted deletion of fibrillin-1 in the mouse eye results in ectopia lentis and other ocular phenotypes associated with Marfan syndrome. Dis Model Mech. 2019; 12(1): dmm037283. [DOI] [PMC free article] [PubMed] [Google Scholar]