

Abstract
In humans, various genetic defects or age-related diseases, such as diabetic retinopathies, glaucoma, and macular degeneration, cause the death of retinal neurons and profound vision loss. One approach to treating these diseases is to utilize stem and progenitor cells to replace neurons in situ, with the expectation that new neurons will create new synaptic circuits or integrate into existing ones. Reprogramming non-neuronal cells in vivo into stem or progenitor cells is one strategy for replacing lost neurons. Zebrafish have become a valuable model for investigating cellular reprogramming and retinal regeneration. This review summarizes our current knowledge regarding spontaneous reprogramming of Müller glia in zebrafish and compares this knowledge to research efforts directed toward reprogramming Müller glia in mammals. Intensive research using these animal models has revealed shared molecular mechanisms that make Müller glia attractive targets for cellular reprogramming and highlighted the potential for curing degenerative retinal diseases from intrinsic cellular sources.
Keywords: zebrafish, retina, epigenetics, growth factors, Notch, cytokines
1. OVERVIEW: RESPONSE OF MÜLLER GLIA TO RETINAL INJURY
Müller glia are a type of radial astroglia present in the retinas of all vertebrates and the predominant non-neuronal cell type in this tissue (Bringmann et al. 2006). Arrayed as radial columns, Müller glia span the thickness of the retina (Figure 1). Müller glia somata reside in the inner nuclear layer and extend cytoplasmic processes that ensheath neuronal somata, axons, dendrites, and synaptic complexes. In vascularized retinas, Müller glia also help to form the blood–retina barrier. Similar to astrocytes in other regions of the brain, Müller glia mediate neurovascular coupling and form an intimate metabolic partnership with photoreceptors and other retinal neurons (Bringmann et al. 2006, Vecino et al. 2016).
Figure 1.
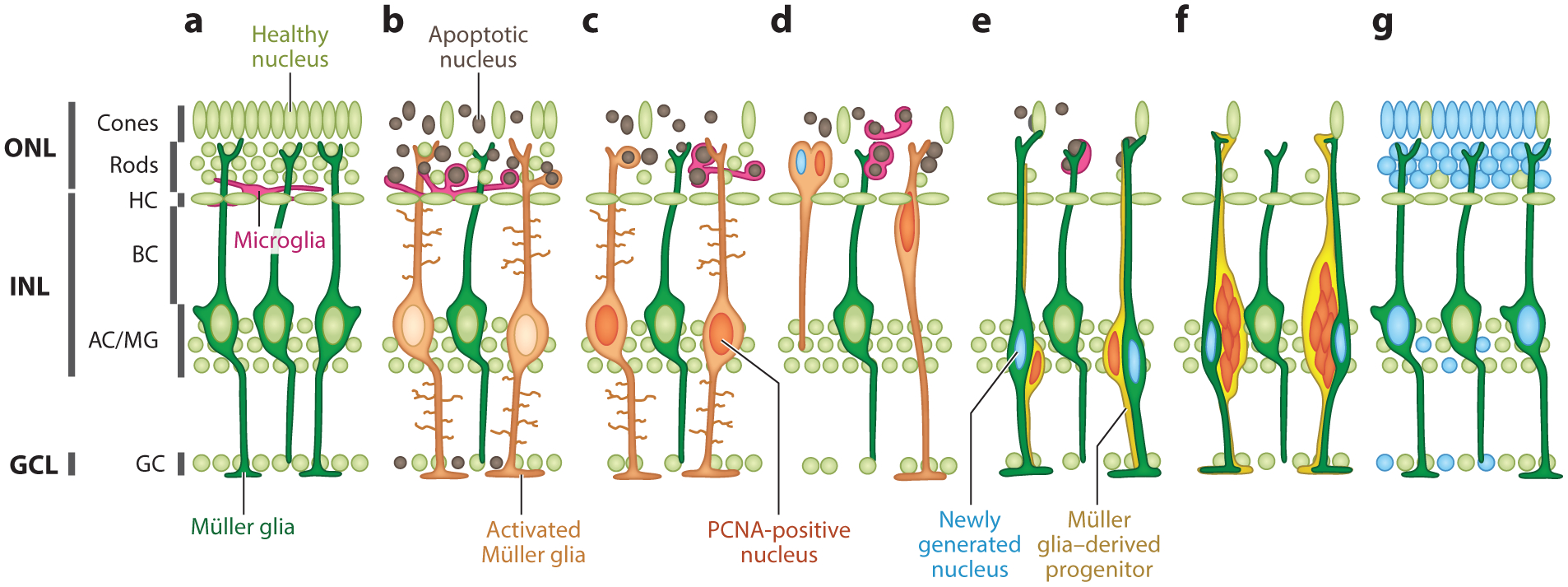
Time course of retinal regeneration in zebrafish. (a) Schematic of an uninjured retina. In the injured retina, (b) apoptotic cells are cleared by microglia and MG, and a subset of MG dedifferentiate or reprogram (light orange), (c) resulting in reentry into the cell cycle, indicated by the upregulation of PCNA. (d) MG undergo interkinetic nuclear migration to the ONL, (e) where they divide into MG-derived progenitors and postmitotic MG that return to the INL. (f) MG-derived progenitors continue to proliferate, generating clusters of progenitor cells that (g) ultimately differentiate into neurons that replace those that were ablated. Abbreviations: AC, amacrine cell; BC, bipolar cell; GC, ganglion cell; GCL, ganglion cell layer; HC, horizontal cell; INL, inner nuclear layer; MG, Müller glia; ONL, outer nuclear layer; PCNA, proliferating cell nuclear antigen. Figure adapted with permission from Lahne & Hyde (2017).
The response of Müller glia to injury or disease has been intensely studied (e.g., Bringmann et al. 2006, Graca et al. 2018, Reichenbach & Bringmann 2013, Subirada et al. 2018). When retinal neurons are stressed or die, Müller glia undergo a complex cascade of structural and molecular changes that are most frequently described as a gliotic response. In mammals, this reactive gliosis is characterized by cellular hypertrophy, increased expression of intermediate filaments (GFAP, Vimentin, Nestin), and activation of extracellularly regulated kinases 1 and 2 (ERK1/2) (Bringmann et al. 2009). In mammals, in the short term, gliosis acts as a wound repair response to maintain a permissive environment for neuronal function and to prolong neuronal survival through the release of neuroprotective factors (Bringmann et al. 2009). However, chronic gliosis results in changes in Müller glia stiffness, decreased expression of enzymes that maintain retinal homeostasis, marked structural remodeling, secretion of proinflammatory cytokines, abnormal proliferation, and formation of a gliotic scar (Bringmann et al. 2009). Paradoxically, in mammals, chronic gliosis can accelerate neuronal death, thereby exacerbating the original insult. In teleost fish, Müller glia also initiate a gliotic response following neuronal death (Thomas et al. 2016); however, in some teleosts, this reactive gliosis is transient and precedes reparative neurogenesis, in which Müller glia acquire characteristics of retinal stem cells and replace ablated neurons that restore functional vision (Bernardos et al. 2007, Fausett & Goldman 2006, Kassen et al. 2007, Sherpa et al. 2008, Vihtelic & Hyde 2000). In this article, we systematically review the literature on retinal regeneration in teleost fish, focusing on the molecular mechanisms that underlie the endogenous reprogramming of zebrafish Müller glia. We then briefly review Müller glia reprogramming in chicks and mammals and report on recent comparative analyses of injury-induced genetic changes in Müller glia in zebrafish, chicks, and mammals.
2. RETINAL REGENERATION IN TELEOST FISH
Teleost fish are a long-standing model for studying retinal development and neuronal regeneration. In teleosts, the embryonic retina is a tiny fraction of its adult size, and retinal growth proceeds both by expansion of the extant tissue and by persistent neurogenesis (Johns 1977, Johns & Fernald 1981). Furthermore, some teleosts have the capacity to fully regenerate ablated retinal neurons from a source intrinsic to the retina (Raymond et al. 1988; see also Maier & Wolburg 1979). Initially, rod progenitors, which give rise to new rod photoreceptors, were suggested as the likeliest origin of regenerated neurons (Raymond et al. 1988). However, it was discovered that proliferating cells in the inner nuclear layer, subsequently identified as Müller glia, give rise to both rod progenitors during persistent neurogenesis and neural progenitors during regenerative neurogenesis (Bernardos et al. 2007, Fausett & Goldman 2006, Hitchcock & Raymond 2004, Kassen et al. 2007, Otteson et al. 2001, Raymond & Rivlin 1987, Raymond et al. 2006, Wu et al. 2001, Yurco & Cameron 2005). These foundational studies led to the immediate realization that unraveling the mechanisms underlying the spontaneous reprogramming in fish Müller glia could provide invaluable insights into approaches to induce reprogramming of mammalian Müller glia and thereby advance the goal of repairing the human retina via intrinsic cellular sources.
In the zebrafish retina, neuronal injury induces Müller glia to dedifferentiate or reprogram and adopt hallmarks of a retinal stem cell, which then reenters the cell cycle and divides asymmetrically to produce a progenitor cell, which continues to divide and differentiate into retinal neurons (Bernardos et al. 2007, Kassen et al. 2007, Nagashima et al. 2013). The response of Müller glia to a retinal injury is a complex, highly regulated, multistep process that requires the expression of specific genetic programs at each step (Gorsuch & Hyde 2014). To undergo reprogramming, Müller glia must sense that neurons are injured or dying. This can occur via molecules released from dying cells or reactive microglia, the phagocytosis of dying neurons by Müller glia themselves, or disruptions in intercellular signaling between Müller glia and nearby neurons (Bailey et al. 2010, Battista et al. 2009, Conner et al. 2014, Nelson et al. 2013, White et al. 2017). The latter could be mediated either by loss of direct cell–cell interactions or contacts or by the absence of soluble signaling mediators such as neurotransmitters (Conner et al. 2014, Rao et al. 2017). Müller glia then integrate these external signaling events into epigenetic and transcriptional changes that accommodate reprogramming (Figures 1 and 2). Transcriptional changes include the upregulation of genes encoding pluripotency factors and cell cycle regulators, and the downregulation of genes that maintain Müller glia in a differentiated and mitotically quiescent state (Conner et al. 2014, Gorsuch & Hyde 2014, Gorsuch et al. 2017). Progress through the cell cycle includes interkinetic nuclear migration, where approximately 85% of Müller glia nuclei move to the outer nuclear layer and undergo a single asymmetric cell division to produce a Müller glia–derived progenitor and a postmitotic Müller glia (Nagashima et al. 2013, Lahne et al. 2015). While the phenomenology of retinal regeneration in zebrafish is well described, many questions remain unanswered regarding the molecular events that underlie reprogramming. Although this is not the topic of this review, many details are also lacking regarding the mechanisms that regulate Müller glia–derived progenitors, e.g., their numerical amplification, fate commitment, differentiation, and integration into extant neuronal circuits.
Figure 2.
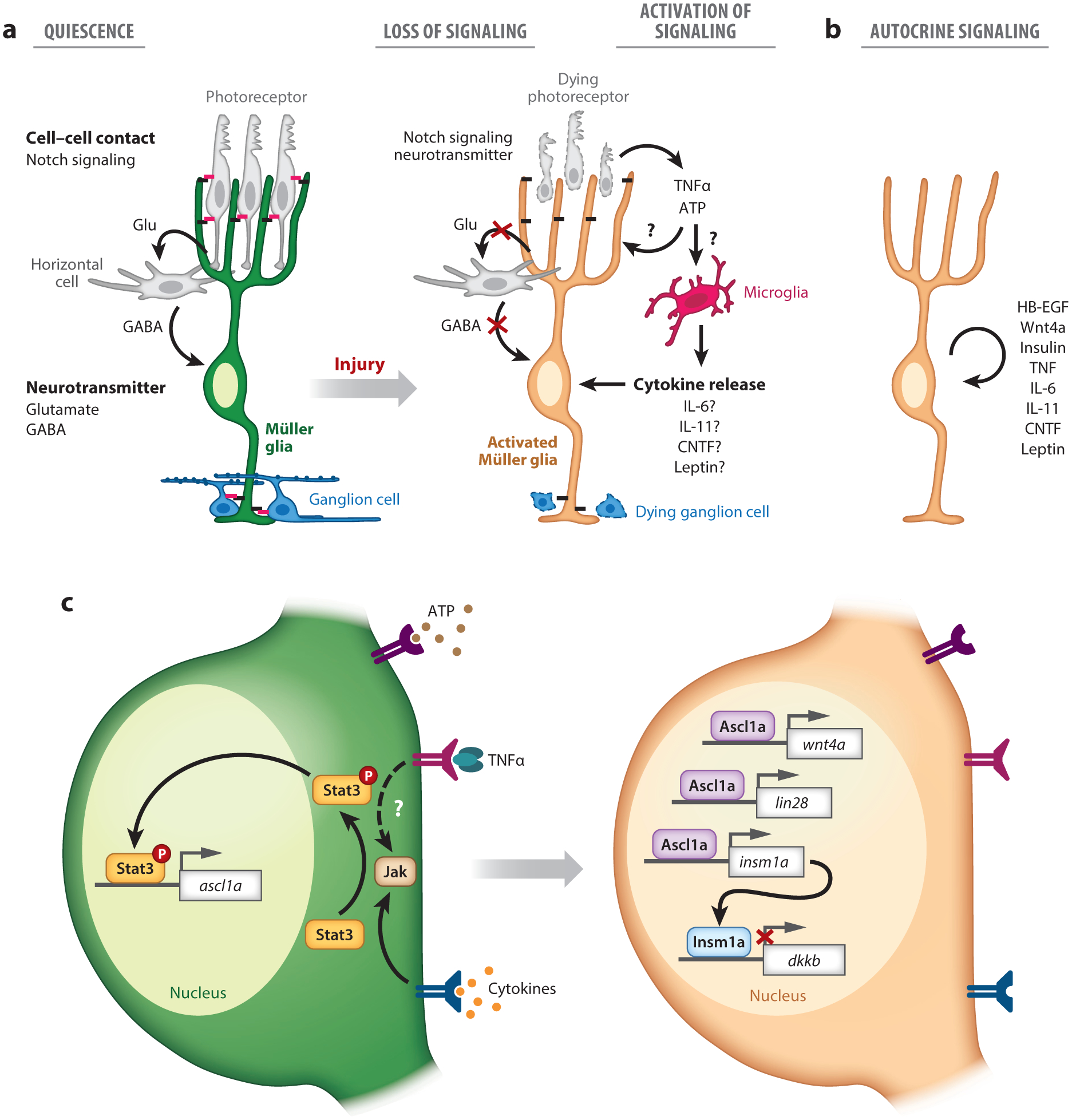
Intercellular and transcriptional regulation of Müller glia reprogramming. (a) In the uninjured retina, neurotransmitter- and cell–cell-contact-mediated signaling through Notch receptors and their ligands maintain Müller glia in a quiescent state (left panel). The loss of neurotransmitter- and cell-contact-mediated signaling, in combination with increased paracrine signaling, initiates the reprogramming of Müller glia (right panel). Tumor necrosis factor alpha (TNFα) and nucleotides released from dying neurons directly act on Müller glia and/or stimulate microglia to release cytokines that induce reprogramming (right panel). (b) Müller glia also release growth factors and cytokines that act in an autocrine fashion. (c) Cytokines including TNFα stimulate Jak-mediated phosphorylation of Stat3, which leads to its nuclear translocation and transcription of its target gene, ascl1a (left). Ascl1a then initiates the expression of insm1a, lin28, and wnt4a, and downstream, Insm1a represses the expression of the Wnt signaling inhibitor, dkkb (right).
3. ACUTE AND CHRONIC INJURY MODELS IN ZEBRAFISH
Various approaches have been employed to induce retinal injury in zebrafish, including (a) intense light damage, which kills photoreceptors but spares other retinal neurons; (b) exposure to neurotoxins, such as NMDA, 6-OHDA or tunicamycin, which kill specific retinal neurons, or ouabain, which induces widespread neuronal cell death; and (c) mechanical injury, such as by inserting a needle through the globe to focally ablate all retinal cell types (Braisted & Raymond 1993, Fimbel et al. 2007, Li & Dowling 2000, Powell et al. 2016, Vihtelic & Hyde 2000). In addition, genetic ablation models have been developed that express the gene encoding Escherichia coli nitroreductase under cell-type-specific promoters, thereby enabling the selective ablation of specific cell types. (Ariga et al. 2010, Fraser et al. 2013, Hagerman et al. 2016, Montgomery et al. 2010, White et al. 2017, Yoshimatsu et al. 2016). Importantly, the cellular events leading to regeneration occur in a similar time frame irrespective of the nature of the injury, thereby allowing results from studies using different injury paradigms to be directly compared.
Retinal regeneration has also been studied in mutant and transgenic lines exhibiting chronic cell death. For example, lines have been isolated that display early onset cone (pde6cw59/w59, aipl2oki6/oki6) or rod (Tg[Xla.Rho:GAP-CFP]q13) photoreceptor cell loss, late onset cone photoreceptor loss (cep290fh297/fh297, syut4+/−), or late ganglion cell death due to increased intraocular pressure (lrp2mw1/mw1) (Iribarne et al. 2017, Lessieur et al. 2019, Morris et al. 2008, Sherpa et al. 2011, Stenkamp et al. 2008, Veth et al. 2011). The lrp2mw1/mw1 mutant and the cone degeneration mutants, except for syut4+/−, display a low level of proliferation in the inner nuclear layer, whereas, following rod photoreceptor degeneration, proliferation occurs predominantly among rod precursors in the outer nuclear layer (Iribarne et al. 2019, Lessieur et al. 2019, Morris et al. 2008, Sherpa et al. 2011, Stenkamp et al. 2008). Together, these genetic models mimic chronic injuries, similar to some inherited retinal diseases in humans, and may provide important insight into regenerative processes in the context of a continuously injured environment.
4. REPROGRAMMING MÜLLER GLIA IN ZEBRAFISH: NOTCH SIGNALING, GROWTH FACTORS, CYTOKINES, AND INFLAMMATION
Müller glia actively monitor the retina’s extracellular environment in support of retinal homeostasis and neuronal function and to maintain the retina’s structural integrity (Bringmann et al. 2006, Insua et al. 2008). In addition to sensing the extracellular environment, cell–cell contacts also allow Müller glia to communicate with their neighbors. Notch signaling—one form of contact-mediated intercellular signaling—occurs between neighboring cells when the Notch receptor binds to its ligands, Delta or Jagged (Figure 2a). This stimulates cleavage of the Notch intracellular domain to form a transcriptional complex that regulates Notch target gene expression (Ho et al. 2019). Pharmacological suppression of Notch signaling in undamaged retinas stimulates a subset of Müller glia to proliferate, indicating that active Notch signaling likely functions to maintain Müller glia in a quiescent state and that decreased Notch signaling is required for reprogramming (Conner et al. 2014). Single-cell RNA-seq analysis of injured zebrafish retinas revealed the downregulation of notch3 in Müller glia; thus, Notch3 could be the receptor that maintains Müller glia in a quiescent state (Hoang et al. 2019). The identity of the ligand and the cell types that express the interacting ligand have not yet been determined. Following retinal injury, Notch ligands, receptors, and downstream target genes are differentially expressed in Müller glia and their progenitors (Raymond et al. 2006, Wan et al. 2012, Yurco & Cameron 2007), suggesting that Notch signaling is dynamically regulated to control the proliferation and differentiation of Müller glia–derived progenitors and to reestablish Müller glia quiescence (Furukawa et al. 2000, Mizeracka et al. 2013, Wan et al. 2012).
Loss of neurotransmitter signaling has also been suggested as a cue that induces Müller glia programming (Figure 2a). In unlesioned retinas, inhibiting signaling through the GABAA receptor is sufficient to stimulate proliferation of Müller glia, and increasing signaling through the GABAA receptor following photoreceptor death suppresses proliferation (Rao et al. 2017). Similarly, inhibition of ionotropic glutamate receptors of the AMPA type stimulates ascl1a-dependent Müller glia proliferation in uninjured retinas, and activation of these receptors reduces proliferation following rod photoreceptor death (Rao et al. 2017). Thus, it has been proposed that, following photoreceptor death, GABA release from horizontal cells is disrupted due to a lack of photoreceptor-mediated glutamate signaling, and the resulting absence of GABA signaling initiates Müller glial reprogramming. These results suggest more broadly that ambient neurotransmitter levels, monitored by the Müller glia, may function to maintain these cells in a quiescent state, and altered neurotransmitter levels are also able to initiate reprogramming.
Following an injury, numerous factors are released into the extracellular environment that activate complex networks of signaling cascades to reprogram Müller glia (Goldman 2014, Gorsuch & Hyde 2014, Hamon et al. 2016, Karl & Reh 2010, Lenkowski & Raymond 2014) (Figure 2a). Dying neurons represent one source of extrinsic signaling factors (Kassen et al. 2009, Qin et al. 2011). Tumor necrosis factor alpha (TNFα) was identified as a signaling molecule produced by dying neurons that is both necessary and sufficient to reprogram Müller glia by upregulating the reprogramming factor genes, ascl1a and stat3 (Nelson et al. 2013). Immunoblot analysis revealed that the active, i.e., cleaved, form of TNFα is present at the peak of cell death, and active TNFα induces reprogramming (Conner et al. 2014, Nelson et al. 2013). Additionally, coinjecting TNFα and a gamma secretase inhibitor, to suppress Notch signaling, into an uninjured retina is sufficient to induce nearly 90% of Müller glia to proliferate, a greater percentage than is normally observed following injury, and the resulting Müller glia–derived progenitors generate a small number of retinal neurons (Conner et al. 2014). This demonstrates that a full regeneration response likely requires both the induction of activating signals and the suppression of inhibitory ones. Interestingly, once reprogramming has commenced, Müller glia also synthesize TNFα, which functions to regulate the number of Müller glia–derived progenitors (Nelson et al. 2013).
It was also suggested that dying neurons release nucleotides, such as ATP or ADP, which activate purinergic P2 receptors (Figure 2a). Following retinal injury, inhibiting the metabotropic ADP receptor, P2RY1, reduces the expression of the reprogramming factors ascl1a and lin28 and proliferation of Müller glia via an unknown mechanism (Battista et al. 2009, Medrano et al. 2017). Future studies will have to establish which cell type expresses P2RY1 receptors.
Microglia, the innate immune cells of the central nervous system, are another potential cellular source of extrinsic reprogramming factors (Craig et al. 2008, White et al. 2017) (Figure 2a). The best evidence for this is that injury-induced proliferation of Müller glia depends on the presence of activated microglia (Conedera et al. 2019, White et al. 2017). However, the mechanisms by which microglia detect retinal injury and the factors released by activated microglia are not well elucidated. Interestingly, molecules released by dying neurons, e.g., TNFα and ATP, can directly act on immune cells (Sieger et al. 2012, Veroni et al. 2010, Yin et al. 2017). Thus, TNFα and/or ATP might not stimulate Müller glia directly, but rather activate microglia that then release factors that induce Müller glia reprogramming. The inflammatory cytokines IL-6, IL-11, CNTF, and leptin play a role in reprogramming Müller glia, although it is currently unknown whether these factors are expressed by microglia (Kassen et al. 2009, Wan et al. 2014). Microglia-mediated phagocytosis of dying neurons likely also modulates the availability of factors released by dying neurons and thus the downstream reprogramming of Müller glia. Finally, acute inflammation is both necessary and sufficient to reprogram radial glia in the forebrain of zebrafish (Kizil et al. 2015, Kyritsis et al. 2012), and immune suppression diminishes microglial activation and proliferation of Müller glia in the retina (White et al. 2017), indicating that inflammation is a required component of reprogramming in Müller glia.
Müller glia themselves rapidly upregulate the expression of both growth factors and cytokines, which activate a core set of signaling cascades, in an autocrine fashion (Figure 2b), that are required for reprogramming and progression through the cell cycle (Goldman 2014; Kassen et al. 2009; Nagashima et al. 2020; Wan et al. 2012, 2014). Binding of the Wnt ligands Wnt4a or Wnt8b to their receptor, Fzd2, inhibits glycogen synthase kinase-3β, thereby preventing β-catenin degradation and allowing the translocation of β-catenin to the nucleus for transcriptional regulation of Wnt target genes (Meyers et al. 2012, Ramachandran et al. 2011). Pharmacological inhibition of glycogen synthase kinase-3β in an unlesioned retina is sufficient to initiate reprogramming of Müller glia, supporting the interpretation that, in an uninjured retina, signals that promote reprogramming of Müller glia are actively suppressed (Ramachandran et al. 2011; see also Conner et al. 2014, Hamon et al. 2019, Rueda et al. 2019). Heparin binding-epidermal growth factor (HB-EGF), which signals through the epidermal growth factor receptor, induces Müller glia proliferation by activating ERK1/2 of the mitogen-activated protein kinase family and the phospho-inositol-3 kinase-Akt signaling pathways (Wan et al. 2012, 2014). These kinase pathways are also activated by insulin, which acts through the insulin receptors Insra and Insrb (Wan et al. 2014). Genes encoding cytokines, such as m17, clcf1, crlf1a, il-11a, il-11b, and the mammalian leptin homologs lepa and lepb, are also expressed during Müller glia reprogramming. IL-6, IL-11, and CNTF activate cytokine receptors that contain the signaling subunit Gp130, and leptin acts via its receptor, Lepr (Zhao et al. 2014). CNTF and leptin activate Jak-Stat3 signaling; however, the signaling pathways downstream of IL-11 and IL-6 are unknown (Zhao et al. 2014). These growth factors and cytokines have been suggested to act synergistically, amplifying paracrine signals released by dying cells and microglia (Wan et al. 2014). This synergy is thought to lower the activation threshold of intracellular signaling cascades within Müller glia (Wan et al. 2014, Zhao et al. 2014). Intracellular signaling in Müller glia also exhibits remarkable crosstalk between the different pathways (Goldman 2014, Wan et al. 2012), which may serve to activate multiple signaling cascades simultaneously to initiate the transcriptional changes underlying reprogramming.
Intravitreal injections of recombinant Sonic Hedgehog protein, SHH-N, increase the number of Müller glia that enter the cell cycle following an injury, whereas inhibiting the Hedgehog pathway reduces proliferation (Thomas et al. 2018). It remains to be determined if SHH-N is sufficient to reprogram Müller glia when injected into an uninjured retina. Interestingly, Shh has been suggested to be normally expressed in the nerve fiber layer (Sherpa et al. 2014), which would indicate that mechanisms are in place or that restricted availability of Shh may prevent Müller glia from reprogramming in the uninjured retina. In support of this, the expression of Hedgehog pathway components increases at the time of Müller glia reprogramming; however, the cell-specific expression patterns remain to be established (Kaur et al. 2018).
5. EPIGENETIC CONTROL OF MÜLLER GLIA REPROGRAMMING
The epigenomic landscape of cells is tightly regulated. Epigenetic changes occur during development but also during reprogramming of induced pluripotent stem cells (Gökbuget & Blelloch 2019). In the injured retina, activation and inhibition of gene expression are necessary to mediate the regenerative response, and thus, transcription has to be tightly regulated. DNA methylation and histone modifications control the accessibility of DNA for transcription by regulating DNA compaction. Both DNA methylation and certain histone marks are associated with compacted, inaccessible chromatin (heterochromatin), while unmethylated DNA, together with different histone marks at promoter regions, allows chromatin decondensation (euchromatin) and thereby permits transcriptional machinery to access promoter binding sites. In the regenerating zebrafish retina, the expression of transgenes driven by ubiquitous promoters, which is silenced in the adult retina, is reactivated due to epigenomic changes in the promoter regions of these transgenes (Thummel et al. 2006). This early observation suggested that epigenetic changes likely also occur in genes that facilitate regeneration.
5.1. Function of DNA Methylation in the Regenerating Retina
Methylation of cytosines within CpG contexts in promoter regions is important for gene silencing (Corso-Diaz et al. 2018). DNA methylation is established by DNA methyltransferases (Dnmts), and demethylation can occur either in a passive manner, i.e., loss of methylation sites during replication in the absence of Dnmt1, or in an active manner driven by enzymes (Gökbuget & Blelloch 2019). Active demethylation is a multistep process involving several enzymes such as DNA dioxygenases, cytidine deaminases, and DNA glycosylases (Corso-Diaz et al. 2018).
Injury-induced changes in DNA methylation of cytosine in the context of CpG were observed in Müller glia (Powell et al. 2013). Interestingly, although 73.6% of the analyzed CpGs showed decreased methylation, the promoters of genes required for Müller glia reprogramming (lin28, mycb, sox2, ascl1a, insm1a; see below) are hypomethylated in zebrafish (and in mice), indicating that these genes are in a poised state (Powell et al. 2013). This raises the question of how the expression of these reprogramming genes is regulated. Furthermore, many bases in the zebrafish retina are differentially methylated, but it is unclear which genes or genomic regions are affected and if these play any role in Müller glia reprogramming. Increased expression of the de novo DNA methylase genes dnmt3ba, dnmt3bb1, and dnmt3bb3 (previously named dnmt7, dnmt4, and dnmt5, respectively) suggests that DNA methylation might play a role in regulating Müller glia reprogramming (Powell et al. 2012, 2013). These Dnmt3s may regulate accessibility of genes that keep Müller glia in a quiescent state in the uninjured retina, and these quiescence-maintaining genes have to be downregulated to allow Müller glia reprogramming following injury. It will be important to know whether this dinucleotide methylation occurs in genes that are downregulated as Müller glia undergo reprogramming (Powell et al. 2013). Dnmt1 is also upregulated following retinal injury, and based on its role in maintaining global methylation states during S-phase, it likely also fulfills this role during DNA synthesis in Müller glia (Du et al. 2015).
Morpholino-mediated knockdown of the cytidine deaminases Apobec2a and Apobec2b reduces the expression of ascl1a and lin28, suggesting that Apobec2a and Apobec2b regulate Müller glia reprogramming. However, it was not assessed whether knockdown of Apobec2a and Apobec2b affects Müller glia proliferation (Powell et al. 2012). Surprisingly, only 0.023% of analyzed CpG regions were differentially methylated in apobec2a and apobec2b morphants relative to controls, suggesting that Apobec2a and Apobec2b do not play a role in demethylating DNA during Müller glia reprogramming (Powell et al. 2013). It is unknown which enzymes mediate the methylation changes in Müller glia. The ten–eleven translocation (tet) gene tet2, but not tet1 or tet3, is expressed in Müller glia, and none of these three genes change expression following injury (M. Lahne, P. Boyd and D.R. Hyde, unpublished data). Interestingly, TET2 is phosphorylated by the Janus kinase, JAK2, downstream of cytokine signaling (Jeong et al. 2019). Given that Jak signaling plays a role in Müller glia reprogramming (Conner et al. 2014, Zhao et al. 2014), phosphorylation by Jaks might result in rapid activation of Tets that subsequently induce DNA demethylation, allowing effective transduction of a retinal injury into a transcriptional response.
5.2. Function of Histone-Modifying Enzymes in the Regenerating Retina
Methylation, acetylation, and phosphorylation of the N-terminal regions of histones regulate the accessibility of DNA for transcription (Corso-Diaz et al. 2018). The histone modifications at transcription start sites that result in active gene transcription are acetylation of lysine 27 at histone H3 (H3K27ac) together with triple methylation of lysine 4 at histone H3 (H3K4me3). In contrast, triple methylation of lysine 9 and/or 27 of histone H3 (H3K9me3 or H3K27me3, respectively) results in repressive marks. In addition, promoter regions are considered poised in the presence of H3K4me3 and H3K27me3 (Corso-Diaz et al. 2018, Voigt et al. 2013). Acetylation is mediated by histone acetyltransferases [also called Kat for lysine (K) acetyltransferase], whereas deacetylation is facilitated by either histone deacetylases (Hdac) or sirtuins. Lysine methyltransferases (Kmt) and lysine demethylases (Kdm) catalyze the methylation and demethylation of histone N-terminals, respectively (Corso-Diaz et al. 2018).
Following an injury, knockdown of Hdac1 expression reduces proliferation among Müller glia (Mitra et al. 2018). Furthermore, global inhibition of Hdac function results in differential expression of genes associated with Müller glia reprogramming; the expression of mycb, ascl1a, and let7 increases, while the expression of the transcriptional repressors insm1a and lin28 decreases. Interestingly, chromatin immunoprecipitation–polymerase chain reaction (ChIP-PCR) established that Hdac binds to Myc binding sites within the lin28 and her4.1 promoters (Mitra et al. 2019). While the global acetylation state of histone H4 was assessed following pharmacological inhibition, it was not determined whether H3K27 acetylation was changed in the lin28 promoter and in any genes that are differentially regulated following these treatments (Mitra et al. 2018). However, downregulation of the kat2b gene, encoding an acetylating enzyme, in Müller glia following photo-injury suggests that deacetylation is a necessary step in reprogramming (M. Lahne, P. Boyd and D.R. Hyde, unpublished data).
The Polycomb repressive complex 2 (PRC2) is an enzyme complex that mediates the trimethylation of H3K27 (O’Meara & Simon 2012). Single-cell RNA-seq revealed upregulation of PRC2 complex components ezh2, eed, and suz12a/b in Müller glia following injury (M. Lahne, P. Boyd and D.R. Hyde, unpublished data). The role of the PRC2 complex in the regenerating zebrafish retina is unknown, but trimethylation of H3K27 results in a repressed transcriptional state and is likely correlated with the deacetylation of the same histone mark, which is critical for Müller glial reprogramming (Mitra et al. 2018). Interestingly, the Wnt signaling inhibitor DKK2 and Notch receptors and ligands (NOTCH4, DLL4) were identified as Polycomb group target genes in human embryonic diploid fibroblasts, and downregulation of these pathways is required for reprogramming (Bracken et al. 2006, Conner et al. 2014, Ramachandran et al. 2011, Wan et al. 2012). Thus, in the context of Müller glia reprogramming, PRC2 might repress the transcription of genes that maintain Müller glia in a quiescent state.
Phosphorylation of histone H3 at serine 10 and 28 (H3S10, H3S28) provides another level of regulation; this phosphorylation, in combination with the acetylation of nearby H3K14 or H3K27 in promoter regions, respectively, is implicated as active histone marks allowing transcription of target genes, such as immediate early genes (Sawicka & Seiser 2012). Interestingly, phosphorylation of H3S28 induces the release of PRC2 from trimethylated H3K27 without altering the methylation status and thereby leading to gene activation of immediate early genes or genes involved in differentiation, depending on the upstream stimulus (Gehani et al. 2010). Mitogen- and stress- activated kinases 1 and 2, which are activated by either ERK1/2 or p38 kinases, have been shown to phosphorylate both H3S10 and H3S28 downstream of growth factor or stress signaling (Duncan et al. 2006, Gehani et al. 2010). As ERK1/2 are activated in Müller glia following injury, extracellular signals might be efficiently relayed via these kinases into a response that renders the epigenomic landscape active, thereby allowing for gene transcription (Wan et al. 2012). In addition to the PRC2 components, genes encoding other methylating (kmt5ab, nsd2, suv39h1b) and demethylating (jarid2b) enzymes increase expression in Müller glia during reprogramming (M. Lahne, P. Boyd and D.R. Hyde, unpublished data), suggesting that histone modifications are tightly regulated and accommodate both the activation and repression of gene expression.
The differential expression of various histone and DNA modifying enzymes following retinal injury suggests that the epigenetic landscape is tightly regulated to facilitate Müller glia reprogramming. While a few enzymes have been functionally examined for their role in retinal regeneration, further analysis of the specific function of DNA modifying enzymes, their target genes, and the upstream mechanisms that facilitate the remodeling of the epigenetic landscape is necessary to obtain a full understanding of epigenetic changes and their role in regulating Müller glia reprogramming.
6. TRANSCRIPTIONAL CONTROL OF MÜLLER GLIA REPROGRAMMING IN ZEBRAFISH
The bHLH transcription factor Ascl1a was the first transcription factor shown to be required for reprogramming Müller glia in zebrafish. In situ hybridization showed that ascl1a is induced in Müller glia by 4 h post-injury, and knocking down Ascl1a expression prevents quiescent Müller glia from entering the cell cycle (Fausett et al. 2008). Ascl1a was identified as a direct transcriptional regulator of the pluripotency factor Lin28 (Ramachandran et al. 2010), an RNA-binding protein highly expressed in embryonic stem cells. In Müller glia, lin28 expression parallels that of ascl1a, and knockdown of Ascl1a reduces lin28 expression. Furthermore, Lin28 blocks the functional maturation of the let7 miRNA, indicating that Lin28 and let7 form a regulatory circuit that governs the balance between pluripotency and fate commitment (Rehfeld et al. 2015). As Müller glia enter the cell cycle following injury, the levels of lin28 and let7 change in opposite directions, and knocking down either Ascl1a or Lin28 completely eliminates the injury-induced suppression of let7 (Ramachandran et al. 2010). These data suggest that Ascl1a functions through a Lin28–let7 network to promote reprogramming, in part by suppressing genetic programs that normally function to promote or maintain cellular differentiation (Goldman 2014). Interestingly, overexpression of either ascl1a or lin28 in an uninjured retina is insufficient to induce Müller glia proliferation, whereas coexpressing these genes activates proliferation (Elsaeidi et al. 2018).
Convincing, indirect evidence shows that the transcription factor gene insm1a is a direct transcriptional target of Ascl1a, and knockdown experiments reveal an Ascl1a–Insm1a regulatory loop and an Insm1a autoregulatory loop (Ramachandran et al. 2012) (Figure 2c). In Müller glia, expression of insm1a is upregulated by 6 h post-injury, but it is suppressed at 24 h post injury, suggesting a complex functional role for this gene. Insm1a was found to repress the expression of the Wnt signaling inhibitor Dkk, which is required for reprogramming (Ramachandran et al. 2011, 2012). Insm1a likely directly regulates dkk1b (Figure 2c). Expression of the Wnt ligand wnt4, and to a lesser extent the receptor fzd2, is also regulated in an Ascl1a-dependent manner (Ramachandran et al. 2011). In contrast, the mechanisms that regulate increased expression of the Wnt signaling effector ctnnb2, which is required for Müller glial proliferation, are currently unknown (Gorsuch et al. 2017). Together, these data reveal that a complex Ascl1a and Insm1a signaling network plays a role in Wnt-mediated reprogramming of Müller glia.
Stat3 is a member of the family of Stat proteins, which function as activatable transcriptional regulators downstream of cytokine and growth factor signaling pathways. Phosphorylation of Stat3 enables its homodimerization and thereby allows binding of Stat3 to specific DNA binding sites (Huynh et al. 2017). Following retinal injury, Stat3 expression is strongly induced in all Müller glia, although only a subset of Müller glia proliferate (Kassen et al. 2007). While Stat3 phosphorylation levels increase during reprogramming (Kassen et al. 2007), the Stat3 phosphorylation state within individual quiescent and proliferating Müller glia has not been established. Nonetheless, knockdown of Stat3 or inhibition of Jaks, which phosphorylate Stat3, demonstrated that Stat3 is required for Müller glia reprogramming (Conner et al. 2014, Nelson et al. 2012, Zhao et al. 2014). Interestingly, following photoreceptor death, induction of stat3 expression precedes ascl1a, and while Stat3 is present in all Müller glia, Ascl1a is restricted to those Stat3-positive Müller glia that enter the cell cycle (Nelson et al. 2012). In support of Stat3 acting upstream of ascl1a, removal of Stat binding sites within the ascl1a promoter diminishes ascl1a promoter–driven green fluorescent protein (GFP) transgene expression (Zhao et al. 2014). Additionally, Jak inhibition reduces the levels of ascl1a and lin28 expression (Zhao et al. 2014). However, knockdown experiments showed that Lin28 is required for the expression of both Ascl1a and Stat3, and Ascl1a is required for Stat3 expression, but only in a subset of Müller glia that enter the cell cycle (Nelson et al. 2012). Taken together, these data suggest that at least two populations of Müller glia exist in the regenerating retina; both upregulate Stat3, but one remains mitotically quiescent, while the other enters the cell cycle (Nelson et al. 2012). Although this model is incomplete, this study confirmed that not all Müller glia respond similarly to cell death and revealed the complex nature of reprogramming among the population of Müller glia.
Similar to Stat3, there are other transcription factors that are directly regulated by signaling pathways activated by retinal injury. For example, Notch signaling regulates the expression of her and hes genes, which subsequently control the expression of Notch-responsive genes (Ho et al. 2019). The only Notch target gene known to be downregulated early in response to retinal injury is hey1, but it is unknown whether, in an uninjured retina, hey1 maintains Müller glia in a quiescent state (Wan & Goldman 2017). Expression of another Notch target gene, her4.1, is upregulated following injury (Mitra et al. 2018). The promoter regions of the reprogramming-associated genes lin28, il-6, il-11b, lepr, lifra, lepa, lepb, and crlf1a contain Her4.1 binding sites, and knockdown of Her4.1 expression in Müller glia–derived progenitors increases expression of these genes (Kaur et al. 2018, Mitra et al. 2018). This implies that Her4.1 represses expression of the genes mentioned above, and as such, that Her4.1 could play a role in maintaining Müller glia quiescence.
Sox2 has a well-established association with neural stem cells in both the embryonic and adult central nervous system and is required for neurogenesis in both tissues. Notably, Sox2 is an essential transcription factor, together with c-Myc, Oct4, and Klf4, which facilitate reprogramming of somatic cells into induced pluripotent stem cells (Takahashi & Yamanaka 2006). In zebrafish, Sox2 is also required to reprogram Müller glia (Gorsuch et al. 2017). Interestingly, and in contrast to many genes induced by injury, Sox2 is constitutively expressed in Müller glia, a feature of all vertebrate retinas studied (Fischer et al. 2010, Gorsuch et al. 2017, Surzenko et al. 2013). However, following injury, Sox2 is strongly upregulated in those Müller glia that will enter the cell cycle. Using loss- and gain-of-function approaches, Gorsuch et al. (2017) showed that induction of Sox2 is necessary for Müller glia to enter the cell cycle following injury, and in an uninjured retina, induced overexpression of Sox2 is sufficient to stimulate proliferation. Furthermore, consistent with its role as a reprogramming factor in somatic cells, Sox2 is near the top of a gene network that regulates the expression of ascl1a and lin28, but not stat3, identifying Sox2 as the first transcription factor that differentially regulates ascl1a/lin28 and stat3. This suggests that independent signaling pathways are likely required for reprogramming Müller glia. It is currently unknown how Sox2 in Müller glia exerts different functions in injured versus uninjured retinas. Interestingly, in human embryonic stem cells, SOX2 interacts with the pluripotency transcription factor OCT4 to regulate the expression of pluripotency genes, while in neural epithelial cells derived from human embryonic stem cells, SOX2 interacts with PAX6 to mediate neuronal differentiation (Zhang et al. 2019). In the retina, a similar switch could explain the context-dependent function of Sox2. In support of this idea, following injury and prior to the onset of proliferation, the expression of pax6b declines in Müller glia (Hoang et al. 2019), whereas the expression of oct4 increases (Ramachandran et al. 2010). Moreover, Oct4 regulates ascl1a, oct4, and sox2 expression during reprogramming; however, the effect of Oct4 knockdown on proliferation of Müller glia has not been assessed (Sharma et al. 2019). Similarly, while expression of the zebrafish paralogs of the pluripotency factor Myc, myca and mycb, increases in the injured retina (Mitra et al. 2019), it is also unknown whether Myc regulates Müller glia proliferation. The upstream mechanisms that regulate the expression of the pluripotency factors sox2, oct4, and myc remain to be identified.
7. COMPARATIVE ANALYSIS
7.1. Chicks
In the chick retina, loss of neuronal cells stimulates the dedifferentiation of Müller glia and their reentry into the cell cycle for a short timeframe after hatching (Fischer & Reh 2001). Proliferating chick Müller glia only undergo a single cell division, thereby producing Müller glia–derived progenitors that do not continue in the cell cycle (Fischer & Reh 2001). While some of the Müller glia–derived progenitors replace a subset of the ablated retinal neurons, the majority of these cells remain in a progenitor state (Fischer & Reh 2001, 2002). In the uninjured chick retina, intravitreal injections of FGF2 are sufficient to induce Müller glia proliferation (Todd & Fischer 2015). A variety of other ligands and their corresponding receptors also play critical roles in mediating proliferation in the injured chick retina, including Sonic Hedgehog, HB-EGF, BMP, retinoic acid, Notch, and Wnt/β-catenin (Gallina et al. 2016; Todd & Fischer 2015; Todd et al. 2015, 2017, 2018). In contrast to FGF2, the aforementioned ligands are insufficient to induce cell cycle reentry in the uninjured retina. Intracellular signaling pathways that are activated in response to injury or exogenous FGF2 include Jak/Stat, ERK1/2, mTOR, and Smad (Fischer et al. 2009, Todd et al. 2016, Zelinka et al. 2016). In addition to activation, repression of signaling pathways such as TGFβ2 is also required for Müller glial reprogramming (Todd et al. 2017). Interestingly, and in contrast to zebrafish, upregulated Notch signaling is required for efficient reprogramming of chick Müller glia (Conner et al. 2014, Ghai et al. 2010, Hayes et al. 2007). In addition to deciphering the similarities and differences in the signaling pathways activated in the chick and zebrafish retina, it will also be critical to understand the road blocks in the chick retina that prevent the amplification of Müller glia–derived progenitors and their efficient differentiation into surviving neurons.
7.2. Mammals
A fundamental insight into the molecular biology of Müller glia in mammals was gained via the discovery that these cells and late-stage neural progenitors share a common gene expression profile (Blackshaw et al. 2004, Gotz et al. 2015). This discovery provided context regarding the proliferative potential of Müller glia in mammals (Dyer & Cepko 2000, Vetter & Moore 2001) and led to the suggestion that Müller glia may be a form of a late-stage retinal progenitor with a latent ability to generate neurons (Jadhav et al. 2009). In mammals, the ability of Müller glia to reenter the cell cycle is variable and can depend on the species or the nature or severity of the injury (Joly et al. 2011, Karl et al. 2008, Lewis et al. 2010, Ooto et al. 2004). For example, in mice, photoreceptor injury induces the expression of cyclin D1 in Müller glia, but this is not accompanied by BrdU uptake, suggesting that, in mice, Müller glia are unable to progress into the S-phase of the cell cycle (Joly et al. 2011). In contrast, following retinal detachment in rabbits or NMDA-induced toxicity in rats, some Müller glia reenter the cell cycle, as evidenced by either phospho-histone 3 or BrdU labeling, suggesting that Müller glia in these animals can progress through the cell cycle (Lewis et al. 2010, Ooto et al. 2004). However, the majority of these BrdU-labeled cells either remain positive for markers of Müller glia or undergo apoptosis (Lewis et al. 2010, Ooto et al. 2004). In the rat, a few BrdU-positive cells express neuronal markers, but the low number of such cells is far from sufficient to replace the ablated neurons (Ooto et al. 2004).
A variety of strategies have been employed to increase the efficiency of neuronal regeneration in mammals. Exogenous application of growth factors (e.g., EGF, FGF1) can induce Müller glia to proliferate in NMDA-injured retinas, and some of these proliferating cells differentiate into amacrine cells (Karl et al. 2008). Targeted overexpression of Ascl1 in mice can reprogram Müller glia into progenitor cells that are competent to regenerate retinal neurons in vivo. This action of ASCL1 is due to its capacity to transform chromatin into an active state at genes that are regulated by ASCL1 during retinal development and that induce a neurogenic program (Pollak et al. 2013). How ASCL1 transforms chromatin is currently unknown. However, Wnt, a transcriptional target of Ascl1a, regulates the expression of histone-modifying enzymes in retinal development (Aldiri et al. 2013), and thus, an ASCL1–WNT signaling axis could potentially link ASCL1 to chromatin remodeling during reprogramming. Additionally, combining targeted expression of Ascl1 with a histone deacetylase inhibitor appears to induce Müller glia in adult mice to bypass a progenitor stage and convert directly to a neuronal fate (Jorstad et al. 2017). This could provide an additional mechanism to regenerate retinal neurons from Müller glia in the mammalian retina, although the effect of losing a significant proportion of Müller glia is yet to be determined.
In an uninjured retina, overexpression of Lin28, β-catenin, and constitutively active Yap (Yap5SA) also stimulates cell cycle reentry among Müller glia. However, it is unclear whether these Müller glia efficiently divide and produce amplifying neural progenitors (Elsaeidi et al. 2018, Hamon et al. 2019, Rueda et al. 2019, Ueki et al. 2015, Yao et al. 2016). Interestingly, in mice, coexpression of Ascl1a and Lin28 in Müller glia induces a modest amount of cell proliferation in uninjured retinas, and this proliferative response is increased by retinal injury (Elsaeidi et al. 2018). In contrast to the zebrafish retina, suppression of Notch signaling does not increase proliferation when Ascl1 and Lin28 are overexpressed (Elsaeidi et al. 2018). Similar to Notch activation in the injured chick retina, exposure of retinal explants to the Notch ligand Jag1 increases Müller glia cell cycle reentry, suggesting that the function of Notch signaling diverges between zebrafish and chicks or mammals (Del Debbio et al. 2010, Ghai et al. 2010, Hayes et al. 2007).
Stat3 and ERK1/2 signaling pathways involved in Müller glia reprogramming in the zebrafish retina are activated by injury in the mammalian retina (Galan et al. 2014, Jiang et al. 2014, Kassen et al. 2007, Kirsch et al. 2010, Nakazawa et al. 2007, Zhao et al. 2014). Following retinal detachment in mice, ERK1/2-dependent upregulation of Cyclin D suggests that ERK1/2 might be required for cell cycle reentry in the mammalian retina in vivo (Kase et al. 2006). In support of this, EGF stimulates Müller glia proliferation in an ERK1/2-dependent manner in retinal explants from juvenile mice (Ueki & Reh 2013). In contrast, the role of STAT3 in reprogramming of mammalian Müller glia is unknown. However, following optic nerve injury, the gliotic response in Müller glia is regulated by STAT3 (Kirsch et al. 2010). It remains to be determined what regulates reprogramming versus gliosis, and transcription factors like STAT3 need to be examined in this context.
The studies discussed above induced reprogramming of Müller glia by either targeted expression of genes or application of exogenous factors. An alternative approach to reprogramming Müller glia is being pioneered through cell fusion–based techniques (Lluis & Cosma 2010; Lluis et al. 2008; Sanges et al. 2013, 2016). Cell–cell fusion, the merging of plasma membranes to integrate intercellular components, is a tightly regulated process that occurs naturally during vertebrate development (Willkomm & Bloch 2015). Following activation of Wnt signaling, a variety of murine and human stem and progenitor cells, when transplanted into NMDA-injured retinas of adult mice, spontaneously fuse with host retinal neurons and Müller glia (Sanges et al. 2013, 2016). This cellular fusion transiently reprograms the fused host cell into a retinal progenitor that can divide and produce regenerated neurons. If transplantation experiments are performed in rd10 mice, in which photoreceptors selectively die, the Wnt-activated stem and progenitor cells transplanted into the eye fuse exclusively with and reprogram Müller glia, which give rise to regenerated photoreceptors (Sanges et al. 2016).
7.3. Comparative Omics Analysis
The ability to reprogram mammalian Müller glia by overexpressing genes identified in zebrafish represents a significant advance toward developing strategies to repair the injured or diseased human retina. However, some genes required for reprogramming Müller glia in zebrafish are involved in the mammalian gliotic response. To successfully develop therapies in human, it is necessary to fully identify the signaling mechanisms and gene regulatory networks that act in species with and without regenerative capacities. To our knowledge, only one study has compared differentially regulated genes in Müller glia isolated from photo-lesioned zebrafish to genes expressed in Müller glia from mouse mutants that display either early- (Pde6brd1/rd1) or late-onset (Rho−/−) photoreceptor degeneration (Sifuentes et al. 2016). This study revealed that metabolic pathway genes are enriched in both species, whereas genes within cytokine signaling, circadian, and pluripotency pathways that were upregulated in zebrafish were reduced or unaltered in mice. However, this study compared the response of Müller glia in an acute injury in zebrafish to chronic photoreceptor degenerations in mice. Additionally, the data sets for the two organisms were acquired using different techniques (RNA-seq for zebrafish and microarray for mice) that possess different sensitivities.
We recently performed both ATAC-Seq and RNA-Seq analysis on FAC-sorted Müller glia isolated from acutely injured zebrafish and mouse retinas that both experienced light-induced outer or NMDA-induced inner retinal injury (Hoang et al. 2019). Changes in chromatin accessibility resulted either in reduced (i.e., a repressed chromatin state) or in rapidly or slowly induced chromatin accessibility. Chromatin accessibility of genes like Rlbp/rlbp1a, Nfix/nfixb, and Pax6/pax6b were repressed in Müller glia from both mouse and zebrafish, and this correlated with reduced expression of these genes in both species. Additionally, we observed (a) genes that upregulated expression in both species, (b) species-specific gene expression changes, and (c) genes that were differentially expressed but for which an ortholog was not present in the other species.
We also performed single-cell RNA-Seq (scRNA-seq) on control and injured zebrafish, chick, and mouse retinas to obtain specific expression profiles of individual cells (Hoang et al. 2019). In our scRNA-Seq data, zebrafish Müller glia from uninjured and injured retinas separated into distinct clusters based on their RNA expression profiles (Hoang et al. 2019). Heterogeneity was also observed between Müller glia within a cluster. For example, proliferating Müller glia at 36 h of photo-injury can be distinguished according to the expression of cell cycle stage–specific cyclins (ccnd1: G1; ccnb1: mitosis). Subcategorizing Müller glia according to cell cycle phase may help identify genes that are necessary for a specific process. For example, genes required to mediate interkinetic nuclear migration would be expected to be expressed in ccnb1-positive cells. Müller glia from injured and uninjured retinas of chicks or mice also accumulated in distinct clusters (resting and activated Müller glia). Analysis revealed 325 genes that were differentially regulated in all three species, and there were also a large number of genes that were regulated in a species-specific manner or that overlapped among only two species. Computational modeling that assessed the ATAC-Seq, bulk RNA-Seq, and scRNA-Seq data for differentially expressed transcription factors or regulators in Müller glia at different injury time points predicted transcriptional networks consisting of gene regulatory modules that implicate the progression of Müller glia through different functional states (Hoang et al. 2019). In mice, Müller glia become gliotic and then return toward a transcriptionally quiescent state (Figure 3), while zebrafish Müller glia progress from a quiescent to a transient gliotic or neurogenic state and subsequently to a proliferative state (Figure 3). In support of this transient intermediate gliotic state, Müller glia persist in a morphologically gliotic state when proliferation is blocked in the damaged zebrafish retina (Thomas et al. 2016). The transcription factors identified in these regulatory modules will require functional testing to determine if they play a role in the predicted cellular processes or functional states in both mice and zebrafish. It will also be of particular interest to examine whether differentially regulated genes that lack an ortholog in mice are instrumental in reprogramming Müller glia in zebrafish. Additionally, it is important to determine if the mouse genes that lack a zebrafish ortholog function to repress regeneration.
Figure 3.
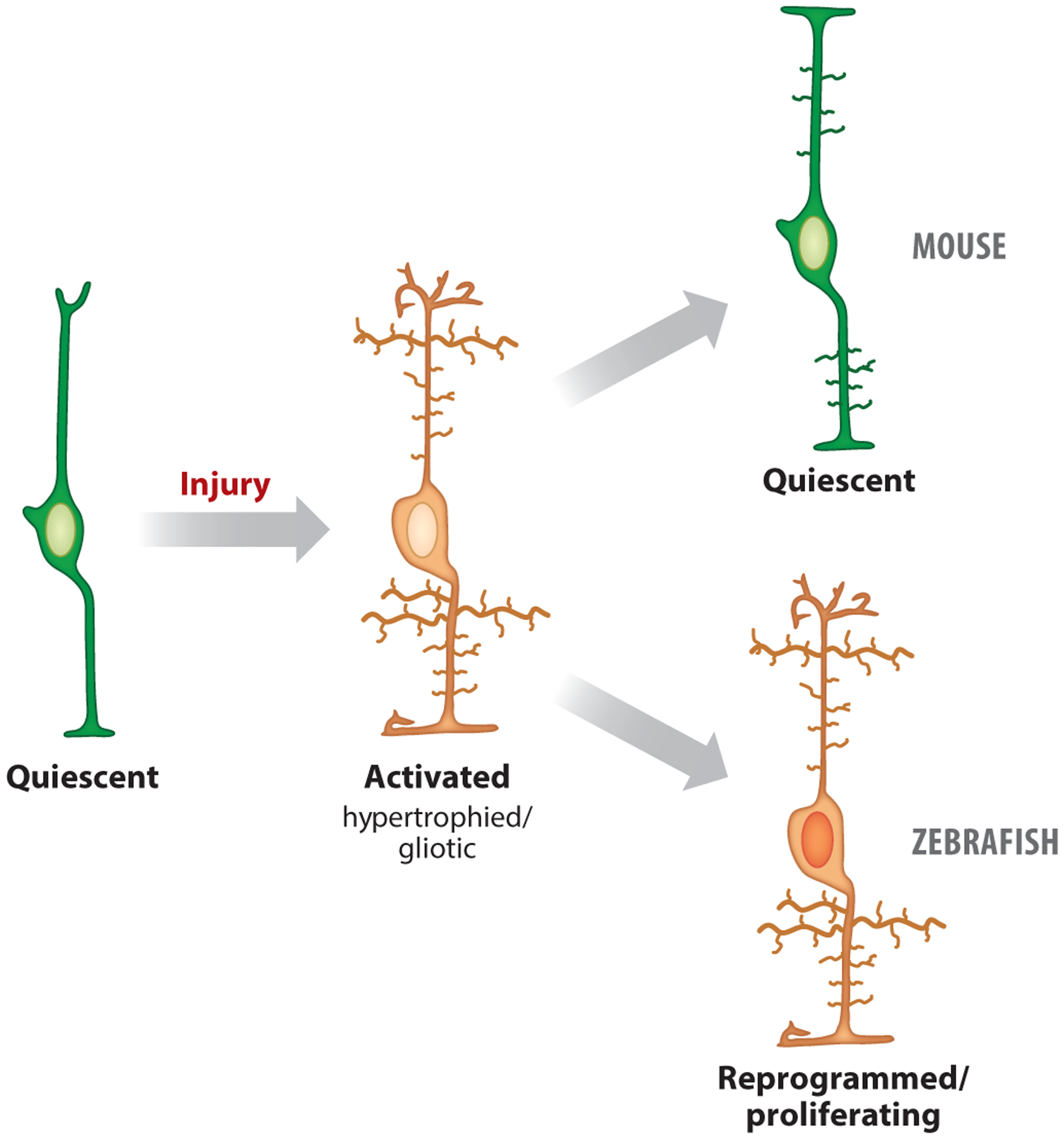
Comparison of the injury response in zebrafish and mice: schematic of a quiescent Müller glia that becomes activated or gliotic following retinal injury, displaying a hypertrophied cell body and processes in both mice and zebrafish. In mice, the activated or gliotic Müller glia returns toward quiescence based on gene expression and morphology, whereas in zebrafish, the activated or gliotic Müller glia reprograms and proliferates.
8. CONCLUSION
The studies reviewed in this article establish that injury-induced reprogramming of zebrafish Müller glia involves changes in paracrine and autocrine signaling, which control multiple signal transduction pathways and gene regulatory networks. Retinal injury alters neurotransmitter signaling and stimulates the release of nucleotides and cytokines from dying cells, and cell death itself likely disrupts cell-contact-mediated signaling; these different events can initiate reprogramming. The release of inflammatory cytokines and growth factors from activated microglia also represents a crucial step in the regulatory cascades leading to reprogramming. Additionally, in response to cell death, Müller glia themselves upregulate and release growth factors and inflammatory cytokines, which likely then act in an autocrine manner. Among the proteins regulated by injury, Stat3 and Sox2 reside near the apex of a gene regulatory network, which engages an Ascl1a–Lin28–let7 regulatory loop. This loop functions to upregulate the expression of genes that are shared with retinal progenitors and suppress the expression of genes associated with cellular differentiation. The output of this regulatory network allows Müller glia to enter the cell cycle and undergo a single asymmetric cell division that generates a multipotent retinal progenitor. Much less is known about the epigenetic mechanisms that underlie reprogramming in Müller glia, but recent studies show that injury alters the DNA methylation status and initiates the differential expression of histone-modifying enzymes in Müller glia. Importantly, in zebrafish, quiescent Müller glia contain open chromatin for pluripotency and other injury-induced transcription factors. In mice, the same genes contain open chromatin, but injury is insufficient to induce their expression. Numerous gaps remain in our knowledge of the molecular mechanisms that allow zebrafish Müller glia to, but prevent mammalian Müller glia from, reprogramming into stem cells. A recent comparative RNA-Seq study revealed that retinal injury activates shared and distinct genetic programs in Müller glia of zebrafish and mice. Mining these data will doubtless reveal new and important avenues for research that will aid in parsing the injury-induced molecular processes that permit or prevent Müller glia reprogramming.
ACKNOWLEDGMENTS
Support was provided by National Institutes of Health grants R01EY07060 (to P.F.H.), R01EY024519 (to D.R.H.), U01EY027267 (to D.R.H.), and P30EYO7003 (to P.F.H.) and by an unrestricted grant from the Research to Prevent Blindness, New York (to P.F.H.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Aldiri I, Moore KB, Hutcheson DA, Zhang J, Vetter ML. 2013. Polycomb repressive complex PRC2 regulates Xenopus retina development downstream of Wnt/beta-catenin signaling. Development 140(14):2867–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga J, Walker SL, Mumm JS. 2010. Multicolor time-lapse imaging of transgenic zebrafish: visualizing retinal stem cells activated by targeted neuronal cell ablation. J. Vis. Exp 43:2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TJ, Fossum SL, Fimbel SM, Montgomery JE, Hyde DR. 2010. The inhibitor of phagocytosis, O-phospho-L-serine, suppresses Muller glia proliferation and cone cell regeneration in the light-damaged zebrafish retina. Exp. Eye Res 91(5):601–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battista AG, Ricatti MJ, Pafundo DE, Gautier MA, Faillace MP. 2009. Extracellular ADP regulates lesion-induced in vivo cell proliferation and death in the zebrafish retina. J. Neurochem 111(2):600–13 [DOI] [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, Raymond PA. 2007. Late-stage neuronal progenitors in the retina are radial Muller glia that function as retinal stem cells. J. Neurosci 27(26):7028–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshaw S, Harpavat S, Trimarchi J, Cai L, Huang H, et al. 2004. Genomic analysis of mouse retinal development. PLOS Biol. 2(9):E247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. 2006. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20(9):1123–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braisted JE, Raymond PA. 1993. Continued search for the cellular signals that regulate regeneration of dopaminergic neurons in goldfish retina. Dev. Brain Res 76(2):221–32 [DOI] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, et al. 2009. Cellular signaling and factors involved in Muller cell gliosis: neuroprotective and detrimental effects. Prog. Retin. Eye Res 28(6):423–51 [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, et al. 2006. Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res 25(4):397–424 [DOI] [PubMed] [Google Scholar]
- Conedera FM, Quintela Pousa AM, Mercader N, Tschopp M, Enzmann V. 2019. Retinal microglia signaling affects Muller cell behavior in the zebrafish following laser injury induction. Glia 67(6):1150–66 [DOI] [PubMed] [Google Scholar]
- Conner C, Ackerman KM, Lahne M, Hobgood JS, Hyde DR. 2014. Repressing Notch signaling and expressing TNFalpha are sufficient to mimic retinal regeneration by inducing Muller glial proliferation to generate committed progenitor cells. J. Neurosci 34(43):14403–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corso-Diaz X, Jaeger C, Chaitankar V, Swaroop A. 2018. Epigenetic control of gene regulation during development and disease: a view from the retina. Prog. Retin. Eye Res 65:1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig SE, Calinescu AA, Hitchcock PF. 2008. Identification of the molecular signatures integral to regenerating photoreceptors in the retina of the zebra fish. J. Ocul. Biol. Dis. Inform 1(2–4):73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Debbio CB, Balasubramanian S, Parameswaran S, Chaudhuri A, Qiu F, Ahmad I. 2010. Notch and Wnt signaling mediated rod photoreceptor regeneration by Muller cells in adult mammalian retina. PLOS ONE 5(8):e12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ. 2015. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol 16(9):519–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan EA, Anest V, Cogswell P, Baldwin AS. 2006. The kinases MSK1 and MSK2 are required for epidermal growth factor-induced, but not tumor necrosis factor-induced, histone H3 Ser10 phosphorylation. J. Biol. Chem 281(18):12521–25 [DOI] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. 2000. Control of Muller glial cell proliferation and activation following retinal injury. Nat. Neurosci 3(9):873–80 [DOI] [PubMed] [Google Scholar]
- Elsaeidi F, Macpherson P, Mills EA, Jui J, Flannery JG, Goldman D. 2018. Notch suppression collaborates with Ascl1 and Lin28 to unleash a regenerative response in fish retina, but not in mice. J. Neurosci 38(9):2246–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, Goldman D. 2006. A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. J. Neurosci 26(23):6303–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, Gumerson JD, Goldman D. 2008. The proneural basic helix-loop-helix gene ascl1a is required for retina regeneration. J. Neurosci 28(5):1109–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimbel SM, Montgomery JE, Burket CT, Hyde DR. 2007. Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. J. Neurosci 27(7):1712–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. 2001. Muller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat. Neurosci 4(3):247–52 [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. 2002. Exogenous growth factors stimulate the regeneration of ganglion cells in the chicken retina. Dev. Biol 251(2):367–79 [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Scott MA, Tuten W. 2009. Mitogen-activated protein kinase-signaling stimulates Müller glia to proliferate in acutely damaged chicken retina. Glia 57(2):166–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AJ, Zelinka C, Scott MA. 2010. Heterogeneity of glia in the retina and optic nerve of birds and mammals. PLOS ONE 5(6):e10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser B, DuVal MG, Wang H, Allison WT. 2013. Regeneration of cone photoreceptors when cell ablation is primarily restricted to a particular cone subtype. PLOS ONE 8(1):e55410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Mukherjee S, Bao ZZ, Morrow EM, Cepko CL. 2000. rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron 26(2):383–94 [DOI] [PubMed] [Google Scholar]
- Galan A, Dergham P, Escoll P, de-la-Hera A, D’Onofrio PM, et al. 2014. Neuronal injury external to the retina rapidly activates retinal glia, followed by elevation of markers for cell cycle re-entry and death in retinal ganglion cells. PLOS ONE 9(7):e101349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina D, Palazzo I, Steffenson L, Todd L, Fischer AJ. 2016. Wnt/beta-catenin-signaling and the formation of Muller glia-derived progenitors in the chick retina. Dev. Neurobiol 76(9):983–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehani SS, Agrawal-Singh S, Dietrich N, Christophersen NS, Helin K, Hansen K. 2010. Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation. Mol. Cell 39(6):886–900 [DOI] [PubMed] [Google Scholar]
- Ghai K, Zelinka C, Fischer AJ. 2010. Notch signaling influences neuroprotective and proliferative properties of mature Muller glia. J. Neurosci 30(8):3101–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gökbuget D, Blelloch R. 2019. Epigenetic control of transcriptional regulation in pluripotency and early differentiation. Development 146:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D 2014. Muller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci 15(7):431–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsuch RA, Hyde DR. 2014. Regulation of Muller glial dependent neuronal regeneration in the damaged adult zebrafish retina. Exp. Eye Res 123:131–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsuch RA, Lahne M, Yarka CE, Petravick ME, Li J, Hyde DR. 2017. Sox2 regulates Muller glia reprogramming and proliferation in the regenerating zebrafish retina via Lin28 and Ascl1a. Exp. Eye Res 161:174–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz M, Sirko S, Beckers J, Irmler M. 2015. Reactive astrocytes as neural stem or progenitor cells: in vivo lineage, in vitro potential, and genome-wide expression analysis. Glia 63(8):1452–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graca AB, Hippert C, Pearson RA. 2018. Muller glia reactivity and development of gliosis in response to pathological conditions. Adv. Exp. Med. Biol 1074:303–8 [DOI] [PubMed] [Google Scholar]
- Hagerman GF, Noel NC, Cao SY, DuVal MG, Oel AP, Allison WT. 2016. Rapid recovery of visual function associated with blue cone ablation in zebrafish. PLOS ONE 11(11):e0166932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon A, Garcia-Garcia D, Ail D, Bitard J, Chesneau A, et al. 2019. Linking YAP to Muller glia quiescence exit in the degenerative retina. Cell Rep. 27(6):1712–1725.e6 [DOI] [PubMed] [Google Scholar]
- Hamon A, Roger JE, Yang XJ, Perron M. 2016. Muller glial cell-dependent regeneration of the neural retina: an overview across vertebrate model systems. Dev. Dyn 245(7):727–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes S, Nelson BR, Buckingham B, Reh TA. 2007. Notch signaling regulates regeneration in the avian retina. Dev. Biol 312(1):300–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock PF, Raymond PA. 2004. The teleost retina as a model for developmental and regeneration biology. Zebrafish 1(3):257–71 [DOI] [PubMed] [Google Scholar]
- Ho DM, Artavanis-Tsakonas S, Louvi A. 2019. The Notch pathway in CNS homeostasis and neurodegeneration. Wiley Interdiscip. Rev. Dev. Biol 9:e358. [DOI] [PubMed] [Google Scholar]
- Hoang T, Wang J, Boyd P, Wang F, Santiago C, et al. 2019. Cross-species transcriptomic and epigenomic analysis reveals key regulators of injury response and neuronal regeneration in vertebrate retinas. bioRxiv 717876. 10.1101/717876 [DOI] [Google Scholar]
- Huynh J, Etemadi N, Hollande F, Ernst M, Buchert M. 2017. The JAK/STAT3 axis: a comprehensive drug target for solid malignancies. Semin. Cancer Biol 45:13–22 [DOI] [PubMed] [Google Scholar]
- Insua MF, Simon MV, Garelli A, de Los Santos B, Rotstein NP, Politi LE. 2008. Trophic factors and neuronal interactions regulate the cell cycle and Pax6 expression in Muller stem cells. J. Neurosci. Res 86(7):1459–71 [DOI] [PubMed] [Google Scholar]
- Iribarne M, Hyde DR, Masai I. 2019. TNFα induces Müller glia to transition from non-proliferative gliosis to a regenerative response in mutant zebrafish presenting chronic photoreceptor degeneration. Front. Cell Dev. Biol 7:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iribarne M, Nishiwaki Y, Nakamura S, Araragi M, Oguri E, Masai I. 2017. Aipl1 is required for cone photoreceptor function and survival through the stability of Pde6c and Gc3 in zebrafish. Sci. Rep 7:45962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav AP, Roesch K, Cepko CL. 2009. Development and neurogenic potential of Muller glial cells in the vertebrate retina. Prog. Retin. Eye Res 28(4):249–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JJ, Gu X, Nie J, Sundaravel S, Liu H, et al. 2019. Cytokine-regulated phosphorylation and activation of TET2 by JAK2 in hematopoiesis. Cancer Discov. 9(6):778–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Wright KL, Zhu P, Szego MJ, Bramall AN, et al. 2014. STAT3 promotes survival of mutant photoreceptors in inherited photoreceptor degeneration models. PNAS 111(52):E5716–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns PR. 1977. Growth of the adult goldfish eye. III. Source of the new retinal cells. J. Comp. Neurol 176(3):343–57 [DOI] [PubMed] [Google Scholar]
- Johns PR, Fernald RD. 1981. Genesis of rods in teleost fish retina. Nature 293(5828):141–42 [DOI] [PubMed] [Google Scholar]
- Joly S, Pernet V, Samardzija M, Grimm C. 2011. Pax6-positive Müller glia cells express cell cycle markers but do not proliferate after photoreceptor injury in the mouse retina. Glia 59(7):1033–46 [DOI] [PubMed] [Google Scholar]
- Jorstad NL, Wilken MS, Grimes WN, Wohl SG, VandenBosch LS, et al. 2017. Stimulation of functional neuronal regeneration from Muller glia in adult mice. Nature 548(7665):103–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl MO, Hayes S, Nelson BR, Tan K, Buckingham B, Reh TA. 2008. Stimulation of neural regeneration in the mouse retina. PNAS 105(49):19508–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl MO, Reh TA. 2010. Regenerative medicine for retinal diseases: activating endogenous repair mechanisms. Trends Mol. Med 16(4):193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kase S, Yoshida K, Harada T, Harada C, Namekata K, et al. 2006. Phosphorylation of extracellular signal-regulated kinase and p27(KIP1) after retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol 244(3):352–58 [DOI] [PubMed] [Google Scholar]
- Kassen SC, Ramanan V, Montgomery JE, Burket CT, Liu CG, et al. 2007. Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Dev. Neurobiol 67(8):1009–31 [DOI] [PubMed] [Google Scholar]
- Kassen SC, Thummel R, Campochiaro LA, Harding MJ, Bennett NA, Hyde DR. 2009. CNTF induces photoreceptor neuroprotection and Muller glial cell proliferation through two different signaling pathways in the adult zebrafish retina. Exp. Eye Res 88(6):1051–64 [DOI] [PubMed] [Google Scholar]
- Kaur S, Gupta S, Chaudhary M, Khursheed MA, Mitra S, et al. 2018. let-7 microRNA-mediated regulation of Shh signaling and the gene regulatory network is essential for retina regeneration. Cell Rep. 23(5):1409–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch M, Trautmann N, Ernst M, Hofmann H. 2010. Involvement of gp130-associated cytokine signaling in Müller cell activation following optic nerve lesion. Glia 58(7):768–79 [DOI] [PubMed] [Google Scholar]
- Kizil C, Kyritsis N, Brand M. 2015. Effects of inflammation on stem cells: together they strive? EMBO Rep. 16(4):416–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyritsis N, Kizil C, Zocher S, Kroehne V, Kaslin J, et al. 2012. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 338(6112):1353–56 [DOI] [PubMed] [Google Scholar]
- Lahne M, Hyde DR. 2017. Live-cell imaging: new avenues to investigate retinal regeneration. Neural Regen. Res 12:1210–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahne M, Li J, Marton RM, Hyde DR. 2015. Actin-cytoskeleton- and Rock-mediated INM are required for photoreceptor regeneration in the adult zebrafish retina. J. Neurosci 35(47):15612–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski JR, Raymond PA. 2014. Muller glia: stem cells for generation and regeneration of retinal neurons in teleost fish. Prog. Retin. Eye Res 40:94–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessieur EM, Song P, Nivar GC, Piccillo EM, Fogerty J, et al. 2019. Ciliary genes arl13b, ahi1 and cc2d2a differentially modify expression of visual acuity phenotypes but do not enhance retinal degeneration due to mutation of cep290 in zebrafish. PLOS ONE 14(4):e0213960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GP, Chapin EA, Luna G, Linberg KA, Fisher SK. 2010. The fate of Muller’s glia following experimental retinal detachment: nuclear migration, cell division, and subretinal glial scar formation. Mol. Vis 16:1361–72 [PMC free article] [PubMed] [Google Scholar]
- Li L, Dowling JE. 2000. Effects of dopamine depletion on visual sensitivity of zebrafish. J. Neurosci 20(5):1893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluis F, Cosma MP. 2010. Cell-fusion-mediated somatic-cell reprogramming: a mechanism for tissue regeneration. J. Cell. Physiol 223(1):6–13 [DOI] [PubMed] [Google Scholar]
- Lluis F, Pedone E, Pepe S, Cosma MP. 2008. Periodic activation of Wnt/beta-catenin signaling enhances somatic cell reprogramming mediated by cell fusion. Cell Stem Cell 3(5):493–507 [DOI] [PubMed] [Google Scholar]
- Maier W, Wolburg H. 1979. Regeneration of the goldfish retina after exposure to different doses of ouabain. Cell Tissue Res. 202(1):99–118 [DOI] [PubMed] [Google Scholar]
- Medrano MP, Bejarano CA, Battista AG, Venera GD, Bernabeu RO, Faillace MP. 2017. Injury-induced purinergic signalling molecules upregulate pluripotency gene expression and mitotic activity of progenitor cells in the zebrafish retina. Purinergic Signal. 13(4):443–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JR, Hu L, Moses A, Kaboli K, Papandrea A, Raymond PA. 2012. β-Catenin/Wnt signaling controls progenitor fate in the developing and regenerating zebrafish retina. Neural Dev. 7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Sharma P, Kaur S, Khursheed MA, Gupta S, et al. 2018. Histone deacetylase-mediated Muller glia reprogramming through Her4.1-Lin28a axis is essential for retina regeneration in zebrafish. iScience 7:68–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Sharma P, Kaur S, Khursheed MA, Gupta S, et al. 2019. Dual regulation of lin28a by Myc is necessary during zebrafish retina regeneration. J. Cell Biol 218(2):489–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizeracka K, DeMaso CR, Cepko CL. 2013. Notch1 is required in newly postmitotic cells to inhibit the rod photoreceptor fate. Development 140(15):3188–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JE, Parsons MJ, Hyde DR. 2010. A novel model of retinal ablation demonstrates that the extent of rod cell death regulates the origin of the regenerated zebrafish rod photoreceptors. J. Comp. Neurol 518(6):800–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AC, Scholz TL, Brockerhoff SE, Fadool JM. 2008. Genetic dissection reveals two separate pathways for rod and cone regeneration in the teleost retina. Dev. Neurobiol 68(5):605–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima M, Barthel LK, Raymond PA. 2013. A self-renewing division of zebrafish Muller glial cells generates neuronal progenitors that require N-cadherin to regenerate retinal neurons. Development 140(22):4510–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima M, D’Cruz TS, Danku AE, Hesse D, Sifuentes C, et al. 2020. Midkine-a is required for cell cycle progression of Müller glia during neuronal regeneration in the vertebrate retina. J. Neurosci 40(6):1232–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Takeda M, Lewis GP, Cho KS, Jiao J, et al. 2007. Attenuated glial reactions and photoreceptor degeneration after retinal detachment in mice deficient in glial fibrillary acidic protein and vimentin. Investig. Ophthalmol. Vis. Sci 48(6):2760–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Ackerman KM, O’Hayer P, Bailey TJ, Gorsuch RA, Hyde DR. 2013. Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. J. Neurosci 33(15):6524–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Gorsuch RA, Bailey TJ, Ackerman KM, Kassen SC, Hyde DR. 2012. Stat3 defines three populations of Muller glia and is required for initiating maximal Muller glia proliferation in the regenerating zebrafish retina. J. Comp. Neurol 520(18):4294–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara MM, Simon JA. 2012. Inner workings and regulatory inputs that control Polycomb repressive complex 2. Chromosoma 121(3):221–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooto S, Akagi T, Kageyama R, Akita J, Mandai M, et al. 2004. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. PNAS 101(37):13654–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otteson DC, D’Costa AR, Hitchcock PF. 2001. Putative stem cells and the lineage of rod photoreceptors in the mature retina of the goldfish. Dev. Biol 232(1):62–76 [DOI] [PubMed] [Google Scholar]
- Pollak J, Wilken MS, Ueki Y, Cox KE, Sullivan JM, et al. 2013. ASCL1 reprograms mouse Muller glia into neurogenic retinal progenitors. Development 140(12):2619–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C, Cornblath E, Elsaeidi F, Wan J, Goldman D. 2016. Zebrafish Muller glia-derived progenitors are multipotent, exhibit proliferative biases and regenerate excess neurons. Sci. Rep 6:24851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C, Elsaeidi F, Goldman D. 2012. Injury-dependent Muller glia and ganglion cell reprogramming during tissue regeneration requires Apobec2a and Apobec2b. J. Neurosci 32(3):1096–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C, Grant AR, Cornblath E, Goldman D. 2013. Analysis of DNA methylation reveals a partial reprogramming of the Muller glia genome during retina regeneration. PNAS 110(49):19814–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Kidd AR 3rd, Thomas JL, Poss KD, Hyde DR, et al. 2011. FGF signaling regulates rod photoreceptor cell maintenance and regeneration in zebrafish. Exp. Eye Res 93(5):726–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Fausett BV, Goldman D. 2010. Ascl1a regulates Muller glia dedifferentiation and retinal regeneration through a Lin-28-dependent, let-7 microRNA signalling pathway. Nat. Cell Biol 12(11):1101–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Zhao XF, Goldman D. 2011. Ascl1a/Dkk/beta-catenin signaling pathway is necessary and glycogen synthase kinase-3beta inhibition is sufficient for zebrafish retina regeneration. PNAS 108(38):15858–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Zhao XF, Goldman D. 2012. Insm1a-mediated gene repression is essential for the formation and differentiation of Muller glia-derived progenitors in the injured retina. Nat. Cell Biol 14(10):1013–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MB, Didiano D, Patton JG. 2017. Neurotransmitter-regulated regeneration in the zebrafish retina. Stem Cell Rep. 8(4):831–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond PA, Barthel LK, Bernardos RL, Perkowski JJ. 2006. Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev. Biol 6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond PA, Reifler MJ, Rivlin PK. 1988. Regeneration of goldfish retina: Rod precursors are a likely source of regenerated cells. J. Neurobiol 19(5):431–63 [DOI] [PubMed] [Google Scholar]
- Raymond PA, Rivlin PK. 1987. Germinal cells in the goldfish retina that produce rod photoreceptors. Dev. Biol 122(1):120–38 [DOI] [PubMed] [Google Scholar]
- Rehfeld F, Rohde AM, Nguyen DT, Wulczyn FG. 2015. Lin28 and let-7: ancient milestones on the road from pluripotency to neurogenesis. Cell Tissue Res. 359(1):145–60 [DOI] [PubMed] [Google Scholar]
- Reichenbach A, Bringmann A. 2013. New functions of Muller cells. Glia 61(5):651–78 [DOI] [PubMed] [Google Scholar]
- Rueda EM, Hall BM, Hill MC, Swinton PG, Tong X, et al. 2019. The Hippo pathway blocks mammalian retinal Muller glial cell reprogramming. Cell Rep. 27(6):1637–49.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanges D, Romo N, Simonte G, Di Vicino U, Tahoces AD, et al. 2013. Wnt/beta-catenin signaling triggers neuron reprogramming and regeneration in the mouse retina. Cell Rep. 4(2):271–86 [DOI] [PubMed] [Google Scholar]
- Sanges D, Simonte G, Di Vicino U, Romo N, Pinilla I, et al. 2016. Reprogramming Muller glia via in vivo cell fusion regenerates murine photoreceptors. J. Clin. Investig 126(8):3104–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka A, Seiser C. 2012. Histone H3 phosphorylation: a versatile chromatin modification for different occasions. Biochimie 94(11):2193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Gupta S, Chaudhary M, Mitra S, Chawla B, et al. 2019. Oct4 mediates Muller glia reprogramming and cell cycle exit during retina regeneration in zebrafish. Life Sci. Alliance 2(5):e201900548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa T, Fimbel SM, Mallory DE, Maaswinkel H, Spritzer SD, et al. 2008. Ganglion cell regeneration following whole-retina destruction in zebrafish. Dev. Neurobiol 68(2):166–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa T, Hunter SS, Frey RA, Robison BD, Stenkamp DL. 2011. Retinal proliferation response in the buphthalmic zebrafish, bugeye. Exp. Eye Res 93(4):424–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa T, Lankford T, McGinn TE, Hunter SS, Frey RA, et al. 2014. Retinal regeneration is facilitated by the presence of surviving neurons. Dev. Neurobiol 74(9):851–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieger D, Moritz C, Ziegenhals T, Prykhozhij S, Peri F. 2012. Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev. Cell 22(6):1138–48 [DOI] [PubMed] [Google Scholar]
- Sifuentes CJ, Kim JW, Swaroop A, Raymond PA. 2016. Rapid, dynamic activation of Muller glial stem cell responses in zebrafish. Investig. Ophthalmol. Vis. Sci 57(13):5148–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkamp DL, Satterfield R, Muhunthan K, Sherpa T, Vihtelic TS, Cameron DA. 2008. Age-related cone abnormalities in zebrafish with genetic lesions in sonic hedgehog. Investig. Ophthalmol. Vis. Sci 49(10):4631–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subirada PV, Paz MC, Ridano ME, Lorenc VE, Vaglienti MV, et al. 2018. A journey into the retina: Muller glia commanding survival and death. Eur. J. Neurosci 47(12):1429–43 [DOI] [PubMed] [Google Scholar]
- Surzenko N, Crowl T, Bachleda A, Langer L, Pevny L. 2013. SOX2 maintains the quiescent progenitor cell state of postnatal retinal Muller glia. Development 140(7):1445–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–76 [DOI] [PubMed] [Google Scholar]
- Thomas JL, Morgan GW, Dolinski KM, Thummel R. 2018. Characterization of the pleiotropic roles of Sonic Hedgehog during retinal regeneration in adult zebrafish. Exp. Eye Res 166:106–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JL, Ranski AH, Morgan GW, Thummel R. 2016. Reactive gliosis in the adult zebrafish retina. Exp. Eye Res 143:98–109 [DOI] [PubMed] [Google Scholar]
- Thummel R, Burket CT, Hyde DR. 2006. Two different transgenes to study gene silencing and re-expression during zebrafish caudal fin and retinal regeneration. Sci. World J 6(Suppl. 1):65–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Fischer AJ. 2015. Hedgehog signaling stimulates the formation of proliferating Muller glia-derived progenitor cells in the chick retina. Development 142(15):2610–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Palazzo I, Squires N, Mendonca N, Fischer AJ. 2017. BMP- and TGFbeta-signaling regulate the formation of Muller glia-derived progenitor cells in the avian retina. Glia 65(10):1640–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Squires N, Suarez L, Fischer AJ. 2016. Jak/Stat signaling regulates the proliferation and neurogenic potential of Muller glia-derived progenitor cells in the avian retina. Sci. Rep 6:35703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Suarez L, Quinn C, Fischer AJ. 2018. Retinoic acid-signaling regulates the proliferative and neurogenic capacity of Muller glia-derived progenitor cells in the avian retina. Stem Cells 36(3):392–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L, Volkov LI, Zelinka C, Squires N, Fischer AJ. 2015. Heparin-binding EGF-like growth factor (HB EGF) stimulates the proliferation of Muller glia-derived progenitor cells in avian and murine retinas. Mol. Cell. Neurosci 69:54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y, Reh TA. 2013. EGF stimulates Muller glial proliferation via a BMP-dependent mechanism. Glia 61(5):778–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y, Wilken MS, Cox KE, Chipman L, Jorstad N, et al. 2015. Transgenic expression of the proneural transcription factor Ascl1 in Muller glia stimulates retinal regeneration in young mice. PNAS 112(44):13717–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. 2016. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res 51:1–40 [DOI] [PubMed] [Google Scholar]
- Veroni C, Gabriele L, Canini I, Castiello L, Coccia E, et al. 2010. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci 45(3):234–44 [DOI] [PubMed] [Google Scholar]
- Veth KN, Willer JR, Collery RF, Gray MP, Willer GB, et al. 2011. Mutations in zebrafish lrp2 result in adult-onset ocular pathogenesis that models myopia and other risk factors for glaucoma. PLOS Genet. 7(2):e1001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter ML, Moore KB. 2001. Becoming glial in the neural retina. Dev. Dyn 221(2):146–53 [DOI] [PubMed] [Google Scholar]
- Vihtelic TS, Hyde DR. 2000. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J. Neurobiol 44(3):289–307 [DOI] [PubMed] [Google Scholar]
- Voigt P, Tee WW, Reinberg D. 2013. A double take on bivalent promoters. Genes Dev. 27(12):1318–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Goldman D. 2017. Opposing actions of Fgf8a on Notch signaling distinguish two Muller glial cell populations that contribute to retina growth and regeneration. Cell Rep. 19(4):849–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Ramachandran R, Goldman D. 2012. HB-EGF is necessary and sufficient for Muller glia dedifferentiation and retina regeneration. Dev. Cell 22(2):334–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Zhao XF, Vojtek A, Goldman D. 2014. Retinal injury, growth factors, and cytokines converge on beta-catenin and pStat3 signaling to stimulate retina regeneration. Cell Rep. 9(1):285–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DT, Sengupta S, Saxena MT, Xu Q, Hanes J, et al. 2017. Immunomodulation-accelerated neuronal regeneration following selective rod photoreceptor cell ablation in the zebrafish retina. PNAS 114(18):E3719–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willkomm L, Bloch W. 2015. State of the art in cell-cell fusion. Methods Mol. Biol 1313:1–19 [DOI] [PubMed] [Google Scholar]
- Wu DM, Schneiderman T, Burgett J, Gokhale P, Barthel L, Raymond PA. 2001. Cones regenerate from retinal stem cells sequestered in the inner nuclear layer of adult goldfish retina. Investig. Ophthalmol. Vis. Sci 42(9):2115–24 [PubMed] [Google Scholar]
- Yao K, Qiu S, Tian L, Snider WD, Flannery JG, et al. 2016. Wnt regulates proliferation and neurogenic potential of Muller glial cells via a Lin28/let-7 miRNA-dependent pathway in adult mammalian retinas. Cell Rep. 17(1):165–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Chen Z, Ouyang Y, Zhang H, Wan Z, et al. 2017. Thrombin-induced, TNFR-dependent miR-181c downregulation promotes MLL1 and NF-κB target gene expression in human microglia. J. Neuroinflamm 14(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimatsu T, D’Orazi FD, Gamlin CR, Suzuki SC, Suli A, et al. 2016. Presynaptic partner selection during retinal circuit reassembly varies with timing of neuronal regeneration in vivo. Nat. Commun 7:10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurco P, Cameron DA. 2005. Responses of Muller glia to retinal injury in adult zebrafish. Vis. Res 45(8):991–1002 [DOI] [PubMed] [Google Scholar]
- Yurco P, Cameron DA. 2007. Cellular correlates of proneural and Notch-delta gene expression in the regenerating zebrafish retina. Vis. Neurosci 24(3):437–43 [DOI] [PubMed] [Google Scholar]
- Zelinka CP, Volkov L, Goodman ZA, Todd L, Palazzo I, et al. 2016. mTor signaling is required for the formation of proliferating Muller glia-derived progenitor cells in the chick retina. Development 143(11):1859–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Bell E, Zhi H, Brown S, Imran SAM, et al. 2019. OCT4 and PAX6 determine the dual function of SOX2 in human ESCs as a key pluripotent or neural factor. Stem Cell Res. Therapy 10(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XF, Wan J, Powell C, Ramachandran R, Myers MG Jr., Goldman D. 2014. Leptin and IL-6 family cytokines synergize to stimulate Muller glia reprogramming and retina regeneration. Cell Rep. 9(1):272–84 [DOI] [PMC free article] [PubMed] [Google Scholar]