

Abstract
Cyclin D1 is an essential regulator of the G1-S cell cycle transition and is overexpressed in many cancers. Expression of cyclin D1 is under tight cellular regulation that is controlled by many signaling pathways. Here we report that PARP14, a member of the Poly(ADP-ribose) polymerase (PARP) family, is a regulator of cyclin D1 expression. Depletion of PARP14 leads to decreased cyclin D1 protein levels. In cells with a functional retinoblastoma (RB) protein pathway, this results in G1 cell cycle arrest and reduced proliferation. Mechanistically, we found that PARP14 controls cyclin D1 mRNA levels. Using luciferase assays, we show that PARP14 specifically regulates cyclin D1 3’UTR mRNA stability. Finally, we also provide evidence that G1 arrest in PARP14-depleted cells is dependent on an intact p53-p21 pathway. Our work uncovers a new role for PARP14 in promoting cell cycle progression through both cyclin D1 and the p53 pathway.
Introduction
Evading growth suppressors is one of the hallmarks of cancer[1]. The retinoblastoma protein (RB) is a canonical tumor suppressor protein that regulates the G1-S phase cell cycle transition and cell growth. In its non-phosphorylated state, RB is active and restricts cell cycle progression by repressing E2F1, a transcription factor that drives the expression of S-phase genes. In responses to upstream activating signals, cyclin D1, in a complex with CDK4/6, mono-phosphorylates RB, leading to RB hyper-phosphorylation by the CDK2-Cyclin E complex[2]. Upon hyper-phosphorylation, RB is inactivated and de-represses E2F1, which leads to the expression of S-phase genes[3] and subsequent cell cycle progression. Dysregulation of the RB pathway, through various mechanisms, is seen across numerous cancers. Germline mutations in RB1, the gene that encodes RB, lead to retinoblastoma, a rare form of eye cancer in children[4], while RB1 loss or mutation is prevalent in osteosarcoma, small cell lung cancer, and at a lower frequency in other tumor types[5]. Overexpression or amplification of the CCND1 gene, which encodes cyclin D1, is seen in 50% of breast cancer patients[6]. This is thought to cause constitutive phosphorylation, and thus inactivation, of RB by CDK4/6, leading to uncontrolled proliferation of breast cancer cells. In 2015, palbociclib was approved as a specific CDK4/6 kinase inhibitor for breast cancer patients after a clinical trial showed a marked improvement in progression-free survival[7]. Two additional CDK4/6 inhibitors, abemaciclib8 and ribociclib[8, 9], were subsequently approved, giving breast cancer patients three new options to treat their disease.
Despite the remarkable progress made in understanding the functions of the RB pathway, many questions remain. While it is well established that RB regulates E2F1 to affect cell growth, recent research has identified many additional functions and targets for RB[10]. Additionally, while cyclin D1 has been widely studied, our understanding of its regulation is incomplete, as many different proteins in numerous pathways can affect cyclin D1 expression[11] to facilitate cell cycle progression. Finally, the role of p21 in regulating RB phosphorylation and cell cycle progression is still controversial. While early work clearly identified p21 as an inhibitor of CDK4/6 and CDK2 kinase activity[12], and thus RB phosphorylation, paradoxically, p21 was later shown to be required for CDK4/6-cyclinD1 complex formation[13]. How p21 can both inhibit and activate CDK4/6 is still not well understood.
One group of proteins whose role in cell cycle progression has not been widely investigated is the Poly(ADP-ribose) polymerase (PARP) family of proteins. The PARP family consists of 17 proteins that are involved in a wide range of biological processes, including DNA repair, transcriptional regulation, apoptosis, and the heat shock response[14]. PARP1 and PARP3, two members of the family, are reported to have functions in cell cycle progression[15, 16], yet what role, if any, the other PARP family members have is still unclear. PARP14 is a PARP family member that has roles in DNA repair, B cell regulation, and focal adhesion[17–19]. Here, we describe a role for PARP14 in facilitating cell cycle progression. We show that PARP14 regulates expression of cyclin D1 to drive the G1-S phase transition in cells with a functional RB pathway. Specifically, we find that PARP14 knockdown or knockout leads to a marked decrease in cyclin D1 mRNA levels. Using luciferase assays, we demonstrate that PARP14 controls cyclin D1 3’UTR mRNA stability. As a consequence of cyclin D1 depletion, PARP14 knockdown results in a decrease in RB phosphorylation and a marked G1 arrest. Additionally, we demonstrate that an intact p53-p21 pathway is required for cell cycle arrest in PARP14-depleted cells. Overall, our work uncovers a new function for PARP14 in regulating cell cycle progression through both cyclin D1 and the p53 pathway.
Results
Impact of PARP14 on cell cycle progression
We first investigated the impact of PARP14 on cell cycle progression in non-transformed cells. We used hTERT-RPE-1 (RPE-1) cells, a chromosomally stable, non-transformed epithelial cell line. In RPE-1 cells, PARP14 knockdown using multiple siRNA oligonucleotides resulted in a marked G1 arrest phenotype (Fig. 1A, 1B). This phenotype was also observed when the PARP14 siRNA was used at lower concentrations, but was not observed upon knockdown of an unrelated gene, namely IPO11 (Supplementary Fig. S1A). To further explore this cell cycle arrest phenotype, we next knocked down PARP14 in RPE-1 cells and then added nocodazole, a microtubule inhibitor that arrests cells in G2/M. While control cells showed an expected arrest in G2/M after nocodazole treatment, strikingly, PARP14-knockdown cells showed almost no progression into G2/M (Fig. 1C, 1D). This G1-cell cycle arrest phenotype was not nocodazole-specific, as treatment with RO3306, a CDK1 inhibitor that prevents mitotic entry, also resulted in G1 arrest in PARP14-knockdown cells (Supplementary Fig. S1B). Additionally, a similar G1 arrest after nocodazole treatment was observed upon PARP14 knockdown in BJ cells, a non-transformed fibroblast cell line (Supplementary Fig. S1C–S1D). As an additional control, knockdown of PARP10, another PARP family member, showed no G1 arrest after nocodazole treatment in these cells. Finally, cell cycle arrest due to PARP14 knockdown caused an expected decrease in proliferation of RPE-1 cells (Fig. 1E). Previously, PARP14 was shown to suppress apoptosis upon IL4 stimulation of mouse B cells [20] and in multiple myeloma cell lines with constitutive JNK2 activation [21]. However, we did not observe an impact of PARP14 depletion on apoptosis in RPE-1 cells grown under normal conditions (Supplementary Fig. S1E), indicating that the reduced proliferation observed upon PARP14 knockdown in RPE-1 cells was likely not due to cell death.
Figure 1. PARP14 is required for cell cycle progression.
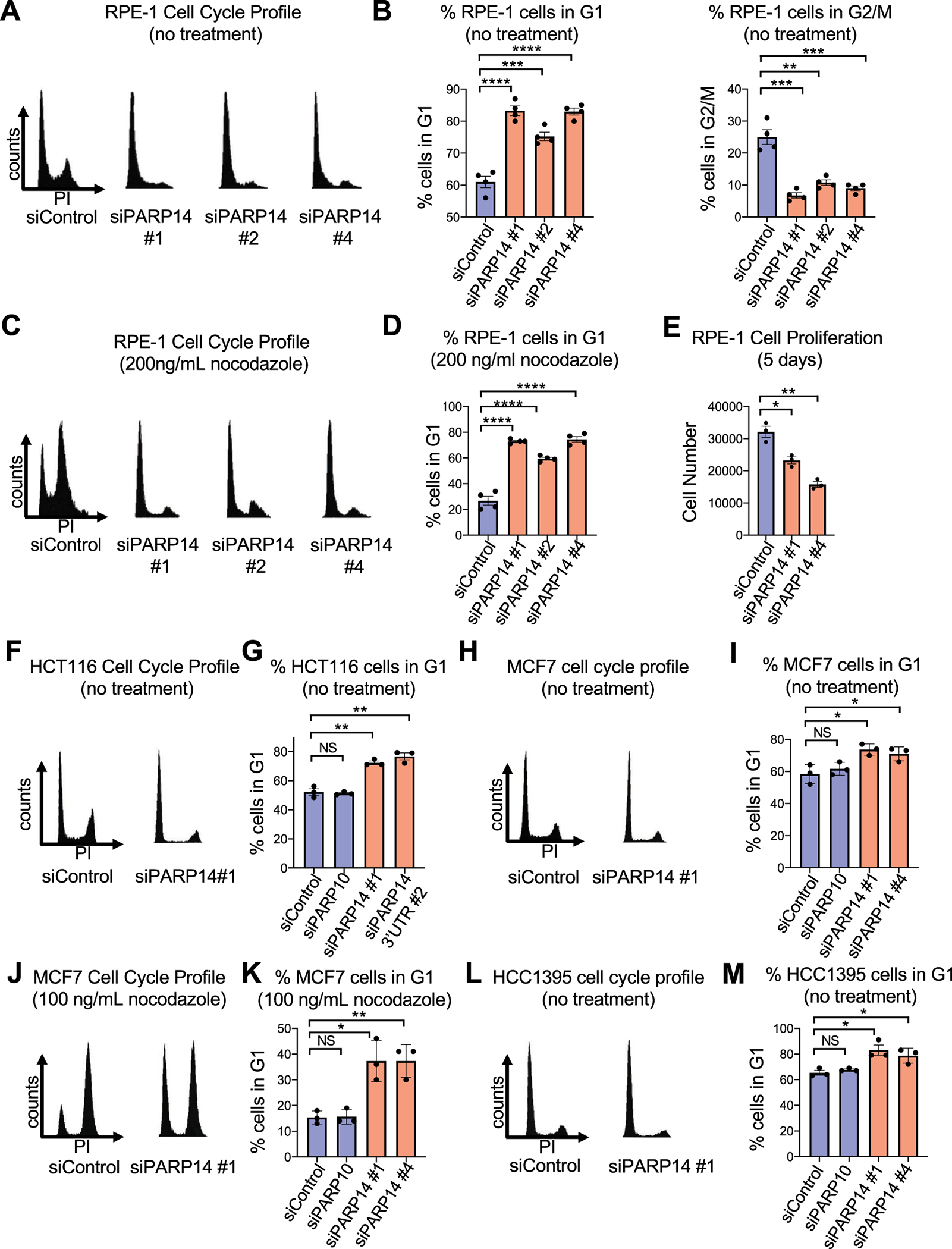
A. Cell cycle profiles of RPE-1 cells after PARP14 knockdown. B. Quantification showing the percent of RPE-1 cells in G1 and G2/M cell cycle phases upon PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired). C. Cell cycle profiles of RPE-1 cells after PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. D. Quantification showing the percent of RPE-1 cells in G1 cell cycle phase upon PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. Bars represent the means ± SEM (t-test, unpaired). E. Proliferation assay in RPE-1 cells showing the impact of PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired). F. Cell cycle profiles of HCT116 cells after PARP14 knockdown. G. Quantification showing the percent of HCT116 cells in G1 cell cycle phase upon PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired). PARP10 knockdown was used as a second control. Bars represent the means ± SEM (t-test, unpaired). H. Cell cycle profiles of MCF7 cells after PARP14 knockdown. I. Quantification showing the percent of MCF7 cells in G1 cell cycle phase upon PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired). J. Cell cycle profiles of MCF7 cells after PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. K. Quantification showing the percent of MCF7 cells in G1 cell cycle phase upon PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. Bars represent the means ± SEM (t-test, unpaired). L. Cell cycle profiles of HCC1395 cells after PARP14 knockdown. M. Quantification showing the percent of HCC1395 cells in G1 cell cycle phase upon PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired).
Having established a cell cycle arrest defect caused by PARP14 knockdown in two non-transformed cell lines, we next determined if PARP14 depletion results in G1 arrest in cancer cells. Palbociclib is a widely used inhibitor in the clinic that arrests cell cycle progression of breast cancer cells specifically in G1. We tested two breast cancer cell lines that were previously shown to be sensitive to palbociclib[22], MCF7 and HCC1395, as well as HCT116 cells, a colorectal carcinoma cell line whose growth is also attenuated by palbociclib treatment[23]. Similar to RPE-1 cells, knockdown of PARP14 in HCT116 cells in the absence of any treatment caused a significant G1 arrest (Fig. 1F–1G, Supplementary Fig. S1F). Additionally, MCF7 cells also underwent G1 arrest due to PARP14 knockdown both under normal growth conditions and after nocodazole treatment (Fig.1H–1K, Supplementary Fig. S1G). Knockdown of PARP14 in MCF7 cells also resulted in attenuated proliferation (Supplementary Fig. S1H). Finally, similar to MCF7 cells, knockdown of PARP14 caused G1 arrest in HCC1395 cells both under normal conditions and after nocodazole addition (Fig.1L–1M, Supplementary Fig. S1I–S1K). Taken together, these results from five different cell lines demonstrate that PARP14 is required for cell cycle progression in both non-transformed epithelial cells and cancer cells.
PARP14 is required for RB phosphorylation and expression of E2F1 target proteins
We next sought to determine in which cell cycle control pathway PARP14 may be functioning. One of the primary pathways that drive cell cycle progression in non-transformed and many breast cancer cell lines cells is the cyclin D1-CDK4/6-RB pathway[24]. In response to growth signals, cyclin D1 and CDK4 form a complex that phosphorylates the retinoblastoma protein (RB). RB in its non-phosphorylated form negatively regulates E2F1, a transcription factor which drives expression of genes required for S-phase progression. After RB phosphorylation, E2F1 and its target genes are fully expressed, facilitating the G1-S transition. Given the pronounced cell cycle defect we observed in both non-transformed epithelial cells and breast cancer cells after PARP14 depletion, we first investigated pRB levels in RPE-1 cells. PARP14 knockdown resulted in a marked decrease in pRB (Ser780) levels without a change in RB expression in RPE-1 cells (Fig. 2A, Supplementary Fig. S2A). The decrease in pRB (Ser780) levels due to PARP14 knockdown was also seen in MCF7 and HCT116 cells (Fig. 2B–2C, Supplementary Fig. S2B–S2C), as well as in HCC1395 cells (Supplementary Fig. S2D). Similar to RPE-1 cells, the decrease in RB phosphorylation was not due to an obvious decrease in overall RB levels in MCF7 cells (Supplementary Fig. S2B). In RPE-1 cells, PARP14 knockdown also caused decreased pRB levels at an additional phosphorylation sites (Thr821/826), and it was accompanied by an expected decrease in E2F1 levels (Fig. 2D, Supplementary Fig. S2E). E2F1 regulates expression of hundreds of genes. We investigated by western blot the expression of four E2F1 targets. These include genes required for cell cycle progression (Cyclin A, Cyclin B, Cdc6) and DNA repair (Chk1). All four of these proteins were expressed at lower levels in PARP14-depleted cells (Fig. 2E, Supplementary Fig. S2F). To further demonstrate that PARP14-mediated regulation of cell cycle progression is dependent on the RB pathway, we used a previously described RPE-1 cell line in which RB was knocked out using CRISPR-Cas9 (Supplementary Fig. S2G) [25]. We treated RPE-1 WT or RPE-1 RB knockout (RBKO) cells with PARP14 siRNA and incubated with nocodazole for 24 hours. While PARP14 knockdown resulted in the previously observed complete G1 arrest in RPE-1 WT cells, RBKO significantly rescued the G1 arrest phenotype due to PARP14 depletion (Fig. 2F–2G, Supplementary Fig. S2H).
Figure 2. PARP14 is required for RB phosphorylation and E2F1-target protein expression.
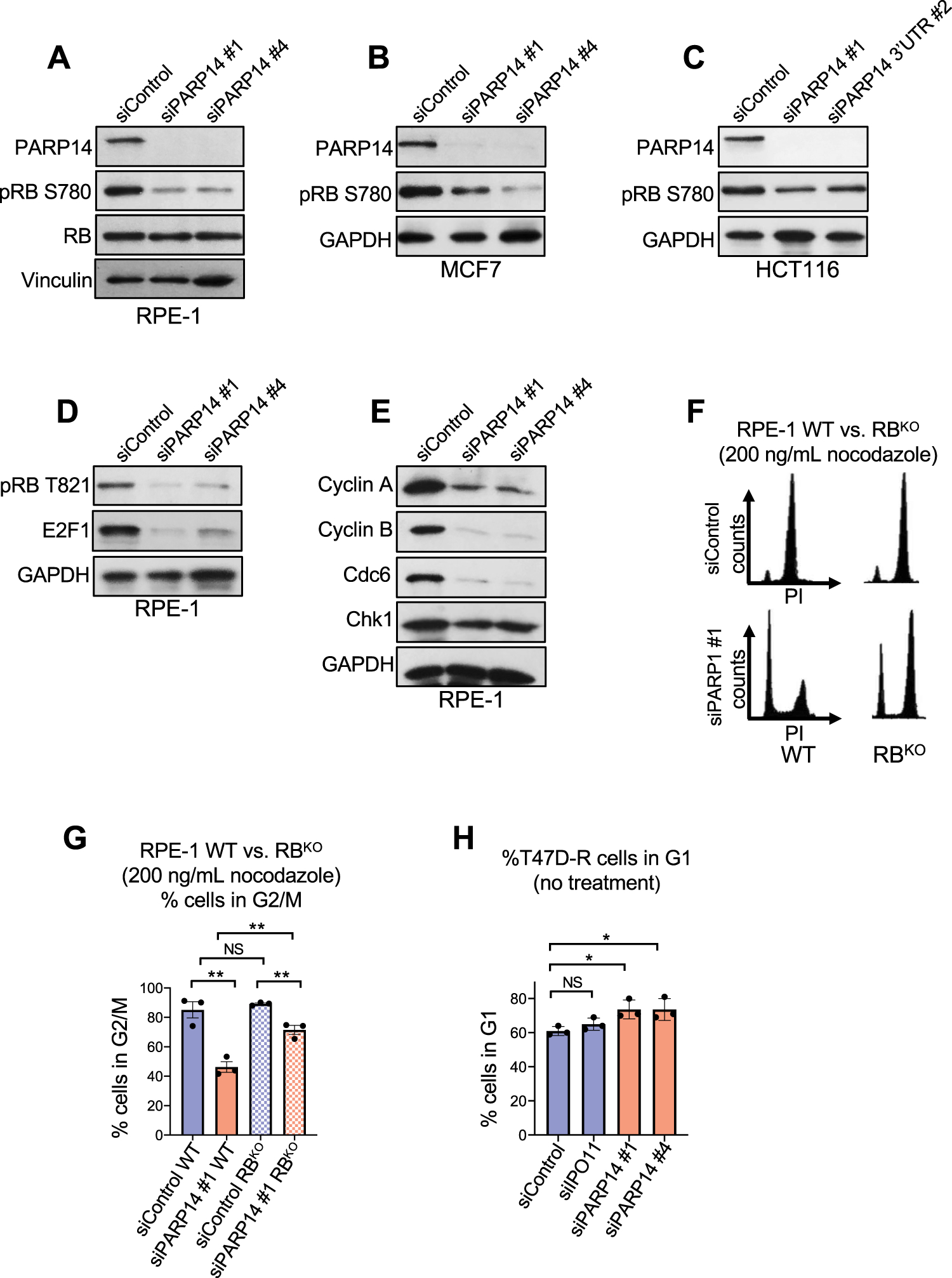
A-C. Western blots showing the levels of phosphorylated RB at Ser780 upon PARP14 knockdown in RPE-1 (A), MCF7 (B) and HCT116 (C) cells. D. Western blots showing the levels of E2F1 and phosphorylated RB at Thr821/826 upon PARP14 knockdown in RPE-1 cells. E. Western blots showing the levels of E2F1 targets Cyclin A, Cyclin B, Cdc6 and Chk1 upon PARP14 knockdown in RPE-1 cells. F. Cell cycle profiles of RPE-1 WT (left) and RBKO (right) cells after PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. G. Quantification showing the percent of RPE-1 WT and RBKO cells in G2/M cell cycle phase upon PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. Bars represent the means ± SEM (t-test, unpaired). H. Quantification showing the percent of T47D palbociclib-resistant cells (T47D-R) cells in G1 cell cycle phase upon PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired).
Given that RB knockout does not completely rescue G1 arrest due to PARP14 knockdown, we next investigated whether PARP14 depletion also causes G1 arrest in cells that are resistant to CDK4/6 inhibitors due to loss of RB. To test this, we employed a previously described T47D breast cancer cell line that was continuously grown in palbociclib and became resistant to palbociclib-mediated G1 arrest as a result of RB loss (T47D-R) [26]. Consistent with our data using the RPE-1 RBKO cells described above, knockdown of PARP14 in the T47D-R cell line caused G1 arrest, suggesting that PARP14 depletion impairs cell cycle progression despite loss of RB in palbociclib-resistant cells (Fig. 2H, Supplementary Fig. S2I).
G1 arrest caused by PARP14 depletion depends on the pocket proteins RB, p107, and p130
RB, p107 and p130 make up the group of “pocket” proteins that are regulated by cyclin D1-CDK4/6 to facilitate the G1-S transition[27, 28]. Having established that RB loss does not completely rescue G1 arrest due to PARP14 knockdown, we next determined if this partial G1 arrest in RBKO cells after PARP14 depletion could be further rescued by knockdown of p107. To test this, we knocked down PARP14 in RBKO cells either alone or in combination with p107 knockdown, and treated cells with nocodazole. While PARP14 knockdown caused partial G1 arrest in RBKO cells, this cell cycle arrest was further rescued by co-depletion of both p107 and PARP14 (Supplementary Fig. S2J–S2K). We conclude that G1 arrest due to PARP14 knockdown is caused by a decrease in both RB phosphorylation and expression of E2F1 target proteins, and this G1 arrest is dependent on both RB and p107.
Given the nearly complete rescue of G1 arrest that we observed in PARP14-knockdown cells that were co-depleted of both RB and p107, we hypothesized that depletion of all three pocket proteins, RB, p107, and p130, would completely rescue the cell cycle arrest phenotype due to PARP14 knockdown. To test this, we used a previously described RPE-1 CRISPR triple knockout cell line (RPE-1 TKO) in which RB, p107 and p130 are all deleted [29]. G1 arrest due to PARP14 knockdown was completely rescued in RPE-1 TKO cells both under normal growth conditions (Fig.3A–3B, Supplementary Fig. S2L) and upon nocodazole treatment (Fig. 3C–3D). Consistent with a lack of cell cycle arrest, we observed no change in the levels of E2F1 or its targets Cyclin A and Cdc6 (Fig. 3E).
Figure 3. Cell cycle arrest due to PARP14 knockdown is not observed in RPE-1 RB/p107/p130 triple knockout cells or HeLa cells.
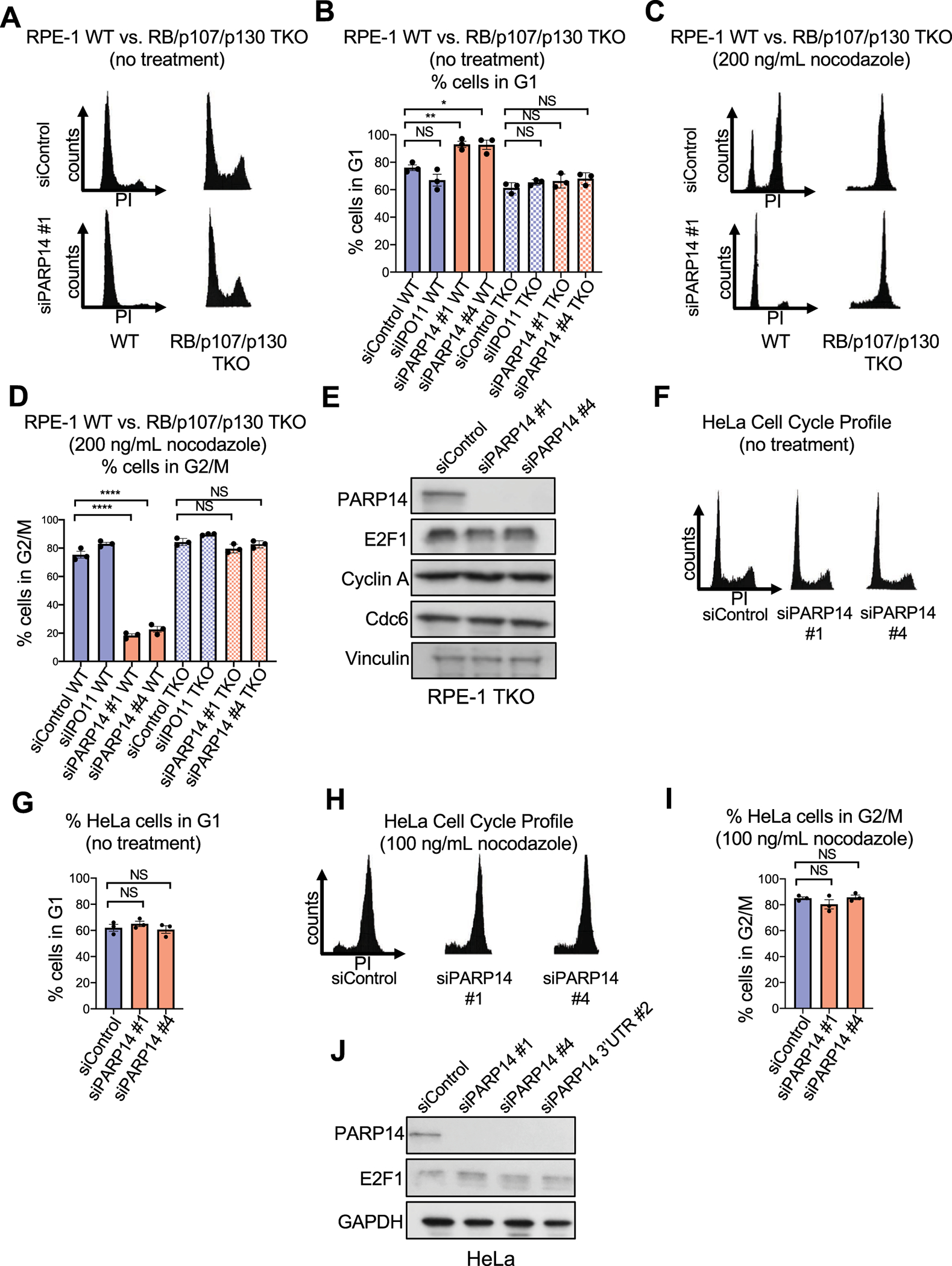
A. Cell cycle profiles of RPE-1 WT and RB/p107/p130 TKO cells after PARP14 knockdown. B. Quantification showing the percent of RPE-1 WT and RB/p107/p130 TKO cells in G1 cell cycle phase upon PARP14 knockdown. IPO11 knockdown was used as a second control. Bars represent the means ± SEM (t-test, unpaired). C. Cell cycle profiles of RPE-1 WT and RB/p107/p130 TKO cells after PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. D. Quantification showing the percent of RPE-1 WT and RB/p107/p130 TKO cells in G2/M cell cycle phase upon PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. Bars represent the means ± SEM (t-test, unpaired). E. Western blots showing the levels of E2F1 and its targets Cyclin A and Cdc6 upon PARP14 knockdown in RPE-1 RB/p107/p130 TKO cells. F. Cell cycle profiles of HeLa cells after PARP14 knockdown. G. Quantification showing the percent of HeLa cells in G1 cell cycle phase upon PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired). H. Cell cycle profiles of HeLa cells after PARP14 knockdown and subsequent treatment with 100ng/mL nocodazole for 24h. I. Quantification showing the percent of HeLa cells in G2/M cell cycle phase upon PARP14 knockdown and subsequent treatment with 100ng/mL nocodazole for 24h. Bars represent the means ± SEM (t-test, unpaired). J. Western blots showing the levels of E2F1 upon PARP14 knockdown in HeLa cells.
The human papillomavirus (HPV) E6 and E7 genes, encoding the E6 and E7 proteins, are the major oncogenes responsible for cervical cancer[30]. While E6 targets p53 for degradation, E7 binds to the pocket proteins and disrupts their interaction with the E2F proteins, leading to de-repression of S-phase genes and uncontrolled cellular proliferation. Based on the RPE-1 TKO results, we hypothesized that PARP14 knockdown in HeLa cells, a cervical cancer cell line, would not result in a G1 arrest phenotype. Consistent with this, knockdown of PARP14 in HeLa cells did not affect the cell cycle profile with or without nocodazole treatment (Fig. 3F–3I). Finally, in line with the lack of a functional RB pathway in HeLa cells due to the HPV E7 oncoprotein, PARP14 knockdown did not cause any change in E2F1 levels in HeLa cells (Fig. 3J). Thus, G1 arrest due to PARP14 knockdown is dependent on RB, p107, and p130.
G1 arrest due to PARP14 knockdown is partially rescued by depletion of p21
Having established that PARP14 regulates RB phosphorylation, we next investigated which canonical proteins in the RB pathway are affected by PARP14 knockdown. In response to DNA damage, p53 levels are increased. This results in increased p21 expression, which can inhibit CDK4 and CDK2 to regulate RB phosphorylation and S-phase entry[31]. An increase in p53 and p21 levels would provide a potential explanation for the decrease in RB phosphorylation due to PARP14 knockdown. However, we observed no substantial change in p21 or p53 levels after PARP14 knockdown in RPE-1 cells (Fig. 4A).
Figure 4. Depletion of p21 or p53 moderately rescues cell cycle arrest caused by PARP14 knockdown.
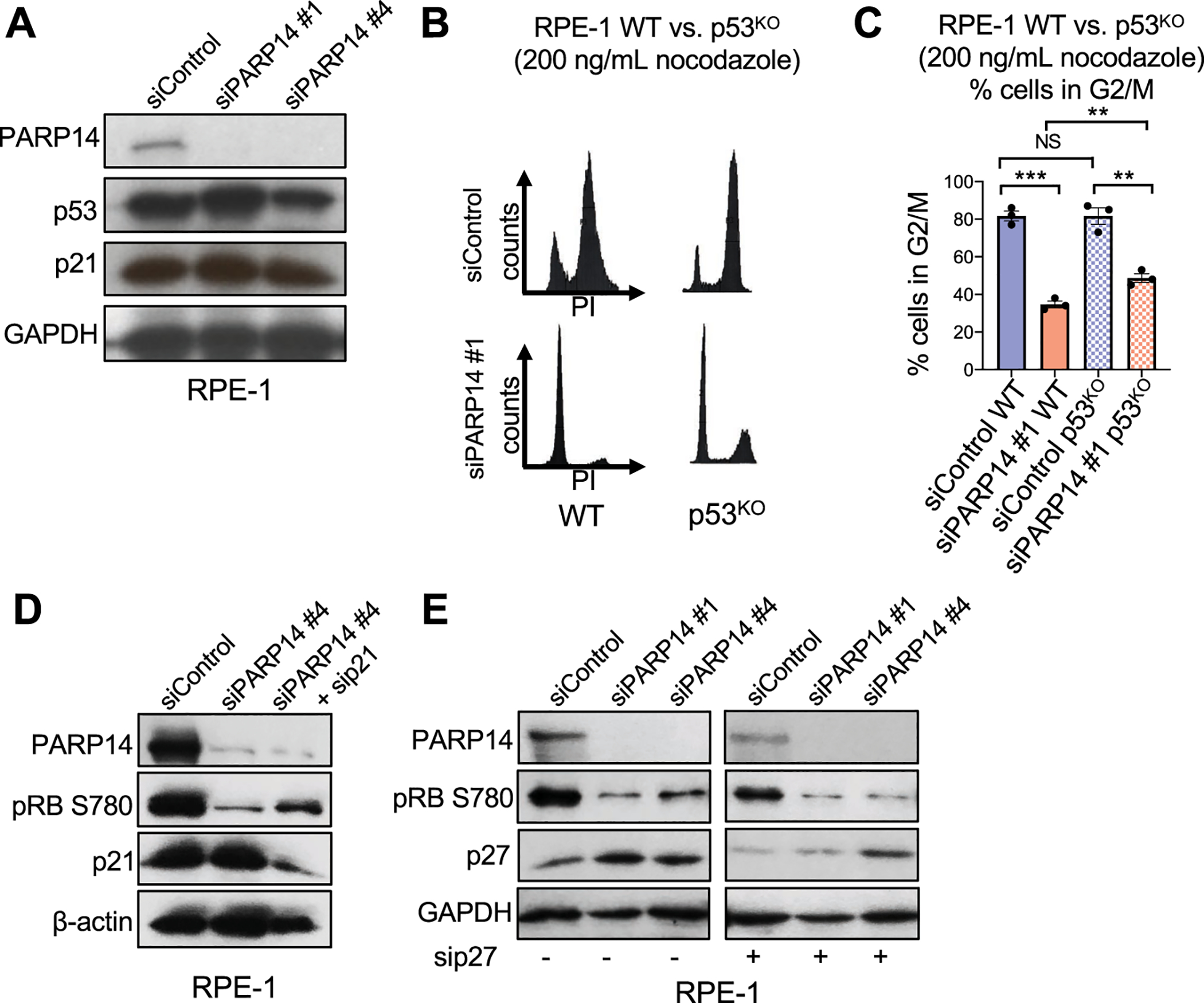
A. Western blots showing the levels of p53 and p21 upon PARP14 knockdown in RPE-1 cells. B. Cell cycle profiles of RPE-1 WT and p53KO cells after PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. C. Quantification showing the percent of RPE-1 WT and p53KO cells in G2/M cell cycle phase upon PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. Bars represent the means ± SEM (t-test, unpaired). D. Western blots showing the levels of phosphorylated RB at Ser780 upon PARP14 knockdown alone or in combination with p21 knockdown in RPE-1 cells. E. Western blots showing the levels of phosphorylated RB at Ser780 upon PARP14 knockdown alone or in combination with p27 knockdown in RPE-1 cells.
We next tested the effect of PARP14 knockdown in previously described p53-knockout RPE-1[32] and HCT116 [33] cell lines. Surprisingly, p53 knockout did moderately rescue G1 cell cycle arrest due to PARP14 knockdown in both RPE-1 (Fig. 4B–4C, Supplementary Fig. S3A) and HCT116 cells (Supplementary Fig. S3B–S3C). We next examined if the partial rescue of cell cycle arrest due to PARP14 knockdown was through loss of p21. Using RPE-1 WT cells, we knocked down either PARP14 alone or in combination with p21 knockdown. PARP14 knockdown alone, as expected, caused a marked decrease in pRB (Ser780) levels without an increase in p21 levels, while co-depletion of p21 and PARP14 partially rescued pRB (Ser780) levels (Fig. 4D, Supplementary Fig. S3D). Finally, we investigated the effect of PARP14 knockdown on p27, which can inhibit CDK4 kinase activity to prevent RB phosphorylation[34]. Knockdown of PARP14 using two separate siRNAs caused a modest increase in p27 levels in RPE-1 WT cells, yet co-depletion of PARP14 and p27 did not rescue the decrease in pRB (Ser780) levels (Fig. 4E). Overall, this indicates that that G1 arrest due to PARP14 knockdown is partially caused by p21, and does not depend on p27.
PARP14 is required for cyclin D1 expression
Since co-depletion of p21 only partially rescued the decreased pRB (Ser780) levels due to PARP14 knockdown, we investigated the possible involvement of other canonical proteins that regulate the RB pathway. Knockdown of PARP14 in both RPE-1 and MCF7 cells resulted in a marked decrease in cyclin D1 protein levels by western blot (Fig. 5A, Supplementary Fig. S4A). To confirm the specificity of the siRNAs, and to ensure that the decrease in cyclin D1 levels was not simply an effect of cell cycle arrest, we knocked down PARP14 in HeLa and RPE-1 TKO cells, two cell lines that do not undergo G1 arrest after PARP14 knockdown. In both HeLa and RPE-1 TKO cells, PARP14 knockdown decreased cyclin D1 levels (Fig. 5A, Supplementary Fig. S4A–S4B). RT-qPCR experiments showed that cyclin D1 mRNA levels were reduced upon PARP14 knockdown in RPE-1, HeLa, and MCF7 cells, indicating that the decrease in protein levels was due to a decrease in mRNA levels (Fig. 5B).
Figure 5. PARP14 regulates Cyclin D1 protein expression and mRNA levels.
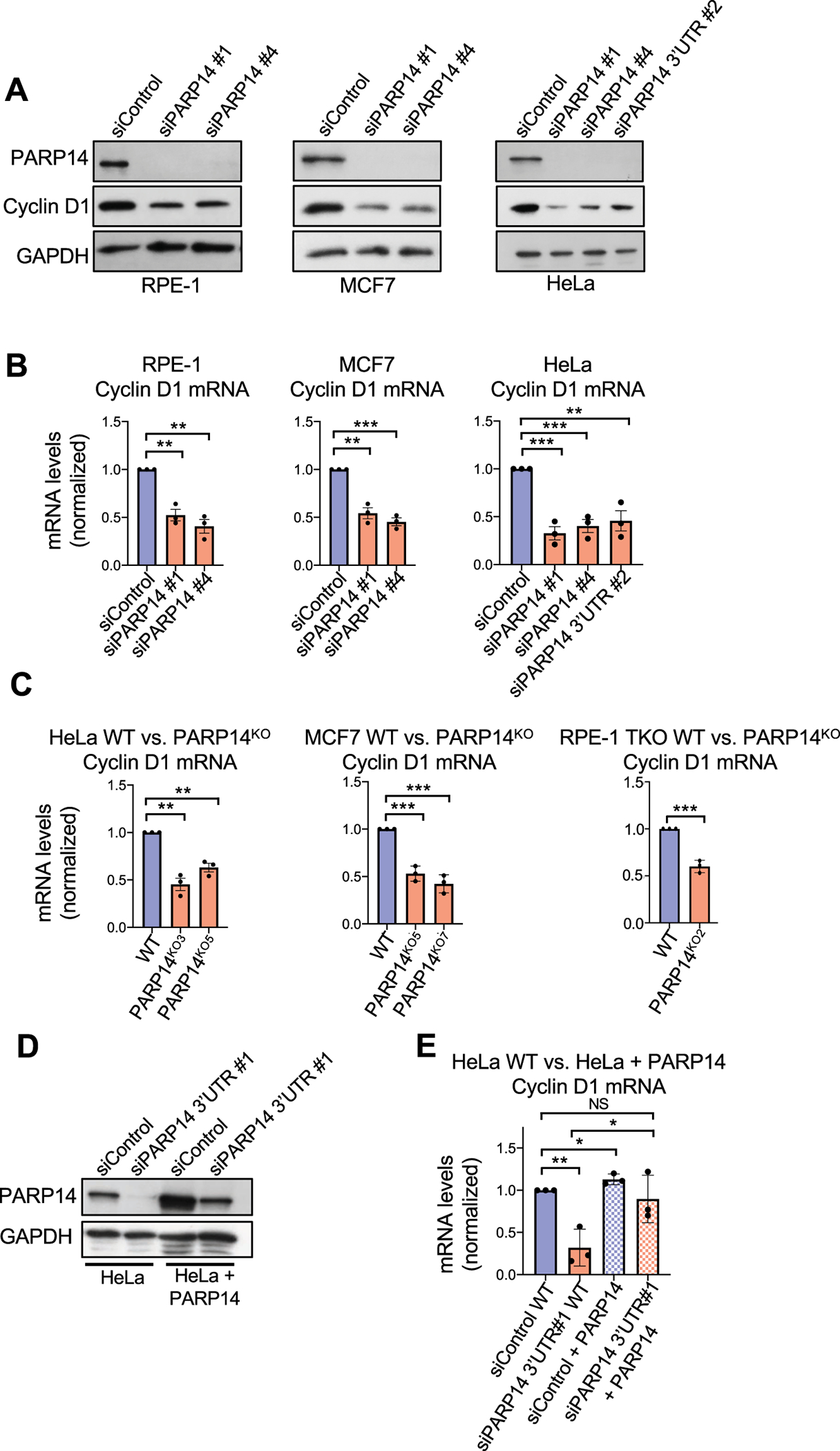
A. Western blots showing Cyclin D1 protein levels upon PARP14 knockdown in RPE-1, MCF7 and HeLa cells. B. RT-qPCR experiments showing Cyclin D1 mRNA levels upon PARP14 knockdown in RPE-1, MCF7 and HeLa cells. Bars represent the means ± SEM (t-test, unpaired). C. RT-qPCR experiments showing Cyclin D1 mRNA levels upon PARP14 knockout in HeLa, MCF7 and RPE-1 RB/p107/p130 TKO cells. Bars represent the means ± SEM (t-test, unpaired). D. Western blots showing PARP14 levels in HeLa WT and PARP14-overexpressing cells upon knockdown of endogenous PARP14 using siRNA targeting the 3’UTR. E. RT-qPCR experiments showing Cyclin D1 mRNA in HeLa WT and PARP14-overexpressing cells upon knockdown of endogenous PARP14 using siRNA targeting the 3’UTR. Bars represent the means ± SEM (t-test, unpaired).
To rule out that the effects observed upon PARP14 knockdown are caused by potential siRNA off-target effects, we generated multiple PARP14-knockout lines in HeLa, MCF7, and RPE-1 TKO cells using CRISPR-Cas9 genome editing (Supplementary Fig. S4C). Consistent with the siRNA results, we observed a significant decrease in cyclin D1 mRNA levels in all PARP14KO cell lines (Fig. 5C). Moreover, we created a HeLa cell line stably expressing PARP14 under control of the SV40 promoter, and depleted endogenous PARP14 using an siRNA that targets the 3’UTR region of the PARP14 mRNA, but has no effect on the exogenously expressed PARP14 (Fig. 5D). In WT cells, PARP14 knockdown resulted in the expected decrease in cyclin D1 mRNA levels. This decrease was almost completely rescued in PARP14-overexpressing cells (Fig. 5E). Altogether, these results further confirm that the effect of PARP14 on cyclin D1 mRNA levels is specific.
Having established that PARP14 specifically regulates cyclin D1, we next employed a previously described cyclin D1-overexpressing RPE-1 cell line [29] to further test if PARP14 regulates cell cycle progression by regulating cyclin D1 expression. In this cell line, we observed no change in cyclin D1 levels due to PARP14 depletion (Supplementary Fig. S4D). While PARP14 knockdown resulted in G1 arrest in RPE-1 WT cells, overexpression of cyclin D1 significantly rescued the G1 arrest phenotype caused by PARP14 knockdown, both under normal growth conditions (Fig. 6A–6B) and upon nocodazole treatment (Fig. 6C–6D, Supplementary Fig. S4E). Consistent with this, cyclin D1 overexpression partially rescued pRB, E2F1, and Cdc6 levels in PARP14 knockdown cells (Fig. 6E, Supplementary Fig. S4F). Thus, PARP14 knockdown causes cell cycle arrest and decreased RB phosphorylation due to, in part, decreased cyclin D1 levels. Given that the cyclin D1-CDK4 kinase is responsible for RB phosphorylation, we hypothesized that PARP14 knockdown would cause a decrease in this kinase activity, which would be restored by overexpression of cyclin D1. To examine this, we knocked down PARP14 in RPE-1 WT and RPE-1 cyclin D1-overexpressing cells and immunoprecipitated cyclin D1. We used the immunoprecipitated cyclin D1-CDK4 complex to perform a kinase assay using a recombinant RB fragment. As expected, in RPE-1 WT cells, the loss of cyclin D1 resulted in almost a complete absence of phosphorylation of the recombinant RB fragment in the kinase assay. However, in the RPE-1 cyclin D1 overexpression cell line, we observed no change in cyclin D1 levels, and RB fragment phosphorylation was comparable to control levels (Fig. 6F, Supplementary Fig. S4G). Altogether, these results indicate that PARP14-mediated regulation of cyclin D1 expression drives RB phosphorylation and E2F1 expression to facilitate the G1-S transition.
Figure 6. Overexpression of Cyclin D1 rescues both the E2F1 protein expression and the CDK4/6 kinase activity defects caused by PARP14 knockdown.
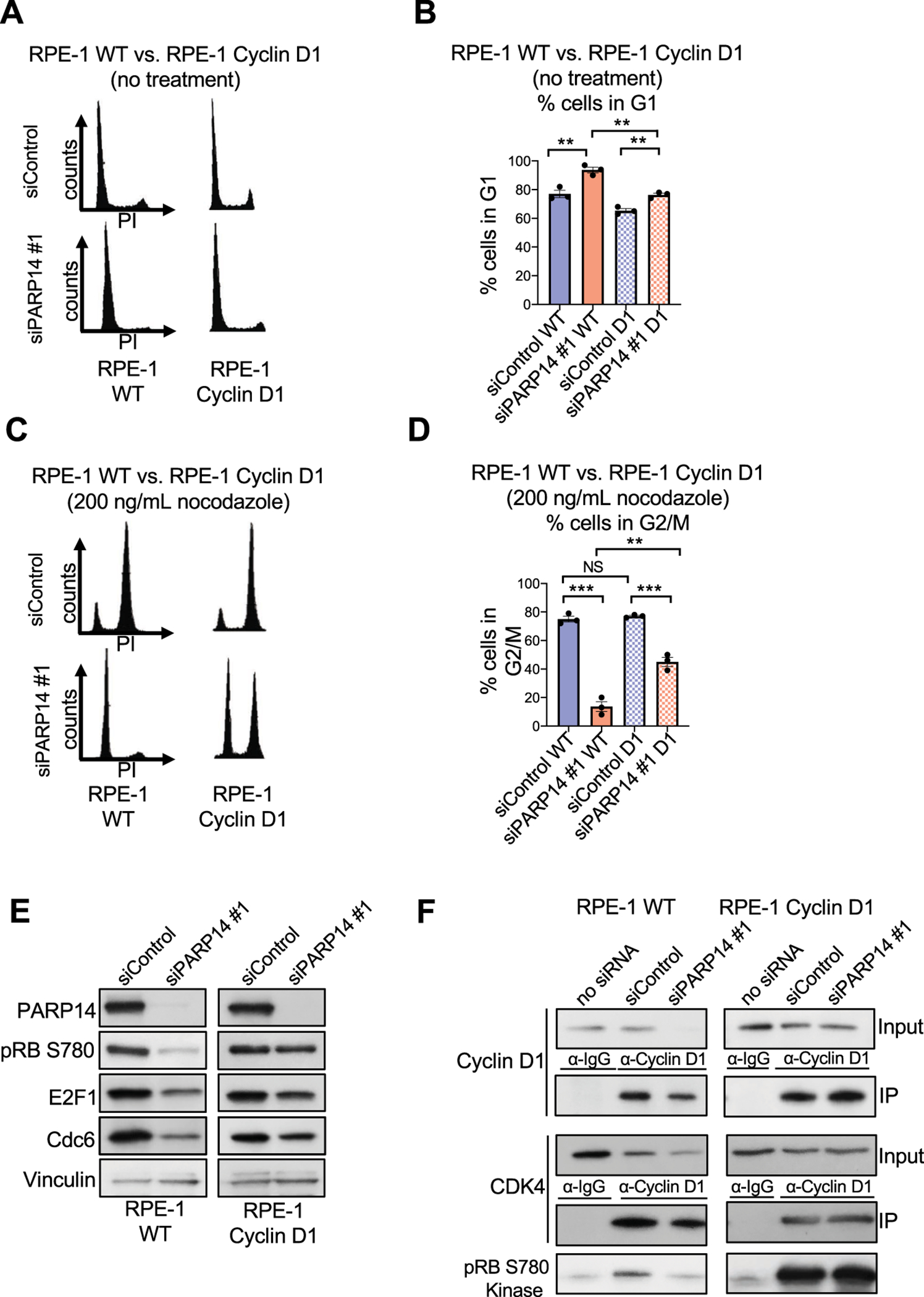
A. Cell cycle profiles of RPE-1 WT and cyclin D1-overexpressing cells upon PARP14 knockdown. B. Quantification showing the percent of RPE-1 WT and cyclin D1-overexpressing cells in G1 cell cycle phase upon PARP14 knockdown. Bars represent the means ± SEM (t-test, unpaired). C. Cell cycle profiles of RPE-1 WT and cyclin D1-overexpressing cells upon PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. D. Quantification showing the percent of RPE-1 WT and cyclin D1-overexpressing cells in G2/M cell cycle phase upon PARP14 knockdown and subsequent treatment with 200ng/mL nocodazole for 24h. Bars represent the means ± SEM (t-test, unpaired). E. Western blots showing the levels of phosphorylated RB at Ser780, E2F1 and Cdc6 upon PARP14 knockdown in RPE-1 WT or cyclin D1- overexpressing cells. To facilitate direct comparison, longer film exposures are shown for the RPE-1 WT samples and shorter film exposures are shown for the RPE-1 cyclin D1-overexpressing samples. F. In vitro kinase assay using recombinant RB as a substrate and Cyclin D1/CDK4 kinase complexes immunoprecipitated from RPE-1 WT or cyclin D1- overexpressing cells upon PARP14 knockdown. To facilitate direct comparison, longer film exposures are shown for the RPE-1 WT samples and shorter film exposures are shown for the RPE-1 cyclin D1-overexpressing samples.
Previously, both the AKT and MEK/ERK pathways have been implicated in regulation of cyclin D1 expression[35, 36]. However, we observed no decrease in p-ERK1/2 or p-AKT levels in PARP14 depleted RPE-1 WT cells (Supplementary Fig. S4H–S4I), indicating that PARP14 is unlikely to function through these pathways to regulate cyclin D1 expression. In fact, the increase in p-AKT levels we observed in PARP14-depleted cells (Supplementary Fig. S4I) is consistent with a recent report that impaired RB phosphorylation activates the AKT pathway [37].
G1 arrest due to PARP14 knockdown in cyclin D1-overexpressing cells is dependent on p53
In cyclin D1-overexpressing cells, PARP14 knockdown had no effect on cyclin D1 levels. However, cyclin D1 overexpression only partially rescued the decrease in RB phosphorylation and G1 cell cycle arrest caused by PARP14 knockdown, indicating the additional involvement of a cyclin D1-independent pathway. We next tested if co-depleting p53 and PARP14 in cyclin D1-overexpressing cells would further alleviate G1 arrest. The partial G1 arrest due to PARP14 knockdown in cyclin D1-overexpressing cells was completely rescued by co-depletion of both PARP14 and p53 (Supplementary Fig. S5A). Moreover, this also rescued the decrease in RB phosphorylation (Supplementary Fig. S5B). We conclude that an intact p53 pathway in cyclin D1-overexpressing cells facilitates the cell cycle arrest induced by PARP14 knockdown.
PARP14 regulates cyclin D1 3’UTR stability
After establishing that PARP14 regulates cyclin D1 mRNA levels, we next asked through what mechanism this was occurring. We used actinomycin D, a potent transcription inhibitor, to determine if PARP14 knockdown affected cyclin D1 mRNA stability. PARP14 knockdown in HeLa cells resulted in a significant decrease in cyclin D1 mRNA levels after actinomycin treatment (Fig. 7A), supporting a role for PARP14 in regulating cyclin D1 mRNA stability. We next investigated if PARP14 controls transcription of cyclin D1. To this end, we performed luciferase reporter assays using a previously described construct in which the cyclin D1 promoter is cloned upstream of the Firefly luciferase gene [38]. We observed no reduction in relative luciferase activity in this assay (Fig. 7B), arguing against a role of PARP14 in promoting cyclin D1 transcription.
Figure 7. PARP14 is required for Cyclin D1 3’UTR mRNA stability.
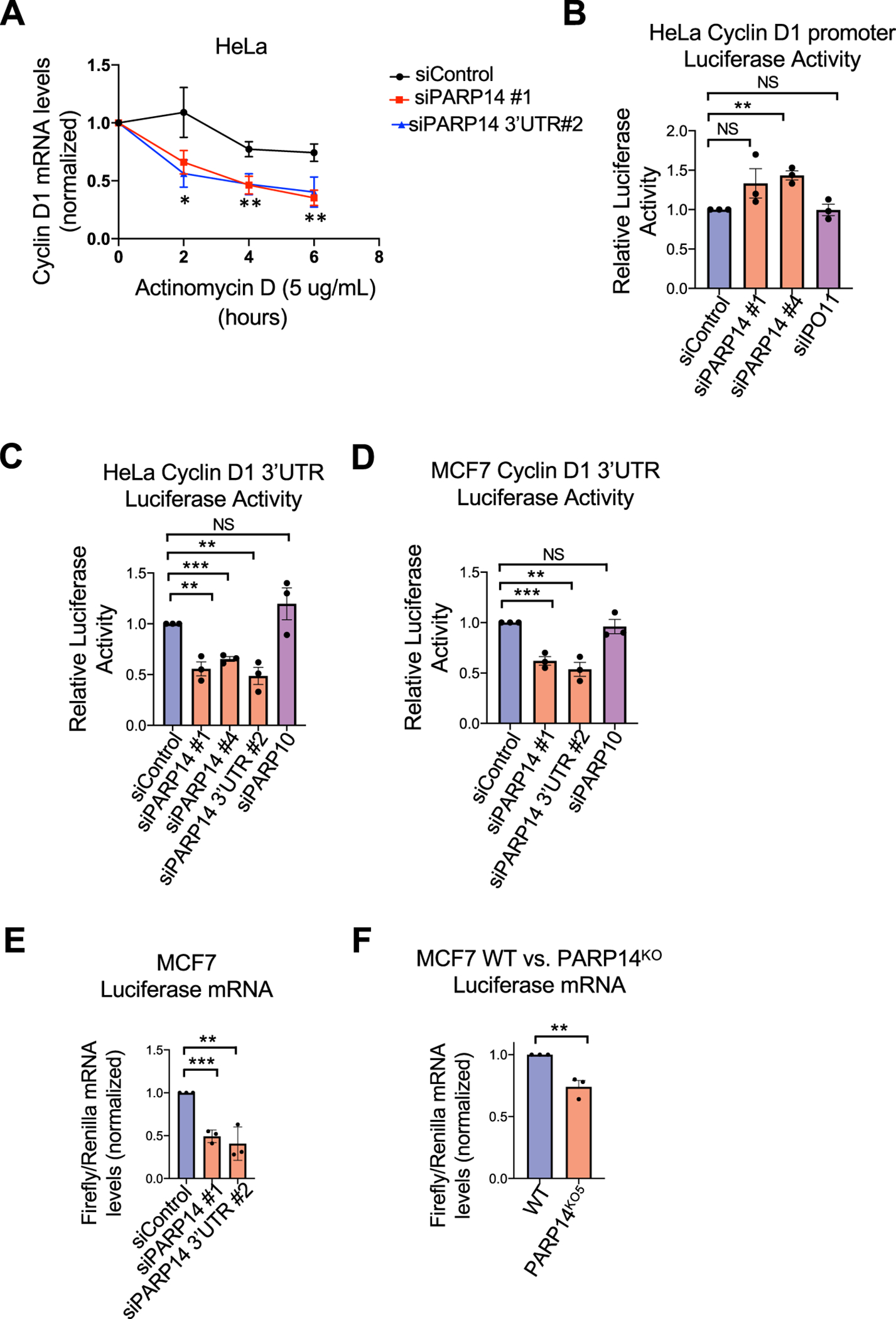
A. Quantification of Cyclin D1 mRNA levels by RT-qPCR, normalized to 18S RNA levels, upon actinomycin treatment of HeLa cells. Bars represent the means ± SEM. Asterisks indicate statistical significance for siPARP14 #1 (t-test, unpaired). B. Cyclin D1 promoter activity luciferase assay in HeLa cells upon PARP14 depletion. IPO11 knockdown was used as an additional negative control. Bars represent the means ± SEM (t-test, unpaired). C, D. Cyclin D1 3’UTR mRNA stability luciferase assays in HeLa (C) and MCF7 (D) cells upon PARP14 depletion. PARP10 knockdown was used as an additional negative control. Bars represent the means ± SEM (t-test, unpaired). E, F. RT-qPCR experiments showing the ratio of Firefly/Renilla mRNA levels in PARP14-knockdown (E) or knockout (F) MCF7 cells. Bars represent the means ± SEM (t-test, unpaired).
We next investigated how PARP14 regulates the stability of cyclin D1 mRNA. mRNA stability is often regulated by the 3’UTR region. To determine if PARP14 affects cyclin D1 mRNA stability, we performed luciferase reporter assays using a previously described construct in which the entire 3’UTR of cyclin D1 was cloned downstream of the Firefly luciferase gene [39]. In both HeLa and MCF7 cells, a marked decrease in luciferase activity was observed upon PARP14 depletion (Fig. 7C–7D). As an additional control, PARP10 knockdown showed no significant change in luciferase activity. To rule out the possibility that the observed effect is caused by a potential impact of PARP14 on mRNA translation, we also quantified the Firefly luciferase mRNA by RT-qPCR. PARP14 knockdown (Fig. 7E) or knockout (Fig. 7F) in MCF7 cells resulted in a significant decrease in Firefly luciferase mRNA. Taken together, these results support the model that PARP14 regulates cyclin D1 mRNA stability via the cyclin D1 3’UTR region (Supplementary Fig. S6).
Discussion
Our work uncovers PARP14 as a novel regulator of cell cycle progression, and suggests that PARP14 acts, at least in part, through stabilization of cyclin D1 mRNA. Through both knockdown and knockout approaches, we show that loss of PARP14 reduces cyclin D1 mRNA levels. PARP14 knockdown reduced both cyclin D1 mRNA and protein levels in multiple cell lines (Fig. 5A, B). PARP14 knockout also reduced cyclin D1 mRNA levels in these cell lines (Fig. 5C), and its impact on protein levels was not examined. Additionally, in luciferase assays, both knockdown and knockout of PARP14 caused reduced cyclin D1 3’UTR mRNA stability (Fig. 7E, F). Importantly, cyclin D1 overexpression markedly rescued both the cell cycle arrest and RB phosphorylation defect caused by PARP14 knockdown (Fig. 6). Overall, these findings suggest that decreased cyclin D1 expression is, at least in part, driving cell cycle arrest in PARP14-deficient cells. Additionally, we demonstrate that both non-transformed epithelial cells and cancer cells depend on PARP14 for the G1-S cell cycle transition (Fig. 1). Knockout of the pocket proteins RB, p107 and p130, completely rescued the cell cycle arrest phenotype caused by PARP14 knockdown, and cancer cells that regulate the G1-S transition independently of pocket proteins do not rely on PARP14 for cell cycle progression (Fig. 3).
While our results indicate a role for cyclin D1 regulation in cell cycle arrest upon loss of PARP14, this does not preclude the possibility that PARP14 also regulates other proteins that are responsible for the G1-S transition, in addition to cyclin D1. In support of this, overexpression of cyclin D1 in RPE-1 cells only partially rescued the cell cycle arrest caused by PARP14 knockdown (Fig. 6). Additionally, RB phosphorylation and expression of E2F1 and its target proteins were only partially restored by cyclin D1 overexpression in PARP14 knockdown cells (Fig. 6). This partial rescue provides a mechanistic explanation for the incomplete cell cycle profile restoration. This also suggests that PARP14 may regulate other cell cycle proteins that are responsible for RB phosphorylation. An obvious candidate was p21, which negatively regulates CDK2, another kinase that phosphorylates RB. An increase in p21 would impair CDK2 kinase activity and thus could partially explain the cell cycle arrest phenotype due to PARP14 knockdown. We observed no change in p21 levels after PARP14 knockdown (Fig. 4). However, we observed that loss of p53 caused a significant, though modest, rescue of cell cycle arrest due to PARP14 knockdown. Additionally, when we specifically depleted p21 with siRNA, we observed a moderate rescue of pRB (Ser780) levels due to PARP14 knockdown on western blot (Fig. 4). Finally, co-depletion of p53 and PARP14 in cyclin D1-overexpressing cells completely alleviated both the G1 arrest phenotype and the decrease in RB phosphorylation due to PARP14 knockdown (Supplementary Fig. S5). While we did not explore the p53/p21-dependent mechanism beyond these experiments, these results warrant further investigation. Our experiments show that loss of p53 has only a minor effect in WT cells depleted of PARP14, but a major effect in cyclin D1-overexpressing cells lacking PARP14. This is likely through the function of p21. How p21 can both positively and negatively regulate CDK4/6 kinase activity and RB phosphorylation is a question that has been studied for decades, without any clear consensus. Indeed, while p27 can function as both an activator and an inhibitor of CDK4/6 kinase activity based on post-translational modifications[34], no such mechanism has currently been reported for p21. Since we observed no change in p53 or p21 levels after PARP14 depletion in our experiments, it was unexpected to observe partial rescue of cell cycle arrest or RB phosphorylation by knocking out p53 or co-depleting p21. Whether PARP14 facilitates a post-translational modification on p21 to facilitate cell cycle progression, or acts on some other pathway that then activates p21, will be the subject of future work.
Our results indicate that that PARP14 affects cyclin D1 expression by regulating its 3’UTR mRNA stability (Fig. 7). Further investigation will determine the exact mechanism through which PARP14 regulates cyclin D1 mRNA. PARP14 has previously been suggested to function as an RNA binding protein [40, 41]. Moreover, PARP14 was shown to impact expression of immune response genes in T helper cells [42]. However, a comprehensive study of PARP14 binding targets or elucidation of its exact functions as an RNA binding protein has not been undertaken. Given that we show that PARP14 regulates the 3’UTR of cyclin D1, and that RNA binding proteins primarily function through their interaction with the 3’UTR of mRNA [43], an intriguing possibility is that PARP14 regulates cyclin D1 levels by specifically binding to the cyclin D1 3’UTR.
Previous work has shown that PARP14 expression correlates with pour prognosis in multiple myeloma [21], pancreatic cancer [44] and hepatocellular carcinoma [45]. Here, we show that PARP14 promotes cell cycle progression, in part through regulating cyclin D1 mRNA stability. Given the importance of the cyclin D1-CDK4/6-RB pathway in tumor growth, and that knockdown of PARP14 impairs breast cancer cell proliferation, further work to determine whether targeting PARP14 in cancer treatment would result in therapeutic efficacy is warranted.
Materials and Methods
Cell Culture
HeLa, BJ, HCT116 WT and p53KO, and all RPE-1 cell lines were grown in DMEM with 10% FBS, pen/strep and glutamine. MCF7 and HCC1395 cells were grown in RPMI with 10% FBS Pen/Strep and glutamine. HeLa cells and BJ cells were obtained from ATCC. RPE-1 WT, D1, and TKO cells were from Hein Te Riele (Netherlands Cancer Institute). RPE-1 p53−/− cells were from Titia de Lange (Rockefeller University). RPE-1 RBKO cells were from Nicholas Dyson (Harvard Medical School). MCF7 cells were from Hong-Gang Wang (Penn State College of Medicine). HCC1395 cells were from Raymond Hohl (Penn State Cancer Institute). HCT116 WT and p53KO cells were from Bert Vogelstein (Johns Hopkins). T47D-R cells were from Khandan Keyomarsi (MD Anderson Cancer Center). To generate the PARP14-knockout lines, the commercially available PARP14 CRISPR/Cas9 KO plasmid was used (Santa Cruz Biotechnology sc-402812). Transfected cells were FACS-sorted into 96-well plates using a BD FACSAria II instrument and resulting monoclonal lines were screened by western blot. HeLa cells stably expressing exogenous PARP14 were obtained upon infection with the lentiviral construct pLV-Puro-SV40>Flag/hPARP14 (Cyagen), constitutively expressing Flag-tagged PARP14 under the control of the SV40 promoter.
For gene knockdowns, cells were transfected using Lipofectamine RNAiMAX (Life Tech) at 40nM final concentration (unless otherwise indicated) for two consecutive days, and harvested for analyses 72 hours after initial siRNA transfection. AllStars Negative Control siRNA (Qiagen, 1027281) was used as control. Oligonucleotide sequences used (Life Tech Stealth, unless otherwise noted) were: siPARP14#1: AGGCCGACUGUGACCAGAUAGUGAA; siPARP14 #2:CGGCACUACACAGUGAACUUGAACA;siPARP14#4: CAAUGCCAAUUAUUCUGCCAAUGAU; siPARP14 3’UTR#1: CAGGAAGGAGAGAAUAACAGUCUUA; siPARP14 3’UTR#2: CCAUCCAAAGUAGGAACUAUCUCUU; siPARP10: GCCUGGUGGAGAUGGUGCUAUUGAU; sip53: CUGGAAGACUCCAGUGGUAAUCUAC; sip21: GAUCUUCUCCAAGAGGAAGCCCUAA; sip27:Santa Cruz, sc-29429; sip107: Santa Cruz, sc-29423; siIPO11: Life Tech Silencer Select s27652.
For cell cycle analyses, cells were fixed in 70% ethanol and stained with propidium iodide. Cell cycle profiles were read on a BD FACS Canto instrument and analyzed using FlowJo software. Apoptosis was measured using the FITC Annexin V kit (Biolegend 640906) according to the manufacturer’s instructions. Cellular proliferation was determined using the CellTiterGlo reagent (Promega).
Protein Techniques
Cell extracts and western blots were performed as previously described [46]. Western blots are representative images of at least two independent experiments. Quantifications of the important western blot results, from multiple independent experiments, using ImageJ software, are presented in the Supplementary Information. Antibodies used were: GAPDH (Santa Cruz, sc-47724), Vinculin (Santa Cruz, sc-73614), Chk1 (Cell Signaling Technology 2360), PARP14 (Santa Cruz, sc-53433), Cyclin D1 (Thermo, MA5–14512; Santa Cruz, sc-8396), Actin (Santa Cruz, sc-4778), pRB Ser780 (BD, 558385; Cell Signaling, 8180T), pRB Thr821/826 (Santa Cruz, sc-271930), RB (Santa Cruz, sc-102), E2F1 (Santa Cruz, sc-251), Erk1/2 (Cell Signaling, 9102S), pErk1/2 (Cell Signaling, 4370S), Akt (Santa Cruz, sc-5298), pAkt Ser473 (Cell Signaling, 9271), Cyclin A (Santa Cruz, sc-239), Cyclin B (Cell Signaling, 4138), Cdc6 (Santa Cruz, sc-9964), CDK4 (Cell Signaling Technology, 12790S), p27 (Santa Cruz, sc-56338), p21 (Cell Signaling Technology, 2947), p53 (Santa Cruz, sc-126), p107 (Cell Signaling Technology, 89798).
For in vitro kinase assays, cells were transfected as indicated and lysed in lysis buffer (50mM HEPES pH 7.5, 150mM NaCl, 1mM EDTA, 1%Triton, 10% glycerol, 10μM MgCl2, with complete Protease inhibitor (Roche), DTT, PMSF, and NaF) for 1 hour at 4°C. Lysates were subjected to immunoprecipitation with Cyclin D1 antibodies (Santa Cruz, sc-8396) and protein A/G resin. After washing, the resin was resuspended in 30μL kinase reaction buffer with phosphatase inhibitors (20mM MOPS, NaF, MgCl2, DTT, glycerol, and sodium orthovanadate); 3μL recombinant RB (Sigma Aldrich, 12–439) and 1μL of 100mM ATP were added, and the mixture was incubated at 30°C for 30 min. Samples were then analyzed by western blot using Cyclin D1 (Thermo, MA5–14512) and pRB Ser780 (Cell Signaling, 8180T) antibodies.
Gene Expression Analyses
Quantification of gene expression by RT-qPCR was performed as previously described [47]. The cDNA of GAPDH gene was used for normalization. Primers used for qPCR were: Cyclin D1 for: AGCTCCTGTGCTGCGAAGTGGAAAC; Cyclin D1 rev: AGTGTTCAATGAAATCGTGCGGGGT; GAPDH for: AAATCAAGTGGGGCGATGCTG; GAPDH rev: GCAGAGATGATGACCCTTTTG.
For 3’UTR mRNA stability luciferase assays, pmirGLO empty (Promega) or pmirGLO Cyclin D1 3’UTR (obtained from Peter Sicinski, Harvard University) was transfected using Lipofectamine Plus with LTX (Life Tech). Firefly and Renilla luciferase activities were measured 24 hours after transfection, using Dual-Glo Luciferase Assay (Promega). The measurements for Firefly luciferase were first normalized to those of Renilla, and then further normalized to empty vector. Transfected cells were also subjected to mRNA extraction and the Firefly and Renilla mRNA levels were detected by RT-qPCR using primer sequences: Firefly for: GTGGTGTTGTGTTCGTGGA; Firefly rev: CGCCCTTCTTGGCCTTAAT; Renilla for: ATGGCTTCCAAGGTGTAC; Renilla rev: TAGTTGATGAAGGAGTCCA.
For cyclin D1 promoter activity luciferase assays, a pGL3Basic plasmid containing the 1748bp cyclin D1 promoter cloned upstream of the Firefly luciferase gene (Addgene #32726) was co-transfected along with pRL-SV40 Renilla Luciferase plasmid. The measurements for Firefly luciferase were normalized to those of Renilla.
For actinomycin experiments, cells were treated as indicated and Cyclin D1 mRNA levels were measured by RT-qPCR. 18S rRNA was used for normalization using the primer sequences: 18S rRNA for: TTCGAACGTCTGCCCTATCAA; 18S rRNA rev: CCACAGTTATCCAAGTAGGAGAGGA.
Statistical Analyses and Data Availability
For all assays, the statistical analysis performed was the t-test (two-tailed, unequal variance). The results presented in all graphs are derived from independent experiments. Statistical significance is indicated for each graph (ns = not significant, for p > 0.05; * for p < 0.05; ** for p < 0.01; *** for p < 0.001; **** for p < 0.0001). All statistical analyses were performed using GraphPad Prism 8 software. All source data underlying each figure, including the values plotted in graphs, the exact p-values, and the uncropped blots are presented in Supplementary Table S1.
Supplementary Material
Acknowledgements
We would like to thank Drs. James Broach, Gregory Yochum, Lisa Shantz, Hein Te Riele, Titia deLange, Nicholas Dyson, Hong-Gang Wang, Raymond Hohl, Myriam Gorospe, Khandan Keyomarsi, and Peter Sicinski for materials and advice, as well as the Penn State College of Medicine Flow Cytometry core for their assistance with flow cytometry experiments. This work was supported by NIH R01ES026184 (to GLM).
Footnotes
Competing Interests
The authors declare no competing interests
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wells J, Graveel CR, Bartley SM, Madore SJ, Farnham PJ. The identification of E2F1-specific target genes. Proc Natl Acad Sci U S A 2002; 99: 3890–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Velez-Cruz R, Johnson DG. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int J Mol Sci 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viatour P, Sage J. Newly identified aspects of tumor suppression by RB. Dis Model Mech 2011; 4: 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 2010; 9: 4153–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016; 375: 1925–1936. [DOI] [PubMed] [Google Scholar]
- 8.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol 2017; 35: 3638–3646. [DOI] [PubMed] [Google Scholar]
- 9.Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N Engl J Med 2019; 381: 307–316. [DOI] [PubMed] [Google Scholar]
- 10.Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev 2016; 30: 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci 2008; 121: 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P et al. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell 1995; 6: 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev 1997; 11: 847–862. [DOI] [PubMed] [Google Scholar]
- 14.Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun 2014; 5: 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carbone M, Rossi MN, Cavaldesi M, Notari A, Amati P, Maione R. Poly(ADP-ribosyl)ation is implicated in the G0-G1 transition of resting cells. Oncogene 2008; 27: 6083–6092. [DOI] [PubMed] [Google Scholar]
- 16.Augustin A, Spenlehauer C, Dumond H, Menissier-De Murcia J, Piel M, Schmit AC et al. PARP-3 localizes preferentially to the daughter centriole and interferes with the G1/S cell cycle progression. J Cell Sci 2003; 116: 1551–1562. [DOI] [PubMed] [Google Scholar]
- 17.Nicolae CM, Aho ER, Choe KN, Constantin D, Hu HJ, Lee D et al. A novel role for the mono-ADP-ribosyltransferase PARP14/ARTD8 in promoting homologous recombination and protecting against replication stress. Nucleic Acids Res 2015; 43: 3143–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goenka S, Boothby M. Selective potentiation of Stat-dependent gene expression by collaborator of Stat6 (CoaSt6), a transcriptional cofactor. Proc Natl Acad Sci U S A 2006; 103: 4210–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vyas S, Chesarone-Cataldo M, Todorova T, Huang YH, Chang P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun 2013; 4: 2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho SH, Goenka S, Henttinen T, Gudapati P, Reinikainen A, Eischen CM et al. PARP-14, a member of the B aggressive lymphoma family, transduces survival signals in primary B cells. Blood 2009; 113: 2416–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbarulo A, Iansante V, Chaidos A, Naresh K, Rahemtulla A, Franzoso G et al. Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene 2013; 32: 4231–4242. [DOI] [PubMed] [Google Scholar]
- 22.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009; 11: R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MS, Helms TL, Feng N, Gay J, Chang QE, Tian F et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016; 7: 39595–39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 2016; 6: 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolay BN, Danielian PS, Kottakis F, Lapek JD Jr., Sanidas I, Miles WO et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev 2015; 29: 1875–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kettner NM, Vijayaraghavan S, Durak MG, Bui T, Kohansal M, Ha MJ et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin Cancer Res 2019; 25: 3996–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stengel KR, Thangavel C, Solomon DA, Angus SP, Zheng Y, Knudsen ES. Retinoblastoma/p107/p130 pocket proteins: protein dynamics and interactions with target gene promoters. J Biol Chem 2009; 284: 19265–19271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farkas T, Hansen K, Holm K, Lukas J, Bartek J. Distinct phosphorylation events regulate p130- and p107-mediated repression of E2F-4. J Biol Chem 2002; 277: 26741–26752. [DOI] [PubMed] [Google Scholar]
- 29.Benedict B, van Harn T, Dekker M, Hermsen S, Kucukosmanoglu A, Pieters W et al. Loss of p53 suppresses replication-stress-induced DNA breakage in G1/S checkpoint deficient cells. Elife 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol 2003; 77: 1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R et al. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene 2005; 24: 2929–2943. [DOI] [PubMed] [Google Scholar]
- 32.Yang Z, Maciejowski J, de Lange T. Nuclear Envelope Rupture Is Enhanced by Loss of p53 or Rb. Mol Cancer Res 2017; 15: 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497–1501. [DOI] [PubMed] [Google Scholar]
- 34.Guiley KZ, Stevenson JW, Lou K, Barkovich KJ, Kumarasamy V, Wijeratne TU et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019; 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muise-Helmericks RC, Grimes HL, Bellacosa A, Malstrom SE, Tsichlis PN, Rosen N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem 1998; 273: 29864–29872. [DOI] [PubMed] [Google Scholar]
- 36.Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta 2007; 1773: 1299–1310. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Xu K, Liu P, Geng Y, Wang B, Gan W et al. Inhibition of Rb Phosphorylation Leads to mTORC2-Mediated Activation of Akt. Mol Cell 2016; 62: 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398: 422–426. [DOI] [PubMed] [Google Scholar]
- 39.Hydbring P, Wang Y, Fassl A, Li X, Matia V, Otto T et al. Cell-Cycle-Targeting MicroRNAs as Therapeutic Tools against Refractory Cancers. Cancer Cell 2017; 31: 576–590 e578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iqbal MB, Johns M, Cao J, Liu Y, Yu SC, Hyde GD et al. PARP-14 combines with tristetraprolin in the selective posttranscriptional control of macrophage tissue factor expression. Blood 2014; 124: 3646–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milek M, Imami K, Mukherjee N, Bortoli F, Zinnall U, Hazapis O et al. DDX54 regulates transcriptome dynamics during DNA damage response. Genome Res 2017; 27: 1344–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riley JP, Kulkarni A, Mehrotra P, Koh B, Perumal NB, Kaplan MH et al. PARP-14 binds specific DNA sequences to promote Th2 cell gene expression. PLoS One 2013; 8: e83127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayr C Regulation by 3’-Untranslated Regions. Annu Rev Genet 2017; 51: 171–194. [DOI] [PubMed] [Google Scholar]
- 44.Yao N, Chen Q, Shi W, Tang L, Fu Y. PARP14 promotes the proliferation and gemcitabine chemoresistance of pancreatic cancer cells through activation of NF-kappaB pathway. Mol Carcinog 2019; 58: 1291–1302. [DOI] [PubMed] [Google Scholar]
- 45.Iansante V, Choy PM, Fung SW, Liu Y, Chai JG, Dyson J et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat Commun 2015; 6: 7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicolae CM, Aho ER, Vlahos AH, Choe KN, De S, Karras GI et al. The ADP-ribosyltransferase PARP10/ARTD10 interacts with proliferating cell nuclear antigen (PCNA) and is required for DNA damage tolerance. J Biol Chem 2014; 289: 13627–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicolae CM, O’Connor MJ, Constantin D, Moldovan GL. NFkappaB regulates p21 expression and controls DNA damage-induced leukemic differentiation. Oncogene 2018; 37: 3647–3656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.