

Abstract
Acute lung injury (ALI) leading to acute respiratory distress syndrome is the major cause of COVID-19 lethality. Cell entry of SARS-CoV-2 occurs via the interaction between its surface spike protein (SP) and angiotensin-converting enzyme-2 (ACE2). It is unknown if the viral spike protein alone is capable of altering lung vascular permeability in the lungs or producing lung injury in vivo. To that end, we intratracheally instilled the S1 subunit of SARS-CoV-2 spike protein (S1SP) in K18-hACE2 transgenic mice that overexpress human ACE2 and examined signs of COVID-19-associated lung injury 72 h later. Controls included K18-hACE2 mice that received saline or the intact SP and wild-type (WT) mice that received S1SP. K18-hACE2 mice instilled with S1SP exhibited a decline in body weight, dramatically increased white blood cells and protein concentrations in bronchoalveolar lavage fluid (BALF), upregulation of multiple inflammatory cytokines in BALF and serum, histological evidence of lung injury, and activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways in the lung. K18-hACE2 mice that received either saline or SP exhibited little or no evidence of lung injury. WT mice that received S1SP exhibited a milder form of COVID-19 symptoms, compared with the K18-hACE2 mice. Furthermore, S1SP, but not SP, decreased cultured human pulmonary microvascular transendothelial resistance (TER) and barrier function. This is the first demonstration of a COVID-19-like response by an essential virus-encoded protein by SARS-CoV-2 in vivo. This model of COVID-19-induced ALI may assist in the investigation of new therapeutic approaches for the management of COVID-19 and other coronaviruses.
Keywords: acute lung injury, COVID-19 murine model, endothelial permeability, SARS-CoV-2, spike protein
INTRODUCTION
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a β-Coronaviridae responsible of the 2020 pandemic. Since its first report in the Wuhan city of China (1), it has infected more than 164,000,000 and provoked the death of over 3.4 million worldwide [COVID-19 Map—Johns Hopkins Coronavirus Resource Center (www.jhu.edu)]. Although vaccination programs have started, significant delays can result from limited stocks, problems with worldwide distribution, and limited compliance. In addition, the emergence of new resistant strains of SARS-CoV-2 could delay the development of herd immunity and further reduce the effectiveness of current vaccines. Despite global efforts toward the development of randomized clinical trials, beneficial pharmaceutical interventions are few (2). It is thus prudent to accelerate investigations of new therapeutic approaches that can reduce mortality worldwide.
The study of SARS-CoV2 pathogenicity is challenging due to limited availability of animal models. Whereas hamsters and monkeys express the angiotensin-converting enzyme 2 receptor (ACE2) (3, 4) that mediates viral cellular entry, wild-type mice, which are more commonly used in basic research, do not (5). Transgenic mice expressing the human ACE2 gene under the control of the cytokeratin 18 promoter (K18-hACE2) have been created and they faithfully reproduce the pathology seen in humans when infected with the live SARS-CoV-2 (5–7). Studies with live SARS-CoV-2 require biosafety level 3 (BSL-3) facilities for personnel protection, thus making them more complicated and available to a limited group of investigators. When infected with live SARS-CoV-2, K18-hACE2 mice exhibit sex- and time-dependent symptomatology and 100% lethality (5–7).
Patients with COVID-19 develop a “cytokine storm” and a sepsis-like condition (8, 9); the contribution of surface elements to the activation of these inflammatory pathways remains unknown. Spike protein (SP) mediates SARS-CoV-2 cell entry by binding to the plasmalemmal ACE2 receptor. SP, a class I viral fusion protein, requires protease cleavage for the activation of its fusion potential (10). SP is composed of two subunits, S1 and S2, that mediate attachment and membrane fusion, respectively (11). Proteases capable of cleaving SP include furin, trypsin, cathepsin, transmembrane protease serine protease-2 (TMPRSS-2), TMPRSS-4, phosphoinositide 5-kinases (PIK5), two pore segment channel 2 (TPC2), and cathepsin L or human airway trypsin-like protease (HAT) (12–15). Following cleavage, S1SP binds ACE2 receptor and promotes the fusion of the viral membrane to the host cell and the subsequent release of genomic material into the cytoplasm (16). The binding to ACE2 also disrupts the renin-angiotensin signaling (17) and causes ACE2 shedding, which is related to acute lung injury (ALI), increased vascular permeability (18), and cytokine production (19).
When injected intraperitoneally, the 2003 SARS-CoV SP exacerbates histologically evident ALI via upregulation of angiotensin II levels (20). Whether SARS-CoV-2 SP or S1SP can induce local or systemic inflammation in vivo, remains unknown. To that end, we have tested the hypothesis that intratracheal instillation of SARS-CoV-2 S1SP in K18-hACE2 mice leads to ALI. For the first time, we report that S1SP, when instilled intratracheally, produces biochemically, immunologically, and histologically evident COVID-19-like ALI, including “cytokine storm.” In agreement with these findings, we also report a direct effect of S1SP on human lung microvascular endothelial cell barrier integrity, in culture. This mouse model is easily reproducible and can be used for the study of new therapeutic approaches to COVID-19 outside BSL3 environments.
MATERIALS AND METHODS
Ethical Statement
All animal studies were approved by the Old Dominion University IACUC and adhere to the principles of animal experimentation as published by the American Physiological Society and under Biosafety Level 2 (BLS-2) and animal BSL2 (Animal Biosafety Level (ABSL)-2) facilities at Frank Reidy Center for Bioelectrics in Norfolk, Virginia.
Materials
Recombinant SARS-CoV-2 Spike Protein, subunit 1 (S1SP) was purchased from RayBiotech (No. 230–011101-100), recombinant SARS-CoV-2 spike protein (SP) was from BEI Resources (No. NR-53257). RIPA (radioimmunoprecipitation assay) buffer and protease inhibitor cocktail were obtained from Sigma-Aldrich Corporation. Socumb (pentobarbital), Anased (xylazine), and Ketaset (ketamine) were supplied by Henry Schein Animal Health. Wright-Giemsa Stain Kit, 10% formaldehyde, 10% SDS, and ammonium persulfate were purchased from Thermo Fisher Scientific; the BCA protein assay kit from Pierce Co.; and EDTA and Western blot membranes from GE Healthcare. ProtoGel (30% acrylamide mix) and tetramethylethylenediamine (TEMED) were from National Diagnostics; Tris–HCl buffer from Teknova; and Protein Dual Color Standards and Tricine Sample Buffer from Bio-Rad Laboratories. All antibodies were purchased from reputable commercial sources and have published immunospecificity data. Rabbit total and phosphorylated IκBα (No. 9242; No. 2859), STAT3 (No. 4904; No. 9145) were obtained from Cell Signaling Technology, Inc.; mouse monoclonal anti-β-actin from Sigma-Aldrich Corporation; and IRDye 800CW Goat anti-rabbit and IRDye 680RD Goat anti-mouse from LI-COR Biosciences.
Animal Model and Treatment Groups
Male C57BL/6J (No. 000664) and K18-hACE2 transgenic (No. 034860) mice (8–10 wk old, 24–28 g body wt; Jackson Laboratories) were housed under pathogen-free conditions.
S1SP (400 µg/kg in 2 mL/kg body wt) was given intratracheally to K18-hACE2 mice. Briefly, mice were anesthetized with intraperitoneal injection of xylazine (6 mg/kg) and ketamine (60 mg/kg). After cleaning and disinfecting the surgical field, a small neck skin incision (∼1 cm) was made, and the salivary glands were separated to visualize the trachea. Mice were suspended vertically from their incisors and a fine (22 G) plastic catheter was advanced from the mouth into the trachea (∼2 cm) in such a way that it could be seen through the walls of the trachea. S1SP was introduced and flushed with 150 µL air as we have previously described (21). The catheter was withdrawn, neck incision closed by surgical adhesive, and the animal was placed in the ventral position in a small chamber on top of a heating pad, under supplemental oxygen (slowly weaned from 100% to 21% O2) and observed for the next 4 h for signs of respiratory distress before being returned to the regular cage. Mice were monitored daily for abnormal physical appearances. Seventy-two hours later, mice were subjected to bronchoalveolar lavage (BAL), analysis of lung tissue and BAL fluid (BALF) and histological evaluation. Controls included administration of SP (400 µg/kg in 2 mL/kg body wt) or saline to K18-hACE2 mice and S1SP to wild-type (WT) mice. They were all examined 72 h after saline, SP, or S1SP administration.
Histopathology and Lung Injury Score
Mice were euthanized and lungs were fixed in upright position. A small transverse incision was made in the middle of the trachea and the lungs were instilled and inflated with 10% formaldehyde solution to a pressure of 15 cmH2O using a 20 G catheter. The trachea was then ligated with suture; the lungs were removed from the thorax and placed in 10% formaldehyde solution for 72 h. Mid-transverse slices were made from the formalin-fixed lung and embedded in paraffin. Sections 5 µm thick were stained with hematoxylin-eosin (H&E). Twenty randomly selected fields from each slide were examined under×10 and ×40 magnification and the lung injury score was computed (22).
Measurement of Total Protein, Cell, and Cytokine Concentration in BALF
BALF was centrifuged at 2,500 g for 10 min, and the supernatant was collected for total protein and cytokine analysis. Total protein concentration was determined using the bicinchoninic acid (BCA) protein assay kit according to manufacturer’s instructions. Cell numbers were measured with a hemocytometer and differential analysis was performed with the Wright-Giemsa stain kit. Quantitative analysis of BALF and serum chemokines and cytokines were performed by the University of Virginia Flow Cytometry Core Facility, using a MILLIPLEX map murine cytokine/chemokine premixed panel (EMD Millipore) using a Luminex MAGPIX system.
Western Blotting
Lung homogenates from frozen lung tissues were prepared by sonication in ice-cold RIPA buffer with protease inhibitor cocktail (100:1 -EMD, Millipore Corporation). Protein lysates were agitated for 3 h at 4°C, and then centrifuged twice at 14,000 g for 10 min. The supernatants were separated from the pellets and total protein concentration was determined by the bicinchoninic acid method (BCA Assay). Equal amounts of protein from the lysates were first mixed with tricine sample buffer 1:1, heat denatured at 95°C for 10 min and separated on a 12% SDS-PAGE gel by electrophoresis. Proteins were then transferred on a nitrocellulose membrane, incubated with primary and secondary antibodies, and detected by digital fluorescence imaging (LI-COR Odyssey CLx). β-actin was used as a loading control. Densitometric quantification of the bands from the blots was performed using ImageJ software (National Institutes of Health).
Ingenuity Pathway Analysis
Chemokine and cytokine genes of interest in BALF were identified using a P < 0.05 threshold as assessed using the unpaired-samples t test. Ingenuity Pathway Analysis (Qiagen) was used for subsequent bioinformatics analysis, which included canonical pathway analysis, disease and function, regulator effects, upstream regulators, and molecular networks. Analyses were restricted accordingly by species (i.e., mouse) and tissue type (i.e., lung).
Endothelial Cell Culture and Barrier Measurements
In‐house harvested and identified human lung microvascular endothelial cells (HLMVECs) were isolated and maintained in M199 media supplemented with 20% FBS and antibiotics/antimycotics as described previously (23). The barrier function of confluent endothelial cell monolayers was estimated using electric cell‐substrate impedance sensing (ECIS) model 1600R ζθ (Applied Biophysics) as previously described (24). Experiments were conducted with cells that had reached a steady‐state resistance of at least 800 Ω.
RESULTS
Spike Protein Induces Alveolar Inflammation and Acute Lung Injury
K18-hACE2 mice instilled with S1SP displayed a rapid and sustained decline in body weight, unlike mice receiving SP or saline. WT type instilled with S1SP exhibited a significant but less pronounced decrease (Fig. 1A). Strong alveolar inflammation was also shown by increased concentrations of leukocytes and proteins in BALF of transgenic mice instilled with the S1SP, and significantly less in WT type receiving S1SP, whereas similar baseline values were observed in K18-hACE2 mice receiving SP or saline (Fig. 1, B and C). Monocytes, macrophages, and neutrophils were primarily increased in S1SP-instilled K18-hACE2, but only neutrophils increased in WT-type mice (Fig. 1D).
Figure 1.
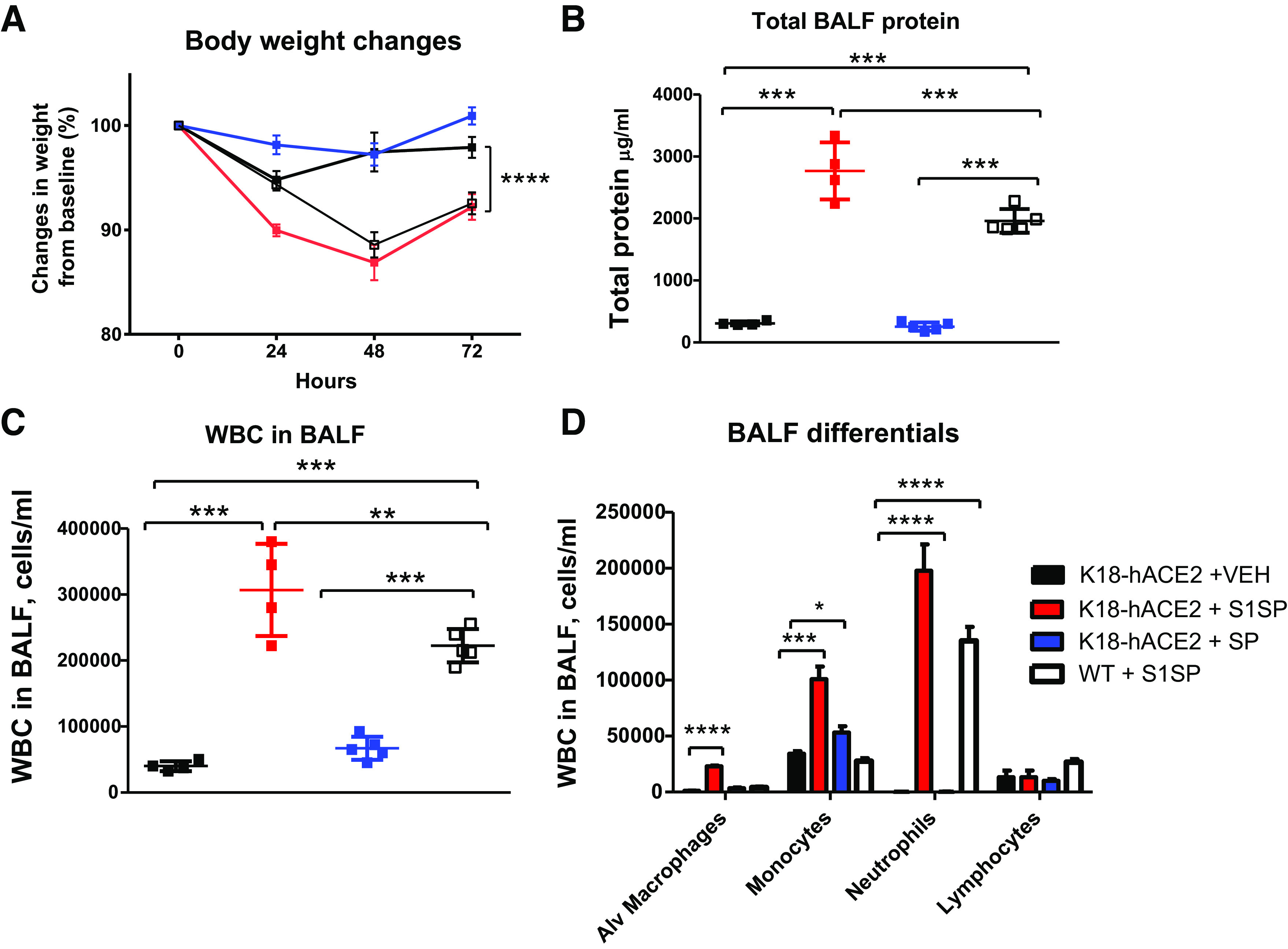
Exposure to SARS-CoV-2 subunit 1 of spike protein (S1SP) induces body weight loss and alveolar inflammation. K18-hACE2 and WT S1SP-instilled mice exhibit weight loss compared with saline controls or SP-instilled K18-hACE2 mice (A). S1SP-instilled mice have elevated protein (B) and leukocyte (C) concentrations in BALF, 72 h after instillation, especially increased monocyte and neutrophil levels that are maximal in the K18-hACE2 strain (D). means ± SE; n = 5 mice/group; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 with one-way ANOVA followed by Tukey’s. BALF, bronchoalveolar lavage fluid; Alv, alveolar; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; VEH, vehicle; SP, spike protein; WBC, white blood cell(s); WT, wild type.
Spike Protein Elicits “Cytokine Storm” in BALF and Serum
Mice instilled with S1SP displayed a cytokine storm in BALF (Fig. 2A) and serum (Fig. 2B), in agreement with the observed neutrophil, monocyte, and macrophage recruitment (Fig. 1D). Minimal cytokine levels were observed in mice exposed to either saline or SP.
Figure 2.
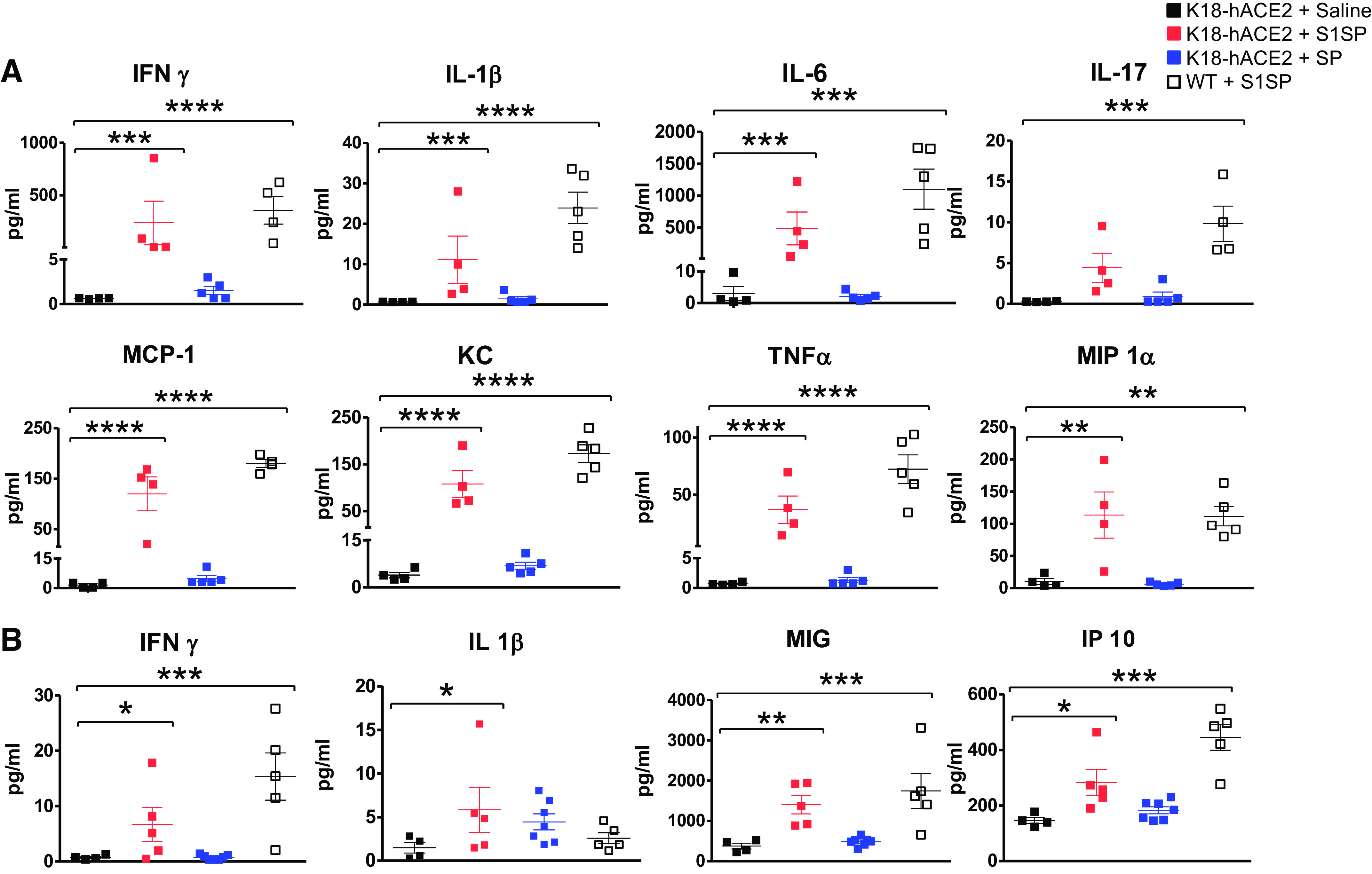
Exposure to S1SP of SARS-CoV-2 induces alveolar and systemic “cytokine storm” in BALF (A) and serum (B) 72 h after instillation.means ± SE; n = 5 mice/group; *P < 0.05; **P < 0.01, ***P < 0.001; ****P < 0.0001 with one-way ANOVA followed by Tukey’s in log-transformed values. BALF, bronchoalveolar lavage fluid; MCP, monocyte chemoattractant protein; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; SP, spike protein; VEH, vehicle; S1SP, subunit 1 of spike protein; KC, keratinocytes-derived chemokine; MIP1α, macrophage inflammatory protein 1-alpha; MIG, gamma interferon; IP 10, interferon gamma-induced protein 10.
Spike Protein Induces Morphologically Evident ALI and Activates the NF-κB and STAT3 Pathways in the Lungs
K18-hACE2 mice instilled with S1SP revealed alveolar septal thickening, alveolar edema, hyaline membranes, and extensive leukocyte infiltrates when compared with saline or SP controls and, to a lesser extent, —WT mice receiving S1SP (Fig. 3A); these observations were confirmed by quantification in the Lung Injury Index (Fig. 3B). Western analysis of lung homogenates demonstrated a dramatical increase in the phosphorylation of STAT3 in K18-hACE2 mice instilled with S1SP compared with all other groups (Fig. 3C), whereas phosphorylation of IκBα (I-kappaB alpha - cytosolic inhibitor of NF-κB) increased in both K18-hACE2 and WT mice instilled with S1SP (Fig. 3C).
Figure 3.
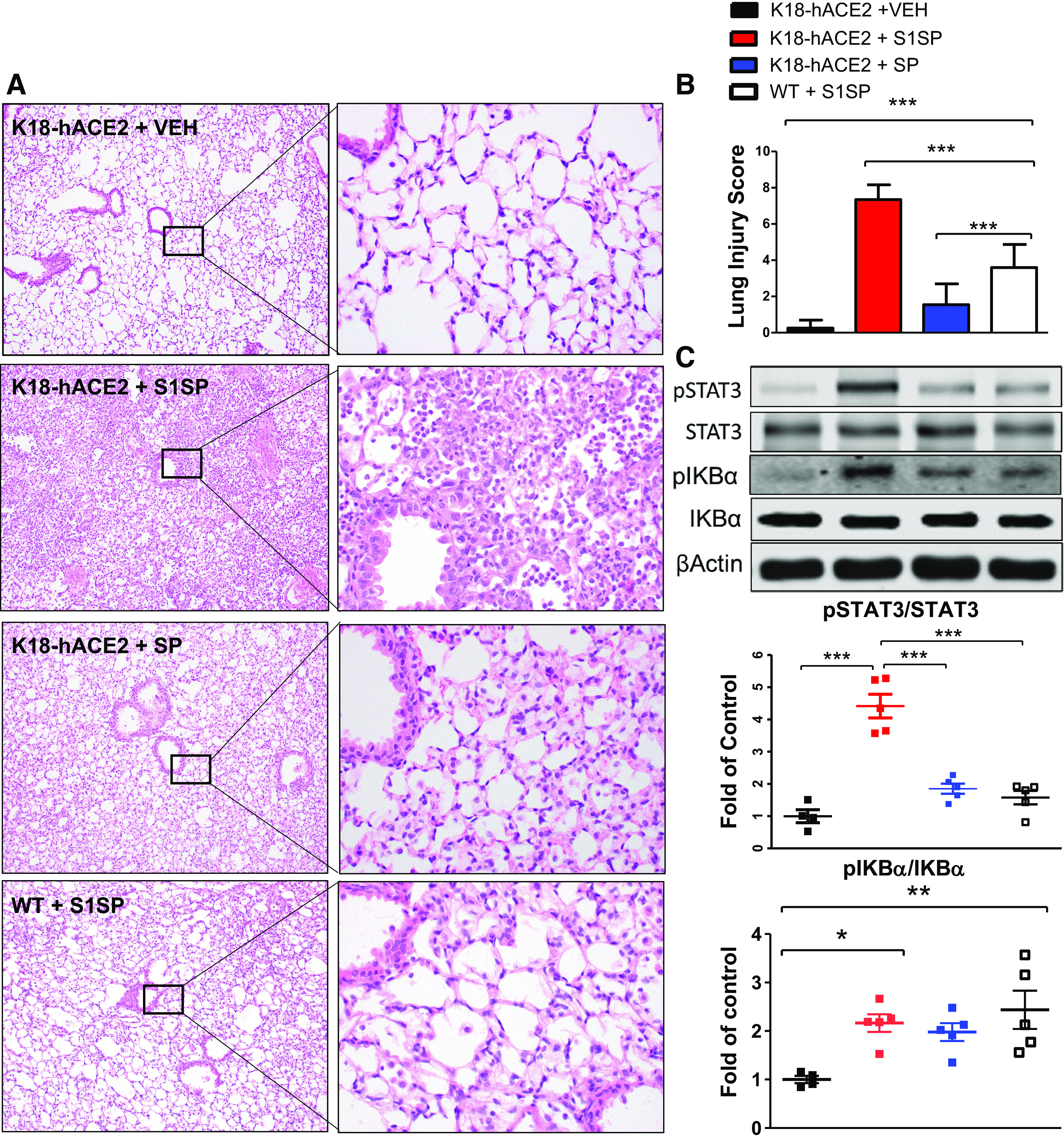
The S1 subunit of the SARS-CoV-2 spike protein (S1SP) causes acute lung injury and the activation of the STAT3 and NF-κB inflammatory pathways 72 h after exposure. A: H&E staining of lung sections demonstrates septal thickening, neutrophil infiltration, and edema in S1SP-instilled K18-hACE2 mice, minimal edema and leucocyte infiltration in S1SP-instilled WT, whereas SP-instilled K18-hACE2 display minimal changes compared with controls. B: the Lung Injury Score quantifies maximal injury in S1SP-instilled K18-hACE2 mice, a milder pathology in S1SP-instilled WT and no significant changes in SP- or saline-instilled K18-hACE2 mice. C: Western blot analysis of lung homogenates revealed increased phosphorylation of I-kappaB alpha (IκBα) and STAT3. Bands were normalized to β-actin and are shown as fold of control. (means ± SE; n = 5 mice/group; *P < 0.05; **P < 0.01; ***P < 0.001 with one-way ANOVA and Tukey’s). H&E, hematoxylin-eosin; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; VEH, vehicle; SP, spike protein; S1SP, subunit 1 of spike protein; WT, wild type.
Ingenuity Pathway Analysis
Ingenuity Pathway Analysis of the significantly upregulated and downregulated cytokines and chemokines in BALF computationally revealed a prominent association of our in vivo model system with diseases and biofunctions aligned with inflammatory disease, and most specifically, lung infection, inflammation, and permeability of microvascular endothelial cells (Fig. 4A).
Figure 4.

A: Ingenuity Pathway Analysis of BALF cytokine and chemokine in mice after S1SP or SP instillation. The statistically significant cytokines were computed into a database for known pathways of inflammation, infection, apoptosis and permeability. B: the S1 subunit of the SARS-CoV-2 spike protein (S1SP) causes a fast concentration- and time-dependent decrease in transendothelial electrical resistance (TER), whereas the intact SP provokes a late and milder decrease in TER of human lung microvascular endothelial cells. n = 4 replicates/time point. ****P < 0.0001 by two-way ANOVA for repeated measures and Tukey’s. Values were normalized to time = 0 for easier comparisons. Starting resistance was >800 Ω. BALF, bronchoalveolar lavage fluid; SP, spike protein; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; CCL, C-C motif chemokine.
Spike Protein Disrupts Pulmonary Microvascular Endothelial Barrier Function
Because S1SP produced alveolar inflammation and edema, we also examined possible direct effects on endothelial barrier dysfunction. In primary human lung microvascular endothelial cells that naturally express ACE2, S1SP—but not the entire, intact SP—produced a profound, concentration-dependent decrease in transendothelial resistance (TER), indicative of concentration dependence increases in permeability and barrier dysfunction (Fig. 4B).
DISCUSSION
The live SARS-CoV-2 can be lethal in K18-hACE2 but not in WT mice and that, like what is observed in patients with COVID-19, infected K18-hACE2 mice exhibit strong inflammation and acute respiratory distress syndrome (ARDS)-like pathology (5). Indeed, critically ill patients with COVID-19 develop a strong inflammatory response, with “cytokine storm” (25). The contribution of individual viral elements to the pathology of COVID-19 remains unclear. The SP of SARS-CoV is proinflammatory in vitro (26). Although inactive by itself, SARS-CoV SP exacerbates lung injury (20). Similarly, S1SP of SARS-CoV-2 is proinflammatory in isolated peritoneal macrophages (27) and in human bronchial epithelial cells (28). However, these data do not necessarily predict COVID-19-like lung injury or systemic inflammation.
Here, we show that the activated (cleaved) form of the SP, S1SP, elicits strong pulmonary and systemic inflammatory responses in K18-hACE2 mice and milder responses in WT mice, whereas the intact SP provokes minimal or no responses.
The cytokine storm observed in this study exhibits critical similarities with the inflammatory response elicited by the live virus in K18-hACE2 mice (5, 29). Hamsters, intratracheally instilled with a pseudovirus expressing SP, display ALI, endothelial dysfunction, and mitochondrial damage mediated by the downregulation of ACE2 (30).
Our results indicate that S1SP can induce a milder form of ALI in WT mice that do not express the human ACE2. Recently, Asandei et al. (31) characterized a non-receptor-mediated mechanism for SARS-CoV-2 S1SP to destabilize cell membranes. Furthermore, the human and mouse peptidase M2 domain of ACE2 that binds S1SP share 85% homology (32). Either or both observations could explain the milder form of COVID-19 induced by S1SP in WT mice. This data suggest that, despite the strong biological activity of S1SP in wild-type animals, the hACE2 receptor is an important requirement for the pathogenesis of COVID-19 and thus, k18-hACE2 mice represent a more valid and trustable model for COVID-19. They further suggest that the SARS-CoV-2 S1SP could play the role of a “toxin” and, when released in the vascular compartment after cell lysis, participate in the pathogenesis of systemic inflammation.
Additional work is required to determine the precise dose-response relationships to S1SP in vivo, define S1SP pathogenicity in different cell lines, and further characterize the diverse pathways of activity between hACE2 expressing and WT mice.
HLMVEC exposed to S1SP displayed a rapid concentration-dependent decrease in TER, whereas the highest dose of SP evoked a late and minimal decrease in resistance. As the SP requires to be cleaved into its subunits to bind the ACE2 receptor, it is not surprising that the intact SP evokes a minimal and late response, which could be delayed due to the time required for the cleavage, and lesser because of the small amounts of proteases present in this in vitro model.
Here, we demonstrate that intratracheal instillation of a single element of SARS-CoV-2, S1SP, in K18-hACE2 transgenic mice induces COVID-19-like local (lung) and systemic inflammatory responses. This model will allow the preclinical investigation of potential countermeasures and of the long-term effects of the inflammatory response provoked by SARS-CoV-2.
GRANTS
This work was supported by the Old Dominion Research Foundation and the Fiske Drug Discovery Laboratory at the University of Virginia.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.S.L., P.E.M., and J.D.C. conceived and designed research; R.M.L.C.B., P.A.S., and E.R.S. performed experiments; R.M.L.C.B., P.A.S., E.R.S., and J.D.C. analyzed data; E.R.S. and J.D.C. interpreted results of experiments; R.M.L.C.B., P.A.S., and E.R.S. prepared figures; R.M.L.C.B. and P.A.S. drafted manuscript; E.R.S., J.S.L., P.E.M., and J.D.C. edited and revised manuscript; R.M.L.C.B., P.A.S., E.R.S., J.S.L., P.E.M., and J.D.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Eastern Virginia Medical School (EVMS), Department of Anatomy and Pathology Laboratory for lung tissue processing and staining. We thank Betsy Gregory for outstanding technical assistance.
REFERENCES
- 1.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270–273, 2020. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horby P, Lim WS, Emberson J, Mafham M, Bell J, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, Chadwick D, Rege K, Fegan C, Chappell LC, Faust SN, Jaki T, Jeffery K, Montgomery A, Rowan K, Juszczak E, Baillie JK, Haynes R, Landray MJ; RECOVERY Collaborative Group. Effect of dexamethasone in hospitalized patients with COVID-19: preliminary report (Preprint). medRxiv, 2020. doi: 10.1101/2020.06.22.20137273. [DOI] [Google Scholar]
- 3.Munster VJ, Feldmann F, Williamson BN, van Doremalen N, Pérez-Pérez L, Schulz J, Meade-White K, Okumura A, Callison J, Brumbaugh B, Avanzato VA, Rosenke R, Hanley PW, Saturday G, Scott D, Fischer ER, de Wit E. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature 585: 268–272, 2020. doi: 10.1038/s41586-020-2324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sia SF, Yan L-M, Chin AWH, Fung K, Choy K-T, Wong AYL, Kaewpreedee P, Perera RAPM, Poon LLM, Nicholls JM, Peiris M, Yen H-L. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583: 834–838, 2020. doi: 10.1038/s41586-020-2342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oladunni FS, Park J-G, Pino PA, Gonzalez O, Akhter A, Allué-Guardia A, et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat Commun 11: 6122, 2020. doi: 10.1038/s41467-020-19891-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583: 830–833, 2020. doi: 10.1038/s41586-020-2312-y. [DOI] [PubMed] [Google Scholar]
- 7.Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, Fox JM, Chen RE, Earnest JT, Keeler SP, Ritter JH, Kang L-I, Dort S, Robichaud A, Head R, Holtzman MJ, Diamond MS. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat Immunol 21: 1327–1335, 2020[Erratum inNat Immunol21: 1470, 2020]. doi: 10.1038/s41590-020-0778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, Zheng M, Sundaram B, Banoth B, Malireddi RKS, Schreiner P, Neale G, Vogel P, Webby R, Jonsson CB, Kanneganti T-D. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184: 149–168.e17, 2021. doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zafer MM, El-Mahallawy HA, Ashour HM. Severe COVID-19 and sepsis: immune pathogenesis and laboratory markers. Microorganisms 9: 159, 2021. doi: 10.3390/microorganisms9010159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ou X, Zheng W, Shan Y, Mu Z, Dominguez SR, Holmes KV, Qian Z. Identification of the fusion peptide-containing region in betacoronavirus spike glycoproteins. J Virol 90: 5586–5600, 2016. doi: 10.1128/JVI.00015-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309: 1864–1868, 2005. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 12.Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci USA 111: 15214–15219, 2014. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann-Winkler H, Thiel V, Pöhlmann S. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87: 6150–6160, 2013. doi: 10.1128/JVI.03372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertram S, Glowacka I, Müller MA, Lavender H, Gnirss K, Nehlmeier I, Niemeyer D, He Y, Simmons G, Drosten C, Soilleux EJ, Jahn O, Steffen I, Pöhlmann S. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J Virol 85: 13363–13372, 2011. doi: 10.1128/JVI.05300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Krämer-Kühl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, von Hahn T, Simmons G, Hofmann H, Pöhlmann S. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol 87: 5502–5511, 2013. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181: 281–292.e6, 2020[Erratum inCell183: 1735, 2020]. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaddam RR, Chambers S, Bhatia M. ACE and ACE2 in inflammation: a tale of two enzymes. Inflamm Allergy Drug Targets 13: 224–234, 2014. doi: 10.2174/1871528113666140713164506. [DOI] [PubMed] [Google Scholar]
- 18.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436: 112–116, 2005. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci USA 105: 7809–7814, 2008. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11: 875–879, 2005. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marinova M, Solopov P, Dimitropoulou C, Colunga Biancatelli RML, Catravas JD. Acute exposure of mice to hydrochloric acid leads to the development of chronic lung injury and pulmonary fibrosis. Inhal Toxicol 31: 147–160, 2019. doi: 10.1080/08958378.2019.1624895. [DOI] [PubMed] [Google Scholar]
- 22.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. ; Acute Lung Injury in Animals Study Group. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catravas JD, Snead C, Dimitropoulou C, Chang ASY, Lucas R, Verin AD, Black SM. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vascul Pharmacol 52: 175–181, 2010. doi: 10.1016/j.vph.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barabutis N, Dimitropoulou C, Gregory B, Catravas JD. Wild-type p53 enhances endothelial barrier function by mediating RAC1 signalling and RhoA inhibition. J Cell Mol Med 22: 1792–1804, 2018. doi: 10.1111/jcmm.13460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rumende CM, Susanto EC, Sitorus TP. The management of cytokine storm in COVID-19. Acta Med Indones 52: 306–313, 2020. [PubMed] [Google Scholar]
- 26.Dosch SF, Mahajan SD, Collins AR. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res 142: 19–27, 2009. doi: 10.1016/j.virusres.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirato K, Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 7: e06187, 2021. doi: 10.1016/j.heliyon.2021.e06187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu AC-Y, Wang G, Reid AT, Veerati PC, Pathinayake PS, Daly K, Mayall JR, Hansbro PM, Horvat JC, Wang F, Wark PA. SARS-CoV-2 Spike protein promotes hyper-inflammatory response that can be ameliorated by Spike-antagonistic peptide and FDA-approved ER stress and MAP kinase inhibitors in vitro (Preprint). bioRxiv, 2020. doi: 10.1101/2020.09.30.317818. [DOI]
- 29.Yinda CK, Port JR, Bushmaker T, Offei Owusu I, Purushotham JN, Avanzato VA, Fischer RJ, Schulz JE, Holbrook MG, Hebner MJ, Rosenke R, Thomas T, Marzi A, Best SM, de Wit E, Shaia C, van Doremalen N, Munster VJ. K18-hACE2 mice develop respiratory disease resembling severe COVID-19. PLoS Pathog 17: e1009195, 2021. doi: 10.1371/journal.ppat.1009195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, Zhang Y, Yin Q, Cho Y, Andrade L, Shadel GS, Hepokoski M, Lei T, Wang H, Zhang J, Yuan JX-J, Malhotra A, Manor U, Wang S, Yuan Z-Y, Shyy JY-J. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res 128: 1323–1326, 2021. doi: 10.1161/CIRCRESAHA.121.318902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asandei A, Mereuta L, Schiopu I, Park J, Seo CH, Park Y, Luchian T. Non-receptor-mediated lipid membrane permeabilization by the SARS-CoV-2 spike protein S1 subunit. ACS Appl Mater Interfaces 12: 55649–55658, 2020. doi: 10.1021/acsami.0c17044. [DOI] [PubMed] [Google Scholar]
- 32.Soldatov VO, Kubekina MV, Silaeva YY, Bruter AV, Deykin AV. On the way from SARS-CoV-sensitive mice to murine COVID-10 model. Res Results Pharmacol 6: 1–7, 2020. doi: 10.3897/rrpharmacology.6.53633. [DOI] [Google Scholar]