

Abstract
Significant differences may exist among different descents, but the current studies are mainly based on European populations. In the present study, we analyzed the population-specific differences of coronary artery disease (CAD) between European and East Asian descents. In stage 1, we identified CAD susceptibility genes by gene-based tests in European and East Asian populations. We identified two novel susceptibility genes for CAD, namely, CUX2 and OAS3. In stage 2, we carried out meta-analyses for the population-specific variants. rs599839 (PSRC1) represented a protective variant for CAD in East Asian populations (ORASN = 0.72. 95% CI: 0.63-0.81) but a risk factor in European populations (OREUR = 1.13, 95% CI: 0.93-1.36). In stage 3, we enriched the risk genes and explored the population-specific differences in Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), regulatory element, tissues, and cell types. In stage 4, in order to predict genes that showed pleiotropic/potentially causal association with CAD, we integrated summary-level data from independent genome-wide association studies (GWAS) and expression quantitative trait loci (eQTLs) by using summary data-based Mendelian randomization (SMR). The results showed that NBEAL1 and FGD6 were population-specific pleiotropic/causal genes. Although some potential mutations and risk genes of CAD are shared, it is still of great significance to elucidate the genetic differences among different populations. Our analysis provides a better understanding of the pathogenic mechanisms and potential therapeutic targets for CAD.
1. Introduction
Coronary artery disease (CAD) remains one of the leading causes of mortality worldwide [1, 2]. Although many efforts have been made to prevent and treat CAD, there is still a long way to go to curb the development of CAD, especially in underdeveloped countries and regions [3]. Epidemiological studies have shown that the occurrence of CAD is caused by both genetic and environmental factors, with gender, age, smoking, drinking, hypertension, dyslipidemia, diabetes, obesity, and mental stress being its potential risk factors [4].
Thanks to genome-wide association studies (GWAS), more than one hundred and sixty CAD susceptibility loci have been identified [5–8]. However, some drawbacks exist using GWAS to identify susceptibility loci. First of all, GWAS only reported the genetic variants significantly correlated with the trait (P < 5E − 08) but seldom considered the variants moderately correlated or uncorrelated with the trait. Secondly, more than 90% of the variants identified by GWAS are located in noncoding regions (introns or intergenic regions) [9, 10]. The function of variants in the regulatory region is still unclear. Thirdly, due to the complex linkage disequilibrium (LD) between pathogenic mutation sites and other SNPs, the genes closest to the lead SNPs in the physical distance are not necessarily the most likely causal genes [11–13]. Finally, GWAS explain only a modest fraction of the missing heritability of human diseases [14, 15]. Therefore, it is far from enough to analyze the risk loci from lead SNPs alone.
Furthermore, significant differences have been found in the risk of CAD among different descents, but the current GWAS is mainly based on European populations [16]. A deeper understanding of the genetic structure of other ethnic groups will lead to the development of drugs for population-specific targets and the precise treatment of patients. Previous studies have used the polygenic risk score (PRS) to assess the differences in clinical risk factors for CAD between Japanese and European populations and found several novel loci [17, 18]. However, European ancestry GWAS from the CARDIoGRAMplusC4D 1000 Genomes meta-analysis of previous studies contains some Asian, Hispanic, and African ancestry, which may confuse population-specific loci [8, 18].
In this study, we used two large-scale GWAS which were made up entirely of European and Japanese ancestry, respectively, to identify risk loci by gene-based tests. By integrating GWAS and eQTL datasets, we comprehensively analyzed population-specific differences of susceptibility loci, genetic variants, tissue, cell lines, regulatory elements, and metabolic pathways. Our study provides new insights into the heterogeneity of CAD in the stratification of different populations.
2. Materials and Methods
2.1. Data Sources
Two large-scale GWAS and 54 eQTLs were used in this study. European ancestry GWAS was obtained from a meta-analysis of 14 GWAS of CAD comprising 22,233 cases and 64,762 controls by the CARDIoGRAMplusC4D Consortium [6]. East Asian ancestry GWAS was obtained from the GWAS Catalog which included 2,808 cases and 7,261 controls [18, 19]. Both of them were each made up of a single population. The corresponding SNPs, effect allele, other allele, effect allele frequency (EAF), beta coefficients (β), standard errors (se), P value, and sample numbers were obtained from the above two datasets. We selected 54 eQTL summary data from the Genotype-Tissue Expression (GTEx, version 7) [13, 20, 21]. The donors were of multiple descents including European (85.3%), African (12.3%), and Asian (1.4%) [21]. All data used in this study are allowed to be available in the public database.
2.2. Identification of Susceptibility Genes
Gene-based association analyses were used to identify risk genes [22]. Compared with the traditional approach based on genome-wide association (P < 5E − 08), the gene-based approach considers the association of traits with all SNPs. Several SNPs in a gene may not be highly associated with traits but may play an important role in traits together. Herein, we defined gene boundaries in this case as ±50 kb of 5′ and 3′UTRs and used VEGAS to calculate the risk statistics of CAD for each gene in GWAS [23]. VEGAS combined information from a full set of SNPs (markers) within a gene and accounts for LD between SNPs by using simulations from the multivariate normal distribution [23].
2.3. Meta-Analysis of Population-Specific Genetic Variants
Polymorphisms of genetic variants were associated with CAD risk. In order to explore population-specific variants, we pooled the published population-specific variants from CAD GWAS with the variants we used the gene-based test to identify. We then reviewed above susceptibility variants in CAD case-control studies in NCBI PubMed. Finally, we carried out meta-analysis and sensitivity tests. Considering the heterogeneity between studies, the I2 statistic was used to evaluate the heterogeneity, and the magnitude of the variation was determined using τ2 [24]. In cases where I2 is greater than 50% and funnel plots were asymmetric, we tended to use random-effects models to combine effect values. The criteria for selecting study variants were that the total number of studies at the variant was greater than five and that β (OR), se, P value, and genetic models were disclosed clearly. The details of studies that met the criteria for meta-analysis are shown in Supplementary Table 1 and Supplementary Table 2.
2.4. Pathway, Regulatory Element, Tissue, and Cell-Type Enrichment Analyses
For susceptibility loci that passed the risk statistics threshold (P < 0.01), we first used hypergeometric distribution test to evaluate whether CAD risk genes (protein-coding genes) were significantly enriched in GO terms and the KEGG pathway [25]. Secondly, in order to test the relationship between highly expressed genes and genetic associations in a specific tissue, gene-property analysis was performed using the average expression of genes per tissue type by MAGMA [26–28]. Thirdly, a gene expression heat map was used to indicate the expression of susceptibility loci in 54 human tissues of GTEx, and the SciPy package of Python helped with displaying hierarchical clustering [21, 28]. Finally, we used GARFIELD (GWAS Analysis of Regulatory or Functional Information Enrichment with LD correction) to enrich the elements in the regulatory region [29]. We assessed the enrichment of association analysis signals in 1005 features extracted from ENCODE, GENCODE, and Roadmap Epigenomics projects, including histone modifications, DNase I hypersensitive sites (DHSs), and transcription factor binding sites [9, 10, 29, 30].
2.5. Identification of Causal Gene Targets
In order to identify genes that showed pleiotropic/causal association with CAD, we integrated summary-level data from independent GWAS and eQTLs to perform summary data-based Mendelian randomization (SMR) [13]. SMR was based on the framework of Mendelian randomization (MR), which could determine whether gene expression (exposure) was related to traits (outcome) [13, 31, 32]. Therefore, the effect of gene expression (x) on trait (y) can be expressed as the ratio of the least-square estimates of y and x on a genetic variant (z), respectively, namely,
| (1) |
In order to test the significance of bxy, the T statistic was designed as
| (2) |
where was the sampling variance of the two-step least-square (2SLS) estimate of bxy and TSMR was a statistic of the approximate χ2 test. For the variants that passed the test threshold, three possible explanations for the association between a trait and gene expression existed, including pleiotropy, causality, and linkage. Since the biological significance of linkage genes might lack value, we used heterogeneity in dependent instruments (HEIDI) to distinguish functional association from linkage subsequently [13]. Rejection of the null hypothesis (PHEIDI < 0.05) indicated that the observed association might be due to two distinct genetic variants in high LD [33]. For each probe in eQTLs, only the top associated cis-eQTL was used as the instrument for the SMR test. Eventually, we analyzed common and specific CAD gene targets in different populations and pinpointed functionally relevant genes adjacent to it on the chromosome.
3. Results
3.1. Novel Susceptibility Loci for CAD
In contrast to the 1000 Genome Project (1KGP) reference panels, LD between SNPs were calculated [34]. By carrying out a gene-based test, 12 and 42 susceptibility genes for CAD passed the FDR threshold (0.05/gene numbers) in the European and East Asian populations, respectively (Supplementary Table 3). Only six of them were shared in different populations (Supplementary Figure 1). The Q − Q plot of the gene-based tests is shown in Supplementary Figure 2. Importantly, we identified a novel locus in the European population (CUX2) and two loci in the East Asian population (CUX2 and OAS3) (Table 1 and Figure 1). CUX2, located on chromosome 12q23.13, participates in the proliferation and differentiation of higher vertebrates [35]. CUX2 is usually expressed in the nervous system, whose disturbance is associated with the occurrence of many neurological diseases [36]. Sinner et al. have shown that CUX2 contributes to atrial fibrillation, which confirms the association between neurological diseases and cardiovascular diseases [37]. In addition to CUX2, which is a shared susceptibility locus in different populations, OAS3 shows its East Asian population specificity (Table 1). OAS3 is one of the key antiviral factors induced by IFN, but it is also related to some characteristic factors of cardiovascular disease [38, 39]. This would help us better understand the complex relationship between CAD and other human diseases.
Table 1.
New loci identified by gene-based test.
| Population | Chr | Gene | nSNPs | Test | Top-SNP | Ref | Alt | EAF | Beta | P value |
|---|---|---|---|---|---|---|---|---|---|---|
| European | 12 | CUX2 | 113 | 1051.29 | rs4766453 | C | T | 0.74 | 0.080 | 1.80E-06 |
| East Asian | 12 | CUX2 | 360 | 6077.64 | rs79105258 | A | C | 0.75 | 0.269 | 6.55E-32 |
| 12 | OAS3 | 336 | 4020.37 | rs3937435 | A | G | 0.65 | -0.138 | 2.04E-12 |
Figure 1.
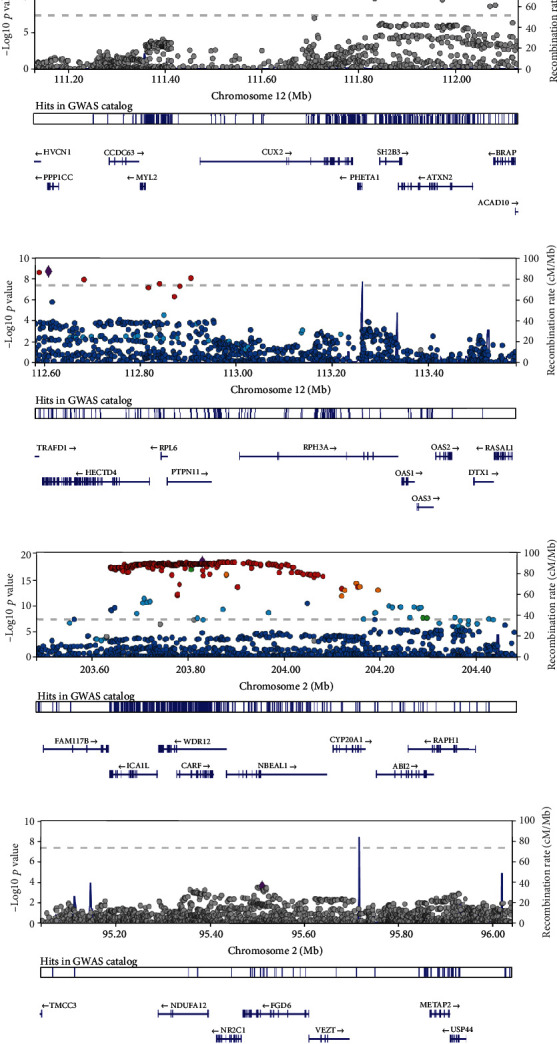
LocusZoom plots for novel loci. The x-axis denotes chromosomal location and the y-axis denotes -log10P value for each SNP. Variants in linkage disequilibrium with the lead variant are shown in red (0.8 < r2 ≤ 1), orange (0.6 < r2 ≤ 0.8), green (0.4 < r2 ≤ 0.6), sky blue (0.2 < r2 ≤ 0.4), light blue (0 < r2 ≤ 0.2), and gray (no r2 data).
3.2. rs599839 Was a Population-Specific Variant of CAD
We integrated the previously published GWAS and our gene-based calculation of Eurasian-specific risk loci, and rs599839 (PSRC1), rs17465637 (MIA3), rs4977574 (CDKN2A/B, ANRIL), and rs1746048 (CXCL12) met the meta-analysis criteria (Supplementary Table 1). We used a total of 28 independent studies from 23 published literatures, all of which tried to use additive models to evaluate the odds ratio (OR) of genetic variants on CAD to minimize heterogeneity. Of the four genetic variants, only rs599839 (PSRC1) showed significant variation among different populations by random-effects models. rs599839 (PSRC1) was a protective variant of CAD in East Asian populations (ORASN = 0.72, 95% CI: 0.63-0.81) but a risk factor for CAD in European populations (OREUR = 1.13, 95% CI: 0.93-1.36) (Figure 2). We found no unacceptable publication bias and heterogeneity through the funnel plot (Supplementary Figure 3). However, the polymorphisms of other variants are positively correlated with the risk of CAD in any population (Supplementary Figure 4, Supplementary Figure 5, and Supplementary Figure 6).
Figure 2.
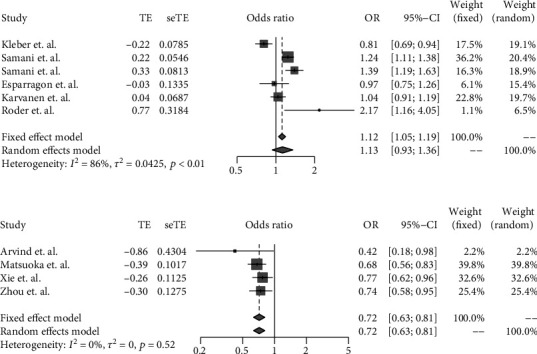
Meta-analysis of the association between rs599839 (PSRC1) and CAD.
3.3. Cholesterol Metabolism Contributed to CAD in Both Populations
We enriched CAD-related genes in the GO and KEGG pathway and found the evidence of differences among different populations from biological process, cellular component, and molecular function. The CAD-related genes of the European population were mainly enriched in plasma lipoprotein and serine activity pathways, while the East Asian population in triglyceride-rich lipoprotein particle pathways (Figure 3). Only cholesterol metabolism contributed to CAD in both populations (Figure 3). High total cholesterol in the blood, high low-density lipoprotein cholesterol (LDL-C), and low high-density lipoprotein cholesterol (HDL-C) were considered to be important risk factors for CAD [40]. For the tissue-specific expression analysis (TSEA), risk genes of the East Asian population were significantly expressed in cervix uteri (Supplementary Figure 7 and Supplementary Figure 8). No specific distributions were enriched in the European population, and the top three were the heart, blood vessel, and brain (Supplementary Figure 7).
Figure 3.
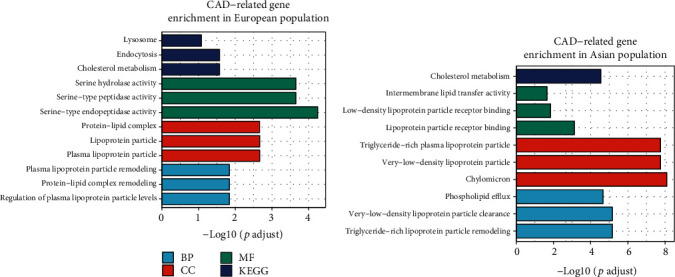
GO and KEGG enrichment of CAD-related genes.
3.4. Regulatory Element Enrichment Analysis in Noncoding Regions
In order to explore how noncoding region variants regulated the occurrence of CAD in different populations, we performed the enrichment of regulatory elements in DHSs, histone modifications, and transcription factor binding sites (Figure 4) [29]. We found that CAD susceptibility sites (P < 0.001) were significantly enriched in DHS of blood cells (OREUR = 2.69, PEUR = 0.036; ORASN = 1.38, PASN = 4.5E − 04) and blood vessels (OREUR = 3.05, PEUR = 0.016; ORASN = 1.40, PASN = 4.8E − 04) in different lineages (Figure 3). Surprisingly, DHSs in skin tissues also seemed to play an important role in the pathogenesis of CAD (OREUR = 6.05, PEUR = 8.0E − 05; ORASN = 1.34, PASN = 4.3E − 04). It was a novel view building a connection between skin and CAD.
Figure 4.
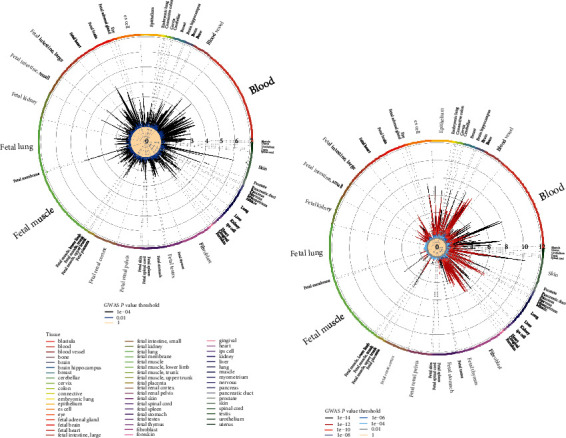
Regulatory element enrichment analysis of CAD susceptibility loci located in noncoding regions: (a) European; (b) Asian. ENCODE and Roadmap Epigenomics DNase I hypersensitive cell lines were enriched. The size of the letters outside the circle is proportional to the number of cell lines, and the different colors inside the circle are the thresholds of different P values of GWAS.
Besides DHSs, blood was also rich in transcription factor binding sites (Supplementary Figure 9). However, other regulatory elements had significant population-specific differences. Genetic annotations showed that CAD variants in the European population were mainly enriched in 3′UTR and 5′UTR, while those in the East Asian population were significantly enriched in exons (Supplementary Figure 10). Generally, the significance of enrichment (P value) in the East Asian population was higher than that in the European population (Supplementary Figure 11). More details were presented in the Supplementary Table 4-5.
3.5. Population-Specific Differences of Causal Genes
We selected five related eQTLs, including artery coronary (22950 probes), artery aorta (22366 probes), blood (19432 probes), heart atrial appendage (21733 probes), and heart left ventricle (20155 probes) tissues, for SMR analysis in the European and East Asian populations. We mapped all the susceptibility loci to eQTL target genes in cis-regions and then linked them to CAD. Manhattan plots of SMR tests for association between gene expression and CAD could be found in Supplementary Figure Figure 12. In order to test the significance of each probe, the FDR threshold was set to 0.05/probe numbers. In the above 10 studies, only NBEAL1 (PSMR = 8.42E − 06, PHEIDI = 0.53) in the European population and FGD6 (PSMR = 5.70E − 06, PHEIDI = 0.20) in the Asian population passed the threshold of the χ2 test and HEIDI test (Figure 5 and Table 2). In other words, NBEAL1 and FGD6 were population-specific pleiotropic/causal genes of CAD (regional plots for them are shown in Figure 1). In addition, PHACTR1, ADAMTS7, RPH3A, ABO, RP11-378J18.8, and RP1-257A7.5 were linkage genes (two distinct causal variants in top-associated cis-eQTL concurrently, one affecting gene expression and the other affecting trait variation).
Figure 5.
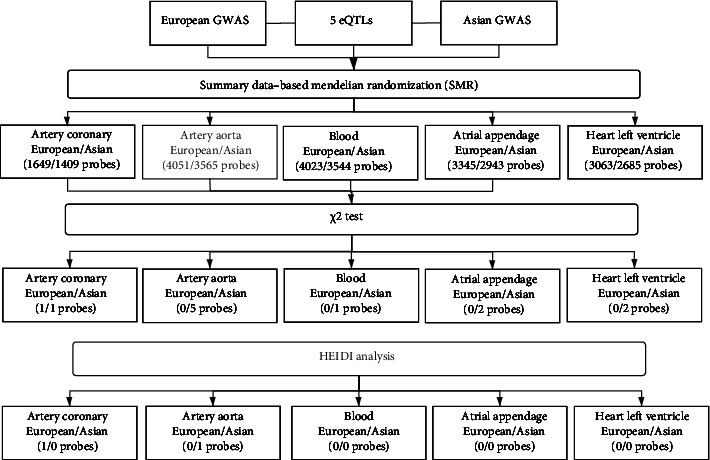
The experimental process of SMR. SMR analyses were performed between 5 eQTLs and GWAS from European and Asian populations, respectively. The probes were screened by significance test and HEIDI analysis.
Table 2.
Genes that are causally associated with the risk of CAD.
| Population | eQTL | Probe ID | CHR | Gene | Top SNP | p_GWAS | p_eQTL | b_SMR | p_SMR | p_HEIDI |
|---|---|---|---|---|---|---|---|---|---|---|
| European | Artery coronary | ENSG00000144426.14 | 2 | NBEAL1 | rs2351524 | 0.4860822 | 2.77E-09 | -0.314 | 8.42E-06 | 0.533 |
| East Asian | Artery aorta | ENSG00000272750.1 | 1 | RP11-378J18.8 | rs2291832 | 3.672E-10 | 2.01E-30 | -0.161 | 3.79E-08 | 4.54E-04 |
| ENSG00000112137.12 | 6 | PHACTR1 | rs9349379 | 1.177E-13 | 1.68E-11 | -0.616 | 6.12E-07 | 1.72E-07 | ||
| ENSG00000272379.1 | 6 | RP1-257A7.5 | rs9349379 | 1.177E-13 | 4.01E-10 | -0.576 | 1.72E-06 | NA | ||
| ENSG00000180263.9 | 12 | FGD6 | rs7954260 | 8.589E-08 | 1.49E-17 | 0.228 | 5.70E-06 | 0.198 | ||
| ENSG00000136378.10 | 15 | ADAMTS7 | rs7173743 | 2.741E-08 | 4.94E-13 | -0.329314 | 1.06E-05 | 0.00590 | ||
| Artery coronary | ENSG00000272750.1 | 1 | RP11-378J18.8 | rs17163358 | 1.854E-09 | 2.78E-10 | -0.202 | 1.32E-05 | 0.00121 | |
| Blood | ENSG00000089169.10 | 12 | RPH3A | rs7979186 | 5.75E-09 | 7.75E-55 | -0.150 | 4.95E-08 | 6.11E-04 | |
| Heart atrial appendage | ENSG00000272750.1 | 1 | RP11-378J18.8 | rs61824331 | 3.534E-08 | 3.07E-20 | -0.172 | 2.20E-06 | 0.0129 | |
| ENSG00000175164.9 | 9 | ABO | rs643434 | 5.315E-08 | 5.00E-22 | 0.170 | 2.20E-06 | 2.07E-05 | ||
| Heart left ventricle | ENSG00000272750.1 | 1 | RP11-378J18.8 | rs2291832 | 3.672E-10 | 1.41E-23 | -0.155 | 1.08E-07 | 3.71E-04 | |
| ENSG00000175164.9 | 9 | ABO | rs657152 | 6.34E-09 | 6.84E-20 | 0.214 | 9.14E-07 | 1.053E-05 |
NBEAL1 was known as a susceptibility locus of CAD, which was highly expressed in the heart and artery [41]. It affected cholesterol metabolism and LDL uptake by regulating the activity of SREBP2 in cells and then affected the pathogenesis of CAD afterwards [42]. However, FGD6 might be a novel CAD susceptibility locus, which regulated the proangiogenic activity in vitro. For NBEAL1 and FGD6, the significant population-specific genes, we further analyzed their regulation mechanism on CAD. Functionally relevant genes and regulatory genes adjacent to it were pinpointed on the chromosome (Supplementary Figure 13). The effect sizes of SNPs showed that the overexpression of NBEAL1 was associated with the increased risk of CAD, while overexpression of FGD6 was associated with decreased CAD level (Supplementary Figure 14). It was worth noting that the top SNP of NBEAL1 (rs2351524) failed to pass the genome-wide association level (P < 5E − 08). Thus, we lent support to the theory that genetic variants and genes with low associations with traits might affect traits as well.
4. Discussion
In this study, the population-specific differences of CAD were revealed from various aspects, including susceptibility loci, risk genetic variants, biological pathways, and regulatory elements in the noncoding region. The differences in CAD between the European population and the East Asian population were obvious from every aspect. We provided new insights from the perspective of coding and noncoding regions at the same time, which were more novel than previous studies.
Sex and ancestry were the potential causes of CAD differences [18, 43–45]. However, previous studies were limited to small sample sizes, which made it difficult to overcome the defects of heterogeneity. Therefore, our study had many advantages. First of all, we selected two summary-level data from independent GWAS, both of which were each composed of the same ancestry. Mixing different lineages together for meta-analysis might blur the differences among different populations. Secondly, we used a gene-based test to obtain the risk loci effectively that were often overlooked in GWAS (P > 5E − 08) and identified two novel susceptibility loci for CAD (CUX2, OAS3). Finally, we fully revealed the population-specific differences of CAD from various perspectives.
However, our study also had certain limitations. The lack of large-scale GWAS in East Asia led to only the Japanese ancestry being used to replace the East Asian ancestry. In addition, the donors of the GTEx project were of multiple descents including European (85.3%), African (12.3%), and Asian (1.4%) [21]. We expect more non-European eQTL studies to complement our deficiencies. Secondly, little evidence showed that the two novel susceptibility loci (CUX2 and OAS3) contributed to CAD in previous studies, so further biological experiment verification was necessary. Finally, SMR analysis had difficulty in distinguishing causal genes from pleiotropic genes. NBEAL1 and FGD6, the population-specific pleiotropic/causal genes for CAD, needed further exploration.
5. Conclusions
In this study, we integrated multiple-omics data to mine the population-specific differences for CAD. We provided a new insight into the genetic mechanism of nonwhite and genetic evidence for CAD precision medicine in different populations. We call for more large-scale GWAS research on CAD with different lineages to achieve accurate treatment of specific gene targets, especially those of non-European lineages that have been previously neglected.
Acknowledgments
We thank the CARDIoGRAMplusC4D Consortium and GWAS Catalog for the GWAS summary statistics. Data on coronary artery disease have been contributed by CARDIoGRAMplusC4D investigators and have been downloaded from http://www.CARDIOGRAMPLUSC4D.ORG/. We thank GTEx for the eQTL dataset resources. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. We acknowledge Dr. Zhou Wenyang for his help on Figure 4. We thank Zhixuan Zhu from the University of Manchester for her help on the manuscript. This work was supported by the National Key R&D Program of China (2017YFC1201201, 2018YFC0910504, and 2017YFC0907503), the Natural Science Foundation of China (61801147 and 82003553), and the Heilongjiang Postdoctoral Science Foundation (LBH-Z6064).
Data Availability
All data generated or analyzed during this study are included in this article.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Yang Hu and Shizheng Qiu contributed equally to this work.
Supplementary Materials
Supplementary Figure 1: Venn diagram of the number of risk genes in the European population and East Asian population. Supplementary Figure 2: Q − Q plot of the gene-based test computed by MAGMA. Supplementary Figure 3: funnel plot of meta-analysis of rs599839 (PSRC1). Supplementary Figure 4: meta-analysis of the association between rs17465637 (MIA3) and CAD. Supplementary Figure 5: meta-analysis of the association between rs4977574 (CDKN2A/B, ANRIL) and CAD. Supplementary Figure 6: meta-analysis of the association between rs1746048 (CXCL12) and CAD. Supplementary Figure 7: relationship between highly expressed genes in a specific tissue and genetic associations. Supplementary Figure 8: gene expression heat map by hierarchical clustering. Supplementary Figure 9: transcription factor binding site enrichment analysis of CAD susceptibility loci. Supplementary Figure 10: genetic annotation analysis of CAD susceptibility loci. Supplementary Figure 11: histone modification enrichment analysis of CAD susceptibility loci. Supplementary Figure 12: Manhattan plots of SMR tests for association between gene expression and CAD. Supplementary Figure 13: prioritizing genes at GWAS loci using SMR analysis. Supplementary Figure 14: effect sizes of SNPs from GWAS plotted against those for SNPs from eQTL studies. Supplementary Table 1: integrating the population-specific genetic variants reported before and identified by a gene-based test in the present study. Supplementary Table 2: meta-analysis of population-specific genetic variants. Supplementary Table 3: identification of CAD risk genes by using VEGAS. Supplementary Table 4: the results of regulatory element enrichment analysis for CAD in the European population. Supplementary Table 5: the results of regulatory element enrichment analysis for CAD in the Asian population.
References
- 1.Roth G. A., Forouzanfar M. H., Moran A. E., et al. Demographic and epidemiologic drivers of global cardiovascular mortality. The New England Journal of Medicine. 2015;372(14):1333–1341. doi: 10.1056/NEJMoa1406656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Writing Group Members, Mozaffarian D., Benjamin E. J., et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 3.Tunstall-Pedoe H., Kuulasmaa K., Mähönen M., Tolonen H., Ruokokoski E., Amouyel P. Contribution of trends in survival and coronary-event rates to changes in coronary heart disease mortality: 10-year results from 37 WHO MONICA Project populations. Lancet. 1999;353(9164):1547–1557. doi: 10.1016/S0140-6736(99)04021-0. [DOI] [PubMed] [Google Scholar]
- 4.Khera A. V., Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nature Reviews Genetics. 2017;18(6):331–344. doi: 10.1038/nrg.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Harst P., Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circulation Research. 2018;122(3):433–443. doi: 10.1161/CIRCRESAHA.117.312086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schunkert H., König I. R., Kathiresan S., et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nature Genetics. 2011;43(4):333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The CARDIoGRAMplusC4D Consortium, DIAGRAM Consortium, CARDIOGENICS Consortium, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature Genetics. 2013;45(1):25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikpay M., Goel A., Won H. H., et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nature Genetics. 2015;47(10):1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernstein B. E., Stamatoyannopoulos J. A., Costello J. F., et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature Biotechnology. 2010;28(10):1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smemo S., Tena J. J., Kim K. H., et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claussnitzer M., Dankel S. N., Kim K. H., et al. FTO obesity variant circuitry and adipocyte Browning in humans. The New England Journal of Medicine. 2015;373(10):895–907. doi: 10.1056/NEJMoa1502214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Z., Zhang F., Hu H., et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nature Genetics. 2016;48(5):481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 14.Tam V., Patel N., Turcotte M., Bosse Y., Pare G., Meyre D. Benefits and limitations of genome-wide association studies. Nature Reviews Genetics. 2019;20(8):467–484. doi: 10.1038/s41576-019-0127-1. [DOI] [PubMed] [Google Scholar]
- 15.Manolio T. A., Collins F. S., Cox N. J., et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes L. O., Raval U., Raftery E. B. First myocardial infarctions in Asian and white men. BMJ. 1989;298(6684):1345–1350. doi: 10.1136/bmj.298.6684.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koyama S., Ito K., Terao C., et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nature Genetics. 2020;52(11):1169–1177. doi: 10.1038/s41588-020-0705-3. [DOI] [PubMed] [Google Scholar]
- 18.Matsunaga H., Ito K., Akiyama M., et al. Transethnic meta-analysis of genome-wide association studies identifies three new loci and characterizes population-specific differences for coronary artery disease. Circulation: Genomic and Precision Medicine. 2020;13(3, article e002670) doi: 10.1161/CIRCGEN.119.002670. [DOI] [PubMed] [Google Scholar]
- 19.Buniello A., MacArthur J. A. L., Cerezo M., et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research. 2019;47(D1):D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium G. T. Erratum: genetic effects on gene expression across human tissues. Nature. 2018;553(7689):p. 530. doi: 10.1038/nature25160. [DOI] [PubMed] [Google Scholar]
- 21.GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neale B. M., Sham P. C. The future of association studies: gene-based analysis and replication. American Journal of Human Genetics. 2004;75(3):353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J. Z., McRae A. F., Nyholt D. R., et al. A versatile gene-based test for genome-wide association studies. American Journal of Human Genetics. 2010;87(1):139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quinn K. L., Shurrab M., Gitau K., et al. Association of receipt of palliative care interventions with health care use, quality of life, and symptom burden among adults with chronic noncancer illness: a systematic review and meta-analysis. JAMA. 2020;324(14):1439–1450. doi: 10.1001/jama.2020.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Research. 2017;45(W1):W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Leeuw C. A., Mooij J. M., Heskes T., Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLOS Computational Biology. 2015;11(4, article e1004219) doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe K., Taskesen E., van Bochoven A., Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nature Communications. 2017;8(1):p. 1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe K., Umicevic Mirkov M., de Leeuw C. A., van den Heuvel M. P., Posthuma D. Genetic mapping of cell type specificity for complex traits. Nature Communications. 2019;10(1):p. 3222. doi: 10.1038/s41467-019-11181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.UK10K Consortium, Iotchkova V., Ritchie G. R. S., et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nature Genetics. 2019;51(2):343–353. doi: 10.1038/s41588-018-0322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thurman R. E., Rynes E., Humbert R., et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davey Smith G., Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Molecular Genetics. 2014;23(R1):R89–R98. doi: 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu S., Cao P., Guo Y., Lu H., Hu Y. Exploring the causality between hypothyroidism and non-alcoholic fatty liver: a Mendelian randomization study. Frontiers in Cell and Development Biology. 2021;9:p. 643582. doi: 10.3389/fcell.2021.643582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu D., Yang J., Feng B., Lu W., Zhao C., Li L. Mendelian randomization analysis identified genes pleiotropically associated with the risk and prognosis of COVID-19. The Journal of Infection. 2021;82(1):126–132. doi: 10.1016/j.jinf.2020.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.1000 Genomes Project Consortium, Abecasis G. R., Altshuler D., et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiefer A. K., Tung J. Y., Do C. B., et al. Genome-wide analysis points to roles for extracellular matrix remodeling, the visual cycle, and neuronal development in myopia. PLoS Genet. 2013;9(2, article e1003299) doi: 10.1371/journal.pgen.1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marin O. The neuron family tree remodelled. Nature. 2012;490(7419):185–186. doi: 10.1038/490185a. [DOI] [PubMed] [Google Scholar]
- 37.Sinner M. F., Tucker N. R., Lunetta K. L., et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. 2014;130(15):1225–1235. doi: 10.1161/CIRCULATIONAHA.114.009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Middelberg R. P., Ferreira M. A., Henders A. K., et al. Genetic variants in LPL, OASL and TOMM40/APOE-C1-C2-C4 genes are associated with multiple cardiovascular-related traits. BMC Medical Genetics. 2011;12(1):p. 123. doi: 10.1186/1471-2350-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The GenOMICC Investigators, The ISARIC4C Investigators, The COVID-19 Human Genetics Initiative, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591(7848):92–98. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 40.Wilson P. W. Changing cholesterol levels and coronary heart disease risk. Circulation. 2016;133(3):239–241. doi: 10.1161/CIRCULATIONAHA.115.020380. [DOI] [PubMed] [Google Scholar]
- 41.Hixson J. E., Jun G., Shimmin L. C., et al. Whole exome sequencing to identify genetic variants associated with raised atherosclerotic lesions in young persons. Scientific Reports. 2017;7(1):p. 4091. doi: 10.1038/s41598-017-04433-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bindesboll C., Aas A., Ogmundsdottir M. H., et al. NBEAL1 controls SREBP2 processing and cholesterol metabolism and is a susceptibility locus for coronary artery disease. Scientific Reports. 2020;10(1):p. 4528. doi: 10.1038/s41598-020-61352-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waheed N., Elias-Smale S., Malas W., et al. Sex differences in non-obstructive coronary artery disease. Cardiovascular Research. 2020;116(4):829–840. doi: 10.1093/cvr/cvaa001. [DOI] [PubMed] [Google Scholar]
- 44.Narasimhan S., McKay K., Bainey K. R. Coronary artery disease in south Asians. Cardiology in Review. 2012;20(6):304–311. doi: 10.1097/CRD.0b013e3182532286. [DOI] [PubMed] [Google Scholar]
- 45.Gijsberts C. M., den Ruijter H. M., Asselbergs F. W., Chan M. Y., de Kleijn D. P. V., Hoefer I. E. Biomarkers of coronary artery disease differ between Asians and Caucasians in the general population. Global Heart. 2020;10(4):301–311. doi: 10.1016/j.gheart.2014.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Venn diagram of the number of risk genes in the European population and East Asian population. Supplementary Figure 2: Q − Q plot of the gene-based test computed by MAGMA. Supplementary Figure 3: funnel plot of meta-analysis of rs599839 (PSRC1). Supplementary Figure 4: meta-analysis of the association between rs17465637 (MIA3) and CAD. Supplementary Figure 5: meta-analysis of the association between rs4977574 (CDKN2A/B, ANRIL) and CAD. Supplementary Figure 6: meta-analysis of the association between rs1746048 (CXCL12) and CAD. Supplementary Figure 7: relationship between highly expressed genes in a specific tissue and genetic associations. Supplementary Figure 8: gene expression heat map by hierarchical clustering. Supplementary Figure 9: transcription factor binding site enrichment analysis of CAD susceptibility loci. Supplementary Figure 10: genetic annotation analysis of CAD susceptibility loci. Supplementary Figure 11: histone modification enrichment analysis of CAD susceptibility loci. Supplementary Figure 12: Manhattan plots of SMR tests for association between gene expression and CAD. Supplementary Figure 13: prioritizing genes at GWAS loci using SMR analysis. Supplementary Figure 14: effect sizes of SNPs from GWAS plotted against those for SNPs from eQTL studies. Supplementary Table 1: integrating the population-specific genetic variants reported before and identified by a gene-based test in the present study. Supplementary Table 2: meta-analysis of population-specific genetic variants. Supplementary Table 3: identification of CAD risk genes by using VEGAS. Supplementary Table 4: the results of regulatory element enrichment analysis for CAD in the European population. Supplementary Table 5: the results of regulatory element enrichment analysis for CAD in the Asian population.
Data Availability Statement
All data generated or analyzed during this study are included in this article.