

Summary
Genomics studies in wild species of wheat have been limited due to the lack of references; however, new technologies and bioinformatics tools have much potential to promote genomic research. The wheat–Haynaldia villosa translocation line T6VS·6AL has been widely used as a backbone parent of wheat breeding in China. Therefore, revealing the genome structure of translocation chromosome 6VS·6AL will clarify how this chromosome formed and will help to determine how it affects agronomic traits. In this study, chromosome flow sorting, NGS sequencing and Chicago long‐range linkage assembly were innovatively used to produce the assembled sequences of 6VS·6AL, and gene prediction and genome structure characterization at the molecular level were effectively performed. The analysis discovered that the short arm of 6VS·6AL was actually composed of a large distal segment of 6VS, a small proximal segment of 6AS and the centromere of 6A, while the collinear region in 6VS corresponding to 230–260 Mb of 6AS‐Ta was deleted when the recombination between 6VS and 6AS occurred. In addition to the molecular mechanism of the increased grain weight and enhanced spike length produced by the translocation chromosome, it may be correlated with missing GW2‐V and an evolved NRT‐V cluster. Moreover, a fine physical bin map of 6VS was constructed by the high‐throughput developed 6VS‐specific InDel markers and a series of newly identified small fragment translocation lines involving 6VS. This study will provide essential information for mining of new alien genes carried by the 6VS·6AL translocation chromosome.
Keywords: wheat–Haynaldia villosa translocation line T6VS·6AL, Chicago long‐range linkage assembly, genome annotation, InDel markers, physical bin map
Introduction
Bread wheat (Triticum aestivum. L) is the most widely cultivated cereal crop worldwide and has made great contributions to food production and security. However, in comparison with other cereal crops, such as rice and maize, wheat is almost the last crop whose complete and accurate reference genome has been revealed, because of its huge ~15 GB genome size, approximately 85% repeat sequence proportion and complex hexaploid characteristics. In the past decade, the rapid development of wheat genome sequencing has benefited from the sequencing of the diploid ancestor species and single chromosomes. The outstanding advantage of these two strategies is that the bottleneck of sequencing and assembly in a large polyploid genome was broken by splitting the large genome to the subgenomic level or at the single‐chromosome level. Individual chromosome flow sorting technology made the most critical contribution to single‐chromosome sequencing, which shows great ability in evaluating chromosome size and reducing the sequencing workload. The first high‐quality individual chromosome sequence assembly of wheat was produced from the 1‐gigabase chromosome 3B of Chinese Spring by BAC library construction and NGS sequencing (Paux et al., 2006). Then, using ditelosomic lines of Chinese Spring as materials, each single‐chromosome arm was flow‐sorted, sequenced and assembled as organized to form the complete wheat genome assembly IWGSC RefSeq v0.4 (IWGSC, 2014). Now, the improved IWGSC RefSeq v1.0 (IWGSC, 2018), generally considered to be the best quality reference sequence version, has also been released. The breakthrough survey sequences provide valuable information about the genomic organization and evolution of the wheat genome. However, due to high variations in genomic structure and wide differences in gene distribution patterns among wheat cultivars, the reference genome is usually limited in the production of whole‐genome assemblies of specific genotypes to reference quality and sometimes incapable of assisting map‐based cloning of genes related to important agronomic traits. For complex genomes such as Triticeae species, it is very urgent to find a way to obtain the target chromosome sequences with high quality in a specific wheat genotype, which will produce huge effects for functional genomic research and evolutionary research based on chromosomal‐scale comparisons.
To improve the assembly quality of large and complex plant genomes, scaffold lengths should be enhanced, which leads to the application of a series of novel long‐read sequencing technologies and computational approaches. For example, long‐read‐type NGS technologies, such as Roche 454 (Brenchley et al., 2012; Tanaka et al., 2014), mate‐pair (Clavijo et al., 2017), PacBio SMRT (Zimin et al., 2017), Nanopore, NRGene, Hi‐C, BioNano and optical map, 10× genomic, BAC/fosmid ends and combining with genetic map (Avni et al., 2017; IWGSC, 2018; Jia et al., 2013; Ling et al., 2013; Luo et al., 2013, 2017; Zhao et al., 2017), have been successfully used. However, all of the above strategies were used in the process of whole‐genome sequencing and assembly to facilitate a better understanding of plant genomic diversity, genetic variation, evolution and vital functional gene mining. The application of long scaffold‐producing strategies has rarely been reported for individual chromosome resequencing. Chicago technology can build a large fragment library based on in vitro recombination of chromatin, and then, a high‐accuracy and long‐fragment superscaffold can be produced with the initial scaffold assembled by NGS. Compared with other long‐read sequencing technologies, Chicago can use a very small amount of DNA prepared even by flow cytometry sorted chromosomes. Thind et al. (2017) reported a TACCA approach (targeted chromosomal‐based cloning via long‐range assembly) for successfully cloning of the broad‐spectrum wheat leaf rust resistance gene Lr22a by Illumina sequencing and Chicago assembly of the sorted Lr22a carrier 2D chromosome. The assembly consisted of large scaffolds with an N50 size of 10–20 Mb, which is almost 500 times longer than the assemblies generated by NGS. A comparative genomic analysis of the chromosome 2D between the two genotypes ‘Chinese Spring’ and ‘CH Campala Lr22a’ at a megabase scale revealed novel large structural variations and increased SNP density, thereby supporting the notion that the physical map based on Chicago assembly was highly reliable (Thind et al., 2018).
The genome of wheat wild relative species is evolutionarily similar to that of common wheat; therefore, the large size and even higher content of repetitive sequences makes the genome sequencing more challenging. However, the abundance of cytogenetic materials has brought new opportunities for genomic research in wild species. Sorting and sequencing of an individual chromosome or chromosome arm carrying excellent genes offer a low‐cost, time‐efficient and high assembly accuracy strategy to effectively promote the genetic research of alien species. For example, 5Mgs from Leymus chinensis was sorted and sequenced, and high‐throughput SNP markers were developed to significantly improve the identification flux and resolution of alien chromatin, which will help clone the stripe rust resistance gene located on 5Mgs (Tiwari et al., 2016). In addition, 4VS from Haynaldia villosa was sorted and sequenced, and then, massive molecular markers were developed for the identification of 4VS chromosomal structural variants, which will promote physical mapping of the yellow mosaic disease resistance gene Wss1 (Wang et al., 2017). However, both sequencing methods used short‐read NGS, which has limited help with whole‐chromosome sequence splicing.
Many useful genes responsible for high yield, disease resistance and plant development have been explored on chromosome 6A of wheat. More than 20 QTLs related to yield traits, such as 1000 grain weight, grain width, grain number per spike, spike length and plant height, were identified (Guo et al., 2017; Guo et al., 2018; Gupta et al., 2006; Lopes et al., 2013; Simmonds et al., 2014; Sun et al., 2009; Tahmasebi et al., 2017; Wu et al., 2012). Based on the association analysis of unit type segments by sequencing and genomic comparison, some key genes on 6A positively related to grain number per ear, 1000 grain weight, heading stage and effective tiller number were confirmed (Zhang et al., 2017a; Zhang et al., 2017b). Regarding the resistance genes, wheat stem rust resistance genes Sr8 (Rohringer et al., 1979), Sr13 (Simons et al., 2011) and Sr26 (Mago et al., 2005), leaf blight resistance gene Stb15 (Arraiano et al., 2007), a major QTL (Qfhs.lfl‐6AL) conferring resistance to Fusarium head blight (Holzapfel et al., 2008), and an aphid resistance gene (Castro et al., 2008) are mapped on chromosome 6A. In addition, a seeding vigour‐related gene related to plant development has been identified (Spielmeyer et al., 2007). Some important genes have been cloned from the 6A chromosome. For example, TaGW2, the homologous gene of rice GW2, was cloned and found to be significantly related to grain width and grain weight by association analysis. Variation in the promoter region of the gene produced the small grain‐type allele TaGW2‐6A‐G and the large grain‐type allele TaGW2‐6A‐A (Su et al., 2011). Wang et al. (2019) cloned three homologous genes of the ADP glucose transporter gene TaBT1 located on chromosomes 6A, 6B and 6D, and proved that they could affect 1000 grain weight by regulating the wheat starch synthesis pathway.
Haynaldia villosa, a diploid wild grass (2n = 14, VV) with numerous traits favourite to wheat, can be crossed with tetraploid wheats and the AABBVV amphiploid can be backcrossed with hexaploid wheat to generate wheat germplasm introgressed with chromatin from Haynaldia villosa (De Pace et al., 2011). In our previous study, the wheat–Haynaldia villosa translocation line T6VS·6AL was developed, in which the short arm of the 6A chromosome of wheat was translocated with the short arm of the 6V chromosome of Haynaldia villosa. A broad‐spectrum wheat powdery mildew resistance gene, Pm21, was identified from 6VS (Chen et al., 1995). The T6VS·6AL was applied as a backbone parent of wheat breeding in China, and more than 40 commercial varieties carrying the 6VS·6AL translocation chromosome have been released and cultivated on a large scale. Even more importantly, the introduction of the 6VS·6AL translocation chromosome and substitution of 6AS in various wheat genetic backgrounds showed no negative effect on the major agronomic traits but exhibited partial positive contributions to 1000 grain weight, spike length and abiotic stress tolerance. It is indicated that there are some new elite alleles located on 6VS and some vital functional genes maintained on 6AL, so precise genomic analysis of this chromosome will help us to deeply explore these genes and to understand how this germplasm has become a backbone parent in breeding. Previously, a high‐quality de novo assembly was generated from flow cytometry sorting of chromosome 6VS·6AL of the wheat–Haynaldia villosa translocation line 92R137 using short‐read sequencing in combination with Chicago long‐range scaffolding. In this study, a cytogenetic bin of 6VS·6AL was generated by using the newly developed cytogenetic stocks and InDel molecular markers. Then, further analysis of the assembly scaffolds was carried out with various recent bioinformatics approaches to obtain a deep understanding of chromosome 6VS·6AL. The results will greatly help to reveal the evolution of 6VS and the molecular mechanism of how 6VS·6AL translocation affects agronomic traits. In addition, Haynaldia villosa has more than 300 accessions worldwide, and at least 6 accessions had been introduced into wheat backgrounds (Chen et al., 1995; Li et al., 2005; Liu et al., 2011; Lukaszewski, 1988; Sears, 1953; Zhang et al., 2018). So, the assembled sequence of 6VS obtained in this study will also help to systematically explore new alleles from other Haynaldia villosa accessions. Furthermore, the established technology in this study, combining single‐chromosome sorting, long‐range assembly, cytogenetic stocks creation and high‐throughput molecular markers developing, will provide a feasible approach to investigate the genomic structure of chromosomes from other wild species.
Results
Chicago assembly, length classification and chromosomal anchoring of scaffolds
The 6VS·6AL chromosome was flow‐sorted and sequenced by the Hiseq 2500 platform, and over 65 GB of effective sequences were obtained, 96% of which were larger than 20 bp. Long‐range assembly was obtained by using Chicago technology combined with NGS de novo assembly. Compared with the results of the NGS de novo assembly, long‐range assembly increased the total length from 537.25 to 574.57 Mb, and the longest scaffolds increased from 210 276 bp to 83 726 258 bp, the scaffold N50 increased from 25.0 kb to 22.39 Mb, and the scaffold N90 increased from 5.3 KB to 5.07 MB, while the number of scaffolds decreased from 45073 to 5546. It was exciting to find that the length of scaffolds N50 and N90 increased approximately 1000 times, and the length of the longest scaffold, Sc18Q1Z_139, reached even to 83.726 Mb (Table 1). The 5546 scaffolds were then classified according to the length as shown in Figure S1a. Although the number of scaffolds 1–2 kb in size reached to 3573, the accumulated length of 28 superscaffolds each longer than 5 Mb covered 83.75% of the total assembled length.
Table 1.
Comparison of NGS sequencing and Chicago assembly results of 6VS·6AL
| NGS sequencing | Chicago assembly | 6VS chromosome | 6AL chromosome | |
|---|---|---|---|---|
| Total length (Mb) | 537.25 | 574.57 | 243.39 | 331.18 |
| Scaffold amount | 45 073 | 5546 | 3769 | 1777 |
| Longest scaffold (bp) | 210 276 | 83 726 258 | 42 845 531 | 83 726 258 |
| Scaffold N50 | 25.0 kb | 22.39 Mb | – | – |
| Scaffold N90 | 5.3 kb | 5.07 Mb | – | – |
Sequence alignment between 6VS·6AL and 6A of the IWGSC RefSeq v1.0 chromosome assembly was performed. The scaffold shorter than 10 kb was used as a complete sequence for comparion with the 6A assembly sequence by BLASTN. The scaffold longer than 10 kb was divided into one or more 10‐kb segments, and the first 1 kb of each 10‐kb size segment was selected for BLAST and anchored to 6A with a threshold identity >90% and a coverage length >800 bp. For example, Sc18Q1Z_4418 was anchored to the physical location of 6A at 285 433 117 bp with a coverage length of 1000 kb, and it was close to the centromere of 6AL among all the anchored scaffolds. After removing 1777 6AL‐anchored scaffolds, the remaining 3769 scaffolds were predicted to be located on 6VS with a total assembly length of 243.39 Mb (Table 1). Among all the scaffolds on the 6VS chromosome, there were 13 superscaffolds each longer than 5 Mb, accounting for 69% of the total assembly length, in which the largest scaffold was Sc18Q1Z_1327, with a size of 42.845 Mb (Figure S1b, Table 1).
Distribution of repeated sequences on chromosome 6VS·6AL
Repetitive sequence analysis found that the transposable elements accounted for 78.27% (449.76 Mb) of the content of 6VS·6AL, of which retrotransposons accounted for 67.75% and transposons for 9.95%. For retrotransposons, long terminal repeats (LTR retroelements) accounted for the most abundant (66.60%), of which Gypsy‐type accounted for 51.04% and Copia‐type accounted for 13.68%. For the transposons, the CACTA superfamily accounted for 8.97%, ranking first (Table S2). Different from the distribution density of annotated genes increasing from centromere to telomere, the distribution density of Gypsy‐type repeat sequences decreased gradually from centromere to telomere, while the distribution density of Copia‐type sequences was uniform along the chromosome. This distribution pattern is essentially consistent with that observed in other sequenced wheat chromosomes (Figure S2).
Gene prediction and annotation
The high‐confidence genes were predicted by ab initio, homologue and RNA‐seq analysis, and then, a total of 5,781 genes were obtained with an average length of 3120.83 bp, an average CDS length of 1054.83 bp, an average exon length of 271.65 bp, an average intron length of 716.60 bp, and an average exon number of 3.88 per gene (Table S3). Among them, 5,343 genes were predicted by ab initio using the existing probability model but with low accuracy in predicting of the cutting sites and UTRs; 4,315 genes were predicted by the homology‐based method by tBLATn with the latest coding protein databases of Aegilops tauschii, Hordeum vulgare, Triticum aestivum, Triticum urartu and Triticum turgidum; and 2,844 genes were predicted by the RNA‐seq method with accurate alternative splicing sites and exon regions (Figure 1). In addition, noncoding RNAs were also identified including 307 tRNAs, 13 rRNAs, 6,850 miRNAs and 264 snRNAs (Table S4).
Figure 1.

Predicted gene set using VENN diagram. Note: De novo: EVM integration of genes supported by de novo prediction; Homologue: EVM integration of genes supported by homology prediction; RNA: EVM integration of genes supported by RNA‐seq. Each line of evidence is based on an overlap larger than 50%.
After inquiring the coding proteins in the databases of NR, Swiss Prot, KEGG, Pfam and GO databases, 86.1% of the predicted genes of the 6VS·6AL translocation chromosome were obtained with clear functional annotations (Table S5). The other proteins are unknown, and need to be further investigated. A total of 2602 genes, accounting for 45.0% of the total predicted genes, were categorized into 43 GO terms in three categories: cellular component, biological process and molecular function (Table S5, Figure S3). Among them, the cell region term in the cellular components category, the activity of structural components term in the molecular category and the metabolic process term in biological process category were enriched and expressed significantly. A total of 3170 genes, accounting for 54.8%, were assigned to 19 KEGG pathways, in which signal transduction and environmental adaptation were significantly abundant (Figure S4).
Comparative analysis of syntenic chromosomes to 6VS.6AL among Triticeae species
Comparative genomics was performed using the annotated genes of 6VS·6AL to compare with those of 6A/6B/6D in Chinese Spring, 6A in Triticum urartu, 6D in Aegilops tauschii and 6H in Hordeum vulgare (Figure 2a). The results showed that the 6AL of T6VS.6AL (6AL‐T) showed an excellent collinear relationship with 6AL in CS (6AL‐CS). Furthermore, comparative analysis was also performed using the assembly scaffolds longer than 1 Mb of 6AL‐T to compare with the reference sequences of 6AL‐CS, and the results also showed a high degree of collinearity (Figure S5). Therefore, the above analysis strongly supported the reliability of the Chicago assembly results.
Figure 2.
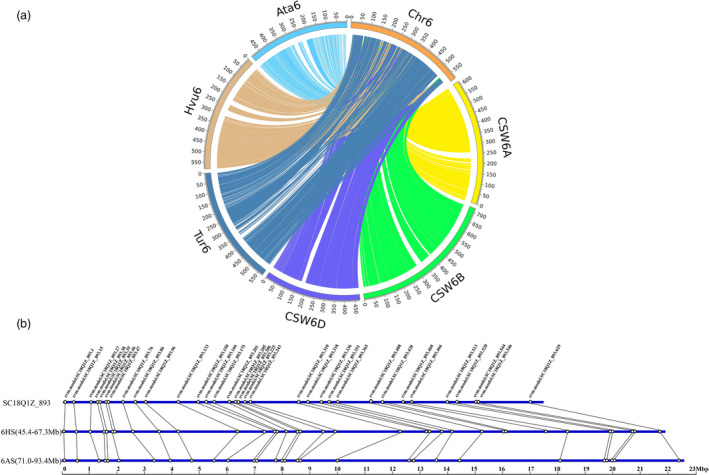
Collinearity analysis between the assembled sequence and the reference sequences. (a) Collinearity analysis between 6VS·6AL and the homoeologous group 6 chromosomes. (Note: Chr6 indicates 6VS·6AL; CSW6A, CSW6B and CSW6D indicate wheat chromosomes from Chinese Spring; Tur6 indicates 6A chromosome from Triticum urartu; Hvu6 indicates 6H chromosome from Hordeum vulgare; and Ata6D indicates 6D chromosome from Aegilops tauschii). (b) Microcollinearity analysis of 33 predicted genes in scaffold‐893 (in length of 17.4 Mb) and their corresponding orthologous genes in the collinear region of 6HS (45.4–67.3 Mb) of Hordeum vulgare and 6AS (71.0–93.4 Mb) of Chinese Spring. The black lines represent the correspondence between genes.
The SNP number between 6AL‐T and 6AL‐CS was compared with the SNP number between 6VS‐T and 6AS‐CS, using the genes with similarity higher than 95% as the analysed targets. For 6AL, 3,803 SNPs were detected in 529 of 2367 genes from 6AL‐T, with a SNP density of 7.19 SNPs per gene. However, 23,882 SNPs were detected in 1012 of 1204 genes from 6VS‐T, with a SNP density of 23.60 SNPs per gene. Actually, the real number of SNPs involved in genes of 6VS‐T is much higher than that estimated because most of the genes on 6VS‐T were not included in the analysis due to the low sequence similarity with orthologues of 6AS‐CS. Therefore, SNP calling also showed that collinearity was better between 6AL‐T and 6AL‐CS than that between 6VS and 6AS‐CS.
It was interesting to find a gap in 6VS corresponding to 230–260 Mb of 6A‐Ta, and the same gap in 6VS was also found in the collinearity region on 310–330 Mb of 6B‐Ta, on 170–190 Mb of 6D‐Ta, on 210–230 Mb of 6D‐Ata and on 205–245 Mb of 6H‐Hv (Figure 2a). According to the predicted positions of the centromeres in IWGSC RefSeq v1.0 (IWGSC, 2018), the gap occurred in 6VS was collinear to the regions on the short arms close to the centromeres of 6A, 6B and 6D, respectively. The annotated genes corresponding to 260–280 Mb of 6AS‐CS were extracted from assembled sequences of 6VS·6AL, and it was found that these genes were actually from 6AS‐Ta. In addition, the collinearity analysis of 6VS‐T with 6AS‐Ta showed better collinearity in the proximal part of 6AS‐Ta than that in the distal part of 6AS‐Ta (Figure 2a). Therefore, it was indicated that the translocation of 6VS·6AL was not produced by Robertsonian translocation, and centromere fusion was not involved in the process of recombination between 6AS and 6VS. Sequence analysis showed that the short arm of the translocated chromosome was composed of a large distal segment of 6VS, a small proximal segment of 6AS and the centromere of 6A (Figure 3). Based on the deletion detected in 6VS, it was proposed that the breakpoint in 6VS and in 6AS did not occur in the corresponding orthologous locus, so the recombination between 6VS and 6AS was not completely complementary.
Figure 3.
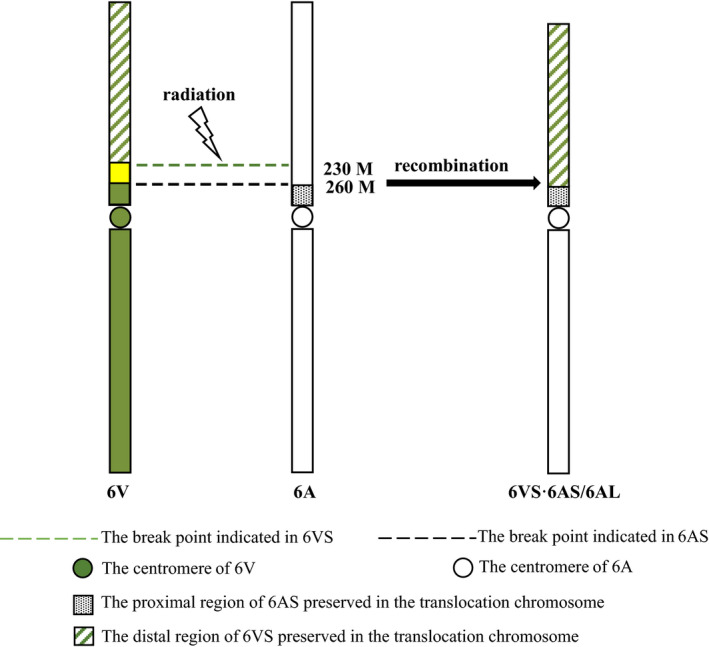
Formation of the translocation chromosome 6VS·6AS/6AL. ( ) The break point indicated in 6VS; (
) The break point indicated in 6VS; ( ) The centromere of 6V; (
) The centromere of 6V; ( ) The proximal region of 6AS preserved in the translocation chromosome; (
) The proximal region of 6AS preserved in the translocation chromosome; ( ) The distal region of 6VS preserved in the translocation chromosome; (
) The distal region of 6VS preserved in the translocation chromosome; ( ) The breakpoint indicated in 6AS; (
) The breakpoint indicated in 6AS; ( ) The centromere of 6A.
) The centromere of 6A.
The microcollinearity was also analysed using Pm21‐located scaffold‐893 with a length of 17.4 Mb. Thirty‐three predicted genes from scaffold‐893 and their orthologous genes, with sequence coverage >95% and identity >80%, in 6AS and 6HS were selected and marked in each genome. In general, good collinear relationships of 6VS with 6AS‐Ta and 6HS‐Hv were observed, and the gene orders matched perfectly with rarely disrupted microcollinearity (Figure 2b). However, 6VS showed the better collinearity with 6HS‐Hv. For example, five genes, including the predicted gene NO. SC18Q1Z_893.428, the cloned powdery mildew resistance regulation gene RLK1‐V, could found orthologues in barley 6HS, but no orthologues in wheat 6AS.
Prediction of transcription factor (TF) genes and NRT genes
Prediction of transcription factors helps to understand the transcriptional regulatory mechanisms for gene expression. In this study, three different websites, PlantTFcat, PlantTFDB and iTAK, were used to predict the transcription factors using different computational approaches in 6VS. As a result, 183 TFs were predicted by PlantTFcat, 93 TFs by PlantTFDB and 95 TFs by iTAK, while 86 TFs were common to the three tools (Figure 4a). A total of 193 TFs predicted by the three tools could be divided into 33 categories, among which C2H2 and Hap3/NF‐YB were the most abundant TFs (Figure 4a). Nearly all the TFs predicted by PlantTFDB could be detected by PlantTFcat and iTAK, while PLATZ, CSD and OFP types could only be detected by iTAK, additionally, some members belonging to C2H2, GRF, Hap3/NF‐YB and WD40‐like types could only be detected by PlantTFcat.
Figure 4.

Identification and classification of functional genes. (a) The classification and number of transcription factors (TFs) predicted by PlantTFcat, PlantTFDB and iTAK. (b) Comparison of NRT homologous genes on 6VS with that on 6AS. (c) Distribution of the predicted NLR genes on chromosomes (Note: 6VS·6AL: translocation chromosome, 6A‐CS: chromosome 6A of wheat cv. CS, 6H‐morex: chromosome 6H of barley cv. Morex. The scale bar indicates the physical position in Mb).
Nitrogen is the most important nutrient element for crop growth and yield formation. The wheat–H. villosa translocation line T6VS·6AL has the characteristics of large grains, long spikes and darker leaf colour, which are usually correlated with a high efficiency of nitrogen utilization. In this study, the genes related to nitrogen utilization on 6VS were investigated, and it was found that there is a family of nitrogen transport genes (NRTs) with multiple members located on 6VS. When using the TaNRT2.1, TaNRT2.2, TaNRT2.3 and TaNRT2.5 to search the reference of 6AS of CS and the long‐range linkage assembly of 6VS, 13 TaNRTs were identified in 6A‐CS located at 15.7–16.4 Mb, while 11 NRT‐V genes were identified from two scaffolds of 6VS. In scaffold 1009, 6 NRT‐V genes were identified with one missing NRT gene and one chromosome inversion, while in scaffold 5277, 5 NRT‐V genes were identified as in 6AS, but with one gene transcribed in the opposite direction (Figure 4b).
Annotation and chromosome‐scale comparison of NLR genes
NLRs are one of the most important resistance gene resources for wheat breeding and undergo rapid evolution to adapt to rapid variations in pathogens. To obtain more information on NLRs located on the 6VS.6AL chromosome, the sequence structure and distribution of NLRs were compared with those on the 6A and 6H chromosomes. A total of 115 NLRs were annotated from 6VS.6AL scaffolds, while 115 NLRs from the barley 6H reference (Morex V2_All_Gene) and 137 NLRs from the wheat 6A reference (IWGSC v1.0_HighConf_LowConf_gene). It was observed from the gene distribution map that NLRs tended to be clustered in the chromosome and located in the telomeric regions (Figure 4c). Fifty‐eight NLRs, accounting for 50.4% of the total annotated NLRs, gathered in four gene clusters, including Sc18Q1Z_3573, Sc18Q1Z_5538, Sc18Q1Z_893 on the 6VS and Sc18Q1Z_3699 on the 6AL. Among them, Sc18Q1Z_3699 was the largest gene cluster carrying 30 NLRs, implying that it might be a rapid adaptive evolutionary locus.
Construction of the 6VS chromosome bin map using InDel markers and introgression lines
A total of 13 scaffolds larger than 5 Mb were selected from 6VS assembly scaffolds,which covered 69% of the assembled 6VS chromosome arm, including Sc18Q1Z_1327 (41.84 Mb), Sc18Q1Z_1707 (22.41 Mb), Sc18Q1Z_2084 (5.45 Mb), Sc18Q1Z_2155 (9.44 Mb), Sc18Q1Z_2204 (6.12 Mb), Sc18Q1Z_3069 (13.2 Mb), Sc18Q1Z_4548 (31.8 Mb), Sc18Q1Z_5541 (5.08 Mb), Sc18Q1Z_5542 (5.01 Mb), and Sc18Q1Z_5544 (14.69 Mb). A total of 1920 primers were designed per 10 Kb, evenly covering all the 13 scaffolds, and each primer was designed based on the insertion/deletion region detected in 6VS sequences compared with the Chinese Spring A/B/D genomes. Then, 1089 (56.7%) primers produced polymorphisms specific to 6VS; the primers from Sc18Q1Z_5541 showed the highest polymorphism rate 74.1%, while those from Sc18Q1Z_5542 showed the lowest polymorphism rate of 38.0% (Table S6, Figure S6).
Using GISH technology to screen the translocation lines in the radiation‐induced progeny of 92R137, 14 different types of translocation lines involving different 6VS segments were obtained, including 9 homozygous and five heterozygous lines (Figure S7). Using 43 molecular markers to identify the breakpoint of the alien 6VS chromosomal segment in the above 6VS structural variant, combined with the physical distribution of the 13 scaffolds, a fine 6VS physical map consisting of at least 28 chromosomal segments and high‐density molecular markers was constructed (Figure 5, Table S7). Bin1, located in the terminal region of 6VS, was 24 Mb in length, containing 6 scaffolds that were longer than 1 Mb: scaffold703, scaffold5277, scaffold3573, scaffold5218, scaffold5538 and scaffold4514. Bin2 was covered by scaffold5542. Bin4 and bin5 were covered by scaffold5544. Bin7, bin8 and bin9 were covered by scaffold893. Bin11 was covered by scaffold5541. Bin13 was covered by scaffold156. Bin15 and bin16 were covered by scaffold3069. Bin18 and bin19 were covered by scaffold1327. Bin20 was covered by scaffold4548. Bin22 and bin23 were covered by scaffold1707. Bin25 was covered by scaffold470. Bin26 was covered by scaffold2155. Bin27 was covered by scaffold2204. Bin28 was covered by scaffold2084 (Figure 6).
Figure 5.
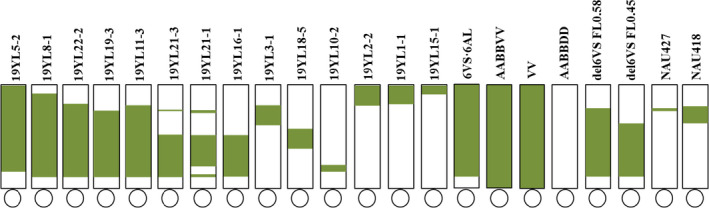
Identification of the chromosome structural variations of the wheat–Haynaldia villosa translocated chromosome 6VS.6AL. Note: AABBVV means T. durum‐Haynaldia villosa amphiploid; VV means Haynaldia villosa; and AABBDD means Chinese Spring.
Figure 6.
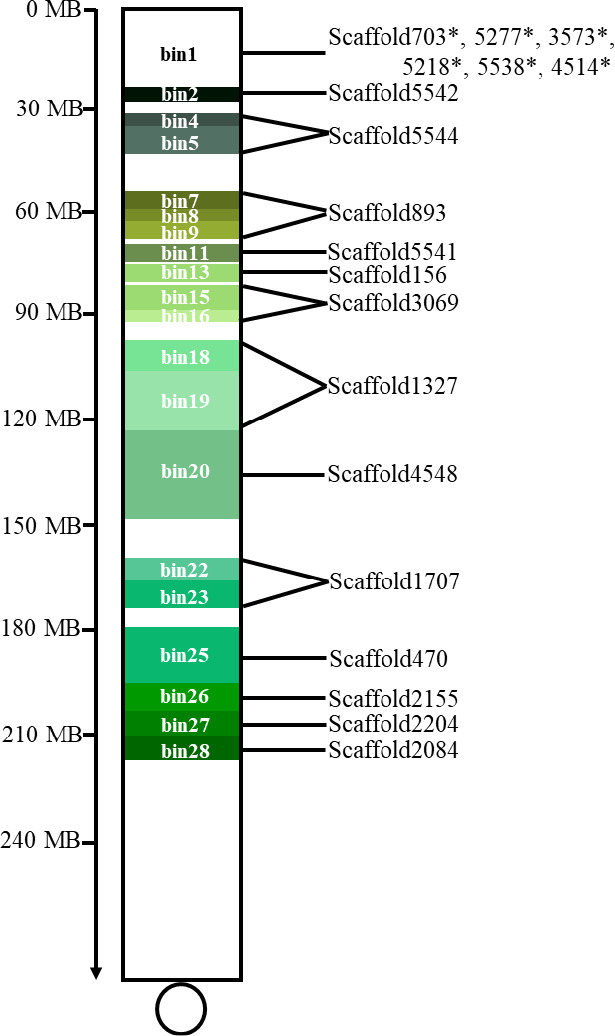
Physical bin map of the 6VS chromosome.
Discussion
Improved genome assembly helped to reveal the real structure of the translocated chromosome 6VS·6AL
Single‐chromosome flow sorting technology has great advantages in analysing large genomes at the subgenome level to reduce the complexity of high‐throughput sequencing and assembly. Previously, this technology has contributed largely to wheat chromosome sequencing by combining with BAC library construction and NGS. Chromosome sorting combined with Chicago technology, a recently developed strategy assisting in assembling superscaffolds, shows extreme potential to prompt the target chromosome genomics. The TACCA technology is the first practice to combine these two approaches to help clone Lr22a from the sorted 2D chromosome, and the physical map based on Chicago assembly was proven to be highly reliable. The N50 value produced by Chicago assembly increased that produced by NGS more than 500 times (Thind et al., 2017). In this study, the N50 value was 22.39 Mb which was approximately 1000 times of that produced by NGS. Therefore, Chicago could really improve the quality of genome assembly.
6VS·6AL was previously considered to be the translocation between the whole short arm of 6V and the whole short arm of 6A. In our previous study, 6VS·6AL chromosome was sorted by flow cytometry, sequenced by NGS and long range assembled by Chicago, which helped in the fine mapping and cloning of the Pm21 gene in the cryptical introgression line NAU427 (Xing et al., 2018). The total length of 6VS·6AL was 574.57 MB, in which 6VS was 243.39 Mb and 6AL was 331.18 Mb, respectively. The total length of the 6A chromosome of Chinese Spring is 618.08 Mb, in which 6AS was approximately 282 Mb and 6AL was approximately 336 Mb. Therefore, the lengths of 6AL‐T and 6AL‐CS were similar, while 6VS‐T was significantly shorter than 6AS‐CS. This result was supported by collinearity analysis, which showed an obvious gap in 6VS corresponding to 230–260 Mb of 6AS. The same gap was also identified when 6V was compared with 6B‐Ta, 6D‐Ta, 6H‐Hv and 6D‐Ata. The grain weight negatively regulated gene TaGW2 was located on 230–260 Mb of 6AS, and the orthologous gene in H. villosa HvGW2‐6V was identified from the 6VS telosome addition line (Xiao et al., 2020); thus, it was proposed that the collinear region corresponding to 230–260 MB of 6AS was originally existed in 6VS but was deleted when translocation occurred after radiation. Therefore, the breaking point occurred in the short arm of 6AS close to the wheat 6A centromere when 6AS and 6VS recombination was generated, and it fused with a large distal segment of 6VS missing approximately 30 Mb of the proximal region collinear to the 230–260 Mb of 6AS. Therefore, the 6VS·6AL chromosome was actually not formed by Robertsonian translocation, and the centromere was complete from the 6A chromosome. The long‐range assembly could reveal the micro disruption which could not be observed previously.
Improved genome assembly facilitated the comprehensive evolutionary study of H. villosa
Based on the analysis of chromosome pairing of intergeneric hybrids, the evolutionary relationship between H. villosa and other diploid Triticeae species has been studied. Chen and Liu (1982) found that the evolutionary distance between the V genome and the D, A or B genome was gradually increased when studying the configuration of F1 pollen mother cells at metaphase I of wheat × H. villosa. However, Blanco et al. (1983) analysed the configuration of F1 pollen mother cells at metaphase I of T. turgidum × H.villosa and concluded that the V genome was closer to the A genome than to the D and B genomes. However, RFLP analysis based on wheat cDNA probe found that the distance between V and A/B/D was relatively far, which was supported by analysis based on rDNA sequences (Monte et al., 1993). Cao et al. (2011) used a barley gene chip to detect Bgt‐responsive genes in H. villosa and cloned several functional genes from the V genome based on the barley sequences, which proposed that the relationship between H. villosa and barley was close. However, the above analysis on a small scale inevitably led to incomplete conclusions. Analysis based on the entire genome sequence is currently the most comprehensive approach to reveal the evolutionary patterns. The perfect collinearity between the 6AL of the translocation line and the 6AL of Chinese spring reflected the high quality and accuracy of the sequence assembly we obtained. Therefore, the long‐range assembly of 6VS provided a great opportunity to study the relationship of 6V with other putative related genomes both at the level of gene identity and at the level of genomic structure.
The miRNAs are the important regulators of gene expression, so the predicted 3451 miRNAs of 6VS were compared with those of 6AS, 6BS and 6DS to identify specific miRNAs on 6VS. A total of 12 families detected over twice were found to be newly occurred in 6VS, 13 families were found to be expanded in 6VS, and 4 families were found to be decreased in 6VS. The target genes of these specific miRNAs need to be identified in the future to elucidate how they participate in the species divergence and how they affect biological processes in Haynaldia villosa.
High‐density InDel markers contributed to identifying structural variants and constructing of the physical bin map of 6VS
Wild species H. villosa harbours many favourable characteristics for wheat improvement; thus, molecular markers are constantly being developed for efficiently tracking alien chromosome fragments. Zhang et al. (2013) developed 5 EST markers on 6V, Zhang et al. (2017c) developed 1624 IT markers on 1V‐7V including 138 on 6V, and Sun et al. (2018) developed 297 markers on 6VL and 1 on 6VS. However, the density of these markers is relatively low and the chromosome distribution is uneven. In this study, the improved 6VS·6AL genome data were used for high‐throughput development of InDel markers, and the average density of the markers reached 6.74/Mb. Most of the markers could produce the same polymorphism bands among VV, AABBVV and 6VS·6AL; however, abnormal amplification results were found for very few markers. For example, 48 markers from SC18Q1Z_4548 produced no specific amplicons in T. durum‐H. villosa amphiploid, which may be due to the deletion of micro alien segments with the amphiploid formation. Therefore, the high‐throughput markers could identify small structural variations efficiently, and the developed markers provided a great help for mapping more favourable genes on 6VS in the future.
In this study, a physical bin map of the 6VS chromosome was constructed using 14 structural variants combined with a high‐density InDel marker. However, the types of translocation lines were not rich enough to build a high‐resolution bin map, and more 6VS chromosome structural variants need to be developed. It should be pointed out that InDel markers were from only 13 large scaffolds covering 69% of the assembled genome, so the uneven distribution of these scaffolds led to a blind area that could not be detected by these markers. Considering that the location arrangements of small scaffolds are not easy to determine using the current structural variants, so the smaller scaffolds have not yet been used to develop more markers.
Transcription factor analysis provided a rich resource for mining the beneficial genes
The predicted TFs were used to search against the full‐length transcriptome sequencing of H. villosa on PacBio platform with a threshold identity >95% and a coverage ratio >95%. A total of 58 members belonging to 15 types were found to be expressed; however, most expressed members belonged to the AP2/EREBP, C2H2, Hap3/NF‐YB, GRF, MYB and WD40‐like types. The AP2 transcription factor family in plants is involved in the abiotic stress tolerance. Among the expressed AP2 transcription factors, one member, designated ERF1‐V, has been cloned and has proven to significantly improve the drought resistance of wheat after being genetically transformed into common wheat (Xing et al., 2017). Another AP2 transcription factor predicted from SC18Q1Z_2084 was found to respond rapidly to drought treatment, as revealed by RNA‐seq analysis in our laboratory. In addition, C2H2, Hap3/NF‐YB, GRF, MYB and WD40‐like were reported to regulate plant development, immunity and abiotic stress tolerance. Therefore, the systematic analysis and functional identification of transcription factors will be helpful for elucidating the molecular mechanism of drought tolerance, disease resistance and other beneficial traits of the T6VS·6AL. Furthermore, the transcriptome analysis of TFs and predicted genes from the 6VS·6AL genome will be helpful for building a gene expression regulation network, and to mining more beneficial genes related to the agronomic traits in the future.
Experimental procedures
Plant materials
Haynaldia villosa (L.) Schur (syn. Dasypyrum villosum (L.) P. Candargy, 2n = 14, VV) line 91C43 was induced from Cambridge plant breeding in the UK and maintained by the Cytogenetic Institute, Nanjing Agricultural University (CINAU) from 1970s. 92R137, a Triticum aestivum‐H. villosa T6VS·6AL translocation line resistant to powdery mildew conferred by Pm21, was developed by CINAU and used for the 6VS·6AL single‐chromosome flow cytometric sorting. T. durum‐H. villosa amphiploid (AABBVV), a powdery mildew‐resistant line del.6VS‐1 (FL0.58), a susceptible deletion line del.6VS‐2 (FL0.45), a resistant terminal translocation line NAU418, and a resistant cryptic introgression line NAU427, were developed by CINAU (Xing et al., 2018). New introgression lines involving small 6VS segments were screened from the offspring generated by the irradiated mature female gametes of the T6VS·6AL with 60CO γ‐rays at a 1600 Rad/M dosage rate (Chen et al. 2013). The 6VS‐introgressed plants were backcrossed as female parents with Chinese Spring for preserving the alien chromosome in the following generation. Fourteen 6VS‐introgression lines were identified, including nine terminal translocation lines 19YL1‐1, 19YL3‐1, 19YL5‐2, 19YL15‐1, 19YL18‐5, 19YL19‐3, 19YL2‐2, 19YL21‐3 and 19YL21‐1; four intercalary translocation line 19YL8‐1, 19YL10‐2, 19YL11‐3 and 19YL22‐2; and one terminal deletion line 19YL16‐1. All the above materials were used to develop molecular markers specific to 6VS and to construct a physical bin map of 6VS.
Genome annotation
Annotations of repeat sequences, structural genes and noncoding RNA (ncRNA) were performed using multiple proprietary software and open databases. Then, the predicted results combined with the transcriptome comparison data were used by the EVidenceModeler (EVM) integration software to integrate the multiple gene sets into a nonredundant complete gene set. Finally, PASA combined with transcriptome assembly structure was used to correct the annotation structure of EVM followed by UTR and alternative splicing addition, and then, the necessary artificial quality control was carried out to obtain the feature gene set. For repeat sequence annotation, the homologous sequence alignment method was used with RepeatMasker software to identify the putative repeat sequences highly similar to the deposited sequences in the repeat sequence database (CLARI‐TE Library).
Homology prediction, de novo prediction and transcriptome sequencing prediction were employed to annotate the gene structure. Homology prediction was performed to compare the genome sequence of 6VS·6AL with the protein database of homologous species, including Aegilops tauschii, Hordeum vulgare, Triticum aestivum, Triticum urartu and Triticum turgidum, and then to predict the gene structures by BLASTX, genewise and other comparison tools. De novo prediction was performed using software programs, including Augustus, GlimmerHMM and SNAP, based on the statistical characteristics of genome sequence data. The NGS and PacBio transcriptome sequencing databases of mixed tissues from Haynadia villosa without or with various stress treatments were generated previously and used in this study.
Noncoding RNA (ncRNA) was searched according to its characteristics, and conserved sequences, such as tRNAs, were found using tRNAscan‐SE software. rRNAs were selected through BLAST with rRNA sequences of related species, and miRNAs and snRNAs were predicted by INFERNAL software using the covariance model of the Rfam family.
Gene function annotation
Gene function was annotated by BLASTP analysis of the gene set with the known protein database, such as NR, Swiss prot, KEGG, Pfam and GO database. After function annotation, GO database and KEGG database were used for gene functional classification. Then, three online websites, planttfcat, planttfdb and itak, were used to predict the transcription factors from the above gene set.
NLRs annotation and comparison
NLRs on the 6VS·6AL chromosome were selected from the above gene set with functional annotation, and were compared with NLRs on the 6A chromosome of Chinese Spring (IWGSCv1.0_HighcConf_LowConf_gene) and the 6H chromosome of barley (Morex v2.0_All_Gene) by drawing a draft physical location map. In silico chromosomal location of the NLRs on the 6VS·6AL chromosome was based on the chromosome collinearity with the 6HS chromosome of barley and the 6AL chromosome of CS, respectively. Renseq of Haynaldia villosa was performed in our previous study (Xing et al., 2018), and 101 complete NLRs found by using NLR‐Annotator and in silico mapped on the 6V chromosome were screened and compared with the 115 NLRs on the 6VS·6AL chromosome. Multiple alignment files were generated, and a phylogenetic tree was created using Geneious version 10.2.2 (Biomatters Ltd., Auckland, New Zealand, USA).
Collinearity analysis
Interspecific comparative genomics was performed for collinearity analysis. The final gene set integrated by EVM on 6VS was aligned with HC genes predicted on 6AS/6BS/6DS of Chinese Spring, on 6AS of Triticum urartu, on 6DS of Aegilops tauschii and on 6HS of Hordeum vulgare by BLAST. The threshold value was set to 10−6. The collinearity figure was drawn by the Circos software (http://circos.ca/software/download/circos/) (Krzywinski et al., 2009) with three prepared documents, including a chromosome information file (karyotype file) for showing the chromosome length and related settings, a label text file (text file) for marking specific gene positions on the chromosome, and a link file output by using JCVI for collinearity analysis. Similarly, the Circos map was generated by collinearity analysis of the predicted gene set on the 6AL genome compared with those on 6AL/6BL/6DL of Chinese Spring, 6AL of Triticum urartu, 6DL of Aegilops tauschii and 6HL of Hordeum vulgare.
Gene order analysis and comparison located on Sc18Q1Z_893
Sequence alignment of 269 predicted positive genes from Sc18Q1Z_893 with the collinear gene sets from the 6A chromosome of Chinese Spring (IWGSCv1.0_HighcConf_LowConf_gene) and the 6H chromosome of barley (Morex v2.0_HignConf_Gene) was performed to identify the microcollinearity at the individual gene level.
Cytogenetic identification
GISH and FISH analyses were performed to identify the introgressed 6VS chromosome fragment in the wheat genetic background using root tip cells at mitotic metaphase following an improved protocol developed by Zhang et al. (2004) and Du et al. (2017). The probe for GISH analysis used total genomic DNA of H. villosa labelled with fluorescein‐12‐dUTP by the nick translation method. The chromosomes were observed using an Olympus BX60 fluorescence microscope with a DP72 CCD camera for image acquisition, and individual chromosomes with 6VS hybridization signals were cropped using Adobe Photoshop software.
Molecular marker development and detection
The assembly scaffold was BLAST searched with the IWGSC RefSeq v1.0 database of Chinese Spring (https://urgi.versailles.inra.fr/blast_iwgsc/) to develop InDel molecular markers according to insertion/deletion segments in the genomic sequences. Primers were designed using Primer5.0 software. The lengths of the primers were ~25 bp, the annealing temperatures were 57–60 °C, and the lengths of the amplification products were 200–2000 bp. A total of 1920 6VS‐specific InDel markers were designed, and 43 were ultimately selected to detect the breakpoints and fragment sizes of the 6VS chromosome (Table S1). The physical bin map of the 6VS chromosome was drawn using Adobe Photoshop software.
Conflicts of interest
The authors declare no competing interests.
Authors' contributions
Aizhong Cao and Liping Xing participated in the design of the experimental plan. Jaroslav Doležel designed the 6VS·6AL sorting and sequence assembling. Lu Yuan and Zengshuai Lv performed the gene annotation. Shuqi Cao and Jiaqian Liu evaluated the resistance of cytogenetic stocks. Chunhong Yin and Qiang Wang participated in molecular marker development. Peidu Chen provided the progeny population of radiation. Ruiqi Zhang and Zhenpu Huang identified the cytogenetic stocks. Miroslava Karafiátová performed chromosome sorting and FISH. Jan Vrána performed flow cytometry. Jan Bartoš performed illumina sequencing and assembly. Liping Xing and Aizhong Cao wrote the manuscript. All authors have read and approved the final manuscript.
Supporting information
Figure S1 Size distribution of scaffolds from 6VS·6AL (a) and from 6VS (b).
Figure S2 Distribution of repeat sequences in the 6VS·6AL chromosome. Note: (a) distribution of genes; (b) density of Copia; and (c) density of Gypsy.
Figure S3 Statistics of GO functional classification.
Figure S4 Gene enrichment analysis by KEGG.
Figure S5 Dot plot of genome comparison between 6AL‐T and 6AL‐CS.
Figure S6 Amplification of InDel markers in T6VS·6AL, AABBVV, VV and AABBDD.
Figure S7 GISH analysis of the 14 wheat–H. villosa introgression lines involved in 6VS. Nine homozygous lines included six terminal translocation lines 19YL1‐1, 19YL3‐1, 19YL5‐2, 19YL15‐1, 19YL18‐5, and 19YL19‐3, and three intercalary translocation lines 19YL8‐1, 19YL10‐2, and 19YL11‐3. Five heterozygous lines included three terminal translocation lines 19YL2‐2, 19YL21‐3, and 19YL21‐1; one intercalary translocation line 19YL22‐2; and one terminal deletion line 19YL16‐1.
Table S1 Primer information used for 6VS fragments detection.
Table S2 Classification and statistics of repeated sequences.
Table S3 Gene prediction by de novo, homologue and RNA‐seq analysis.
Table S4 Noncoding RNAs.
Table S5 Gene function annotation.
Table S6 Statistics of Indel markers designed on the scaffold.
Table S7 Marker analysis of the chromosome structural variations of wheat–Haynaldia villosa translocated chromosome 6VS·6AL.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 31771779, 31671685, 31971933), the Important National Science & Technology Specific Projects of Transgenic Research (Grant No. 2018ZX0800905B), the Fundamental Research Funds for the National Central Universities (Grant No. KYYJ202006) and supported by the Bioinformatics Center of Nanjing Agricultural University. We thank Zdeňka Dubská, Romana Šperková and Jitka Weiserová for the assistance with chromosome sorting and Dr. Hana Šimková for preparation of HMW DNA. Jaroslav Doležel, Miroslava Karafiátová, Jan Vrána and Jan Bartoš were supported from ERDF project ‘Plants as a tool for sustainable global development’ (No. CZ.02.1.01/0.0/0.0/16_019/0000827).
Xing, L. , Yuan, L. , Lv, Z. , Wang, Q. , Yin, C. , Huang, Z. , Liu, J. , Cao, S. , Zhang, R. , Chen, P. , Karafiátová, M. , Vrána, J. , Bartoš, J. , Doležel, J. and Cao, A. (2021) Long‐range assembly of sequences helps to unravel the genome structure and small variation of the wheat–Haynaldia villosa translocated chromosome 6VS.6AL. Plant Biotechnol. J., 10.1111/pbi.13570
Data Availability
The submission of ‘wheat–Haynaldia villosa translocation chromosome T6VS·6AL’ is available in the NCBI with No. PRJNA700793. The transcriptome of H. villosa is available in the NCBI with No. SRP132108.
References
- Arraiano, L.S. , Chartrain, L. , Bossolini, E. , Slatter, H.N. , Keller, B. and Brown, J.K.M. (2007) A gene in European wheat cultivars for resistance to an African isolate of Mycosphaerella graminicola . Plant Pathol. 56, 73–78. [Google Scholar]
- Avni, R. , Nave, M. , Barad, O. , Baruch, K. , Twardziok, S.O. , Gundlach, H. , Hale, I. et al. (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science, 357, 93–97. [DOI] [PubMed] [Google Scholar]
- Blanco, A. , Simeone, R. , Tanzarella, O.A. and Greco, B. (1983) Morphology and chromosome pairing of a hybrid between Triticum durum Desf. and Haynaldia villosa (L.) Schur. Theor Appl Genet. 64, 333–337. [DOI] [PubMed] [Google Scholar]
- Brenchley, R. , Spannagl, M. , Pfeifer, M. , Barker, G.L.A. , D’Amore, R. , Allen, A.M. , McKenzie, N. et al. (2012) Analysis of the bread wheat genome using whole‐genome shotgun sequencing. Nature, 491, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, A. , Xing, L. , Wang, X. , Yang, X. , Wang, W. , Sun, Y. , Qian, C. et al. (2011) Serine/threonine kinase gene Stpk‐V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc. Natl Acad Sci. USA, 19, 7727–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro, A.M. , Vasicek, A. , Manifiesto, M. , Giménez, D.O. , Tacaliti, M.S. , Dobrovolskaya, O. , Röder, M.S. et al. (2008) Mapping antixenosis genes on chromosome 6A of wheat to greenbug and to a new biotype of Russian wheat aphid. Plant Breeding, 124, 229–233. [Google Scholar]
- Chen, P. and Liu, D. (1982) Cytogenetic studies of hybrid progenies between Triticum aestivum and Haynaldia villosa. Journal of Nanjing Agricultural College 4, 1–16. [Google Scholar]
- Chen, P.D. , Qi, L.L. , Zhou, B. , Zhang, S.Z. and Liu, D.J. (1995) Development and molecular cytogenetic analysis of wheat‐Haynaldia villosa 6VS/6AL translocation lines specifying resistance to powdery mildew. Theor. Appl. Genet. 91, 1125–1128. [DOI] [PubMed] [Google Scholar]
- Chen, P. , You, C. , Hu, Y. , Chen, S. , Zhou, B. , Cao, A. and Wang, X. (2013) Radiation‐induced translocations with reduced Haynaldia villosa chromatin at the Pm21 locus for powdery mildew resistance in wheat. Mol. Breeding, 31, 477–484. [Google Scholar]
- Clavijo, B.J. , Venturini, L. , Schudoma, C. , Accinelli, G.G. , Kaithakottil, G. , Wright, J. , Borrill, P. et al. (2017) An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 27, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pace, C. , Vaccino, P. , Cionini, G. , Pasquini, M. , Bizzarri, M. and Qualset, C.O. (2011) Dasypyrum. In Wild Crop Relatives: Genomic and Breeding Resources, Cereals, Vol 1, chapter 4 ( Kole, C. , ed), pp. 185–292.Heidelberg: Springer. [Google Scholar]
- Du, P. , Zhuang, L. , Wang, Y. , Yuan, L. , Wang, Q. , Wang, D. , Dawadondup et al. (2017) Development of oligonucleotides and multiplex probes for quick and accurate identification of wheat and Thinopyrum bessarabicum chromosomes. Genome, 60, 93–103. [DOI] [PubMed] [Google Scholar]
- Guo, J. , Shi, W. , Zhang, Z. , Cheng, J. , Sun, D. , Yu, J. , Li, X. et al. (2018) Association of yield‐related traits in founder genotypes and derivatives of common wheat (Triticum aestivum L.). BMC Plant Biol. 18, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z. , Chen, D. , Alqudah, A.M. , Roder, M.S. , Ganal, M.W. and Schnurbusch, T. (2017) Genome‐wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat. New Phytol. 214, 257–270. [DOI] [PubMed] [Google Scholar]
- Gupta, P.K. , Rustgi, S. and Kumar, N. (2006) Genetic and molecular basis of grain size and grain number and its relevance to grain productivity in higher plants. Genome, 49, 565–571. [DOI] [PubMed] [Google Scholar]
- Holzapfel, J. , Voss, H.H. , Miedaner, T. , Korzun, V. , Haberle, J. , Schweizer, G. , Mohler, V. et al. (2008) Inheritance of resistance to Fusarium head blight in three European winter wheat populations. Theor. Appl Genet. 117, 1119–1128. [DOI] [PubMed] [Google Scholar]
- IWGSC . (2014) A chromosome‐based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science, 345, 1251788. [DOI] [PubMed] [Google Scholar]
- IWGSC . (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 361, 7191. [DOI] [PubMed] [Google Scholar]
- Jia, J. , Zhao, S. , Kong, X. , Li, Y. , Zhao, G. , He, W. , Appels, R. et al. (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature, 496, 91–95. [DOI] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , Jones, S.J. et al. (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Chen, X. , Xin, Z.Y. , Ma, Y.Z. , Xu, H.J. , Chen, X.Y. and Jia, X. (2005) Development and identification of wheat–Haynaldia villosa T6DL.6VS chromosome translocation lines conferring resistance to powdery mildew. Plant Breed. 124, 203–205. [Google Scholar]
- Ling, H.Q. , Zhao, S. , Liu, D. , Wang, J. , Sun, H. and Al, E. (2013) Draft genome of the wheat A‐genome progenitor Triticum urartu . Nature, 496, 87–90. [DOI] [PubMed] [Google Scholar]
- Liu, C. , Qi, L. , Liu, W. , Zhao, W. , Wilson, J. , Friebe, B. and Gill, B.S. (2011) Development of a set of compensating Triticum aestivum–Dasypyrum villosum Robertsonian translocation lines. Genome, 54, 836–844. [DOI] [PubMed] [Google Scholar]
- Lopes, M.S. , Reynolds, M.P. , McIntyre, C.L. , Mathews, K.L. , Jalal Kamali, M.R. , Mossad, M. , Feltaous, Y. et al. (2013) QTL for yield and associated traits in the Seri/Babax population grown across several environments in Mexico, in the West Asia, North Africa, and South Asia regions. Theor. Appl. Genet. 126, 971–984. [DOI] [PubMed] [Google Scholar]
- Lukaszewski, A.J. (1988) A comparison of several approaches in the development of disomic alien addition lines of wheat. In: Proceedings of the 7th International Wheat Genetics Symposium, vol 1 ( Miller, T.E. and Koebner, R.M.D. , eds) , pp. 363–367. Cambridge, UK: Institute of Plant Sciences Research. [Google Scholar]
- Luo, M.C. , Gu, Y.Q. , You, F.M. , Deal, K.R. , Ma, Y. , Hu, Y. , Huo, N. et al. (2013) A 4‐gigabase physical map unlocks the structure and evolution of the complex genome of Aegilops tauschii, the wheat D‐genome progenitor. Proc. Natl Acad. Sci. USA, 110, 7940–7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M.C. , Gu, Y.Q. , Puiu, D. , Wang, H. , Twardziok, S.O. , Deal, K.R. , Huo, N. et al. (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii . Nature, 551, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mago, R. , Bariana, H.S. , Dundas, I.S. , Spielmeyer, W. , Lawrence, G.J. , Pryor, A.J. and Ellis, J.G. (2005) Development of PCR markers for the selection of wheat stem rust resistance genes Sr24 and Sr26 in diverse wheat germplasm. Theor. Appl. Genet. 111, 496–504. [DOI] [PubMed] [Google Scholar]
- Monte, J.V. , McIntyre, C.L. and Gustafson, J.P. (1993) Analysis of phylogenetic relationships in the Triticeae tribe using RFLPs. Theor. Appl. Genet. 86, 649–655. [DOI] [PubMed] [Google Scholar]
- Paux, E. , Roger, D. , Badaeva, E. , Gay, G. , Bernard, M. , Sourdille, P. and Feuillet, C. (2006) Characterizing the composition and evolution of homoeologous genomes in hexaploid wheat through BAC‐end sequencing on chromosome 3B. Plant J. 48, 463–474. [DOI] [PubMed] [Google Scholar]
- Rohringer, R. , Kim, W.K. and Samborski, D.J. (1979) A histological study of interactions between avirulent races of stem rust and wheat containing resistance genes Sr5, Sr6, Sr8, or Sr22 . Canadian J. Bot. 57, 324–331. [Google Scholar]
- Sears, E.R. (1953) Addition of the genome of Haynaldia villosa to Triticum aestivum . Am. J. Bot. 40, 168–174. [Google Scholar]
- Simmonds, J. , Scott, P. , Leverington‐Waite, M. , Turner, A.S. , Brinton, J. , Korzun, V. , Snape, J. et al. (2014) Identification and independent validation of a stable yield and thousand grain weight QTL on chromosome 6A of hexaploid wheat (Triticum aestivum L.). BMC Plant Biol. 14, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, K. , Abate, Z. , Chao, S. , Zhang, W. , Rouse, M. , Jin, Y. , Elias, E. et al. (2011) Genetic mapping of stem rust resistance gene Sr13 in tetraploid wheat (Triticum turgidum ssp. durum L.). Theor. Appl. Genet. 122, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmeyer, W. , Hyles, J. , Joaquim, P. , Azanza, F. , Bonnett, D. , Ellis, M.E. , Moore, C. et al. (2007) A QTL on chromosome 6A in bread wheat (Triticum aestivum) is associated with longer coleoptiles, greater seedling vigour and final plant height. Theor. Appl. Genet. 115, 59–66. [DOI] [PubMed] [Google Scholar]
- Su, Z. , Hao, C. , Wang, L. , Dong, Y. and Zhang, X. (2011) Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 122, 211–223. [DOI] [PubMed] [Google Scholar]
- Sun, H. , Song, J. , Lei, J. , Song, X. , Dai, K. , Xiao, J. , Yuan, C. et al. (2018) Construction and application of oligo‐based FISH karyotype of Haynaldia villosa . J. Genet Genomics, 45, 463–466. [DOI] [PubMed] [Google Scholar]
- Sun, X.Y. , Wu, K. , Zhao, Y. , Kong, F.M. , Han, G.Z. , Jiang, H.M. , Huang, X.J. et al. (2009) QTL analysis of kernel shape and weight using recombinant inbred lines in wheat. Euphytica, 165, 615–624. [Google Scholar]
- Tahmasebi, S. , Heidari, B. , Pakniyat, H. and McIntyre, C.L. (2017) Mapping QTLs associated with agronomic and physiological traits under terminal drought and heat stress conditions in wheat (Triticum aestivum L.). Genome, 60, 26–45. [DOI] [PubMed] [Google Scholar]
- Tanaka, Y. , Tsuda, M. , Yasumoto, K. , Terachi, T. and Yamagishi, H. (2014) The complete mitochondrial genome sequence of Brassica oleracea and analysis of coexisting mitotypes. Curr Genet. 60, 277–284. [DOI] [PubMed] [Google Scholar]
- Thind, A.K. , Wicker, T. , Simkova, H. , Fossati, D. , Moullet, O. , Brabant, C. , Vrana, J. et al. (2017) Rapid cloning of genes in hexaploid wheat using cultivar‐specific long‐range chromosome assembly. Nat Biotechnol. 35, 793–796. [DOI] [PubMed] [Google Scholar]
- Thind, A.K. , Wicker, T. , Muller, T. , Ackermann, P.M. , Steuernagel, B. , Wulff, B.B.H. , Spannagl, M. et al. (2018) Chromosome‐scale comparative sequence analysis unravels molecular mechanisms of genome dynamics between two wheat cultivars. Genome Biol. 19, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari, V.K. , Wang, S. , Danilova, T. , Koo, D.H. and Gill, B.S. (2016) Exploring the tertiary gene pool of bread wheat: sequence assembly and analysis of chromosome 5Mg of Aegilops geniculata . Plant J. 84, 733–746. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Dai, K. , Xiao, J. , Yuan, C. , Zhao, R. , Dolezel, J. , Wu, Y. et al. (2017) Development of intron targeting (IT) markers specific for chromosome arm 4VS of Haynaldia villosa by chromosome sorting and next‐generation sequencing. BMC Genom. 18, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Hou, J. , Liu, H. , Li, T. , Wang, K. , Hao, C. , Liu, H. et al. (2019) TaBT1, affecting starch synthesis and thousand kernel weight, underwent strong selection during wheat improvement. J. Exp. Bot. 70, 1497–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. , Chang, X. and Jing, R. (2012) Genetic insight into yield‐associated traits of wheat grown in multiple rain‐fed environments. PLoS One, 7, e31249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, J. , Wan, W. , Li, M. , Yu, Z. , Zhang, X. , Liu, J. , Holušová, K. et al. (2020) Targeted sequencing of the short arm of chromosome 6V of a wheat relative Haynaldia villosa for marker development and gene mining. (Preprint). 10.21203/rs.2.22109/v1 [DOI]
- Xing, L. , Di, Z. , Yang, W. , Liu, J. , Li, M. , Wang, X. , Cui, C. et al. (2017) Overexpression of ERF1‐V from Haynaldia villosa can enhance the resistance of wheat to powdery mildew and increase the tolerance to salt and drought stresses. Front. Plant Sci. 8, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, L. , Hu, P. , Liu, J. , Witek, K. , Zhou, S. , Xu, J. , Zhou, W. et al. (2018) Pm21 from Haynaldia villosa encodes a CC‐NBS‐LRR protein conferring powdery mildew resistance in wheat. Mol Plant, 11, 874–878. [DOI] [PubMed] [Google Scholar]
- Zhang, P. , He, Z. , Tian, X. , Gao, F. , Xu, D. , Liu, J. , Wen, W. et al. (2017a) Cloning of TaTPP‐6AL1 associated with grain weight in bread wheat and development of functional marker. Mol. Breed. 37, 78. [Google Scholar]
- Zhang, P. , Li, W. , Fellers, J. , Friebe, B. and Gill, B.S. (2004) BAC‐FISH in wheat identifies chromosome landmarks consisting of different types of transposable elements. Chromosoma, 112, 288–299. [DOI] [PubMed] [Google Scholar]
- Zhang, R. , Fan, Y. , Kong, L. , Wang, Z. , Wu, J. , Xing, L. , Cao, A. et al. (2018) Pm62, an adult plant powdery mildew resistance gene introgressed from Dasypyrum villosum chromosome arm 2VL into wheat. Theor. Appl. Genet. 131, 2613–2620. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Zhang, R. , Feng, Y. , Bie, T. and Chen, P. (2013) Distribution of highly repeated DNA sequences in Haynaldia villosa and its application in the identification of alien chromatin. Chinese Sci. Bull. 8, 890–897. [Google Scholar]
- Zhang, X. , Ma, L. and Zheng, J. (2017b) Characteristics of genes selected by domestication and iIntensive breeding in crop plants. Acta Agronomica Sinica, 2, 157–170. [Google Scholar]
- Zhang, X. , Wei, X. , Xiao, J. , Yuan, C. , Wu, Y. , Cao, A. , Xing, L. et al. (2017c) Whole genome development of intron targeting (IT) markers specific for Dasypyrum villosum chromosomes based on next‐generation sequencing technology. Mol. Breed. 37. [Google Scholar]
- Zhao, G. , Zou, C. , Li, K. , Wang, K. , Li, T. , Gao, L. , Zhang, X. et al. (2017) The Aegilops tauschii genome reveals multiple impacts of transposons. Nat. Plants, 3, 946–955. [DOI] [PubMed] [Google Scholar]
- Zimin, A.V. , Puiu, D. , Hall, R. , Kingan, S. , Clavijo, B.J. and Salzberg, S.L. (2017) The first near‐complete assembly of the hexaploid bread wheat genome, Triticum aestivum . Gigascience 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Size distribution of scaffolds from 6VS·6AL (a) and from 6VS (b).
Figure S2 Distribution of repeat sequences in the 6VS·6AL chromosome. Note: (a) distribution of genes; (b) density of Copia; and (c) density of Gypsy.
Figure S3 Statistics of GO functional classification.
Figure S4 Gene enrichment analysis by KEGG.
Figure S5 Dot plot of genome comparison between 6AL‐T and 6AL‐CS.
Figure S6 Amplification of InDel markers in T6VS·6AL, AABBVV, VV and AABBDD.
Figure S7 GISH analysis of the 14 wheat–H. villosa introgression lines involved in 6VS. Nine homozygous lines included six terminal translocation lines 19YL1‐1, 19YL3‐1, 19YL5‐2, 19YL15‐1, 19YL18‐5, and 19YL19‐3, and three intercalary translocation lines 19YL8‐1, 19YL10‐2, and 19YL11‐3. Five heterozygous lines included three terminal translocation lines 19YL2‐2, 19YL21‐3, and 19YL21‐1; one intercalary translocation line 19YL22‐2; and one terminal deletion line 19YL16‐1.
Table S1 Primer information used for 6VS fragments detection.
Table S2 Classification and statistics of repeated sequences.
Table S3 Gene prediction by de novo, homologue and RNA‐seq analysis.
Table S4 Noncoding RNAs.
Table S5 Gene function annotation.
Table S6 Statistics of Indel markers designed on the scaffold.
Table S7 Marker analysis of the chromosome structural variations of wheat–Haynaldia villosa translocated chromosome 6VS·6AL.
Data Availability Statement
The submission of ‘wheat–Haynaldia villosa translocation chromosome T6VS·6AL’ is available in the NCBI with No. PRJNA700793. The transcriptome of H. villosa is available in the NCBI with No. SRP132108.